

Abstract
Nutritional metal ions play critical roles in many important immune processes. Hence, effective modulation of metal ions may open up new forms of immunotherapy, termed as metalloimmunotherapy. Here, we demonstrate a prototype of cancer metalloimmunotherapy using cyclic dinucleotide (CDN) stimulator of interferon genes (STING) agonists and Mn2+. We screened various metal ions and discovered specific metal ions augmented STING agonist activity, wherein Mn2+ promoted a 12- to 77-fold potentiation effect across the prevalent human STING haplotypes. Notably, Mn2+ coordinated with CDN STING agonists to self-assemble into a nanoparticle (CDN-Mn2+ particle, CMP) that effectively delivered STING agonists to immune cells. CMP administered either by local intratumoral or systemic intravenous injection initiated robust anti-tumor immunity, achieving remarkable therapeutic efficacy with minute doses of STING agonists in multiple murine tumor models. Overall, CMP offers a new platform for local and systemic cancer treatments, and this work underscores the great potential of coordination nanomedicine for metalloimmunotherapy.
Immunotherapy is revolutionizing cancer treatment1–3; however, only a small subset of patients respond to immunotherapies4. The limited patient response rate has been attributed to poor anti-tumor immunity in “cold” tumors, characterized by a low frequency of pro-inflammatory immune cells and immunosuppressive network in the tumor microenvironment (TME)5. Recent studies have shown that the stimulator of interferon genes (STING) pathway plays critical roles in the initiation of anti-tumor immunity and conversion of “cold” tumor to “hot” tumor6–9. Briefly, cyclic GMP-AMP synthase (cGAS) detects damage-associated double-stranded DNA in the cytosol and catalyzes the generation of cyclic [G(2’,5’)pA(3’,5’)p] (cGAMP), which serves as the second messenger to activate STING and induce type-I interferons (IFNs)6,10,11. Preclinical studies with STING agonists have shown promising anti-tumor efficacy7,8. Yet, because of their metabolic instability, limited cellular permeability, and poor drug-like properties, conventional CDN-based STING agonists are administered intratumorally (I.T.)12,13. However, the I.T. route of administration is not applicable for treating metastasis, and I.T. injection of CDN-based STING agonists have produced disappointing results in clinical trials12,13. While new STING agonists based on non-CDN structures have been recently reported for systemic administration14–17, their toxicity profiles and efficacy are yet to be studied in clinical trials. Alternatively, nanoparticles, such as those based on polymers and liposomes, could augment the local and systemic therapeutic effects of CDN-based STING agonists9,18–22, thus underscoring the potential and utility of nanomedicine-based delivery of STING agonists.
Emerging evidence has indicated essential roles of metal ions in immune regulation23,24, including T-cell activation (Ca2+)25,26 and stemness (K+)27,28, activation of inflammasome (K+, Ca2+, Na+)29–31, pathogen-host interaction (Fe2+/3+, Zn2+, Mn2+, Cu2+)32,33, and cGAS-STING signaling (Zn2+, Mn2+)34,35. “Metalloimmunotherapy” may harness the immune modulatory functions of metal ions for disease treatment. For example, Mg2+ increases NKG2D expression and restores cytotoxicity of NK and T-cells for EBV infection treatment36. Potassium (K+) preserves T-cell stemness and increases persistence and potency of T-cells28. In particular, recent studies have shown that Mn2+ sensitizes the cGAS-STING pathway to double-stranded DNA during DNA virus infection34 and synergizes with immune checkpoint inhibitors37, chemotherapy38, in-situ vaccine39, and photodynamic therapy40. However, despite their promise, it remains largely unknown how to systemically develop an effective metalloimmunotherapy and deliver them in appropriate pharmaceutical forms.
Here, we have developed metalloimmunotherapy based on coordination nanomedicine as a new form of cancer immunotherapy (Fig. 1). Briefly, we screened various metal ions for potential synergy with STING agonists and discovered that Mn2+ and Co2+ could significantly augment type-I IFN (IFN-I) activity of STING agonists. As Mn2+ is an essential inorganic trace element required for the immune system34,41 and is used in FDA-approved pharmaceuticals42–44, we focused on the combination of Mn2+ and STING agonists. We report that Mn2+ markedly increases the type-I IFN activities of STING agonists in multiple human STING haplotypes. We also demonstrate that Mn2+ self-assembles with CDN STING agonists to form a coordination nanoparticle (CDN-Mn2+ Particle, CMP) that elicits robust anti-tumor immunity after local or systemic administration (Fig. 1a–b). Using c-di-AMP (CDA) as an example, we show that CMPCDA administered via either intratumoral (I.T.) or intravenous (I.V.) route significantly increased STING activation, reversed immunosuppression in the TME, and exerted remarkable anti-tumor efficacy (Fig. 1c). Overall, CMP, a coordination nanomedicine composed of bioactive metal ions and STING agonists, is a promising novel platform for metalloimmunotherapy.
Figure 1. Amplifying STING activation with CDN-Manganese particles (CMP) for cancer metalloimmunotherapy.
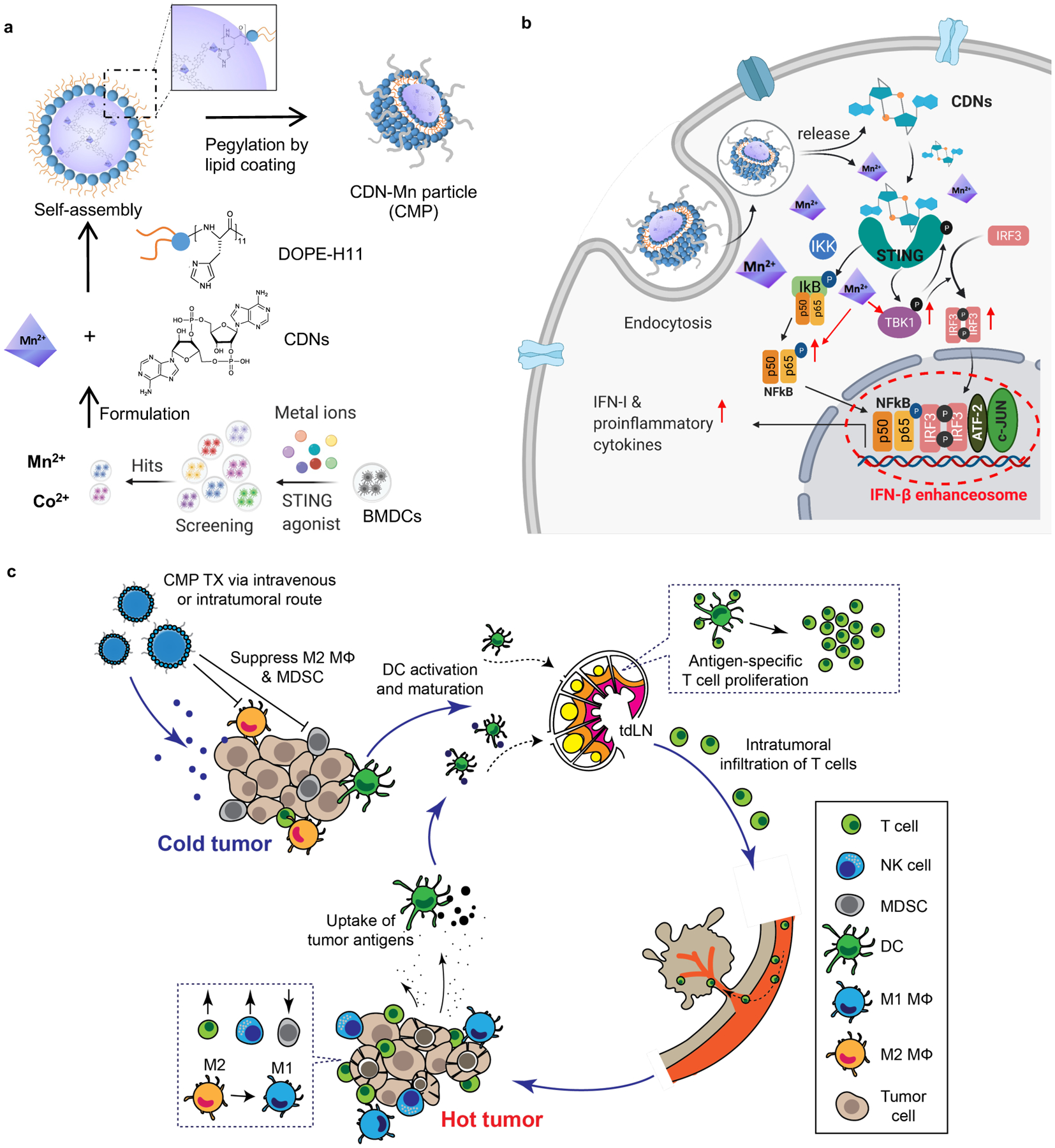
a) CMP is composed of cyclic di-nucleotides (CDNs), manganese ions (Mn2+), phospholipid-histidine11 (DOPE-H11), and a PEG-lipid layer (DOPC: cholesterol: DSPE-PEG5000). Mn2+ potentiates type-I IFN activities of STING agonists. Mn2+ and CDNs self-assemble into coordination polymer. CDN-Mn2+ coordination polymer was coated with DOPE-H11 via Mn-histidine coordination to form CDN-Mn@DOPE, followed by PEGylation with PEG-lipid layer, resulting in the formation of CMP. b) CMP boosts STING activation: 1) CMP promotes cellular uptake of CDNs and Mn2+; 2) Mn2+ augments CDN-induced STING activation via STING-independent TBK1 and p65 phosphorylation, STING-dependent IRF3 phosphorylation, and assembly of the IFN-β transcriptional enhanceosome. c) CMP exerts potent anti-tumor efficacy after intratumoral (I.T.) or intravenous (I.V.) administration. CMP reverses immunosuppressive tumor microenvironment while activating T-cells, natural killer (NK) cells, and dendritic cells (DCs). Figure 1a and 1b were created using BioRender.com.
Mn2+ potentiates STING agonist activity and IFN-I response
Specifically, we examined the cGAS-STING-type-I IFN pathway and screened various nutritional metal ions (e.g., Zn2+, K+, Mg2+, Mn2+, Ca2+, Al3+, Cu2+, Fe3+, Fe2+, and Co2+) for potentiating STING agonists as a new form of metalloimmunotherapy. To our surprise, adding either Mn2+ or Co2+ to cGAMP dramatically increased the type-I IFN production in murine bone marrow-derived dendritic cells (BMDCs) (Fig. 2a) and in human monocyte-like THP1 cells in a dose-dependent manner (Supplementary Fig. 1a, Fig. 2b). Given the previous examples of Mn2+-based pharmaceuticals42–44, we further investigated the combination of Mn2+ and STING agonists. We examined the impact of Mn2+ on human STING haplotypes known to exhibit distinct response profiles to STING agonists. The addition of Mn2+ to various concentrations of cGAMP significantly amplified the type-I IFN responses in THP1 cells expressing hSTINGR232, hSTINGH232, or hSTINGHAQ, achieving 77-fold, 14-fold, and 12-fold dose-sparing effect, respectively (Fig. 2c–e). The allele frequencies of hSTINGR232, hSTINGH232, and hSTINGHAQ in humans are 57.9%, 20.4%, and 13.7%, respectively7. Even the insensitive hSTINGH232, which did not respond to as high as 500 μM cGAMP, exhibited a strong IFN-β response when Mn2+ was added (Fig. 2e), suggesting that Mn2+ offers a widely applicable strategy that covers > 90% allele frequency of human STING variants. In addition, Mn2+ in doses ranging from 500 μM down to 62.5 μM amplified the type-I IFN-inducing activities of other CDN-based STING agonists, including CDA45, ADU-S1007, 2’3’-cGAM(PS)2 (Rp/Sp)46, as well as a non-CDN STING agonist, diABZI14 (Supplementary Fig. 1b–e, 2a–e). These results indicate that Mn2+-mediated potentiation of STING agonists is a general phenomenon independent of STING variants and STING agonist structures.
Figure 2. Mn2+ augments type-I IFN activity of STING agonists.
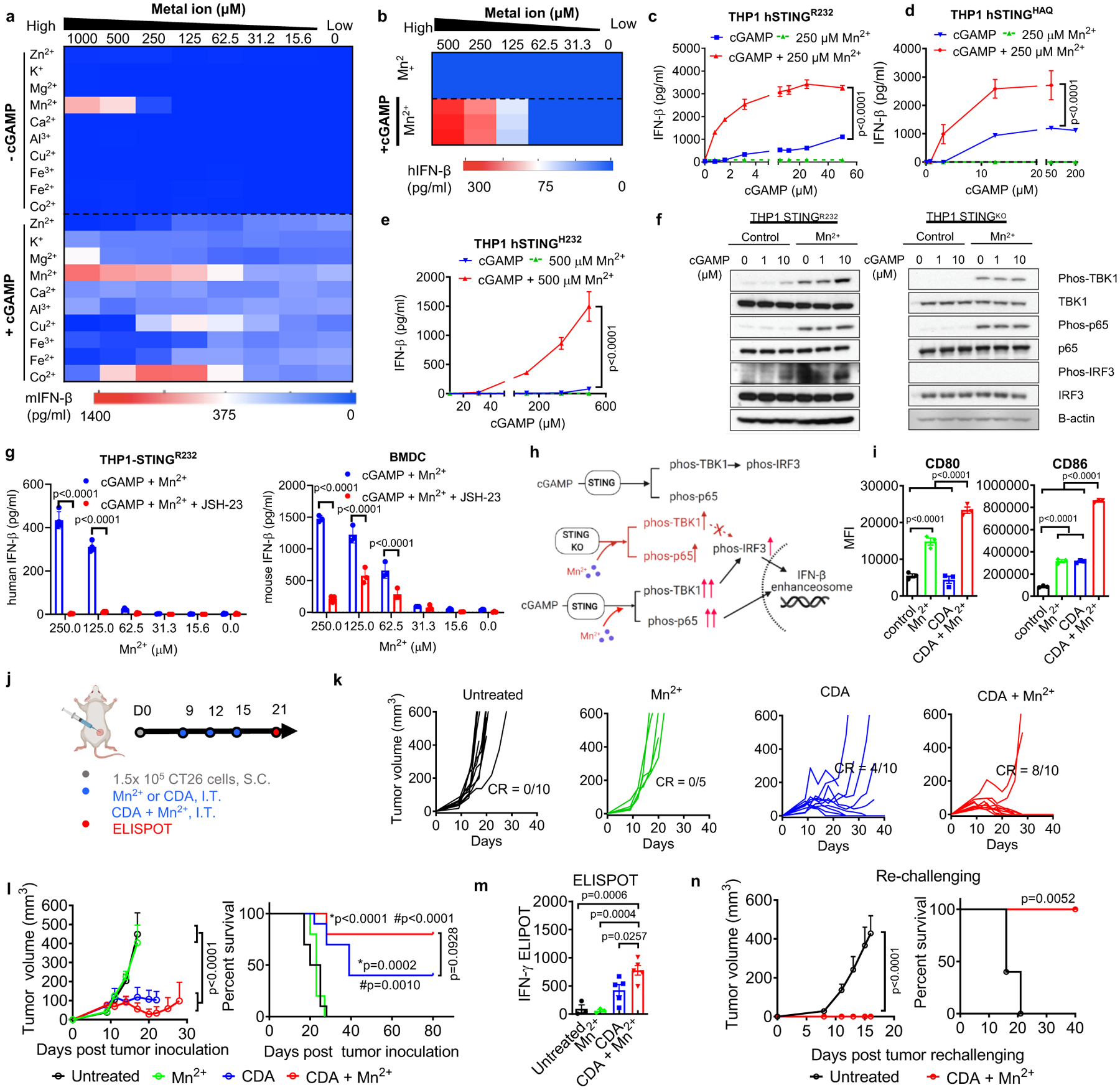
a) BMDCs or b) THP1 cells were incubated with various concentrations of metal ions ± 5 μM cGAMP, and after 24 h, IFN-β secretion was quantified. c–e) THP1 cells expressing c) hSTINGR232, d) hSTINGHAQ, or e) hSTINGH232 were treated for 24 h with cGAMP ± Mn2+, followed by quantification of IFN-β production. f) THP-1 STINGR232 or THP-1 STINGKO cells were incubated with increasing concentrations of cGAMP ± 250 μM Mn2+ for 6 hours, followed by Immunoblotting for marker proteins in the STING-IFN-I pathway. Shown are representative data from two independent experiments with similar results. g) Pharmacological inhibition of p65 nucleus translocation inhibited Mn2+-potentiated IFN-β production. h) Proposed mechanism of Mn2+-mediated potentiation of STING agonist via STING-independent TBK1 and p65 phosphorylation and STING-dependent IRF3 phosphorylation. Activation of p65 and IRF3 further facilitates assembly of the IFN-β transcriptional enhanceosome. i) BMDCs treated with 5 μM CDA, 250 μM Mn2+, or their combination for 24 h were analyzed for activation by flow cytometry. j–n) CT26 tumor-bearing BALB/c mice were treated by I.T. administration with 20 μg CDA, 17.5 μg Mn2+, or their combination on days 9, 12, and 15 (j). Mice were monitored for tumor growth (k, l) and survival (l). m) AH1-specific T-cells among PBMCs were assessed by ELISPOT on day 21. n) Survivors from CDA + Mn2+ group were re-challenged with CT26 cells on day 80. Data represent mean ± SEM, from a representative experiment from 2–3 independent experiments with n = 3–4 (c–e, g, i) and n = 5–10 (k–n) biologically independent samples. Data were analyzed by (i, m) one-way ANOVA or (c–e, g, l, n) two-way ANOVA with Bonferroni’s multiple comparisons test, or (l, n) log-rank (Mantel-Cox) test. *p and #p in (j) denote statistical significance relative to the untreated or Mn2+ group, respectively. Figure 2h and 2j were created using BioRender.com.
To characterize the mechanism of Mn2+-amplified STING activation, we first performed a thermal shift assay of STING (both hSTINGR232 and hSTINGH232) binding to various STING agonists. However, regardless of the STING agonists and STING variants of choice, Mn2+ did not increase the binding affinity between STING and STING agonists (Supplementary Fig. 3). Thus, we examined the impact of STING agonist + Mn2+ on the downstream of the STING-IFN-I signaling pathway. Maximal transcription of IFN-I genes depends on the formation of an enhanceosome, which contains phosphorylated IRF3 and p6547–49. Hence, we focused on these two transcription factors. The combination of cGAMP + Mn2+ potently enhanced the levels of phosphorylated TBK1, IRF3, and p65 in hSTINGR232 THP1 cells (Fig. 2f). Interestingly, Mn2+ alone without cGAMP still induced phosphorylation of TBK1 and p65 (Fig. 2f). Notably, in STING-knockout THP1 cells and STING-deficient murine STINGgt/gt BMDCs, Mn2+ triggered STING-independent phosphorylation of TBK1 and p65, but not IRF350 (Fig. 2f, Supplementary Fig. 4–5). In hSTINGR232 THP1 cells, hSTINGH232 THP1 cells, and murine WT BMDCs, addition of JSH-23, an inhibitor of p65 nucleus translocation51, abrogated IFN-β production promoted by cGAMP + Mn2+ (Fig. 2g, Supplementary Fig. 5). Taken together, Mn2+ has STING-independent immune activating potential by inducing phosphorylation of TBK1 and p65, which is further augmented and translated to IRF3 phosphorylation in the presence of STING agonists, resulting in amplification of the STING signaling cascade and production of type-I IFNs (Fig. 2h).
We evaluated the effects of STING agonist + Mn2+ on DCs. While CDA or Mn2+ promoted BMDC maturation as single agents, the CDA + Mn2+ combination significantly up-regulated CD80 and CD86 on BMDCs (Fig. 2i, Supplementary Fig. 6). We examined the therapeutic efficacy of CDA + Mn2+ in vivo. BALB/c mice were inoculated with CT26 colon carcinoma cells subcutaneously (S.C.) on day 0, and 20 μg CDA, 17.5 μg Mn2+ (in 40 μg MnCl2), or their combination was administered I.T. on days 9, 12, and 15 (Fig. 2j). CDA + Mn2+ eradicated CT26 tumors in 80% of mice (Fig. 2k–l). In contrast, CDA monotherapy eliminated tumors in only 40% of mice, while MnCl2 treatment showed no benefit (Fig. 2k–l). We assessed CD8+ T-cell responses against CT26 cells by performing IFN-γ ELISPOT assay with peripheral blood mononuclear cells (PBMCs) re-stimulated with AH1 epitope (H-2Ld–restricted SPSYVYHQF, the immunodominant MHC-I minimal epitope of CT26 gp7052). Mice treated with CDA + Mn2+ exhibited significantly elevated antigen-specific T-cell response, compared with either CDA or Mn2+ monotherapy (Fig. 2m). In addition, 100% of survivors from the CDA+ Mn2+ treatment group were resistant to CT26 tumor re-challenge performed on day 80 (Fig. 2n). These results show that Mn2+ potentiates STING agonist activity and induces robust anti-tumor T-cell response with long-term memory.
CDN-Mn2+ self-assembled into CMP amplify STING activation
Despite these promising results, the free admixture of CDNs and Mn2+ has a number of limitations, including their poor metabolic stability, cellular permeability, as well as potential safety concerns17. We sought to address these issues by developing a delivery system that can co-deliver STING agonists and Mn2+ and achieve dose-sparing with minimal side effects (Fig. 1). We discovered that Mn2+ mixed with various CDNs in methanol, including CDA, CDG, and cGAMP, coordinated their self-assembly into coordination polymers ranging from nanometers to micrometers in diameter (Fig. 3a). We also observed the formation of coordination polymers when CDA was mixed in water with either Mn2+ or Zn2+, but not with Ca2+, manganese phosphate or calcium phosphate (Supplementary Fig. 7). Based on Mn2+-mediated potentiation of type-I IFN response and the fact that STING agonists in clinical trials are mainly derived from CDA53, we focused on the CDA + Mn2+ combination for the remainder of studies. Isothermal titration calorimetry (ITC) analysis indicated that the CDA-Mn2+ interaction in methanol fitted a multiple-site binding model with a Ka1 of 9.367E8 M−1 and Ka2 of 1.206E7 M−1 while the interaction of CDA-Mn2+ interaction in water fitted an independent binding model with a KD of 1 mM (Supplementary Fig. 8). However, in a physiological condition, CDA-Mn2+ interaction was unstable, leading to a rapid dissolution in PBS. Thus, to stabilize the CDA-Mn2+ coordination polymer, we added dioleoyl-sn-glycero-3-phosphoethanolamine-N-[histidine]11 (DOPE-H11) (Supplementary Fig. 9) that served as an additional coordination ligand and promoted the formation of hydrophobic core termed CDA-Mn@DOPE. To allow for aqueous suspension, we coated CDA-Mn@DOPE with an outer PEG-lipid layer by resuspension in a mixture solution of DOPC: cholesterol: DSPE-PEG5000 (4:1:1 molar ratio), followed by solvent evaporation and rehydration. The resulting CDA-Mn2+ particles, termed CMPCDA, exhibited a uniform spherical morphology with an average hydrodynamic diameter of 118 ± 41 nm, a polydispersity index of 0.107, and a neutral surface charge (Fig. 3c–d). CDA and Mn2+ were efficiently loaded into CMPCDA with the loading efficiency of 39.6% and 25.3% and the loading capacity (wt/wt) of 13.2% and 6.72% for CDA and Mn2+, respectively.
Figure 3. CMP co-delivering Mn2+ and STING agonist amplifies STING activation.
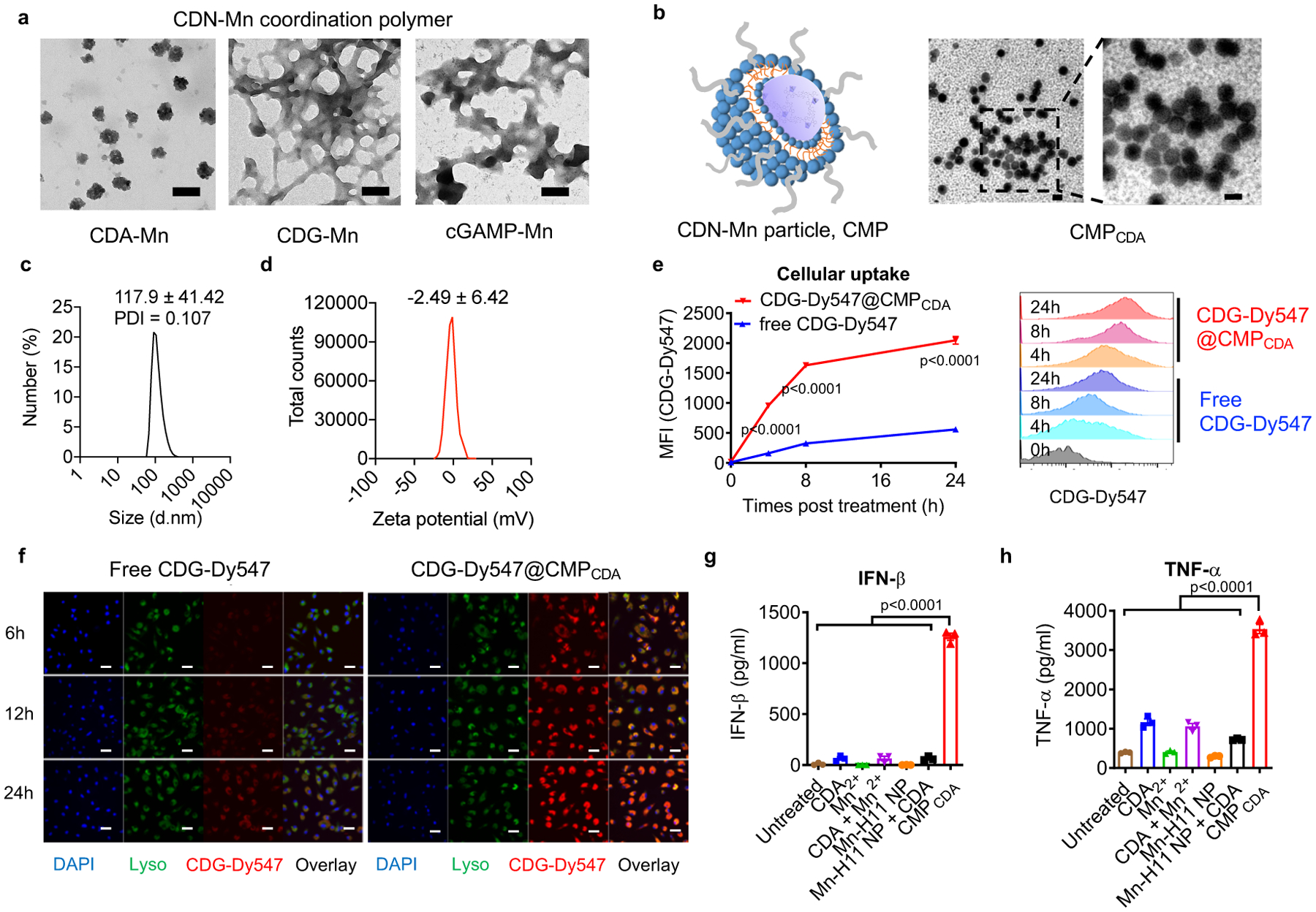
a) Self-assembly behavior of CDNs and Mn2+. TEM images of CDN-Mn coordination polymers formed by mixing CDA, CDG, or cGAMP with Mn2+ (10:1, n/n) for 1h. Scale bars = 100 nm. b) TEM images showed homogenous CMPCDA formed by coating CDA-Mn coordination polymers with a PEGylated lipid layer. Scale bar = 100 nm. c) Dynamic light scattering and d) zeta potential analyses of CMPCDA. e–f) CMPCDA increased cellular uptake of STING agonist. BMDCs were incubated with free CDG-Dy547 or CDG-Dy547@CMPCDA for 6, 12, or 24 h, followed by analyses by e) flow cytometry and f) confocal microscopy. Scale bars = 10 μm. g–h) CMPCDA increased STING activation and cytokine production. BMDCs were treated for 24h with 2.5 μM CDA and/or 15.6 μM Mn2+ in free form, blank nanoparticle without CDA (Mn-H11 NP), or CMPCDA, followed by quantification of g) IFN-β and h) TNF-α secretion by ELISA. Shown TEM images (a–b) and confocal microscopy images (f) are representative data from two independent experiments with similar results. Data represent mean ± SEM, from a representative experiment from 2 independent experiments with n = 3 (e, g–h) biologically independent samples. Data were analyzed by (g–h) one-way ANOVA or (e) two-way ANOVA with Bonferroni’s multiple comparisons tests.
We employed CDG-Dy547, a fluorophore-labeled CDN, to track cellular uptake of STING agonists by BMDCs. Soluble CDG-Dy547 was poorly internalized by BMDCs (Fig. 3e–f). In stark contrast, CMPCDA carrying CDG-Dy547 exhibited significantly increased cellular uptake, with a 6.3-fold improvement at 4 h (P < 0.0001, Fig. 3e). Confocal microscopic images of BMDCs showed cytosolic localization of CMP with gradually reduced overlap with Lysotracker signal over time (Fig. 3f), suggesting CMP-mediated trafficking of CDN to the cytosol where STING is expressed. CMPCDA increased IFN-β secretion by BMDCs by > 20-fold, compared with free CDA, Mn2, or their admixture (Fig. 3g). CMP-mediated co-delivery of CDA and Mn2+ was crucial for robust STING activation as Mn2+-particles alone or Mn2+-particles admixed with free CDA induced a weak IFN-β response (Fig. 3g). We also observed similar responses with TNF-α secretion (Fig. 3h). Taken together, CMP significantly augmented cellular uptake of CDA, STING activation, and IFN-β response in vitro.
Local CMP administration eliminates established tumours
Next, we evaluated the therapeutic efficacy of CMP in vivo. CT26 tumor-bearing BALB/c mice were treated on days 9, 12, and 15 by I.T. administration of CDA and Mn2+ in CMP or soluble formulation (Fig. 4a). Whereas 20 μg CDA and 17.5 μg Mn2+ were used in Fig. 2j–n, here we decreased their doses to 5 μg CDA and 2.5 μg Mn2+ to examine the dose-sparing effect of CMPCDA. Intratumoral injection of CMPCDA led to significantly improved immune activation, as shown by elevated levels of IFN-β, TNF-α, CXCL-10, and CCL-2, compared with free CDA + Mn2+ admixture (Fig 4b). CMPCDA also induced robust AH1-specific CD8+ T-cell response (Fig 4c, Supplementary Fig. 10). Importantly, CMPCDA eradicated 78% of established tumors, compared with 30% response rate for the soluble CDA + Mn2+ group (P < 0.05, Fig 4d–e). In addition, 100% of survivors from the CMPCDA group were resistant to the CT26 tumor re-challenge performed on day 145 (Fig. 4f). Remarkably, even with a minute dose of 1 μg CDA + 0.5 μg Mn2+, CMPCDA induced strong immune activation and eliminated established tumors in 40% of mice, compared with 0% response rate for the soluble CDA + Mn2+ group (Fig. 4g, Supplementary Fig. 11). We also tested CMP I.T. therapy on untreated, distal tumors in a two-tumor model (Supplementary Figure 12). As reported previously for free STING agonist53, while high dose of 20 μg CMP was better at inhibiting the primary tumor growth, lower doses of CMP (either 1 μg or 5 μg CDA) exerted robust abscopal effect against distal tumors, significantly outperforming free CDA injections, without any overt body weight change.
Figure 4. Local intratumoral administration of CMPCDA eliminates established tumors.
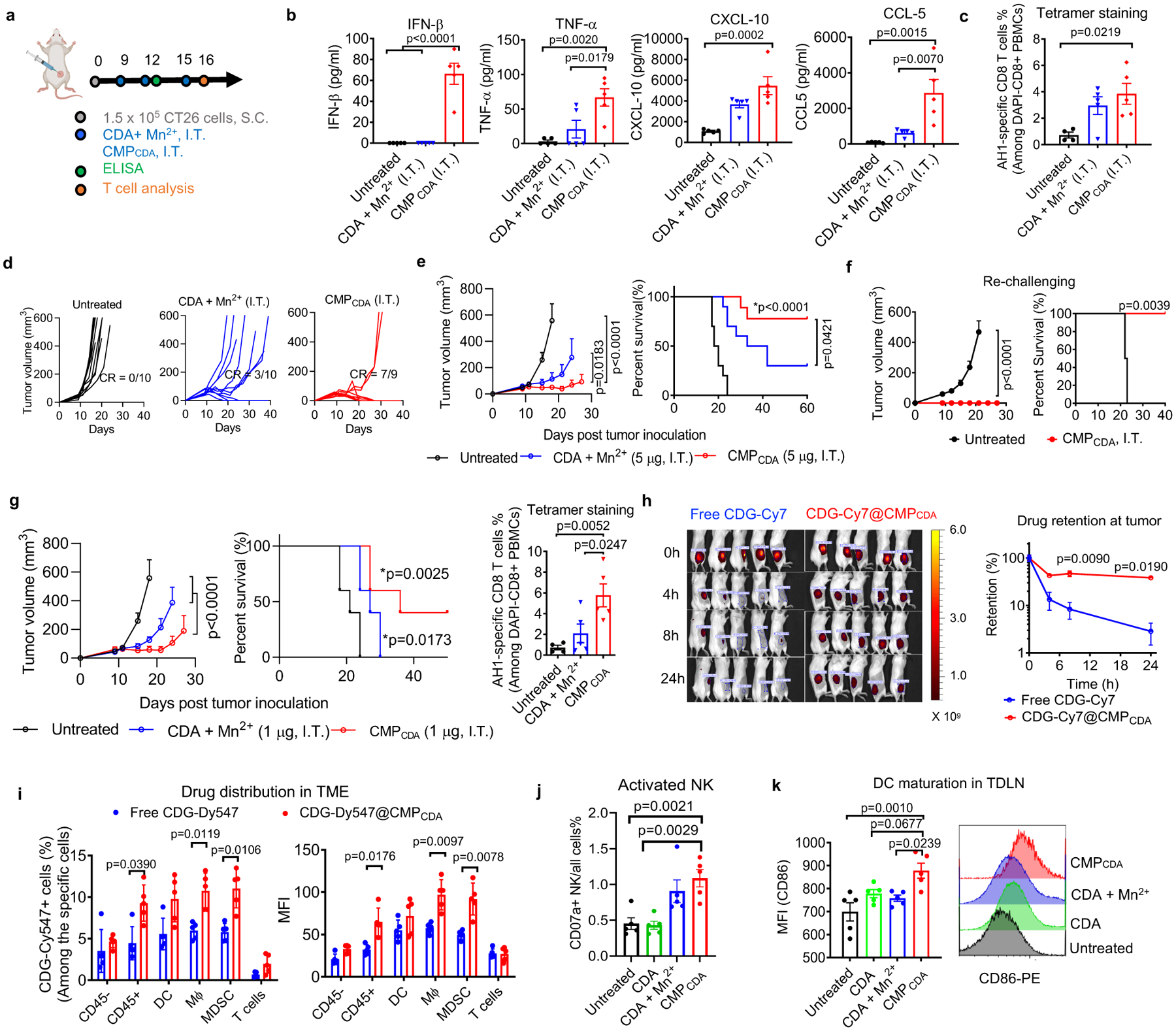
a–f) BALB/c mice were inoculated with 1.5 × 105 CT26 tumor cells at the S.C. flank and treated with CDA+ Mn2+ or CMPCDA, I.T., containing 5 μg CDA and 2.5 μg Mn2+, on days 9, 12, and 15. b) Serum cytokines were measured by ELISA at 6 h post the second dose. c) Antigen-specific T-cell response in PBMCs was analyzed by AH1 tetramer staining on day 16. d–e) Tumor growth and animal survival were monitored over time. f) Survivors were re-challenged with CT26 tumor cells on day 145. g) BALB/c mice were inoculated with 1.5 × 105 CT26 tumor cells at the S.C. flank and treated with CDA+ Mn2+ or CMPCDA, I.T., containing 1 μg CDA and 0.5 μg Mn2+, on day 9, 12, and 15. Tumor growth and survival were monitored, and antigen-specific T-cell response was analyzed on day 16. h) CDG-Cy7 either in free or CMPCDA form was administrated I.T., and retention of STING agonist retention within the TME was quantified by in-vivo imaging. i) CDG-Dy547 either in free or CMPCDA form was administrated I.T., and after 6 h, CDG-Dy547 signal among immune cells within the TME was analyzed by flow cytometry. j–k) CT26 tumor-bearing mice were treated as in g) and analyzed on day 16 by flow cytometry for the frequency of CD107a+ NK cells within tumors (j) and CD86 expression on DCs in tumor-draining lymph nodes (TDLNs) (k). Data represent mean ± SEM, from a representative experiment from 2 independent experiments with n = 4–5 (b–c, f–k), and n = 9–10 (d–e), analyzed by (b–c, g, j–k) one-way ANOVA or (e–h) two-way ANOVA with Bonferroni’s multiple comparisons test, or (i) two-tailed multiple t-tests with Bonferroni-Dunn correction, or (e–g) log-rank (Mantel-Cox) test. *p in (e, g) denote statistical significance relative to the untreated group. Figure 4a was created using BioRender.com.
Next, we examined how CMP potentiates STING activation. First, we measured the retention and distribution of STING agonists delivered via CMPCDA in the TME. Within 24 h of I.T. administration, free CDG-Cy7 was rapidly cleared from the TME, whereas 13.4-fold higher CDG-Cy7 signal was detected for the CDG-Cy7@CMPCDA group (Fig. 4h). CDG-Dy547@CMPCDA significantly improved cellular uptake of CDG by CD11c+ DCs, F4/80+ macrophages, and Ly6C+ myeloid-derived suppressor cells (MDSCs) (Fig. 4i, Supplementary Figure 13–14), but not among CD45- tumor cells and CD3+ T-cells. We also analyzed changes in the activation status of immune cells after CMPCDA treatment. Compared with free CDA with or without Mn2+, CMPCDA treatment promoted activation of intratumoral NK cells (Fig. 4j, Supplementary Figure 15) and DCs in tumor-draining lymph nodes (TDLNs) (Fig. 4k). Taken together, CMPCDA efficiently modulated the TME, in part by promoting tissue retention of STING agonists and their uptake by local immune cells, leading to Mn2+-mediated potentiation of STING agonists and anti-tumor immune response.
Systemic CMP therapy exerts potent anti-tumour effects
Due to rapid enzymatic degradation and poor drug-like properties, most STING agonists in clinical trials are administered directly into tumors; however, I.T. treatment is not applicable for metastatic tumors. To address this issue, we evaluated the therapeutic effect of CMP after I.V. administration. CT26 tumor-bearing BABL/c mice were treated I.V. on days 9, 12, and 15 with 20 μg CDA and 10 μg Mn2+ either in CMPCDA or soluble form. Compared with the soluble control group, CMPCDA promoted accumulation of Mn2+ and CDN in TME and significantly increased the serum levels of IFN-β, TNF-α, CXCL-9, and CXCL-10 (Fig 5b). As shown by IFN-y ELISPOT assay performed on PBMCs, CMPCDA administered I.V. significantly enhanced AH1-specific CD8+ T-cell response, compared with the soluble CDA + Mn2+ control (Fig. 5c, Supplementary Figure 17). Importantly, CMPCDA administered I.V. significantly decreased CT26 tumor growth and eliminated established tumors in 50% of mice (P < 0.0001, Fig. 5d–f), whereas soluble CDA + Mn2+ treatment had 0% response rate. Notably, even increasing the dose of free CDA I.V. therapy to 100 μg could not control tumor growth, whereas 20 μg CMPCDA I.V. therapy regressed established tumors (Supplementary Fig. 18). Flow cytometric analysis performed on day 23 showed that CMPCDA I.V. therapy significantly expanded CD8+ T-cell subsets with CD44+CD62L+ central memory and CD44+CD62L- effector memory phenotypes (Supplementary Fig. 19). The survivors from the CMPCDA treatment group were largely resistant to CT26 tumor re-challenge performed on day 105 (Fig. 5g). CMPCDA I.V. therapy significantly increased the drug uptake by intratumoral CD45+ immune cells, especially F4/80+ macrophages and Ly6C+ MDSCs (Fig. 5h, Supplementary Figure 13, 20). TME analysis revealed that CMPCDA I.V. therapy significantly reduced intratumoral MDSCs and promoted M2-to-M1 re-polarization of intratumoral macrophages (Fig. 5i–j, Supplementary Figure 15), while also inducing robust DC maturation in TDLNs (Fig. 5k). We also validated our results in a second tumor model. In C57BL/6 mice B16F10 melanoma, CMPCDA I.V. therapy exerted significantly enhanced therapeutic efficacy, compared with CDA + Mn2+ mixture (P < 0.001, Fig. 5l–n, Supplementary Fig. 21). Overall, CMPCDA administered I.V. induces robust anti-tumor immune responses and exhibits potent anti-tumor efficacy.
Figure 5. Systemic I.V. administration of CMPCDA eliminates established tumors.
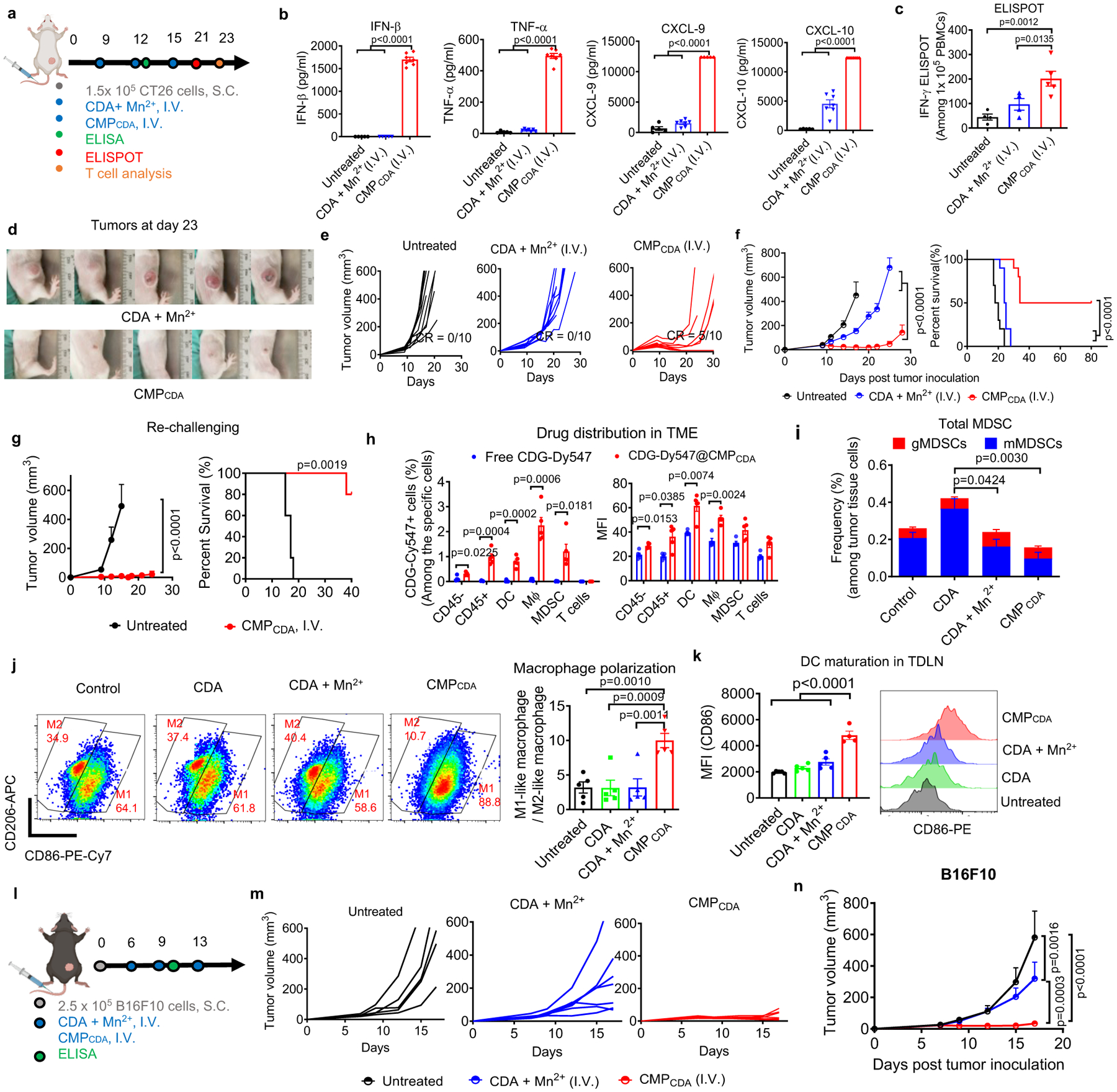
a–i) Therapeutic effects of CMPCDA on CT26 tumors after I.V. administration. a) CT26 tumor-bearing BALB/c mice were treated with CDA+ Mn2+ or CMPCDA, I.V., containing 20 μg CDA and 10 μg Mn2+, on days 9, 12, and 15. b) Serum cytokines were measured by ELISA at 6 h post the second dose. c) Antigen-specific T-cell response was analyzed on day 21 by re-stimulating PBMCs with AH1 peptide, followed by IFN-γ ELISPOT assay. d–f) Tumor growth (d–e) and animal survival (f) were monitored over time. g) Survivors re-challenged with CT26 tumor cells on day 145 were monitored for tumor growth and survival. h) CDG-Dy547 either in free form or CMPCDA was administrated I.V., and after 24 h, CDG-Dy547 signal among immune cells within the TME was analyzed by flow cytometry. i–k) CT26 tumor-bearing mice were treated as in a) and analyzed on day 17 by flow cytometry for the frequency of granulocytic and monocytic MDSCs (i), M1-like and M2-like macrophages (j) within the TME and CD86 expression on DCs in TDLNs (k). l–n) Therapeutic effects of CMPCDA on B16F10 tumors after I.V. administration. B16F10 tumor-bearing C57BL/6 mice were treated with CDA+ Mn2+ or CMPCDA, I.V., containing 20 μg CDA and 10 μg Mn2+, on days 6, 9, and 13 (l), and tumor growth was monitored over time (o–p). Data represent mean ± SEM, from a representative experiment from 2 independent experiments with n = 5 (b–c, g–k), n = 5–7 (m–n), and n = 10 (e–f). Data were analyzed by (b, c, j, k) one-way ANOVA or (f, g, i, n) two-way ANOVA with Bonferroni’s multiple comparisons test, or (h) two-tailed multiple t-tests with Bonferroni-Dunn correction, or (f, g) log-rank (Mantel-Cox) test. Figure 5a and 5l were created using BioRender.com.
Benchmarking and validation in multiple tumor models
To further evaluate the potency of CMP, we performed head-to-head comparison studies between CMPCDA and other STING-activating formulations. C57BL/6 mice were inoculated at S.C. flank with 3×105 B16F10 tumors cells, and we administered three doses of CMPCDA on three-day intervals via either I.T. route when the average tumor volume reached 153 ± 17 mm3 (Fig. 6a–e) or via I.V. route when the average tumor volume reached 63 ± 7 mm3. (Fig. 6f–j). We compared CMPCDA with the equivalent dose of four other STING-activating therapeutics, which included CDA-loaded liposomes18; CZP particle system formed by replacing Mn2+ with Zn2+ in CMP (Supplementary Figure 7); ADU-S100, a leading CDN STING agonist tested in clinical trials7; and diABZI, a leading non-CDN STING agonist (used as an I.V. formulation, currently in clinical trials14). After I.T. therapy, CMPCDA significantly delayed the tumor growth, eliminated B16F10 tumors in 30% animals, and prolonged animal survival, whereas other control groups (except for diABZI) showed significantly reduced anti-tumor effects (Fig. 6a–e). Importantly, in the setting of I.V. therapy, CMPCDA also exerted remarkable anti-tumor efficacy, slowing the tumor growth and prolonging animal survival with 20% complete response rate (Fig. 6f–j). In stark contrast, all other control groups (including diABZI) had only minor anti-tumor effects in this difficult-to-treat tumor model. Interestingly, even though CMP and diABZI generated comparable anti-tumor responses after I.T. therapy, CMP significantly outperformed diABZI after I.V. injection (Fig. 6f–j). Moreover, our data showing superiority of CMP to both CZP and CDA-liposomes demonstrates the indispensable role of Mn2+-mediated potentiation of STING agonists as well as the advantages of our coordination-based STING agonist delivery system.
Figure 6. Robust therapeutic effect of CMPCDA in multiple tumor models.
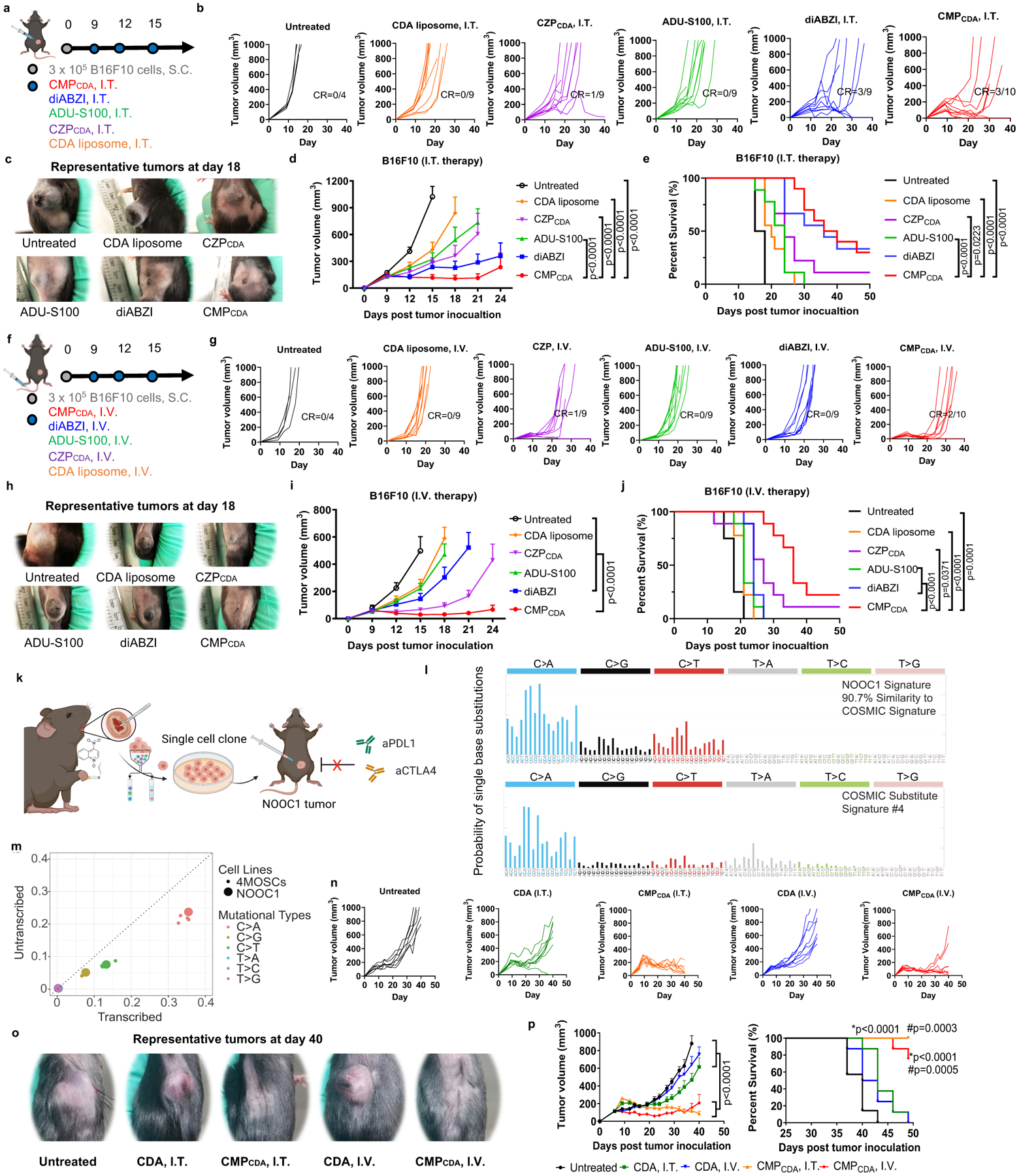
a–j) Therapeutic effect of CMPCDA was compared with other CDA formulations and other STING agonists in an established B16F10 tumor model. a–e) Tumor-bearing C57BL/6 mice were treated with CMPCDA, CDA-Zn particle (CZPCDA), CDA liposome), ADU-S100, or diABZI (all 5 μg doses of STING agonists, I.T.) on the indicated time points (a). Shown are b) the individual tumor growth, c) representative photos of tumors, d) average tumor growth and e) survival. f–j) Tumor-bearing C57BL/6 mice were treated with the indicated regimens (all 20 μg doses, I.V.) (f). Shown are g) the individual tumor growth, h) representative photos of tumors, i) average tumor growth, and j) survival. k–p) Therapeutic effect of CMPCDA in an immune checkpoint blocker (ICB)-resistant tobacco-associated tumor model (NOOC1). k) NOOC1 single-cell clone were isolated from the visible oral squamous cell carcinoma lesions of C57BL/6J mice treated with 4NQO-containing drinking water for 16 weeks. l) Mutational signatures indicate NOOC1 tumors with high fidelity to human cancers. m) Mutational profile of NOOC1 in comparison to other 4NQO-induced murine squamous cell carcinoma cell lines (4MOSCs). n–p) NOOC1 tumor-bearing C57BL/6 mice were treated with CDA in CMPCDA or free form via I.T. (5 μg dose) or I.V. route (20 μg dose) on days 9, 12, 16, and 20 post tumor inoculation. Shown are n) the individual tumor growth, o) representative photos of tumors, p) average tumor growth and survival. Data represent mean ± SEM, from a representative experiment from 2 independent experiments with n = 4–10 (d–e, i–j) and n = 7–8 (n-p). Data were analyzed by two-way ANOVA (d, i, p) with Bonferroni multiple comparisons post-test. Survival of (e, j, p) was analyzed by Kaplan–Meier survival analysis with log-rank (Mantel-Cox) test. *p and #p in (p) denote statistical significance relative to the untreated and CDA groups, respectively. Figure 6a, 6fk, and 6k were created using BioRender.com.
Lastly, we examined the therapeutic efficacy of CMP in a novel tobacco carcinogen-associated syngeneic squamous cell carcinoma model that is completely refractory to high doses of ICB therapy (Fig. 6k–p, Supplementary Fig. 22). Epithelial malignancies, such as the squamous cell carcinomas of the head and neck, only show a modest response to immunotherapy, typically < 15% in the clinics54. To model a cold epithelial malignancy, C57BL/6J mice were given 4NQO-containing (50 μg/mL) drinking water for 16 weeks, and visible oral squamous cell carcinoma lesions were isolated to produce single cell clones, which were then screened in vitro and in vivo. We identified a cell clone (4-NQO-induced Oral Cancer 1, NOOC1) that stably produced tumors when implanted in syngeneic C57BL/6J hosts (Fig. 6k). Whole exome sequencing revealed that the mutational signatures of NOOC1 bore 90.7% similarity to the COSMIC signature #4, which is driven by smoking-associated mutations in human cancers (Fig. 6l). The mutation profile of NOOC1 was highly similar to that of 4MOSCs, a recently reported 4-NQO-induced cell line, thus validating its tobacco-association (Fig. 6l–m). Notably, NOOC1 was refractory to high doses (200 μg × 6 doses) of ICB therapy, including anti-PD-L1 and anti-CTLA4 (Supplementary Fig. 22). To evaluate CMP in this ICB-resistant epithelial malignancy model, mice were inoculated with 2 × 106 NOOC1 tumor cells, and when the average tumor volume reached > 100 mm3, animals were treated on days 9, 12, 16, and 20 with CMPCDA or free CDA. We employed the equivalent CDA dose of 5 μg for I.T. therapy and 20 μg for I.V. therapy. NOOC1 was also refractory to free CDA treatments, regardless of the administration routes. In stark contrast, both CMP I.T. and I.V. therapy exerted robust anti-tumor efficacy, regressing established NOOC1 tumors (P < 0.0001) and extending animal survival (P < 0.001) (Fig. 6n–p).
Notably, each injection dose of Mn2+ in CMPCDA employed in our I.T. (Fig. 4) and I.V. (Fig. 5) treatment studies was 2.5 μg and 10 μg (0.13 mg/kg and 0.5 mg/kg), respectively. For comparison, the LD50 of MnCl2 in mice is 1715 mg/kg55, and average adults on typical Western diets consume up to 10 mg manganese per day56. CMPCDA I.V. therapy transiently increased the serum levels of IFN-β, TNF-α, and IL-6 peaking at 6 hours while inducing a 13.5% maximum body weight loss; however, animals quickly recovered within 3 days, and multiple treatments of CMPCDA or CDA + Mn2+ mixture were generally well-tolerated, as indicated by the serum chemistry and neurotoxicity marker (Supplementary Fig. 24–25). In addition, histological analysis of major organs by a pathologist in a blinded manner showed no abnormal histological conditions (Supplementary Fig. 26).
Conclusions
In summary, we have developed a self-assembled coordination nanomedicine based on Mn2+ and CDN-based STING agonists. CMP represents a major technological advancement to amplify the potency of STING agonists. The combination of Mn2+ and STING agonists dramatically augmented STING activation (Fig. 2). CMP effectively delivered Mn2+ and STING agonists and amplified the type-I IFN responses (Fig. 3–6, Supplementary Fig. 16). CMP administered I.T. produced strong anti-tumor efficacy, achieving drug dose-sparing with minimal side effects (Fig. 4, 6, Supplementary Fig. 11–12). Moreover, systemic treatments with CMP exerted remarkable therapeutic efficacy in multiple difficult-to-treat murine tumor models (Fig. 5–6, Supplementary Fig. 18). Our work presents the concept of “metalloimmunotherapy” and demonstrate, for the first time, powerful potential of nanomedicine-based cancer metalloimmunotherapy. As nutritional metal ions play crucial roles in various immune processes, metalloimmunotherapy may be broadly applicable for other immune-related diseases.
Materials and Methods
Assessing metal ions for modulation of IFN-I response of STING agonists in vitro
Mouse bone marrow-derived dendritic cells (BMDCs) were isolated and cultured as reported previously57. Human monocyte cell line THP1 cells expressing hSTINGHAQ were purchased from ATCC (Manassas, VA) and cultured according to ATCC’s instruction. THP1 cells expressing hSTINGR232 (WT), hSTINGH232 (REF) were purchased from Invivogen and cultured according to Invivogen’s instruction. To screen for metal ions for modulating IFN-I response of STING agonists, we seeded 0.1 million BMDCs or THP1 cells per well in 96-well plate, and metal ions (e.g., ZnCl2, KCl, MgCl2, MnCl2, CaCl2, Al2(SO4)3, CuCl2, FeCl2, FeCl3, and CoCl2) (Sigma-Aldrich) at various concentrations ranging 0–500 μM were added with or without 5 μM cGAMP (Invivogen). After 24 h incubation at 37 °C, 5% CO2, the supernatants were collected for IFN-β ELISA assay (R&D). To evaluate the effect of MnCl2 on IFN-I response of STING agonists in various human STING variants, the indicated concentrations of MnCl2 and STING agonists, including cGAMP, CDA (Invivogen), 2’3’-cGAM(PS)2 (Rp/Sp) (Invivogen), ADU-S100 (MedChemExpress) and diABZI (MedChemExpress), were added to 0.1 million THP1 reporter cells in 96-well plate. After 24 h incubation at 37 °C, 5% CO2, the supernatants were collected and assessed for IFN-β by ELISA.
Synthesis and characterization of CDN-Mn/Zn coordination polymer, CMPCDA, CZPCDA, and CDA liposomes
c-di-AMP, c-di-GMP, or cGAMP (Invivogen) were dissolved in methanol in 1 mg/ml. MnCl2 or ZnCl2 (Sigma-Aldrich) was dissolved in methanol to prepare 100 mM stock solution. In a typical synthesis reaction, MnCl2 or ZnCl2 (Sigma-Aldrich) solution was added to 1 mg/ml CDNs solution in a 10:1 (n/n) ratio under vigorous stirring. The mixture was sonicated for 1 min and stirred for another 1 h at room temperature. The resulting CDN-Mn was centrifuged at 20000 × g for 10 min to remove free CDNs and metal ions, followed by washing with methanol.
CMP was synthesized according to a method adapted from previous reports58–60. First, dioleoyl-sn-glycero-3-phosphoethanolamine-N-[histidine]11 (DOPE-H11) was synthesized by reaction of DOPE-NHS and H11 (2 eq) in DMF, purified by dialysis using 2KD MWCO dialysis tubes, and characterized by HPLC. A mixture containing 1ml of 1 mg/ml CDA in methanol, 0.14 ml of 100 mM MnCl2 in methanol and 2 ml of 2 mg/ml DOPE-H11 in ethanol was sonicated and then vortexed overnight, followed by centrifugation at 20000 × g for 10 min. The resulting CDA-Mn@DOPE was resuspended in ethanol containing DOPC: cholesterol: DSPE-PEG5000 (4:1:1), sonicated, and added into a solution of 30% (v/v) ethanol/H2O. Lastly, CMP was obtained by evaporating the organic solvent under reduced pressure and washing with 10% sucrose using 100KD (MWCO) centrifugal ultrafiltration. CZP was synthesized using the same method except for replacing MnCl2 with ZnCl2. CDA liposomes were synthesized as reported previously18.
Loading of CDA in CMP, CZP, and CDA liposomes was quantified by UV-absorbance at 260 nm, followed by verification by HPLC. Loading of Mn2+ in CMP was quantified by inductively coupled plasma-mass spectrometry (The Perkin-Elmer Nexion 2000 ICP-MS) and verified by thermogravimetric analysis (Discovery TGA, TA Instrument, New Castle, DE). The size and surface charge of CMP were measured by Zetasizer (Nano ZSP, Malvern, UK). The morphology of CDN-Mn was observed by transmission electron microscopy (TEM). All images were acquired on JEM 1200EX electron microscope (JEOL USA, Peabody, MA) equipped with an AMT XR-60 digital camera (Advanced Microscopy Techniques Corp. Woburn, MA).
In vitro evaluation of BMDC activation, cellular uptake, and STING activation
BMDCs were prepared as described previously57. Briefly, bone marrow was harvested and plated in bacteriological Petri dishes with GM-CSF containing culture media. The cell culture media were refreshed on days 3, 6, and 8. After 8 days of differentiation, BMDC were harvested for use. To observe BMDC activation by CDNs and Mn2+, 1×106/well BMDCs seeded in 12-well plate were incubated with 5 μM CDA and/or 250 μM Mn2+ for 24 h. Treated BMDCs were harvested, washed with FACS buffer (1% BSA in PBS), incubated with anti-CD16/32 at room temperature, and then stained on ice with fluorophore-labeled antibodies against CD11c, CD80, and CD86. Cells were then washed twice by FACS buffer, resuspended in 2 μg/ml DAPI solution, and analyzed by flow cytometry (Ze 5 with Everest Software v.3.0.75., Bio-Rad, USA). Data were processed by FlowJo v.10.5.
To visualize and quantify cellular uptake of STING agonist, fluorophore-labeled CDN, CDG-Dy547 (Biolag, Bremen, German), was admixed with CDA (1:10, n/n) to prepare CDG-Dy547@CMPCDA following the same synthesis procedure as CMPCDA mentioned above. Loading of CDG-Dy547 in CMP was quantified by absorbance at 550 nm. BMDCs were seeded at 1 × 106 cells on 35 mm Petri dishes (MatTek Corp., Ashland, MA) and incubated with CDG-Dy547 in free form or in CDG-Dy547@CMPCDA for 6, 12, or 24 h. For confocal imaging, cells were washed 3 times with PBS, incubated with 50 nM LysoTracker® green DND-99 (Invitrogen) for 30 min at 37 °C to stain lysosomes, and then imaged using a confocal microscope (Nikon A1). For cellular uptake quantification, cells were harvested and washed with FACS buffer (1% BSA in PBS). The fluorescence of CDG-Dy547 was analyzed by flow cytometry (Ze 5 with Everest Software v.3.0.75., Bio-Rad, USA) and data were processed by FlowJo v.10.5.
To measure STING activation of CDA and/or Mn2+ in free form or in CMPCDA, 1 × 105/well BMDCs were seeded in 96-well plate and incubated with CDA and/or Mn2+ in free form or in CMPCDA (containing 2.5 μM CDA or/and 15.6 μM Mn2+). After 24 h incubation at 37 °C, 5% CO2, the supernatants were collected for ELISA assay of cytokines at the Cancer Center Immunology Core of the University of Michigan.
In vivo cancer immunotherapy
All animals were cared for following federal, state, and local guidelines. All work performed on animals was in accordance with and approved by the Institutional Animal Care & Use Committee (IACUC) at the University of Michigan, Ann Arbor. For CT26 murine tumor model, female BALB/c mice of age 6–8 weeks (Jackson Laboratories) were inoculated with 1.5 × 105 CT26 colon cancer cells subcutaneously on the right back flank. For CT26 two-tumor model, 3 × 105 and 1 × 105 CT26 tumor cells were inoculated on the right (primary) and left (distal) flank, respectively. For the B16F10 tumor model, C57BL/6 mice (Jackson Laboratory) were inoculated with indicated number of B16F10 cells subcutaneously on the right flank. Tumor-bearing mice were randomly assigned to different treatment groups. Indicated drugs or formulations were administrated via indicated route at indicated time points. Tumor size and survival were monitored every 2–4 days. Tumor size was calculated based on equation: volume = length × width2 × 0.5. Animals were euthanized when the tumor reached 1.5 cm in diameter or when animals became moribund with severe weight loss or un-healing ulceration. At indicated time points, the cytokine levels in serum were measured by ELISA assay in the Cancer Center Immunology Core of the University of Michigan. The percentages of tumor antigen-specific CD8α+ T cells among PBMC were analyzed using the tetramer staining assay as described previously with AH1 peptide-MHC tetramer (H-2Kb-restricted AH1 (SPSYVYHQF)) (the NIH Tetramer Core Facility, Atlanta, GA). ELISPOT assay was performed with PBMCs from the treated mice, as described previously61.
NOOC1 was maintained in the IMDM media (Gibco cat#12440053). To make 1L of growth media for NOOC1, 626 ml IMDM base was mixed with 313 ml F-12 nutrient mix (Gibco cat#11765054), 50 ml FBS (Hyclone cat#SH3039603), 10 ml Pen Strep (Thermo Fisher cat#15-140-122), 1.25ml of 4mg/ml insulin (Invitrogen cat#12585014), 200 μl of 200 μg/ml hydrocortisone (Sigma-Aldrich cat#H0888–1G), and 50 μl of 100 μg/ml EGF (EMD Millipore cat#01–107). For in vivo implantation, Matrigel (Thermo Fisher cat#CB-40230) was thawed overnight at 4°C. On the day of injection, NOOC1 was washed once with PBS and mixed with Matrigel to reach a density of 2×107 cells/ml. Each mouse was inoculated subcutaneously with 2×106 cells (100 μl). Tumor size and survival were monitored every 2 or 3 days as indicated above.
In vivo immune response analysis
T cell phenotypic and functional assessment in PBMC were analyzed. Briefly, PBMCs were collected after removing red blood cells by ACK lysis buffer. Fc receptor of PBMCs was blocked with CD16/32 antibody for 10 min. To analyze tumor antigen-specific CD8+ T cells response and T cell memory phenotype, cell pellets were first stained with PE-tagged AH1 peptide-MHC tetramer, and then further stained with APC-CD8a rat anti-mouse (Clone: 53–6.7(RUO), BD Biosciences), with FITC-CD44 rat anti-human/mouse (Clone: IM7, eBioscience) and PE-Cy7-CD62L monoclonal antibody (Clone: MEL-14, eBioscience). Stained cells were incubated with DAPI prior to flow cytometry analysis (Ze5 with Everest Software v.3.0.75., Bio-Rad).
For the IFN-γ ELISPOT assay, ELISPOT plate was coated with IFN-γ capture antibody for 24 h and blocked with DMEM + 10% FBS for 2 h. PBMCs obtained from treated mice were added to 96-well plate with a fixed number of alive cells/well. SPSYVYHQF peptide (20 μg/mL) was added to stimulate PBMCs. Ionomycin and PMA were employed as positive control. After 18 h, IFN-γ spots were detected with biotinylated detection antibody, followed by streptavidin-HRP and AEC substrate kit. The IFN-γ spot number and size were measured in the Cancer Center Immunology Core at the University of Michigan.
Tumor tissues were analyzed as described before52. Tumor tissues were excised at preset time points and cut into small pieces, which were treated additionally with collagenase type IV (1 mg/ml) and DNase I (0.1 mg/ml) under gentle shaking. After 30 min, the cell suspension was filtered through a 70-μm strainer. The cells were washed with fluorescence-activated cell sorting (FACS) buffer and blocked with CD16/32 antibody. Cells were then stained with designated antibodies panels: APC-Cy7-Anti-mouse CD45 Antibody (Clone: 30-F11, BioLegend), Pacific Blue-Anti-mouse CD8a Antibody (Clone: 53–6.7, BioLegend), FITC Rat Anti-mouse CD49b (Clone: DX5, BioLegend), BV786-Anti-CD3 Antibody (Clone: 17A2, BD Biosciences), APC-Anti-mouse CD107a Antibody (1D4B (RUO), BD Biosciences), PE-Cy7- Anti-mouse CD11c Antibody (Clone: HL3 (RUO), BD Biosciences), FITC-Anti-MHC-II Antibody (Clone: M5/114.15.2, eBioscience), BV605-Anti-mouse CD86 Antibody (Clone: GL1, BD Bioscience); Pacific Blue-Anti-mouse CD45 Antibody (Clone: 30-F11, BioLegend), APC-Cy7-Anti-mouse CD3 Antibody (Clone: 17A2, BD Bioscience), APC-Anti-mouse CD8a Antibody (Clone: 53–6.7(RUO), BD Biosciences), FITC Rat Anti-mouse CD49b (Clone: DX5, BioLegend), BV786-Anti-CD3 Antibody (Clone: 17A2, BD Biosciences), PE-Cy7- Anti-mouse CD11c Antibody (Clone: HL3 (RUO), FITC-Anti-MHC-II Antibody (Clone: M5/114.15.2, eBioscience), BV605-Anti-mouse/human CD11b (Clone: M1/70, BioLegend), PE-Anti-mouse F4/80 (Clone: BM8, BioLegend), APC-Anti-mouse CD206 (MMR, Clone: C068C2, BioLegend), PE-Cy7-Anti-mouse CD86 Antibody (Clone: GL1, BD Bioscience), FITC-Anti-mouse Ly6C Antibody (Clone: AL-21, BD Bioscience); BV421-Anti-mouse Ly6G Antibody (Clone: RB6–8C5, BioLegend); BV605-Anti-mouse CD45 Antibody (Clone: 30-F11, BioLegend), APC-Cy7-Anti-mouse/human CD11b (Clone: M1/70, BioLegend), PE-Anti-mouse F4/80 (Clone: BM8, BioLegend), APC-Anti-mouse CD206 (MMR, Clone: C068C2, BioLegend), PE-Cy7-Anti-mouse CD86 Antibody (Clone: GL1, BD Bioscience), BV421-Anti-mouse Ly6G Antibody (Clone: RB6–8C5, BioLegend), FITC-Anti-mouse Ly6C Antibody (Clone: AL-21, BD Bioscience). Stained cells were analyzed by cytometry (MoFlo Astrios Cell Sorter, Beckman). Data were processed by FlowJo v.10.5.
In vivo drug distribution analysis
To analyze in-vivo biodistribution of STING agonist, CDG-Dy547 (Biolag, Bremen, German) or CDG-Cy7 (Biolag, Bremen, German), was admixed with CDA (1:10, n/n) to prepare CDG-Dy547@CMPCDA or CDG-Cy7@CMPCDA following the same synthesis procedure as CMPCDA mentioned above. Loading of CDG-Dy547 and CDG-Cy7 were quantified by absorbance at 550 nm or 750 nm. To quantify drug retention at the tumor site after intratumoral injection, CDG-Cy7 in free form or in CMPCDA were injected into tumors. Mice were imaged by IVIS at different time points (0 h, 4 h, 8 h, 24 h), and the fluorescence signal of CDG-Cy7 in tumors was measured accordingly. Drug retention was calculated by normalizing the remaining fluorescence signal of CDG-Cy7 in tumor on the indicated time point by that of injected CDG-Cy7 at 0 h. To quantify the biodistribution of CMP after I.V. administration, CDG-Cy7 in free form or in CMPCDA was injected I.V. Mice were euthanized 24 h post-injection, and the fluorescence intensity in major organs was measured accordingly. To quantify the biodistribution of Mn, tissues were microwave digested, and the amount of Mn was measured using ICP-MS by Michigan Elemental Analysis Laboratory.
To analyze drug distribution in TME, CDG-Dy547@CMPCDA was injected via I.T. or I.V. routes. On the indicated time point, cells were harvested from TME and stained, as mentioned above. The amount of phagocytosed CDG-Dy547 in different cell populations was analyzed by FACS. Cells were then stained with designated antibodies panels: Pacific Blue-anti-mouse CD45 antibody (Clone: 30-F11, BioLegend), BV605-anti-mouse CD3 antibody (Clone: 17A2, BioLegend), APC-anti-mouse CD11c antibody (Clone: N418, BioLegend), FITC-anti-MHC-II antibody (Clone: M5/114.15.2, eBioscience), BV605-anti-mouse/human CD11b (Clone: M1/70, BioLegend), APC-anti-mouse F4/80 (Clone: BM8, BioLegend), FITC-anti-mouse Ly6C antibody (Clone: AL-21, BD Bioscience). Stained cells were analyzed by cytometry (MoFlo Astrios Cell Sorter, Beckman). Data were processed by FlowJo v.10.5.
Statistical analysis.
The results are expressed as means ± SEM. A one-way or two-way ANOVA, followed by Bonferroni’s multiple comparisons post hoc test, or two-tailed multiple t-tests with Bonferroni-Dunn correction, was used for testing differences among groups. Data were approximately normally distributed, and variance was similar between the groups. Experiments were repeated multiple times as independent experiments as indicated in the figure captions. Shown in the figure is a complete dataset from one representative, independent experiment. No samples were excluded from analysis. GraphPad Prism 8.0 (GraphPad Software, La Jolla, CA) was used for statistical analyses.
Supplementary Material
Acknowledgments
This work was supported in part by NIH (R01AI127070, R01CA210273, U01CA210152, R01DK108901, R01DE026728, R01DE030691), the University of Michigan Rogel Cancer Center Support Grant (P30CA46592), and the University of Michigan, Michigan Drug Discovery (MDD21102). J.J.M. is supported by NSF CAREER Award (1553831). L.W. was supported in part by NIH U24CA232979 and R01CA255242. X.S. is supported by Rackham International Student Fellowship and Rackham Predoctoral Fellowship. We acknowledge Justin Hong for aiding with isothermal titration calorimetry (ITC) analysis, Angela Dial at Michigan Element Analysis Lab for Mn biodistribution analysis, Krishnapriya Chinnaswamy at University of Michigan Center for Structural Biology for aiding with protein thermal shift assay, Joel Whitfield at the University of Michigan Cancer Center Immunology Core for ELISA analysis, Dr. Carlson Heather at University of Michigan for molecular dynamic analysis, and Qiaoling Zheng at Fujian Medical University Union Hospital for histological analysis. We also thank the University of Michigan Flow Cytometry Core, the ULAM (Unit for Laboratory Animal Medicine) In Vivo Animal Core (IVAC), and the University of Michigan Microscope Imaging Core for technical assistants. We acknowledge the NIH Tetramer Core Facility (contract HHSN272201300006C) for the provision of MHC-I tetramers.
Footnotes
Competing Interests Statement
A patent application (WO2020014644A1) for CMP-based metalloimmunotherapy has been filed, with J.J.M. and X.S. as inventors. The remaining authors declare no competing interests.
Additional Information
Supplementary Information is available.
Data Availability Statement
The authors declare that data supporting the findings of this study are available within the article and its Supplementary Information files. All relevant data can be provided by the authors upon reasonable request.
REFERENCES
- 1.Couzin-Frankel J Cancer immunotherapy. Science 342, 1432–1433 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Gubin MM et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 515, 577–581 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee DW et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. The Lancet 385, 517–528 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Syn NL, Teng MW, Mok TS & Soo RA De-novo and acquired resistance to immune checkpoint targeting. The Lancet Oncology 18, e731–e741 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Duan Q, Zhang H, Zheng J & Zhang L Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends Cancer 6, 605–618, doi: 10.1016/j.trecan.2020.02.022 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Sun L, Wu J, Du F, Chen X & Chen ZJ Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 339, 786–791 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Corrales L et al. Direct activation of STING in the tumor microenvironment leads to potent and systemic tumor regression and immunity. Cell reports 11, 1018–1030 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flood BA, Higgs EF, Li S, Luke JJ & Gajewski TF STING pathway agonism as a cancer therapeutic. Immunol Rev 290, 24–38, doi: 10.1111/imr.12765 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shae D et al. Endosomolytic polymersomes increase the activity of cyclic dinucleotide STING agonists to enhance cancer immunotherapy. Nat Nanotechnol 14, 269–278, doi: 10.1038/s41565-018-0342-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schadt L et al. Cancer-Cell-Intrinsic cGAS Expression Mediates Tumor Immunogenicity. Cell Rep 29, 1236–1248 e1237, doi: 10.1016/j.celrep.2019.09.065 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Nicolai CJ et al. NK cells mediate clearance of CD8(+) T cell-resistant tumors in response to STING agonists. Sci Immunol 5, doi: 10.1126/sciimmunol.aaz2738 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harrington KJ et al. Preliminary results of the first-in-human study of MK-1454, an agonist of stimulator of interferon genes (STING), as monotherapy or in combination with Pembrolizumab (Pembro) in patients with advanced solid tumors or lymphomas. ESCO, Abstract 5475 (2018). [Google Scholar]
- 13.Meric-Bernstam F et al. Phase I dose-finding study of MIW815 (ADU-S100), an intratumoral STING agonist, in patients with advanced solid tumors or lymphomas. SITC, Poster 309 (2018). [Google Scholar]
- 14.Ramanjulu JM et al. Design of amidobenzimidazole STING receptor agonists with systemic activity. Nature 564, 439 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Chin EN et al. Antitumor activity of a systemic STING-activating non-nucleotide cGAMP mimetic. Science 369, 993–999, doi: 10.1126/science.abb4255 (2020). [DOI] [PubMed] [Google Scholar]
- 16.Pan BS et al. An orally available non-nucleotide STING agonist with antitumor activity. Science 369, doi: 10.1126/science.aba6098 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Gajewski TF & Higgs EF Immunotherapy with a sting. Science 369, 921–922, doi: 10.1126/science.abc6622 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Koshy ST, Cheung AS, Gu L, Graveline AR & Mooney DJ Liposomal Delivery Enhances Immune Activation by STING Agonists for Cancer Immunotherapy. Adv Biosyst 1, doi: 10.1002/adbi.201600013 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan YS et al. Mitigating SOX2-potentiated Immune Escape of Head and Neck Squamous Cell Carcinoma with a STING-inducing Nanosatellite Vaccine. Clinical cancer research : an official journal of the American Association for Cancer Research 24, 4242–4255, doi: 10.1158/1078-0432.CCR-17-2807 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y et al. An inhalable nanoparticulate STING agonist synergizes with radiotherapy to confer long-term control of lung metastases. Nature communications 10, 5108, doi: 10.1038/s41467-019-13094-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He Y et al. Self-assembled cGAMP-STINGDeltaTM signaling complex as a bioinspired platform for cGAMP delivery. Sci Adv 6, eaba7589, doi: 10.1126/sciadv.aba7589 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li S et al. Prolonged activation of innate immune pathways by a polyvalent STING agonist. Nat Biomed Eng, doi: 10.1038/s41551-020-00675-9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaigne-Delalande B & Lenardo MJ Divalent cation signaling in immune cells. Trends Immunol 35, 332–344, doi: 10.1016/j.it.2014.05.001 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang C, Zhang R, Wei X, Lv M & Jiang Z Metalloimmunology: The metal ion-controlled immunity. Adv Immunol 145, 187–241, doi: 10.1016/bs.ai.2019.11.007 (2020). [DOI] [PubMed] [Google Scholar]
- 25.Macian F NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol 5, 472–484, doi: 10.1038/nri1632 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Shi X et al. Ca2+ regulates T-cell receptor activation by modulating the charge property of lipids. Nature 493, 111–115, doi: 10.1038/nature11699 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Chandy KG & Norton RS Immunology: Channelling potassium to fight cancer. Nature 537, 497–499, doi: 10.1038/nature19467 (2016). [DOI] [PubMed] [Google Scholar]
- 28.Vodnala SK et al. T cell stemness and dysfunction in tumors are triggered by a common mechanism. Science 363, doi: 10.1126/science.aau0135 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Munoz-Planillo R et al. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 38, 1142–1153, doi: 10.1016/j.immuni.2013.05.016 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rossol M et al. Extracellular Ca2+ is a danger signal activating the NLRP3 inflammasome through G protein-coupled calcium sensing receptors. Nat Commun 3, 1329, doi: 10.1038/ncomms2339 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Scambler T et al. ENaC-mediated sodium influx exacerbates NLRP3-dependent inflammation in cystic fibrosis. Elife 8, doi: 10.7554/eLife.49248 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hood MI & Skaar EP Nutritional immunity: transition metals at the pathogen-host interface. Nat Rev Microbiol 10, 525–537, doi: 10.1038/nrmicro2836 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bessman NJ et al. Dendritic cell-derived hepcidin sequesters iron from the microbiota to promote mucosal healing. Science 368, 186–189, doi: 10.1126/science.aau6481 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang C et al. Manganese increases the sensitivity of the cGAS-STING pathway for double-stranded DNA and is required for the host defense against DNA viruses. Immunity 48, 675–687. e677 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Du M & Chen ZJ DNA-induced liquid phase condensation of cGAS activates innate immune signaling. Science 361, 704–709, doi: 10.1126/science.aat1022 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chaigne-Delalande B et al. Mg2+ regulates cytotoxic functions of NK and CD8 T cells in chronic EBV infection through NKG2D. Science 341, 186–191, doi: 10.1126/science.1240094 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lv M et al. Manganese is critical for antitumor immune responses via cGAS-STING and improves the efficacy of clinical immunotherapy. Cell Res 30, 966–979, doi: 10.1038/s41422-020-00395-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hou L et al. Manganese-Based Nanoactivator Optimizes Cancer Immunotherapy via Enhancing Innate Immunity. ACS nano 14, 3927–3940, doi: 10.1021/acsnano.9b06111 (2020). [DOI] [PubMed] [Google Scholar]
- 39.Chen C et al. Cytosolic Delivery of Thiolated Mn-cGAMP Nanovaccine to Enhance the Antitumor Immune Responses. Small 17, e2006970, doi: 10.1002/smll.202006970 (2021). [DOI] [PubMed] [Google Scholar]
- 40.Yang X et al. Converting primary tumor towards an in situ STING-activating vaccine via a biomimetic nanoplatform against recurrent and metastatic tumors. 38, 101109 (2021). [Google Scholar]
- 41.Aschner JL & Aschner M Nutritional aspects of manganese homeostasis. Mol Aspects Med 26, 353–362, doi: 10.1016/j.mam.2005.07.003 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang C Mangafodipir trisodium (MnDPDP)-enhanced magnetic resonance imaging of the liver and pancreas. Acta Radiol Suppl 415, 1–31 (1998). [PubMed] [Google Scholar]
- 43.Takagi Y et al. Evaluation of indexes of in vivo manganese status and the optimal intravenous dose for adult patients undergoing home parenteral nutrition. Am J Clin Nutr 75, 112–118, doi: 10.1093/ajcn/75.1.112 (2002). [DOI] [PubMed] [Google Scholar]
- 44.Pan D, Schmieder AH, Wickline SA & Lanza GM Manganese-based MRI contrast agents: past, present and future. Tetrahedron 67, 8431–8444, doi: 10.1016/j.tet.2011.07.076 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jin L et al. MPYS is required for IFN response factor 3 activation and type I IFN production in the response of cultured phagocytes to bacterial second messengers cyclic-di-AMP and cyclic-di-GMP. J Immunol 187, 2595–2601, doi: 10.4049/jimmunol.1100088 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Li L et al. Hydrolysis of 2’3’-cGAMP by ENPP1 and design of nonhydrolyzable analogs. Nat Chem Biol 10, 1043–1048, doi: 10.1038/nchembio.1661 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thanos D & Maniatis T Virus induction of human IFN beta gene expression requires the assembly of an enhanceosome. Cell 83, 1091–1100, doi: 10.1016/0092-8674(95)90136-1 (1995). [DOI] [PubMed] [Google Scholar]
- 48.Wang J et al. NF-kappa B RelA subunit is crucial for early IFN-beta expression and resistance to RNA virus replication. J Immunol 185, 1720–1729, doi: 10.4049/jimmunol.1000114 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ting JP, Duncan JA & Lei Y How the noninflammasome NLRs function in the innate immune system. Science 327, 286–290, doi: 10.1126/science.1184004 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu S et al. Phosphorylation of innate immune adaptor proteins MAVS, STING, and TRIF induces IRF3 activation. Science 347, aaa2630, doi: 10.1126/science.aaa2630 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Shin HM et al. Inhibitory action of novel aromatic diamine compound on lipopolysaccharide-induced nuclear translocation of NF-kappaB without affecting IkappaB degradation. FEBS Lett 571, 50–54, doi: 10.1016/j.febslet.2004.06.056 (2004). [DOI] [PubMed] [Google Scholar]
- 52.Kuai R et al. Elimination of established tumors with nanodisc-based combination chemoimmunotherapy. Sci Adv 4, eaao1736, doi: 10.1126/sciadv.aao1736 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sivick KE et al. Magnitude of Therapeutic STING Activation Determines CD8(+) T Cell-Mediated Anti-tumor Immunity. Cell Rep 25, 3074–3085 e3075, doi: 10.1016/j.celrep.2018.11.047 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Luo X et al. HPV16 drives cancer immune escape via NLRX1-mediated degradation of STING. J Clin Invest 130, 1635–1652, doi: 10.1172/JCI129497 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lewis R & Tatken R Registry of Toxic Effects of Chemical Substances, Vol. 1. US Department of Health and Human Services, National Institute for Occupational Safety and Health, Washington, DC, 252 (1980). [Google Scholar]
- 56.Greger JL Nutrition versus toxicology of manganese in humans: evaluation of potential biomarkers. Neurotoxicology 20, 205–212 (1999). [PubMed] [Google Scholar]
- 57.Lutz MB et al. An advanced culture method for generating large quantities of highly pure dendritic cells from mouse bone marrow. J Immunol Methods 223, 77–92, doi: 10.1016/s0022-1759(98)00204-x (1999). [DOI] [PubMed] [Google Scholar]
- 58.Liu D, Poon C, Lu K, He C & Lin W Self-assembled nanoscale coordination polymers with trigger release properties for effective anticancer therapy. Nature communications 5, 4182, doi: 10.1038/ncomms5182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu J et al. Light-controlled drug release from singlet-oxygen sensitive nanoscale coordination polymers enabling cancer combination therapy. Biomaterials 146, 40–48, doi: 10.1016/j.biomaterials.2017.09.007 (2017). [DOI] [PubMed] [Google Scholar]
- 60.Yang Y et al. One-pot synthesis of pH-responsive charge-switchable PEGylated nanoscale coordination polymers for improved cancer therapy. Biomaterials 156, 121–133, doi: 10.1016/j.biomaterials.2017.11.038 (2018). [DOI] [PubMed] [Google Scholar]
- 61.Kuai R et al. Subcutaneous Nanodisc Vaccination with Neoantigens for Combination Cancer Immunotherapy. Bioconjug Chem 29, 771–775, doi: 10.1021/acs.bioconjchem.7b00761 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that data supporting the findings of this study are available within the article and its Supplementary Information files. All relevant data can be provided by the authors upon reasonable request.