

Abstract
Despite efforts to achieve tissue selectivity, the majority of systemically administered drug delivery systems (DDSs) are cleared by the mononuclear phagocyte system (MPS) before reaching target tissues regardless of disease/injury pathology. Previously, we showed that while tartrate-resistant acid phosphatase (TRAP) binding peptide (TBP)-targeted polymeric nanoparticles (TBP-NP) delivering a bone regenerative Wnt agonist improved NP fracture accumulation and expedited healing compared to controls, there was also significant MPS accumulation. Here we show that TBP-NPs are taken up by liver, spleen, lung, and bone marrow macrophages Mϕ), with 76 ± 4%, 49 ± 11%, 27 ± 9%, and 92 ± 5% of tissue-specific Mϕ positive for NP. Clodronate liposomes (CLO) significantly depleted liver and spleen Mϕ, resulting in 1.8- and 3-fold lower liver and spleen and 1.3-and 1.6-fold greater fracture and naïve femur accumulation of TBP-NP. Interestingly, depletion and saturation also resulted in significantly higher TBP-NP accumulation at lungs and kidneys potentially through compensatory clearance mechanisms. A 10-fold higher NP dose resulted in greater TBP-NP accumulation at naïve bone tissue however, other MPS tissues (i.e., heart and lungs) exhibited greater TBP-NP accumulation, suggesting uptake by other cell types. Most importantly, neither Mϕ depletion nor saturation strategies improved fracture site selectivity of TBP-NPs possibly due to a reduction of Mϕ-derived osteoclasts, which deposit the TRAP epitope. Altogether, these data support that MPS-mediated clearance is a key obstacle in robust and selective fracture accumulation for systemically administered bone-targeted DDS and motivates the development of more sophisticated approaches to further improve fracture selectivity of DDS.
Keywords: bone targeting, macrophages, clodronate-liposomes, nanoparticles, biodistribution
2. Introduction
Drug delivery systems (DDSs) improve solubility, mitigate degradation, and overcome rapid renal excretion of drugs (1, 2). Since regulatory approval of Doxil in 1995, 28 nanoparticle (NP)-based DDS have been approved to treat myriad diseases (3). Despite these successes, an average of only 0.7% of injected DDS dose reaches the tissues of interest (4, 5). To improve tissue specific localization of DDS, active targeting of DDS via introduction of targeting ligands (e.g., peptide, antibody, small molecule, carbohydrate, etc.) has emerged as the next generation of DDSs (1). However, targeted DDSs suffer from similar systemic delivery challenges to their untargeted counterparts, which has resulted in only modest improvement in target tissue delivery to ~ 0.9% of injected dose (4, 5). The marginal success of DDS at achieving tissue specificity despite longstanding investigations of various DDS designs to improve these parameters (i.e., size, surface charge, shape, surface chemistry, etc.) suggests that a greater understanding of factors that impact DDS biodistribution is warranted.
Clearance by the mononuclear phagocyte system (MPS) is a barrier to tissue-specific delivery (1, 6). The MPS is comprised of mononuclear cells (i.e., monocytes, neutrophils, dendritic cells) in systemic circulation and within the liver, spleen, lung, lymph nodes, and bone marrow (6). Though important for defending the body against foreign substances, the MPS also limits NP-based drug delivery through foreign body recognition and clearance mechanisms. MPS manipulation has been used to improve DDS tissue specific delivery (7–13). Depleting phagocytic MPS cells including macrophages reduces off-target accumulation of untargeted gold NPs but only provides a slight increase in tumor biodistribution from 0.7% to 2% of injected dose (8, 14). Furthermore, delivery of high doses of DDSs to saturate MPS improves tumor biodistribution by up to 30% of injected dose per tumor weight for liposomes and gold nanoparticles (15, 16). However, neither of these approaches are clinically translatable due to innate immune system manipulation and potential toxicities associated with high doses of DDSs. Rather, these studies highlight the need to overcome MPS clearance to improve DDS efficiency. Notably, the aforementioned studies focused on passive (e.g., untargeted) DDS delivery within tumor models rather than targeted counterparts and other disease indications.
Although MPS is a universal barrier in drug delivery, very little is known about its function in bone delivery despite bone marrow as an MPS organ. Considering poor bone biodistribution of systemically administered drugs (<1%) (17) and the economic impact of orthopaedic injuries, osteoporosis, and hematologic and bone-metastasizing cancers, improving drug delivery to bone is highly significant. Previously, we developed a bone-targeted DDS that improved bone regeneration owing to robust delivery of a Wnt agonist to fracture sites (18). Specifically, tartrate-resistant acid phosphatase (TRAP) binding peptides (TBP) were introduced to enable bone targeting (16) of poly(styrene-alt-maleic anhydride)-b-poly(styrene) (PSMA-b-PS) nanoparticles (NPs), which improve drug solubility and stability (19, 20). Notably, there was significant off-target liver and spleen accumulation of TBP-NPs, which limits the potential of this bone-targeting platform (18). In the current study, liver, spleen, and lung accumulation of TBP-NPs was investigated to identify macrophage involvement in systemic TBP-NP accumulation. Next, macrophage (Mϕ) depletion was investigated to improve delivery efficiency of TBP-NPs in both uninjured and fractured mice. Finally, high doses of TBP-NP were used to saturate MPS and evaluate subsequent TBP-NP fracture biodistribution to interrogate the role of MPS in delivery of targeted bone-targeted DDS.
3. Materials and Methods
3.1. Materials
All chemicals were supplied by Sigma-Aldrich and Alfa Aesar unless stated otherwise and solvents used were spectroscopic grade. Distilled/deionized water (ddH2O) with resistivity of 18 MΩ or greater (ultrapure) was used for all studies.
3.2. Animals
Male C57BL/6J and female BALB/c mice (6–8-week-old) from Jackson Laboratories (Bar Harbor, ME) were used for uninjured and fracture studies, respectively, to maintain continuity and enable comparisons with previous studies (18, 21). Animals were maintained in the University of Rochester Vivarium according to standard housing conditions. Analgesics (0.05 mg/kg Buprenorphine (IP)) were provided for pain management throughout fracture study experiments. Anesthesia (60 mg/kg ketamine and 4 mg/kg xylazine (intraperitoneal injection, IP)) was provided during retroorbital injections (5 mg/kg and 50 mg/kg NPs) and mice were sacrificed using carbon dioxide followed by cervical dislocation. All animal experiments were approved by the University of Rochester Institutional Animal Care and University Committee of Animal Resources (UCAR).
3.3. Peptide and nanoparticle synthesis, conjugation and characterization
Peptide synthesis has been detailed previously in the following publications (18, 21, 22). Briefly, tartrate-resistant acid phosphatase (TRAP) binding peptides (TBP) (sequence: TPLSYLKGLVTVG) were generated using microwave-assisted solid-phase peptide synthesis (CEM Corp, Liberty1 synthesizer) and Fluorenylmethyloxycarbonyl chloride (FMOC)-protected amino acids (AAPPTec and Peptides International). Amino acid coupling was achieved with an activator mix of 0.5 M O-(benzotriazole-1-yl)-N, N, N’, N’-tetramethyluronium hexafluorophosphate (HBTU) in DMF and an activator base mix of 2 M N, N – Diisopropylethylamine (DIEA) in 1-methyl-2-pyrrolidinone (NMP) while deprotection of amino acids was achieved with 5% piperazine in dimethylformamide (DMF). Cleavage from Fmoc-Gly-Wang resin (Millipore, MA) was achieved via 92.5% trifluoroacetic acid (TFA), 2.5% H2O, 2.5% 3, 6-dioxa-1,8-ocatanedithiol (DODT), and 2.5% triisopropylsilane (TIPS) for 2.5 hours followed by precipitation in ice-cold diethyl ether. Peptide was vacuum filtered and molecular weight was validated using matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF) (Brüker Autoflex III).
Development of poly(styrene-alt-maleic anhydride)-b-poly(styrene) (PSMA-b-PS) based amphiphilic diblock copolymers formed into NPs for these studies has been detailed in previous publications (18–21, 23). Briefly, one-pot reversible addition-fragmentation chain transfer agent (RAFT) polymerization was used (18–21, 23). Distilled styrene (99%, ACS grade) and maleic anhydride recrystallized from chloroform ([STY]:[MA] = 5:1) were added to chain transfer agent, 4-cyano-4-dodecylsulfanyltrithiocarbonyl sulfanyl pentanoic acid (DCT) ([STY]:[CTA] = 100:1) and the radical initiator, 2,2’-Azo-bis(isobutylnitrile) (AIBN), recrystallized from methanol ([CTA]:[AIBN] = 5:1) in dioxane (50% w/w). Diblocks were analyzed using gel permeation chromatography as previously documented (16–18). NPs were formed by dissolving diblock copolymers (200 mg) in DMF (20 mL) and adding ddH2O (31 mL) via a syringe pump set at 24.4 μL/min. NPs were dialyzed against ddH2O for 72 hours using 6–8 kDa MW cut-off dialysis tubing (16–18) (Spectrum Laboratories, CA) and filtered using 0.2 μm cellulose acetate filters (VWR international, PA) prior to storage in 4 C. Gravimetric determination of NP concentrations was performed via lyophilization.
Peptide coupling to NPs has been described previously (18, 21). Briefly, 1-ethyl-3-(3-dimethylamino)propyl carbodiimide (EDC, Thermo Fisher) ([EDC]:[polymer] = 10:1), 5 mM N-hydroxysulfosuccinimide (sulfo-NHS, Thermo Fisher), and TBP ([TBP]:[polymer] = 10:1 in 0.1 M sodium phosphate buffer (pH 7.4)) were combined and mixed overnight. Samples were dialyzed against ddH2O for 72 hours (MWCO 6–8 kDa) to remove unconjugated peptide and conjugation efficiency was determined using fluoraldehyde o-Phthaldialdehyde (OPA, Thermo Fisher, Ex/EM = 360 nm/455 nm) and nanoparticle tracking analysis (NTA, Nanosight NS300). NP physiochemical properties such as size and surface charge were analyzed using dynamic light scattering (DLS) and a Malvern Zetasizer.
3.4. Femur fracture model
Mid-diaphyseal femur fractures were established as previously detailed (18). Briefly, an 8 mm skin incision was used to expose the femur midshaft and a fracture was made using a rotary Dremel with a diamond blade attachment. Following fracture, a 25-gauge needle was inserted into the medullary canal to stabilize the femur.
3.5. Clodronate treatments
PBS and clodronate liposomes (CLO) (5 mg/mL) were purchased from Liposoma BV (Netherlands). Uninjured mice were retro-orbitally injected (100 μL) every other day for four doses with PBS and CLO. For fracture studies, mice were retro-orbitally injected every other day for three doses prior to fracture and injected one day after fracture.
3.6. Nanoparticle injections and biodistribution
Cy5 or Cy7 amine labeling of TBP-NPs was achieved by mixing molar ratios of EDC, dye and polymer (10:10:1), 5 mM sulfo-NHS and 0.1 M sodium phosphate buffer (pH 7.4) overnight. Samples were dialyzed against ddH2O for 72 hours (MWCO 6–8 kDa) to remove unconjugated dye prior to use. NP physiochemical properties such as size and surface charge were analyzed using dynamic light scattering (DLS) and a Malvern Zetasizer.
To investigate fracture accumulation of TBP-NPs, 5 mg/kg dose Cy7-TBP-NP, Cy5-TBP-NP, and 50 mg/kg Cy5-TBP-NPs were retro-orbitally injected a day after the final dose of PBS or CLO liposomes in uninjured mice and three (3) days after fracture in fracture models (16). Twenty-four (24) hours following TBP-NP injections, mice were sacrificed, perfused with PBS, and imaged using the XENOGEN/IVIS imaging system (PerkinElmer) (Ex/E, = 640/800 for Cy5 and Ex/Em = 745/800 nm for Cy7) quantification of NP biodistribution in anatomized tissues. Free draw contour regions of interest (ROIs) were placed around individual organs to determine the total radiance efficiency (TRE) using Living Image software (PerkinElmer). The TRE of each organ was normalized to the sum of TRE to calculate the percent of total TRE for each organ. Control organs from untreated mice were used for background subtractions due to tissue autofluorescence
3.7. Tissue Processing and Immunohistochemistry
Tissues isolated from mice injected with Cy5-TBP-NPs 24 hours prior were stored in neutralbuffered formalin (NBF) for three days and submerged in a daily sucrose gradient (10%, 20%, 30% sucrose in PBS) prior to being embedded in Cryomatrix™ (Thermo Scientific, Cat #: 6769006) on dry ice. Cryomolds were sectioned using a Leica CM1850 cryostat (Germany) and sections were stored in -20 °C prior to staining. To stain F4/80 positive cells in tissue samples, slides were dried in a 37 °C oven overnight, fixed in cold acetone (-20 °C) for 10 minutes, and washed three times in phosphate buffered solution (PBS) for 5 minutes. Sections were then washed twice in 1X TBS/0.3% Triton X-100 for 5 minutes. Slides were incubated for 30 minutes at room temperature in blocking solution (10% normal goat serum in PBS, Cat #: 50–062Z) to reduce non-specific primary antibody staining. Primary antibody (Cat #: ab6640) was added to the slide and sample stored in a humidified chamber at 4 °C overnight. Slides were washed three times in 1X TBS/0.3% Triton X-100 for 5 minutes each and secondary antibody (Cat #: ab150160) was added to the slide in a humidified chamber at room temperature for 30 minutes protected from light. Samples were washed three times in PBS for 5 minutes each and then mounted using DAPI containing mounting media (Cat #: ab104139). Slides were imaged using a Nikon A1R HD laser scanning confocal microscope. Quantification was performed using Imaris Image Visualization & Analysis Software (Oxford Instruments).
3.8. Flow cytometry
Cy5-TBP-NP injected mice were sacrificed 24 hours later, and bone marrow tissue was harvested for cell isolation by flushing with PBS + 2% FBS (FACS buffer) using a 25- gauge needle (16). Liberated marrow cells were resuspended in 1 mg/mL collagenase type IV (Sigma), 2 mg/mL dispase (Sigma) and 10 U/mL DNase (Thermo Fisher Scientific) dissolved in Hank’s Balanced Salt Solution (HBSS) for 45 mins at 37 °C. Red blood cell (RBC) lysis buffer (156 mM NH4Cl, 127 μM EDTA, and 12 mM NaHCO3) was used to lyse RBCs for 5 minutes at room temperature. Antibody incubations were performed for 30 min on ice in the dark using PE-Cy7 labeled TER-119 (Biolegend, Cat # 116221), PE-labeled CD51 (Thermo Fisher Scientific, Cat #: 12–0512-81), PE-Dazzle 594 labeled CD31 (Biolegend, Cat #: 102429), Brilliant Violet (BV) 510 labeled Ly6G (Biolegend, Cat #: 127633), SuperBright 702 labeled F4/80 (Thermo Fisher Scientific, Cat #: 67–4801-80), BV 785 labeled CD206 (Biolegend, Cat #: 108123), PerCP-Cy5.5 labeled Ly6A/Ly-6E (Biolegend, Cat #: 108123), and APC-Cy7 labeled CD45 (BD Biosciences, Cat #: 561037). Samples were resuspended in 100 μL FACS buffer and analyzed on an LSRII flow cytometer: 3 lasers, 355 nm, 488 nm, and 633 nm (BD Biosciences). 74,000 – 310,000 total events were collected for fractured and unfractured bone, respectively.
In a subset of studies, liver tissue was processed for flow cytometry using 10 mL Roswell Park Memorial Institute 1640 (RPMI 1640, Hyclone) containing 0.1% type IV collagenase, spleen tissue was processed using PBS + 2% FBS containing 10 U/mL DNAse, and lung tissue was digested in HBSS containing 1 mg/mL DNase/5 mg/mL type I collagenase. Briefly, livers were minced, placed in digestion media at 37 C for 30 mins, strained through 70 μm filters after centrifugation, and incubated in RBC lysis buffer for 5 mins at 4 C to remove red blood cells. Spleens were crushed through 70 μm cell strainers and into digestion media, incubated at 37 C for 30 mins, and incubated in RBC lysis buffer for 5 mins at 4 C. Lungs were incubated in digestion media for 30 mins at 37 C, tissue was disrupted using a 18-gauge needle, cells were passed through a 70 μm cell strainer prewetted with 0.5% BSA in PBS and incubated in ACK lysis buffer for 5 mins at 4 C. All cells were centrifuged at 300 x g’s for 5 minutes at 4 C between digestion, straining, and ACK lysis steps. Cell suspensions were incubated with APC labeled F/480 (eBioscience/17–4801-82/BM8) and 50 μg/mL propidium iodine in FACs buffer for flow cytometry.
3.9. Statistical analysis
Prism software (GraphPad Version 6.0) was used for all statistical analysis. Comparisons between two groups were performed using unpaired t-tests. Multiple comparisons were performed using one-way or two-way ANOVA followed by Tukey’s post-hoc analysis or Dunnett’s post-hoc analysis to determine significance (p-values are noted in figure legends).
4. Results
4.1. MPS cells uptake bone targeted PSMA-b-PS NPs
Previous studies that exploited TRAP-binding peptide targeted PSMA-b-PS NPs (TBP-NPs) (Schematic 1) to deliver small molecule Wnt agonists revealed substantial liver and spleen accumulation despite improved localization and healing of fractures (18). Based on this observation, in vivo NP uptake by tissue resident Mϕ was investigated 24 hours after TBP-NP injection. Histological analysis of liver, spleen, and lung Mϕ revealed robust TBP-NP uptake (Fig. 1A–C). Quantification of histological images was performed to identify the percent of total cells positive for NPs, NP positive (NP+) Mϕ per total NP+ cells, and NP+ Mϕ per total Mϕ, as shown in Fig. 1 D–F. Of all cells analyzed in liver, spleen, and lung, 21 ± 1%, 14 ± 1%, and 9 ± 2% were positive for NPs, respectively (Fig. 1D). Furthermore, of the total NP+ cells in liver, spleen, and lung, 12 ± 0.8%, 1.5 ± 0.4%, and 1.8 ± 0.7% (Fig. 1E) were Mϕ, as measured by analysis of histological sections, suggesting significant NP uptake in Mϕ and also non-Mϕ cell types. Of all F4/80+ Mϕ in the liver, spleen, and lung, 76 ± 4%, 49 ± 11%, and 27 ± 9% were NP+ (Fig. 1F). Varying degrees of tissue Mϕ uptake corroborate other studies that have shown that liver resident Mϕ, known as Kupffer cells, are primarily responsible for systemically administered NP clearance, followed by splenic Mϕ and, to a lesser extent, lung Mϕ (6). Since the intention of TBP-NP is to deliver drugs to bone, cellular uptake of NPs in naïve and fractured femurs was also quantified. There were no differences in the percent of cells positive for NPs, Mϕ per total NP+ cells, and NP+ Mϕ per total Mϕ between naïve and fractured femurs (23 ± 7% and 21 ± 9%) (Fig. 1G–I). Of the total NP+ cells isolated from naïve and fractured bone, 0.19 ± 0.04% and 0.6 ± 0.3% were Mϕ, suggesting involvement of other cells besides Mϕ in TBP-NP uptake (Fig. 1H). Of total Mϕ from naïve bone and fractures, 99.6 ± 0.4% and 92 ± 5% were NP+, respectively, highlighting that Mϕ are highly phagocytic of TBP-NPs even at the target tissue (Fig. 1I). Of note, neutrophils, osteoblasts, and endothelial cells at naïve and fractured bone were also observed to be NP+ (Fig.S3). Overall, this study showed that Mϕ, within MPS tissue play a major role in clearance of systemically injected bone-targeted NPs and are also responsible for significant NP uptake at the target site.
Schematic 1:
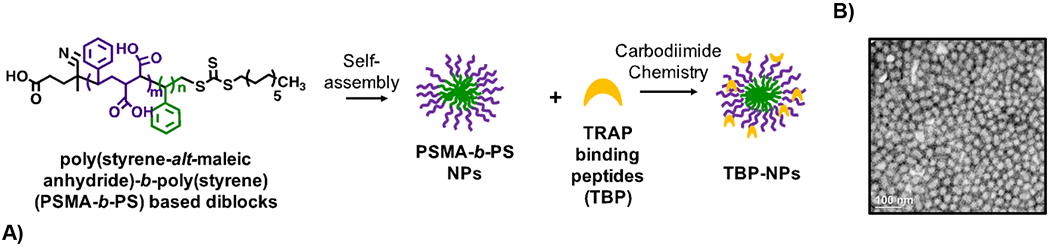
Poly(styrene-alt-maleic anhydride)-b-poly(styrene) (PSMA-b-PS) NP functionalization with TRAP binding peptide (TBP-NPs). Scale bar = 100 nm in B.
Figure 1: Mϕ in the liver, spleen, lungs, and bone take up TBP-NPs.
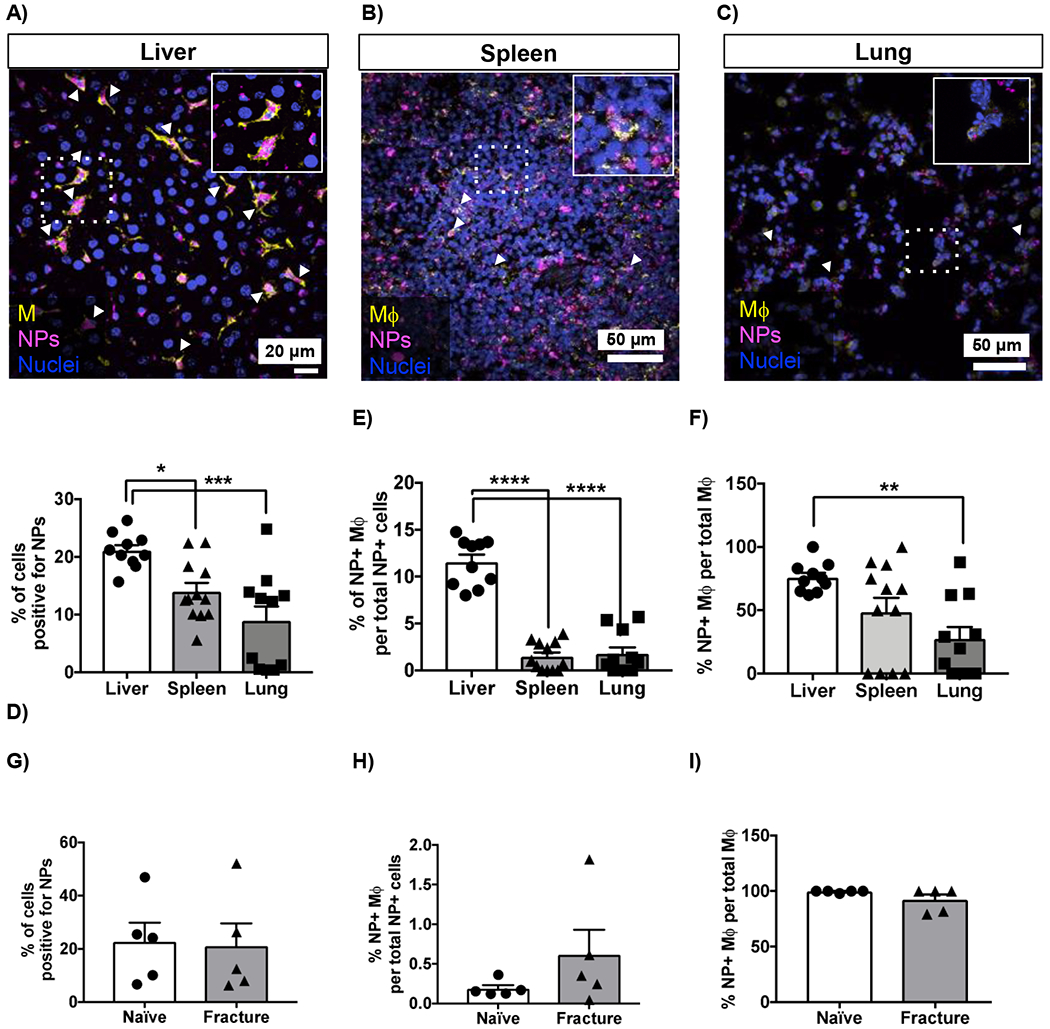
Representative histological images of A) liver, B) spleen and C) lung. Scale bar – 20 μm (liver). Scale bar – 50 μm (spleen and lung). D-F) Imaris was used to quantify cell nuclei, NP signal and Mϕ as measured by F4/80 expression. Data represents mean ± standard error mean (n = 5 per group). D) shows the % of cells positive for NPs. * p < 0.05 and *** p < 0.001 represents significant differences between groups using one-way ANOVA followed by Tukey’s multiple comparisons test. E) shows the % of F4/80+/NP+ cells. **** p < 0.0001 represents difference between groups using one-way ANOVA followed by Tukey’s multiple comparisons test. F) Quantification of the percent of Mϕ positive for NPs. ** p < 0.01 represents difference between groups using one-way ANOVA followed by Tukey’s multiple comparisons test. G-I) Flow cytometry analysis of Mϕ (CD45+/F4/80+/Gr-1-) in naïve and fractured bone. Data represents mean ± standard error mean (n = 5).
4.2. Systemically depleting Mϕ improves bone accumulation
Based on data showing that Mϕ play a significant role in bone-targeted PSMA-b-PS NP clearance (Fig. 1), we postulated that Mϕ depletion would improve bone targeting efficiency. Clodronate liposomes (CLO) were injected every other day for four total doses to systemically deplete Mϕ prior to TBP-NP injection (Fig. 2A) (24). Qualitative histological analysis performed 48 hours after the final CLO dose showed nearly complete loss of Mϕ in the liver (Fig. 2B), which was corroborated via flow cytometry analysis showing a significant reduction of 52% Mϕ (Fig. S2). In the spleen and lung, flow cytometry of Mϕ corroborated histology that showed a decrease in spleen Mϕ and no difference in lung Mϕ (Fig. S2). After CLO Mϕ depletion, NPs were injected into uninjured mice (Fig. 2A). Twenty-four hours after CLO treatment, in vivo live imaging system (IVIS) analysis revealed a significant 5.6-fold increase in integrated NP signal within the collected bones (tibia, femur, radius, and ulna) of CLO depleted compared to non-depleted mice. In contrast, there was a significant 2-fold decrease of NP in liver. Though not statistically significant due to variability in the analysis, a 2.6-fold reduction of NP was also observed in the spleen (Fig. 2C, D). Thus, increased accumulation of TBP-NP at bone and decreases at liver and spleen are consistent with reduced clearance of TBP-NP by Mϕ, which maintains greater concentrations of TBP-NP systemically to enable more robust extravasation to bone. However, increases in NP signal were also observed for lungs, heart, and kidney of 4, 7, and 3-fold versus non-depleted mice, indicating a shift in biodistribution due to CLO-reduction of Mϕ (Fig. 2C, D).
Figure 2: Clodronate treatment significantly improved bone accumulation of TBP-NPs in uninjured mice.
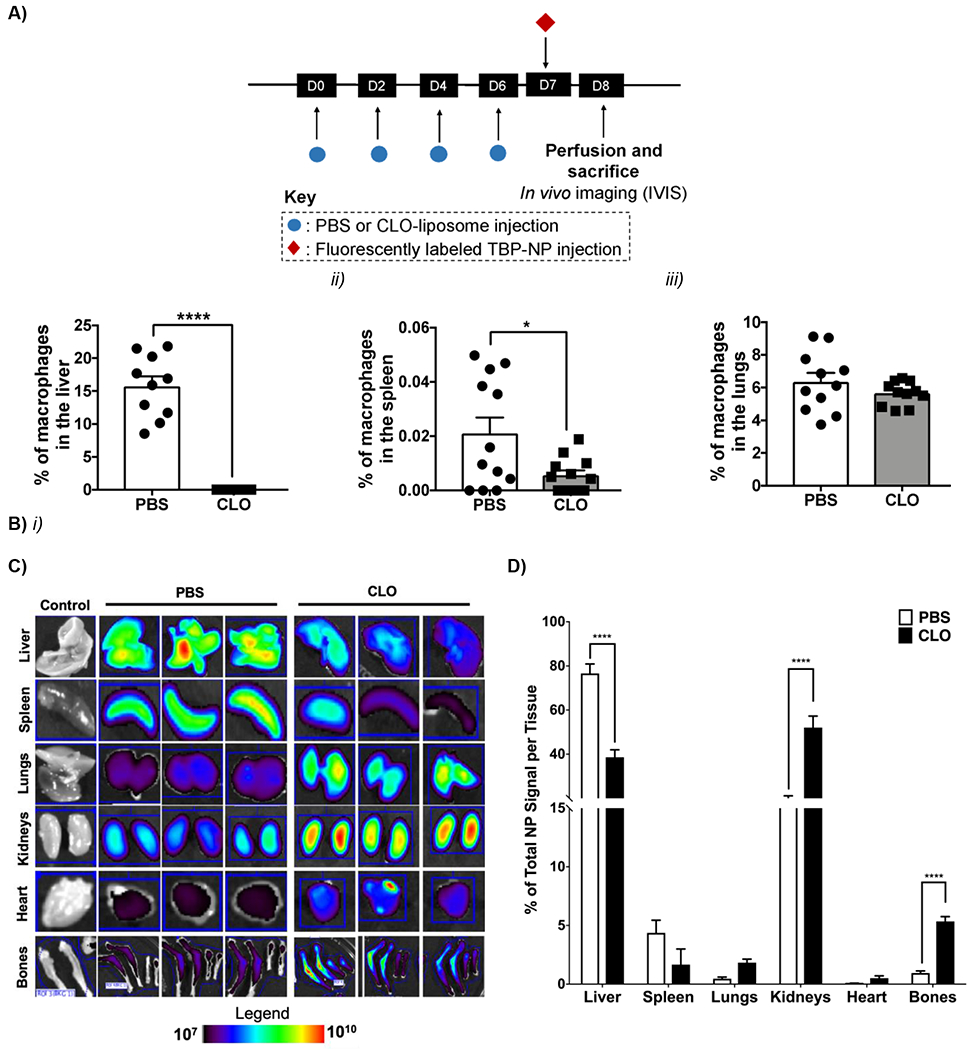
A) Timeline showing injection of PBS, clodronate liposomes (CLO) and fluorescently labeled TBP-NPs. B) Percent of F4/80 positive cells in the liver, spleen, and lung via histological analysis. Data represents mean ± standard error mean (n = 8). *p < 0.05, **** p < 0.001 represent differences between groups via unpaired t-tests. C) IVIS images. Bones = femur, tibia, radius, and ulna. D) Quantification of IVIS images showing the percent of total TBP-NP signal per tissue. Data represents mean ± standard deviation (n = 3). **** p < 0.001 represents comparisons performed using unpaired t-tests between PBS and CLO in uninjured mice.
4.3. Depleting Mϕ improves bone-targeted NP accumulation in naïve bone but not fractures
With significant improvement in bone biodistribution observed following Mϕ depletion in unfractured mice, NP biodistribution was investigated post-fracture. To ensure that Mϕ would not recover during the fracture healing process, three CLO injections were administered every other day prior to femur fracture and one dose after fracture (Fig. 3A). Three days after fracture, when TRAP deposition is elevated, TBP-NPs were injected, as in previous studies (18). Similar to uninjured mice, CLO treatment resulted in a significant decrease of NP signal in the liver by 1.8- fold (Fig. 3B) and a 1.7- and 4-fold increase in kidney and lung accumulation. However, unlike uninjured mice, there was a significant 3-fold reduction of NP signal in the spleen. Additionally, a significant 1.6-fold increase of TBP-NP was observed in contralateral femurs, while fractures exhibited an insignificant 1.3-fold increase in TBP-NP signal (Fig. 3B). Overall, these data suggest that reducing Mϕ is an effective way to improve bone accumulation, albeit only for uninjured bone tissue and not fractures.
Figure 3: Clodronate treatment significantly improved naïve contralateral bone accumulation of TBP-NPs in fractured mice.
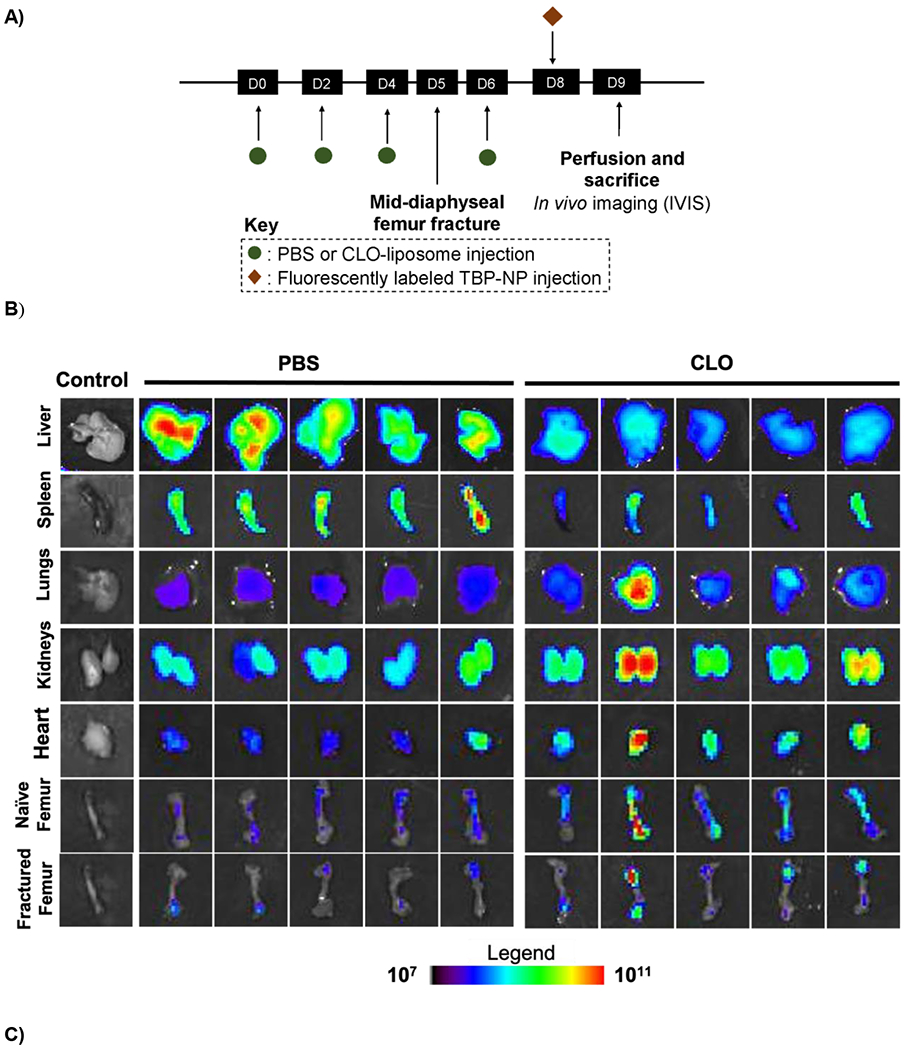
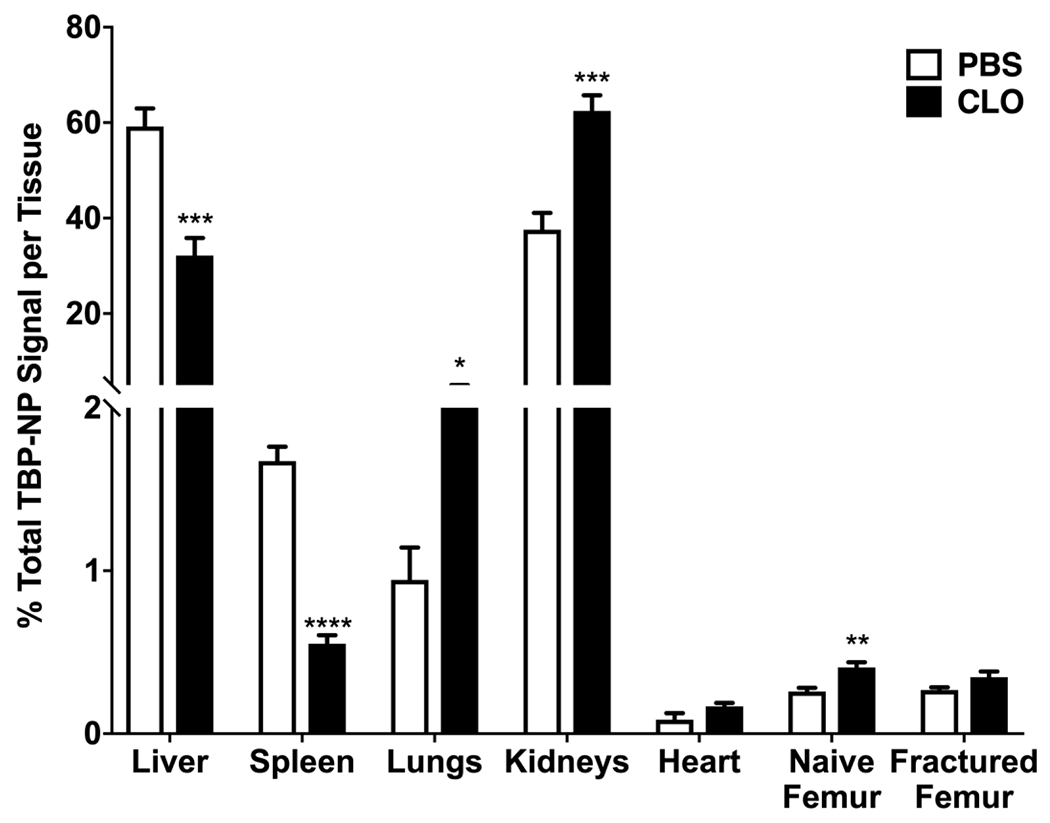
A) Timeline of clodronate and NP treatments. B) IVIS images of tissue after TBP-NPs injection three (3) days after fracture. C) Quantification of IVIS images. Data represents mean ± standard error mean (n = 5). * p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001 represents statistical differences noted using unpaired t-tests between PBS and CLO mice.
4.4. NP dose influences biodistribution via MPS saturation
To test whether MPS could be saturated or overwhelmed to increase bone accumulation, TBP-NP dose was increased from 5 to 50 mg/kg in both clodronate-treated and untreated mice. Data are shown as percent of total tissue signal, resulting in normalization to total dose accumulated in tissues on a per mouse basis. Generally, CLO treatment at the high dose did not change liver, spleen, or lung accumulation but increased heart, naïve, and fractured femur accumulation, 2-fold, 2.6-fold, and 1.9-fold respectively (Fig. 4). However, 50 mg/kg doses resulted in 3-fold increases in NP signal in naïve and fractured femurs in non-depleted mice compared to the 5 mg/kg dose (Fig. 4A). Interestingly, there were no differences in liver accumulation between the low and high dose treatments in non-depleted mice, suggesting that liver saturation was achieved even at the 5 mg/kg dose (Fig. 4B). In other MPS organs, i.e., spleen and lungs, though, there were significant, 2.9-fold and 22-fold increases in accumulation in the high versus low dose in non-depleted mice (Fig. 4B). In Mϕ depleted mice, greater accumulation was found in the spleen (9.8-fold versus low dose) but less so in the lungs (5-fold versus low dose) (Fig. 4B). Higher doses in Mϕ depleted mice resulted in 5-fold increases in both naïve femur and fracture accumulation compared to the low dose, suggesting that combined effects of depleting Mϕ and MPS saturation improve tissue selectivity (Fig. 4B). Overall, these data demonstrated that high dose TBP-NP combined with reducing Mϕ in MPS tissues was an effective method to improve bone accumulation, although these manipulations generally increased overall levels of TBP-NP accumulation irrespective of tissue and did not selectively improve TBP-NP accumulation at fractures compared to uninjured contralateral femurs.
Figure 4: NP biodistribution is dose dependent.
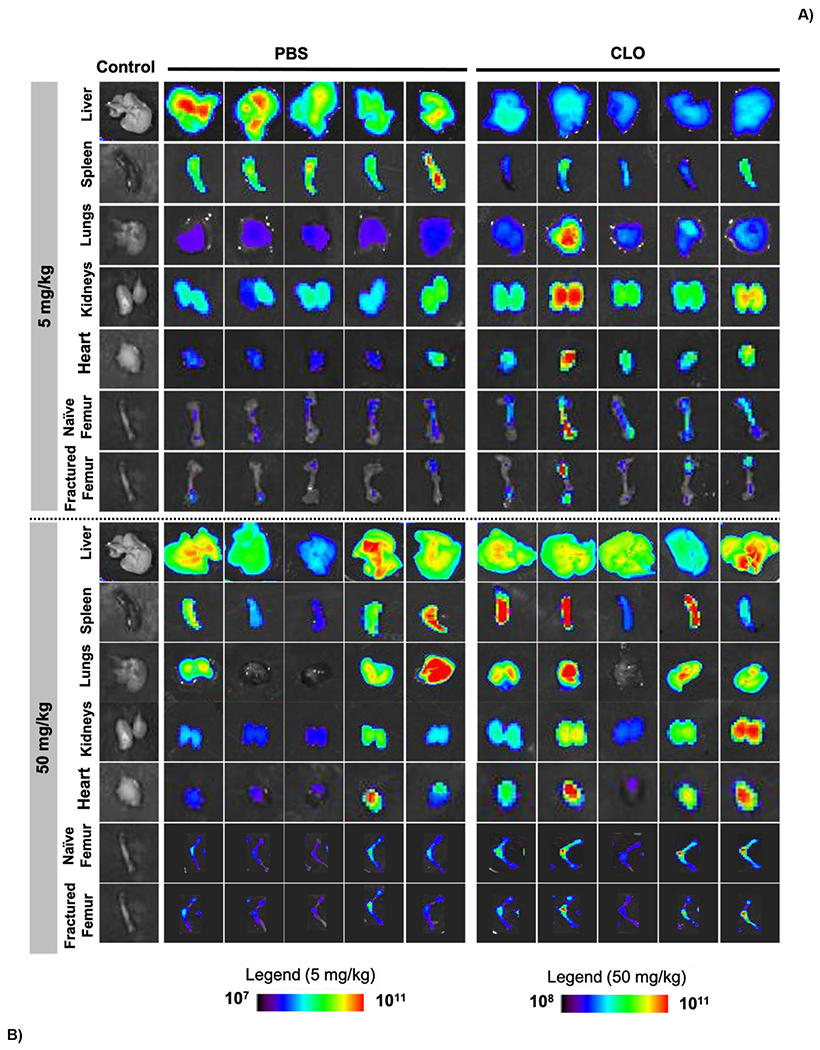
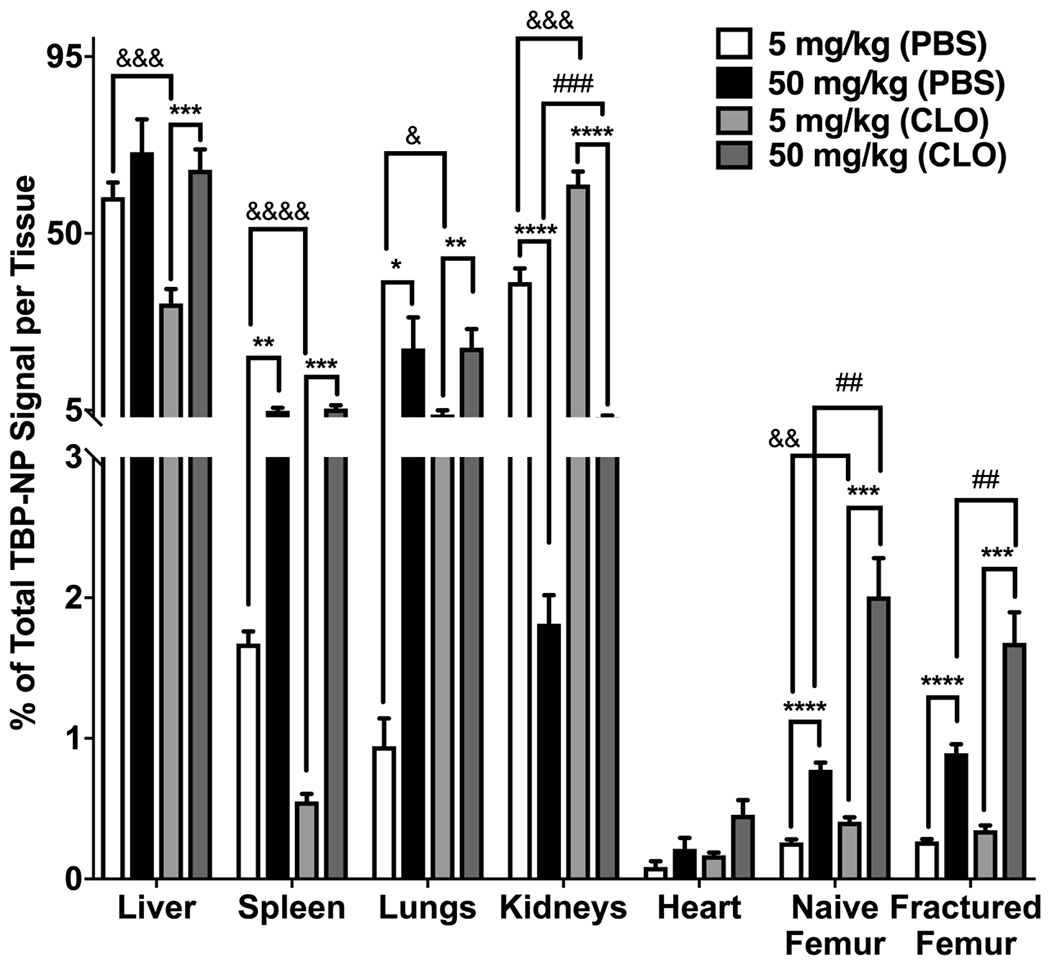
A) IVIS images showing the accumulation showing tissues after low and high doses in PBS and CLO treated mice. B) Quantification of TBP-NP accumulation after low and high dose treatments in fractured mice. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 represents statistical differences noted using one-way ANOVA followed by Tukey’s multiple comparisons test. && p < 0.01, &&& p < 0.001, &&&& p < 0.0001 represents statistical differences using t-test comparisons between PBS and CLO treated with 5 mg/kg. ## p < 0.01, ### p < 0.001 represents differences between PBS and CLO treated with 50 mg/kg. Data represents mean ± standard error mean (n = 5).
5. Discussion
Despite initial excitement of the concept of tissue-specific targeting and potential for revolutionary improvements in drug delivery (25), the development of DDS ‘magic bullets’ remains elusive. Increasingly, evidence shows that MPS clearance hinders efficient DDS-mediated drug delivery irrespective of introduction of tissue-targeting ligands (6). Previously, we showed that despite ~6-fold greater fracture accumulation and expedited fracture healing via Wnt agonist delivery by tartrate-resistant acid phosphatase (TRAP) binding peptide (TBP)-targeted PSMA-b-PS NPs, substantial off-target accumulation occurs, which may limit further DDS development (18). Herein, we aimed to investigate the role of MPS in bone-targeted PSMA-b-PS NP accumulation in bone versus off-target tissues. Clodronate liposomes (CLO) were used to systemically deplete Mϕ in control (uninjured) and fractured mice, leading to a significant reduction in liver accumulation and improved localization at uninjured but not fractured bone. In the lung and kidneys, clodronate treatment significantly increased NP accumulation, while a significant reduction in spleen accumulation was observed but only in fractured mice. Mϕ saturation using 10-fold higher doses of TBP-NP in non-depleted and Mϕ depleted mice also increased fracture accumulation of NP, although this improvement was not specific to fractures versus contralateral femurs. In the liver, spleen, and lung, higher NP doses did not impact accumulation in non-depleted and Mϕ depleted mice while NP accumulation was significantly increased in kidneys and heart between non-depleted and Mϕ depleted mice. Overall, our studies show that Mϕ depletion and saturation strategies alter TBP-NP biodistribution but fail to improve fracture targeting, which motivates materials-focused strategies rather than biological manipulation to improve bone targeting.
Similar to tumors, fractured bone is characterized by increased inflammation and vascular permeability during healing, resulting in enhanced permeability and retention (EPR) mediated accumulation of NPs (18, 26). EPR enables DDS escape from systemic circulation via leaky vasculature associated with tumors, injury, and inflammation (27). Nevertheless, EPR does not guarantee tumor accumulation due to MPS clearance mechanisms (7, 8). Here, the role of MPS was investigated for limiting tissue-specific accumulation of fracture-targeted DDS. Our data show that naïve contralateral bone accumulation of NP was increased in the Mϕ depleted fracture model and even more so in the uninjured model, suggesting systemic inflammation and increased vascular permeability due to the fracture resulted in increased overall widespread bone extravasation in conjunction with reduced clearance of TBP-NP. Similarly, CLO reduces MPS uptake of liposomal NPs, silver NPs, silica NPs, and gold NPs, which subsequently increases NP circulation time and improves delivery to tumors (7, 8). In our studies, increasing NP dose 10-fold in non-depleted fractured mice increased naïve bone accumulation, but did not improve fracture accumulation, similar findings to those in a tumor-bearing model (16). Even combining Mϕ depletion and saturation did not improve selective fracture accumulation compared to naïve bone, which suggests an important role of TRAP in the TBP-NP DDS. While TBP-NPs accumulate passively at fractures due to EPR, TBP binding of TRAP deposited by osteoclasts during resorption results in NP persistence (18). This may be due to the depletion of Mϕ, which are osteoclast progenitors, subsequently reducing osteoclast-deposited TRAP, as previously described (24, 28). These data highlight a limitation of Mϕ depletion in the context of fracture site accumulation of TBP-NPs and motivate other strategies to maintain tissue selectivity while evading Mϕ.
To enable direct comparisons with our previous data (18, 21), and broaden the applicability of these findings, different sex and strain mice were used in our uninjured and fracture models. Interestingly, Mϕ depletion in both models resulted in decreased liver and spleen NP accumulation coupled with increased lung, kidney, heart, and naïve bone accumulation. Biodistribution observed in our studies are complementary to others that show DDS accumulation shifts from liver to other MPS organs including lung, kidney, and even lymph nodes after CLO treatment (7, 8). Similar to our findings, studies in tumor and non-tumor models show CLO-mediated depletion of Mϕ, which are responsible for DDS clearance after systemic administration (6), reduced but did not completely abrogate liver accumulation regardless of DDS or disease pathology (7, 8, 11, 14, 29). Thus, Mϕ depletion is insufficient to eliminate all liver accumulation, likely due to uptake by residual Mϕ as well as hepatocytes, hepatic B cells, and other cells in liver (29, 30). Our studies show that ~43% of NP+ cells in the liver were not Mϕ, further supporting the notion that other cells in the liver were responsible for NP accumulation. In our studies and others, spleen Mϕ were depleted using CLO, yet, unlike tumor studies that use liposomes, gold NPs, and silica NPs (7, 8), we observed decreased TBP-NP accumulation in the spleen after CLO treatment, which may suggest differential NP-immune system interactions based on DDS type (31). Conversely, exosomes and polymeric NPs (11, 14) exhibit reduced spleen accumulation in Mϕ depleted non-diseased mice, suggesting a contribution of disease pathology on NP accumulation at spleen. Findings of decreased NP accumulation in liver and spleen emphasize the role of Mϕ in off-target tissue accumulation, but also underscore the involvement of other cell types in NP clearance.
Off-target NP accumulation at tissues, such as kidney and lung, are impacted by NP physiochemical properties (i.e., size, surface charge, shape, surface chemistry, etc.). Here, kidney and lung exhibited the most interesting changes in biodistribution after Mϕ depletion (32, 33). Based on our findings of increased kidney biodistribution, it is likely that an inflammatory milieu promoted NP extravasation through leaky vessels due to kidney tubular injury caused by Mϕ depletion (34), though kidney Mϕ depletion was not directly analyzed in our studies since significant NP kidney accumulation was not observed previously (18). In the lungs, studies report increased NP accumulation regardless of NP type (7, 8). However, NP accumulation in lung is more extensive in epithelial and endothelial cells versus Mϕ (33). In conjunction with our data that shows lung Mϕ depletion was elusive, it is, therefore, not surprising that CLO treatment did not decrease NP lung accumulation. Increasing NP dose 10-fold in non-depleted mice resulted in significant increases in all off-target organs except for kidneys. At high NP doses, Mϕ depletion did not decrease NP accumulation in the liver and spleen, as had been observed in low dose studies, likely due to compensatory uptake of NPs by other cells besides Mϕ. Interestingly, NP accumulation in the lung was also unchanged in Mϕ depleted and saturated conditions, which further supports NP accumulation mechanisms independent of Mϕ. Overall, these studies, combined with prior reports (7, 8), emphasize that the magnitude of accumulation is dependent on injury and/or disease (7, 8). Most importantly, the fracture data highlighted the limitations of Mϕ depletion and saturation strategies on improving site specific delivery.
The findings that Mϕ depletion impacts naïve bone accumulation but not in fractures suggest significant hurdles remain for tissue-targeted DDS, namely, to successfully evade clearance by Mϕ. The cascade of events leading up to tissue accumulation involve, 1) protein adsorption to DDS, including opsonins, 2) significant phagocytic clearance from circulation, 3) extravasation through leaky vasculature, and, finally, 4) binding to target tissue (6, 35–37). While reducing Mϕ and saturating MPS are useful for fundamental investigations, these approaches are not clinically translatable due to risk of immune system compromise, potential off-target toxicities (8), and disruptions in the fracture healing cascade (28), beyond lack of efficacy observed in these studies. Therefore, further modifications of the TBP-NP platform must be developed such that MPS is avoided, the fracture healing cascade is not impacted, and access of targeting ligands are not compromised. Current approaches to achieve these design criteria include PEGylation and zwitterionic surface chemistries that prolong the circulation time of NPs through Mϕ evasion (38, 39). Other notable approaches involve using CD47 mimicking peptides to reduce recognition by Mϕ (40). Nevertheless, these Mϕ evasive approaches are not without challenges. For example, PEGylation has been associated with complement activation, adverse immune reactions, and can impact target ligand accessibility (41, 42). These limitations motivate fundamental studies, such as those undertaken here, to investigate how MPS interact with various nanomaterials based on material properties and disease/injury type. Though our studies did not definitively identify a route to improve TBP-NP fracture site accumulation due to the relationship of Mϕ and targeting epitope (TRAP), the findings inform future design approaches, including Mϕ evading functionalities, for the next generation of fracture targeted NPs.
6. Conclusion
MPS was discovered to play a major role in limiting bone delivery efficiency of TRAP binding peptide (TBP) – PSMA-b-PS NPs, despite improved fracture site accumulation. Here, we showed that Mϕ were the predominant cells responsible for bone-targeted NP clearance. When Mϕ were systemically depleted in uninjured and fractured mice, decreased liver and splenic NP accumulation and increased bone accumulation was observed. Interestingly, there were also increases in lung, kidney, and heart accumulation after reducing Mϕ, which suggested compensatory mechanisms of clearance within MPS. When a high dose of TBP-NPs was used to saturate liver MPS, there was a significant increase NP accumulation in all tissues except for kidneys. These studies confirm the need to develop approaches evade Mϕ to improve delivery efficiency. Furthermore, the circulation time of TBP-NPs and how it relates to tissue accumulation should be investigated to gain a greater appreciation for the DDS and the lessons learned from this work should be applied for developing other bone-targeted DDS.
Supplementary Material
Table 1:
TBP-NP physiochemical properties
| Size (nm) | Surface Charge (mV) | Peptides per NP |
|---|---|---|
| 86 ± 16 | −35 ± 3 | 19,000 ± 1,500 |
Acknowledgements
The authors gratefully thank Karen Bentley, M.S., (University of Rochester Medical Center Electron Microscopy Shared Laboratory) for EM image acquisition and Dr. James McGrath (University of Rochester) for equipment use.
Funding
Funding for this study was provided by the National Science Foundation (CBET1450987 (CAREER Award) and DMR2103553 (D.S.W.B.)), National Institutes of Health (NIH) P30 AR069655, R01 AR064200, R01 AR056696 (D.S.W.B.), F31 CA228391 (M.A.F.), University of Rochester CTSA award number UL1 TR002001 (D.S.W.B.), University Research Award (D.S.W.B.) and Drug Discovery Grant (D.S.W.B.).
Footnotes
Conflict of Interest
The authors have no conflict of interest to declare.
References
- 1.Bertrand N, Wu J, Xu X, Kamaly N, Farokhzad OC. Cancer nanotechnology: the impact of passive and active targeting in the era of modern cancer biology. Adv Drug Deliv Rev. 2014;66:2–25. Epub 2013/11/26. doi: 10.1016/j.addr.2013.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Whitehead KA, Langer R, Anderson DG. Knocking down barriers: advances in siRNA delivery. Nature Reviews Drug Discovery. 2009;8(2):129–38. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Anselmo AC, Mitragotri S. Nanoparticles in the clinic: An update. Bioeng Transl Med. 2019;4(3):e10143. Epub 2019/10/02. doi: 10.1002/btm2.10143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng Y-H, He C, Riviere JE, Monteiro-Riviere NA, Lin Z. Meta-Analysis of Nanoparticle Delivery to Tumors Using a Physiologically Based Pharmacokinetic Modeling and Simulation Approach. ACS Nano. 2020;14(3):3075–95. doi: 10.1021/acsnano.9b08142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wilhelm S, Tavares AJ, Dai Q, Ohta S, Audet J, Dvorak HF, Chan WCW. Analysis of nanoparticle delivery to tumours. Nature Reviews Materials. 2016;1(5). doi: 10.1038/natrevmats.2016.14. [DOI] [Google Scholar]
- 6.Blanco E, Shen H, Ferrari M. Principles of nanoparticle design for overcoming biological barriers to drug delivery. Nat Biotechnol. 2015;33(9):941–51. Epub 2015/09/09. doi: 10.1038/nbt.3330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ohara Y, Oda T, Yamada K, Hashimoto S, Akashi Y, Miyamoto R, Kobayashi A, Fukunaga K, Sasaki R, Ohkohchi N. Effective delivery of chemotherapeutic nanoparticles by depleting host Kupffer cells. Int J Cancer. 2012;131(10):2402–10. Epub 2012/03/01. doi: 10.1002/ijc.27502. [DOI] [PubMed] [Google Scholar]
- 8.Tavares AJ, Poon W, Zhang YN, Dai Q, Besla R, Ding D, Ouyang B, Li A, Chen J, Zheng G, Robbins C, Chan WCW. Effect of removing Kupffer cells on nanoparticle tumor delivery. Proc Natl Acad Sci U S A. 2017;114(51):E10871–E80. Epub 2017/12/07. doi: 10.1073/pnas.1713390114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu T, Choi H, Zhou R, Chen IW. RES blockade: A strategy for boosting efficiency of nanoparticle drug. Nano Today. 2015;10(1):11–21. doi: 10.1016/j.nantod.2014.12.003. [DOI] [Google Scholar]
- 10.Wolfram J, Nizzero S, Liu H, Li F, Zhang G, Li Z, Shen H, Blanco E, Ferrari M. A chloroquine-induced macrophage-preconditioning strategy for improved nanodelivery. Sci Rep. 2017;7(1):13738. Epub 2017/10/25. doi: 10.1038/s41598-017-14221-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wan Z, Zhao L, Lu F, Gao X, Dong Y, Zhao Y, Wei M, Yang G, Xing C, Liu L. Mononuclear phagocyte system blockade improves therapeutic exosome delivery to the myocardium. Theranostics. 2020;10(1):218–30. doi: 10.7150/thno.38198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ellens H, Mayhew E, Rustum Y. Reversible depression of the reticuloendothelial system by liposomes. Biochimica et Biophysica Acta (BBA) - General Subjects. 1982;714(3):479–85. doi: 10.1016/0304-4165(82)90157-x. [DOI] [PubMed] [Google Scholar]
- 13.Sun X, Yan X, Jacobson O, Sun W, Wang Z, Tong X, Xia Y, Ling D, Chen X. Improved Tumor Uptake by Optimizing Liposome Based RES Blockade Strategy. Theranostics. 2017;7(2):319–28. doi: 10.7150/thno.18078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hao J, Han T, Wang M, Zhuang Q, Wang X, Liu J, Wang Y, Tang H. Temporary suppression the sequestrated function of host macrophages for better nanoparticles tumor delivery. Drug Deliv. 2018;25(1):1289–301. Epub 2018/06/06. doi: 10.1080/10717544.2018.1474965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Proffitt RT, Williams LE, Presant CA, Tin GW, Uliana JA, Gamble RC, Baldeschwieler JD. Liposomal blockade of the reticuloendothelial system: improved tumor imaging with small unilamellar vesicles. Science. 1983;220(4596):502–5. Epub 1983/04/29. doi: 10.1126/science.6836294. [DOI] [PubMed] [Google Scholar]
- 16.Ouyang B, Poon W, Zhang YN, Lin ZP, Kingston BR, Tavares AJ, Zhang Y, Chen J, Valic MS, Syed AM, MacMillan P, Couture-Senecal J, Zheng G, Chan WCW. The dose threshold for nanoparticle tumour delivery. Nat Mater. 2020. Epub 2020/08/12. doi: 10.1038/s41563-020-0755-z. [DOI] [PubMed] [Google Scholar]
- 17.Low SA, Galliford CV, Yang J, Low PS, Kopecek J. Biodistribution of Fracture-Targeted GSK3beta Inhibitor-Loaded Micelles for Improved Fracture Healing. Biomacromolecules. 2015;16(10):3145–53. doi: 10.1021/acs.biomac.5b00777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Y, Newman MR, Ackun-Farmmer M, Baranello MP, Sheu TJ, Puzas JE, Benoit DSW. Fracture-Targeted Delivery of beta-Catenin Agonists via Peptide-Functionalized Nanoparticles Augments Fracture Healing. ACS Nano. 2017;11(9):9445–58. Epub 2017/09/08. doi: 10.1021/acsnano.7b05103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Baranello MP, Bauer L, Benoit DS. Poly(styrene-alt-maleic anhydride)-based diblock copolymer micelles exhibit versatile hydrophobic drug loading, drug-dependent release, and internalization by multidrug resistant ovarian cancer cells. Biomacromolecules. 2014;15(7):2629–41. doi: 10.1021/bm500468d. [DOI] [PubMed] [Google Scholar]
- 20.Baranello MP, Bauer L, Jordan CT, Benoit DSW. Micelle Delivery of Parthenolide to Acute Myeloid Leukemia Cells. Cell Mol Bioeng. 2015;8(3):455–70. doi: 10.1007/s12195-015-0391-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ackun-Farmmer MA, Soto CA, Lesch ML, Byun D, Yang L, Calvi LM, Benoit DSW, Frisch BJ. Reduction of leukemic burden via bone-targeted nanoparticle delivery of an inhibitor of C-chemokine (C-C motif) ligand 3 (CCL3) signaling. FASEB J. 2021;35(4):e21402. Epub 2021/03/17. doi: 10.1096/fj.202000938RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Newman MR, Russell SG, Schmitt CS, Marozas IA, Sheu TJ, Puzas JE, Benoit DSW. Multivalent Presentation of Peptide Targeting Groups Alters Polymer Biodistribution to Target Tissues. Biomacromolecules. 2018;19(1):71–84. Epub 2017/12/12. doi: 10.1021/acs.biomac.7b01193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ackun‐Farmmer MA, Alatise KL, Cross G, Benoit DSW. Ligand Density Controls C‐Type Lectin‐Like Molecule‐1 Receptor–Specific Uptake of Polymer Nanoparticles. Advanced Biosystems. 2020. doi: 10.1002/adbi.202000172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Selander KS, Monkkonen J, Karhukorpi EK, Harkonen P, Hannuniemi R, Vaananen HK. Characteristics of clodronate-induced apoptosis in osteoclasts and macrophages. Mol Pharmacol. 1996;50(5):1127–38. [PubMed] [Google Scholar]
- 25.Ehrlich P Croonian lecture.—On immunity with special reference to cell life. Proceedings of the Royal Society of London. 1900;66(424–433):424–48. doi: 10.1098/rspl.1899.0121. [DOI] [Google Scholar]
- 26.Chen KH, Lundy DJ, Toh EK, Chen CH, Shih C, Chen P, Chang HC, Lai JJ, Stayton PS, Hoffman AS, Hsieh PC. Nanoparticle distribution during systemic inflammation is size-dependent and organ-specific. Nanoscale. 2015;7(38):15863–72. Epub 2015/09/12. doi: 10.1039/c5nr03626g. [DOI] [PubMed] [Google Scholar]
- 27.Sindhwani S, Syed AM, Ngai J, Kingston BR, Maiorino L, Rothschild J, MacMillan P, Zhang Y, Rajesh NU, Hoang T, Wu JLY, Wilhelm S, Zilman A, Gadde S, Sulaiman A, Ouyang B, Lin Z, Wang L, Egeblad M, Chan WCW. The entry of nanoparticles into solid tumours. Nat Mater. 2020;19(5):566–75. doi: 10.1038/s41563-019-0566-2. [DOI] [PubMed] [Google Scholar]
- 28.Lin HN, O'Connor JP. Osteoclast depletion with clodronate liposomes delays fracture healing in mice. J Orthop Res. 2017;35(8):1699–706. doi: 10.1002/jor.23440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Samuelsson E, Shen H, Blanco E, Ferrari M, Wolfram J. Contribution of Kupffer cells to liposome accumulation in the liver. Colloids and Surfaces B: Biointerfaces. 2017;158:356–62. doi: 10.1016/j.colsurfb.2017.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang YN, Poon W, Tavares AJ, McGilvray ID, Chan WCW. Nanoparticle-liver interactions: Cellular uptake and hepatobiliary elimination. J Control Release. 2016;240:332–48. Epub 2016/01/18. doi: 10.1016/j.jconrel.2016.01.020. [DOI] [PubMed] [Google Scholar]
- 31.Cataldi M, Vigliotti C, Mosca T, Cammarota M, Capone D. Emerging Role of the Spleen in the Pharmacokinetics of Monoclonal Antibodies, Nanoparticles and Exosomes. Int J Mol Sci. 2017;18(6). Epub 2017/06/13. doi: 10.3390/ijms18061249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Du B, Yu M, Zheng J. Transport and interactions of nanoparticles in the kidneys. Nature Reviews Materials. 2018;3(10):358–74. doi: 10.1038/s41578-018-0038-3. [DOI] [Google Scholar]
- 33.Dhand C, Prabhakaran MP, Beuerman RW, Lakshminarayanan R, Dwivedi N, Ramakrishna S. Role of size of drug delivery carriers for pulmonary and intravenous administration with emphasis on cancer therapeutics and lung-targeted drug delivery. RSC Adv. 2014;4(62):32673–89. doi: 10.1039/c4ra02861a. [DOI] [Google Scholar]
- 34.Kim MG, Su Boo C, Sook Ko Y, Young Lee H, Yong Cho W, Kyu Kim H, Jo SK. Depletion of kidney CD11c+ F4/80+ cells impairs the recovery process in ischaemia/reperfusion-induced acute kidney injury. Nephrology Dialysis Transplantation. 2010;25(9):2908–21. doi: 10.1093/ndt/gfq183. [DOI] [PubMed] [Google Scholar]
- 35.Nel AE, Madler L, Velegol D, Xia T, Hoek EMV, Somasundaran P, Klaessig F, Castranova V, Thompson M. Understanding biophysicochemical interactions at the nano-bio interface. Nat Mater. 2009;8(7):543–57. doi: 10.1038/Nmat2442. [DOI] [PubMed] [Google Scholar]
- 36.Tenzer S, Docter D, Kuharev J, Musyanovych A, Fetz V, Hecht R, Schlenk F, Fischer D, Kiouptsi K, Reinhardt C, Landfester K, Schild H, Maskos M, Knauer SK, Stauber RH. Rapid formation of plasma protein corona critically affects nanoparticle pathophysiology. Nat Nanotechnol. 2013;8(10):772–U1000. [DOI] [PubMed] [Google Scholar]
- 37.Xiao W, Gao H. The impact of protein corona on the behavior and targeting capability of nanoparticle-based delivery system. Int J Pharm. 2018;552(1–2):328–39. Epub 2018/10/12. doi: 10.1016/j.ijpharm.2018.10.011. [DOI] [PubMed] [Google Scholar]
- 38.Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 2001;53(2):283–318. [PubMed] [Google Scholar]
- 39.Yoo J-W, Chambers E, Mitragotri S. Factors that Control the Circulation Time of Nanoparticles in Blood: Challenges, Solutions and Future Prospects. Current Pharmaceutical Design. 2010;16(21):2298–307. doi: 10.2174/138161210791920496. [DOI] [PubMed] [Google Scholar]
- 40.Rodriguez PL, Harada T, Christian DA, Pantano DA, Tsai RK, Discher DE. Minimal "Self" Peptides That Inhibit Phagocytic Clearance and Enhance Delivery of Nanoparticles. Science. 2013;339(6122):971–5. doi: 10.1126/science.1229568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verhoef JJ, Carpenter JF, Anchordoquy TJ, Schellekens H. Potential induction of anti-PEG antibodies and complement activation toward PEGylated therapeutics. Drug Discov Today. 2014;19(12):1945–52. Epub 2014/09/11. doi: 10.1016/j.drudis.2014.08.015. [DOI] [PubMed] [Google Scholar]
- 42.Suk JS, Xu Q, Kim N, Hanes J, Ensign LM. PEGylation as a strategy for improving nanoparticle-based drug and gene delivery. Advanced Drug Delivery Reviews. 2016;99:28–51. doi: 10.1016/j.addr.2015.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.