

INTRODUCTION
Neuromyelitis optica spectrum disorder (NMOSD) is a rare immune-mediated disease of the central nervous system (CNS), associated, in the majority of cases, with autoantibodies directed against the astrocytic water channel aquaporin-4 (AQP4).(1) AQP4-IgG+ NMOSD presents predominantly with episodes of optic neuritis (ON) and longitudinally extensive transverse myelitis. Unlike other neuro-inflammatory diseases, such as multiple sclerosis (MS), where a progressive disease course has been well established, AQP4-IgG+ NMOSD is considered a primarily relapsing disorder and disease progression independent of attack-associated incidents is considered exceedingly rare.(1, 2) A limited number of studies have investigated the presence of ongoing disease activity independent of relapses in NMOSD and data are conflicting.
Cross-sectional studies utilizing retinal optical coherence tomography (OCT) have suggested that retinal ganglion cell damage may occur in NMOSD eyes in the absence of a history of ON, as evidenced by lower thickness of the inner retinal layers when compared to healthy controls (HC).(3–5) To date, few longitudinal studies have explored whether there is subclinical visual pathway involvement outside of ON incidents in NMOSD. While some studies have suggested that progressive visual evoked potentials (VEP) latency delay and progressive GCIPL atrophy may occur in NMOSD even in the absence of ON attacks, existing evidence is conflicting.(6–10) Utilizing OCT, the objective of our study was to examine whether AQP4-IgG+ NMOSD patients exhibit progressive retinal neuro-axonal loss, independently of ON attacks.
METHODS
Standard protocol approvals, registrations, and patient consents
The study was approved by the Johns Hopkins University Institutional Review Board. All participants provided written informed consent.
Study design and participants
Patients were recruited from the Johns Hopkins Neuromyelitis Optica Clinic between 2008 and 2020, and fulfilled the 2015 International Panel for Neuromyelitis Optica Diagnosis criteria for AQP4-IgG+ NMOSD (retroactively applied to patients recruited prior to 2015).(1) AQP4-IgG+ NMOSD patients underwent serial retinal imaging with OCT and visual function assessments at their clinical visits. HC were recruited by convenience sampling, and were invited for annual OCT scans.
Patients with a clinical history of ON less than six months prior to baseline were excluded, because acute ON events are expected to lead to significant inner retinal atrophy. Existing literature supports that the vast majority of GCIPL and pRNFL thinning, as well as maximal visual recovery, has already occurred by six months.(11–13) Data of subjects who developed ON during follow-up were censored at the last available scan prior to ON. Since chiasmal/optic tract involvement is common in ON in AQP4-IgG+ NMOSD, both eyes were excluded or censored in the analyses, even in cases of apparent unilateral ON during follow-up.(1) Only eyes with at least six months of OCT follow-up were included.
Subjects with history of diabetes mellitus, uncontrolled hypertension, or other significant ophthalmological/neurological disorders were excluded from the study.
OCT
Retinal imaging was performed with spectral-domain OCT (Cirrus HD-OCT, Model 5000; Carl Zeiss Meditec, Dublin, CA), as previously described.(14) Briefly, peripapillary retinal nerve fiber layer (pRNFL) thicknesses were derived by the conventional Cirrus HD-OCT software. Macular ganglion cell+inner plexiform layer (GCIPL) was automatically segmented and thickness was calculated within an annulus centered at the fovea, with an internal diameter of 1mm and an external diameter of 5mm. The scans underwent rigorous quality control in accordance with the OSCAR-IB criteria; scans with signal strength below 7/10, with artifact, or with segmentation errors were excluded from the study.(15)
Visual Function
Monocular visual acuity (VA) was assessed using standardized retro-illuminated eye charts (Precision Vision, La Salle, IL) with habitual correction. High-contrast (100%) letter acuity (HCLA) was assessed with Early Treatment Diabetic Retinopathy Study (ETDRS) charts (at 4m), and low-contrast (2.5% and 1.25%) letter acuity (LCLA) with Sloan Letter charts (at 2m). The maximum score for each chart is 70 letters, corresponding to a Snellen visual acuity of 20/10.
Clinically significant VA worsening was defined as a decrease in monocular letter acuity ≥5 letters for HCLA, and ≥7 letters for LCLA, in accordance with prior studies in MS.(16, 17)
Statistical Methods
Statistical analyses were performed using Stata version 16 (StataCorp) and R version 4.0.2. Baseline comparisons were performed with mixed-effects linear regression models with subject-specific random intercepts. Rates of change of retinal layer thicknesses and letter acuity scores were analyzed using mixed-effects linear regression models, with subject-specific and eye-specific random intercepts and random slopes in time, using time from baseline visit as a continuous variable. All analyses were adjusted for age, sex, and race. Statistical significance was defined as p<0.05.
RESULTS
Cohort
32 AQP4-IgG+ NMOSD patients (61 eyes) met inclusion criteria (Figure E1) and we also included 48 HC (95 eyes), approximately matched to the NMOSD group for age, sex, and race. Demographics and clinical characteristics are summarized in Table 1. Median OCT follow-up was 4.3 years in NMOSD and 4.0 years in HC.
Table 1.
Demographic and clinical characteristics at baseline
| HC | AQP4-IgG+ NMOSD | P-values | |
|---|---|---|---|
| Participants, n | 48 | 32 | |
| Age in years, mean (SD) | 40.5 (11.3) | 45.5 (12.3) | 0.063 |
| Female, n (%) | 38 (79%) | 27 (84%) | 0.77 |
| Race, n (%) | 0.50 | ||
| Caucasian-American | 29 (60%) | 15 (47%) | |
| African-American | 18 (38%) | 16 (50%) | |
| Asian-American | 1 (2%) | 1 (3%) | |
| Relapse history, n (%) | - | - | |
| ON & TM | 13 (41%) | ||
| ON & TM & Brainstem attack | 6 (19%) | ||
| Isolated TM | 8 (25%) | ||
| Isolated ON | 2 (6%) | ||
| Brainstem attack & TM | 3 (9%) | ||
| Treatment during follow-up, patient-years (%) | - | - | |
| No treatment | 11.3 (8.7%) | ||
| Rituximab | 86.3 (66.7%) | ||
| Mycophenolate Mofetil | 15.9 (12.3%) | ||
| Azathioprine | 4.7 (3.6%) | ||
| Eculizumab | 3.8 (2.9%) | ||
| Eculizumab + Mycophenolate Mofetil | 6.0 (4.6%) | ||
| Inebilizumab | 1.4 (1.1%) | ||
| Eyes with ON history, n (%) | - | 34 (55%)a | - |
| Disease duration; median (IQR) | - | 6 (1–9) | - |
| Follow-up, median (IQR) | 4.0 (1.8 – 7.5) | 4.3 (2.6 – 7.5) | 0.89 |
8 NMOSD-ON eyes had microcystic macular pathology (MMP)
HC: healthy controls; AQP4-IgG+ NMOSD: aquaporin-4-IgG seropositive Neuromyelitis Optica Spectrum Disorder; SD: standard deviation; TM: transverse myelitis; ON: optic neuritis; IQR: interquartile range
For a subset of NMOSD patients (n=30) and HC (n=42), VA assessments were available cross-sectionally. Additionally, 20 NMOSD patients (63%) had available serial VA assessments (median follow-up 3.0 years, IQR: 1.0 – 7.6).
Baseline OCT analysis
At baseline, GCIPL and pRNFL thicknesses were markedly lower in NMOSD eyes with a prior ON history (NMOSD-ON) compared to HC (adjusted mean differences — GCIPL:−20.29μm; pRNFL: −27.80μm; p<0.001 for both; Table 2).
Table 2.
Cross-sectional OCT measures and visual acuity scores in AQP4-IgG+ NMOSD and HC eyes and comparisons between groups
| HC vs. NMOSD-nonON | HC vs. NMOSD-ON | ||||||
|---|---|---|---|---|---|---|---|
| HC (n=95 eyes) | NMOSD-nonON (n=30 eyes) | NMOSD-ON (n=31 eyes) | Difference (95% CI)a | P-value | Difference (95% CI)a | P-value | |
| OCT measures, μm, mean (SD) | |||||||
| GCIPL | 76.9 (5.2) | 71.6 (4.8) | 56.7 (8.2) | −5.38 (−8.16 to −2.60) | <0.001 | −20.29 (−23.37 to −17.20) | <0.001 |
| pRNFL | 93.9 (9.2) | 91.2 (8.2) | 67.3 (13.1) | −4.06 (−8.78 to 0.66) | 0.09 | −27.80 (−32.62 to −22.98) | <0.001 |
| Letter acuity scores b , median (IQR) | |||||||
| 100% contrast (HCLA) | 61.5 (59–65) | 59 (51.5–63) | 44.5 (7–54) | −2.42 (−5.24 to 0.40) | 0.10 | −23.00 (−28.92 to −17.07) | <0.001 |
| 2.5% contrast (LCLA) | 34 (29–38) | 24 (16–33) | 0 (0–16.5) | −8.15 (−12.15 to −4.15) | <0.001 | −23.95 (−27.93 to −19.96) | <0.001 |
| 1.25% contrast (LCLA) | 20 (14–25) | 6.5 (5–18) | 0 (0–1.5) | −8.66 (−13.24 to −4.08) | <0.001 | −16.97 (−20.90 to −13.05) | <0.001 |
Derived from mixed effects linear regression models, including age, sex and race
Available for 42 HC and 30 NMOSD patients
OCT: optical coherence tomography; AQP4: aquaporin-4; NMOSD: Neuromyelitis Optica Spectrum Disorder; HC: healthy controls; ON: optic neuritis; CI: confidence interval; SD: standard deviation; GCIPL: ganglion cell + inner plexiform layer; pRNFL: peri-papillary retinal nerve fiber layer; IQR: interquartile range; HCLA: high-contrast letter acuity; LCLA: low-contrast letter acuity
NMOSD eyes without a prior ON history (NMOSD-nonON) also exhibited significantly lower GCIPL thickness, albeit less pronounced relative to NMOSD-ON eyes, as compared to HC (adjusted mean difference: −5.38μm, p<0.001). pRNFL thickness was also lower in NMOSD-nonON eyes compared to HC, but this difference did not attain statistical significance (adjusted mean difference: −4.06μm, p=0.09; Table 2).
Longitudinal OCT analysis
Mean annualized rates of change in GCIPL and pRNFL thicknesses in NMOSD and HC participants are reported in Table 3 and individual eye trajectories are shown in Figure 1. NMOSD patients exhibited faster rates of pRNFL thinning compared to HC (−0.45μm/year vs. −0.21μm/year, β=−0.25μm/year, 95%CI:−0.45 to −0.05, p=0.014). Similarly, GCIPL thinning was also accelerated in NMOSD patients (−0.29μm/year vs. −0.20μm/year, β=−0.09μm/year, 95%CI:−0.17 to 0.00, p=0.05). These differences correspond to 119% faster pRNFL and 45% faster GCIPL thinning in NMOSD compared to HC eyes.
Table 3.
Annualized change in retinal layer thickness in AQP4-IgG+ NMOSD and HC eyes and comparisons between groups
| Annualized Rate of Change, μm/year | HC vs. NMOSD | HC vs. NMOSD-nonON | HC vs. NMOSD-ON | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HC (n=95 eyes) | NMOSD (n=61 eyes) | NMOSD-nonON (n=30 eyes) | NMOSD-ON (n=31 eyes) | Difference in Annualized Rate of Change (95% CI)a, μm/year | P-value | Difference in Annualized Rate of Change (95% CI)a, μm/year | P-value | Difference in Annualized Rate of Change (95% CI)a, μm/year | P-value | |
| GCIPL | −0.20 (−0.25 to −0.15) | −0.29 (−0.36 to −0.22) | −0.35 (−0.44 to −0.26) | −0.21 (−0.30 to −0.12) | −0.09 (−0.17 to 0.00) | 0.05 | −0.15 (−0.25 to −0.05) | 0.005 | −0.01 (−0.11 to 0.10) | 0.90 |
| pRNFL | −0.21 (−0.33 to −0.08) | −0.45 (−0.61 to −0.30) | −0.64 (−0.86 to −0.43) | −0.28 (−0.48 to −0.07) | −0.25 (−0.45 to −0.05) | 0.014 | −0.43 (−0.67 to −0.19) | <0.001 | −0.07 (−0.31 to 0.16) | 0.53 |
Derived from mixed effects linear regression models, including age, sex and race
AQP4: aquaporin-4; NMOSD: Neuromyelitis Optica Spectrum Disorder; HC: healthy controls; ON: optic neuritis; CI: confidence interval; GCIPL: ganglion cell + inner plexiform layer; pRNFL: peri-papillary retinal nerve fiber layer
Figure 1.
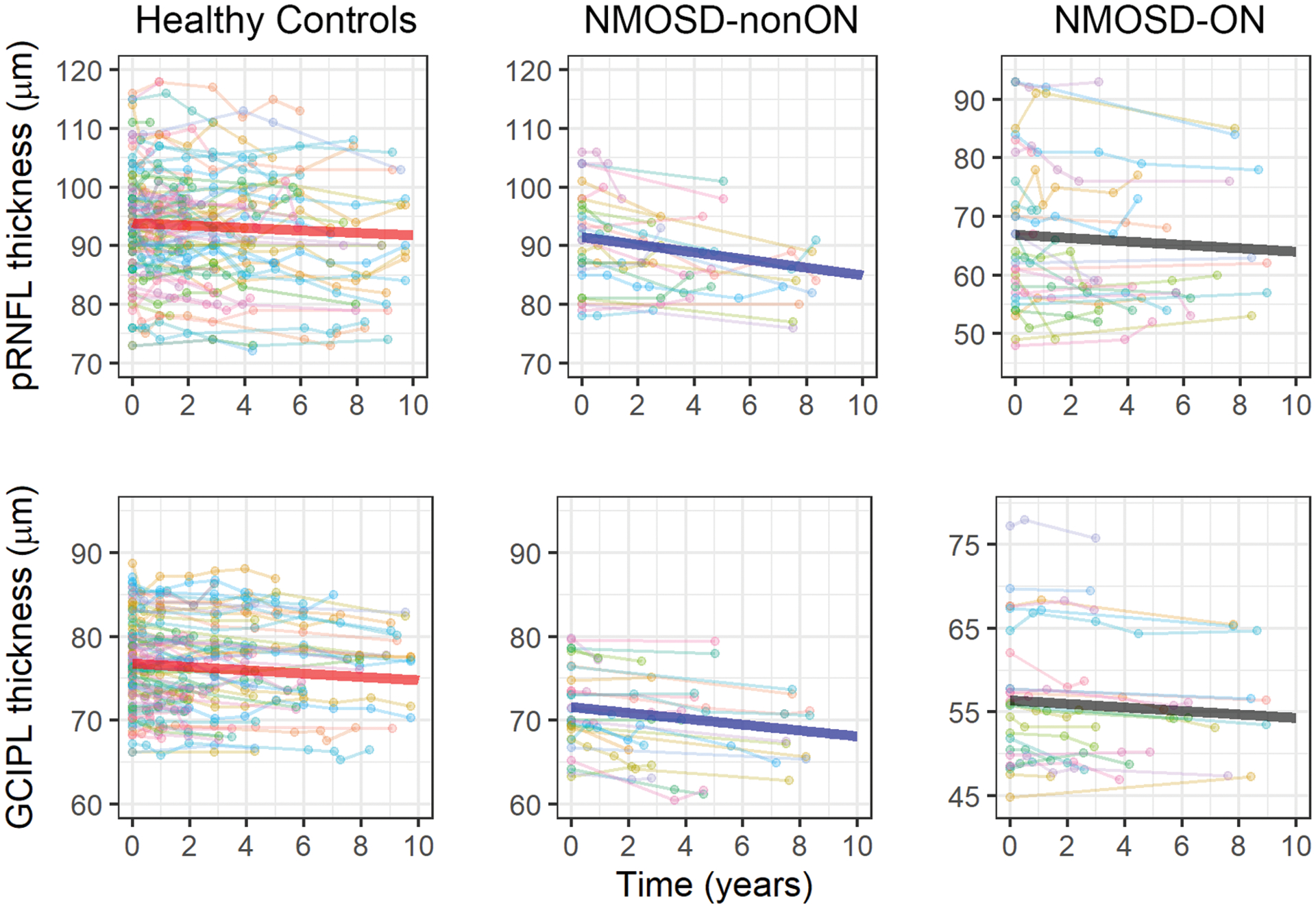
Longitudinal changes in pRNFL and GCIPL thickness
Longitudinal trajectories of GCIPL and pRNFL thickness for individual eyes are shown, separately for healthy controls, AQP4-IgG+ NMOSD eyes without a history of ON (NMOSD-nonON) and with a history of ON (NMOSD-ON). The thicker lines represent the average trajectory, as fitted by mixed-effects models. The y-axis range differs for the NMOSD-ON group (given lower GCIPL and pRNFL thicknesses), but the slopes are comparable across panels since the scales of the y-axes identical.
NMOSD: neuromyelitis optica spectrum disorder; ON: optic neuritis; GCIPL: ganglion cell + inner plexiform layer; pRNFL: peri-papillary retinal nerve fiber layer
This was driven by faster pRNFL and GCIPL thinning in NMOSD-nonON eyes compared to HC (pRNFL: −0.64μm/year vs. −0.21μm/year, β=−0.43μm/year, 95%CI:−0.67 to −0.19, p<0.001; GCIPL: −0.35μm/year vs. −0.20μm/year, β=−0.15μm/year, 95%CI:−0.25 to −0.05, p=0.005). In contrast, NMOSD-ON eyes did not demonstrate significant differences in rates of pRNFL or GCIPL thinning in comparison to HC (pRNFL: −0.28μm/year vs. −0.21μm/year, β=−0.07μm/year, 95%CI:−0.31 to 0.16, p=0.53; GCIPL: −0.21μm/year vs. −0.20μm/year, β=−0.01μm/year, 95%CI:−0.11 to 0.10, p=0.90). However, when restricting analyses to NMOSD-ON eyes with baseline pRNFL thickness ≥ 70μm (lower limit of pRNFL thickness range in the HC cohort; n=12 NMOSD-ON eyes), we found faster rates of pRNFL thinning as compared to HC (−0.57μm/year vs −0.21μm/year, β=−0.36μm/year, 95%CI:−0.57 to −0.21, p=0.034), consistent with a potential floor effect. The rate of GCIPL thinning was also faster in NMOSD-ON eyes with baseline pRNFL thickness ≥ 70μm compared to HC, although this finding was not statistically significant (−0.21μm/year vs −0.32μm/year, β=−0.11μm/year, 95%CI: −0.27 to 0.05, p=0.16).
In order to exclude effects due to chiasmal or optic tract involvement in patients with a prior history of contralateral ON, we compared rates of retinal atrophy between NMOSD patients who had never experienced ON in either eye (n=11 patients) and HC and the results were similar (pRNFL: −0.62μm/year vs. −0.21μm/year, β=−0.41μm/year, 95%CI:−0.68 to −0.15, p=0.002; GCIPL: −0.31μm/year vs. −0.20μm/year, β=−0.11μm/year, 95%CI:−0.22 to 0.00, p=0.05).
Finally, we explored the effects of non-ON relapses during follow-up on rates of pRNFL and GCIPL thinning. Ten patients had relapses during follow-up (9 transverse myelitis, 1 area postrema syndrome). Patients with relapses did not exhibit significant differences in rates of GCIPL (−0.27μm/year vs. −0.32μm/year, β=0.05μm/year, 95%CI:−0.10 to 0.20, p=0.51) or pRNFL thinning (−0.44μm/year vs. −0.51μm/year, β=0.08μm/year, 95%CI:−0.28 to 0.43; p=0.67), compared to those who were clinically stable.
Baseline VA analysis
At baseline, as expected, HCLA and LCLA were markedly worse in NMOSD-ON eyes compared to HC (adjusted mean differences — HCLA: −23.00 letters; 2.5% LCLA: −23.95 letters; 1.25% LCLA: −16.97 letters, p<0.001 for all; Table 2).
HCLA did not differ between NMOSD-nonON and HC eyes (adjusted mean difference: −2.42 letters, p=0.10), however NMOSD-nonON eyes exhibited worse 2.5% and 1.25% LCLA compared to HC (adjusted mean differences: 2.5% LCLA: −8.15 letters; 1.25% LCLA: −8.66 letters, p<0.001 for both; Table 2).
Longitudinal VA analysis
Of the 61 NMOSD eyes included in this study, 40 eyes had serial visual function assessments. Of these 40 eyes, 6 eyes (15%) exhibited clinically significant HCLA worsening by the end of the follow-up period, 4 eyes (10%) exhibited clinically significant 2.5% LCLA worsening and 5 eyes (13%) exhibited clinically significant 1.25% LCLA worsening. Nine eyes (23%) exhibited HCLA and/or LCLA worsening.
Rates of GCIPL and pRNFL thinning were compared between eyes with clinically significant VA worsening by the end of the follow-up period and eyes with stable VA. We did not find significant differences in these rates between eyes with VA worsening compared to those with stable VA (GCIPL: −0.28μm/year vs. −0.33μm/year, β=0.05μm/year, 95%CI:−0.11 to 0.22, p=0.51; pRNFL: −0.63μm/year vs. −0.51μm/year, β=−0.12μm/year, 95%CI:−0.55 to 0.32, p=0.6).
We also calculated the annualized rates of change of HCLA and LCLA eyes in NMOSD eyes, and did not observe a significant change in visual function over time (Table 4). There was no difference in the rate of change of HCLA and LCLA between NMOSD-ON and NMOSD-nonON eyes.
Table 4.
Annualized change in visual acuity in AQP4-IgG+ NMOSD eyes
| Annualized Rate of Change, letters/year | NMOSD-nonON vs. NMOSD-ON | ||||
|---|---|---|---|---|---|
| NMOSD (n=40 eyes) | NMOSD-nonON (n=20 eyes) | NMOSD-ON n=20 eyes) | Difference in Annualized Rate of Change 95% CI)a, letters/year | P-value | |
| 100% contrast (HCLA) | −0.04 (−0.38 to 0.29) | −0.10 (−0.54 to 0.33) | 0.03 (−0.51 to 0.57) | 0.13 (−0.56 to 0.83) | 0.71 |
| 2.5% contrast (LCLA) | 0.08 (−0.39 to 0.55) | −0.12 (−0.71 to 0.48) | 0.20 (−0.51 to 0.91) | 0.31 (−0.62 to 1.24) | 0.51 |
| 1.25% contrast (LCLA) | 0.02 (−0.31 to 0.34) | 0.12 (−0.30 to 0.54) | −0.08 (−0.59 to 0.42) | −0.20 (−0.86 to 0.45) | 0.55 |
Derived from mixed effects linear regression models, including age, sex and race
AQP4: aquaporin-4; NMOSD: Neuromyelitis Optica Spectrum Disorder; ON: optic neuritis; CI: confidence interval; HCLA: high-contrast letter acuity; LCLA: low-contrast letter acuity
DISCUSSION
In this longitudinal study, we observed accelerated pRNFL and GCIPL thinning in AQP4-IgG+ NMOSD eyes clinically unaffected by ON. This finding suggests that subclinical progressive retinal neuro-axonal loss occurs in AQP4-IgG+ NMOSD. This has important implications for our understanding of the disease mechanisms of NMOSD, since it provides evidence that there is ongoing insidious disease activity independent of clinical relapses. Importantly, our results remained unaltered in sensitivity analyses that were restricted to patients who had never experienced ON in either eye, suggesting that our findings are not the consequence of a symptomatic ON event but they likely reflect a separate pathophysiologic process.
Our findings are in agreement with prior work by Oertel et al. reporting faster GCIPL thinning in AQP4-IgG+ NMOSD eyes, independently of ON attacks.(6) Furthermore, in a multi-center longitudinal VEP study, an increase of P100 latencies and a decline of amplitudes was observed in AQP4-IgG+ NMOSD eyes without ON during follow-up.(7) Together, these findings support that the visual pathway may be a site of subclinical involvement in AQP4-IgG+ NMOSD. In our study, these phenomena appeared to be independent, not only of ON attacks, but also of other non-ON attacks, since pRNFL and GCIPL atrophy rates did not differ between patients who experienced relapses during follow-up and patients who were clinically stable. In a cohort of 27 NMOSD patients, Pisa et al. reported accelerated pRNFL thinning in patients with disease activity during follow-up, but not in stable patients.(18) In contrast, in the study by Oertel et al, patients who experienced attacks during follow-up had pRNFL thickening.(6) Further larger studies are likely needed to investigate the retinal phenomena that occur during inflammatory attacks and reconcile these conflicting results.
Interestingly, we observed progressive pRNFL and GCIPL atrophy in AQP4-IgG+ NMOSD eyes without a prior history of ON, but not in ON eyes. This is presumably due to a floor effect, since ON associated with AQP4-IgG+ NMOSD results in marked retinal ganglion cell and axonal loss which may limit detection of further decreases in pRNFL or GCIPL thicknesses.(19, 20) Notably, subgroup analyses restricted to NMOSD-ON eyes with baseline pRNFL thickness ≥ 70μm revealed faster rates of pRNFL thinning compared to HC, consistent with this hypothesis.
Even though the pathoetiology of the observed progressive retinal neuro-axonal loss could not be established in the current study, several hypotheses need to be considered. AQP4 is a water channel protein that is highly expressed in the retina by astrocytes and more abundantly by a unique type of glial cell, Müller cells. Müller cells regulate vital retinal homeostatic mechanisms, including release of neurotropic factors and degradation of glutamate. It is therefore conceivable that the observed retinal changes are a consequence of direct retinal damage due to anti-AQP4 antibody activity.(21) Interestingly, it has been previously reported that Müller cells are richly expressed in the foveal region, located in the center of the macula, and foveal thinning has been reported in AQP4-IgG+ eyes without a history of ON; it has been postulated that this may represent the occurrence of a primary retinal astrocytopathy.(21, 22) In a pathological retinal study, AQP4-IgG+ NMOSD was characterized by loss of AQP4 immunoreactivity on Müller cells; it is likely that alterations in the dynamics of astrocyte and Müller cell function may be responsible for subsequent neurodegeneration.(23) Alternatively, our findings may be the result of subclinical involvement of the anterior or posterior visual pathway. A study by Hokari et al. reported pathological evidence of optic nerve involvement in AQP4-IgG+ NMOSD eyes with no clinical ON attacks.(23) Moreover, gadolinium enhancement of the optic nerve has been reported at the time of attacks involving other anatomical locations, in the absence of visual symptoms.(24) Retrograde trans-synaptic degeneration due to involvement of the posterior visual pathway is another consideration. This is a less likely explanation however, since optic radiation (OR) lesions are not as common in NMOSD relative to MS.(25) Moreover, development of new asymptomatic brain lesions in clinically stable AQP4-IgG+ NMOSD patients appears to be rare.(26) Furthermore, we did not observe any cases of homonymous GCIPL thinning (which would be expected to be observed in unilateral posterior visual pathway involvement), although this may be difficult to assess in the presence of pre-existing optic neuropathy and/or bilateral posterior visual pathway involvement.
Our VA findings are also of interest, since we found that NMOSD-nonON eyes had worse performance in LCLA charts compared to HC. This suggests that some level of vision impairment may be present in AQP4-IgG+ NMOSD eyes, even without a clinical history of ON, and while the etiology of this finding is not clear, considerations include prior subclinical ON and/or primary retinal astrocytopathy, as outlined above. We also found that 23% of NMOSD eyes had worsening visual function during follow-up, based on cut-offs that have been previously proposed for MS eyes.(16) However, rates of retinal thinning did not differ between eyes with VA worsening compared to those with stable VA and, on a group level, we did not observe a significant change in visual function over time. Even though this finding may suggest that there is no direct correlation between OCT measures and functional outcomes, this negative finding should be interpreted with caution, given our relatively small sample size and the fact that VA assessments were available for only a subset of our cohort. Alternatively, this result may suggest that OCT is a more sensitive indicator of anterior visual pathway involvement and retinal neuro-axonal loss may precede visual deficits.
One limitation of our study is the relatively small sample size, which is expected given the rarity of the studied condition. Furthermore, longitudinal visual acuity data were not available for the majority of the healthy control participants. It is likely that our study was underpowered to detect potential associations between neuro-axonal loss and vision loss, due to insufficient follow-up time and/or sample size. Moreover, our study was retrospective and brain and optic nerve MRI scans were not routinely performed in all patients. Therefore, we could not assess for the presence of lesions along the visual pathway. Finally, the vast majority of participants were on treatment with rituximab during follow-up, and we were therefore unable to investigate any potential associations between different disease-modifying treatments and OCT measures. To conclude, our findings support that subclinical progressive retinal axonal loss may occur in AQP4-IgG+ NMOSD. Further studies are warranted to validate these findings and to elucidate their clinical relevance, since the presence of insidious pathology of the visual system has important implications for our understanding of the disease mechanisms of NMOSD and for monitoring treatment response.
Supplementary Material
Figure E1. Study flowchart
AQP4: aquaporin-4; NMOSD: Neuromyelitis Optica Spectrum Disorder; OCT: Optical Coherence Tomography; ON: optic neuritis
Funding
This study was funded by the Caring Friends NMO Research fund, National MS Society (FP-1607-24999 to ESS, RG-1606-08768 to SS; TA-1805-31136 to KCF), NIH/NINDS (R01NS082347 to PAC, K23NS117883 to ESS), NIH/NIMH (K01MH121582 to KCF).
Disclosures
Angeliki Filippatou, Eleni Vasileiou, Yufan He, Grigorios Kalaitzidis, Kathryn Fitzgerald, Jeffrey Lambe, and Yihao Liu report no disclosures.
Maureen Mealy is an employee of Viela Bio.
Michael Levy has received research support from: National Institutes of Health, Maryland Technology Development Corporation, Sanofi, Genzyme, Alexion, Alnylam, Shire, Acorda and Apopharma. He has also received personal compensation for consultation with Alexion, Acorda, and Genzyme and he serves on the scientific advisory boards for Alexion, Acorda and Quest Diagnostics.
Jerry Prince is a founder of Sonovex, Inc. and serves on its Board of Directors. He has received consulting fees from JuneBrain LLC and is PI on research grants to Johns Hopkins from Biogen.
Ellen Mowry has grants from Biogen and Genzyme, is site PI for studies sponsored by Biogen, has received free medication for a clinical trial from Teva and receives royalties for editorial duties from UpToDate.
Shiv Saidha has received consulting fees from Medical Logix for the development of CME programs in neurology and has served on scientific advisory boards for Biogen, Genzyme, Genentech Corporation, EMD Serono, and Celgene. He is the PI of investigator-initiated studies funded by Genentech Corporation and Biogen, and received support from the Race to Erase MS foundation. He has received equity compensation for consulting from JuneBrain LLC, a retinal imaging device developer. He is also the site investigator of a trial sponsored by MedDay Pharmaceuticals.
Peter Calabresi has received consulting fees from Disarm Therapeutics and Biogen and is PI on grants to JHU from Biogen and Annexon.
Elias Sotirchos has has served on scientific advisory boards for Viela Bio, Genentech and Alexion and has received speaker honoraria from Viela Bio and Biogen.
REFERENCES
- 1.Wingerchuk DM, Banwell B, Bennett JL, Cabre P, Carroll W, Chitnis T, de Seze J, Fujihara K, Greenberg B, Jacob A, Jarius S, Lana-Peixoto M, Levy M, Simon JH, Tenembaum S, Traboulsee AL, Waters P, Wellik KE, Weinshenker BG. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology. 2015; 85(2):177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wingerchuk DM, Pittock SJ, Lucchinetti CF, Lennon VA, Weinshenker BG. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology. 2007; 68(8):603–605 [DOI] [PubMed] [Google Scholar]
- 3.Tian D, Su L, Fan M, Yang J, Zhang R, Wen P, Han Y, Yu C, Zhang C, Ren H, Shi K, Zhu Z, Dong Y, Liu Y, Shi F. Bidirectional degeneration in the visual pathway in neuromyelitis optica spectrum disorder (NMOSD). Mult Scler. 2018; 24(12):1585–1593 [DOI] [PubMed] [Google Scholar]
- 4.Sotirchos ES, Saidha S, Byraiah G, Mealy MA, Ibrahim MA, Sepah YJ, Newsome SD, Ratchford JN, Frohman EM, Balcer LJ, Crainiceanu CM, Nguyen QD, Levy M, Calabresi PA. In vivo identification of morphologic retinal abnormalities in neuromyelitis optica. Neurology. 2013; 80(15):1406–1414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Filippatou AG, Vasileiou ES, He Y, Fitzgerald KC, Kalaitzidis G, Lambe J, Mealy MA, Levy M, Liu Y, Prince JL, Mowry EM, Saidha S, Calabresi PA, Sotirchos ES. Evidence of subclinical quantitative retinal layer abnormalities in AQP4-IgG seropositive NMOSD. Mult Scler. 2020:1352458520977771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Oertel FC, Havla J, Roca-Fernández A, Lizak N, Zimmermann H, Motamedi S, Borisow N, White OB, Bellmann-Strobl J, Albrecht P, Ruprecht K, Jarius S, Palace J, Leite MI, Kuempfel T, Paul F, Brandt AU. Retinal ganglion cell loss in neuromyelitis optica: a longitudinal study. J Neurol Neurosurg Psychiatry. 2018; 89(12):1259–1265 [DOI] [PubMed] [Google Scholar]
- 7.Ringelstein M, Harmel J, Zimmermann H, Brandt AU, Paul F, Haarmann A, Buttmann M, Hümmert MW, Trebst C, Schroeder C, Ayzenberg I, Kleiter I, Hellwig K, Havla J, Kümpfel T, Jarius S, Wildemann B, Rommer P, Weber MS, Pellkofer H, Röpke L, Geis C, Retzlaff N, Zettl U, Deppe M, Klotz L, Young K, Stellmann J, Kaste M, Kermer P, Marouf W, Lauda F, Tumani H, Graf J, Klistorner A, Hartung H, Aktas O, Albrecht P. Longitudinal optic neuritis-unrelated visual evoked potential changes in NMO spectrum disorders. Neurology. 2020; 94(4):e407–e418 [DOI] [PubMed] [Google Scholar]
- 8.Oertel FC, Zimmermann H, Paul F, Brandt AU. Optical coherence tomography in neuromyelitis optica spectrum disorders: potential advantages for individualized monitoring of progression and therapy. EPMA J. 2018; 9(1):21–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouyon M, Collongues N, Zéphir H, Ballonzoli L, Jeanjean L, Lebrun C, Chanson J, Blanc F, Fleury M, Outteryck O, Defoort S, Labauge P, Vermersch P, Speeg C, De Seze J. Longitudinal follow-up of vision in a neuromyelitis optica cohort. Mult Scler. 2013; 19(10):1320–1322 [DOI] [PubMed] [Google Scholar]
- 10.Manogaran P, Traboulsee AL, Lange AP. Longitudinal Study of Retinal Nerve Fiber Layer Thickness and Macular Volume in Patients With Neuromyelitis Optica Spectrum Disorder. J Neuroophthalmol. 2016; 36(4):363–368 [DOI] [PubMed] [Google Scholar]
- 11.Kupersmith MJ, Garvin MK, Wang J, Durbin M, Kardon R. Retinal Ganglion Cell Layer Thinning within One Month of Presentation for Optic Neuritis. Mult Scler. 2016; 22(5):641–648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Syc SB, Saidha S, Newsome SD, Ratchford JN, Levy M, Ford E, Crainiceanu CM, Durbin MK, Oakley JD, Meyer SA, Frohman EM, Calabresi PA. Optical coherence tomography segmentation reveals ganglion cell layer pathology after optic neuritis. Brain. 2012; 135(Pt 2):521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Andorrà M, Alba-Arbalat S, Camos-Carreras A, Gabilondo I, Fraga-Pumar E, Torres-Torres R, Pulido-Valdeolivas I, Tercero-Uribe AI, Guerrero-Zamora AM, Ortiz-Perez S, Zubizarreta I, Sola-Valls N, Llufriu S, Sepulveda M, Martinez-Hernandez E, Armangue T, Blanco Y, Villoslada P, Sanchez-Dalmau B, Saiz A, Martinez-Lapiscina EH. Using Acute Optic Neuritis Trials to Assess Neuroprotective and Remyelinating Therapies in Multiple Sclerosis. JAMA Neurol. 2020; 77(2):234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sotirchos ES, Filippatou A, Fitzgerald KC, Salama S, Pardo S, Wang J, Ogbuokiri E, Cowley NJ, Pellegrini N, Murphy OC, Mealy MA, Prince JL, Levy M, Calabresi PA, Saidha S. Aquaporin-4 IgG seropositivity is associated with worse visual outcomes after optic neuritis than MOG-IgG seropositivity and multiple sclerosis, independent of macular ganglion cell layer thinning. Mult Scler. 2019:1352458519864928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tewarie P, Balk L, Costello F, Green A, Martin R, Schippling S, Petzold A. The OSCAR-IB consensus criteria for retinal OCT quality assessment. PLoS ONE. 2012; 7(4):e34823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Talman LS, Bisker ER, Sackel DJ, Long DA, Galetta KM, Ratchford JN, Lile DJ, Farrell SK, Loguidice MJ, Remington G, Conger A, Frohman TC, Jacobs DA, Markowitz CE, Cutter GR, Ying G, Dai Y, Maguire MG, Galetta SL, Frohman EM, Calabresi PA, Balcer LJ. Longitudinal study of vision and retinal nerve fiber layer thickness in multiple sclerosis. Ann Neurol. 2010; 67(6):749–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balcer LJ, Baier ML, Pelak VS, Fox RJ, Shuwairi S, Galetta SL, Cutter GR, Maguire MG. New low-contrast vision charts: reliability and test characteristics in patients with multiple sclerosis. Mult Scler. 2000; 6(3):163–171 [DOI] [PubMed] [Google Scholar]
- 18.Pisa M, Ratti F, Vabanesi M, Radaelli M, Guerrieri S, Moiola L, Martinelli V, Comi G, Leocani L. Subclinical neurodegeneration in multiple sclerosis and neuromyelitis optica spectrum disorder revealed by optical coherence tomography. Mult Scler. 2019:1352458519861603 [DOI] [PubMed] [Google Scholar]
- 19.Costello F Optical Coherence Tomography in Neuro-ophthalmology. Neurol Clin. 2017; 35(1):153–163 [DOI] [PubMed] [Google Scholar]
- 20.Filippatou AG, Mukharesh L, Saidha S, Calabresi PA, Sotirchos ES. AQP4-IgG and MOG-IgG Related Optic Neuritis-Prevalence, Optical Coherence Tomography Findings, and Visual Outcomes: A Systematic Review and Meta-Analysis. Front Neurol. 2020; 11:540156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.You Y, Zhu L, Zhang T, Shen T, Fontes A, Yiannikas C, Parratt J, Barton J, Schulz A, Gupta V, Barnett MH, Fraser CL, Gillies M, Graham SL, Klistorner A. Evidence of Müller Glial Dysfunction in Patients with Aquaporin-4 Immunoglobulin G-Positive Neuromyelitis Optica Spectrum Disorder. Ophthalmology. 2019; 126(6):801–810 [DOI] [PubMed] [Google Scholar]
- 22.Oertel FC, Kuchling J, Zimmermann H, Chien C, Schmidt F, Knier B, Bellmann-Strobl J, Korn T, Scheel M, Klistorner A, Ruprecht K, Paul F, Brandt AU. Microstructural visual system changes in AQP4-antibody-seropositive NMOSD. Neurol Neuroimmunol Neuroinflamm. 2017; 4(3):e334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hokari M, Yokoseki A, Arakawa M, Saji E, Yanagawa K, Yanagimura F, Toyoshima Y, Okamoto K, Ueki S, Hatase T, Ohashi R, Fukuchi T, Akazawa K, Yamada M, Kakita A, Takahashi H, Nishizawa M, Kawachi I. Clinicopathological features in anterior visual pathway in neuromyelitis optica. Ann Neurol. 2016; 79(4):605–624 [DOI] [PubMed] [Google Scholar]
- 24.Aksoy D, Gokce E, Kurt S, Cevik B, Demir HD. Subclinical Optic Neuritis in Neuromyelitis Optica. Journal of Neuro-Ophthalmology. 2013; 33(2):205–207 [DOI] [PubMed] [Google Scholar]
- 25.Shen T, You Y, Arunachalam S, Fontes A, Liu S, Gupta V, Parratt J, Wang C, Barnett M, Barton J, Chitranshi N, Zhu L, Fraser CL, Graham SL, Klistorner A, Yiannikas C. Differing Structural and Functional Patterns of Optic Nerve Damage in Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorder. Ophthalmology. 2019; 126(3):445–453 [DOI] [PubMed] [Google Scholar]
- 26.Lee MY, Yong KP, Hyun J, Kim S, Lee S, Kim HJ. Incidence of inter-attack asymptomatic brain lesions in NMO spectrum disorder. Neurology. 2020 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure E1. Study flowchart
AQP4: aquaporin-4; NMOSD: Neuromyelitis Optica Spectrum Disorder; OCT: Optical Coherence Tomography; ON: optic neuritis