

Abstract
Zebrafish (Danio rerio) have become a highly-utilized model system in the field of regenerative biology because of their endogenous ability to regenerate many tissues and organs, including the retina. The vast majority of previous research on retinal regeneration in adult zebrafish utilizes acute methodologies for retinal damage. Acute retinal cell death triggers a reactive gliosis response of Müller glia (MG), the resident macroglia of the retina. In addition, each activated MG undergoes asymmetric cell division to produce a neuronal progenitor, which continues to divide and ultimately gives rise to new retinal neurons. Studies using these approaches have uncovered many crucial mechanisms by which MG respond to acute damage. However, they may not adequately mimic the chronic neuronal degeneration observed in many human retinal degenerative diseases. The current study aimed to develop a new long-term, chronic photoreceptor damage and degeneration model in adult zebrafish. Comparing the subsequent cellular responses to that of the commonly-used acute high-intensity model, we found that low, continuous light exposure damaged the outer segments of both rod and cone photoreceptors, but did not result in significant apoptotic cell death, MG gliosis, or MG cell-cycle re-entry. Instead, chronic light nearly completely truncated photoreceptor outer segments and resulted in a recruitment of microglia to the area. Together, these studies present a chronic photoreceptor model that can be performed in a relatively short time frame (21 days), that may lend insight into the cellular events underlying non-regenerative photoreceptor degeneration observed in other model systems.
Keywords: Müller glia, stem cell, gliosis, regeneration, zebrafish, phototoxic lesion
1. INTRODUCTION
Zebrafish (Danio rerio) have become a highly utilized model system in the field of regenerative biology because of their endogenous ability to regenerate many tissues and organs, including the heart, spinal cord, and retina (Becker and Becker, 2008; Becker et al., 1997; Lenkowski and Raymond, 2014; Poss, 2007; Poss et al., 2002; Vihtelic and Hyde, 2000). Each of these regenerative events are mediated by a resident cell that is triggered by massive tissue damage to re-enter the cell cycle and act as a source of new progenitors. For example, following damage to the zebrafish ventricle, peripheral cardiomyocytes re-enter the cell cycle to produce progenitors that will replace the lost cardiac muscle (Kikuchi et al., 2011; Lepilina et al., 2006). Similarly, following severe damage to retinal neurons, Müller glial cells - the resident macroglia of the retina - undergo asymmetric cell division to produce a neuronal progenitor; this cell continues to divide symmetrically to produce a pool of progenitors that ultimate give rise to new retinal neurons (Bernardos et al., 2007; Fausett and Goldman, 2006; Fimbel et al., 2007; Lenkowski and Raymond, 2014; Thummel et al., 2008b). This is significant because human retinas also possess resident Müller glial cells, and yet these cells exhibit a reactive scar-like response following retinal damage (Bringmann et al., 2006; Bringmann and Wiedemann, 2012). Because the zebrafish retina possesses the same major retinal cell types as humans (rods, cones, retinal ganglion cells, bipolar, amacrine, horizontal, and Müller glia cells), arranged in a nearly identical laminar pattern, it is an increasingly popular model to study possible recovery options for a wide variety of human retinal degenerative diseases and injuries, including glaucoma, macular degeneration, and traumatic retinal injuries (Angueyra and Kindt, 2018; Massoz et al., 2021; Noel et al., 2021).
The vast majority of previous research on retinal regeneration in adult zebrafish utilizes various acute, extreme models for damage. For example, retinal punctures that span the full thickness of the retina have been used to damage retinal neurons in all three nuclear layers in single foci (Fausett and Goldman, 2006; Zhang et al., 2016). And neurotoxic compounds such as NMDA and Ouabain have been utilized to selectively damage subsets of retinal neurons (Fimbel et al., 2007; Lahne et al., 2020; Luo et al., 2019; McGinn et al., 2019; Sherpa et al., 2008; Thomas et al., 2016). The most common method, however, is to use acute high intensity light exposure to destroy rod and cone photoreceptors, leaving the inner retina intact. Extensive previous studies using this model have defined windows of photoreceptor degeneration, Müller glia cell cycle re-entry, neuronal progenitor cells amplification, and differentiation of new rod and cone photoreceptors (Kassen et al., 2007; Kassen et al., 2008; Lahne and Hyde, 2016; Nagashima et al., 2013; Thummel et al., 2008a; Vihtelic and Hyde, 2000). In addition, this phototoxic model, combined with morpholino- or pharmacological protein inhibition, has revealed many genetic pathways required for proper retinal regeneration in zebrafish (Gorsuch and Hyde, 2014; Lenkowski and Raymond, 2014).
In each of the acute damage models utilized to elicit retinal damage, only Müller glia within and immediately surrounding retinal cell death respond to produce new neuronal progenitors. As such, many researchers have suggested a “threshold” of retinal damage and signaling to Müller glia is needed to elicit their cell cycle re-entry (Ahmad et al., 2011; Iribarne et al., 2019; Lenkowski and Raymond, 2014; Wan and Goldman, 2017). This theory is consistent with previous work that compared two acute models of phototoxicity and found that the more toxic model resulted in greater numbers of proliferating Müller glia (Thomas et al., 2012). Thus, to ensure robust regenerative responses, all current literature on phototoxic light lesions in adult zebrafish retina use various degrees of acute, high intensity light treatment meant to quickly destroy large numbers of photoreceptors. While these informative studies have uncovered mechanisms by which Müller glia respond to acute, strong damage, they may not adequately mimic the chronic photoreceptor degeneration response observed in many human retinal degenerative diseases.
The current study aimed to develop a new method of modeling more long-term, chronic photoreceptor damage and degeneration in adult zebrafish. In particular, we asked whether gradual photoreceptor damage would elicit Müller glia cell cycle re-entry and/or gliosis. For this, we compared our new chronic low-light model to that of the commonly-used acute high-intensity model at multiple time points previously established as peaks of photoreceptor degeneration and regeneration. We found that low, but continuous light exposure damaged the outer segments of both rod and cone photoreceptors without significant apoptotic cell death as typically observed in the acute high-intensity model. Outer segment damage in the chronic model resulted in a recruitment of microglia to the area, but did not elicit Müller glia cell-cycle re-entry. Furthermore, only minimal signs of Müller glia gliosis were observed in this model. Together, these studies present a chronic photoreceptor model that can be performed in a relatively short time frame (21 days), results in photoreceptor outer segment truncation and microglial response, but does not reach the threshold of damage to “trigger” MG cell cycle re-entry.
2. MATERIALS AND METHODS
2.1. Zebrafish Maintenance
Adult albino zebrafish (Danio rerio) were used for these experiments. Fish were maintained on a closed water circulation system (Aquaneering, LLC) equipped with a biofilter, UV light purifier, and a self-dosing monitoring system to control water pH and conductivity. Fish were maintained in a standard circadian light cycle (14 h light: 10 h dark) and were fed a combination of flake food and brine shrimp three times daily (Westerfield, 1995). All procedures were approved by the Institutional Animal Care and Use Committee (IACUC).
2.2. Light Lesion Protocols
Two different light lesion protocols were used in this study. The acute high-intensity light paradigm involved 9–12 months old zebrafish, dark-adapted for 10 days, subjected to a 30-minute photolesion using the filament light source of a fluorescent microscope (~100,000 lux), followed by continuous light exposure from four 200W halogen lights (~8,000 lux) for four days (Ranski et al., 2018). After 4 days of halogen light exposure, the fish were returned to normal light and dark conditions. The chronic low-light paradigm involved 24 hour dark-adaption, no 30 min high-intensity light exposure, and continuous exposure from only two 200W halogen lights (~4000 lux) for 21 days. For both light treatments, N=5–6 animals per time point were euthanized for tissue collection at the following time points: a non-light treated control group (0 hpL), 18 hours post-light onset (hpL), 48 hpL, 96 hpL, and 21 days post-light onset (dpL).
2.3. Immunohistochemistry
For tissue collection, whole eyes were removed from euthanized animals using forceps and placed in 9:1 ethanolic formaldehyde solution (100% ethanol: 37% formaldehyde) overnight at 4°C. The following day, eyes were cryoprotected with washes in 5% sucrose/1X Phosphate Buffered Saline (PBS) at room temperature (RT) for 1–2 hours followed by an overnight incubation at 4°C in 30% sucrose/1XPBS. Finally, eyes were washed in a 1:1 solution of 30% sucrose/1XPBS:Tissue Freezing Medium (TFM; General Data Healthcare cat# TFM-Color) overnight at 4°C, embedded in 100% TFM, and stored at −80°C. Blocks of embedded eyes were cryosectioned to a thickness of 16µm, sections were transferred to a glass microscope slide, dried on a slide warmer for 1–2 hours at 55°C, and stored at −80°C.
On the day of immunohistochemistry, selected slides were re-warmed to 55°C for 20 min and the tissue was outlined with a hydrophobic PapPen barrier. Next, the tissue was rehydrated in 1XPBS for 20 min, and then blocked for 1 hour at RT in a solution of 2% normal goat serum, 0.2% Triton X-100, 1% dimethyl sulfoxide (DMSO) in 1XPBS. Following blocking, primary antibodies were diluted in blocking solution and incubated on the tissue overnight at RT. The primary antibodies used in this experiment were as follows: Zpr-3 (Zebrafish International Resource Center, 1:200), Zpr-1 (Zebrafish International Resource Center, 1:200), 4c4 (courtesy of Peter Hitchcock laboratory, 1:250), mouse anti-Glial fibrillary acidic protein (Gfap; Chemicon MAB360, 1:500), and mouse-anti-Proliferating Cell Nuclear Antigen (PCNA; Sigma Aldrich clone PC10, 1:1,000). The following day, the tissue was washed at room temperature with 1xPBT (1XPBS, 0.05% Tween-20), incubated 1 hr at RT with secondary antisera (goat-antimouse AlexaFluor488; Invitrogen; 1:500) and TO-PRO-3 (Invitrogen; 1:750) in PBT, washed with PBT, and mounted using Prolong Gold Antifade Reagent (Cell Signaling Technology).
2.4. Terminal Deoxynucleotidyl Transferase dUTP Nick End Labeling (TUNEL) Analysis
TUNEL was performed as previously described using tissue sections processed for standard immunohistochemistry (Ranski et al., 2018). Briefly, tissue sections were washed in 1xPBS for 20 minutes, permeabilized with ice-cold buffer of 0.1% NaCitrate/0.1% Triton X-100/1XPBS for 2 minutes, and washed in 1xPBS for 5 minutes at RT. Next, sections were incubated in 100µL labeling buffer (ApoAlert DNA fragmentation kit; Clontech International) for 10 minutes at room temperature, followed by humidified incubation with 50µL of labeling mix (48µL labeling buffer, 1µL of 1mM biotinylated dNTPs (New England Biolabs) and 1µL of TdT enzyme (45U/µL; ApoAlert DNA fragmentation kit; Clontech International), at 37°C, 1–2 hours. The reaction was stopped with a 15 min RT wash with 150 µL of 2xSSC. Tissue sections were washed in 1xPBS and then incubated with StrepTavidin conjugated to AlexaFluor 488 (Invitrogen 1:200) and TO-PRO-3 (1:750) diluted 1XPBT for 60 minutes in the dark, washed with PBT, and mounted with Prolong Gold Antifade Reagent (Cell Signaling Technology).
2.5. Confocal Microscopy and Quantification of Immunohistochemical Pixel Density
Immunohistochemistry and confocal imaging for each antisera was performed on tissue sections collected at each timepoint for a given treatment paradigm (acute or chronic) to maintain internal consistency with staining and imaging (i.e. in a single experiment, GFAP immunohistochemistry was performed on all 5 timepoints in the acute light treatment and then confocal images from all 5 timepoints were collected in a single imaging session). For all samples in a given experiment, single-plane confocal images were acquired from the central dorsal retina using identical confocal settings on a Leica TCS SP8 confocal microscope. Next, unaltered confocal images were processed in ImageJ to isolate the green channel containing the fluorescence of the target protein of interest, followed by calculation of the raw pixel density values within the linear distance of the central dorsal retina (~300 microns; identical regions of interest [ROIs] were used for each measurement within a group). Significant differences between each time point of a given antisera/treatment paradigm was determined by a one-way ANOVA followed by a post-hoc Tukey test (p<0.05 was used as a significant cut-off). Finally, pixel intensities were converted into percent change using the 0 hpL control value as a 100% baseline and graphed using Microsoft Excel. Subsequent hand counts were performed on the same raw images for PCNA and 4c4 immunolocalization in order to quantify the number of immunolabeled cells at each timepoint within the ~300 micron linear distance of the central dorsal retina. Significant differences between each time point of a given antisera/treatment paradigm was determined by a one-way ANOVA followed by a post-hoc Tukey test (p<0.05 was used as a significant cut-off) and graphed using Microsoft Excel.
2.6. Histopathology
Eyes from euthanized fish were fixed with 2.5 % glutaraldehyde (Ted Pella, Inc.) in a 1:1:1 mixture of 0.1M phosphate buffer (pH 7.4), 0.2M phosphate buffer and water overnight at 4°C. Samples were subsequently transferred to a fixative containing 3% osmium tetroxide (Electron Microscopy Sciences, Hatfield, PA), 2.5% glutaraldehyde and 0.2M phosphate buffer in a 1:1:1 ratio on ice for 2 x 1.5 hrs. Samples were rinsed with 0.1M phosphate buffer 2 × 15 min, and then dehydrated in increasing gradients of ethanol followed by propylene oxide (Sigma-Aldrich). Samples were infiltrated and embedded in Epon-Araldite (Electron Microscopy Sciences). Sections (1.0 µm) were cut on a Leica EM UC6 microtome (LeicaMicrosystems, Inc. Buffalo Grove, IL) and stained with a 2:1 mixture of 1% Toluidine Blue (Electron Microscopy Sciences) and 1% Pyronin B (Hopkins & Williams Chadwell Health, Essex England). Toluidine Blue is a basic thaizine metachromatic dye with a high affinity for DNA and RNA and stains the retinal nuclei dark blue and the cytoplasm a light blue, and Pyronin B is a basic xanthene dye that stains the cytoplasmic and RNA components of the retina a pink-red (Sullivan-Brown et al., 2011). Sections were imaged with a Leica DM4000B microscope (LeicaMicrosystems, Inc.) and images adjusted for brightness/contrast in Adobe Photoshop using identical levels of adjustment for each image.
3. RESULTS
3.1. Characterization of the chronic low-light paradigm
All of the current literature describing phototoxic light lesions to the adult zebrafish utilize acute, high intensity light exposure aimed at rapidly destroying photoreceptors. Here, we utilized an established acute model that involved 10 days of dark adaptation followed by two tiers of acute light exposure: 30 min of exposure to 100,000 lux light followed by 4 days of exposure to 8,000 lux halogen lamps (Fig. 1A). Although acute phototoxic exposure can vary slightly from model to model, the pathological result of each model follows a highly-characterized, robust, and step-wise regenerative response within the first 96 hours of light exposure: 1) photoreceptor apoptosis, 2) acute M ller glia MG reactivity and cell-cycle re-entry, and 3) progenitor cell amplification. Therefore, we first sought to test whether the chronic low-light paradigm resulted in a similar “classic” response. To accomplish this, we compared the established acute model to a new chronic damage model that only involved 1 day of dark adaptation followed by continual low-light exposure of 4,000 lux for 21 days (Fig. 1B–C).
Figure 1. Graphic comparison of the acute and chronic light models.
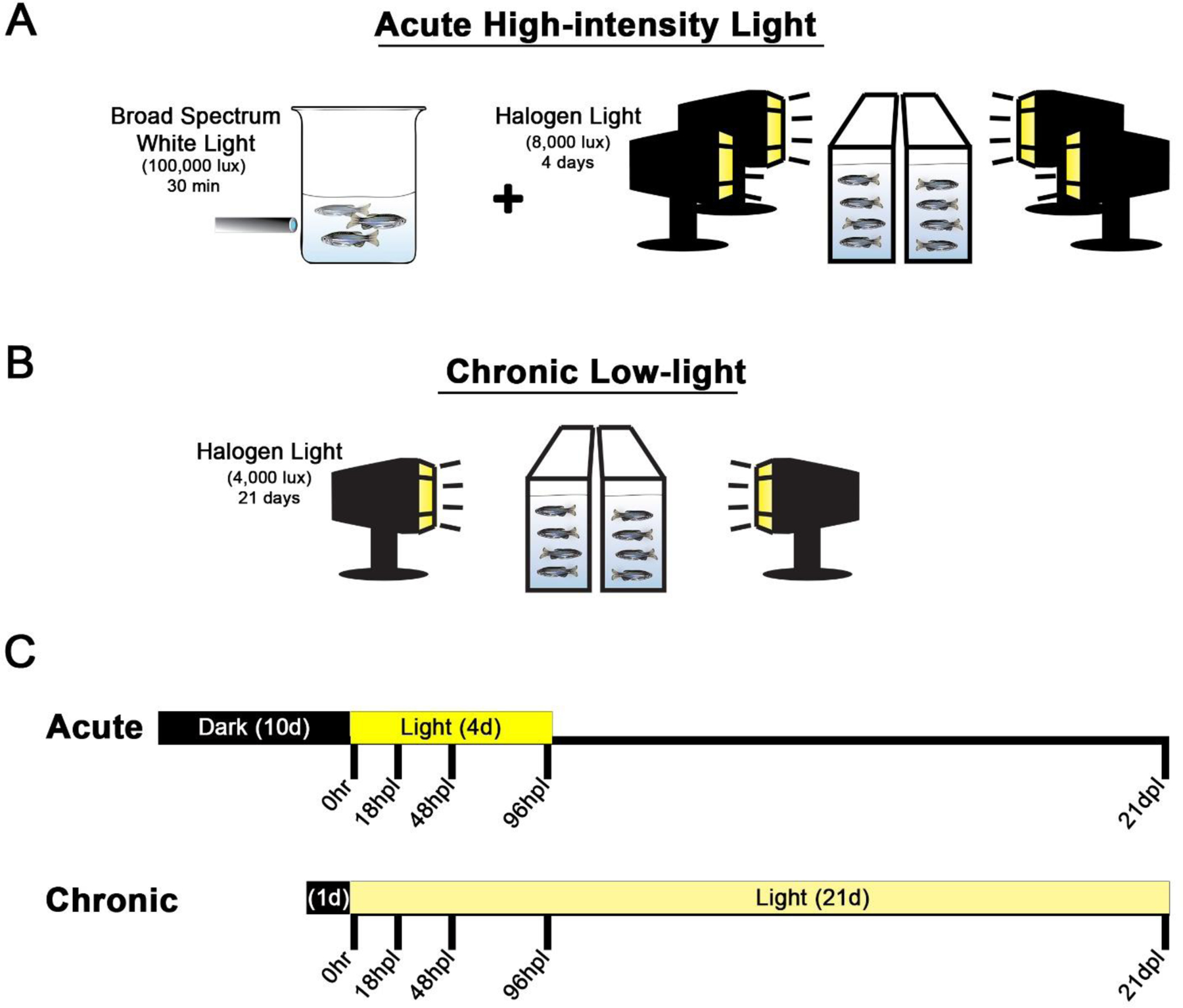
In the acute model (A), adult albino fish are dark-adapted for 10 days, subjected to a 30-minute photolesion using the filament light source of a fluorescent microscope (~100,000 lux), followed by continuous light exposure from four 250W halogen lights (~8,000 lux) for four days (Ranski et al., 2018). After 4 days of halogen light exposure, the fish were returned to normal light and dark conditions. The chronic low-light paradigm (B) involved 1 day dark-adaption, no 30 min high-intensity light exposure, and continuous exposure from only two 250W halogen lights (~4000 lux) for 21 days. For both models (C), tissue collection occurred on the same 5 time points post-light onset (0 hr, 18 hpL, 48 hpL, 96 hpL, and 21 dpL).
3.2. The chronic low-light paradigm resulted in no detectable apoptosis via TUNEL analysis
In acute high-intensity phototoxic lesion models, TUNEL analysis has shown that the peak time of photoreceptor apoptosis is 18–24 hours post light onset (Thummel et al., 2008a). Here, we tested whether the chronic low-light paradigm elicited a similar cell death response. Specifically, we performed TUNEL analysis on retinal sections from both the acute high-intensity and chronic low-intensity paradigms at the following timepoints: 0 hours post light onset (hpL), 18 hpL, 48 hpL, 96hpL and 21 days post light onset (dpL). Consistent with previous findings, in the acute group, we first observed large numbers of TUNEL-positive apoptotic cells in the outer retina at 18 hpL (Fig. 2B). A few TUNEL-positive cells were still present in these retinas at 48 hpL (Fig. 2C), but returned to baseline control levels by 96 hpL and remained TUNEL-negative through 21dpL (Fig. 2D–E). In contrast, we observed no evidence of cell death at any time point in the chronic group (Fig. 2H–K). This is consistent with the observation that the ONL nuclei remained intact throughout the time course of chronic low-light exposure (Fig. 2H–K). Together, these data indicate that the acute high-intensity phototoxic lesion model resulted in a significant increase in apoptosis soon after light onset, but that the chronic low-intensity light treatment did not result in significant levels of apoptosis throughout the 21-day time course (Fig. 2F, L).
Figure 2. Cell death analysis during acute and chronic light treatments.
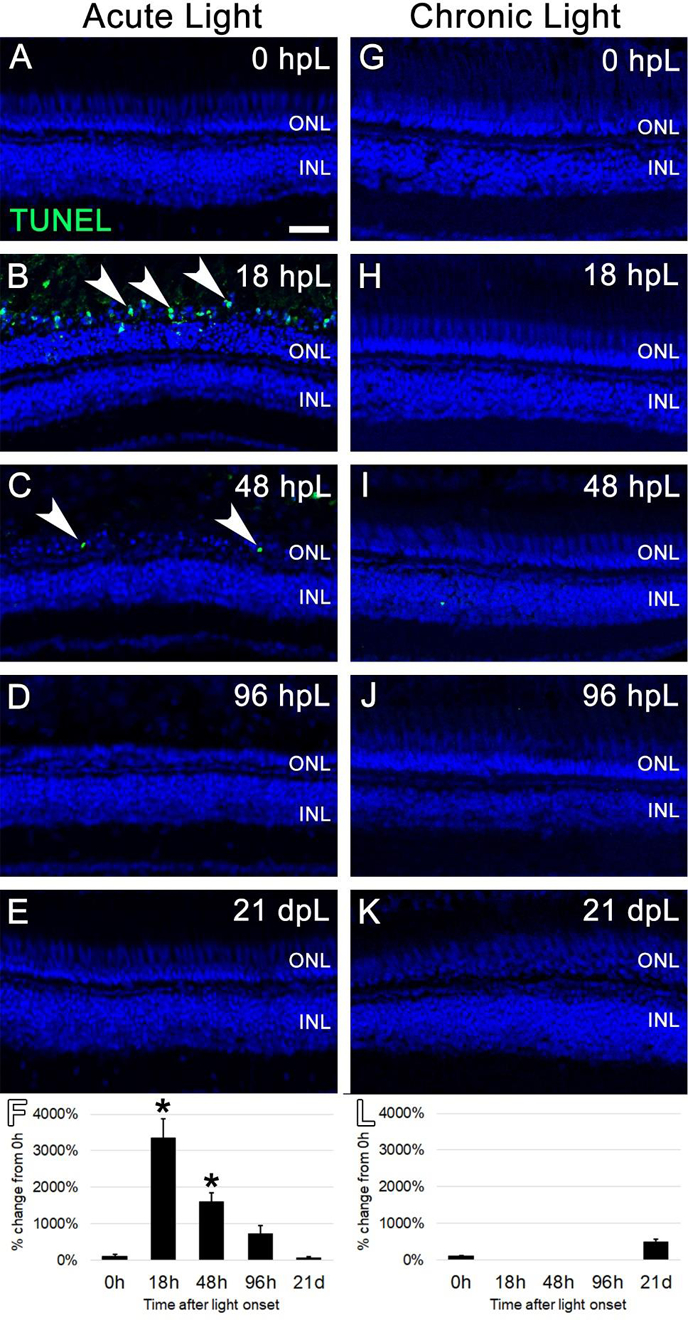
In the acute group (A-E), TUNEL-positive apoptotic cells (green; arrowheads) were observed in the outer retina at 18 hpL (B) and 48 hpL (C), at the initial stages of light damage. Graphic representation of the percent-change in TUNEL immunostaining from 0 hpL at each time point (F). In the chronic group (G-K), significant numbers of TUNEL-positive cells were not observed at any time point. Graphic representation of the percent-change in TUNEL immunostaining from 0 hpL at each time point (L). hpL: hours post light onset; ONL: outer nuclear layer; INL: inner nuclear layer; asterisk: significantly different from 0 hpL (p<0.05) as determined by post-hoc Tukey test from one-way ANOVA (N=5–6 retinas per time point). Scale bar=25 microns.
3.3. The chronic low-light paradigm did not elicit a robust gliotic or regenerative response from resident Müller glia
Next, we tested whether the chronic low-light paradigm resulted in MG reactive gliosis, cell-cycle re-entry, and subsequent progenitor cell amplification. In acute, high-intensity phototoxic lesion models, MG in proximity to the lesion undergo cellular hypertrophy and an upregulation of their intermediate filament protein Glial Acidic Fibrillary Protein (GFAP) (Thomas et al., 2016). This response is part of a larger phenotypic change on the part of MG, termed “reactive gliosis,” as an initial attempt to upregulate neuroprotective pathways in order to protect adjacent neurons from further damage (Qin et al., 2011; Thomas et al., 2016). In addition, in each of the acute damage models, the rapid loss of large numbers of retinal neurons elicits MG cell cycle re-entry followed by daughter cell proliferation, which produces large pools of amplifying progenitors to replace the lost neurons (Lenkowski and Raymond, 2014).
To determine whether the chronic low-light paradigm elicited a similar response, an anti-GFAP antibody was used to immunolabel MG at all aforementioned time points during the acute and chronic light exposures. In undamaged retinas (0 hpL), a near absence of GFAP expression was observed (Fig. 3A, G), consistent with previous reports in zebrafish (Thomas et al., 2016). In the acute group, an increase in GFAP immunolocalization was first observed at 18 hpL as puncta in MG appendages immediately adjacent to rod photoreceptor nuclei in the outer nuclear layer (ONL; Fig. 3B; arrows). This expression pattern persisted at 48 hpL (Fig. 3C; arrows). In addition, very weak GFAP localization was observed throughout the MG cell body, spanning from the ganglion cell layer (GCL) to the outer limiting membrane in the ONL (Fig. 3C). At 96 hpL, this expression pattern intensified, with strong expression in MG endfeet (Fig. 3D; asterisks) and within the inner nuclear layer (Fig. 3D; arrowheads). At 21 dpL in the acute group, GFAP immunolocalization was again nearly completely absent, and returned to a level and morphological state more comparable to the baseline (Fig. 3E). Quantification of the mean fluorescence intensity of GFAP expression at each time point did not pick up the subtle, morphological changes in GFAP expression at 18 hpL and 48 hpL, but did confirm a significant increase in expression at 96 hpL, followed by a decrease at 21 dpL to above undamaged baseline levels (Fig. 3F). In the chronic group, we observed a slight (but insignificant) increase in GFAP expression in MG cell endfeet at 18 hpL that persisted throughout the time course (Fig. 3H–L; asterisks). While this response may represent a type of gliosis, at no point did we observe the classic response of an increase in GFAP expression in the outer retina near the photoreceptor nuclei or a hypertrophy of Müller glial cell bodies in the inner retina (Fig. 3H–L).
Figure 3. Analysis of Müller glial response to photoreceptor degeneration/regeneration during acute and chronic light treatments.
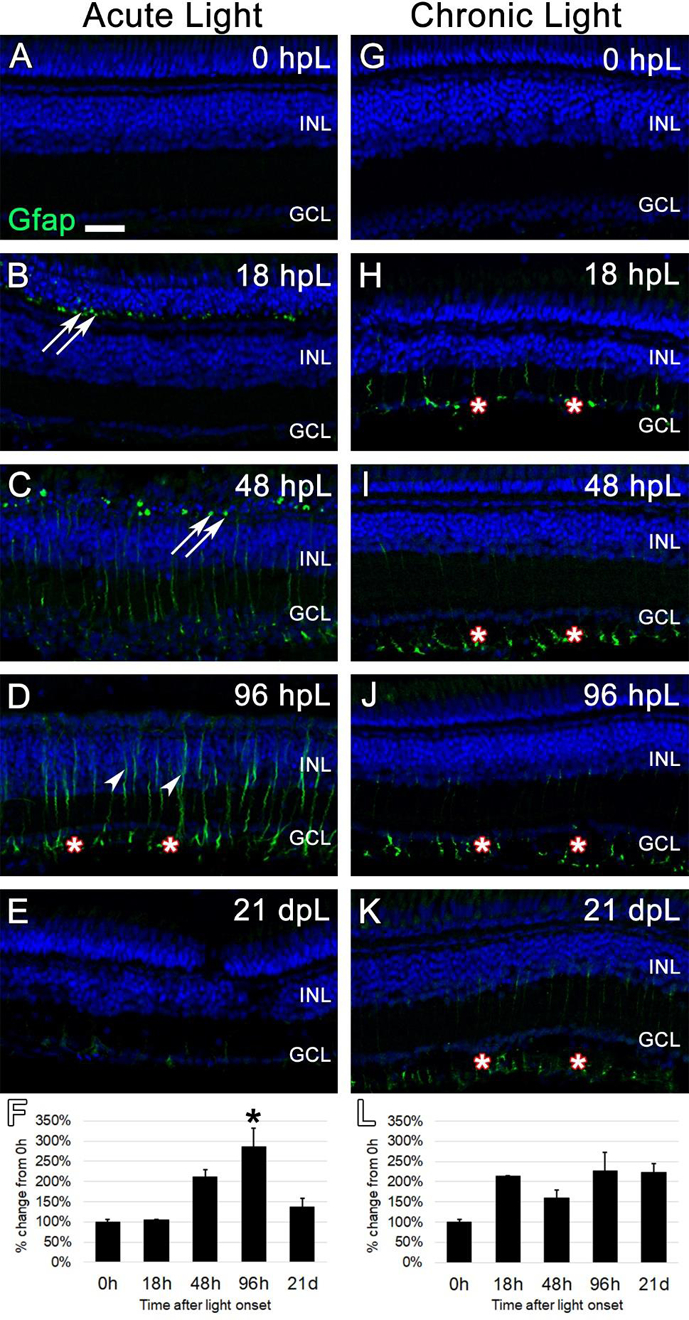
In undamaged retinas (A and G), Glial Fibrillary Acid Protein (Gfap) immunostaining was not observed, as Müller glia remain in a non-reactive state. In the acute group (A-E), Gfap immunolabeled (green) reactive Müller glial processes among dying photoreceptors in the outer nuclear layer, as indicated by Gfap-positive puncta (arrows). These puncta increased in number through 48 hpL (C), and by 96 hpL (D), outlines of entire hypertrophied Müller glial cells were observed (arrowheads), including strong labeling of Müller glial endfeet (asterisks). At 21 dpL (E), Gfap-positive Müller glia had returned to near baseline levels, although weak staining of Müller glia endfeet was still present in places. Graphic representation of the percent-change in Gfap immunostaining from 0 hpL at each time point (F). In the chronic group (G-K) at 18 hpL (H), Gfap-positive Müller glia endfeet were observed (asterisks). This expression pattern persisted at 48 hpL (I), 96 hpL (J), and 21 dpL (K); however, the overall intensity of staining was not significantly higher than 0 hpL retina (L). In addition, hypertrophied Müller glia cell bodies were never observed in the chronic group. Graphic representation of the percent-change in Gfap immunostaining from 0 hpL at each time point (L). hpL: hours post light onset; INL: inner nuclear layer; GCL: ganglion cell layer; asterisks in F and L: significantly different from 0 hpL (p<0.05) as determined by post-hoc Tukey test from one-way ANOVA (N=5–6 retinas per time point). Scale bar=25 microns.
Next, we utilized the G1/S-phase cell cycle marker, Proliferating Cell Nuclear Antigen (PCNA), to immunolabel dividing cells during both acute and chronic light treatments. In untreated (0 hpL) retinas, minimal evidence of proliferation was observed (Fig. 4A, G). An occasional PCNA-positive rod precursor was observed in the ONL (Fig. 4A; arrow), which is consistent with the continual neurogenesis of the adult zebrafish retina (Bernardos et al., 2007). In the acute group, this low level of PCNA expression was also present at 18 hpL (Fig 4B). At 48 hpL, numerous PCNA-positive cells were observed in the inner nuclear layer (Fig. 4C; arrowheads), which is consistent with multiple reports of asymmetrically-dividing MG at this time point (Gorsuch and Hyde, 2014; Kassen et al., 2007; Nagashima et al., 2013; Ranski et al., 2018; Thummel et al., 2008a). At 96 hpL, clusters of PCNA-positive progenitor cells were observed migrating from the INL to the ONL (Fig. 4D: arrowheads). At 21 dpL, PCNA expression returned to baseline levels, with a few residual PCNA positive cells in the INL and ONL (Fig. 4E, arrow and arrowhead, respectively). Quantification of the mean fluorescence intensity of PCNA expression at each time point confirmed a significant increase in expression at 96 hpL, followed by a decrease at 21 dpL to undamaged baseline levels (Fig. 4F). Hand counts of the number of PCNA+ nuclei in the INL and ONL at each time point confirmed these findings (Suppl. Fig. 1). In the chronic group, we never observed a robust proliferation response from MG or clusters of amplifying progenitors, but only an occasional PCNA-positive cell observed in the INL (Fig. 4K; arrowhead). Instead, we observed a slow build-up of individual PCNA-positive cells in the ONL from 18 hpL to 21 dpL (Fig. 4H–K). Based location and morphology, these cells were likely rod precursors, which are involved in the constant neurogenesis of rod photoreceptors, and are known to be highly sensitive to retinal damage (Bernardos et al., 2007; Stenkamp, 2011). Quantification of the mean fluorescence intensity of PCNA expression at each time point showed no significant increase in PCNA expression at any time point and was not sensitive enough to detect the gradual increase in PCNA-positive progenitors in the ONL (Fig. 4L). However, hand counts of the number of PCNA+ nuclei in the INL and ONL at each time point confirmed a significant increase in PCNA+ nuclei in the ONL at 21 dpL (Suppl. Fig. 1). Together, these data suggest that the chronic low-light treatment failed to elicit a robust proliferation response from MG, but did elicit a proliferative response from rod precursors in the ONL.
Figure 4. Analysis of Müller glia and progenitor cell proliferation during acute and chronic light treatments.
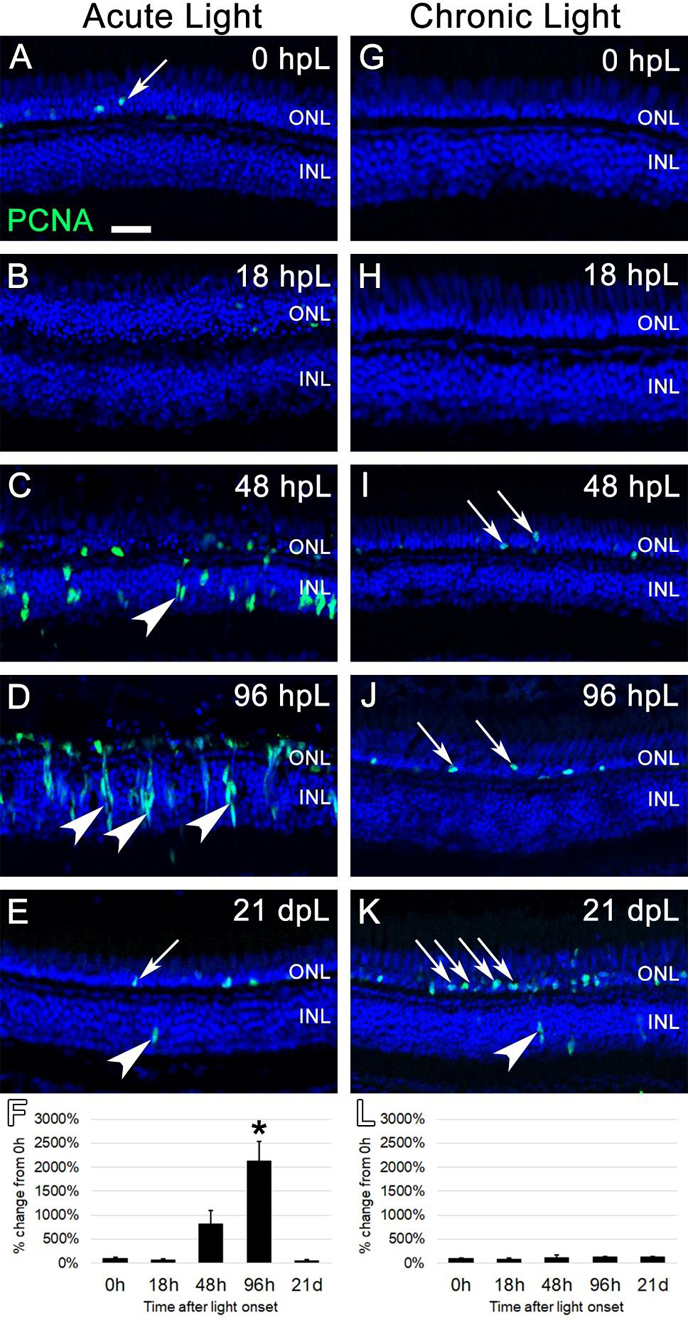
In undamaged retinas (A and G), Proliferating Cell Nuclear Antigen (PCNA) immunostaining (green) was only observed in an occasional rod precursor cell in the outer nuclear layer (A, arrow), contributing to the constant neurogenesis of rod photoreceptors. In the acute group (A-E), significant numbers of individual PCNA-positive cells were first observed at 48 hpL (C), representing Müller glia cells that have re-entered the cell cycle (arrowhead). By 96 hpL (D), larger columns of progenitor cells have formed (arrowheads), and the migration of PCNA-positive progenitor cells to the depleted outer nuclear layer was evident. At 21 dpL (E), PCNA expression had returned to near baseline levels, with the occasional observance of PCNA-positive rod precursors (arrow) and Müller glia (arrowhead). Graphic representation of the percent-change in PCNA immunostaining from 0 hpL at each time point (F). In the chronic group (G-K), large numbers of PCNA-positive Müller glia or progenitors were never observed. Qualitatively, there appeared to be a gradual increase in PCNA-positive rod precursor cells (arrows) starting at 48 hpL (I) and increasing at 96 hpL (J) and 21 dpL (K); however, this expression change did not reach significance based on the overall intensity of staining (L). Graphic representation of the percent-change in PCNA immunostaining from 0 hpL at each time point (L). hpL: hours post light onset; ONL: outer nuclear layer; INL: inner nuclear layer; GCL: ganglion cell layer; asterisks in F and L: significantly different from 0 hpL (p<0.05) as determined by post-hoc Tukey test from one-way ANOVA (N=5–6 retinas per time point). Scale bar=25 microns.
3.4. The chronic low-light paradigm resulted in gradual truncation of rod and cone photoreceptor outer segments
Because TUNEL analysis suggested that the chronic low-light paradigm did not result in photoreceptor apoptosis, we next asked if this light paradigm led to any morphological changes to the structure of the outer segments. For this, the Zpr3 monoclonal antibody was used to label rod photoreceptor outer segments (ROS) on retinal sections harvested at the aforementioned time points during both the acute and chronic light treatments. At 0 hpL, Zpr3 immunolocalization was clearly limited to the ROS (Fig 5A, G). In the acute group, at 18 hpL, Zpr3 expression remained in degenerating ROS; in addition, protein immunolocalization was observed perinuclear to the rod photoreceptor nuclei in the ONL (Fig. 5B). By 48 hpL, the perinuclear expression disappeared as ONL nuclei were destroyed; instead, Zpr3 expression remained in a debris field of ROS (Fig. 5C). At 96 hpL, the Zpr3-positive ROS debris was largely cleared (Fig. 5D). Finally, at 21 dpL, both ONL nuclei and rod outer segments had been restored (Fig. 5E). Quantification of the mean fluorescence intensity of Zpr3 expression at each time point confirmed a significant decrease at 96 hpL, followed by a restoration at 21 dpL (Fig. 5F). In the chronic group, no apparent differences in Zpr3 expression were observed between the 0 hpL and 18 hpL timepoints (Fig 5G–H). However, at 48 hpL, the ROS appeared slightly truncated and weak Zpr3 expression shifted to rod inner segments (RIS) and the perinuclear regions of the ONL (Fig. 5I). At 96 hpL, disorganized and truncated ROS were clearly visible, and now strong Zpr3 expression was observed in the RIS and ONL (Fig. 5J). At 21 dpL, the majority of ROS were visually truncated to the level of the RIS (Fig. 5K). Zpr3 immunolocalization was now largely restricted to the RIS and to the perinuclear space of the still-intact nuclei in the ONL (Fig. 5K). Despite these clear morphological changes to ROS, the mean fluorescence intensity of Zpr3 did not significantly change during the time course (Fig. 5L), again indicating that this method is not sensitive enough to detect morphological changes, only changes in overall fluorescence intensity.
Figure 5. Analysis of rod photoreceptor degeneration/regeneration during acute and chronic light treatments.
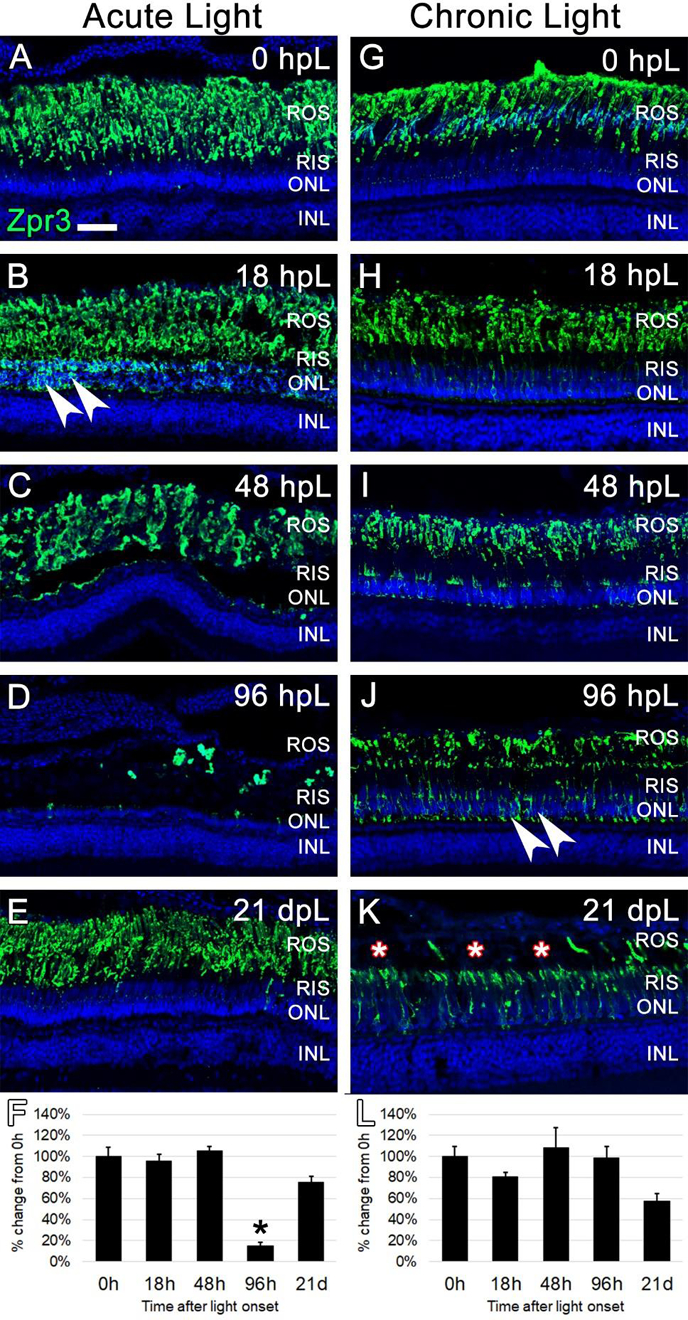
In undamaged retinas (A and G), Zpr3 immunocytochemistry (green) labeled rod photoreceptor outer segments (ROS). In the acute group (A-E), Zpr3 additionally localized to the rod inner segments (RIS) and the perinuclear space of rod photoreceptor nuclei in the ONL (arrowheads) at 18 hpL (B). At 48 hpL (C), ONL nuclei visually decreased in number and ROS were observed in a debris field in the outer retina. At 96 hpL (D), ROS were largely absent. At 21 dpL (E), Zpr3-positive ROS were restored. Graphic representation of the percent-change in Zpr3 immunostaining from 0 hpL at each time point (F). In the chronic group (G-K), Zpr3-positive ROS largely remained intact through 48 hpL (G-I). At 96 hpL (J), Zpr3 localized to the RIS and perinuclear space of rod photoreceptor nuclei in the ONL (arrowheads), and the beginning of outer segment disorganization and truncation was observed. At 21 dpL (K), ROS were visually truncated/absent (asterisks), and Zpr3 immunolocalization was restricted to the intact RIS and the perinuclear space of rod photoreceptor nuclei in the still-intact ONL. Graphic representation of the percent-change in Zpr3 immunostaining from 0 hpL at each time point (L). hpL: hours post light onset; ROS; rod outer segments; RIS: rod inner segments; ONL: outer nuclear layer; INL: inner nuclear layer; asterisks in F and L: significantly different from 0 hpL (p<0.05) as determined by post-hoc Tukey test from one-way ANOVA (N=5–6 retinas per time point). Scale bar=25 microns.
Next, we used the anti-Arr3a Zpr1 monoclonal antibody to label red/green double cones at the aforementioned time points in both acute high intensity and chronic low intensity light treatments. As with all of our immunohistochemical analysis utilizing the mean fluorescence intensity measurements, gain and laser power settings that avoided signal saturation were first established. In the case of Zpr1 immunolabeling, this meant that Zpr1 visualization in undamaged (0 hpL) retinas was predominantly restricted to the strongest area of mRNA and protein expression of Arr3a: the perinuclear/inner segment domain of red/green double cones (Fig. 6A, G) (Renninger et al., 2011; Song et al., 2020). In the acute group at 18 hpL, Zpr1 expression shifted so that it was visualized throughout the cone cell body, including in hypertrophied and degenerating cone outer segments (Fig. 6B, asterisks). Following degeneration and clearing, Zpr1 immunolabeling was not detected at either 48 hpL or 96 hpL (Fig. 6C–D). At 21 dpL, the regenerated cone photoreceptors exhibited Zpr1 immunolocalization in the perinuclear/inner segment domain of red/green double cones, with very weak expression in outer segments (Fig. 6E). Quantification of the mean fluorescence intensity of Zpr1 expression at each time point confirmed a significant decrease in expression at 48 hpL and 96 hpL, followed by a restoration at 21 dpL (Fig. 6F). In the chronic group at 18hpL, Zpr1 expression was visibly weakened in the perinuclear domain, but intensified in the cone outer segments, allowing for a clear visualization of their cone-shaped morphology (Fig. 6H). This pattern of Zpr1 immunostaining persisted through 48 hpL (Fig. 6I). At 96 hpL, Zpr1 expression again changed its location, with new Zpr1 immunolocalization to the cone cell pedicles (Fig. 6J, arrowheads). In addition, Zpr1 immunolocalization was visualized in increasingly-truncated outer segments with intense staining in the perinuclear domain (Fig. 6J). At 21 dpL, Zpr1 was detected in what appeared to be nearly-completely truncated cone outer segments (Fig. 6K, asterisks), with otherwise strong expression throughout the remaining cell body (Fig. 6K). Quantification of the mean fluorescence intensity of Zpr1 expression at each time point was not as revealing as the morphological changes in expression pattern, but confirmed a significant decrease in expression at 18 hpL and 48 hpL, as Zpr1 expression moved to the outer segments (Fig. 6L). Together, these data suggest that the chronic low intensity light paradigm resulted in dynamic changes to Arr3a immunolocalization, and ultimately resulted in the truncation of the red/green double-cone photoreceptor outer segments.
Figure 6. Analysis of cone photoreceptor degeneration/regeneration during acute and chronic light treatments.
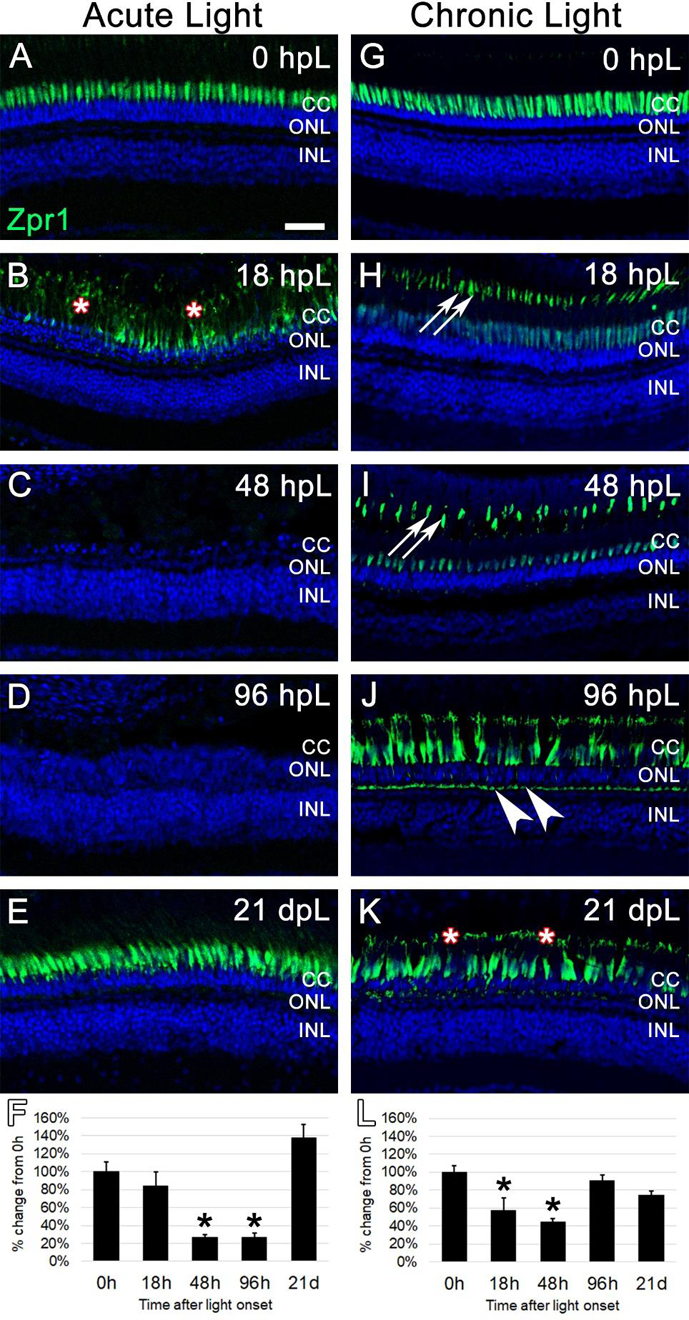
In undamaged retinas, Zpr1 immunostaining was observed in the perinuclear domain of red/green double cones (A and G). In the acute group (A-E), Zpr1 immunolabeling was observed throughout the cytoplasm of hypertrophied and degenerating cones at 18 hpL (B, asterisks). Nearly all double cone photoreceptors were degenerated and Zpr1 immunolabeling was not detected at 48 hpL and 96 hpL (C, D). At 21 dpL, newly regenerated red/green double cones were observed by the return of Zpr1 immunostaining. Graphic representation of the percent-change in Zpr1 immunostaining from 0 hpL at each time point (F). In the chronic group (G-H), hypertrophy of cone photoreceptors was observed at 18 dpL (H), concomitant with a down-regulation of Zpr1 immunostaining in the perinuclear domain and an upregulation of immunostaining of the cone outer segments (H, arrowheads). This expression pattern persisted through 48 hpL (I). At 96 hpL, cone hypertrophy was readily apparent, with Zpr1 immunolabeling throughout the cone cytoplasm, including the cone pedicles (arrowheads). At 21 dpL, cone photoreceptor hypertrophy persisted, and Zpr1 immunolabeling was suggestive of cone outer segment truncation/degeneration (K, asterisks; compare outer segments in H and I to K). Graphic representation of the percent-change in Zpr1 immunostaining from 0 hpL at each time point (L). hpL: hours post light onset; CC; cone cell nuclei; ONL: outer nuclear layer; INL: inner nuclear layer; asterisks in F and L: significantly different from 0 hpL (p<0.05) as determined by post-hoc Tukey test from one-way ANOVA (N=5–6 retinas per time point). Scale bar=25 microns.
3.5. The chronic low-light paradigm elicited a slow build-up of 4c4+ microglia/macrophages to the location of degenerating rod photoreceptor outer segments
The debris-clearing role of 4c4-positive microglia in the regeneration process has previously been characterized in the acute, high-intensity light damage model (Ranski et al., 2018). In that model, the number of 4c4-positive cells in the retina rapidly increases with the destruction of rod and cone photoreceptors, followed by a clearing of the field of photoreceptor debris in the outer retina (Ranski et al., 2018). Given that the chronic low-light paradigm resulted in relatively less damage to the photoreceptors, we next characterized the microglial response in this paradigm. The 4c4 antibody, an established marker of microglia in zebrafish (White et al., 2017), was used to label microglia in the retina at each time point in both the acute and chronic light paradigms. Prior to light treatment (0 hpL), resident 4c4+ cells were observed in very low numbers and exhibited the ramified morphology of resting, inactivated microglia (Fig. 7A, G). In the acute group, 4c4+ cells with activated, amoeboid morphology were observed at 18 hpL and 48 hpL and were primarily restricted to the distal tips of the ROS (Fig. 7B, C; arrowheads). At 96 hpL, the entire outer retina was filled with amoeboid-shaped 4c4+ cells (Fig. 7D). At 21 dpL, the numbers of 4c4+ cells were significantly reduced, but remained above baseline levels (Fig. 7E). Quantification of the mean fluorescence intensity of 4c4 expression at each time point confirmed a significant increase in expression at 96 hpL, followed by a decrease at 21 dpL (Fig. 7F). Hand counts of the number of 4c4+ nuclei in the INL and ONL confirmed these finding (Suppl. Fig. 2). In the chronic group, 4c4+ cells with activated, amoeboid morphology were observed at 18 hpL and 48 hpL; similar to the acute group, these cells were primarily restricted to the distal tips of the ROS (Fig. 7H, I). At 96 hpL, an accumulation of 4c4+ cells with amoeboid morphology were observed in the outermost retina (Fig. 7J), but not nearly to the extent as the acute light-treated retinas at this same time point (Chronic: 22.75 +/− 2.2; Acute: 72.6 +/− 2.7). At 21 dpL, this expression profile persisted (Fig. 7K). Quantification of the mean fluorescence intensity of 4c4 expression at each time point confirmed a significant increase in expression from 48 hpL through 21 dpL, but never to the extent as the 96 hpL peak in the acute group (Fig. 7L). Hand counts of the number of 4c4+ nuclei in the INL and ONL confirmed these findings (Suppl. Fig. 2). Thus, the chronic light damage model resulted in a gradual and persistent build-up in the number of 4c4+ microglia that were restricted to the distal tips of the ROS.
Figure 7. Analysis of microglial response to photoreceptor degeneration/regeneration during acute and chronic light treatments.
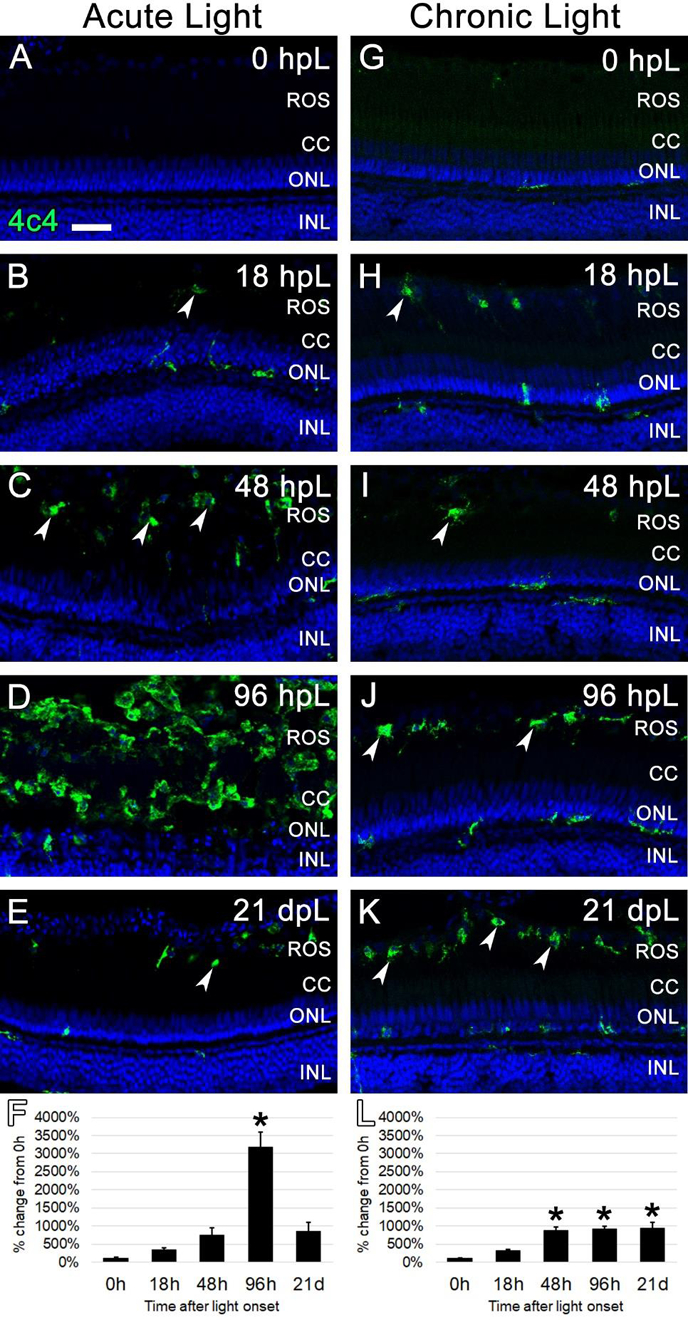
In undamaged retinas (A and G), 4c4 immunocytochemistry (green) labeled occasional quiescent microglia with ramified morphology. In the acute group (A-E), 4c4 immunolabeled occasional activated microglia with amoeboid morphology in the ROS starting at 18 hpL (B, arrowheads). These activated microglia increased in number through 48 hpL (C). At 96 hpL (D), a large field of amoeboid microglia were present in an area that corresponded to rhodopsin photoreceptor debris. At 21 dpL (E), 4c4+ microglia had returned to near baseline levels, although a few remained at the interface of the rod outer segments and the retinal pigmented epithelium (arrowhead). Graphic representation of the percent-change in 4c4 immunostaining from 0 hpL at each time point (F). In the chronic group (G-K), 4c4+ microglia with amoeboid morphology were observed in the area of ROS starting at 18 hpL (H, arrowheads). These increased in number by 48 hpL (I) and then were maintained at the ROS/RPE interface through 96 hpL (J, arrowheads) and 21 dpL (K, arrowheads). Graphic representation of the percent-change in 4c4 immunostaining from 0 hpL at each time point (L). hpL: hours post light onset; ROS; rod outer segments; CC; cone cell nuclei; ONL: outer nuclear layer; INL: inner nuclear layer; asterisks in F and L: significantly different from 0 hpL (p<0.05) as determined by post-hoc Tukey test from one-way ANOVA (N=5–6 retinas per time point). Scale bar=25 microns.
These data suggested that the chronic low-light paradigm induced a gradual truncation of the outer segments of rod and cone photoreceptors, followed by a build-up of 4c4+ microglia to the distal tips of the ROS by 96 hpL. In order to confirm the structural changes to the photoreceptors and test whether the microglia were actively phagocytosing debris, we performed traditional thin-section histology at 0 hpL and 96 hpL in both acute and chronic light treatment paradigms (Fig. 8). In untreated (0 hpL) retinas from both treatment groups, darkly-stained rod photoreceptor nuclei were clearly visible in the ONL and the characteristic discs of rhodopsin were visualized in the ROS (Fig. 8A, B, E, F). In the acute group at 96 hpL (Fig. 8C, D), the multiple rows of darkly-stained ONL nuclei were replaced by a new, single layer of lightly-stained progenitor cell nuclei (Fig. 8C). Posterior to these nuclei was a field of ROS debris (Fig. 8D, asterisk) and amoeboid-shaped cells with characteristic microglial morphology and associated small vacuoles/phagosomes (Fig. 8D, arrowheads). In the chronic group at 96 hpL, nuclei of the rod and cone photoreceptors were still present (Fig. 8G), but both the rod and cone outer segments were nearly absent (Fig. 8G–H). Instead, in the ROS/RPE interface, a row of amoeboid-shaped cells with characteristic microglial morphology were observed with very large vacuoles/phagosomes (Fig. 8H, arrowheads). This finding is consistent with the 4c4+ microglia observed in this location at this time point (Fig. 7J). Of note, these cells contained much larger vacuoles/phagosomes than the amoeboid cells observed in the acute light treatment (compare Fig 8C–D to G–H).
Figure 8. Histological sections of photoreceptor degeneration and microglial localization during acute and chronic light treatments.
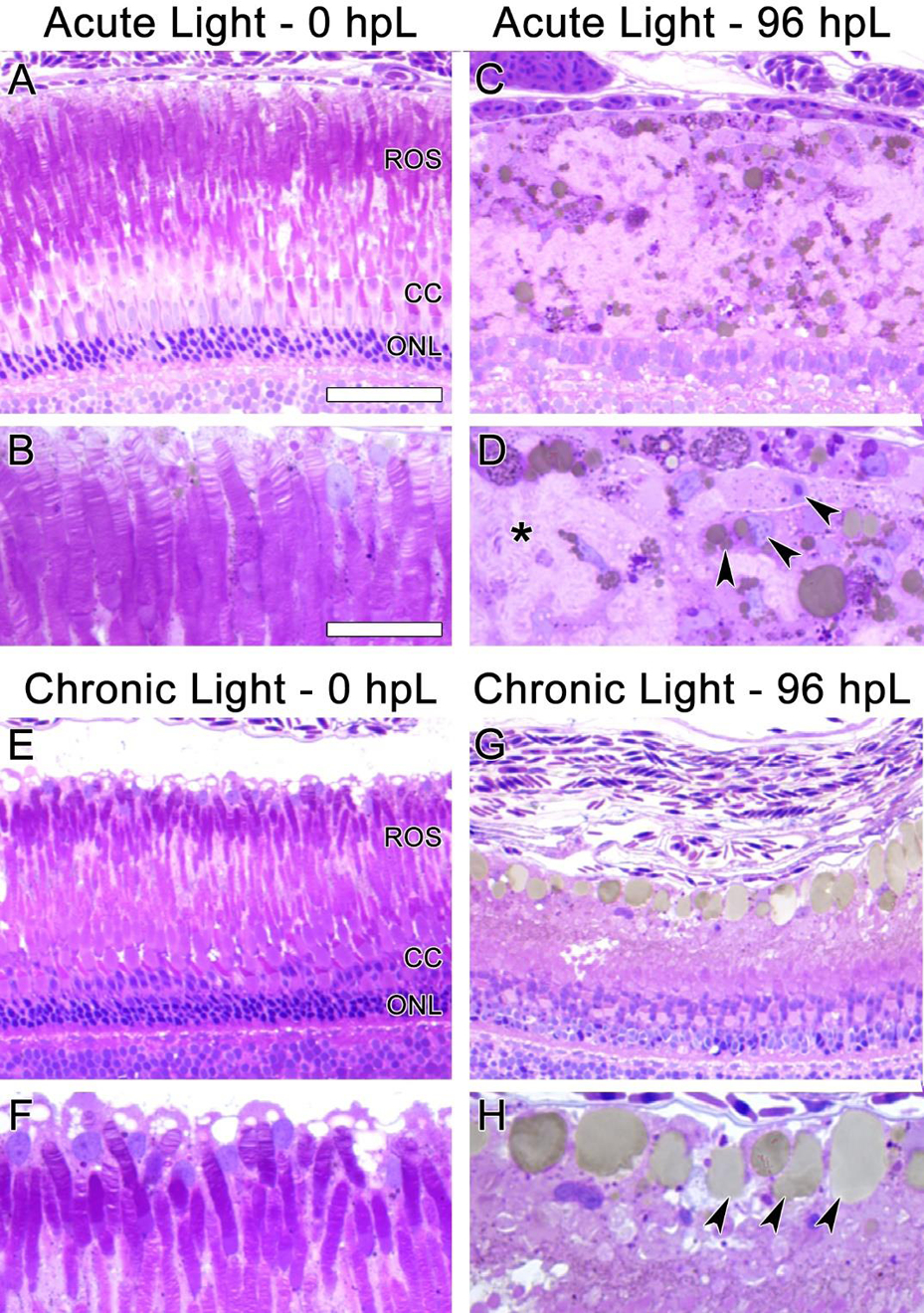
In undamaged retinas (A, B, E, and F) the disc-like structures of ROS were clearly visible. In the acute group at 96 hpL (C, D), rod and cone photoreceptors were completely absent. The resultant cellular debris field (D, asterisk) contained many cells with amoeboid morphology containing small vacuoles/phagosomes (D, arrowheads), likely microglia. In the chronic group at 96 hpL (G, H), ROS and cone outer segments appeared to be absent, but both rod and cone photoreceptor nuclei were still present (G). Near the RPE/ROS interface, cells with amoeboid morphology containing large vacuoles/phagosomes were present (H, arrowheads). Based on location and morphology, these were also likely microglia.
Together, these data provide strong evidence that the chronic low-light paradigm resulted in a gradual truncation of rod and cone photoreceptor outer segments accompanied by the accumulation of microglia containing large vacuoles/phagosomes. However, these morphological changes did not reach the threshold to cause photoreceptor apoptosis, and thus did not elicit the classic characteristics of an acute MG-mediated regeneration response. To our knowledge, this is the first report of the use of light exposure to cause chronic and widespread photoreceptor degeneration without cell death in zebrafish retina.
4. DISCUSSION
In the two decades following the discovery that adult zebrafish have the capacity to regenerate their retinas, the vast majority of the studies have utilized acute retinal damage models. These include needle punctures through the entire eye, intense light exposures, and intraocular injection of neurotoxins such as Ouabain or NMDA (Fausett and Goldman, 2006; Fimbel et al., 2007; Lahne et al., 2020; Luo et al., 2019; McGinn et al., 2019; Sherpa et al., 2008; Thomas et al., 2016). In all cases, these models result in rapid retinal cell death that triggers adjacent MG to respond. In at least two of these models - intense light damage and Ouabain injections - MG initially respond with characteristics of classic reactive gliosis, including cellular hypertrophy, an upregulation of GFAP, and a release of neurotrophic factors (Thomas et al., 2016). This is followed by MG asymmetric cell cycle re-entry, whereby a single daughter cell continues to divide symmetrically to produce pools of retinal progenitors (Nagashima et al., 2013). In all of these acute models, the severe amount of initial retinal damage is so rapid that MG re-enter the cell cycle between 1–5 days post onset of damage (Fimbel et al., 2007; Lahne et al., 2020; Thummel et al., 2008a). Finally, depending on the location of the initial retinal damage (i.e. outer retina, inner retina, or all laminar layers), these progenitors have the capacity to migrate to the area of damage and replace the lost neurons (Lenkowski and Raymond, 2014).
In contrast to the large number of studies that utilize acute models of retinal damage, only a handful of studies have described models with a chronic loss of neurons. Morris et al., described two mutant/transgenic lines in zebrafish that resulted in photoreceptor loss early in retinal development (Morris et al., 2008; Morris et al., 2005). Interestingly, the line that resulted in loss of cone photoreceptors elicited a regenerative response from MG, but the line that resulted in loss of rod photoreceptors did not (Morris et al., 2008; Morris et al., 2005). In contrast, Iribarne et al., described a different zebrafish developmental mutant that resulted in rod and cone degeneration by 3 weeks in development that did not elicit a regenerative response (Iribarne et al., 2019). Although these conflicting findings are worth further investigation, interpretation of the phenotypes in each of these mutant models is likely complicated by their very early onset in retinal development. In adult zebrafish, Stenkamp et al., showed that aged fish with a single copy of Shh (syu +/−) exhibited age-related cone photoreceptor loss and abnormalities (Stenkamp et al., 2008). More recently, work from the Perkins lab has described chronic cone photoreceptor degeneration in a zebrafish model of Bardet-Beidl syndrome (BBS) using the zebrafish bbs2(−/−) line (Song et al., 2020). In this model, they describe a robust response from resident microglia that is associated with cone cell degeneration. However, in neither of these chronic models of adult cone cell degeneration did MG re-enter the cell cycle; they only did so if these animals were further subjected to acute light damage (Song et al., 2020; Stenkamp et al., 2008). These chronic models support a relatively recent hypothesis in the retinal regeneration field; namely, that a combination of signals from dying retinal neurons must reach a certain “threshold” in order to elicit a regenerative response from MG.
Each of the previously-described chronic models of adult photoreceptor loss or damage involve analysis at 7–24 months of age. Here, we describe a model of photoreceptor damage using low levels of chronic light exposure within 21 days of light onset. We show that this model does not result in photoreceptor apoptosis, but does cause a significant truncation of photoreceptor outer segments (Figs. 2, 5–6, 8). In addition, we observed changes to Zpr1 and Zpr3 immunolocalization within the damaged photoreceptors. Within rod photoreceptors, Zpr3 immunolocalization changed from degenerating ROS to RIS (Fig. 5I, J). This finding is consistent with Rhodopsin mislocalization to the RIS and ONL in mouse models of light-induced photoreceptor degeneration (Kong et al., 2006; Li et al., 2020). In cone photoreceptors, Zpr1 immunolocalization changed from cone inner segments to degenerating outer segments as well as the innermost cone pedicles (Fig. 6H–J). The immunolocalization of Zpr1 to the outer segment likely represents a light-dependent subcellular translocation of Arrestin 3 to the cone outer segment, as is well-established in light-saturated mammalian retinas (Haire et al., 2006; Zhang et al., 2003; Zhu et al., 2002). In addition, Arrestin 3 immunolocalization to the cone pedicle is well-established in both adult zebrafish and mouse retinas (Renninger et al., 2011; Zhu et al., 2002).
Using the ImageJ-based quantification method to measure immunolabeled pixel density did not always result in significant changes that were revealed using traditional hand-counting measurements. For example, pixel density measurements did not show an increase in PCNA+ nuclei in the ONL at 21 dpL in the chronic group (Fig. 4K), but subsequent hand counting did reveal a significant increase (Suppl. Fig. 1B). In addition, the morphological changes we observed in the photoreceptors did not always result in significant changes to immunolabeled pixel density. However, given the NIH’s recent emphasis on rigor and reproducibility, we sought to implement the pixel density method as an unbiased method for quantification. This required that all of our images be captured at the same confocal settings throughout the timecourse. In addition, since ImageJ was used to detect pixel density, we were cautious to use gain and laser settings that did not oversaturate any of the confocal images. Interestingly, this rigid approach to imaging allowed us to observe the subtle changes to Zpr1 and Zpr3 immunolocalization within damaged photoreceptors, even if ImageJ did not reflect these changes. Therefore, although not perfect, going forward, we feel that this approach could be used to complement the hand-counting method typically employed by the field.
Besides differences in photoreceptor responses, we also observed distinct differences in the MG response between the acute and chronic light treatments. In the acute light treatment, we observed an initial gliotic response as indicated by an upregulation/localization of GFAP to the outer retina (Fig. 3B). This preceded MG cell cycle re-entry, but persisted as MG re-entered the cell cycle (Figs. 3C, 4C). This is consistent with previous reports that zebrafish MG re-enter the cell cycle amidst a gliotic environment Thomas et al., 2016. We further observed a “delayed” gliotic response as indicated by an upregulation/localization of GFAP throughout the MG cytoplasm starting at 48 hpL and persisting through 96 hpL (Fig. 3C–D). This later gliotic response, which occurred at the peak of progenitor amplification, has not been previously reported. By 21 dpL, both gliosis and proliferation had returned to near baseline levels (Fig. 3E). In the chronic group, we never observed an upregulation of GFAP in the outer retina (Fig. 2G–K). Instead, starting at 18 hpL, very weak but discernible GFAP expression was observed in MG endfeet (Fig. 3H). This expression profile persisted throughout the timecourse at ~2-fold higher than baseline levels, but not enough to achieve significance with the ImageJ method of quantification (Fig. 3L). MG proliferation was also not observed throughout the chronic light treatment timecourse (Fig. 4G–K). Instead, a slow build-up of PCNA+ cells in the ONL was observed (Fig. 4I–K). Based on location and morphology, these were likely rod precursors, which are known to be highly sensitive to retinal damage (Stenkamp, 2011).
The immune response to photoreceptor degeneration has become a recent focus of interest in the zebrafish community following the demonstration of a role for microglia in adult retinal regeneration (White et al., 2017). Since this report, multiple groups have reported that retinal microglia respond to retinal cell death by migrating to the area of damage, proliferating in number, and clearing debris from dying cells (Ranski et al., 2018). More recent work used “omics” approaches to reveal a “transcriptional signature” of retinal microglia during regeneration (Issaka Salia and Mitchell, 2020; Mitchell et al., 2019). Here we show distinct responses of resident microglia to acute and chronic photoreceptor damage. In the acute group, we observed a 3000-fold increase in 4c4 immunodetection at 96 hpL (Fig. 7F), which was associated with a large number of ameboid microglia with relatively small intracellular vacuoles (Fig. 8C–D). These vacuoles were presumably phagosomes or phagolysosomes, but were not immuno-positive for either Zpr3 or Zpr1 (Figs. 7, 8). In the chronic group, we observed a ~800-fold increase in 4c4 immunodetection from 96 hpL through 21 dpL (Fig. 7L), which was associated with a single layer of ameboid microglia with relatively large vacuoles located at the interface of the rod outer segments and the retinal pigmented epithelium. Future work will be needed to determine if these microglia play a functional role by phagocytosing and clearing the slowly truncated photoreceptor outer segments and whether this dampens the overall inflammatory response in the chronic light model.
3.1. Conclusions
In summary, here we present a chronic photoreceptor model that can be performed in a relatively short time frame (21 days), results in photoreceptor outer segment truncation, but does not reach the threshold of damage to “trigger” MG cell cycle re-entry. Many open questions remain about the utility of this model, which we feel warrants future exploration. First, it is unclear whether MG in this model were truly unresponsive to the photoreceptor loss. We previously showed that inhibiting MG cell cycle re-entry resulted in growth factor release and photoreceptor neuroprotection (Thomas et al., 2016). Future studies using this model should determine whether MG are responding with protective gliotic responses. Transcriptional approaches on isolated MG from acute and chronic models could also be utilized to reveal differences in MG response to these damage paradigms. Second, it is unclear whether continued chronic light treatment would eventually result in sufficient photoreceptor damage to trigger MG proliferation and/or more discernible signs of MG reactivity. Similarly, it is unclear what would happen if the light treatment was stopped at 21 dpL and the retinas were allowed to “recover”. It is possible that outer segments could be regrown in this case, but it is also possible that persistent inflammation and resident microglia would prevent such a response. Finally, it was recently shown that Semper cells in the Drosophila eye are analogous to vertebrate MG (Charlton-Perkins et al., 2017). Given that fruit flies are susceptible to light-induced photoreceptor degeneration (Harris and Stark, 1977; Wang and Montell, 2007), future comparative analyses of glial responses to photoreceptor damage could reveal exciting findings about the conserved roles of retinal macroglia. In summary, we feel that these future experiments will reveal whether this novel damage paradigm is of value to the greater retinal regeneration community, especially as we collectively search for molecular signals that induce MG to switch from gliotic to regenerative potentials.
Supplementary Material
Suppl. Fig. 1. Quantification of PCNA+ cells in acute and chronic light models. A) Graphic representation of the average number of PCNA+ cells in the inner nuclear layer (INL) and outer nuclear layer (ONL) within a linear distance of 300 microns of the central dorsal retina of acute light-treated retinas. B) Graphic representation of the average number of PCNA+ cells in the INL and ONL within a linear distance of 300 microns of the central dorsal retina of chronic light-treated retinas. Note that scales are different between panel A and B. Asterisks represent significant difference from 0 hr control based on one-way ANOVA analysis followed by a post-hoc Tukey’s test using the total number of PCNA+ cells INL + ONL at each time point * = p<0.05; ** = p<0.01).
Suppl. Fig. 2. Quantification of 4c4+ cells in acute and chronic light models. A) Graphic representation of the average number of 4c4+ cells in the inner nuclear layer (INL) and outer nuclear layer (ONL) within a linear distance of 300 microns of the central dorsal retina of acute light-treated retinas. B) Graphic representation of the average number of 4c4+ cells in the INL and ONL within a linear distance of 300 microns of the central dorsal retina of chronic light-treated retinas. Note that scales are different between panel A and B. Asterisks represent significant difference from 0 hr control based on one-way ANOVA analysis followed by a post-hoc Tukey’s test using the total number of 4c4+ cells INL + ONL at each time point ** = p<0.01).
Highlights.
Chronic low light exposure can be performed in a relatively short time frame (21 days)
Chronic low light exposure damaged the outer segments of both rod and cone photoreceptors
Chronic low light exposure but did not result in significant apoptotic cell death, Muller glia gliosis, or Muller glia cell-cycle re-entry
Chronic low light exposure resulted in a recruitment of microglia to the area
This new model may lend insight into the cellular events underlying the non-regenerative photoreceptor degeneration
Acknowledgements
The authors would like to thank Xixia Luo for excellent zebrafish husbandry and technical assistance, and for resources used in the NEI Vision Core [P30EY004068 (PI: Linda Hazlett, Ph.D.)].
Funding Sources
This work was supported by grants from the National Institutes of Health (NEI) [R21EY031526 (to RT); R01EY026551 (to RT); F30EY031142 (to ACK); P30EY004068 (NEI Core Grant to Linda Hazlett, Ph.D.)] and an unrestricted grant from Research to Prevent Blindness to the Department of Ophthalmology, Visual and Anatomical Sciences at WSU SOM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Ahmad I, Del Debbio CB, Das AV, Parameswaran S, 2011. Muller glia: a promising target for therapeutic regeneration. Investigative ophthalmology & visual science 52, 5758–5764. [DOI] [PubMed] [Google Scholar]
- Angueyra JM, Kindt KS, 2018. Leveraging Zebrafish to Study Retinal Degenerations. Frontiers in cell and developmental biology 6, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker CG, Becker T, 2008. Adult zebrafish as a model for successful central nervous system regeneration. Restorative neurology and neuroscience 26, 71–80. [PubMed] [Google Scholar]
- Becker T, Wullimann MF, Becker CG, Bernhardt RR, Schachner M, 1997. Axonal regrowth after spinal cord transection in adult zebrafish. The Journal of comparative neurology 377, 577–595. [DOI] [PubMed] [Google Scholar]
- Bernardos RL, Barthel LK, Meyers JR, Raymond PA, 2007. Late-stage neuronal progenitors in the retina are radial Muller glia that function as retinal stem cells. The Journal of neuroscience : the official journal of the Society for Neuroscience 27, 7028–7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, Osborne NN, Reichenbach A, 2006. Muller cells in the healthy and diseased retina. Progress in retinal and eye research 25, 397–424. [DOI] [PubMed] [Google Scholar]
- Bringmann A, Wiedemann P, 2012. Muller glial cells in retinal disease. Ophthalmologica. Journal international d’ophtalmologie. International journal of ophthalmology. Zeitschrift fur Augenheilkunde 227, 1–19. [DOI] [PubMed] [Google Scholar]
- Charlton-Perkins MA, Sendler ED, Buschbeck EK, Cook TA, 2017. Multifunctional glial support by Semper cells in the Drosophila retina. PLoS genetics 13, e1006782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fausett BV, Goldman D, 2006. A role for alpha1 tubulin-expressing Muller glia in regeneration of the injured zebrafish retina. The Journal of neuroscience : the official journal of the Society for Neuroscience 26, 6303–6313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fimbel SM, Montgomery JE, Burket CT, Hyde DR, 2007. Regeneration of inner retinal neurons after intravitreal injection of ouabain in zebrafish. The Journal of neuroscience : the official journal of the Society for Neuroscience 27, 1712–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorsuch RA, Hyde DR, 2014. Regulation of Muller glial dependent neuronal regeneration in the damaged adult zebrafish retina. Experimental eye research 123, 131–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haire SE, Pang J, Boye SL, Sokal I, Craft CM, Palczewski K, Hauswirth WW, Semple-Rowland SL, 2006. Light-driven cone arrestin translocation in cones of postnatal guanylate cyclase-1 knockout mouse retina treated with AAV-GC1. Investigative ophthalmology & visual science 47, 3745–3753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris WA, Stark WS, 1977. Hereditary retinal degeneration in Drosophila melanogaster. A mutant defect associated with the phototransduction process. The Journal of general physiology 69, 261–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iribarne M, Hyde DR, Masai I, 2019. TNFalpha Induces Muller Glia to Transition From Non-proliferative Gliosis to a Regenerative Response in Mutant Zebrafish Presenting Chronic Photoreceptor Degeneration. Frontiers in cell and developmental biology 7, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issaka Salia O, Mitchell DM, 2020. Bioinformatic analysis and functional predictions of selected regeneration-associated transcripts expressed by zebrafish microglia. BMC genomics 21, 870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassen SC, Ramanan V, Montgomery JE, C, T.B., Liu CG, Vihtelic TS, Hyde DR, 2007. Time course analysis of gene expression during light-induced photoreceptor cell death and regeneration in albino zebrafish. Developmental neurobiology 67, 1009–1031. [DOI] [PubMed] [Google Scholar]
- Kassen SC, Thummel R, Burket CT, Campochiaro LA, Harding MJ, Hyde DR, 2008. The Tg(ccnb1:EGFP) transgenic zebrafish line labels proliferating cells during retinal development and regeneration. Molecular vision 14, 951–963. [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K, Gupta V, Wang J, Holdway JE, Wills AA, Fang Y, Poss KD, 2011. tcf21+ epicardial cells adopt non-myocardial fates during zebrafish heart development and regeneration. Development 138, 2895–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong L, Li F, Soleman CE, Li S, Elias RV, Zhou X, Lewis DA, McGinnis JF, Cao W, 2006. Bright cyclic light accelerates photoreceptor cell degeneration in tubby mice. Neurobiology of disease 21, 468–477. [DOI] [PubMed] [Google Scholar]
- Lahne M, Brecker M, Jones SE, Hyde DR, 2020. The Regenerating Adult Zebrafish Retina Recapitulates Developmental Fate Specification Programs. Frontiers in cell and developmental biology 8, 617923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahne M, Hyde DR, 2016. Interkinetic Nuclear Migration in the Regenerating Retina. Advances in experimental medicine and biology 854, 587–593. [DOI] [PubMed] [Google Scholar]
- Lenkowski JR, Raymond PA, 2014. Muller glia: Stem cells for generation and regeneration of retinal neurons in teleost fish. Progress in retinal and eye research 40, 94–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepilina A, Coon AN, Kikuchi K, Holdway JE, Roberts RW, Burns CG, Poss KD, 2006. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell 127, 607–619. [DOI] [PubMed] [Google Scholar]
- Li H, Lian L, Liu B, Chen Y, Yang J, Jian S, Zhou J, Xu Y, Ma X, Qu J, Hou L, 2020. KIT ligand protects against both light-induced and genetic photoreceptor degeneration. eLife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZW, Wang HT, Wang N, Sheng WW, Jin M, Lu Y, Bai YJ, Zou SQ, Pang YL, Xu H, Zhang X, 2019. Establishment of an adult zebrafish model of retinal neurodegeneration induced by NMDA. International journal of ophthalmology 12, 1250–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massoz L, Dupont MA, Manfroid I, 2021. Zebra-Fishing for Regenerative Awakening in Mammals. Biomedicines 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinn TE, Galicia CA, Leoni DC, Partington N, Mitchell DM, Stenkamp DL, 2019. Rewiring the Regenerated Zebrafish Retina: Reemergence of Bipolar Neurons and Cone-Bipolar Circuitry Following an Inner Retinal Lesion. Frontiers in cell and developmental biology 7, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell DM, Sun C, Hunter SS, New DD, Stenkamp DL, 2019. Regeneration associated transcriptional signature of retinal microglia and macrophages. Scientific reports 9, 4768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AC, Scholz TL, Brockerhoff SE, Fadool JM, 2008. Genetic dissection reveals two separate pathways for rod and cone regeneration in the teleost retina. Developmental neurobiology 68, 605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris AC, Schroeter EH, Bilotta J, Wong RO, Fadool JM, 2005. Cone survival despite rod degeneration in XOPS-mCFP transgenic zebrafish. Investigative ophthalmology & visual science 46, 4762–4771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima M, Barthel LK, Raymond PA, 2013. A self-renewing division of zebrafish Muller glial cells generates neuronal progenitors that require N-cadherin to regenerate retinal neurons. Development 140, 4510–4521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noel NCL, MacDonald IM, Allison WT, 2021. Zebrafish Models of Photoreceptor Dysfunction and Degeneration. Biomolecules 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poss KD, 2007. Getting to the heart of regeneration in zebrafish. Seminars in cell & developmental biology 18, 36–45. [DOI] [PubMed] [Google Scholar]
- Poss KD, Wilson LG, Keating MT, 2002. Heart regeneration in zebrafish. Science 298, 2188–2190. [DOI] [PubMed] [Google Scholar]
- Qin Z, Kidd AR 3rd, Thomas JL, Poss KD, Hyde DR, Raymond PA, Thummel R, 2011. FGF signaling regulates rod photoreceptor cell maintenance and regeneration in zebrafish. Experimental eye research 93, 726–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranski AH, Kramer AC, Morgan GW, Perez JL, Thummel R, 2018. Characterization of retinal regeneration in adult zebrafish following multiple rounds of phototoxic lesion. PeerJ 6, e5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renninger SL, Gesemann M, Neuhauss SC, 2011. Cone arrestin confers cone vision of high temporal resolution in zebrafish larvae. The European journal of neuroscience 33, 658–667. [DOI] [PubMed] [Google Scholar]
- Sherpa T, Fimbel SM, Mallory DE, Maaswinkel H, Spritzer SD, Sand JA, Li L, Hyde DR, Stenkamp DL, 2008. Ganglion cell regeneration following whole-retina destruction in zebrafish. Developmental neurobiology 68, 166–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song P, Fogerty J, Cianciolo LT, Stupay R, Perkins BD, 2020. Cone Photoreceptor Degeneration and Neuroinflammation in the Zebrafish Bardet-Biedl Syndrome 2 (bbs2) Mutant Does Not Lead to Retinal Regeneration. Frontiers in cell and developmental biology 8, 578528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenkamp DL, 2011. The rod photoreceptor lineage of teleost fish. Progress in retinal and eye research 30, 395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenkamp DL, Satterfield R, Muhunthan K, Sherpa T, Vihtelic TS, Cameron DA, 2008. Age-related cone abnormalities in zebrafish with genetic lesions in sonic hedgehog. Investigative ophthalmology & visual science 49, 4631–4640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JL, Nelson CM, Luo X, Hyde DR, Thummel R, 2012. Characterization of multiple light damage paradigms reveals regional differences in photoreceptor loss. Experimental eye research 97, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JL, Ranski AH, Morgan GW, Thummel R, 2016. Reactive gliosis in the adult zebrafish retina. Experimental eye research 143, 98–109. [DOI] [PubMed] [Google Scholar]
- Thummel R, Kassen SC, Enright JM, Nelson CM, Montgomery JE, Hyde DR, 2008a. Characterization of Muller glia and neuronal progenitors during adult zebrafish retinal regeneration. Experimental eye research 87, 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thummel R, Kassen SC, Montgomery JE, Enright JM, Hyde DR, 2008b. Inhibition of Muller glial cell division blocks regeneration of the light-damaged zebrafish retina. Developmental neurobiology 68, 392–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vihtelic TS, Hyde DR, 2000. Light-induced rod and cone cell death and regeneration in the adult albino zebrafish (Danio rerio) retina. J Neurobiol 44, 289–307. [DOI] [PubMed] [Google Scholar]
- Wan J, Goldman D, 2017. Opposing Actions of Fgf8a on Notch Signaling Distinguish Two Muller Glial Cell Populations that Contribute to Retina Growth and Regeneration. Cell reports 19, 849–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Montell C, 2007. Phototransduction and retinal degeneration in Drosophila. Pflugers Archiv : European journal of physiology 454, 821–847. [DOI] [PubMed] [Google Scholar]
- Westerfield, 1995. The Zebrafish Book: A guide for the laboratory use of zebrafish (Danio rerio). Univ. of Oregon Press., Eugene, OR. [Google Scholar]
- White DT, Sengupta S, Saxena MT, Xu Q, Hanes J, Ding D, Ji H, Mumm JS, 2017. Immunomodulation-accelerated neuronal regeneration following selective rod photoreceptor cell ablation in the zebrafish retina. Proceedings of the National Academy of Sciences of the United States of America 114, E3719–E3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Cuenca N, Ivanova T, Church-Kopish J, Frederick JM, MacLeish PR, Baehr W, 2003. Identification and light-dependent translocation of a cone-specific antigen, cone arrestin, recognized by monoclonal antibody 7G6. Investigative ophthalmology & visual science 44, 2858–2867. [DOI] [PubMed] [Google Scholar]
- Zhang S, Mu Z, He C, Zhou M, Liu D, Zhao XF, Goldman D, Xu H, 2016. Antiviral Drug Ganciclovir Is a Potent Inhibitor of the Proliferation of Muller Glia-Derived Progenitors During Zebrafish Retinal Regeneration. Investigative ophthalmology & visual science 57, 1991–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Li A, Brown B, Weiss ER, Osawa S, Craft CM, 2002. Mouse cone arrestin expression pattern: light induced translocation in cone photoreceptors. Molecular vision 8, 462–471. [PubMed] [Google Scholar]
- Bernardos RL, Barthel LK, Meyers JR, Raymond PA, 2007. Late-stage neuronal progenitors in the retina are radial Muller glia that function as retinal stem cells. The Journal of neuroscience : the official journal of the Society for Neuroscience 27, 7028–7040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stenkamp DL, 2011. The rod photoreceptor lineage of teleost fish. Progress in retinal and eye research 30, 395–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan-Brown J, Bisher ME, Burdine RD, 2011. Embedding, serial sectioning and staining of zebrafish embryos using JB-4 resin. Nature protocols 6, 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Suppl. Fig. 1. Quantification of PCNA+ cells in acute and chronic light models. A) Graphic representation of the average number of PCNA+ cells in the inner nuclear layer (INL) and outer nuclear layer (ONL) within a linear distance of 300 microns of the central dorsal retina of acute light-treated retinas. B) Graphic representation of the average number of PCNA+ cells in the INL and ONL within a linear distance of 300 microns of the central dorsal retina of chronic light-treated retinas. Note that scales are different between panel A and B. Asterisks represent significant difference from 0 hr control based on one-way ANOVA analysis followed by a post-hoc Tukey’s test using the total number of PCNA+ cells INL + ONL at each time point * = p<0.05; ** = p<0.01).
Suppl. Fig. 2. Quantification of 4c4+ cells in acute and chronic light models. A) Graphic representation of the average number of 4c4+ cells in the inner nuclear layer (INL) and outer nuclear layer (ONL) within a linear distance of 300 microns of the central dorsal retina of acute light-treated retinas. B) Graphic representation of the average number of 4c4+ cells in the INL and ONL within a linear distance of 300 microns of the central dorsal retina of chronic light-treated retinas. Note that scales are different between panel A and B. Asterisks represent significant difference from 0 hr control based on one-way ANOVA analysis followed by a post-hoc Tukey’s test using the total number of 4c4+ cells INL + ONL at each time point ** = p<0.01).