

Abstract
Fluorescent probes have gained profound use in biotechnology, drug discovery, medical diagnostics, molecular and cell biology. The development of methods for the translation of fluorophores into fluorescent probes continues to be a robust field for medicinal chemists and chemical biologists, alike. Access to new experimental designs has enabled molecular diversification and led to the identification of new approaches to probe discovery. This review provides a synopsis of the recent lessons in modern fluorescent probe discovery.
Keywords: chemical biology, fluorophores, fluorescence, molecular probes, sensors
Graphical Abstract
Fluorescent probes play fundamental roles in nearly all aspects of molecular science. This review provides an overview of the methods used to discover fluorescent natural products, beginning with classical synthetic approaches. Here, lessons are presented that provide modern examples for classical methods and identify new classics that chart novel avenues for probe discovery. The central focus of this work embodies an understanding of the fundamental properties that guide the development of fluorescent probes and the challenges within their design.
1. Introduction
Fluorescence is a form of light emission exhibited by certain substances as a result of exposure to external radiation of a shorter wavelength. It is believed to be first observed in 1565 by Monardes[1] as a blue tinge in the infusion Lignum nephriticum, derived from a Mexican medicinal tree Eysenhardtia polystachya. The same tree is believed to have been used by Aztec healers, who also observed a blue color in an infusion used to treat urinary disorders.[2] Although the investigation of this plant revealed no fluorescent compounds, it was recently found that oxidation of one of its flavonoids gives rise to a polycyclic fluorophore, matlaline (Figure 1a), which fluoresces sky-blue in aqueous solutions.[3]
Figure 1.

Exemplary fluorophores and fluorescent probes. a) Structure of matlaline, the presumptive first observed fluorescent compound. b) Fluorophore-conjugated phalloidin. Early work by Wieland in 1979 was one of the first studies on semi-synthetic preparation of a fluorescent derivative of phalloidin, where the sphere represents the position of the fluorescent tag. c) Structure of dansyl chloride and its amino acid conjugates.
Since these sixteenth-century observations, fluorescence has been investigated over the centuries by Boyle, Newton, Herschel, among others.[4] Fluorescence-based techniques have transformed virtually all areas of basic and applied life sciences, including genomics, proteomics, medical diagnoses, drug discovery, microscopy and many others.[5] In comparison to other analytical methods[6] such as high-performance liquid chromatography, mass spectroscopy, capillary electrophoresis or electrochemical assays, fluorescent techniques display high sensitivity, low detection limit, fast response, and great potential for visualizing biochemical processes in cells and bioimaging in living organisms.[7,8]
Fluorescent probes are molecules that can change their fluorescence in response to binding with a specific target, chemical reaction, or change in their immediate environment. Historically, core fluorescent motifs (fluorophores) have originated from synthetic studies of organic dyes as well as those derived from natural sources.[9] Examples include fluorescein, rhodamine, sulfonamides, quinine, coumarins, naphthalimides, and the BODIPY dyes. As illustrated in one of the first efforts to prepare a fluorescent probe, the fluorophore fluorescein was conjugated with phalloidin (Figure 1b), first isolated by Lynen and Wieland in 1938[10] from the poisonous mushroom Amanita phalloides and belonging to a class of toxins known as phallotoxins.[11] Fluorescent phallotoxin probes are widely used to localize actin filaments in fixed cells, permeabilized cells and tissues. Actin is a family of multifunctional proteins that form microfilaments and are found in all eukaryotic cells. Here, phalloidin selectively binds to filamentous actin (F-actin) and prevents its depolymerization.[12]
Another elegant example of a small organic fluorophore is 1-dimethylaminonaphthalene-5-sulfonyl chloride commonly known as dansyl chloride (Figure 1c). Introduced by Weber,[13] the dansyl fluorophore gains its utility due to its high fluorescence quantum yields and large stokes shifts.[14] It is well known for amino acid modifications and specifically for protein sequencing and amino acid analysis.[15] One of the notable applications of dansyl chloride is the formation of dansyl amino acids, which exhibit a bright yellow fluorescence.[16]
A successful fluorescent probe is selective towards a chosen analyte in the environment dictated by the nature of its selected target. Even small changes in the probe structure can be crucial in determining its applicability. In order to design an efficient probe, the precise control of absorption and emission wavelengths is of critical importance. Also, such spectral properties such as large extinction coefficients and high quantum yield are particularly desirable and cannot be over-looked. This review critically discusses various design considerations that go into the discovery of successful fluorescent probes and provides instructive examples from the recent literature. Each chapter focuses on a specific approach to fluorescent probe discovery. Throughout this manuscript, we have provided spheres next to each structure providing the color of each fluorescent probe, as illustrated by blue, green, and chartreuse in Figure 1. Black spheres denote non-fluorescent materials (see DiFMUP, Figure 2).
Figure 2.

Asian scorpion Mesobuthus martensii fluoresces under UVA light due to accumulated coumarins, such as 4-methylumbelliferone. The latter natural product has evolved to synthetic derivatives, such as Marina Blue, as a fluorescent reporter. Its phosphorylated derivative, DuFMUP is used as a probe for phosphatase activity. The image of the scorpion was used without editing and with permission from the authors under Creative Commons Attribution-NonCommercial-ShareAlike 3.0.[20]
2. Natural Product-Based Fluorescent Probe Discovery
The natural selection process has endowed natural products with large and unique structural diversity that has been ideally optimized for interactions with biomolecules. They have historically been an important source of inspiration for the design of small molecules with specific biological functions. This has had a tremendous impact on drug discovery, where natural products, in addition to serving as approved drugs themselves, have also given rise to fluorescently derived probes that enable the discovery of new therapeutic targets.[17] Natural products have further been an important source of molecules that modulate and are used to study biological processes.[18] In contrast, the “occasional” fluorescence exhibited by natural products has been primarily treated with curiosity by the scientific community. In this section, we describe how natural product fluorescence provides an excellent starting point for developing fluorescent probes.[18a]
2.1. An example of a coumarin probe creatively designed on the basis of an animal-derived fluorescent molecule
Although functional molecules based on the coumarin (benzo-pyran-2-one) framework have been broadly utilized for biological and pharmaceutical applications, it is not commonly appreciated that these compounds constitute a ubiquitous class of plant-derived secondary metabolites with over 1400 members.[19] These metabolites find their way into herbivorous insects preyed upon by higher animals such as scorpions. Among these, an Asian scorpion Mesobuthus martensii, strongly fluoresces under UV light, and this fluorescence is attributable to the presence of 4-methylumbelliferone in the scorpion’s exoskeleton (Figure 2).[18a] Data suggest that the scorpions acquire 4-methylcoumarin from herbivorous insects and detoxify it by converting to fluorescent 7-hydroxylated derivative 4-methylumbelliferone using cytochrome P450 metabolism.[21]
Such 7-hydroxylated derivatives have found utility as fluorescent probes, but their applications are limited due to the protonated nature of the phenolic hydroxyl at pH ≤ 7 as the fluorophore is maximally fluorescent in the ionized form.[18a,21] For example, glycosidases and phosphatases have optimal turnover rate at pH 7 or below and, thus, the sensitivity of assays based on 7-hydroxycoumarin-based fluorescent conjugate is low. To increase the acidity of the coumarin probe, a new analogue was designed incorporating fluorine atoms flanking the phenolic hydroxyl in Marina Blue (Figure 2). This decreased the pKa value from 7.8 for 4-methylumbelliferone to 4.7 for Marina Blue. The incorporation of fluorine substituents also improved photostability and quantum yield.[21]
The performance of Marina Blue relative to 4-methylumbelliferone was tested in a phosphatase activity assay.[22] The starting coumarins were phosphorylated to create fluorogenic substrates, 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP, Figure 2) and 4-methylumbelliferyl phosphate (MUP, not shown), and at the preferred acid phosphatase reaction pH of 5.0, DiFMUP was found to yield fluorescence signals that were 10-fold higher than those of MUP.
2.2. A design of difluoroboron derivatized curcumin as a near-infrared probe for in vivo detection of amyloid-β deposits
Amyloid-β deposits are viewed as a pathological hallmark of Alzheimer’s disease (AD) and their molecular imaging represents a promising approach for the detection of early-stage asymptomatic AD.[23] Of many techniques, near-infrared imaging (NIR) is most promising due to acceptable tissue penetration depth, non-invasiveness, and inexpensive instrumentation.[24] It was reported that curcumin (Figure 3) could be used as a histological staining agent for senile plaques and could decrease amyloid deposits in vivo.[25] Curcumin is a brightly colored spice, a key component of Indian curry that has been used for thousands of years in Asia and reported to have anticancer, anti-inflammatory, and antioxidant properties.[26] Although curcumin has considerable specificity for amyloid plaques and binds to amyloid-β aggregates with high affinity (Kd value of 0.2 nM), it is not a practical probe for in vivo NIR imaging primarily because of its short emission wavelength.
Figure 3.

Advancement from yellow curcumin to red rosocyanine and the NIR probe CRANAD-2. The color change from yellow (curcumin) to red (rosocyanine) could be explained by π→π* transition from oxygen to the empty orbital on boron. In addition, the replacement of phenolic hydroxyls in rosocyanine by N,N’-dimethyl groups in GRANAD-2 enabled a further red shift in emission.
To design a useful near IR (NIR) probe, it was considered that curcumin reacts with boric acid to form a red fluorophore, known as rosocyanine (Figure 3). The color change from curcumin’s yellow to rosocyanine’s red is explained by the π to π* transition from oxygen to the empty orbital on boron. It was thus hypothesized that the introduction of a boron atom could result in a red shift with emission in the 650–900 nm range. Furthermore, it is known that the introduction of a difluoroboronate ring into a dipyrromethene system results in the common red-shifted BODIPY fluorophores.[27] Finally, the phenolic hydroxyls were replaced by N,N’-dimethyl groups, a change also known to enable a red shift in emission. With these structural modifications, a NIR probe CRANAD-2 (Figure 3) was invented with emission at 760 nm in MeOH compared to 560 nm for curcumin.
Although only a weak fluorescence was observed for the probe alone in phosphate-buffered saline (pH 7.4), a remarkable 70-fold increase in fluorescence intensity was registered in the presence of amyloid-β aggregates, indicating that the probe is “turned on” in the presence of substrate. Additionally, CRANAD-2 was able to detect the plaques in vitro when the brain sections from a 12-month-old APP-PS1 transgenic mouse were stained. High contrast staining of plaques in the tissue was observed to colocalize with that observed from the standard thioflavin, indicating high specificity of the CRANAD-2 probe.
2.3. A combination of a natural substrate and a fluorophore precursor in the design of a probe that monitors cyclooxygenase-2 activity in live cells
Cyclooxygenase (COX) is a family of enzymes responsible for producing prostaglandins from a lipid precursor, arachidonic acid. Two isoforms, COX-1 and COX-2, have been identified. COX-2 expression is generally induced in a particular tissue type compared with COX-1 that is constitutively active. The selective activity of COX-2 plays an important role in inflammation, neurodegenerative disorders and cancer.[28] Generally, strategies for detecting COX-2 in living systems are based on selective inhibitors of COX-2 appended to fluorophores for fluorescent imaging[29] and thus provide information only on whether COX-2 is present, rather than on whether it is catalytically active. In an alternative strategy, one would monitor selective chemical reactivity rather than the enzyme’s concentration, enabling a more accurate read-out of COX-2 contribution to the biological processes.[30]
In a breakthrough contribution in this area, a probe, named CoxFluor, consisting of 3,7-dihydroxyphenoxazine (reduced form of resorufin) linked to arachidonic acid through a cleavable amide bond was designed with the idea that the lipid tail would serve as a substrate in a manner similar to natural compound (Figure 4).[30] After the probe was bound at the cyclooxygenase activity site it was hypothesized to undergo deoxygenation and cyclization to give the CoxFluor-PGG2 intermediate. Translocation and oxidation of the fluorophore at the peroxidase active site would then facilitate amide hydrolysis and release of the fluorophore resorufin and PGG2. Because COX-1 is unable to accommodate large groups at the substrate site, it was thought that the bulky fluorophore would impart selectivity for COX-2 possessing a larger substrate pocket.
Figure 4.

Proposed mechanism of COX-2-catalyzed resorufin production. The arachidonic acid tail serves as a substrate in a manner similar to the natural compound. Amide hydrolysis then releases the fluorophore resorufin and PGG2. Because COX-1 is unable to accommodate large groups at the substrate site, the bulky fluorophore imparts selectivity for COX-2 possessing a larger substrate pocket.
CoxFluor was applied for the detection and imaging of intracellular COX-2 activity within RAW 264.7 macrophage cells (Figure 4). A 1.2-fold increase in fluorescence was observed in lipopolysaccharide (LPS)-stimulated live cells relative to the control using confocal microscopy. This fluorescence enhancement was abolished in cells co-treated with indomethacin, a known COX-2 inhibitor.
2.4. Advantages
Affinity for biological molecules
Facile crossing of biological membranes
High structural diversity
Stability in biological media
The world of fluorescent probes continues to be dominated by synthetic fluorophores despite their limited structural diversity and lack of biological relevance. The combination of fluorescent properties with affinity for biological macromolecules makes natural products a unique starting point for fluorescent probe discovery. In addition, biological relevance associated with natural products manifests itself in favorable physicochemical characteristics, such as the ability to cross cell membranes or blood-brain barrier. Even with non-fluorescent natural products, their clever combination with synthetic fluorophores, such as the one described in Section 2.3, should be an important consideration in probe design.
2.5. Disadvantages
Few literature reports and few examples
Complex structures create significant synthetic challenges
Generally low isolation and/or total synthesis yields
The exploration of the natural product community has been primarily focused on the pharmacological potential of these secondary metabolites, whereas their fluorescent properties have not been scrutinized thoroughly.[21a] Natural product scientists are encouraged to pay closer attention to this useful property if it is observed in newly isolated natural molecules and correctly record it.
3. Rational Design of New Synthetic Fluorescent Motifs
The application of designed synthetic fluorescent motifs is another very attractive and potent research tool that has gained significant popularity in medicinal chemistry and chemical biology areas. The optimization of physicochemical and photophysical properties such as absorption (λabs) and emission wavelengths (λem), molar absorptivity (ɛ), and quantum yield (ΦF) plays a key role in the design of a fluorescent scaffold. Therefore, molecular-level understanding of the relationship between the chemical structure and photophysical properties of a given fluorophore provides a fundamental design principle in the development of novel fluorescent probes.
Many examples of rationally designed synthetic fluorescent scaffolds have been reported. However, the majority still suffer from the inherent drawbacks of narrow pH range of activity, poor sensitivity and selectivity, low water solubility and complexity of synthetic preparations.[31] Development of simple molecular probes that are cost-effective, rapid-acting, and highly sensitive is still in great demand. In this section, we highlight the progress in the rational design of synthetic fluorescent probes.
3.1. Seoul-Fluor: a synthetic fluorescent scaffold as a ratiometric pH sensor and lipid droplet bioprobe
The new fluorescent scaffold “Seoul-Fluor” (9-aryldihydropyrrolo [3,4-b]indolizin-3-one) was first synthesized as part of combinatorial chemistry efforts by Park and co-workers in 2008 (Figure 5).[32] The synthesis was achieved by an intramolecular 1,3-dipolar cycloaddition reaction of azomethine ylides with substituted olefins in the presence of a base using a one-pot process (Figure 6). A few years later, the same group developed another efficient synthetic approach to Seoul-Fluor scaffolds using a silver- and gold-catalyzed intramolecular 1,3-dipolar cyclization of terminal alkynes with substituted azomethine ylides and a subsequent Pd-catalyzed C–H activation (Figure 6).[33]
Figure 5.

The structure-photophysical relationship (SPPR) in the Seoul-Fluor skeleton showing the intramolecular charge transfer (ICT) in the excited state (blue to red sites in the molecule). The structure of one example SF44,[32a] is provided as an illustration of the push-pull system wherein an electron-donating amine is conjugated to an electron-withdrawing acetyl group.
Figure 6.

Retrosynthetic scheme for the Seoul-Fluor scaffold includes two synthetic routes that involve the assembly of three fragments. The first route (blue) begins by assembly of bromoacetyl bromide (1) and pyridines 2 to give substituted pyridines that react with reductive amination product from the coupling of cinnamaldehyde and a primary amine. The second route (red) begins by assembly of 1, pyridines 2, and propargyl amines 3 to generate an amide intermediate, which subsequently undergoes a 1,3-dipolar cycloaddition to produce the Seoul-Flour skeleton.
Before a systematic synthesis of Seoul-Fluor analogues was complete, the team conducted a series of density functional theory calculations. This theoretical study led to the rational design of the substituents bearing different electronic properties. For example, the variation in substituents located in PeT donor and PeT acceptor (blue and red, Figure 5) was shown to influence photophysical properties, such as λem. This allowed the team to prepare compounds covering the full spectrum of visible colors (445–613 nm).
An increase of the PeT-donor properties (blue, Figure 5) along with a corresponding decrease of the PeT-acceptor (red, Figure 5) induce a bathochromic shift of λem as exemplified in SF44 (Figure 5). It is noteworthy that most of the reported donor-acceptor fluorophores are not full color-tunable and require a considerable change in their chemical structures.[34] Of interest, the nitrogen substituent of the lactam ring does not have an effect on the fluorescent properties of Seoul-Fluor and can be used for conjugation with small molecules or proteins without changing the fluorescent properties of the fluorophore. To summarize, these novel fluorophores have been observed to have excellent photophysical and photochemical properties, large Stokes shifts, excellent quantum yields, pH independent fluorescence, drug-like lipophilicity for membrane permeability, and resistance to photobleaching.
The understanding of tunability (λem) of Seoul-Fluor was utilized to develop ratiometric fluorescent pH sensors. It was hypothesized that changing the pH would result in different photophysical changes depending on the type and the location of pH responsive functional groups on the Seoul-Fluor scaffold. Accordingly, Seoul-Fluor analogues, which can undergo either hypsochromic or bathochromic shifts of λem were designed (SF44, Figure 5). For example, the diethyl amino group in SF44 would undergo protonation and switch from an electron-rich to an electron-poor moiety, resulting in a hypsochromic shift of λem upon lowering of the solution pH. In contrast, a carboxylic acid group in the red area will undergo protonation resulting in increased electron-withdrawing strength and a corresponding bathochromic shift. The experimental studies of analogues containing such judiciously positioned pH-responsive groups indeed identified the Seoul-Fluor system as a versatile scaffold for fluorescent ratiometric pH sensors.
A useful application of the Seoul-Fluor probe is as a lipid droplet (LD) bioprobe.[35] The LD is an intracellular hydrophobic organelle, surrounded by a phospholipid monolayer that sequesters cellular fat.[36] Recent studies identify its essential role in regulating lipid metabolism, which is closely related to various metabolic diseases. The Park research group proposed that SF44 selectively stains the LDs in a fluorogenic manner. According to one possible mechanism for this phenomenon, the hydrophobic nature of LDs makes SF44 selectively turn on its fluorescence via the internal charge transfer (ICT) process in LDs, but not in untargeted hydrophilic cellular compartments.
The cellular imaging experiments suggest that the presence of lipophilic substituents at the lactam ring nitrogen position increases cLogP value of the probe and thus induces a higher accumulation of SF44 analogues in the hydrophobic LDs inside the cells, which results in the enhancement of the fluorescence signal for specific LD monitoring.
3.2. Dihydropyridine-based fluorescent probes for protein tyrosine phosphatase and mitochondrial staining
The dihydropyridine (DHP) motif was introduced over a century ago (in 1882) when Hantzsch published the synthesis of 1,4-dihydropyridines.[37] Among the DHPs, 1,4-dihydropyridines (1,4-DHPs) are well studied chemically (4 in Figure 7), and their appropriately substituted derivatives are well known to exhibit blue fluorescence.[38] However, 1,2-DHPs are relatively unexplored for their biological applications, making their conjugated derivatives promising candidates for the design of novel fluorescent probes. Lankalapalli and co-workers have reported a clever design and short synthetic route towards new fluorescent 1,2-DHPs (5 in Figure 7).[39] The synthesis of 1,2-DHP-based fluorophores was achieved by a facile one-pot four-component condensation reaction using dienaminodioates 6, aldehydes 7, and in situ-generated hydrazones 8 mediated by CF3CO2H at ambient temperature (Figure 8).
Figure 7.
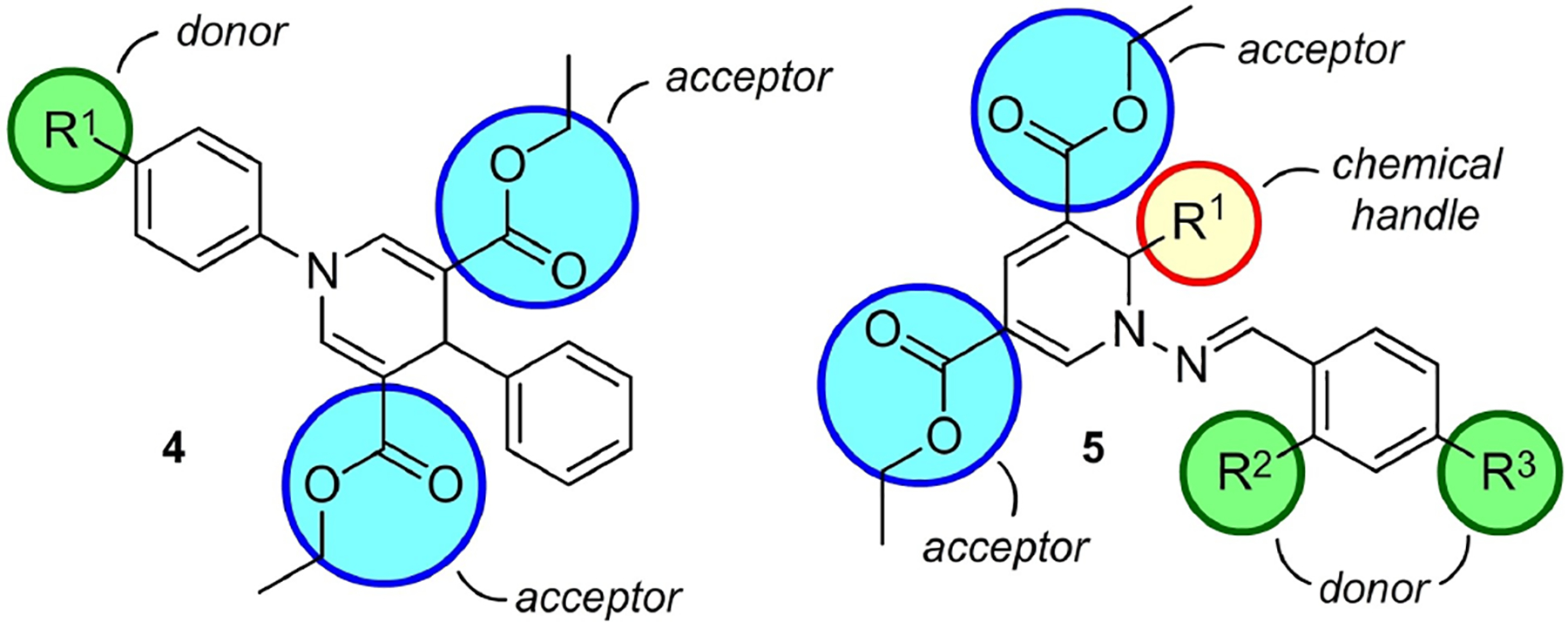
A schematic representation of DHP-based fluorophores. The 1,4-DHP fluorophore 4 has electron rich groups at 1-position and electron deficient groups at the 3- or 5-positions and generally exhibits blue fluorescence. The design of 1,2-dihydropyridine (DHP)-based fluorophore 5 includes arms for bioconjugation, chemical handle R1 and hydrolysable ester groups. The strategy of 1,2-DHP skeleton involves a push-pull system with different electron donor groups.
Figure 8.

One-pot multicomponent synthesis of 1,2-DHP (5) derivatives from dienaminodioates (6), aldehydes (7) and in-situ generated hydrazones (8).
1,4-DHPs are known to fluoresce intensely when there is an electron-donating group at the 1-position and electron-withdrawing groups at positions 3 and 5 (4 in Figure 7). Higher Stokes shifts are observed when an electron-donating aromatic system is present at the 4-position of the 1,4-DHP scaffold attributed to an ICT in the excited state between the two π-systems. The 1,2-DHP-based probe design involves elaborating donor-π-acceptor or push-pull systems using the N-benzylideneamine moiety to modulate the photophysical properties of the fluorophore. For example, substituents possessing electron-donating character lead to increased ICT properties and significant bathochromic shifts. The 1,2-dihydropyridines synthesized by Lankalapalli and co-workers exhibit maximum absorption wavelengths (λmax) between 396 and 448 nm in methanol and emit in the long wavelength region of 500–600 nm.
The application of these fluorophores was demonstrated for selective mitochondrial staining in HeLa cells.[39] The selectivity is based on the push-pull system in 1,2-DHPs (Figure 7), making the nitrogen within a ring attain a significant positive charge. The co-staining experiment with MitoTracker Red chloromethyl-Xrosamine (CMXRos), a commercially available mitochondria stain, confirmed the localization of 1,2-DHPs in the mitochondria, which is supported by Pearson’s correlation coefficient in the range of 0.75–0.89. 1,2-DHPs 5a, 5b, and 5c (Figure 9) were found to exhibit high-intensity fluorescence staining.
Figure 9.

Structures of 1,2-dihydropyridines synthesized using a multicomponent one-pot reaction. The 6-position in N-benzylideneamine is the conjugation site. 1,2-DHPs 5a–e emit in the long wavelength region of 500–600 nm.
Finally, the 6-position has little effect on photophysical properties and is useful for conjugation with small molecules and biomolecules for tagging or modulation of physicochemical characteristics, such as water solubility. Thus, DHP derivative 5d (Figure 9) was designed as an optimal fluorophore suitable for its potential application as a small molecule probe in aqueous medium. Additionally, 1,2-DHP 5d exhibited 6-fold enhanced emission intensity than its phosphorylated analogue 5e in the long wavelength region (λem ≈ 600 nm), which made it a suitable bioprobe for protein tyrosine phosphatases, whose activity was demonstrated in L6 muscle cell lysate.[39]
3.3. Anthracene-BODIPY dyads as fluorescent sensors for biocatalytic Diels-Alder reactions
The third example in this section deals with the rational design and synthesis of water-soluble anthracene-BODIPY dyads as fluorescent probes for biocatalytic Diels-Alder reactions.[40] These reactions are poorly understood but, due to the potential synthetic utility, it is of great interest to design fluorescent probes to monitor such processes. Such cycloaddition reactions are catalyzed by Diels-Alderase enzymes (Figure 10), including ribozymes. The ideal fluorescent probe should distinguish between free substrate, bound substrate, bound product, and free product in order to gain insight into the key species of the catalytic cycle.
Figure 10.

Ribozyme-catalyzed Diels-Alder reaction between 9-hydroxymeth-ylanthracene (9) and N-pentylmaleimide (10).· The development of fluorescent probes for catalytic Diels-Alder reactions involves the optimization of such characteristics as sufficient solubility in an aqueous medium, diene-based reactivity identical to the substrate in the uncatalyzed Diels-Alder reactions, acceptance as a substrate by the ribozymes, the Diels-Alder reaction-based change in photophysical properties, as well as fluorescent changes brought about by binding to the ribozyme to observe and distinguish the key species of the catalytic cycle.
Inspired by “energy-transfer cassettes” phenomena, developed by Burgess,[41] Jäschke and co-workers investigated anthracene-BODIPY dyads to study the Diels-Alder reaction between anthracenes and N-substituted maleimides.[40] Three different anthracene-BODIPY fluorescent probes, 12, 13, and 14 (Figure 11), were synthesized using combinations of coupling reactions.
Figure 11.

Design of fluorescent probe for the investigation of Diels-Alder reactions: substrate-analogue probes 13, 14 and product-analogue probe 12.
These probes consist of anthracene and sulfonated BODIPY scaffolds fused by conjugated phenylacetylenyl bridges. It was hypothesized that upon the Diels-Alder reaction, the conjugated π-system of anthracene would lose its aromatic character leading to an increased fluorescence emission. Indeed, it was found to increase 20-fold. Furthermore, binding in the catalytic pocket of an enzyme (Diels-Alderase ribozyme) was found to yield a ~2-fold increase in fluorescence intensity of between the anthracene-BODIPY probe 14 and the Diels-Alder product 12 confirming that fluorescence-based distinction was possible.
3.4. Advantages
Computational approaches help streamline probe discovery
Potential to discover new powerful fluorophores with numerous future applications
Not limited by the class of fluorophores
Fluorophores can be selected on the basis of synthetic accessibility
Robust synthetic strategies can allow one to meet the general requirements of new synthetic fluorescent motifs such as emission intensity, longer wavelength, water-solubility, photostability, and cell permeability. We have indeed seen one-pot processes or short practical synthetic routes applied to access a potential fluorescent probe for biological applications. Practical synthetic chemistry combined with computational work can indeed help one consider novel fluorophores with optical or chemical properties that appeared too daunting just a few years ago. With the increase of computational power and constant improvements in synthetic methodology, more and more challenging fluorophores will continue to be discovered.
3.5. Disadvantages
Computational predictive power can be limited
Keeping synthetic sequences short due to the risk, may hamper the installation of the desired fluorescent characteristics
Lack of biological relevance
The disadvantages will of course stem from a possible insufficient predictive power of computational resources and a lack of efficient synthetic methods that could be utilized to access fluorophores predicted to have the desired optical and physicochemical properties. Since the structures of natural products are not considered in computational work, the predicted fluorophores will lack biological relevance and may have poor biological properties.
4. Synthetic Expansion and Modification of Known Fluorescent Scaffolds
Advances in synthetic chemistry have led to our ability to manipulate the structures of available fluorescent scaffolds to improve their biological and physicochemical properties, such as reduction of toxicity, improved water solubility or ability to penetrate the blood brain barrier. These synthetic expansions of well-known scaffolds have offered additional levels of target selectivity. In this section, we will show examples of how fluorescent probes possessing specific functionality have been developed from established fluorophore scaffolds to improve on a particular aspect of probe performance.
4.1. Development of a rhodamine-based fluorescent probe for osteoclast activity sensing
In our first example, Kikuchi and co-workers developed a rhodamine-based red fluorescent small molecular probe, RedpHocas, possessing rapidly reversible pH sensing, bone-targeting properties (Figure 12).[42] This red-pHocas probe was useful for the multicolor imaging of acidic environments and analysis of osteoclast proton pump dynamics in mice. Osteoclasts play a vital role in bone functioning, where their imbalances lead to elevation or reduction of bone mass associated with osteopetrosis and osteoporosis.[43] This team designed a series of rhodamine spirolactams possessing an N-alkyl group to rationally control fluorescence response under acidic conditions (Figure 12) and thus enable imaging analysis of the dynamics of osteoclast proton pumps in GFP-mice. To improve the aqueous solubility of rhodamine and strengthen the probe’s affinity for bone tissues, bisphosphonate groups were introduced onto the rhodamine skeleton.
Figure 12.

Schematic design of a Red-pHocas (pH activatable red fluorescent probe) for detecting the areas of bone acidification. This probe was used to demonstrate the direct involvement of osteoclast proton pumps in bone acidification.
In this study, Kikuchi and co-workers reported that activation could be triggered by simply modifying the N-alkyl substituents on the rhodamine-spirolactam skeleton.[43] The introduction of the 2,2,2-trifluoroethyl group into the N-alkyl amide substituent (Figure 12) significantly enhanced the fluorescence activation response time. The electron-withdrawing effects of trifluoroethyl group caused the destabilization of the intermediates and increased the fluorescence turn-on response. The fluorescence activation mainly proceeded via protonation on the lactam amide, followed by the spirolactam ring opening.
RedpHocas (Figure 12) was studied as an imaging probe to demonstrate the direct involvement of osteoclast proton pumps in bone acidification. This imaging approach helped observe drug efficacies in intact tissues and provided a useful tool for developing drugs against bone diseases and inhibitors of trafficking pathways of proton pumps with a high temporal resolution. Also, the reversible quick pH response of RedpHocas allowed the intravital imaging of acidic regions.
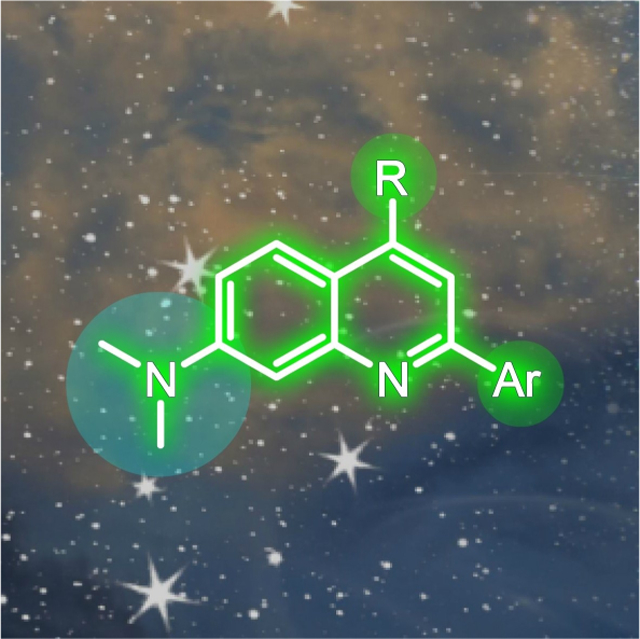
4.2. Dimethylaminoquinoline (DMAQ) fluorescent scaffold for live cell imaging
In a second example, the Chenoweth group recently reported a quinoline based DMAQ (2,4-dichloro-N,N-dimethylquinolin-7-amine) scaffold for live cell imaging (Figure 13).[44] The design of a highly tunable quinoline-based scaffold includes three strategic domains (I, II, III), which can be diversified and optimized to achieve desirable photophysical features for synthetically expanded derivatives. Specifically, these were allotted for compound polarization, tuning of photophysical properties, structural diversity and/or late-stage chemical modification.
Figure 13.

A design strategy of dimethylaminoquinoline (DMAQ) fluorophore consisted of three domains, one polarization domain and two tuning domains. The retrosynthetic approach for DMAQ synthesis using cyclo-condensation of 3-(dimethylamino)aniline (15) and diethylmalonate followed by chlorination with phosphoryl chloride. Box: The structure of an exemplary DMAQ derivative 17.
The electron-donating dimethylamino group at the 7-position of quinoline pushes electron density into the core of DMAQ and serves the role of a polarizer (Figure 13). The 2- and 4-positions of DMAQ are used as tuning domains, where electronically and structurally diverse groups can be installed to result in push or pull effects. For example, the push-pull systems (D-π-A) would be expected to give more red-shifted emissions.
The synthesis of the DMAQ scaffold was achieved through regioselective Pd-catalyzed processes. Using combinatorial techniques, this team was able to synthesize an extensive library and develop an understanding the structure photophysical relationships that guided the intrinsic properties of the DMAQ scaffold.
The two-stage fluorescence response to intracellular pH and better cell permeability made these probes suitable for multicolor live cell imaging. Additionally, these probes existed as amorphous or crystalline solids, indicating the potential for applications as organic materials. It was envisioned that the optimizable fluorescent probes could function in convenient, continuous assays, thereby providing useful tools to study various biological processes.
4.3. Structural design of sulfonated carbofluorescein probes for voltage imaging
Recently, Millan and co-workers developed sulfonated carbofluoresceins as probes for voltage imaging.[45] Prior to their studies, voltage imaging predominantly relied on photoinduced electron transfer (PET).[46] Here, changes in voltage could be measured through fluorescent probes whose spectral properties were modified when voltage changed.[47] Voltage sensing fluorescent probes are also known as voltage-fluors. These voltage-fluors find use in measuring neural activity within the nervous systems and have been demonstrated in multiple organisms. The first known popular parent fluorophore of this family was carboxyfluorescein (Figure 14), widely used to study the blood ocular barriers.[48]
Figure 14.

Structures of fluorescein (18), carbofluorescein (19), a sulfonic acid analogue of carbofluorescein (20), and CarboVoltageFluor (21).
In this study, it was found that a new carbofluorescein scaffold (Figure 14) was more suitable for voltage imaging, however, the carboxy-substitution presented a problem because of the tendency of carboxy groups to undergo cyclization, which converts them into a nonfluorescent motif. The optimization and synthetic expansion finally involved the replacement of the carboxy group with the sulfonate and the addition of a phenylenevinylene ‘molecular wire’ for membrane localization.
Developing on this concept, the synthesis of a new class of sulfonate carbofluoresceins was achieved in 7 steps, depicted retrosynthetically by the conversion of 22 and 23 to 21 (Figure 15). The installation of the sulfonic acid group on the meso aromatic ring of 21 was found to drive the correct alignment of the voltage-sensing phenylenevinylene molecular wire within the plasma membrane. In this design (Figure 15), the geminal dimethyl carbon replaces oxygen at the bridge-head position of the xanthene scaffold, providing an excitation at > 560 nm. The introduction of the sulfonate was useful in preventing the non-fluorescent spirocyclization and the addition of chlorine reduced the pKa by 2 units to 5.2. The new CarboVoltageFluor (21) displayed voltage sensitivity of (> 30% ΔF/F per 100 mV) in HEK cells. It showed excitation and emission spectra of > 560 nm, and readily reports on action potentials in mammalian neurons. The incorporation of the phenolic oxygen also provided a handle to incorporate additional functionality.
Figure 15.

Retrosynthesis of CarboVoltageFluor (21). The assembly begins by synthesis of carbofluorescein precursor 24 using 3-chloro-4-methoxybenzoyl chloride (22) and 4-bromo-2-chloro-1-methoxybenzene (23) followed by the installation of the dimethylcarbon. Nucleophilic addition of an aryllithium species gave 26, which was followed by the Heck coupling of the phenylenevinylene ‘molecular wire’ in 22.
4.4. Advantages
Focus on property optimization rather than a search for a totally new pharmacophore from scratch
Modern synthetic methodology allows the precise modification of structure
Functional group modifications can be aligned to reduce non-fluorescent states
The main advantage of synthetic expansion of known scaffolds is the large amount of knowledge that has accumulated over the years on the performance of classical fluorophores. Thus, synthetic expansion primarily focuses on property optimization rather than a search for fluorescent properties from scratch. Here, one can apply modern synthetic methods to enable access to vitally needed probes from classical motifs.
4.5. Disadvantages
Lack of structural novelty reduces the overall advancement in our access to new fluorescent motifs
Minor modifications may not always fundamentally change a desired property
Challenging synthetic chemistry as the classical fluorophore needs to have points of diversification in the desired positions
Some of drawbacks include the difficulty to achieve specificity for a particular biological analyte, as many of the classical fluorescent materials were designed to lack biological activity. In this case one would need to start with a skeleton, known to display a certain level of specificity. The length of synthetic sequences can be longer as one needs to build the scaffold first with the necessary points of diversification and then perform the introduction of substituents.
5. Target Oriented Fluorescent Probe Library Approach
Fluorescent probes often display molecular properties necessary to unravel complex biological problems. However, our ability to rationally design probes typically requires a high-resolution structure of its target such as an X-ray crystal structure. Furthermore, the utilization of rationally designed probes presents a challenge stemming from significant differences between in vitro and in vivo environments. To this end, the difficulties associated with rational probe design and our inability to precisely predict probe requirements can be overcome by applying combinatorial approaches. In this section, we illustrate recent examples where a target-oriented library-based design can be used to craft molecular probes by adapting established molecular recognition elements in a high-throughput combinatorial process.
5.1. Photoinduced electron transfer sensors: monitoring of saccharides in aqueous systems using boronic acids
Our first example explores the development of boronic acid-based fluorescent probes for detecting saccharides (Figure 16).[49] It can be viewed as a classic example that demonstrates how the advances in library-based methods of fluorescent probe design can lead to the discovery of selective sensors and bioimaging agents with a broad scope of in vivo applications.
Figure 16.

Boronic acid-based fluorescent probes for saccharide detection. Probes are designed by quenching the emission of fluorophores attached to an electron-donating molecular recognition element (photoinduced electron transfer) in order to achieve fluorophores that display enhanced fluorescence when bound to a saccharide.
Early reports by the Wulff[50] and Shinkai groups[51] demonstrate the ability of boronic acids to form reversible cyclic boronate esters with diols of a saccharide. Here, fluorophores were attached to boronic acid-based molecular recognition elements that would exhibit electron-donating properties upon binding to a sugar diol (bottom, Figure 16). By proper positioning on a fluorophore, the team demonstrated that this concept of electron transfer could be used to enhance fluorescence upon saccharide binding, as shown in Figure 16.
The modularity of this system allows one to rapidly assemble libraries of fluorescent boronate probes. This eventually results in a system with three points of diversity.[49] As shown in Figure 17, selective modifications within each site (diversity 1–3) were tuned to adjust the selectivity towards specific saccharides.[52] Utilization of polymer support or solid-phase synthesis,[53] click chemistry,[54] and peptide linkers[55] led to the assembly of saccharide-selective probes. These boronic acid probes have found wide-spread applications, including: applications to the analysis of glycoproteins by Hindsgaul and co-workers;[56] pattern-based saccharide sensing array systems by Anslyn and co-workers;[57] and fluorescence visualization of tumors.[58] The latter, shown in Figure 18, was achieved in vivo by the selective labeling of sialyl Lewis X, a saccharide-based carcinoma biomarker.
Figure 17.

Modular design of fluorescent sensory systems containing two boronic acid residues for detection of specific saccharides. When bound to a saccharide, the photoinduced electron transfer from amino group to the aryl unit within the probe is hindered thus resulting in a fluorescent response. Modular design allows for fine-tuning of selectivity and localization in biological systems.
Figure 18.

Optical imaging of tumors through selective labeling of glycoproteins. Boronic acid-functionalized peptide-based fluorescent sensors are water-soluble and serve as biocompatible tools for in situ recognition and differentiation of cell surface carcinoma biomarkers in vivo.
5.2. Peptide-based probes with controlled in vivo localization for zinc detection
A guiding principle for the design of a selective fluorescent probe lies in engineering a receptor with electronic, geometric and polar characteristics carefully matched to the chosen target, like a lock and a key. The power of this approach is well illustrated by biological systems. Natural receptors incorporate exquisite binding sites that display near perfect selectivity in a competitive biomolecular environment. Accordingly, efficient synthetic probes can be prepared by mimicking this natural tactic. For that purpose, the use of peptides with their inherent modularity and automatable synthesis is very appealing. In this section, we will illustrate this approach by showing examples of using peptide-based fluorescent probe libraries for localized in vivo detection of Zn2+ ion.
One of the first examples of utilizing peptides libraries for development of fluorescent probes was reported by Imperiali and co-workers (Figure 19).[59] In this work, molecular recognition of Zn2+ ion was achieved by implementing a zinc finger domain. The library of dansyl-modified peptides was used in order to detect Zn2+ through a change in emission properties caused by the binding event (Figure 19a). The fluorescence of the covalently attached fluorophore remote from the metal-binding site was affected by metal-induced conformational changes of the peptide.
Figure 19.

Peptide-based Zn2+ fluorescent probes. a) Increase in fluorophore emission due to binding-induced peptide conformational regulation. Protein scaffold shields the fluorophore from solvent interactions. b) Structure of the Palm-ZP1 (34), which was used for localized Zn detection in live HeLa cells. The peptide contains a 3-residue polyproline helix, 2 sequential Asp residues and a C-terminal Lys to serve as the point of attachment for the Zn2+ probe.
In comparison to small-molecule fluorescent probes, a unique attractiveness of peptide-based probes lies in the controlled and predictable localization of such probes in biological environments. As such, the group of Lippard[60] has shown that peptide-based fluorescent probes (Figure 19b) can be directed to the extracellular side of plasma membranes of living cells in order to investigate zinc release from cells. This example illustrates how using combinatorial approach sensors can be tailored to address specific questions in Zn2+ ion biology.
Defects in zinc metabolism in various subcellular regions are associated with a number of diseases, such as type 2 diabetes, neurological disorders and hypertension.[61] The focus of recent research on the development of the fluorescent sensors for zinc ions with controlled intracellular localization is of immense interest. Genetically encoded fluorescent sensors, which include a complex protein consisting of a sensing polypeptide fused with a fluorescent protein, are a very attractive tool. For instance, the use of fluorescent probe library based on cpGFP (circularly permutated green fluorescent proteins) and zinc finger sensing domains allowed Qin and co-workers[62] to develop a Zn2+ ion sensor and a parallel set of imaging tools that specifically targeted the mitochondrial matrix and the mitochondrial intermembrane in live cells. This probe was further modified[63] to achieve sensitivity for Zn2+ ion at sub-nanomolar concentrations that resulted in the discovery of new pools of Zn2+ ion signals in primary hippocampal neurons that play an important role in neuronal function.
5.3. Positional scanning substrate combinatorial library approach for activity-based probe discovery
Enzymes are gaining an increased attention as fluorescent probe design targets due to their role in countless physiological processes.[64] Dysregulation of enzyme activity is linked directly to the development of many human pathologies, including cancer. Thus, monitoring enzymatic processes in living cells is of particular importance in diagnosis and treatment assessment.[65] Due to their high sensitivity and real-time analysis capabilities, fluorescent probes have become an efficient tool for detection and imaging of enzymatic processes in the complex environment of a cell. Most probes rely on interaction with the active sites of their targeted enzyme.[66]
For proteolytic enzymes, the interaction specificity is determined by the amino acid sequence of their target proteins. The accurate knowledge of such substrate preference is crucial for the development of protease selective probes. One of the most common methodologies for defining the protease substrate preference is the Positional Scanning Substrate Combinatorial Library[67] approach (Figure 20), in which libraries of peptides are conjugated to fluorescent leaving groups that are released and able to emit fluorescence following the protease hydrolysis. The libraries are tested by dividing mixtures of peptides into sublibraries. Each sublibrary contains equimolar concentrations of proteins for which one or more positions is fixed with a certain amino acid whereas the remaining positions contain varied amino acids. As shown in Figures 20 and 21, the proper positioning of the cleavage site allows one to screen a diversity of peptide sequences in parallel.
Figure 20.

Analysis of proteolytic enzyme specificity through the release of conjugated fluorescent leaving group in the course of Positional Scanning Substrate Combinatorial Library method. Substrate specificity is assessed by measuring fluorescent response following the protease cleavage of a particular peptide bond.
Figure 21.

Probe discovery process. Schematic representation of the probe discovery process illustrating how probes in libraries P1–P3 are prepared and screened to deliver an active probe 35.
Drag and co-workers have shown that unnatural amino acids can be used to enhance the substrate preferences of closely related proteases.[68] This Hybrid Combinatorial Substrate Library approach was used to determine the extended substrate specificity and develop activity-based fluorescent probes for Mucosa-associated lymphoid tissue (MALT) lymphoma translocation protein 1,[69] a cysteine protease which can be an important target for the treatment of MALT lymphoma.[70] In another study, selective visualization of cathepsin L was achieved in living breast cancer cells in presence of cysteine cathepsins.[71] The ability of the probe to distinguish the individual protease from other human cysteine cathepsins with overlapping substrate preference is important for establishing its role in normal and pathogenic processes, and demonstrates the power of combinatorial approach to probe development.
5.4. Advantages
Detailed understanding of a target’s biology is not required
High selectivity
In vivo compatibility has been demonstrated
Expeditious probe discovery
High-throughput analysis
When the molecular recognition mechanism is known for a target analyte, a very high degree of selectivity can be achieved by using combinatorial approach to fine-tune the probe structure. Mimicking natural binding sites can ensure high sensitivity in complex environments and in the presence of multiple competing species. High-throughput analysis is a key to the fast “trial and error” probe generation process. Compared to many other strategies, the search for new probes through generation of fluorescent libraries is typically less labor intensive. New fluorophores can be discovered quickly by taking advantage of expedited synthetic methodologies.
5.5. Disadvantages
Known molecular recognition mechanism is required
Limited synthetic methods
Low success rate due to combinatorial nature of the study
Some probe structures will be missed
In order to implement the target-oriented library-based fluorescent probe discovery, the knowledge of receptors or binding elements of selected analytes is necessary. Methods described in this section are based on derivatization of known molecular recognition elements and fluorophores. The use of combinatorial approach often limits the structural variety of probes by imposing strict requirements on synthetic methods used. Desired probe structures are accessible through methods such as automated peptide synthesis, coupling reactions, or click chemistry. In addition, quick and efficient purification methods are required in order to make library synthesis feasible. Low success rate and highly random character of probe candidate selection are inherent limitations of all combinatorial studies. Even though useful probes can be found through library-based methodologies, some interesting structures will inevitably be missed.
6. Diversity-Oriented Fluorescent Libraries
In the previous section, we discussed how a combinatorial approach can be successfully applied to develop selective and sensitive fluorescent probes by taking advantage of known molecular recognition elements. However, the discovery of recognition elements and fluorophore scaffolds through rational design remains challenging. In some cases, no knowledge of molecular recognition mechanism for a particular analyte is available or known binding motifs are inadequate for complex biological environments thus limiting the scope of targets and hampering probe development.
In an effort to overcome these obstacles and hasten probe discovery, target-oriented approaches are complemented by the advances in diversity-oriented strategies. These diversity-oriented fluorescent probes take advantage of extensive chemical variability in order to find new probes through unbiased screening. Such a process can be applied to an assortment of probes and targets of very diverse chemical nature and these libraries can be tested for multiple applications. In addition, advances in high-throughput imaging and analysis technologies[72] made in vivo screening of large fluorescent libraries a feasible method for identifying new live cell probes and imaging agents. We begin with a series of unbiased screens.
6.1. Unbiased screening of diversity-oriented fluorescent libraries
In cases when there is no known molecular recognition mechanism for a particular analyte, the development of new binding motifs would typically be the first step towards discovery of a probe. The diversity-oriented fluorescent library approach initially developed in the Chang laboratory[73] was an efficient tool for resolving this problem from scratch. Large libraries consisting of thousands of small molecule probes can be assembled and tested in a systematic and unbiased manner against a wide variety of biologically relevant targets (Figure 22a).
Figure 22.

Systematic screening of diversity-oriented fluorescent libraries. a) An example heat map plot of change in emission intensity after interaction of a probe with a target organized by probe. b) Selected fluorescent probes discovered by Chang and co-workers through unbiased screening of diversity-oriented fluorescent libraries.
For example, in 2011[74] the library consisting of 317 BODIPY probes was prepared and systematically tested against a collection of 94 biomolecules with a broad range of chemical diversity. Different categories of analytes for this study included peptides, metal cations, single strand DNAs, double strand DNAs, pesticides, reduction-oxidation related small molecules (such as H2O2 and L-glutathione) in the range of their endogenous or effective concentrations. Unbiased high-throughput screening of the fluorescent response allowed for the identification of fluorescent sensors with a sharp change in emission spectra upon binding to selected targets. As a result, this study helped to reveal general trends for fluorescent response of BODIPY probes and two new sensors were found, for bovine serum albumin (BSA) and dopamine.
The power of this approach is in its expeditious discovery of fluorescent probes for a broad range of biological molecules. For instance, in 2016[75] a diversity-oriented library of several thousand fluorescent probes was tested against bacterial biofilms formed by Pseudomonas aeruginosa. A novel fluorescent probe CDy11 (Figure 22b) that targets bacterial amyloids was identified and further confirmed to be suitable for in vivo biofilm imaging. In 2018,[76] identification of tumor initiating cells associated with a variety of epithelial cancers was achieved using a small-molecule fluorescent probe, which was found through the screening of a diversity-oriented fluorescent library. Interestingly, the discovered probe also displayed biological activity against tumor initiating cells along with low toxicity towards other types of cells. In an effort to develop an imaging probe for the pancreatic β cell that are important for diabetes diagnostics,[77] Chang and co-workers tested a diverse collection of > 3000 probes. Consequently, a rosamine-based sensor PiF (Figure 22b) allowed for intraoperative fluorescent imaging and positron emission tomography of pancreatic islets.
6.2. Solid-phase diversity-oriented fluorescent probe libraries and array sensing
There are several ways in which the construction of diversity oriented fluorescent libraries can be facilitated through solid-phase protocols. Firstly, assembly of many types of molecules, for example peptides, can greatly benefit from using automatic synthesis and purification procedures through the utilization of solid support. Additionally, libraries of fluorescent probes can be immobilized on different surfaces, such as cellulose or various nanomaterials, for on-bead screening. Combination of such methods can lead to significant simplification of fluorescent probe discovery process as well as convenient transition towards practical applications.
For instance, recently Praneenararat and co-workers[78] demonstrated that probe discovery and evaluation can be expedited through macroarray immobilization of fluorophores. Metal ion sensors were constructed using filter paper as a solid support through the synthesis resulting in covalently bound probes (Figure 23). Such immobilized sensors can be readily applied as convenient paper-based analytical tools capable of detecting nanomolar concentrations of analyte. In addition, newly discovered fluorescent scaffolds were shown to retain their efficiency when used as unbound molecules.
Figure 23.

Solid-supported diversity-oriented fluorescent probe libraries and array sensing. Synthesis of metal ion sensors supported on filter paper by in situ amination and carbodiimide coupling. Simultaneous response from multiple sensors can discriminate up to 12 metal ions. Newly discovered sensors can be utilized as unbound molecules or can be used as paper-based sensing devices.
The use of bead-based assays is often associated with sensing arrays, such as single-molecule detestation arrays pioneered by Walt.[79] Such pattern-based sensing approach is inspired by biological olfactory systems. It complements conventional chemical sensing approach, which requires a highly tuned sensor for each target. Each sensor within the array may not be highly selective towards a specific analyte, instead recognition is achieved by synergistically using signals from multiple probes.
A recent example of utilizing diversity-oriented fluorescent library approach for construction of sensing arrays on solid support was shown by Hu and co-workers (Figure 24).[80] A library of 36 conjugated polymer nanoparticles containing different combinations of monomers and terminal groups was applied on disposable paper cards which can be subjected to a wide range of volatile analytes. The change in fluorescence of sensory array is recorded through optical imaging and analyzed in order to identify a variety of analytes, including drugs, explosive or toxins by pattern-based recognition.
Figure 24.

Fingerprinting model. a) Sensing arrays and pattern-based recognition. Multiple sensors can be utilized synergistically to generate multi-dimensional response and eliminate the need for highly specific sensors for each target. b) Conjugated polymer nanoparticle sensing arrays for the identification of volatile compounds provide an unique visible light response pattern after exposure to each analyte and UV light irradiation.
6.3. Advantages
Knowledge of target binding motifs is not required
Multianalyte screening
Expedited probe discovery
No target scope limitations
Demonstrated in vivo compatibility
Pattern-based recognition of array sensing does not require designing probes for each analyte
Large libraries of probes with a high degree of structural and chemical diversity can be constructed using combinatorial chemistry and then screened against any collection of targets in order to identify new probes without prior knowledge of the associated molecular recognition mechanism for a given analyte. In addition, array sensing does not require a high degree of selectivity towards a particular target and takes advantage of simultaneously recording signals from multiple probes for pattern-base recognition. Such methods significantly broaden the scope of targets accessible for analysis through the use of diverse fluorescent motifs. This approach also provides a rapid timeline for probe discovery.
6.4. Disadvantages
A large library of probes is required
Limited synthetic methods
Low success rate due to combinatorial nature of the study
Some probe structures will be missed
Limited selectivity of array sensing
The success rate of unbiased screenings is limited by the size and diversity of a fluorescent probe library, thus a large library of probes is a prerequisite for conducting diversity-oriented fluorescent library screening. Chemical variety of libraries obtained through combinatorial methods is often limited by synthetic protocols used. Inherently random nature of studies conducted through this method leads to a low success rate and unpredictable results. Nevertheless, initial screening results may identify lead structures that can be further modified in order to improve sensitivity and selectivity. Array sensing methods are intrinsically limited in selectivity towards a specific target that needs to be distinguished from other closely related species.
7. Target-Guided Probe Discovery
For many systems, developing a general probe to a protein is not sufficient.[81] Here one could require a probe that targets a specific protein within a pathway. In such cases, the fact that the substrates are often very similar requires the probe design to specifically target a given enzymatic pocket. Other studies require probes that not only select a single protein motif, but also can do so by selecting different isoforms of that protein or splice variants. In this section, we provide an overview of the recent development of target-guided probe discovery. As described in this section of the review, this approach applies a given target, whether protein, gene, or other molecule and uses that target to directly guide probe discovery.
7.1. Domain selective probes that target dehydratase domains within complex multimodular synthases
Probe design is guided by three primary factors: photophysical properties, a viable response element, and selectivity. One can argue that probes only gain utility when all three are met. The latter, selectivity, is often the most challenging to obtain as there are many different levels of selectivity. The first described in this pathway involves the selection of a specific protein within an ensemble of other proteins. Often referred to as affinity or activity-based selection,[82] the general theme behind these approaches lies in the development of functionality within a probe that specifically targets a protein of interest within a set or subset of biomolecules.
An elegant example of the complexity within this task was developed by the Burkart laboratory at UC San Diego.[83] Here, the researchers were challenged with designing a molecular probe to specifically recognize dehydratase domains within fatty acid and polyketide biosynthetic systems. As shown in Figure 25, adaptation of the alkynylketone motif developed by Bloch[84] into a vinyl-sulfone delivered a probe that could undergo selective recognition of the active site residue within a dehydratase (DH). In this study and follow up efforts,[85] this approach has been shown to allow one to selectively target DH proteins within a proteome as well as DH domains within multimodular synthases.
Figure 25.

Activity-based protein targeting. Schematic representation of the action of a dehydratase (DH) alkynyl-sulfone probe upon binding with the DH domain’s active site. As shown, a conserved active site His-residue within a DH domain deprotonates probe 37 resulting in the formation of a highly-reactive allenylsulfone 38, which in turn undergoes Michael addition to 39 and isomerization to stably-trapped vinylogous sulfonamide 40. The R group represents the position where a 7-dimethylaminocoumarin-4-acetic acid (DMACA) fluorophore was attached. The labeling of the DH domain was identified by fluorescent SDS-PAGE gel analyses.
Most recently, the team furthered these probes by developing a method to mask the reactivity using a novel silylcyanohydrin mask.[86] In this example, probes were designed, tested for their DH selection and optimized through iterative improvements by analogue development, rescreening and prioritization. Ultimately, this team was able to use these probes to develop crosslinkers and apply them to elucidate the structures of protein·protein complexes associated with polyketide[87] and fatty acid biosynthesis.[88]
7.2. Reactivity-based turn-on probes of histone deacetylase enzymes
In the first example, selectivity was identified as a protein within a proteome or a domain within a protein within a proteome. The use of biosynthetic systems provided an excellent example of how this selectivity is required within the probe design. Our second example turns to a theme that often complicates drug discovery efforts, namely the appearance of multiple, often redundant protein isoforms.[89] We turned to histone deacetylases (HDAC),[90] as they provide an excellent example.
HDACs are enzymes that catalyze the removal of acetyl groups from ɛ-amino groups in lysine residues within proteins. While initially identified in histone proteins, HDACs have been shown to act in regulating a wide array of proteins bearing a post-translational acetylated lysine residue.[91] In humans there are 18 HDACs[92] distributed over four different classes, given by: Class I or Rp3d-like (HDAC1, HDAC2, HDAC3 and HDAC8), Class II or Had-1-like (HDAC4, HDAC5, HDAC6, HDAC7, HDAC9, HDAC10), Class III or Sir-2-like (SIRT1-SIRT7) and Class IV (HDAC11).
In 2012, the Kikuchi laboratory, inspired by prior studies by Kapoor and Li[93] described a transesterification switch for the detection of HDAC activity.[94] As shown in Figure 26, their team developed a fluorogenic probe, K4(Ac)-CCB (41) that contained a histone H3 peptide with an acetyl-Lys and a coumarin fluorophore with a carbonate. When presented to an HDAC enzyme activity, deacetylation of the amide was detected by spontaneous intramolecular transesterification, which renders the coumarin probe (43) fluorescent. This study was one of the first reports that demonstrated as a tool for monitoring of HDAC activity by a one-step procedure.
Figure 26.
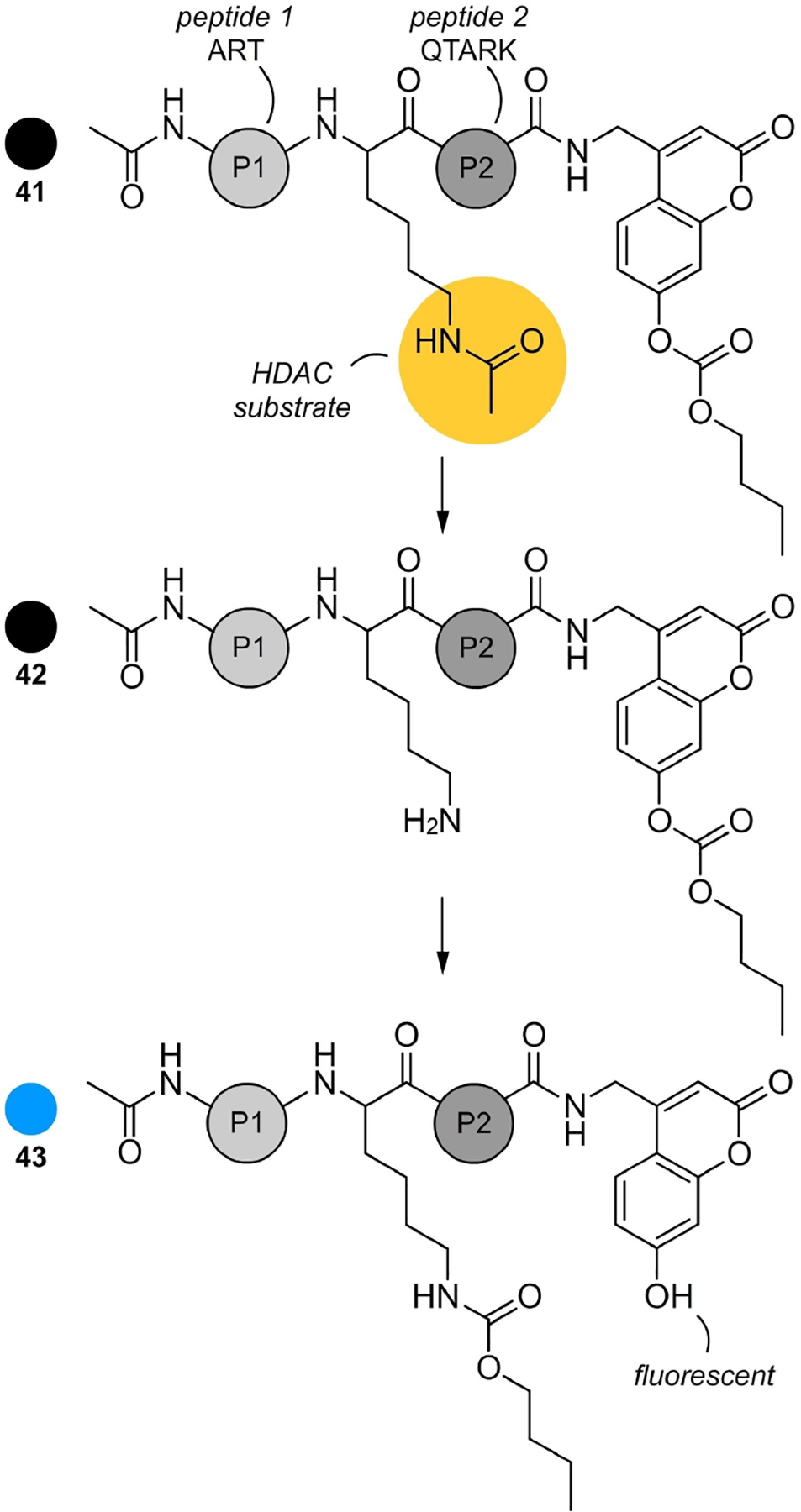
Trans-esterification HDAC probes. This K4(Ac)-CBB probe design contains acetylated peptide that acts as an HDAC substrate and a fluorescent response element appended to its C-terminus. When exposed to an HDAC, hydrolysis results in release of a primary amine, which can undergo an intramolecular trans-esterification with a carbonate ultimately releasing the coumarin 43, which now becomes fluorescent due to increased charge transfer between the free hydroxyl group and the coumarin.
While not refined for HDAC selectivity, the Kikuchi system provided a critical spark that has since led to the development of single step methods for HDAC proteomic profiling. Here, efforts such as that by Sun and co-workers have established facile methods to detect and profile HDAC and related amidases within complex proteomes.[95] While single HDAC selective probes still remain on the horizon, advances such as the transesterification process enable one to expand their ability to identify selectivity. As evident in Figure 26, one can rapidly envision how different HDAC substrates can be prepared, perhaps with different color fluorescent response elements, to reach this selectivity.
7.3. Fluorescent probe discovery via affinity-guided mass spectrometry and chromatography
The prior two examples outlined approaches with a refined target and applied design principles to identify probes. Over the last decade, techniques have now advanced that also allow one to approach probe discovery with a higher degree of autonomy. As shown in Figure 27, affinity based capture methods can be used as a means to guide probe selection. In this example, probe matrixes were developed and used to compare the isolation of specific targets by establishing concentration based inhibitor screening.[96] Here a team at Cellzome demonstrated how one can chemoproteomically profile HDAC inhibitors and reveal their selectivity towards specific HDAC complexes. Using a panel of more specific HDAC leads, the team was able to develop a remarkable profile of the selectivity of commercial inhibitors, tacedinaline and romidepsin (structures not shown). Here, the key to this assay design arose in the generation of matrixes (resins) bearing a probe motif and applying competition assays for their application.
Figure 27.

Methods to map HDAC drug target complexes using a chemoproteomics competition-binding assay to profile HDAC inhibitor target complexes. This process begins by preparing a probe matrix by derivatizing Sepharose with analogues of nonselective HDAC inhibitors for SAHA in a) and givinostat in b).
This study is one of many recent examples of affinity-based methods to expand one’s ability to identify new target protein·ligand pairs.[97] Here, teams, such as the Cravatt laboratory at Scripps Research, applied photoreactive groups and latent affinity handles as fully functionalized probes for integrated phenotypic screening and target identification. In an excellent example[98] they demonstrate how this system can be united with the Ugi-Azide multi-component reaction to identify novel targets using a combination of gel-based and MS-based protein-probe interaction mapping.
The method begins by incubating cell extracts with vehicle or inhibitor (44 or 45, Figure 27) over a range of concentrations. The ‘free’ inhibitor competes with the immobilized probes for drug-binding sites on target-protein complexes. Captured proteins are trypsinized and each peptide mixture is tagged with a distinct isobaric tandem mass tag (TMT). Tagged samples are pooled and analyzed by LC–MS/MS. Each peptide gives rise to six reporter signals in the MS/MS spectrum. When free drug outcompetes protein capture, signal intensities relative to the vehicle control decrease for each peptide. Complexes formed by the target and their associated proteins are then validated their IC50 values as well as by exploring their effects on downstream process.
In addition to competition approaches, methods have been developed that enable one to use specific target proteins directly as a vehicle for hit screening.[99] Examples from the Chapman laboratory at the University of Arizona demonstrate a technique for isolating small molecules from natural product extracts. In these studies, they demonstrated the ability to isolate three molecules that target different sites on the AAA+ chaperone p97.[100] Here, the team demonstrates that one can use a protein on a resin for probe isolation. Whether the matrix contains a probe as illustrated in the examples by the Cravatt laboratory and Cellzome, or the protein target on a resin, these methods clearly demonstrate how affinity capture offers an important tool when united with advanced analytical methods including fluorescent protein electrophoresis, ultrasensitive LC–MS and capillary NMR methods.
7.4. Protein-catalyzed Paal-Knorr reaction for the development of fluorescent cyclooxygenase probes
We have recently demonstrated that fluorescent probes for important protein targets can be prepared using the concept of target-guided synthesis (TGS). Specifically, we found that the active site of cyclooxygenase-2 (COX-2), an important anti-inflammatory drug target, provides a favorable environment for a chemical reaction that results in fluorescent products. As shown in Figure 28, the Paal-Knorr pyrrole synthesis, the condensation of 1,4-diketones and anilines, is an excellent example by providing fluorescent pyrroles (Figure 28).[101] The choice of this classical transformation was based on its important biological significance through the involvement in human physiological processes,[102] as well as the fact that it often proceeds under mild reaction conditions. Furthermore, work by Khanna and co-workers[103] demonstrated that 1,2-diaryl pyrroles could be potent selective COX-2 inhibitors indicating that the starting 1,4-diketones and anilines could be properly positioned at the COX-2 active site to undergo the Paal-Knorr process. We also expected that 1,2-diaryl pyrroles 46a–c (Figure 29) would be fluorescent due to their highly conjugated aromatic structures.
Figure 28.
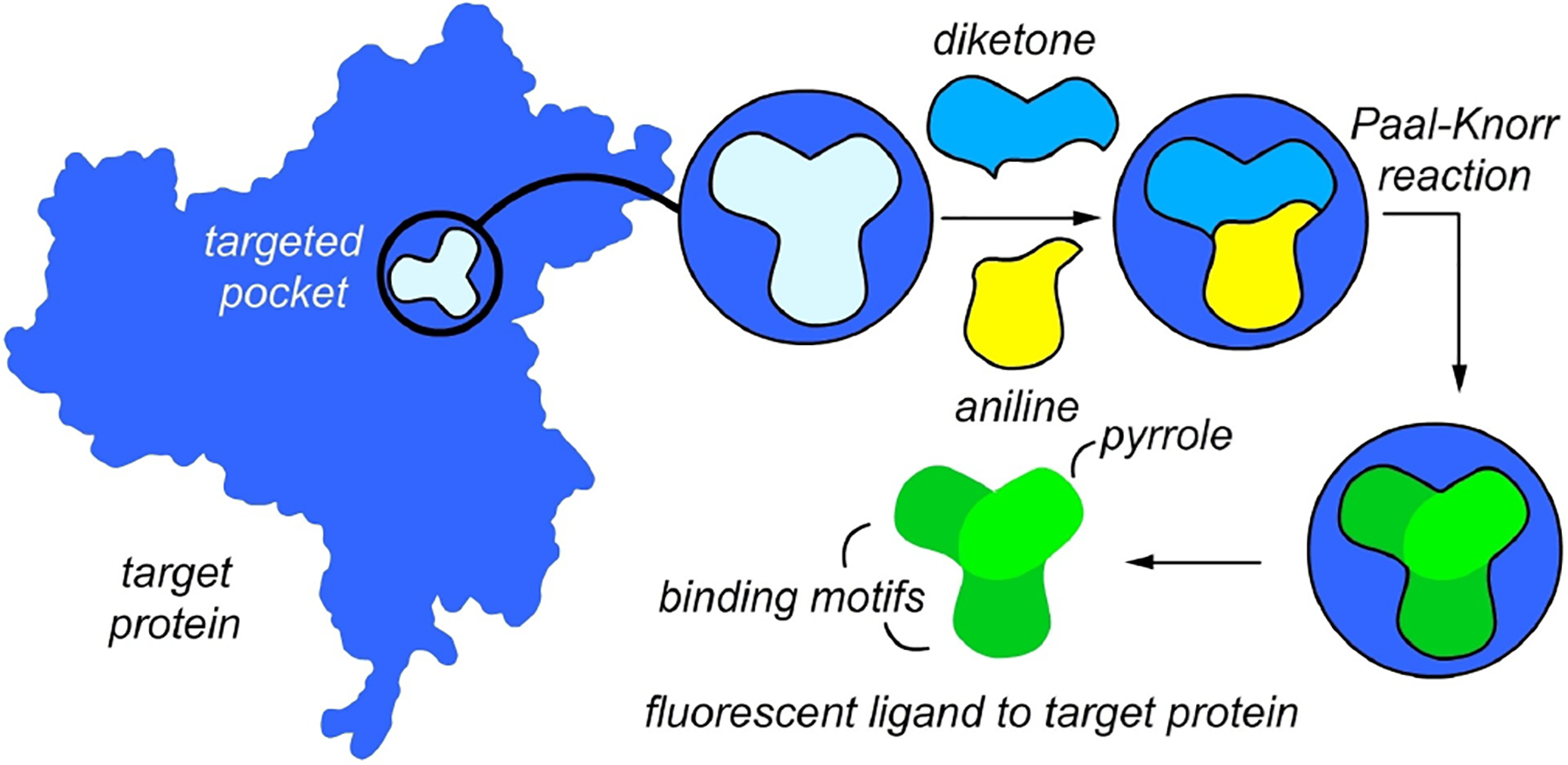
Illustration of the Paal-Knorr target-guided synthesis strategy. The binding of an aniline (yellow) and 1,4-diketone (blue) within the active site of COX-2 enables an in situ Paal-Knorr reaction that delivers a pyrrole product whose structure can be tailored to be fluorescent by the use of aryl-substituted diketones and aromatic amines, as depicted in Figure 29.
Figure 29.

Structure of fluorescent pyrroles 46a, 46b, and 46c. Pyrrole 46a was synthesized at the COX-2 active site using aniline 47 and diketone 48. The latter was generated from the Stetter reaction of aldehyde 49 with methylvinyl ketone (50).
Our team utilized the Stetter reaction[104] to synthesize the required 1,4-diketones (similar to 48 in Figure 29). Then, we screened a 13×13 array of the 1,4-diketones against selected anilines using COX-2 in PBS pH 7.2 containing 5% MeCN. Fluorescence spectra for each combination were compared to the corresponding reactions performed in the absence of COX-2 and the data were processed into a heat map of the fluorescence change identifying the combinations leading to a maximal change in fluorescence. Using these combinations, we prepared a 4×4 array and tested them for selectivity toward COX-2. Indeed, we found only small changes in fluorescence in reactions utilizing COX-1 and human serum albumin (HSA) compared to the COX-2 control.
Finally, we validated these results by NMR. Indeed, we observed the formation of pyrroles from reactions producing fluorescent changes, whereas a combination without fluorescent response did not produce Paal-Knorr reactivity. No reactivity was also observed in the presence of celecoxib, an established COX-2 inhibitor, providing additional evidence that the reaction occurred within the active site of COX-2. Overall, our work lays the foundation to the utilization of target-guided processes for the development of fluorescent probes.
7.5. Advantages
Direct link of biomolecule to probe
Binding guides selection
High-throughput accessible
Low detection
Target-guided approaches often play a vital role for programs with a defined goal in mind. As shown in the examples provided, these approaches can either focus on a single enzymatic pocket (example 1) or expand their complexity to select members of a class or family of proteins (example 2). Advantageously, these methods allow one through iterations of assay-guided optimization to translate a design into a selective agent. In a more autonomous fashion, one can use matrixes (solid surfaces or resins) to geographically isolate one of the two materials (probe or biomolecular target) and use it to affinity capture its partner (bimolecular target or probe) from solution. Overall, both of these strategic choices offer the user a means to directly link their probe to a specific biomolecule of interest ad identify it using high-throughput accessible low detection analyses. While the examples provided in this section were demonstrated for proteins, comparable approaches have been applied to search for probes to RNA/DNA and other biomolecules.[105,106]
7.6. Disadvantages
One often needs to be able to prepare the target in pure form
This process typically requires a de novo understanding of the target’s biology
It can be difficult to develop clear controls
Data can be cluttered with non-specifics responses
While target-guided approaches are enabling access to probes with levels of selectivity that may not be readily obtained by other approaches, these discoveries do come with a cost. First, they are often very labor intensive. One typically has to spend months to years designing and synthesizing the probe if it is used as the starting point for target discovery. Alternatively, one needs to prepare the target either by recombinant protein expression for protein targets, peptide synthesis for peptides, oligonucleotide synthesis for RNA/DNA targets or chemical synthesis/semi-synthesis for other small molecules. One needs to complete this task before one begins the study.
Once engaged, additional complexities also arise. The most complex and often limiting problem arises due to non-specific binding events. While pan-assay interference compounds or PAINS have been recognized as issues in target guided drug discovery, they also present large hurdles for probe development. Pan binding is not just limited to small molecules but is frequently observed in proteins that bind ligands of diverse sizes, shapes and compositions.[107] Comparable issues also exist within peptide[108] and RNA[109] applications of affinity selection. This complexity requires the development of sophisticated analytics that can return a large number of potential hits. This often requires the development of informatics tools that can selectively filter the viable hits from these sets. Further analytical challenges also exist, such as the fact that one does not typically run these experiments with stoichiometric equality between the molecules to be screened. Here a delicate balance exists between the concentration of the target and its affinity to and on/off rate with its associated probe. While improved analytical methods and informatics suggest a solution to these problems, the overall complexity of these studies can also hinder their ability to deliver a probe.
8. Tandem Target and Probe Discovery
For a large part probe discovery begins either with a reporter molecule and develops it as a tool to probe a specific biological event or target. Alternatively, one begins with a target and screens for a molecular probe. Recent efforts suggest a more agnostic approach that allows one to identify probe and its corresponding biological function and/or target in a single workflow. The following sections provide an overview of concepts that have helped enable one to unite target and probe discovery into a single process.
8.1. Streamlining probe discovery through tandem molecular and cellular studies
One of the fundamental advances adopted over the last decade is the discovery of centralized tools that enable both cellular and molecular studies to be conducted in one workflow. Understanding the activity of a fluorophore requires a unique balance between charting its molecular targets and cellular response.
For decades, the majority of the studies published focused on adopting a new method either for molecular target identification or for cellular phenotype analyses. From these studies, one identified a new feature given to a specific molecule, and then applied further studies, typically cellular ones, for target identification, or target identification efforts for cellular phenotypic work. While these efforts often focused on tool development, a recent shift has turned towards enabling methods that allow both molecular (target and pathway analyses) and cellular (localization, selectivity and phenotypic modulation) to be conducted in a single workflow, as shown in Figure 30.[110]
Figure 30.

Chemical proteomics. Comparable to conventional mode of action (MOA) workflows, the advance of chemical proteomics has enabled rapid elucidation of small molecule targets in cells and organisms. Schematically, this process begins with the addition of a Click handle (illustrated by alkyne) to the compound (orange sphere) this is followed by appendage of a covalently reactive group (when possible). a) The probe is then presented to the cell, tissue or organism and in cell Click chemistry can be used to track the cellular up take and subcellular localization. b) The molecular targets are then identified by appending either an affinity (A, green), photoaffinity (P, purple) or fluorescence (F) tag and isolating the proteins chromatographically using affinity resins or 2D SDS-PAGE gels.
While effective, often the tags used for cellular localization studies (Figure 30a) and target identification efforts (Figure 30b) are not the same, requiring one to assume that the structural difference between a fluorescent, affinity and/or photoaffinity tag would contribute no difference activity between the different probes. This is further complicated by the fact that many of these studies use different linker lengths or constructs to enable each approach, a fact that often leads to the incidental isolation of unwanted affinity (off-target binding), unwanted reactivity (reactivity with non-targeted active sites) or ineffective reactivity (lack of tag attachment) as shown on the bottom of Figure 30.
In 2006, a study on the mode of action (MOA) of the phorboxazole natural products[111] led us to carefully identify the salient factors associated with uniting image analyses (cellular uptake and localization of a molecular probe) and target identification studies. Here, we developed a system that used a single fluorescent tag to synthetically replace the bromoalkene terminus of 33-methoxyphorboxazole B (Figure 31a). The resulting probe 51 was then compared against controls that directly mimicked the structure of the tag and the linker used. In addition, this team expanded their SAR around the tag to further ensure that the observations did not arise from the tag, rather were correctly assigned to the natural product core (Figure 31b). This example provides a critically important lesson, namely, the need to present controls that direct mirror the functionality within the tag. All too often, studies compare vastly different tags for cellular imaging and affinity methods, leaving one to assume that the two probes with vastly different structures bear comparable activity. Unfortunately, this is not usually the case and one’s comparison between cellular studies (Figure 31a) and molecular targeting (Figure 31b) are often clouded with off-target events.
Figure 31.

Importance of controls in centralized workflows. The development or uniform linker and controls are critical to studies that explore the cellular and molecular mode and mechanisms of a compound’s activity. a) In this study, a fluorescent coumarin analogue 51 was prepared as a replacement of the alkene terminus of 33-OMe-phorboxazole B (52). b) Both cellular localization and molecular targeting was identified by comparing the activity of 53 to a fluorescent linker control 51. Here, different control analogues were included to expand the SAR of the controls to ensure that the fluorescent tag and its conjugation within51 do not present off-target activity.
Most critically, this and subsequent studies[112] demonstrated the importance of using the same probe for cellular and molecular studies. Since this work, the development of anti-fluorophore monoclonal antibodies (mAbs),[113] now allow direct use of a fluorophore as an affinity reporter. While more conventional interactions such as avidin-streptavidin are commonly used for target identifications in chemical genomics, the fact remains that it is very difficult to compare these data with their cellular counterparts (obtained by clicking with a fluorescent tag instead of the biotin tag used for affinity studies).
8.2. Fluorescent probe discovery through a bidirectional affinity approach
The next strategy explores the concept of using affinity systems as a tool to simultaneously isolate natural products and identify their modes and mechanisms of action. First introduced in 2008, a strategy was developed that used proteomes as tools to isolate natural products from test fractions (whole extracts or fractionated extracts).[114]
As shown in Figure 32, cell lysis (ideally different methods in parallel) was used to prepare proteomes of a targeted cell. The resulting collection of proteins was then attached to a resin by using a succinimidyl-ester resin (Affi-Gel 10) or hydrazide resin (Affi-Gel Hz) (Step 1, Figure 32). The resulting resins were presented to natural product extracts or fractionated extracts and the bound materials were eluted using EtOH (Step 2, Figure 32). Next, the EtOH fractions, containing proteome binding natural products (Step 3, Figure 32), were then labeled using fluorescent tag that has a corresponding mAb for affinity selection, referred to as an immunoaffinity fluorescent (IAF) tag.[113]
Figure 32.

Bidirectional affinity natural product Isolation. A five-step protocol has been reported that enables one to agnostically-discover natural products and their associated protein targets (step 1). The procedure begins by preparation of proteomic resins by lysing targeted cells and covalently-attaching the resulting proteins to reactive resins such as Affi-Gel 10. Step 2: The resulting proteomic resins are then used to as affinity matrixes to isolate natural products from crude or fractionated extracts. Step 3: The proteome bound natural products where then tagged with an immunoaffinity fluorescent (IAF) tag (54) and purified using resins bearing an antibody against the IAF tag (55). Step 4: The resulting probes were then used for uptake and subcellular localization studies and (step 5) target immunoprecipitation using resin bound antibodies against the IAF tag.
Once purified, the fluorescently labeled proteome binding probes were presented to their targeted cell and imaged (step 4, Figure 32). Here, high-resolution imaging was used to determine the rate of cellular uptake and identify potential subcellular targets. The resulting cells were then lysed and immunoprecipitated (IP) using the anti-IAF tag antibody to isolate the bound probes and their targets (step 5, Figure 33). To date, this process has been used to identify the binding of sceptrin to bacterial actin equivalent MreB,[114] aplysqualenol A binding to the light chain of dynein type 1 (DYNLL1),[115] and a glycosphingolipid coniferoside.[116] The first study used an E. coli proteome while the latter two applied human proteomes from the HCT-116 colon tumor cell line. In the latter study, the team applied subcellular fractionated proteomes. Here, the resolution of the system was improved by comparing the efficacy of the affinity purification over the different subcellular fractions. Ideally, this could be conducted in parallel with imaging methods (step 4, Figure 32).
Figure 33.

Inverse drug discovery strategy. Recently a team at Scripps Research described an approach to drug discovery that united covalently reactive compounds, click tags for detection, proteomic analyses and structural biology to identify binding pockets for therapeutic discovery. In this example a reactive fluorosulfate is used to append a clickable tag for SDS PAGE proteomic analyses. One protein GSTO01 was identified and the structure of its covalent adduct was determined by X-ray crystallography. A clicked fluorescent Click tag enabled the detection and isolation of the targeted protein. While the structure of its tag was not critical to the effort, the use of this tag illustrates an important application for probe development.
8.3. The concept of inverse drug discovery
Recently, a team at Scripps Research described a strategy called Inverse Drug Discovery[117] in which compounds bearing electro-philic residues were used to identify their selectivity to proteins within cells or cell lysates. As shown in Figure 33, the strategy begins by synthesizing an analogue of the motif bearing an alkyne tag for Click modification and arylfluorosulfates,[118] an underexplored class of sulfur(VI) halides that are generally unreactive unless activated by protein binding. Here, they juxtaposed the arylfluorosulfate and alkyne on the molecule so the reactive unit (arylfluorosulfate) would point into the pocket and the alkyne tag would be directed outward. Using this motif, the team applied structure-activity data to enhance the selectivity of conjugate formation. Here, the covalent probe can be used as a competitor to develop non-covalent drug candidates.
In one example of this approach study (Figure 33),[119] their team identified and validated covalent ligands for 11 different human proteins, two of which were discovered for the first time. Unlike the bidirectional system (Figure 32), which can suffer from the need for high affinity or slow off rates within its ligand binding, the use of covalent reactivity and generality offers a rapid new strategy to guide the identify ligand binding domains.
8.4. Enabling probe discovery through nanoscale structure elucidation and affinity ranking
In addition to affinity-based approaches, recent efforts have demonstrated an important shift towards nanoscale reactivity. While synthetic programs typically are not limited to scales less than microcales (μmol of compound), natural product programs are often challenged by material access. Over a decade ago,[120] natural product chemists realized the importance of developing nanoscale methods for compound isolation and purification.[121]
Studies described below illustrate how nanomole scale processes can be used to isolate complex polyketides, such as the phorbasides G–I from the sponge Phorbas sp.[122] or the trioxazole macrolides from the nudibranch Hexabrachus sanguineus.[123] The key to these studies arises through access to 1.7 mm cryo-NMR probes that can collect high resolution datasets on 20–50 μL volumes often with less than 50 μg of material.[124] This, along with the development of ultraclean isolation techniques, now enables one to isolate natural products from small sample sizes (often ≤ 100 mg of a specimen).
Nanoscale methods have been adapted to streamline medicinal chemical lead discovery programs. In 2018, a team led by Cernak[125] coupled high-throughput chemical synthesis with a label-free affinity mass spectrometric bioassay to enable the primary synthesis and testing steps required to advance lead 57 from 56 (Figure 34). In their studies, they were able to demonstrate nanoscale synthesis typically conducting each reaction with 50 μg of material. These crude reaction products were screened in line using affinity mass spectroscopy to identify materials with affinity to a protein target. For example, as shown in Figure 34, this resulted in the discovery of one analogue 58 that was prepared in 85% yield and had an IC50 value of 23 nM against CHK1 kinase.
Figure 34.

Nanoscale synthesis and affinity ranking (nanoSAR). A schematic representation of the reaction and chemical space prepared and biologically assayed directly to identify potent inhibitors and reaction conditions, simultaneously. Nu represents building blocks that were coupled to 56 by a Suzuki and C–N couplings. A total of 170 reactions were conducted to prepare and screen a library of 20 CHK1 inhibitors. A total of 8.8 mg of 56 was used for this study. Reaction conditions included a catalyst (tBuBrettPhos Pd G3), base (7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene or MTBD) in 1-methyl-2-pyrrolidinone (NMP, solvent). Each reaction was run at nanomol scales and leads were rescaled at the 50 μmol scale. While the structure of its fluorophore was not critical to the effort, this study illustrates an excellent application for future probe designs.
Overall, the ability to find and prepare lead molecules at nanomolar scales has enabled one to effectively repurpose discovery workflows to provide a more effective union between the chemical and biological aspects of small molecule discovery. While the example described in Figure 34 is not directly applied to production of a fluorescent compound, the strategy developed through this program is readily amenable to fluorescent probe development as described in the previous sections of this review.
8.5. Fluorescent probe discovery through dual affinity screening
Recently, our team demonstrated how nanoscale methods can be applied to the discovery of fluorescent materials.[126] As shown in Figure 35, this approach adapted a high-throughput fluorescence binding assay (CONA)[127] and functional chromatography (FC),[128] two protein-resin technologies, enabling the discovery and isolation of fluorescent natural product probes that target proteins independently of biochemical function.
Figure 35.

Target protein-guided hit prioritization and probe identification. Two different resins, a) a scanning confocal microscopy (CONA) resin and b) a functional chromatographic (FC) resin, are applied to screen extracts A–B for protein binders and isolate them. The CONA resin us used to prioritize the extracts at high-throughput and the second or FC resin is used to isolate the target protein binding compounds. Using 1.7 mm capillary NMR the team was able to conduct this dual-resin affinity screen using to discover probes as low as 5 μg of material.
The tandem application of a CONA screen to identify potential hit extracts (extract prioritization) and functional chromatography to isolate these materials enabled the rapid isolation and elucidation of fluorescent probes. The use of 1.7 mm capillary NMR allowed this team to conduct this process at nanoscale. The resulting probes also suggest targetable pockets for lead discovery. As shown in Figure 35, this method was demonstrated by the discovery of members of the prodiginine family as fluorescent probes to the cancer target, survivin. The strategy of using different resins for screening and isolation in tandem not only improved the efficiency of the screening process, but also, identified a vector to directly transit from screen to active material within one workflow.
8.6. Photoaffinity probes through centralized crosslinking strategy
In addition to affinity resin-based systems, profound advances have arisen in the development of technologies to improve the ability to confirm binding between a small molecule and its target. While direct affinity tools can often be sufficient, success is directly regulated by the potency and rate of these interactions, wherein off-rates (koff) are often critical for any affinity capture system to operate in a practical sense. As one is not always looking for a lead with a slow Koff, the installation of covalent modifying groups offers a means to covalently-trap intermediate stages. Over the recent year tools such as diazirine-based photoaffinity probes (PAs) have been shown to facilitate the ligand·target discovery process by covalently capturing transient molecular interactions[129] both at the small molecule - protein interaction level or at the interface between proteins.[130]
While more commonly used to discover the interaction of non-fluorescent materials, recent efforts have enabled approaches to fluorogenic photoaffinity labeling (f-PAL), light induced labeling with a fluorescent probe.[131] As shown in Figure 36, a team led by Tomohiro report a novel technique for fluorescently-labeling at crosslinked positions of an interactive protein by photochemical induction. Using biotin as their ligand, the process begins by photoactivation of the diazirine and insertion of the incipient carbene into a OH bond within the protein pocket (black, Figure 36). The resulting complex enables the phenol to undergo an intramolecular esterification, releasing the biotin ligand and generating a fluorescently labeled target protein.
Figure 36.

Isotope-coded fluorogenic crosslinking, a schematic representation of the fPAL strategy. A two-step process involves carbene activation and photochemical target capture results in a fluorescent adduct 61 from 59. Intramolecular attack by a proximal phenol within 60 results in the formation of a fluorescent coumarin motif, which is covalently attached to an amino acid residue within the binding pocket of the targeted protein.
8.7. Fluorescent protein labeling through the concept of tag-transfer
The next set of studies explores a concept in transferring a tag between a small molecule ligand and its biomolecular target. In the example in Figure 36, the ligand, biotin, served the purpose of binding within a specific pocket within the targeted protein. Once bound, light induced covalent reactivity and subsequent cyclization resulted in loss of the biotin ligand. This concept of using a ligand as a part of a chemical reaction has recently gained attention as a means to transfer a tag from a probe to a protein pocket of interest. Here, two examples are provided to outline this reactivity.
The strategy in the first example developed around applying amidation with a phenolic ester as a tool to site selectively label within a protein. As shown in Figure 37a, a fluorophore was attached as esters to the phenolic groups to afford the fluorophore-transfer cassette 62. Binding of 62 to its target resulted in the transfer of the fluorescent tag to actin within lysates of the HCT-116 (human colon cancer) cell line. This report in 2009,[132] was completed in parallel with the isolation of this natural product, marinopyrrole A (64 in Figure 37b)[133] and illustrates the efficiency and versatility of the fluorophore transfer concept.
Figure 37.

Acyl fluorophore transfer concept. a) A schematic representation of the strategy, wherein a fluorophore-transfer cassette 62. provides both a fluorophore and ligand (a natural product). Binding of a protein target results in a reaction between a nucleophilic residues in the active site of the bound complex 63. This results in the loss of the natural product (a leaving group) and fluorescent tagging of the target protein within its binding pocket. b) Structures of marinopyrrole A (65) and the fluorophore-transfer cassette 66 prepared in this study.
Also in 2009, a team lead by Hamachi[134] described the use of tosyl chemistry as a general tag-transfer concept. Unlike the phenolic ester approach in Figure 37, this team developed fluorophore-transfer cassette that could be coupled to both a ligand and fluorescent (or affinity) tag (Figure 38). Here, the cassette 67 served as an alkylating agent reacting with nucleophilic residues within the targeted protein’s binding pocket. This reaction resulted in an SN2-type reaction where the ligand acted as a leaving group. One example, cassette 70 was used to fluorescently label human carbonic anhydrase II both in vitro and ex vivo.
Figure 38.

Tosyl chemistry for labeling endogenous proteins in living native cells. a) Schematic illustration of the strategy. The method allows for the labeling of a target protein with a fluorescent (or affinity) tag within its binding pocket. b) Probe 70 contained a central fluorophore-cassette that was linked to a fluorophore and a ligand. In this example the ligand was designed to target carbonic anhydrase.
8.8. Advantages
Versatile, can be adapted to many biological studies
Diverse, incorporation of a target in harmony with small molecule discovery offers a non-biased approach to lead identification.
Rigorous, methods such as the dual affinity approach adapt experimentation in two directions that each of which controls for the other.
Innovative, concepts such as the tag-transfer or dual-affinity approaches open new avenues to identify and selectively mark protein targets.
Over the last decade, it is becoming increasingly clear that the next generation of probe discovery efforts will fuse biological studies in unison with chemical preparation and modification efforts. The strategies provided within this section provided a strong foundation to synergistically-unite small molecule and biomolecular discover within a single workflow. While early in its development approaches such as those described within this section offer ‘game-changing’ potential for a wide array of molecular and cellular biological queries.
8.9. Disadvantages
Complex, one requires a detailed understanding of chemical synthesis to prepare the reactive and tagged materials described in this section.
High selectivity precludes generality, as one develops probes with improved selectivity, the designs and structural modifications required typically reduce the generality of the approach. Here, the fluorophore is appended at an electro-philic site of a small-molecule ligand to allow nucleophilic attack by a protein residue is suggested. If the ligand can act as a leaving group, such as marinopyrrole, then fluorophore transfer occurs.
While attractive concepts, application of the approaches in this section are limited by their generality. In many cases, specific functionality was required for the strategy, which for many materials could not be used. For other approaches, the uses of resins or tags are described in a manner that would not be applicable to all proteins or small molecules due to strict activity guidelines. While innovative, many of the strategies described in this section can require highly skilled chemical biologists with experience in both chemical synthesis, molecular and cellular biology.
9. Reaction-Based Fluorescent Probes
Traditionally, fluorescent probes are developed based on receptor-analyte non-covalent interactions or metal-coordination bonding. However, many analytes undergo molecular transformation in biological systems. Thus, an emerging bio-inspired strategy for molecular imaging was based on exploiting the differences in molecular reactivity, rather than the traditional lock-and-key type of molecular recognition.[135] Fluorescent probes that work through making or breaking (example in section 8.7) of covalent bonds can provide higher selectivity with larger spectroscopic changes due to the more significant structural changes resulting from chemical conversion. In addition, the reaction-based sensing can provide specificity that is bioorthogonal to the endogenous biological reactivity of the analyte to be studied. Of course, in the ideal case the reaction should be unperturbed by interfering species in the biological milieu and proceed with adequate kinetics in water at the physiological pH, high salt content and reactive nucleophilic thiols, such as glutathione and cysteine.
9.1. The use of an oxidative cycloaddition reaction for bioimaging of nitric oxide
Cycloaddition reactions have been widely used for the development of novel fluorescent probes because they can lead to the formation of heterocycles conjugated to a fluorescent scaffold significantly altering its optical properties. In an original contribution, Nagano and co-workers showed that aromatic diamines can react with nitric oxide in the presence of oxygen to form triazoles.[136] This reaction has led to the discovery of diaminofluoresceins (DAFs, 76, Figure 39), fluorescent probes for the detection of nitric oxide in living cells. DAFs are not highly fluorescent because of the photoinduced electron transfer quenching from the electron-rich amino groups. The transformation of the diamino moiety to the triazole reduces the electron-donating ability of the nitrogen atoms, reducing quenching of the fluorophore moiety and resulting in a turn-on emission (Figure 39). This process has been used with various fluorophore scaffolds, such as cyanine 71,[137] BODIPY 72,[138] and rhodamine 75 (Figure 39),[139] possessing different degrees of sensitivity and photostability. DAFs have been applied to study nitric oxide-mediated processes in cells, tissue slices and even the whole organisms elucidating the roles of this small molecule in the cardiovascular system, cancer, immune response, neurotransmission and fertilization.[140]
Figure 39.

Conversion or aromatic diamine to triazole through a reaction with nitric oxide in the presence of oxygen. Diamines 71 and 72 are not fluorescent because of the photoinduced electron transfer quenching from the electron-rich amino groups. The transformation of the diamino moiety to the triazole reduces the electron-donating ability of the nitrogen atoms reducing fluorescence quenching of the fluorophore moiety and resulting in a turn-on emission. The technology has been applied to cyanine 71 and BODIPY 72 scaffolds as well as diaminorhodamine 75 and diaminofluorescein 76. Circles for 71 and 73 provide IR wavelengths numerically in nm.
9.2. Detection of protease activity using a covalent assembly probe design
The most common approach to covalent probe design involves a reversible protection of a functional group on an organic fluorophore, such as an amino or hydroxyl group of a coumarin or xanthene fluorophore.[141] However, such probes often exhibit poor stability in biological media and high background fluorescence due to incomplete quenching of the masked fluorophore. These drawbacks correspondingly lead to insufficient selectivity and sensitivity toward the target analyte. An approach that circumvents these deficiencies involves the design of reactive fluorescence turn-on probes based on a covalent-assembly principle. Here, the reaction with the target analyte would trigger the in situ formation of a fluorophore from a caged bifunctional precursor, often through biocompatible domino processes. In a salient example of this strategy, Debieu and Romieu reported the design of a covalent-assembly probe involving the in situ formation of a pyronin fluorophore for fluorescent protease sensing.[142]
The probe 77,[142] designed to work with leucine aminopeptidase (LAP), was projected to undergo enzyme-mediated amide bond hydrolysis to release aniline 78 (Figure 40). Intramolecular cyclization to yield intermediates 79, and 80, would be followed by the departure of the hydroxyl to give pyronin 81, emitting in the green-yellow region of the spectrum. Indeed, a gradual increase of green-yellow fluorescence intensity at 545 nm (excitation at 525 nm) was observed when probe 77, was incubated with LAP. The presence of pyronin fluorophore 81, was confirmed in the enzymatic mixtures by RP-HPLC analyses.
Figure 40.

Proposed mechanism involved in the detection of protease activity based on the covalent-assembly approach. Leucine aminopeptidase 77 (LAP) hydrolyses the amide bond and releases aniline. Cyclization followed by the hydroxyl departure gives fluorescent pyronin 81.
9.3. Fluorescent detection of Hg2+ ions using a metal-mediated desulfurization reaction
While the traditional approach to the development of metal sensors involves coordination chemistry based on molecular recognition and binding, the utilization of intrinsic and distinct reactivity profiles associated with metal ions offers a powerful alternative strategy. Thus, strong thiophilic reactivity of mercury ions has given rise to a significant number of fluorescent probes involving optical changes associated with desulfurization reactions, such as hydrolysis,[143] eliminations[144] and cyclizations.[145] To use desulfurization chemistry for the detection of Hg2+ ions, Tae and co-workers[146] utilized the known spirolactam to ring-opened amide equilibrium of rhodamine derivatives, in which the latter was fluorescent.[147] This equilibrium was coupled with an irreversible Hg2+-promoted cyclization of thiosemicarbazides to 1,3,4-oxadiazoles that leads to the formation of rhodamine derivative 83 from 82 (Figure 41). When mercury ions are added to a PBS solution of 82, the latter instantaneously turns from colorless to pink and becomes strongly fluorescent with the emission maximum at 557 nm. The probe was also shown to be highly successful in in vivo applications in living cells and vertebrate organisms.[148]
Figure 41.

Hg2+-promoted cyclization of thiosemicarbazides to 1,3,4-oxadiazoles leads to irreversible conversion of spirolactam 82 to fluorescent ring-opened rhodamine derivative 83. When mercury ions are added to a PBS solution of 82, the latter instantaneously turns from colorless to pink and becomes strongly fluorescent.
9.4. Cyanine conjugates as a theranostic for cancer
The discovery of fluorescent probes covalently conjugated to a therapeutic agent for simultaneous tumor imaging and therapy has attracted a great deal of attention recently. Often, such agents possess intrinsic tumor targeting might lead to the personalized treatment of cancer patients in the clinic. A recently discovered[149] near-infrared (NIR) heptamethine fluorophore IR-780 (84 in Figure 42a), was found to accumulate predominantly in the mitochondria of tumor cells. Like other NIR fluorophores, IR-780 (84) was characterized by high extinction coefficients, large Stokes’ shifts and thus emission wavelengths of 700–1000 nm, which can be detected from deep within the tissues in a non-invasive manner.
Figure 42.

Near IR fluorophores. a) Structure of IR-780, a NIR fluorophore with significant accumulation in mitochondria of tumor cells. b) Structure of IR-780/nitrogen mustard theranostic agent showing excellent accumulation in tumor xenografts in vivo. Circles for 84 and 85 provide IR wavelengths numerically in nm.
Due to its mitochondrial targeting and excellent NIR imaging properties, IR-780 was investigated as a potential theranostic agent.[150] Nitrogen mustards are highly effective DNA damaging agents and inducers of apoptotic cell death cancer cells. However, their poor ability to differentiate between normal and cancerous cells leads to severe side effects.[151] Thus, a covalent construct IR-780-nitrogen mustard was synthesized (85 in Figure 42b) and its tumor targeting ability was demonstrated using rTDMCs tumor xenografts. Indeed, fluorescent signals were detected for the rTDMCs tumors grown subcutaneously with low background interference. The results also suggest a significant nitrogen mustard accumulation within the tumors.
9.5. Advantages
Higher selectivity due to more significant structural modifications
Larger spectroscopic changes due to more significant structural modifications
Improved signal to noise ratio due to more significant structural modifications
Opportunities for catalytic processes due to utilization of chemical reactions
Bioorthogonal specificity due to differences in chemical reactivity rather than binding properties
Possibilities for spatially resolved bioimaging due to covalent reactivity toward biological macromolecules
The design of covalent fluorescent probes is a rapidly growing field that can have a major impact on our understanding of biological processes on molecular level. Fluorescent probes whose discovery involves thorough understanding of organic, organometallic and inorganic reactivity principles offer an alternative strategy that provides higher selectivity with larger spectroscopic changes due to the more significant structural modifications resulting from chemical transformation. Currently, most of the reported reaction-based probes are based on irreversible stoichiometric processes. Even more promising are strategies that utilize reversible chemoselective as well as catalytic processes involving signal amplification. These discoveries could profoundly improve signal-to-noise ratio and dynamic imaging. Finally, reaction-based fluorescent probes offer the possibilities of spatially resolved bioimaging through targeted delivery to specific tissue or organs as well as subcellular localization.
9.6. Disadvantages
Consideration of competing reactive analytes often present in larger concentrations than the desired target
The probe must have biocompatible reactivity
Reaction needs to proceed with an adequate rate at physiological pH and high salt concentration
Consideration of reactive thiols
Reaction should not interfere with endogenous biochemical processes
Reaction should not generate toxic products
Because of complexity of biological systems, generally containing large numbers of competing analytes, reaction-based fluorescent probes must be designed with consideration of a number of stringent requirements. Indeed, the competing analytes may have functional groups with similar reactivity and be present at higher concentrations. The probe should be biocompatible and the proposed reaction should proceed at an adequate rate at physiological pH in the presence of high salt concentration and reactive thiols. Bioorthogonality is another important consideration. The reaction should not interfere with endogenous biochemical processes or generate toxic by-products. Thus, generally, the reaction-based bioimaging is a significant challenge requiring the consideration of fundamental organic, organometallic and inorganic reactivity of the probe together with the intrinsic reactivity of the target analyte to achieve sufficient chemoselectivity and bioorthogonality.
10. Conclusion
Ever since the first reports of fluorescence,[1,2] and subsequent elucidation of its structural principles, we continue to challenge the mechanisms in which its action can be used to specifically track molecular events visually. In this review, we have presented several of the key principles within fluorescent probe design. Through specific instructive examples, an illustration of concept and its associated design emerge that unite the presence of a fluorescent signal with a specific molecular transition. Over the last two decades, we have not only understood how to genetically address fluorescence but also, and perhaps most critically, defined some remarkable concepts to aid and inspire molecular design.
With the advent of artificially intelligent algorithms and highly-digitized optical instrumentation, it is very likely that fluorescent systems will not only play a key role in biology, but also, offer a robust future that allows one to rapidly unite algorithms with molecular transitions. It is not unreasonable to expect that uniting computational systems with optical sensor may one day enable fluorescent design to be done through artificial intelligence. It is also not hard to imagine a device comprised of digitally optic arrays, molecular reagents and processors that could design its own fluorescent probes, but until then, we leave the reader with this review of lessons in probe designs.
Acknowledgments
This effort was supported by NIH grants 5R21GM131717 and 1R15CA227680.
Biographies

Sachin B. Wagh obtained his M.Sc. in Chemistry at Pune University in India (2012) and then joined CSIR-National Chemical Laboratory, Pune, as a research assistant in the laboratory of Dr. D. S. Reddy. He received his Ph.D. in 2018 from the National Tsing-Hua University (Taiwan) under the supervision of Prof. R. S. Liu. He continued his postdoctoral studies in the same research group. In 2019, he moved to the United States, where he is currently a postdoctoral research associate at Texas State University.

Vladimir A. Maslivetc completed his B.Sc. in 2011 and his M.Sc. in 2013 at the Perm State University, Russia. He received his Ph.D. in chemistry from the University of Kansas under the supervision of Prof. Michael Rubin in 2019. He is currently a postdoctoral research fellow at Texas State University. His research interests include synthetic methodology for compounds with biological importance and transition metal catalysis.

James J. La Clair received his Ph.D. degree in Chemistry from SUNY Buffalo under the tutelage of Prof. Peter T. Lansbury. His studies continued through a postdoctoral fellowship with Prof. Gilbert Stork at Columbia University. He then undertook a position at the Scripps Research Institute. In 1999, he went independent in order to focus his time on research including positions at UC San Diego, the Salk Institute and XRI.

Alexander Kornienko studied at Mendeleev University in Moscow and then moved to the United States, where he received his Ph.D. in synthetic organic chemistry at Tufts University. After a postdoctoral fellowship at the University of Montreal, he started his independent career at New Mexico Tech in 2001 and then moved to Texas State University in 2012, where he is now Professor of Chemistry.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
References
- [1].Monardes N in Historia medicinal de las cosas que se traen de nuestras Indias Occidentales, Seville, 1565. [Google Scholar]
- [2].Sahagún B in 1499–1590, Florentine Codex: General History of the Things of New Spain, 2nd ed., Revised, Santa Fe, N. M.: The School Of American Research; Salt Lake City, Utah, University Of Utah, 1970. [Google Scholar]
- [3].a) Acuña AU, Amat-Guerri F, Morcillo P, Liras M, Rodriguez B, Org. Lett 2009, 11, 3020–3023; [DOI] [PubMed] [Google Scholar]; b) Tomalia DA, Klajnert-Maculewicz B, Johnson KA-M, Brinkman HF, Janaszewska A, Hedstrand DM, Prog. Polym. Sci 2019, 90, 35–117. [Google Scholar]
- [4].Acuña AU, Amat-Guerri F, in Fluorescence of Supermolecules, Polymers, and Nanosystems, Springer, 2007, pp. 3–20. [Google Scholar]
- [5].Specht EA, Braselmann E, Palmer AE, Annu. Rev. Physiol 2017, 79, 93–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].a) Ercal N, Yang P, Aykin N, J. Chromatogr. Biomed. Appl 2001, 753, 287–292; [DOI] [PubMed] [Google Scholar]; b) Ryant P, Dolezelova E, Fabrik I, Baloun J, Adam V, Babula P, Kizek R, Sensors 2008, 8, 3165–3182; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Rusin O, St Luce NN, Agbaria RA, Escobedo JO, Jiang S, Warner IM, Dawan FB, Lian K, Strongin RM, J. Am. Chem. Soc 2004, 126, 438–439; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Guan X, Hoffman B, Dwivedi C, Matthees DP, J. Pharm. Biomed. Anal 2003, 31, 251–261; [DOI] [PubMed] [Google Scholar]; e) Potesil D, Petrlova J, Adam V, Vacek J, Klejdus B, Zehnalek J, Trnkova L, Havel L, Kizek R, J. Chromatogr. A 2005, 1084, 134–144; [DOI] [PubMed] [Google Scholar]; f) Sato Y, Iwata T, Tokutomi S, Kandori H, J. Am. Chem. Soc 2005, 127, 1088–1089; [DOI] [PubMed] [Google Scholar]; g) Zhou Y, Yoon J, Chem. Soc. Rev 2012, 41, 52–67. [DOI] [PubMed] [Google Scholar]
- [7].a) Lavis LD, Raines RT, ACS Chem. Biol 2008, 3, 142–155; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wang L, Frei MS, Salim A, Johnsson K, J. Am. Chem. Soc 2018, 141, 2770–2781; [DOI] [PubMed] [Google Scholar]; c) Lei Z, Sun C, Pei P, Wang S, Li D, Zhang X, Zhang F, Angew. Chem 2019, 131, 8250–8255; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed 2019, 58, 8166–8171. [DOI] [PubMed] [Google Scholar]
- [8].Chen X, Zhou Y, Peng X, Yoon J, Chem. Soc. Rev 2010, 39, 2120–2135. [DOI] [PubMed] [Google Scholar]
- [9].a) Garfield S in Mauve: How One Man Invented a Color That Changed the World, WW Norton & Company, 2002; [Google Scholar]; b) Christie R in Colour Chemistry, Royal Society Of Chemistry, 2014; [Google Scholar]; c) Kaufman TS, Rúveda EA, Angew. Chem. Int. Ed 2005, 44, 854–885; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2005, 117, 876–907. [Google Scholar]
- [10].Lynen F, Wieland U, Justus Liebigs Ann. Chem 1938, 533, 93–117. [Google Scholar]
- [11].a) Wulf E, Deboben A, Bautz FA, Faulstich H, Wieland T, Proc. Natl. Acad. Sci. USA 1979, 76, 4498–4502; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Anderl J, Echner H, Faulstich H, Beilstein J. Org. Chem 2012, 8, 2072–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].a) Cooper JA, J. Cell Biol 1987, 105, 1473–1478; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Barden JA, Miki M, Hambly BD, Dos Remedios CG, Eur. J. Biochem 1987, 162, 583–588. [DOI] [PubMed] [Google Scholar]
- [13].a) Weber G, Biochem. J 1952, 51, 155–167; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Weber G, Biochem. J 1952, 51, 145–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].a) Xie P, Guo F, Wang L, Yang S, Yao D, Yang G, J. Fluoresc 2015, 25, 319–325; [DOI] [PubMed] [Google Scholar]; b) Xi L-L, Guo X-F, Wang C-L, Wu W-L, Huang M-F, Miao J-Y, Zhao B-X, Sens. Actuators B 2018, 255, 666–671; [Google Scholar]; c) Wang P, Zhou D, Chen B, Spectrochim. Acta Part A 2019, 207, 276–283. [DOI] [PubMed] [Google Scholar]
- [15].a) Walker JM, Basic Protein Pept. Protoc 1994, 321–328; [Google Scholar]; b) Walker JM, Basic Protein Pept. Protoc 1994, 329–334. [Google Scholar]
- [16].a) Gray WR, Hartly BS, Biochem. J 1963, 89, 59; [DOI] [PubMed] [Google Scholar]; b) Sandhu SS, Robbins CR, J. Soc. Cosmet. Chem 1989, 40, 287–296. [Google Scholar]
- [17].Newman DJ, Cragg GM, J. Nat. Prod 2020, 83, 770–803. [DOI] [PubMed] [Google Scholar]
- [18].a) Duval R, Duplais C, Nat. Prod. Rep 2017, 34, 161–193; [DOI] [PubMed] [Google Scholar]; b) Hong J, Curr. Opin. Chem. Biol 2011, 15, 350–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Venugopala KN, Rashmi V, Odhav B, BioMed Res. Int 2013, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cao Z, Yu Y, Wu Y, Hao P, Di Z, He Y, Chen Z, Yang W, Shen Z, He X, Sheng J, Xu X, Pan B, Feng J, Yang X, Hong W, Zhao W, Li Z, Huang K, Li T, Kong Y, Liu H, Jiang D, Zhang B, Hu J, Hu Y, Wang B, Dai J, Yuan B, Feng Y, Huang W, Xing X, Zhao G, Li X, Li Y, Li W, Nat. Commun 2013, 4, 2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].a) Sun WC, Gee KR, Haugland RP, Bioorg. Med. Chem. Lett 1998, 8, 3107–3110; [DOI] [PubMed] [Google Scholar]; b) Hickey SM, Nitschke SO, Sweetman MJ, Sumby CJ, Brooks DA, Plush SE, Ashton TD, J. Org. Chem 2020, 85, 7986–7999. [DOI] [PubMed] [Google Scholar]
- [22].Gee KR, Sun W-C, Bhalgat MK, Upson RH, Klaubert DH, Latham KA, Haugland RP, Anal. Biochem 1999, 273, 41–48. [DOI] [PubMed] [Google Scholar]
- [23].Klunk WE, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, Bergström M, Savitcheva I, Huang G, Estrada S, Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc 2004, 55, 306–319. [DOI] [PubMed] [Google Scholar]
- [24].Tong H, Lou K, Wang W, Acta Pharm. Sin. B 2015, 5, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, J. Biol. Chem 2005, 280, 5892–5901. [DOI] [PubMed] [Google Scholar]
- [26].Hewlings SJ, Kalman DS, Food 2017, 6, 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].a) Ulrich G, Ziessel R, Harriman A, Angew. Chem. Int. Ed 2008, 47, 1184–1201; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2008, 120, 1202–1219; [Google Scholar]; b) Si G, Zhou S, Xu G, Wang J, Wu B, Zhou S, Dyes Pigm. 2019, 163, 509–515. [Google Scholar]
- [28].Zarghi A, Arfaei S, Iran J. Pharm Res: IJPR 2011, 10, 655. [PMC free article] [PubMed] [Google Scholar]
- [29].Uddin MJ, Crews BC, Blobaum AL, Kingsley PJ, Gorden DL, McIntyre JO, Matrisian LM, Subbaramaiah K, Dannenberg AJ, Piston DW, Cancer Res 2010, 70, 3618–3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Yadav AK, Reinhardt CJ, Arango AS, Huff HC, Dong L, Malkowski MG, Das A, Tajkhorshid E, Chan J, Angew. Chem. Int. Ed 2020, 59, 3307–3314; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2020, 132, 3333–3340. [Google Scholar]
- [31].a) Jiang X, Wang L, Carroll SL, Chen J, Wang MC, Wang J, Antioxid. Redox Signaling 2018, 29, 518–540; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Xing P, Zhang Z, Niu Y, Qi Y, Dong L, Wang C, Chem. Commun 2018, 54, 9889–9892; [DOI] [PubMed] [Google Scholar]; c) Collot M, Fam TK, Ashokkumar P, Faklaris O, Galli T, Danglot L, Klymchenko AS, J. Am. Chem. Soc 2018, 140, 5401–5411. [DOI] [PubMed] [Google Scholar]
- [32].a) Kim E, Lee S, Park SB, Chem Comm 2012, 48, 2311–2313; [DOI] [PubMed] [Google Scholar]; b) Kim E, Koh M, Ryu J, Park SB, J. Am. Chem. Soc 2008, 130, 12206–12207. [DOI] [PubMed] [Google Scholar]
- [33].Choi EJ, Kim E, Lee Y, Jo A, Park SB, Angew. Chem. Int. Ed 2014, 53, 1346–1350; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2014, 126, 1370–1374. [Google Scholar]
- [34].a) Kim E, Park SB, Chem. Asian J 2009, 4, 1646–1658; [DOI] [PubMed] [Google Scholar]; b) Lee Y, Cho W, Sung J, Kim E, Park SB, J. Am. Chem. Soc 2018, 140, 974–983. [DOI] [PubMed] [Google Scholar]
- [35].Kim E, Lee Y, Lee S, Park SB, Acc. Chem. Res 2015, 48, 538–547. [DOI] [PubMed] [Google Scholar]
- [36].Thiam AR, Farese RV Jr., Walther TC, Nat. Rev. Mol. Cell Biol 2013, 14, 775–786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hantzsch AR, Justus Liebigs Ann. Chem 1882, 215, 1–82. [Google Scholar]
- [38].a) Sueki S, Takei R, Zaitsu Y, Abe J, Fukuda A, Seto K, Furukawa Y, Shimizu I, Eur. J. Org. Chem 2014, 2014, 5281–5301; [Google Scholar]; b) Nair V, Offerman RJ, Turner GA, J. Am. Chem. Soc 1986, 108, 8283–8285; [Google Scholar]; c) Pávez P, Encinas MV, Photochem. Photobiol 2007, 83, 722–729; [DOI] [PubMed] [Google Scholar]; d) Pinrat O, Boonkitpatarakul K, Paisuwan W, Sukwattanasinitt M, Ajavakom A, Analyst 2015, 140, 1886–1893; [DOI] [PubMed] [Google Scholar]; e) Nair V, Offerman RJ, Turner GA, J. Am. Chem. Soc 1986, 108, 8283–8285; [Google Scholar]; f) Peng J, Liu Y, Wang M, Huang S, Liu M, Zhou Y, Gao W, Huang X, Wu H, Dyes Pigm 2020, 174, 108094–10914. [Google Scholar]
- [39].Jamsheena V, Mishra RK, Veena KS, Sini S, Jayamurthy P, Lankalapalli RS, ACS Omega 2018, 3, 856–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].a) Nierth A, Kobitski AY, Nienhaus GU, Jäschke A, J. Am. Chem. Soc 2010, 132, 2646–2654; [DOI] [PubMed] [Google Scholar]; b) Wombacher R, Keiper S, Suhm S, Serganov A, Patel DJ, Jäschke A, Angew. Chem. Int. Ed 2006, 45, 2469–2472; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2006, 118, 2529–2533; [Google Scholar]; c) Ghattas W, Mahy JP, Réglier M, Simaan AJ, ChemBioChem 2021, 22, 443–459. [DOI] [PubMed] [Google Scholar]
- [41].Ueno Y, Jose J, Loudet A, Pérez-Bolívar C, Anzenbacher P Jr., Burgess K, J. Am. Chem. Soc 2011, 133, 51–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Minoshima M, Kikuta J, Omori Y, Seno S, Suehara R, Maeda H, Matsuda H, Ishii M, Kikuchi K, ACS Cent. Sci 2019, 5, 1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].a) Maeda H, Kowada T, Kikuta J, Furuya M, Shirazaki M, Mizukami S, Ishii M, Kikuchi K, Nat. Chem. Biol 2016, 12, 579–585; [DOI] [PubMed] [Google Scholar]; b) Teitelbaum SL, Science 2000, 289, 1504–1508; [DOI] [PubMed] [Google Scholar]; c) Boyle WJ, Simonet WS, Lacey DL, Nature 2003, 423, 337–342; [DOI] [PubMed] [Google Scholar]; d) Mundy GR, Nat. Rev. Cancer 2002, 2, 584–593; [DOI] [PubMed] [Google Scholar]; e) Matsuura Y, Kikuta J, Kishi Y, Hasegawa T, Okuzaki D, Hirano T, Minoshima M, Kikuchi K, Kumanogoh A, Ishii M, Ann. Rheum. Dis 2018, 77, 1219–1225; [DOI] [PubMed] [Google Scholar]; f) Mütze J, Iyer V, Macklin JJ, Colonell J, Karsh B, Petrášek Z, Schwille P, Looger LL, Lavis LD, Harris TD, Biophys. J 2012, 102, 934–944; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Kowada T, Kikuta J, Kubo A, Ishii M, Maeda H, Mizukami S, Kikuchi K, J. Am. Chem. Soc 2011, 133, 17772–17776. [DOI] [PubMed] [Google Scholar]
- [44].Jun JV, Petersson EJ, Chenoweth DM, J. Am. Chem. Soc 2018, 140, 9486–9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ortiz G, Liu P, Naing SHH, Muller VR, Miller EW, J. Am. Chem. Soc 2019, 141, 6631–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].a) Kulkarni RU, Yin H, Pourmandi N, James F, Adil MM, Schaffer DV, Wang Y, Miller EW, ACS Chem. Biol 2017, 12, 407–413; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) de Silva AP, Gunaratne HQN, Habib-Jiwan J, McCoy CP, Rice TE, Soumillion J, Angew. Chem. Int. Ed. Engl 1995, 34, 1728–1731; [Google Scholar]; c) Miller EW, Lin JY, Frady EP, Steinbach PA, Kristan WB, Tsien RY, Proc. Natl. Acad. Sci. USA 2012, 109, 2114–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Peterka DS, Takahashi H, Yuste R, Neuron 2011, 69, 9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].a) Grimes PA, Stone RA, Laties AM, Li W, Arch. Ophthalmol 1982, 100, 635–639; [DOI] [PubMed] [Google Scholar]; b) Blair NP, Rusin MM Graefe’s Arch. Clin. Exp. Ophthalmol 1986, 224, 419–422. [DOI] [PubMed] [Google Scholar]
- [49].a) Fang G, Wang H, Bian Z, Sun J, Liu A, Fang H, Liu B, Yao Q, Wu Z, RSC Adv 2018, 8, 29400–29427; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhai W, Sun X, James TD, Fossey JS, Chem. Asian J 2015, 10, 1836–1848; [DOI] [PubMed] [Google Scholar]; c) Zhang X, Liu G, Ning Z, Xing G, Carbohydr. Res 2017, 452, 129–148. [DOI] [PubMed] [Google Scholar]
- [50].Wulff G, Pure Appl. Chem 1982, 54, 2093–2102. [Google Scholar]
- [51].James TD, Sandanayake KRAS, Shinkai S, Angew. Chem. Int. Ed. Engl 1996, 35, 1910–1922. [Google Scholar]
- [52].a) James TD, Sandanayake KRAS, Iguchi R, Shinkai S, J. Am. Chem. Soc 1995, 117, 8982–8987; [Google Scholar]; b) Yang W, Fan H, Gao X, Gao S, Karnati VVR, Ni W, Hooks WB, Carson J, Weston B, Wang B, Chem. Biol 2004, 11, 439–448. [DOI] [PubMed] [Google Scholar]
- [53].a) Xu S, Sedgwick AC, Elfeky SA, Chen W, Jones AS, Williams GT, Jenkins ATA, Bull SD, Fossey JS, James TD, Front. Chem 2020, 14, 112–116; [Google Scholar]; b) Wei J, Ni Y, Zhang W, Zhang Z, Zhang J, Anal. Chim. Acta 2017, 960, 110–116; [DOI] [PubMed] [Google Scholar]; c) Stones D, Manku S, Lu X, Hall DG, Chem. Eur. J 2004, 10, 92–100. [DOI] [PubMed] [Google Scholar]
- [54].Scrafton DK, Taylor JE, Mahon MF, Fossey JS, James TD, J. Org. Chem 2008, 73, 2871–2874. [DOI] [PubMed] [Google Scholar]
- [55].Pal A, Bérubé M, Hall DG, Angew. Chem. Int. Ed 2010, 49, 1492–1495; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2010, 122, 1534–1537. [Google Scholar]
- [56].Sørensen MD, Martins R, Hindsgaul O, Angew. Chem. Int. Ed 2007, 46, 2403–2407; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2007, 119, 2455–2459. [Google Scholar]
- [57].a) Edwards NY, Sager TW, McDevitt JT, Anslyn EV, Am J, Chem. Soc 2007, 129, 13575–13583; [DOI] [PubMed] [Google Scholar]; b) Bojanowski NM, Bender M, Seehafer K, Bunz UHF, Chem. Eur. J 2017, 23, 12253–12258. [DOI] [PubMed] [Google Scholar]
- [58].a) Xu XD, Cheng H, Chen WH, Cheng SX, Zhuo RX, Zhang XZ, Sci. Rep 2013, 3, 2679; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chu Y, Wang D, Wang K, Liu Z, Weston B, Wang B, Bioorg. Med. Chem. Lett 2013, 23, 6307–6309; [DOI] [PubMed] [Google Scholar]; c) Das RK, Mohapatra S, J. Mater. Chem. B 2017, 5, 2190–2197. [DOI] [PubMed] [Google Scholar]
- [59].a) Walkup GK, Imperiali B, J. Am. Chem. Soc 1996, 118, 3053–3054; [Google Scholar]; b) Pearce DA, Jotterand N, Carrico IS, Imperiali B, J. Am. Chem. Soc 2001, 123, 5160–5161. [DOI] [PubMed] [Google Scholar]
- [60].a) Radford RJ, Chyan W, Lippard SJ, Chem. Sci 2013, 4, 3080–3084; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chabosseau P, Woodier J, Cheung R, Rutter GA, Metallomics 2018, 10, 229–239. [DOI] [PubMed] [Google Scholar]
- [61].a) Sladek R, Rocheleau G, Rung J, Dina C, Shen L, Serre D, Boutin P, Vincent D, Belisle A, Hadjadj S, Nature 2007, 445, 881–885; [DOI] [PubMed] [Google Scholar]; b) Perez Y, Shorer Z, Liani-Leibson K, Chabosseau P, Kadir R, Volodarsky M, Halperin D, Barber-Zucker S, Shalev H, Schreiber R, Brain 2017, 140, 928–939; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhao L, Oliver E, Maratou K, Atanur SS, Dubois OD, Cotroneo E, Chen C-N, Wang L, Arce C, Chabosseau PL, Nature 2015, 524, 356–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Fudge DH, Black R, Son L, LeJeune K, Qin Y, ACS Chem. Biol 2018, 13, 1897–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Minckley TF, Zhang C, Fudge DH, Dischler AM, LeJeune KD, Xu H, Qin Y, Nat. Commun 2019, 10, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Liu H-W, Chen L, Xu C, Li Z, Zhang H, Zhang X-B, Tan W, Chem. Soc. Rev 2018, 47, 7140–7180. [DOI] [PubMed] [Google Scholar]
- [65].a) Hanash S, Nature 2003, 422, 226–232; [DOI] [PubMed] [Google Scholar]; b) Singh H, Tiwari K, Tiwari R, Pramanik SK, Das A, Chem. Rev 2019, 119, 11718–11760; [DOI] [PubMed] [Google Scholar]; c) Chyan W, Raines RT, ACS Chem. Biol 2018, 13, 1810–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Dunaway-Mariano D, Structure 2008, 16, 1599–1600. [DOI] [PubMed] [Google Scholar]
- [67].Poręba M, Szalek A, Kasperkiewicz P, Drąg M, Caspases, Paracaspases, and Metacaspases, Springer, 2014, pp. 41–59. [Google Scholar]
- [68].Kasperkiewicz P, Poreba M, Snipas SJ, Parker H, Winterbourn CC, Salvesen GS, Drag M, Proc. Natl. Acad. Sci. USA 2014, 111, 2518–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kasperkiewicz P, Kołt S, Janiszewski T, Groborz K, Poręba M, Snipas SJ, Salvesen GS, Drąg M, Sci. Rep 2018, 8, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Vucic D, Dixit VM, J. Exp. Med 2009, 206, 2309–2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Poreba M, Rut W, Vizovisek M, Groborz K, Kasperkiewicz P, Finlay D, Vuori K, Turk D, Turk B, Salvesen GS, Chem. Sci 2018, 9, 2113–2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].a) Pepperkok R, Ellenberg J, Nat. Rev. Mol. Cell Biol 2006, 7, 690–696; [DOI] [PubMed] [Google Scholar]; b) Chessel A, Carazo Salas RE, Essays Biochem 2019, 63, 197–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].a) Lee J-S, Kim YK, Vendrell M, Chang Y-T, Mol. BioSyst 2009, 5, 411–421; [DOI] [PubMed] [Google Scholar]; b) Kim J-Y, Sahu S, Yau Y-H, Wang X, Shochat SG, Nielsen PH, Dueholm MS, Otzen DE, Lee J, Delos Santos MMS, J. Am. Chem. Soc 2016, 138, 402–407. [DOI] [PubMed] [Google Scholar]
- [74].Lee JS, Kim HK, Feng S, Vendrell M, Chang YT, Chem. Commun 2011, 47, 2339–2341. [DOI] [PubMed] [Google Scholar]
- [75].Kim J-Y, Sahu S, Yau Y-H, Wang X, Shochat SG, Nielsen PH, Dueholm MS, Otzen DE, Lee J, Delos Santos MMS, J. Am. Chem. Soc 2016, 138, 402–407. [DOI] [PubMed] [Google Scholar]
- [76].Lee YA, Kim JJ, Lee J, Lee JHJ, Sahu S, Kwon HY, Park SJ, Jang SY, Lee JS, Wang Z, Tam WL, Lim B, Kang NY, Chang YT, Angew. Chem. Int. Ed 2018, 57, 2851–2854; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2018, 130, 2901–2904. [Google Scholar]
- [77].Kang N-Y, Lee JY, Lee SH, Song IH, Hwang YH, Kim MJ, Phue WH, Agrawalla BK, Wan SYD, Lalic J, J. Am. Chem. Soc 2020, 142, 3430–3439. [DOI] [PubMed] [Google Scholar]
- [78].Promchat A, Wongravee K, Sukwattanasinitt M, Praneenararat T, Sci. Rep 2019, 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].a) Albert KJ, Lewis NS, Schauer CL, Sotzing GA, Stitzel SE, Vaid TP, Walt DR, Chem. Rev 2000, 100, 2595–2626; [DOI] [PubMed] [Google Scholar]; b) Rissin DM, Kan CW, Campbell TG, Howes SC, Fournier DR, Song L, Piech T, Patel PP, Chang L, Rivnak AJ, Nat. Biotechnol 2010, 28, 595–599; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Cohen L, Walt DR, Annu. Rev. Anal. Chem 2017, 10, 345–363. [DOI] [PubMed] [Google Scholar]
- [80].a) Zhao P, Wu Y, Feng C, Wang L, Ding Y, Hu A, Anal. Chem 2018, 90, 4815–4822; [DOI] [PubMed] [Google Scholar]; b) Yang T, Feng C, Zhao P, Wu Y, Ding Y, Wang G, Hu A, J. Mater. Chem. C 2020, 8, 2500–2506. [Google Scholar]
- [81].Marshall AP, Shirley JD, Carlson EE, Curr. Opin. Chem. Biol 2020, 57, 155–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].a) Deng H, Lei Q, Wu Y, He Y, Li W, Eur. J. Med. Chem 2020, 191, 112151; [DOI] [PubMed] [Google Scholar]; b) Gardner SH, Reinhardt CJ, Chan J, Angew. Chem. Int. Ed 2021, 60, 5000–5009; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2021, 133, 5052–5062. [Google Scholar]
- [83].a) Finzel K, Lee DJ, Burkart MD, ChemBioChem 2015, 16, 528–547; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Chen R N Re A, Burkart MD, Nat. Prod. Rep 2018, 35, 1029–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Johnson DS, Weerapana E, Cravatt BF, Future Med. Chem 2010, 2, 949–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].a) Ishikawa F, Haushalter RW, Burkart MD, J. Am. Chem. Soc 2012, 134, 769–772; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ishikawa F, Haushalter RW, Lee DJ, Finzel K, Burkart MD, J. Am. Chem. Soc 2013, 135, 8846–8849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Konno S, La Clair JJ, Burkart MD, Angew. Chem. Int. Ed 2018, 57, 17009–17013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Chen R N Re A, Burkart MD, Nat. Prod. Rep 2018, 35, 1029–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].a) Mindrebo JT, Misson LE, Johnson C, Noel JP, Burkart MD, Biochemistry. 2020, 59, 3626–3638; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Mindrebo JT, Patel A, Kim WE, Davis TD, Chen A, Bartholow TG, La Clair JJ, McCammon JA, Noel JP, Burkart MD, Nat. Commun 2020, 11, 1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].a) Liu SY, Qu RY, Li RR, Yan YC, Sun Y, Yang WC, Yang GF, Anal. Chem 2020, 92, 9205–9913; [DOI] [PubMed] [Google Scholar]; b) Yang LL, Wang HL, Yan YH, Liu S, Yu ZH, Huang MY, Luo Y, Zheng X, Yu Y, Li GB, Eur. J. Med. Chem 2020, 192, 112201; [DOI] [PubMed] [Google Scholar]; c) Hedison TM, Hay S, Scrutton NS, Front. Biosci 2018, 23, 1874–1888; [DOI] [PubMed] [Google Scholar]; d) Manal M, Chandrasekar MJ, Gomathi Priya J, Nanjan MJ, Bioorg. Chem 2016. 67, 18–42. [DOI] [PubMed] [Google Scholar]
- [90].a) Luo Y, Li H, Int. J. Mol. Sci 2020, 21, 8828; [Google Scholar]; b) Ho TCS, Chan AHY, Ganesan A, J. Med. Chem 2020, 63, 12460–12484. [DOI] [PubMed] [Google Scholar]
- [91].Bahl S, Seto E, Cell. Mol. Life Sci 2021, 78, 427–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].a) Milazzo G, Mercatelli D, Di Muzio G, Triboli L, De Rosa P, Perini G, Giorgi FM, Genes 2020, 11, 556; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Witt O, Deubzer HW, Milde T, Oehme I, Cancer Lett 2009, 277, 8–21; [DOI] [PubMed] [Google Scholar]; c) de Ruijter AJ, van Gennip AH, Caron HN, Kemp S, van Kuilenburg AB, Biochem. J 2003, 370, 737–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Li X, Kapoor TM, J. Am. Chem. Soc 2010, 132, 2504–2505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Baba R, Hori Y, Mizukami S, Kikuchi K, J. Am. Chem. Soc 2012, 134, 14310–14313. [DOI] [PubMed] [Google Scholar]
- [95].Xie Y, Ge J, Lei H, Peng B, Zhang H, Wang D, Pan S, Chen G, Chen L, Wang Y, J. Am. Chem. Soc 2016, 138, 15596–15604. [DOI] [PubMed] [Google Scholar]
- [96].Bantscheff M, Hopf C, Savitski MM, Dittmann A, Grandi P, Michon A-M, Schlegl J, Abraham Y, Becher I, Bergamini G, Boesche M, Delling M, Dümpelfeld B, Eberhard D, Huthmacher C, Mathieson T, Poeckel D, Reader V, Strunk K, Sweetman G, Kruse U, Neubauer G, Ramsden NG, Drewes G, Nat. Biotechnol 2011, 29, 255. [DOI] [PubMed] [Google Scholar]
- [97].a) Saxena C, Expert Opin. Drug Discovery 2016, 11, 1017–1025; [DOI] [PubMed] [Google Scholar]; b) Lenz T, Fischer JJ, Dreger M, J. Proteomics 2011, 75, 100–115. [DOI] [PubMed] [Google Scholar]
- [98].Kambe T, Correia BE, Niphakis MJ, Cravatt BF, J. Am. Chem. Soc 2014, 136, 10777–10782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Lau EC, Mason DJ, Eichhorst N, Engelder P, Mesa C, Wijeratne EMK, Gunaherath GMKB, Gunatilaka AAL, La Clair JJ, Chapman E, Org. Biomol. Chem 2015, 13, 2255–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Kang M, Wu T, Wijeratne EMK, Lau EC, Mason DJ, Mesa C, Tillotson J, Zhang DD, Gunatilaka AAL, La Clair JJ, ChemBioChem 2014, 15, 2125–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Wagh SB, Maslivetc V, La Clair JJ, Kornienko A, RSC Adv 2020, 10, 37035–37039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].a) Kornienko A, La Clair JJ, Nat. Prod. Rep 2017, 34, 1051–1060; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zarganes-Tzitzikas T, Neochoritis CG, Dömling A, ACS Med. Chem. Lett 2019, 10, 389–392; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Werner-Allen JW, DuMond JF, Levine RL, Bax A, Angew. Chem. Int. Ed 2016, 55, 7374–7378; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2016, 128, 7500–7504. [Google Scholar]
- [103].Khanna IK, Weier RM, Yu Y, Collins PW, Miyashiro JM, Koboldt CM, Veenhuizen AW, Currie JL, Seibert K, Isakson PC, J. Med. Chem 1997, 40, 1619–1633. [DOI] [PubMed] [Google Scholar]
- [104].Stetter H, Schreckenberg M, Angew. Chem. Int. Ed. Engl 1973, 12, 81; [Google Scholar]; Angew. Chem 1973, 85, 89. [Google Scholar]
- [105].Ma B, Shatsky M, Wolfson HJ, Nussinov R, Protein Sci 2009, 11, 184–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Jenson JM, Xue V, Stretz L, Mandal T, Keating AE, Proc. Natl. Acad. Sci. USA 2018, 115, E10342–E10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].a) Rizvi NF, Howe JA, Nahvi A, Klein DJ, Fischmann TO, Kim H-Y, McCoy MA, Walker SS, Hruza A, Richards MP, ACS Chem. Biol 2018, 13, 820–831; [DOI] [PubMed] [Google Scholar]; b) Baell JB, Nissink JWM, ACS Chem. Biol 2018, 13, 36–44; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Baell JB, J. Nat. Prod 2016, 79, 616–628. [DOI] [PubMed] [Google Scholar]
- [108].a) Fosgerau K, Hoffmann T, Drug Discovery Today. 2015, 20, 122–128; [DOI] [PubMed] [Google Scholar]; b) Bobone S, Stella L, Adv. Exp. Med. Biol 2019, 1117, 175–214. [DOI] [PubMed] [Google Scholar]
- [109].a) Morris RJ, Curr. Opin. Plant Biol 2018, 43, 1–7; [DOI] [PubMed] [Google Scholar]; b) Karunanayake Mudiyanselage APKK, Wu R, Leon-Duque MA, Ren K, Yo M, Methods 2019, 161, 24–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].a) Zhu T, Chen C, Wang S, Zhang Y, Zhu D, Li L, Luo J, Kong L, Chem. Commun 2019, 55, 8231–8234; [DOI] [PubMed] [Google Scholar]; b) Sun W, Huo Y, Mei Y, Zhou Q, Zhao S, Zhuang M, ACS Chem. Biol 2020, 15, 39–43; [DOI] [PubMed] [Google Scholar]; c) Wang K, Yang T, Wu Q, Zhao X, Nice EC, Huang C, Expert Rev. Proteomics 2012, 9, 293–310; [DOI] [PubMed] [Google Scholar]; d) La Clair JJ, Nat. Prod. Rep 2010, 27, 969–995. [DOI] [PubMed] [Google Scholar]
- [111].Forsyth CJ, Ying L, Chen J, La Clair JJ, J. Am. Chem. Soc 2006, 128,3858–3859. [DOI] [PubMed] [Google Scholar]
- [112].a) Hughes CC, MacMillan JB, Gaudêncio SP, Fenical W, La Clair JJ, Angew. Chem. Int. Ed 2009, 48, 728–732; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2009, 121, 742–746; [Google Scholar]; b) Farnaes L, La Clair JJ, Fenical W, Org. Biomol. Chem 2014, 12, 418–423; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Álvarez-Micó X, Rocha DD, Guimarães LA, Ambrose A, Chapman E, Costa-Lotufo LV, La Clair JJ, Fenical W, ChemBioChem 2015, 16, 2002–2006; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Trzoss L, Fukuda T, Costa-Lotufo LV, Jimenez P, La Clair JJ, Fenical W, Proc. Natl. Acad. Sci. USA 2014, 111, 14687–14692; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Ambrose AJ, Santos EA, Jimenez PC, Rocha DD, Wilke DV, Beuzer P, Axelrod J, Kumar Kanduluru A, Fuchs PL, Cang H, Costa-Lotufo LV, Chapman E, La Clair JJ, ChemBioChem 2017, 18, 506–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Yu WL, Guizzunti G, Foley TL, Burkart MD, La Clair JJ, Nat. Prod 2010, 73, 1659–1666. [DOI] [PubMed] [Google Scholar]
- [114].Rodríguez AD, Lear MJ, La Clair JJ, J. Am. Chem. Soc 2008, 130, 7256–7258. [DOI] [PubMed] [Google Scholar]
- [115].Vera B, Rodríguez AD, La Clair JJ, Angew. Chem. Int. Ed 2011, 50, 8134–8138; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2011, 123, 8284–8288. [Google Scholar]
- [116].La Clair JJ, Rodríguez AD, Bioorg. Med. Chem 2011, 19, 6645–6653. [DOI] [PubMed] [Google Scholar]
- [117].Mortenson DE, Brighty GJ, Plate L, Bare G, Chen W, Li S, Wang H, Cravatt BF, Forli S, Powers ET, Sharpless KB, Wilson IA, Kell JW, J. Am. Chem. Soc 2018, 140, 200–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].a) Martín-Gago P, Olsen CA, Angew. Chem. Int. Ed 2019, 58, 957–966; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem 2019, 131, 969–978; [Google Scholar]; b) Chen W, Dong J, Plate L, Mortenson DE, Brighty GJ, Li S, Liu Y, Galmozzi A, Lee LP, Hulce JJ, Cravatt BF, Saez E, Powers ET, Wilson IA, Sharpless KB, Kell JW, J. Am. Chem. Soc 2016, 138, 7353–7364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Brighty GJ, Botham RC, Li S, Nelson L, Mortenson DE, Li G, Morisseau C, Wang H, Hammock BD, Sharpless KB, Kelly JW, Nat. Chem 2020, 12, 906–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Molinski TF, Curr. Opin. Drug Discovery Dev 2009, 12, 197. [PubMed] [Google Scholar]
- [121].Dalisay DS, Molinski TF, Org. Lett 2009, 11, 1967–1970. [DOI] [PubMed] [Google Scholar]
- [122].Dalisay DS, Molinski TF, J. Nat. Prod 2010, 73, 679–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Dalisay DS, Rogers EW, Edison AS, Molinski TF, J. Nat. Prod 2009, 72, 732–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].a) Dalisay DS, Molinski TF, J. Nat. Prod 2009, 72, 739–744; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Yoo H-D, Nam S-J, Chin Y-W, Kim M-S, Arch. Pharmacal Res 2016, 39, 143–153. [DOI] [PubMed] [Google Scholar]
- [125].Gesmundo NJ, Sauvagnat B, Curran PJ, Richards MP, Andrews CL, Dandliker PJ, Cernak T, Nature. 2018, 557, 228–232. [DOI] [PubMed] [Google Scholar]
- [126].Ambrose AJ, Pham J Sivinski NT, Guimarães L, Mollasalei N, Jimenez P, Abad MA, Jeyaprakash AA, Shave S, Costa-Lotufo LB, La Clair JJ, Auer M, Chapman E, RSC Chem. Biol 2021, 2, 181–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].a) Pérez-Pi I, Evans DA, Horrocks MH, Pham NT, Dolt KS, Koszela J, Kunath T, Auer M, Anal. Chem 2019, 91, 5582–5590; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Koszela J, Pham NT, Evans D, Mann S, Perez-Pi I, Shave S, Ceccarelli DFJ, Sicheri F, Tyers M, Auer M, BMC Biol 2018, 16, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Kang MJ, Wu T, Wijeratne EMK, Lau EC, Mason DJ, Mesa C, Tillotson J, Zhang DD, Gunatilaka AAL, La Clair JJ, ChemBioChem 2014, 15, 2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].a) Hill JR, Robertson AAB, J. Med. Chem 2018, 61, 6945–6963; [DOI] [PubMed] [Google Scholar]; b) Guo H, Li Z, MedChemComm 2017, 8, 1585–1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].a) Murale DP, Hong SC, Haque MM, Lee J-S, Proteome Sci 2016, 15, 14; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Smith E, Collins I, Future Med. Chem 2015, 7, 159–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].a) Masuda S, Tomohiro T, Yamaguchi S, Morimoto S, Hatanaka Y, Bioorg. Med. Chem. Lett 2015, 25, 1675–1678; [DOI] [PubMed] [Google Scholar]; b) Wen B, Doneanu CE, Gartner CA, Roberts AG, Atkins WM, Nelson SD, Biochemistry. 2005, 44, 1833–1845. [DOI] [PubMed] [Google Scholar]
- [132].Hughes CC, Yang Y-L, Liu W-T, Dorrestein PC, La Clair JJ, Fenical W, J. Am. Chem. Soc 2009, 131, 12094–12096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Hughes CC, Prieto-Davo A, Jensen PR, Fenical W, Org. Lett 2008, 10, 629–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].a) Tsukiji S, Hamachi I, Curr. Opin. Chem. Biol 2014, 21, 136–143; [DOI] [PubMed] [Google Scholar]; b) Tsukiji S, Miyagawa M, Takaoka Y, Tamura T, Hamachi I, Nat. Chem. Biol 2009, 5, 341–343. [DOI] [PubMed] [Google Scholar]
- [135].a) Chan J, Dodani SC, Chang CJ, Nat. Chem 2012, 4, 973–984; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jun ME, Roy B, Ahn KH, Chem. Commun 2011, 47, 7583–7601. [DOI] [PubMed] [Google Scholar]
- [136].Kojima H, Nakatsubo N, Kikuchi K, Kawahara S, Kirino Y, Nagoshi H, Hirata Y, Nagano T, Anal. Chem 1998, 70, 2446–2453. [DOI] [PubMed] [Google Scholar]
- [137].Sasaki E, Kojima H, Nishimatsu H, Urano Y, Kikuchi K, Hirata Y, Nagano T, J. Am. Chem. Soc 2005, 127, 3684–3685. [DOI] [PubMed] [Google Scholar]
- [138].Gabe Y, Urano Y, Kikuchi K, Kojima H, Nagano T, J. Am. Chem. Soc 2004, 126, 3357–3367. [DOI] [PubMed] [Google Scholar]
- [139].Kojima H, Hirotani M, Nakatsubo N, Kikuchi K, Urano Y, Higuchi T, Hirata Y, Nagano T, Anal. Chem 2001, 73, 1967–1973. [DOI] [PubMed] [Google Scholar]
- [140].a) Beija M, Afonso CAM, Martinho JMG, Chem. Soc. Rev 2009, 38, 2410–2433; [DOI] [PubMed] [Google Scholar]; b) Zheng H, Zhan X-Q, Bian Q-N, Zhang X-J, Chem. Commun 2013, 49, 429–447. [DOI] [PubMed] [Google Scholar]
- [141].Tang Y, Lee D, Wang J, Li G, Yu J, Lin W, Yoon J, Chem. Soc. Rev 2015, 44, 5003–5015. [DOI] [PubMed] [Google Scholar]
- [142].Debieu S, Romieu A, Org. Biomol. Chem 2017, 15, 2575–2584. [DOI] [PubMed] [Google Scholar]
- [143].Zhang G, Zhang D, Yin S, Yang X, Shuai Z, Zhu D, Chem. Commun 2005, 2161–2163. [DOI] [PubMed] [Google Scholar]
- [144].Liu B, Tian H, Chem. Commun 2005, 3156–3158. [DOI] [PubMed] [Google Scholar]
- [145].Ros-Lis J, Marcos MD, Martinez-Manez R, Soto J, Angew. Chem. Int. Ed 2005, 44, 4405–4407; [DOI] [PubMed] [Google Scholar]; Angew. Chem 2005, 117, 4479–4482. [Google Scholar]
- [146].a) Yang Y-K, Yook K-J, Tae J, J. Am. Chem. Soc 2005, 127, 16760–16761; [DOI] [PubMed] [Google Scholar]; b) Ko S-K, Yang Y-K, Tae J, Shin I, J. Am. Chem. Soc 2006, 128, 14150–14155. [DOI] [PubMed] [Google Scholar]
- [147].Kwon JY, Jang YJ, Lee YJ, Kim KM, Seo MS, Nam W, Yoon J, J. Am. Chem. Soc 2005, 127, 10107–10111. [DOI] [PubMed] [Google Scholar]
- [148].Zhang C, Liu T, Su Y, Luo S, Zhu Y, Tan X, Fan S, Zhang L, Zhou Y, Cheng T, Biomaterials 2010, 31, 6612–6617. [DOI] [PubMed] [Google Scholar]
- [149].Zhang E, Luo S, Tan X, Shi C, Biomaterials 2014, 35, 771–778. [DOI] [PubMed] [Google Scholar]
- [150].Yaren H, Mollaoglu H, Kurt B, Korkmaz A, Oter S, Topal T, Karayilanoglu T, Res. Vet. Sci 2007, 83, 116–122. [DOI] [PubMed] [Google Scholar]
- [151].Yang Y, Zhao Q, Feng W, Li F, Chem. Rev 2013, 113, 192–270. [DOI] [PubMed] [Google Scholar]