

Summary
Mutations in mitochondrial genes impairing energy production cause mitochondrial diseases (MDs), and clinical studies have shown that MD patients are prone to bacterial infections. However, the relationship between mitochondrial (dys)function and infection remains largely unexplored, especially in epithelial cells, the first barrier to many pathogens. Here, we generate an epithelial cell model for one of the most common mitochondrial diseases, Leigh syndrome, by deleting surfeit locus protein 1 (SURF1), an assembly factor for respiratory chain complex IV. We use this genetic model and a complementary, nutrient-based approach to modulate mitochondrial respiration rates and show that impaired mitochondrial respiration favors entry of the human pathogen Listeria monocytogenes, a well-established bacterial infection model. Reversely, enhanced mitochondrial energy metabolism decreases infection efficiency. We further demonstrate that endocytic recycling is reduced in mitochondrial respiration-dependent cells, dampening L. monocytogenes infection by slowing the recycling of its host cell receptor c-Met, highlighting a previously undescribed role of mitochondrial respiration during infection.
Keywords: mitochondria, metabolism, respiration, mitochondrial disease, infection, Listeria monocytogenes, endocytic recycling, Rab11, 13C isotopologue profiling
Graphical abstract
Highlights
-
•
Enhanced mitochondrial respiration decreases L. monocytogenes infection
-
•
Bacterial entry is affected by the host cell metabolism
-
•
Mitochondrial respiration restricts host cell receptor recycling and thus infection
Spier et al. show that the cellular energy metabolism affects infection of epithelial cells by L. monocytogenes. Mitochondrial respiration modulates L. monocytogenes entry by limiting endocytic recycling of receptors such as c-Met back to the plasma membrane, leading to decreased bacterial load in cells with high respiratory activity.
Introduction
Eukaryotic cells use the following two main pathways to generate the energy they need to function: glycolysis and mitochondrial respiration. In the cytosol, glycolysis generates the universal energy carrier ATP through the oxidation of glucose (Glc) to pyruvate. Glycolysis followed by lactic acid fermentation is often observed under low oxygen conditions or in highly proliferative cells (such as cancer cells, stem cells, or activated macrophages), as it supports accelerated cellular proliferation (Cairns et al., 2011). However, most cells couple glycolysis to mitochondrial respiration, a slower process of energy production that far exceeds glycolysis in terms of efficiency (Schmidt-Rohr, 2020). Mitochondrial energy production can be viewed as a two-step process, as follows: in a first step occurring in the mitochondrial matrix, the tricarboxylic acid (TCA) cycle generates oxidized cofactors, which fuel ATP production by oxidative phosphorylation (OXPHOS) during a second step taking place in the inner mitochondrial membrane. Beyond their crucial role in energy production, mitochondria are multifaceted organelles that are involved in multiple key cellular functions that include calcium buffering, apoptosis, and innate immunity (Nunnari and Suomalainen, 2012).
Impairment of mitochondrial energy production is one of the most frequent defects found in mitochondrial diseases, which are caused by mutations in mitochondrial genes encoded by the mitochondrial or nuclear genomes. Their clinical phenotypes are highly heterogeneous, often leading to progressive neuropathy or myopathy. Depending on the mutation, symptoms can arise early or late in development and affect any organ or tissue, notably those with high energy demands (reviewed in Alston et al., 2017; Gorman et al., 2016). Although mitochondrial diseases are classified as rare disorders, the population incidence of these currently incurable diseases is estimated at 1 in 4,300, making them the most common inherited metabolic disorders (Gorman et al., 2015). The most common childhood onset mitochondrial disease is Leigh syndrome, a clinically and genetically heterogeneous neurodegenerative disorder (Darin et al., 2001; Ng et al., 2021). Pathogenic variants in several different nuclear-encoded and mitochondrial DNA (mtDNA)-encoded genes can result in various energy metabolism defects affecting one or more OXPHOS complexes (Lake et al., 2016). Pathogenic variants in the nuclear gene encoding surfeit locus protein 1 (SURF1) constitute the most common cause of complex-IV-deficient Leigh syndrome (Wedatilake et al., 2013) and among the first nuclear-encoded genes associated with complex IV deficiency (Tiranti et al., 1998; Zhu et al., 1998).
Clinical and scientific literature link mitochondrial diseases with a higher susceptibility to infection, in particular respiratory and enterobacterial infection (McKee et al., 2000), and with an increased risk of crises and disease progression (Morgan-Hughes, 1986; Edmonds, 2004; Varghese et al., 2011; Al-Zubeidi et al., 2014; Gaudó et al., 2020). Strikingly, disease progression is not reverted after the pathogen has been cleared (Edmonds et al., 2002). One such retrospective clinical study that addressed the occurrence and severity of infections in mitochondrial disease patients reported serious and recurrent infections in 42% of the cohort examined, with most infections caused by bacterial pathogens (Walker et al., 2014). It is currently unknown whether alterations in mitochondrial energy metabolism caused by mitochondrial diseases directly affect cellular susceptibility to bacterial infection and how the latter develops in mitochondrially diseased cells.
Here, we addressed these questions using Listeria monocytogenes, a Gram-positive enterobacterium and a well-established model of intracellular bacterial infection. L. monocytogenes causes the human food-borne disease listeriosis in immunocompromised individuals, with a case fatality rate of up to 30% (de Noordhout et al., 2014). Several virulence factors allow L. monocytogenes to invade non-phagocytic cells, escape from its internalization vacuole to replicate in the cytosol, and then spread to neighboring cells, effectively eluding the humoral immune response (Stavru et al., 2011). Intracellular L. monocytogenes relies mainly on carbon sources derived from host cell glycolysis (Grubmüller et al., 2014). Comparative 13C-isotopologue studies revealed that L. monocytogenes strongly increases the glycolytic activity of host cells (Gillmaier et al., 2012), a phenomenon that has been proposed for several other intracellular bacteria, including Mycobacterium tuberculosis (Gleeson et al., 2016; Lachmandas et al., 2016) and Legionella pneumophila (Escoll et al., 2017). We chose to focus on L. monocytogenes, as we have previously shown that L. monocytogenes induces transient fragmentation of the mitochondrial network and a drop in the mitochondrial membrane potential, which is mediated by its pore-forming toxin listeriolysin O (LLO) (Stavru et al., 2011, 2013). Yet, it remains largely unknown how mitochondrial respiration affects L. monocytogenes infection. An interesting study by Gillmaier and colleagues showed that murine macrophage-like J774 cells, which are highly glycolytic, have higher infection rates than primary mouse macrophages (Gillmaier et al., 2012). However, the study compared cells that were genomically and phenotypically different. We thus investigated the interdependence between host cell metabolism and L. monocytogenes infection by using isogenic but metabolically distinct epithelial cells to focus on the contribution of the mitochondrial energy metabolism to infection.
By manipulating mitochondrial respiratory function in epithelial cells, either through nutrient supply to the host cell or by genetic manipulation, we discovered that enhanced mitochondrial respiration negatively impacts the intracellular L. monocytogenes burden. Mechanistically, we revealed that mitochondrial respiration modulates bacterial entry by interfering with the recycling of host cell receptors such as c-Met, which are exploited by L. monocytogenes, to the plasma membrane, thereby uncovering a previously undescribed link between energy metabolism and endocytic recycling in epithelial cells.
Results
Mitochondrial respiration significantly impairs L. monocytogenes infection efficiency
To study the role of host cell energy metabolism and in particular mitochondrial respiration in bacterial infection, we used the L. monocytogenes infection model. We used two approaches to manipulate host cell energy metabolism in HCT116 cells, which is a human intestinal epithelial cell line efficiently infected by L. monocytogenes (Stavru et al., 2013; Carvalho et al., 2020; Figure 1A). In a nutrient-based approach, we replaced glucose (Glc) in the cell culture medium with galactose (Gal). Gal is catabolized by the Leloir pathway (reviewed in Frey, 1996) and thereby enters glycolysis at a significantly lower rate than Glc (Eagle et al., 1958). Thus, instead of glycolysis, cells maintained in Gal-containing medium rely mainly on mitochondrial energy metabolism through glutaminolysis, followed by OXPHOS (Reitzer et al., 1979; Rossignol et al., 2004). In a genetic approach, we performed CRISPR-Cas9-mediated gene editing to deplete cells of the mitochondrial protein SURF1 (SURF1−/− cells; Figures 1A and S1B) to directly test for the impact of mitochondrial dysfunction in infection. SURF1 is an assembly factor for the cytochrome c oxidase (COX) (Zhu et al., 1998; Tiranti et al., 1999), also known as complex IV, the terminal component of the mitochondrial respiratory chain. SURF1 is commonly mutated in mitochondrial diseases, which leads to the neurodegenerative disorder Leigh syndrome (Wedatilake et al., 2013). Its ablation has been shown to reduce but not abolish complex IV activity and O2 consumption in vitro and in vivo (Dell’agnello et al., 2007; Kovářová et al., 2012; Wedatilake et al., 2013; Da-Rè et al., 2014), thereby presenting a viable model with a respiratory defect. HCT116 SURF1−/− cells, which display a respiration defect, were functionally complemented by stable SURF1 re-expression (SURF1−/− + SURF1 or complemented SURF1−/− cells; Figures 1A, S1A, and S1B). We chose the clone re-expressing SURF1 at a level that was most similar to that of wild-type (WT) cells (Figure S1A), which express slightly higher SURF1 levels than the parental HCT116 WT cells from which SURF1−/− cells were generated (Figure S1B).
Figure 1.

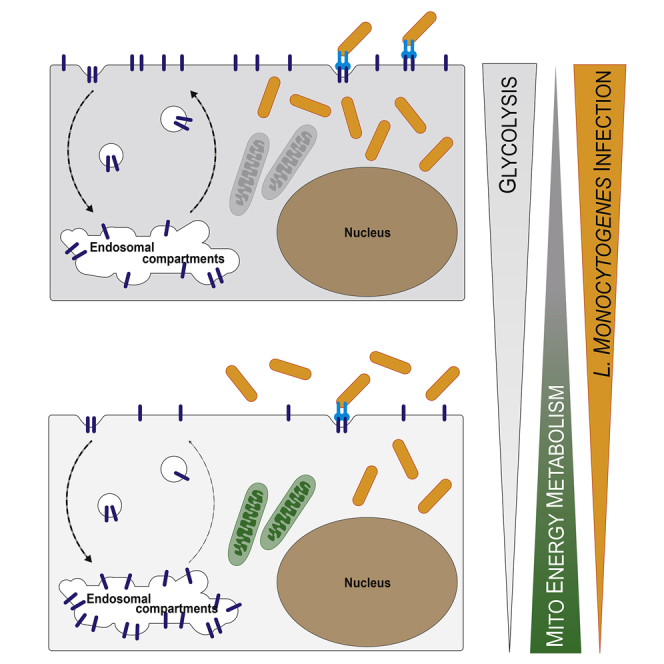
Mitochondrial energy metabolism affects L. monocytogenes infection
(A) Overview of the approaches used to generate cellular models with increased or decreased mitochondrial energy metabolism. HCT116 cells grown in galactose (Gal)-containing medium are energetically dependent on mitochondrial respiration (green mitochondria), whereas cells grown in glucose (Glc)-containing medium are mainly glycolytic and rely less on mitochondrial respiration (light gray mitochondria). Cells depleted for the mitochondrial protein SURF1 (SURF1−/− cells) show decreased mitochondrial respiration (red mitochondria), which can be rescued by reintroducing the SURF1 gene (SURF1−/− + SURF1 cells, dark gray mitochondria). The wedge indicates the level of dependency on mitochondrial respiration for energy production for each cell model.
(B and C) Oxygen consumption rate (OCR; in picomoles per minute) of HCT116 Glc and Gal cells (B) and of HCT116 WT, SURF1−/−, and SURF1−/− + SURF1 cells (C) monitored in a Seahorse XFe96 analyzer. Three independent experiments were performed and data from one representative experiment with six biological replicates per condition are shown as mean ± standard deviation (SD) for each time point. The rates of basal respiration, respiration coupled to ATP production, and maximal respiration were statistically evaluated by two-tailed t tests (B) and one-way ANOVA with Dunnett’s post hoc test (C) (∗∗∗p < 0.001).
(D and E) Intracellular bacterial load in HCT116 Glc and Gal cells (D) and in HCT116 WT, SURF1−/−, and SURF1−/− + SURF1 infected with WT L. monocytogenes EGDe (MOI, 20). The left panel shows values for Gal cells relative to Glc cells and values for SURF1−/− and SURF1−/− + SURF1 cells relative to WT cells, and the right panel shows the absolute quantification (CFU/mL), for each time point. Three independent experiments were performed, and for both panels, one representative experiment with three biological replicates is shown as mean ± SD. Statistical significances were calculated by multiple t tests (D) and one-way ANOVA with multiple comparisons (E); both were further evaluated by the false discovery rate approach of Benjamini, Krieger ,and Yekutieli, with Q = 1% (ns, not significant; ∗∗p < 0.01; ∗∗∗p < 0.001).
As a readout for mitochondrial respiratory capacity, we assessed the metabolic state of our cellular models and monitored mitochondrial respiratory activity by measuring oxygen consumption rates (OCRs) over time. Basal-respiration-associated OCR levels were significantly higher in Gal cells than those in Glc cells (Figure 1B), confirming the expected metabolic shift toward increased mitochondrial respiration. The addition of oligomycin, a specific inhibitor of the mitochondrial F1FO ATPase, caused a larger OCR reduction with respect to basal OCR levels in Gal cells than that of Glc cells, indicating that mitochondrial respiration coupled to ATP production is enhanced in Gal cells. The addition of the uncoupler carbonyl cyanide p-(tri-fluoromethoxy)phenylhydrazone (FCCP), which transports protons across membranes, was used to trigger maximal respiratory capacity; however, FCCP induced maximal respiration in Gal cells but not in Glc cells. Although Glc cells are more glycolytic and Gal cells rely mainly on mitochondrial respiration, the cells did not show any significant differences in ATP levels (Figure S1C).
We then compared the mitochondrial respiratory activity of SURF1−/− cells with the parental WT and with the complemented SURF1−/− cells. As expected, respiration was reduced in SURF1−/− cells, which consumed around 45% less oxygen under basal conditions (Figure 1C). This finding is consistent with previous studies in human fibroblasts derived from SURF1-deficient patients (Kovářová et al., 2012; Lee et al., 2012; Wedatilake et al., 2013). In contrast, complemented SURF1−/− cells showed significantly higher basal OCR levels than WT cells (Figure 1C), which is possibly due to higher protein levels of SURF1 in the SURF1−/− + SURF1 cells than those in WT cells (Figure S1B). The same trend was observed for respiration coupled to ATP production and maximal respiration, with SURF1−/− cells showing significantly lower and complemented SURF1−/− cells significantly higher OCR levels than WT cells. As expected, SURF1−/− cells compensated their impaired mitochondrial respiratory activity by a significantly enhanced glycolysis rate (Figure S1D). This result is corroborated by the observation that there is no difference in total ATP levels between WT, SURF1−/−, and complemented SURF1−/− cells (Figure S1E).
We thus successfully established three cellular models that show alterations in mitochondrial respiration, as follows: (1) Gal cells that depend strongly on mitochondrial respiration, (2) Glc cells that rely on both glycolysis and mitochondrial respiration, and (3) SURF1−/− cells that have a strong respiratory defect, which is rescued in complemented SURF1−/− cells.
These cellular models were then used to study the impact of mitochondrial respiration on L. monocytogenes infection. We first infected Glc and Gal cells and performed gentamicin protection assays to quantify intracellular L. monocytogenes at different time points after infection. The bacterial load in Gal cells was consistently lower than that in Glc cells (Figure 1D), with a difference of 35% ± 11% at 1 h post-infection that remained stable for the next 5 h. The similar slope of the bacterial growth curves observed in Glc and Gal cells (Figure 1D) suggests that there is no shortage of intracellular metabolites required for the intracellular growth of L. monocytogenes. The difference in bacterial load was not limited to intestinal cells or a single L. monocytogenes strain, as we observed the same trend in HeLa cells and primary human fibroblasts and upon infection with different L. monocytogenes strains (EGDe and EGD) (Figures S1F and S1G). Conversely, in SURF1−/− cells, which display low mitochondrial respiratory activity (Figure 1C), the bacterial burden was consistently higher (+36% ± 11%) than WT cells (Figure 1E). This phenotype was reverted by functional complementation of SURF1 in SURF1−/− cells (Figure 1E), where mitochondrial respiration is restored (Figure 1C). These data argue that impaired oxygen consumption of host cells can promote bacterial infection.
We reported previously that L. monocytogenes, and in particular its pore-forming toxin LLO, induces mitochondrial fragmentation (Stavru et al., 2011). To address whether the enhanced intracellular bacterial pool in SURF1−/− cells induces stronger mitochondrial fragmentation, we performed confocal imaging. We revealed no obvious differences of mitochondrial morphology between WT and SURF1−/− cells, neither under mock nor under infected conditions (Figure S2A). In addition, treatment with recombinant LLO, the bacterial effector responsible for the changes in mitochondrial morphology, induced a similar fragmented mitochondrial network in both WT and SURF1−/− cells (Figure S2B).
To exclude the possibility that the alteration of the host cell energy metabolism impacts the infection outcome by increasing the likelihood of cell death, we directly assessed cell death levels in L. monocytogenes-infected cells by flow cytometry. The percentage of dead cells was low (<8%) and similar between infected and non-infected cells, Glc and Gal cells (Figures S2C and S2D), and WT and SURF1−/− cells (Figure S2E). These observations indicate that cell death does not account for the observed differences in L. monocytogenes infection efficiencies across the different cellular models. Collectively, our results indicate that L. monocytogenes infection is affected by mitochondrial respiration, i.e., potentiated in cells with decreased mitochondrial respiration and high glycolytic activity and restricted in cells that strongly rely on mitochondrial respiration.
13C isotopologue profiling of host cells reveals changes in glycolytic and TCA cycle activity upon nutrient-based manipulation or SURF1 depletion
To assess the effects of nutrient-based manipulation and SURF1 depletion on the activity of glycolysis and TCA cycle, we performed 13C isotopologue profiling to obtain a sensitive and direct readout of the metabolic processes occurring in these cellular models (Eisenreich et al., 2015). Cells grown in medium containing a mixture of unlabeled Glc or Gal and 20% [U-13C6]-labeled Glc or Gal were analyzed for 13C incorporation into protein-derived amino acids obtained after acidic hydrolysis of the cells, particularly those produced by transamination from pyruvate (alanine) or TCA cycle intermediates (aspartate and glutamate). Expectedly, Glc cells displayed intense Glc metabolization by glycolysis, resulting in substantial levels of 13C excess in pyruvate/alanine (2.7%) and then fueling into the TCA cycle intermediates α-ketoglutarate/glutamate (2.4%) and oxaloacetate/aspartate (1.8%) (Figure 2A, gray bars). In contrast, cells growing in presence of [U-13C6]Gal barely showed 13C incorporation (<0.5%) in these amino acids (Figure 2A, green bars), which is in line with the previously reported slow glycolytic metabolism of Gal by the Leloir pathway (Frey, 1996).
Figure 2.

Characterization of changes in mitochondrial respiration induced by medium formulation or genetic ablation of the mitochondrial protein SURF1
(A) 13C-Excess in proteinogenic amino acids from HCT116 Glc and Gal cells labeled with [U-13C6]Glc or [U-13C6]Gal, respectively, as quantified by gas chromatography-mass spectrometry (GC-MS). Shown are the mean ± SD (cumulative biological and technical errors) of one experiment with two biological replicates, where each one is analyzed three times in technical replicates. The differences between HCT116 Glc and Gal cells were statistically evaluated by two-tailed t tests (∗∗∗p < 0.001).
(B) 13C-Excess in cytosolic metabolites from HCT116 Glc and Gal cells labeled with [U-13C5]glutamine as quantified by GC-MS. Experimental setup and statistical analysis were performed as in (A) (∗∗∗p < 0.001).
(C) 13C-Excess in cytosolic metabolites from HCT116 WT and SURF1−/− cells labeled with [U-13C6]Glc as quantified by GC-MS. Experimental setup and statistical analysis were performed as in (A) (∗∗p < 0.01; ∗∗∗p < 0.001).
(D) Intracellular bacterial load in HCT116 Glc and Gal cells infected with L. monocytogenes EGDe ΔC3Δhpt (MOI, 20). The left panel shows values for Gal cells relative to Glc cells, and the right panel shows the absolute quantification (CFU/ml) for each time point. Three independent experiments were performed, and for both panels, data from one representative experiment with three biological replicates are shown as mean ± SD. Statistical significances were determined by multiple t tests and evaluated by the false discovery rate approach of Benjamini, Krieger, and Yekutieli, with Q = 1% (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
To assess the degree to which glutaminolysis is exploited under the two conditions, we grew Glc and Gal cells in the presence of [U-13C5]glutamine (Figure 2B). Here, we focused on cytosolic metabolites obtained through mechanical disruption of the cells and especially TCA cycle intermediates (fumarate, succinate, malate, and citrate) to directly evaluate the extent of glutaminolysis and its effect on the TCA cycle. Compared to Glc cells, Gal cells displayed a 1.5-fold increase in 13C incorporation into fumarate, succinate, malate, and citrate, as well as fatty acids (myristate) (Figure 2B). This finding indicates an increase in TCA cycle activity in Gal cells, leading to enhanced NADH and FADH2 production, which are required as electron donors to complexes I and II, respectively, thereby promoting increased respiration. These data thus confirm the strong dependence of Gal cells on glutaminolysis and their more pronounced OXPHOS activity than Glc cells. In addition, cytosolic alanine and lactate showed a 2.8- and 12-fold increase, respectively, in 13C excess compared with Glc cells (Figure 2B), which is due to cataplerosis, as Gal cells have a decreased glycolytic flux but a highly active TCA cycle that is fed through glutaminolysis by [U-13C5]glutamine (see above). Our data are in line with a previous study that showed that the energy deficit caused by decreased glycolysis rates in Gal cells is compensated through an enhanced mitochondrial energy metabolism based on glutaminolysis (Reitzer et al., 1979).
Next, we analyzed the metabolites of SURF1−/− cells grown in the presence of [U-13C6]Glc and observed levels of 13C incorporation for lactate comparable to those in parental WT cells (Figure 2C). Increased glycolytic activity of SURF1−/− cells was apparent from the isotopologue composition of lactate (Figure S3A). Here, the relative abundance of fully labeled lactate carrying three 13C-atoms (M+3) was higher in SURF1−/− cells than that in the WT cells. This isotopologue arises through glycolytic conversion of fully labeled Glc to pyruvate and further reduction to lactate, hence indicating glycolytic activity. A similar effect was observable in alanine (Figure S3A). Here, the overall 13C-incorporation was significantly lower in the SURF1−/− cells, probably due to increased uptake of unlabeled alanine from the medium. However, the M+3 isotopologue in alanine was again more abundant than in the WT cells, thereby showing higher glycolytic activity in SURF1−/− cells.
Downstream of pyruvate, the respiratory deficiency of SURF1−/− cells became apparent as we detected reduced 13C incorporation into fatty acids and TCA cycle intermediates in comparison to WT cells. This result is likely due to the reduced complex IV activity caused by SURF1 depletion, which is essential to sustain the TCA cycle. In particular, succinate showed a 2-fold decrease in the mutant cells (5.1% versus 10.7%, respectively), implying decreased utilization by succinate dehydrogenase, which is the only enzyme that participates in both the TCA cycle and in the respiratory chain (complex II). 13C Isotopologue profiling thus demonstrated that the primary respiratory defect caused by SURF1 depletion also leads to altered TCA cycle activity.
In conclusion, 13C isotopologue profiling allowed us to confirm the metabolic reprogramming induced by nutrient-based metabolic manipulation or SURF1 depletion and also to reveal substantial differences in the extent of glutaminolysis and turnover of TCA metabolites.
Our characterization of Glc and Gal cells indicated that glycolysis rates are decreased in Gal cells (Figure 2A). Because previous studies have shown that L. monocytogenes preferentially uses the glycolytic intermediates Glc-6-phosphate and glycerol as carbon sources (Chico-Calero et al., 2002; Eylert et al., 2008; Joseph et al., 2008; Sauer et al., 2019), we tested whether a shortage of these glycolytic intermediates are responsible for the decreased L. monocytogenes intracellular burden. For this test, we performed infections with a L. monocytogenes mutant that is unable to metabolize Glc-6-phosphate and glycerol (ΔC3Δhpt; Grubmüller et al., 2014). This mutant (Figure 2D) behaved similarly to the WT parental strain (Figure 1D), and there was no observed difference in bacterial replication rate in Glc and Gal cells over 6 h. These data further suggest that Glc-6-phosphate and glycerol availability are not limiting for bacterial replication in Gal cells and that differences in L. monocytogenes infection occur during bacterial entry and establishment of the replicative niche.
Mitochondrial respiration significantly impairs L. monocytogenes entry into host cells
As increased mitochondrial respiration impacted L. monocytogenes infection already at 1 h post-infection (Figures 1D and 1E), we investigated whether the host cell energy metabolism affects the early infection steps, such as bacterial adhesion and/or entry to host cells. We infected our cellular models with GFP-expressing L. monocytogenes and performed differential staining to assess the number of intracellular versus extracellular (adhered after one wash) bacteria (Figures S4A and S4B; Kühbacher et al., 2014). Although the total number of bacteria (i.e., adhered and intracellular bacteria) per host cell was comparable in Glc and Gal cells (Figure S4C), we observed significant differences in both bacterial adhesion and entry. Gal cells showed a significant increase (+33%) in the average number of adhered bacteria per cell compared to Glc cells (Figure 3A). In contrast, the average number of intracellular bacteria per cell decreased by half (−53%; Figure 3B), reflecting less efficient internalization of adhered bacteria. The higher average number of adhered bacteria could therefore represent bacteria that are unable or taking longer to invade. In the case of SURF1−/− cells, we counted similar average numbers for total (Figure S4D) and adhered (Figure 3C) bacteria in WT, SURF1−/−, and complemented SURF1−/− cells. However, SURF1−/− cells displayed a slightly but significantly higher average number of intracellular bacteria than both WT and complemented SURF1−/− cells (+23%; Figure 3D), again pointing to a more efficient bacterial internalization in cells with decreased mitochondrial respiration.
Figure 3.

Mitochondrial respiration restricts L. monocytogenes entry into host cells
(A–D) Quantification of L. monocytogenes EGD (MOI, 20; 1 h) adhered to the surface (A and C) of and internalized by (B and D) HCT116 Glc and Gal cells (A and B) or HCT116 WT, SURF1−/−, and SURF1−/− + SURF1 cells (C and D) by immunofluorescence. Results are representative of four (A and B) and three (C and D) independent experiments and are displayed as box and whiskers plot with absolute numbers of bacteria per cell (with n > 570 cells per condition) and mean indicated by +. Given the non-Gaussian distribution, statistical significance was determined using a two-tailed Mann Whitney test (A and B) or a Kruskal-Wallis test followed by Dunn’s multiple comparisons test (C and D) (∗p < 0.05; ∗∗∗p < 0.001).
(E and F) Quantification of intracellular L. monocytogenes EGD in HCT116 Glc and Gal cells (E) or HCT116 WT, SURF1−/−, and SURF1−/− + SURF1 cells (F) after infection with WT or InlA-deficient (ΔinlA), InlB-deficient (ΔinlB), or LLO-deficient (Δhly) bacterial strains for 1 h. Three independent experiments were performed, and data from one representative experiment with three biological replicates are shown as relative CFU/mL with values normalized to the control condition (Glc or WT cells). Statistical significance was determined by two-tailed t tests (E) or one-way ANOVAs with Dunnett’s post hoc test (F) (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001).
L. monocytogenes enters into non-phagocytic cells, such as epithelial cells, by a zipper mechanism that is induced by the direct interaction between bacterial surface proteins and the host cell receptors. Two different internalization pathways are described, as follows: one relying on the interaction of the bacterial protein InlA with the host cell surface protein E-cadherin (Mengaud et al., 1996) and the second one relying on the interaction of bacterial InlB with the host receptor c-Met (Shen et al., 2000). Both pathways result in bacterial uptake by receptor-mediated endocytosis (Veiga and Cossart, 2005; Bonazzi et al., 2008). As HCT116 cells express both E-cadherin and c-Met (Bradley et al., 2016; Benthani et al., 2018), we investigated whether the InlA-mediated and/or the InlB-mediated entry pathways are affected by changes in the host cell energy metabolism. We infected our cellular models with L. monocytogenes mutants lacking either InlA or InlB. As expected, the intracellular bacterial load was highest when cells were infected with WT L. monocytogenes, whereas the infection efficiency of the mutant strains decreased in the order ΔinlB > ΔinlA (Figures S4E and S4F), which is in line with previous results in other epithelial cell lines (Dramsi et al., 1995). This tendency was observed in all our cellular models, indicating that changes in mitochondrial respiration do not induce a switch between the two host cell invasion mechanisms engaged by L. monocytogenes. The difference in infection efficiency between Glc and Gal cells was maintained after infection with either the ΔinlA or the ΔinlB mutant (Figure 3E). This finding suggested that mitochondrial respiration might affect a common mechanism to which both the InlA/E-cadherin- and InlB/c-Met-dependent pathways converge, such as endocytosis. Next, we observed that infection with WT or any of the ΔinlA and ΔinlB L. monocytogenes strains resulted in higher bacterial loads in SURF1−/− cells than those in WT cells (+48% ± 8%), whereas complemented SURF1−/− cells consistently displayed a lower infection rate (−48% ± 9%) (Figure 3F). These results are in line with the data obtained by nutrient-based manipulation.
Given the previously described role of LLO as the driver of mitochondrial fragmentation during L. monocytogenes infection (Stavru et al., 2011), we wondered whether LLO differentially impacts the infection of cells that rely to different extents on mitochondrial respiration. We infected the cells with LLO-deficient L. monocytogenes (Δhly) and observed that the differences in bacterial load between Glc and Gal cells (−45% in Gal cells; Figure 3E) as well as between WT, SURF1−/−, and complemented SURF1−/− cells (+36% in SURF1−/− cells, −45% in complemented SURF1−/− cells) (Figure 3F) were comparable to the ones obtained upon infection with WT L. monocytogenes. This result suggests that LLO does not contribute to the differences in infection efficiency observed between the metabolically distinct cells.
Collectively, our observations show that receptor-mediated L. monocytogenes entry is promoted in cells with decreased mitochondrial respiration, and increased respiration restricts the host-cell-invading potential of L. monocytogenes. Moreover, mitochondrial respiratory activity affects equally InlA/E-cadherin and InlB/c-Met-dependent bacterial entry pathways.
Increased mitochondrial respiratory activity slows the host cell endocytic recycling pathway
Infection of cells with L. monocytogenes mutants lacking InlA or InlB suggests that mitochondrial respiration affects a common downstream node onto which both bacterial entry pathways converge, such as clathrin-mediated endocytosis (Pizarro-Cerdá and Cossart, 2018). We thus investigated the impact of mitochondrial respiration on clathrin-mediated endocytosis, focusing on our nutrient-based manipulation model (Glc and Gal cells). To monitor endocytosis dynamics in these cells, we performed the fluorescence-microscopy-based transferrin uptake assay, which is classically used to study clathrin-mediated endocytosis (Mayle et al., 2012). For this investigation, cellular internalization was stalled by incubation on ice while in the presence of a transferrin receptor (TfR) antibody. Uptake of the stalled receptor was initiated by placing cells at 37°C; and at multiple time points, cells were again placed on ice to stop internalization, washed to remove extracellular TfR, and fixed to determine intracellular TfR signal intensity with the data displayed as the percent of the 1-min time point. As a control, we quantified TfR signal intensity at 10 min between Dynasore-treated (to prevent TfR uptake) and unwashed cells (total signal) to show the post-wash signal was intracellular TfR (Figure S5A). At early time points (1–5 min), the increase and subsequent decrease at 5 min, marking the internalization of stalled receptor and the turnover of intracellular TfR signal, in Glc and Gal cells was similar (Figure 4A), thus pointing to no detectable effect of the host cell metabolism on the endocytic uptake of the receptor. However, at later time points (10–30 min), intracellular TfR accumulated specifically in Gal cells (Figure 4A), suggesting a decreased rate of endocytic recycling, which ensures the transport of the receptors back to the plasma membrane, and is thus crucial for efficient bacterial uptake. This conclusion is also reflected by the higher 1-min levels of intracellular TfR in Gal cells (Figure 4A).
Figure 4.

Enhanced mitochondrial respiration slows endocytic recycling, leading to reduced L. monocytogenes infection levels
(A and B) Measurement of internalized levels of transferrin receptor (TfR) (A) and c-Met (B) in HCT116 Glc and Gal cells. The total internal TfR and c-Met fluorescence at each time point was quantified by confocal microscopy, and data from one representative experiment are shown as mean field intensity (MFI) per cell from a randomly chosen field of view (n ≥ 5 randomly chosen fields of view).
(C and D) Intracellular c-Met levels in uninfected (mock, same data as displayed in B) and L. monocytogenes EGDe-infected (MOI, 20) HCT116 Glc and Gal cells at 60 min as imaged by confocal microscopy. (C) Representative images of c-Met and cell nuclei in red and blue, respectively. Scale bars, 10 μm. (D) MFI values of the c-Met signal per cell are shown as mean ± SD from one representative experiment. Unpaired, two-tailed t tests were performed to determine statistical significance.
(E) Quantification of intracellular L. monocytogenes EGDe (MOI, 20) in HCT116 Glc and Gal cells transfected with control plasmid (pEGFP-N1) or a plasmid expressing constitutively active Rab11b (Rab11bCA) at 1 h post-infection. Three independent experiments were performed, and data from one representative experiment with three biological replicates are shown as % intracellular bacteria (mean ± SD). Statistical significance was determined by unpaired t tests (∗∗∗p < 0.001).
Next, we investigated the recycling of host cell receptors relevant for L. monocytogenes infection. Because entry exclusively by the InlB/c-Met pathway led to a more pronounced difference in intracellular bacterial load between Glc and Gal cells (Figure 3E; infection with ΔinlA), we focused on c-Met endocytosis. Our observations on endocytic uptake and recycling of the c-Met antibody (Figures 4B and S5B) were similar to the ones made with TfR, i.e., receptor recycling was impaired in Gal cells. Importantly, steady-state c-Met protein levels were similar between Glc and Gal cells (Figure S5C). To study whether infection enhances the observed difference in receptor recycling efficiency, we also monitored intracellular c-Met fluorescence signal upon L. monocytogenes infection. We detected no significant changes in c-Met antibody accumulation in both Glc and Gal cells upon infection (Figure 4C-D), indicating that infection did not alter c-Met receptor recycling dynamics.
Altogether, these data show that Gal cells have slower c-Met recycling back to the plasma membrane, limiting efficient L. monocytogenes infection of Gal, than Glc cells. A hallmark of endocytosed receptors is their association with recycling endosomes marked by Rab GTPases Rab4 and Rab11, which regulate their export back to the plasma membrane. Here, the receptor is either directly transported back to the plasma membrane byRab4 isoforms (fast recycling) or through a concurrent endosome recycling compartment by Rab11 isoforms (slow recycling) (O’Sullivan and Lindsay, 2020). Interestingly, it was found that RNAi depletion of the recycling Rab GTPase Rab11b repressed L. monocytogenes infection in cells maintained in Glc-containing medium (Kühbacher et al., 2015). Because c-Met recycling was reduced in Gal cells (Figure 4B), we hypothesized that nutrient-based metabolic manipulation affected endocytic recycling and thus L. monocytogenes infection. Immunoblot analysis of mock and infected Glc and Gal cells revealed no obvious differences in the expression levels of Rab11 (Figure S5D), indicating that steady-state levels of Rab11 do not link increased mitochondrial respiration to changes in endocytic recycling in Gal cells. Because it would be difficult to strictly increase receptor trafficking and return to the plasma membrane in Gal cells, we choose to repress c-Met return to the plasma membrane in Glc cells by Rab11b. To this end, we overexpressed a GTP-bound and thus constituently active mutant of Rab11b (Rab11bCA), which has been previously shown to impair receptor trafficking back to the plasma membrane. Before L. monocytogenes infection, Glc and Gal cells were transiently transfected with either the Rab11bCA mutant or the control plasmid. Strikingly, in contrast to the control plasmid (Figure 4E; −56% ± 1% decreased bacterial load in Gal cells), overexpression of Rab11bCA reduced L. monocytogenes burden in Glc cells to the level of Gal cells and completely abolished the difference between the intracellular bacterial load of Glc and Gal cells (Figure 4E). These data strengthen our hypothesis that the infection defect observed in Gal cells is indeed due to deficient endocytic recycling, which is in turn caused by nutrient-based manipulation of the host cell energy metabolism.
In order to evaluate the impact of mitochondrial respiration on bacterial infection in general, we expanded our study to Salmonella enterica serovar Typhimurium and Shigella flexneri, which are two other intracellular bacteria, are common causes of food-borne diseases, and infect colonic epithelial cells. Similar to our observations of infections with L. monocytogenes, Gal cells showed a lower S. Typhimurium load than Glc cells (−49% ± 6%), with the same difference observed up to 6 h post-infection (Figure S5E). On the other hand, numbers of intracellular S. flexneri were comparable in Glc and Gal cells (Figure S5F). This result indicates that S. Typhimurium but not S. flexneri infection is reduced in cells with increased respiratory activity. Entries of both S. Typhimurium and S. flexneri into non-phagocytic cells have been classically described as trigger mechanisms involving macropinocytosis-like processes (Cossart and Sansonetti, 2004). However, the bacterial entry mechanisms of S. Typhimurium and S. flexneri are quite dissimilar. S. Typhimurium infection induces the transport of exocytic vesicles to plasma membrane sites of bacterial engulfment (Nichols and Casanova, 2010), a process which uses the same machinery as receptor recycling. In contrast, S. flexneri does not hijack pre-existing host endocytic vesicles during the invasion step (Weiner et al., 2016). We thus hypothesize that increased mitochondrial respiration favors infection by bacteria that exploit cellular endocytic recycling to promote their invasion.
Overall, these results show that enhanced mitochondrial respiration triggered by nutrient-based metabolic manipulation restricts L. monocytogenes entry through slowing the endocytic recycling of the host cell receptors such as c-Met, which mediates L. monocytogenes invasion.
Discussion
Clinical studies on mitochondrial diseases have linked the impairment of mitochondrial respiration to increased bacterial infections (Walker et al., 2014). However, the cellular basis and mechanisms driving these observations are poorly understood. Moreover, the contribution of mitochondrial energy metabolism toward bacterial infection of epithelial cells, a primary host defense barrier to infection, remains to be comprehensively investigated. Indeed, the link between infection and energy metabolism has been primarily studied in macrophages (Escoll and Buchrieser, 2018). Here, we used two different and complementary approaches (nutrient-based and genetic) to manipulate the host cell metabolism to examine the role of mitochondrial respiration in bacterial infection. As intracellular bacteria can crosstalk directly with mitochondria (Spier et al., 2019; Tiku et al., 2020), we chose the well-established intracellular human pathogen L. monocytogenes as a model. We have previously shown that L. monocytogenes infection affects mitochondrial dynamics and function, and in turn, it is affected by these processes (Lebreton et al., 2015).
In this study, we show that human epithelial cells with decreased mitochondrial respiration and high glycolysis rates are subject to more efficient infection by L. monocytogenes. Infection was favored in a cell model of mitochondrial disease in which we ablated the COX assembly factor SURF1. We obtained similar results by using a nutrient-based approach to manipulate mitochondrial respiration, providing complementary evidence that mitochondrial respiration plays an important role in infectivity at the cellular level. Further investigation revealed that the infection differences observed occurred at bacterial entry into the host cell. We discovered that the host cell metabolism alters the recycling kinetics of receptors such as c-Met back to the plasma membrane, leading to decreased L. monocytogenes burden in cells with high respiratory activity. Interfering with endocytic recycling led us to propose a model in which mitochondrial energy metabolism restricts L. monocytogenes infection at the entry step, through yet-to-be discovered molecular mechanisms, which appear to involve Rab11b and other players.
Our present study linking mitochondrial energy metabolism with L. monocytogenes infection in epithelial cells supports our previous hypothesis that L. monocytogenes benefits from the bioenergetic slowdown it induces in host cells (Stavru et al., 2011, 2013). In agreement with this hypothesis, we demonstrate here that inhibition of mitochondrial respiration is advantageous for invading L. monocytogenes, whereas enhanced mitochondrial respiratory activity diminishes L. monocytogenes infection.
Interestingly, in vitro and in vivo studies using macrophages and mouse models demonstrated that other intracellular pathogens such as Legionella pneumophila or S. Typhimurium also replicate more rapidly upon inhibition of mitochondrial function (Francione et al., 2009; Garaude et al., 2016; Escoll et al., 2017). However, it is still unclear whether this is a general rule, as contrasting data exist, for example for the obligate intracellular pathogen Chlamydia spp. Chlamydia trachomatis was shown to require mitochondrial ATP production for its growth (Chowdhury et al., 2017). On the other hand, impairment of mitochondrial function through mutation or silencing of the mitochondrial ATP synthase in fibroblasts and Hep-2 cells results in an increase in C. pneumoniae growth (Käding et al., 2017). This warrants deeper investigation to decipher which essential cellular processes are regulated by host metabolism in the context of bacterial pathogen lifestyles.
For the present study, we created a new and highly tractable cellular model for Leigh disease (SURF1−/− cells), whose metabolic characteristics were analyzed in depth through 13C isotopologue and respirometry analysis, providing a useful tool to understand the relationship between mitochondrial respiration and infection by different pathogens. Several clinical studies point toward infection as a trigger for an increased risk of metabolic crises, lactic acidosis, and irreversible disease progression (Morgan-Hughes, 1986; Edmonds, 2004; Varghese et al., 2011; Al-Zubeidi et al., 2014). It would therefore be of interest to assess the effects of infection on host cell metabolism in SURF1−/− cells. However, such experiments require high infection rates, which are not achievable at the short time points focused on in this work. In the future, it will be essential to assess whether mouse models of mitochondrial disease such as SURF1−/− mice (Dell’agnello et al., 2007) are more susceptible to bacterial infection.
Our data obtained by nutrient-based manipulation are consistent with a previous study, which showed a decrease of ∼42% in L. monocytogenes load after 3.5 h of infection, with another intestinal epithelial cell line (colon cancer HT29 cells) cultured in Gal-containing instead of Glc-containing medium (Velge et al., 1997). However, the authors did not link their findings to mitochondrial respiration. Based on metabolic activity measurements with AlamarBlue, the authors concluded that the main parameter influencing L. monocytogenes infection rates in their model was cell proliferation and differentiation, rather than cell metabolism. Later studies showed that AlamarBlue is a redox indicator, which is rather suited for monitoring cell viability and proliferation (Rampersad, 2012); thus, it is plausible that the results obtained by Velge et al. (1997) are also linked to cellular energy metabolism. Supporting this idea, it has been well described that nutrient-based manipulation through medium supplemented with Gal instead of Glc induces a metabolic switch toward increased mitochondrial respiration (Reitzer et al., 1979) in multiple cell types (Robinson et al., 1992; Rossignol et al., 2004; Aguer et al., 2011).
We thus specifically investigated the role of mitochondrial respiration in infection, focusing on short infection time points, and detected changes in intracellular L. monocytogenes levels already at 1 h post-infection, both in our nutrient-based and in our genetic model for mitochondrial disease (SURF1−/− cells). These findings indicated that the observed differences were likely caused by a perturbation in the early steps of cellular infection by L. monocytogenes, such as adhesion or entry. To investigate the mechanism underlying our observations, we used L. monocytogenes mutants and discovered that both InlA- and InlB-mediated entry pathways are affected, pointing to a process that occurs downstream and is common to both receptors. One such process is endocytosis-mediated receptor uptake, and our data suggest that nutrient-based manipulation of the cellular energy metabolism does not impact the uptake of the receptors by endocytosis, but likely affects receptor recycling. In contrast, several studies in macrophages have shown that the levels of glycolysis and phagocytosis are correlated (Newsholme et al., 1987; Venter et al., 2014; Pavlou et al., 2017), and drug-induced mitochondrial uncoupling was found to trigger massive endocytosis (MEND) in BHK fibroblasts (Hilgemann et al., 2013).
The balance between endocytosis and recycling controls the composition of the plasma membrane, enabling the return of endocytosed material (e.g., receptors) back to the plasma membrane. Receptor recycling can occur by rapid or slow pathways. The slow recycling pathway can be measured experimentally and involves the transport from early endosomes to the endocytic recycling compartment (ERC) and back the plasma membrane (reviewed in Grant and Donaldson 2009). We hypothesize that increased mitochondrial respiration could slow down receptor recycling to the plasma membrane; this would in turn result in reduced numbers of receptors at the cell surface for which L. monocytogenes can engage for its entry. To test this hypothesis, we chose to decrease the trafficking activity from the ERC to the plasma membrane by overexpressing a GTP-locked mutant version of the small GTPase Rab11b (Rab11bCA) (Ullrich et al., 1996; Ren et al., 1998), a key regulator of receptor recycling. Indeed, overexpressing the Rab11bCA mutant decreased L. monocytogenes load in Glc cells and abolished the infection difference between Glc and Gal cells. This report is, to our knowledge, the first one showing that increased mitochondrial respiration by nutrient-based metabolic manipulation triggers a decrease of receptor recycling in epithelial cells, which significantly impacts bacterial uptake. Future work will aim at elucidating the signaling cascades linking mitochondrial respiration with endocytic recycling, and further experiments should focus on Rab11b activity levels in cells with altered mitochondrial respiration.
In the context of L. monocytogenes infection, two recent studies manipulated Rab11 activity and assessed changes in L. monocytogenes infection (albeit at later time points of infection), obtaining opposite results. A small interfering RNA (siRNA) screen showed that downregulation of Rab11a increased L. monocytogenes infection at 5 h post-infection, whereas RNAi of Rab11b repressed infection (Kühbacher et al., 2015). Strikingly, expression of a dominant-negative (GDP-bound) form of Rab11a was shown to decrease bacterial cell-cell spread and thus infection efficiency at 6 h post-infection (Dowd et al., 2020). Furthermore, Dowd et al. (2020) showed that exocytosis is upregulated in L. monocytogenes-containing protrusions, which is a process partially associated with recycling Rab GTPases. Interestingly, another recent study with intestinal organoids showed that L. monocytogenes hijacks Rab11a-dependent E-cadherin recycling to translocate across the intestinal epithelium (Kim et al., 2020), pointing toward another important role of endocytic recycling in in vivo L. monocytogenes infections.
At the mechanistic level, the only connection between Rab11 and mitochondria to date described in the literature involves cytoskeletal rearrangements upon viral activation of Src family kinases, leading to Rab11a-mediated and Drp1-dependent mitochondrial fragmentation, followed by redistribution of the organelles in the vicinity of polarized actin structures (Landry et al., 2014). L. monocytogenes is also known to (1) activate Src kinase to remodel actin during invasion (Van Langendonck et al., 1998; Sousa et al., 2007) and (2) induce transient mitochondrial fragmentation, along with a loss in the mitochondrial inner membrane potential. However, the mitochondrial fragmentation induced by L. monocytogenes is Drp1 independent and does not result in mitochondrial redistribution to polarized structures (Stavru et al., 2011, 2013). Interestingly, a recent report showed that endocytosis-dependent relocalization of the small GTPases RalA and RalB to depolarized mitochondria plays an important role in innate immune signaling (Pollock et al., 2019). Whether L. monocytogenes interferes with similar mechanisms is currently unclear, and future work will be needed to characterize the interplay between mitochondrial energy metabolism, endocytic recycling, and infection. In particular, investigations into the contributions of the three Rab11 isoforms are of interest, as the view emerges that they have distinct functions in endocytic recycling (Ferro et al., 2021).
In conclusion, our results show at the cellular level that decreased mitochondrial respiration results in increased infection by the food-borne pathogen L. monocytogenes. This result might contribute to the clinically observed predisposition of mitochondrial disease patients to bacterial infections (Walker et al., 2014) and suggests that mitochondrial disease patients, similar to immunocompromised individuals or pregnant women, may benefit from decreasing their exposure to food-borne pathogens. Beyond this finding, our data will stimulate research to determine whether host-directed metabolic interventions can have antimicrobial effects or synergize with antibiotic therapies. This information could be particularly relevant in the context of mitochondrial diseases, where finding ways to decrease antibiotic use is critical given that several of these compounds display mitochondrial toxicity (Kalghatgi et al., 2013).
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit polyclonal anti-L. monocytogenes R11 | Dramsi et al., 1998 | N/A |
| mouse monoclonal anti-β-actin clone AC-15 | Sigma-Aldrich | Cat# A1978, RRID:AB_476692 |
| rabbit monoclonal anti-c-Met | Abcam | Cat# ab51067, RRID:AB_880695 |
| rabbit polyclonal anti-SURF1 | Proteintech | Cat# 15379-1-AP, RRID:AB_2239968 |
| rabbit polyclonal anti-Rab11 | Invitrogen | Cat# 71-5300, RRID:AB_2533987 |
| rabbit polyclonal anti-Transferrin receptor | Abcam | Cat# ab84036, RRID:AB_10673794 |
| anti-rabbit Alexa Fluor 488 | Thermo Fisher Scientific | Cat# A-11034, RRID:AB_2576217 |
| anti-rabbit Alexa Fluor 594 | Thermo Fisher Scientific | Cat# A-11012, RRID:AB_2534079 |
| anti-rabbit Alexa Fluor 658 | Thermo Fisher Scientific | Cat# A-11011, RRID:AB_143157 |
| Bacterial and virus strains | ||
| Listeria monocytogenes EGDe (wt strain) | BUG1600, Institut Pasteur | N/A |
| Listeria monocytogenes EGDeΔC3Δhpt (strain deleted by lmo1293, lmo0347/8, lmo2695/6 and uhpt) | Grubmüller et al., 2014 | N/A |
| Listeria monocytogenes EGDe-cGFP (wt strain expressing green fluorescent protein) | BUG2538, Institut Pasteur | N/A |
| Listeria monocytogenes EGD (wt strain) | BUG600, Institut Pasteur | N/A |
| Listeria monocytogenes EGDΔInlA (InlA deleted strain) | BUG947, Institut Pasteur | N/A |
| Listeria monocytogenes EGDΔInlB (InlB deleted strain) | BUG1047, Institut Pasteur | N/A |
| Listeria monocytogenes EGDΔhly (hly deleted strain) | BUG3650, Institut Pasteur | N/A |
| Listeria monocytogenes EGD-cGFP (wt strain expressing GFP) | BUG2539, Institut Pasteur | N/A |
| Salmonella enterica serovar typhimurium 12023 GFP (wt strain expressing GFP) | Institut Pasteur | N/A |
| Shigella flexneri M90T (wt strain) | Centre d’Immunologie de Marseille-Luminy | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| Brain heart infusion (BHI) medium | BD Difco | Cat#237500 |
| McCoy’s 5A medium (Modified) | GIBCO | Cat#26600 |
| McCoy’s 5A medium (Modified) (costum-made based on #26600, GIBCO without glucose and glutamine) | GIBCO | Costum-made |
| DMEM, no glucose | GIBCO | Cat#11966 |
| D-(+)-Glucose | Sigma-Aldrich | Cat#G8270 |
| D-(+)-Galactose | Sigma-Aldrich | Cat#G0625 |
| L-glutamine | GIBCO | Cat#25030 |
| Non-essential amino acids | GIBCO | Cat#11140 |
| L-sodium pyruvate | GIBCO | Cat#11360 |
| Fetal bovine serum (FBS) | BioWest | Cat#S181G |
| [U-13C6]glucose (99% labeled) | Sigma-Aldrich | Cat#389374 |
| [U-13C6]galactose (99% labeled) | Sigma-Aldrich | Cat#605379 |
| [U-13C5]glutamine (98% labeled) | Cortecnet | Cat#CC1050P01 |
| Gentamicin | Sigma-Aldrich | Cat#G1397 |
| Penicillin-Streptomycin | GIBCO | Cat#15140-122 |
| Chloramphenicol | Sigma-Aldrich | Cat#C0378 |
| G418 | InvivoGen | Cat#108321-42-2 |
| Triton X-100 (TX-100) | Sigma-Aldrich | Cat#11332481001 |
| jetPRIME | Polypus Transfection | Cat#114-01 |
| oligomycin A | Sigma-Aldrich | Cat#75351 |
| carbonyl cyanide p-(tri-fluromethoxy)phenyl-hydrazone (FCCP) | Sigma-Aldrich | Cat#C2920 |
| carbonyl cyanide m-chlorophenyl hydrazone (CCCP) | Sigma-Aldrich | Cat#C2759 |
| rotenone | Sigma-Aldrich | Cat#R8875 |
| antimycin A | Sigma-Aldrich | Cat#A8674 |
| 2-Deoxy-D-glucose (2DG) | Sigma-Aldrich | Cat#D6134 |
| dynasore hydrate | Sigma-Aldrich | Cat#D7693 |
| Phalloidin 647 | Thermo Fisher Scientific | Cat#A22287 |
| MitoTracker Deep Red | Thermo Fisher Scientific | Cat#M22426 |
| Hoechst 33342 | Thermo Fisher Scientific | Cat#62249 |
| NucBlue Live ReadyProbes Reagent | Thermo Fisher Scientific | Cat#R37605 |
| 4–20% Mini-PROTEAN TGX Stain-Free Protein Gels | Bio-Rad | Cat#4568096 |
| Critical commercial assays | ||
| Protein Assay Dye Reagent Concentrate | Bio-Rad | Cat##5000006 |
| Amersham ECL Prime | Thermo Fisher Scientific | Cat#10308449 |
| Clarity Western ECL substrate | Bio-Rad | Cat# #1705060 |
| ATPlite kit | Perkin-Elmer | Cat#6016943 |
| LIVE/DEAD Fixable Red Dead Cell Staining dye | Thermo Fisher Scientific | Cat# L34971 |
| Deposited data | ||
| GC-MS data | this manuscript | XXX |
| Experimental models: Cell lines | ||
| Human: HCT116 wt | ATCC | CCL-247 |
| Human: HCT116 SURF1−/− | this manuscript | N/A |
| Human: HeLa | ATCC | CCL-2 |
| Human: primary skin fibroblasts EF10 + tel | Tissue repository of the Association Française contre les Myopathies (AFM) | N/A |
| Human: primary skin fibroblasts KB5 + tel | Tissue repository of the Association Française contre les Myopathies (AFM) | N/A |
| Oligonucleotides | ||
| SURF1 fwd (for CRISPR/Cas9 deletion of SURF1) 5′ caccgCGCCTGGAGGAGCGTCCTCA 3′ | this manuscript | N/A |
| SURF1 rev (for CRISPR/Cas9 deletion of SURF1) 5′ aaacTGAGGACGCTCCTCC AGGCGc 3′ |
this manuscript | N/A |
| SURF1 fwd (for complementation of SURF1−/− cells) 5′ gatccccaagctt ggcctgacaggccTCACAC ACCAGGTGTCCCAC 3′ |
this manuscript | N/A |
| SURF1 rev (for complementation of SURF1−/− cells) 5′ actaccccaagc tggcctctgagTTGGTACCGAGC TCGGATC 3′ |
this manuscript | N/A |
| Recombinant DNA | ||
| Plasmid: pSpCas9(BB)-2A-GFP (PX458) | Ran et al., 2013 | Addgene plasmid #48138 |
| Plasmid pSpCas9(BB)2A-GFP containing the sgRNA for SURF1 | this manuscript | N/A |
| Plasmid: pcDNA3.1+C-(hSURF1)DYK | GenScript | CloneID OHu20778 |
| Plasmid: pSBbi-Neo | Kowarz et al., 2015 | Addgene plasmid #60525 |
| Plasmid pSBbi-Neo-SURF1 | this manuscript | N/A |
| Plasmid: pCMV (CAT)T7-SB100 | Mátés et al., 2009 | Addgene plasmid #34879 |
| Plasmid: pEGFP-N1 | Clontech Laboratories | #6085-1 |
| Plasmid: pC1-EGFP-Rab11bCA | Dr. Matthew Lawrenz, University of Louisville | N/A |
| Software and algorithms | ||
| Prism (version 8) | GraphPad | RRID:SCR_002798 |
| Seahorse Wave (version 2.6.1) | Agilent | RRID:SCR_014526 |
| LabSolutions | Shimadzu | RRID:SCR_018241 |
| MetaMorph (version 7.7.9.0) | Molecular Devices | RRID:SCR_002368 |
| Fiji (version 2.0.0) | ImageJ | RRID:SCR_002285 |
| Image Lab (version 6.0.1) | Bio-Rad | RRID:SCR_014210 |
| FlowJo (version 10) | BD Biosciences | RRID:SCR_008520 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Timothy Wai (timothy.wai@pasteur.fr).
Materials availability
Accession numbers are listed in the Key resources table. All plasmids generated in this study are available from the lead contact upon request.
Experimental model and subject details
Bacterial strains
Information about the bacterial strains used in this work are provided in the Key resources table. Listeria monocytogenes strains were grown at 200 rpm at 37°C in brain heart infusion (BHI) medium (Difco, BD) supplemented with 7 μg/mL chloramphenicol when required. Salmonella enterica serovar Typhimurium and Shigella flexneri strains were cultured in Lysogeny broth (LB).
Cell lines and growth conditions
The details about the cell lines used in this study are provided in the Key resources table and comprise HCT116 (human colorectal adenocarcinoma; ATCC CCL-247), HeLa (human cervical adenocarcinoma; ATCC, CCL-2) and primary skin fibroblasts (from the tissue repository of the AFM (Association Française contre les Myopathies)). The cells were maintained in cell culture media supplemented with D-glucose (Sigma-Aldrich) or D-galactose (Sigma-Aldrich) as well as L-glutamine (GIBCO), fetal bovine serum (BioWest) and other supplements as detailed in the table. The cells were incubated at 37°C with 10% CO2 in a humidified atmosphere and maintained at least 14 days in the respective growth medium prior to experiments.
| Cell line | Base medium | Supplements |
|---|---|---|
| HCT116 wt/Glc (maintenance) | McCoy’s 5A medium (Modified, #26600, GIBCO), contains 3 g/L D-glucose + 1.5 mM L-glutamine | 1 mM non-essential amino acids |
| 10 (v/v) % FBS | ||
| HCT116 wt/Gal (maintenance) | McCoy’s 5A medium (Modified, costum-made based on #26600, GIBCO), contains no D-glucose or L-glutamine | 3 g/L D-galactose |
| 1.5 mM L-glutamine | ||
| 1 mM non-essential amino acids | ||
| 10 (v/v) % FBS | ||
| HCT1116 SURF1−/− (maintenance) | McCoy’s 5A medium (Modified, #26600, GIBCO), contains 3 g/L D-glucose + 1.5 mM L-glutamine | 1 mM non-essential amino acids |
| 10 (v/v) % FBS | ||
| HCT116 SURF1−/−+ SURF1 (maintenance) | McCoy’s 5A medium (Modified, #26600, GIBCO), contains 3 g/L D-glucose + 1.5 mM L-glutamine | 1 mM non-essential amino acids |
| 10 (v/v) % FBS | ||
| Infection and starvation medium for HCT116 cells | McCoy’s 5A medium (Modified, costum-made based on #26600, GIBCO), contains no D-glucose or L-glutamine | / |
| HeLa Glc (maintenance) | DMEM, no glucose (#11966, GIBCO), contains 4 mM L-glutamine | 2 g/L D-glucose |
| 10 (v/v) % FBS | ||
| HeLa Gal (maintenance) | DMEM, no glucose (#11966, GIBCO), contains 4 mM L-glutamine | 2 g/L D-galactose |
| 10 (v/v) % FBS | ||
| Infection medium for HeLa cells | DMEM, no glucose (#11966, GIBCO), contains 4 mM L-glutamine | / |
| Primary skin fibroblasts Glc (maintenance) | DMEM, no glucose (#11966, GIBCO), contains 4 mM L-glutamine | 2 g/L D-glucose |
| 1 mM sodium pyruvate | ||
| 10 (v/v) % FBS | ||
| Primary skin fibroblasts Gal (maintenance) | DMEM, no glucose (#11966, GIBCO), contains 4 mM L-glutamine | 2 g/L D-galactose |
| 1 mM sodium pyruvate | ||
| 10 (v/v) % FBS | ||
| Infection medium for primary skin fibroblasts | DMEM, no glucose (#11966, GIBCO), contains 4 mM L-glutamine | / |
CRISPR/Cas9 deletion of the SURF1 gene in HCT116 cells
Single guide RNAs (sgRNAs) targeting the second exon of SURF1 were designed using the GPP sgRNA Designer tool (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). Information about the oligonucleotides are provided in the Key resources table. The oligonucleotides were annealed and cloned into the Cas9 expression plasmid pSpCas9(BB)-2A-GFP (PX458) (Ran et al., 2013). pSpCas9(BB)-2A-GFP (PX458) was a gift from Feng Zhang (Addgene plasmid #48138). HCT116 wt cells were seeded into 6-well plates at a density of 5 × 105 cells/well and cultured for 24 h prior to transfection. The transfection was carried out using jetPRIME (Polypus Transfection) according to the manufacturer’s instructions. 24 h later single GFP-positive HCT116 cells were sorted into 96-well plate wells by flow cytometry, into cell culture medium supplemented with 25% conditioned medium, 100 I. U./mL penicillin and 100 μg/mL streptomycin (GIBCO). The clones were expanded and SURF1 knockout efficiency was verified by immunoblotting.
Complementation of HCT116 SURF1−/− cells
Stable complementation of HCT116 SURF1−/− cells with SURF1 was achieved by using the Sleeping Beauty transposon system, which achieves genomic integration through the transposase SB100X (Mátés et al., 2009; Kowarz et al., 2015). SURF1 was PCR amplified from the plasmid pcDNA3.1+C-(hSURF1)DYK (CloneID OHu20778, GenScript) using the oligonucleotides provided in the Key resources table. The amplicons were inserted by sequence- and ligation-independent cloning (SLIC) (Jeong et al., 2012) into the SfiI site of pSBbi-Neo, yielding pSBbi-Neo-SURF1. pSBbi-Neo was a gift from Eric Kowarz (Addgene plasmid #60525, (Kowarz et al., 2015). HCT116 SURF1−/− cells were plated into 6-well plates at a density of 5 × 105 cells/well. The following day the cells were co-transfected with the SB100X transposase enzyme expressing plasmid pCMV (CAT)T7-SB100 and pSBbi-Neo-SURF1, using the jetPRIME (Polypus Transfection) according to the manufacturer’s instructions. pCMV(CAT)T7-SB100 was a gift from Zsuzsanna Izsvak (Addgene plasmid #34879; (Mátés et al., 2009). After a 24 h incubation the cell culture medium was changed to medium containing 3 g/L D-galactose instead of D-glucose and supplemented with 400 μg/mL G418 (InvivoGen) to select for SURF1-expressing cells. After one week of selection, individual cells were sorted by flow cytometry in a 96-well plate wells and further expanded. The complementation was verified at the protein level by immunoblotting against SURF1; the clone re-expressing SURF1 at a level that was most similar to that of wild-type SURF1 was used for further experiments.
Method details
Cell transfection
For transient protein overexpression, cells were seeded in a 12-well plate and transfected with 0.6 μg of plasmid DNA using jetPRIME (Polyplus Transfection) according to the manufacturer’s instructions. The medium was changed the following day, and the cells were assayed 48 h post transfection. The plasmid pC1-EGFP-Rab11bCA (kind gift from Dr. Matthew Lawrenz, University of Louisville) was used to express constitutively active Rab11b. Control cells were transfected with pEGFP-N1 (Clontech Laboratories).
Cell infection
Gentamicin protection assays were performed in a 24-well plate format for immunofluorescence, in a 12-well plate format for colony forming unit (CFU) quantification or in a 6-well plate format for flow cytometry. Cells were seeded one or two days before the assay. For the cell infections, overnight cultures of the L. monocytogenes strains, S. Typhimurium at OD600 1.5-2 and S. flexneri at OD600 0.5-0.7 were used. The bacterial cultures were washed 3x in Dulbecco’s phosphate-buffered saline (DPBS, GIBCO) and resuspended in serum-free mammalian cell culture medium without D-glucose or D-galactose to achieve the desired multiplicity of infection (MOI; bacteria/cell). For immunofluorescence and cell death assays, cells were infected with GFP-expressing L. monocytogenes strains. A fixed volume was then added to each well and the cells were centrifuged for 1 min (L. monocytogenes) or 5 min (S. Typhimurium, S. flexneri) at 1,000 rpm to synchronize the infection. The cells were incubated with the bacteria for 30 min or 1 h at 37°C. Following this incubation, the cells were washed 2x with DBPS, then cell growth medium containing D-glucose or D-galactose, and supplemented with 10 (v/v) % FBS (BioWest) and gentamicin (Sigma-Aldrich) was added to kill extracellular bacteria. Gentamicin was used at 20 μg/ml for infections with L. monocytogenes and at 50 μg/ml for 1 h followed by 20 μg/ml for infections with S. Typhimurium and S. flexneri. At the time point indicated in each figure, the cells were washed 3x with DPBS before further processing. To quantify intracellular bacterial load by CFU count, infected cells and inocula were lysed in cold 0.1% (v/v) Triton X-100 (Sigma-Aldrich)/water, serially diluted in DBPS and dropped on agar plates (BHI for L. monocytogenes and LB for S. Typhimurium and S. flexneri) as described in Kühbacher et al., 2014. After overnight incubation at 37°C, CFUs were counted in the dilution displaying a reasonable number (20-300) of well-separated colonies and bacterial numbers were if applicable normalized to the inoculum concentration.
Mitochondrial respiration and glycolysis measurements
Mitochondrial respiration and glycolysis were assessed by measuring oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) using a Seahorse XFe96 analyzer (Agilent Technologies), essentially following the Seahorse XF Cell Mito Stress Test or Glycolysis Stress Test protocols. Briefly, HCT116 cells were plated 48 h before the assay in a Seahorse XF96 cell culture microplate (Agilent Technologies), at a density of 1 × 104 cells/well (six wells per condition). Before the assay, the medium was replaced by Seahorse XF base medium (Agilent Technologies) supplemented with 1.5 mM L-glutamine (GIBCO) and for the Mito Stress Test also with 3 g/L D-glucose (Sigma-Aldrich) or D-galactose (Sigma-Aldrich) and 1 mM sodium pyruvate (GIBCO) (adjusted to pH 7.4), and cells were incubated for 1 h at 37°C in a non-CO2 incubator. During the Mito Stress Test, the OCR was measured under basal conditions as well as upon sequential addition of drugs impacting on mitochondrial respiration. We sequentially added A) oligomycin A (Sigma-Aldrich), an inhibitor of the mitochondrial F1FO-ATPase; B) FCCP or CCCP (Sigma-Aldrich), ionophores that uncouple the mitochondrial membrane potential by transporting H+ across the mitochondrial inner membrane; and C) a mixture of rotenone and antimycin A (Sigma-Aldrich), inhibitors of the respiratory chain complexes I and III, respectively. Each of the drugs was injected to reach a final concentration in the well of 1 μM. During the Glycolysis Stress Test, the ECAR was measured and we sequentially added A) 10 mM D-glucose (Sigma-Aldrich) to trigger glycolysis; B) 1 mM oligomycin A (Sigma-Aldrich) to inhibit mitochondrial respiration, and C) 50 mM 2DG (Sigma-Aldrich) to inhibit glycolysis. Data were analyzed using the Wave software (Agilent Technologies) and normalized to cell numbers counted upon Hoechst 33342 (Thermo Fisher Scientific) staining with a Cytation 5 microplate reader (Biotek).
13C-Labeling experiments
HCT116 cells were seeded in 10 cm culture plates at a density of 1.3 × 106 cells/dish and incubated for 24 h. After 30 min of incubation in medium without D-glucose, D-galactose, L-glutamine and FBS, the cells were washed 1x with DPBS and maintained in medium supplemented with 20 μg/mL gentamicin and [U-13C6]glucose (99% labeled, Sigma), [U-13C6]galactose (99% labeled, Sigma-Aldrich) or [U-13C5]glutamine (98% labeled, Cortecnet). By addition of unlabeled forms, [U-13C6]glucose and [U-13C6]galactose were diluted and accounted for 20/33% of the overall glucose or galactose pool in the medium. Glutamine was only present in the fully labeled form in the respective experiment. 17.5 h later cells were washed with DPBS and lysed in ice-cold 0.1% Triton X-100/distilled water for 10 min. The lysate was vigorously vortexed and centrifuged at 15,000 g for 10 min at 4°C. The pellet and supernatant fractions were snap-frozen in liquid nitrogen and stored overnight at −80°C. The pellet fraction was re-suspended in 500 μL DPBS, snap-frozen in liquid nitrogen again and stored at −80°C until metabolites were extracted.
Metabolite extraction procedure
For the analysis of cytosolic metabolites, about 5 mg of the freeze-dried supernatant or pellet fraction were dissolved in ice-cold methanol (≥99.8%, VWR). 800 mg of glass beads (ø 0.25-0.5 mm, Carl Roth) were added, and mechanical cell lysis was performed for 1 × 20 s at 4.0 ms-1 and for 4 × 20 s at 6.5 ms-1 using a ribolyser system (Hybaid). Afterward, the sample was centrifuged at 3,200 g for 10 min and the supernatant was dried under a stream of nitrogen. For derivatization 50 μL of anhydrous acetonitrile (Sigma-Aldrich) as well as 50 μL of N-(tert-butyldimethylsilyl)-N-methyl-trifluoroacetamide (Sigma-Aldrich) were added and the mixture was incubated at 70°C for 1 h. The resulting tert-butyldimethylsilyl-derivatives (TBDMS) were analyzed via GC-MS.
Protein hydrolysis
For analysis of protein-bound amino acids about 2 mg of the freeze-dried supernatant or pellet fractions were resuspended in 1 mL of 6 N hydrochloric acid (VWR) and hydrolysed for 15 h at 105°C. The reaction mixture was dried under a stream of nitrogen at 70°C. The residue was suspended in 200 μL of 50% acetic acid (VWR) using an ultrasonic bath for 3 min. The solution was applied onto a small column of Dowex 50W X8 (7 × 10 mm; 200-400 mesh, 34-74 μm, H+-form, Sigma-Aldrich). The column was first washed with 1.6 mL H2O, then eluted with 1 mL 4 M aqueous ammonia solution (VWR). The ammonia eluate was dried under a stream of nitrogen at 70°C. The residue was treated with 50 μL of N-(tert-butyldimethylsilyl)-N-methyltrifluoroacetamide containing 1% tert-butyldimethylsilylchloride (Sigma-Aldrich) and 50 μL of anhydrous acetonitrile (Sigma-Aldrich) at 70°C for 30 min. The TBDMS-derivatives of amino acids were then analyzed by GC-MS. Due to degradation during acid hydrolysis, tryptophan, methionine and cysteine could not be analyzed with this method. Furthermore, acid hydrolysis leads to conversion of glutamine and asparagine to glutamate and aspartate, respectively. Therefore, results given for aspartate and glutamate correspond to cumulative values of asparagine/aspartate and glutamine/glutamate, respectively. Due to inefficient derivatization, TBDMS-arginine could not be detected in sufficient amounts for isotopologue profiling.
Gas chromatography-mass spectrometry analysis
GC-MS analysis was performed with a QP2010 Plus gas chromatograph/mass spectrometer (Shimadzu) equipped with a fused silica capillary column (Equity TM-5; 30 m 0.25 mm, 0.25 μm film thickness; Supelco) and a quadrupole detector working with electron impact ionization at 70 eV. An aliquot (0.1 to 6 μl) of the derivatized samples was injected in 1:5 split mode at an interface temperature of 260°C and a helium inlet pressure of 70 kPa. Selected ion monitoring (SIM) was used with a sampling rate of 0.5 s and LabSolutions software (Shimadzu) was used for data collection and analysis. For the measurement of cytosolic metabolites, the column was kept at 100°C for 2 min after sample injection. Afterward, a first temperature gradient of 3°C min-1 was applied until a final temperature of 234°C. Subsequently, a second temperature gradient of 1°C min-1 until a final temperature of 237°C, and a third temperature gradient of 3°C min-1 to a final temperature of 260°C were performed.
For the measurement of proteinogenic amino acids, the column was kept at 150°C for 3 min after sample injection. Subsequently, the column was developed with a gradient of 7°C min-1 to a final temperature of 280°C, which was held for 3 min. Isotopologue calculations were performed with m/z [M-57]+.
All samples were measured three times for technical replicates. 13C-excess values and isotopologue compositions were calculated as previously described (Eylert et al., 2008). This comprises (i) the detection of GC-MS spectra of unlabeled derivatized metabolites, (ii) determination of the absolute mass of isotopologue enrichments and distributions of labeled metabolites of the experiment, and (iii) correction of the absolute 13C incorporation by subtracting the heavy isotopologue contributions due to the natural abundances in the derivatized metabolites to calculate the isotopologue enrichments and distributions (Lee et al., 1991; Eylert et al., 2008).
Differential bacterial staining
To perform differential inside-out bacterial staining as described in Kühbacher et al. (2014), cells were seeded two days prior to the assay in 24-well plates. Infection was carried out as described above, using GPF-expressing L. monocytogenes EGD bacteria. After 1 h of incubation with the bacteria and a subsequent wash with DPBS, cells grown on glass coverslips were processed for immunofluorescence. The cells were fixed for 10 min at RT in 4% (v/v) paraformaldehyde (PFA, Euromedex)/DPBS and extracellular bacteria were stained using rabbit polyclonal anti-L. monocytogenes R11 antibody (1:500, produced in-house; Dramsi et al., 1998). Labeling with primary and fluorophore-conjugated secondary antibodies or dyes was performed in blocking buffer (1% [w/v] BSA + 10% [v/v] goat serum/PBS) for 1 h at RT in a moist dark chamber. Cells were washed 3x with PBS and stained with secondary anti-rabbit antibody conjugated to Alexa Fluor 568 dye (Thermo Fisher Scientific) (1:500). Then cells were permeabilized for 5 min in 0.5% (v/v) Triton X-100/PBS, blocked for 20 min in blocking buffer and incubated with the dyes Hoechst 33342 (Thermo Fisher Scientific) (1:2,000) and Phalloidin 647 (Thermo Fisher Scientific) (1:100) to stain DNA and actin, respectively. Coverslips were mounted onto microscope slides with FluoroMount-G mounting medium (Interchim), dried at RT overnight, and stored in the dark at 4°C. Given that we used GFP-expressing L. monocytogenes for infection, all bacteria were stained in green (GFP), while only extracellular L. monocytogenes displayed an additional red staining (resulting in a yellow signal). Cells were imaged with a Zeiss AxioObserver.Z1 inverted microscope (Carl Zeiss AG) equipped with a high-speed CSU-X1 spinning-disk confocal system (Yokogawa) and an Evolve electron-multiplying charge-coupled-device (EM-CCD) camera (Photometrics), run by MetaMorph software (version 7.7.9.0). Seven focal planes were captured across multiple wavelength channels using an EC Plan-Neofluor 100x/1.3 Oil Ph3 M27 oil objective, covering a total range of 4.5 nm. Fiji was used for further image analysis and for quantification of bacterial adhesion and entry.
Immunoblotting
Expression levels of the cellular proteins SURF1, c-Met and Rab11 were assessed by immunoblotting. To study the effect of infection on total protein levels, cells were infected with L. monocytogenes EGDe (MOI 20, 6 h) as described in the section cell infection. For harvest and lysis, cells were incubated for 30 min in RIPA buffer (1 M Tris pH 7.4, 150 mM Sodium Chloride, 1% [v/v] Triton X-100, 0.05% [v/v] Sodium Deoxycholate, 0.1% [v/v] SDS, 1 mM EDTA) on ice. After centrifugation (16,000 g, 4°C, 10 min), the total protein concentrations of the supernatants were estimated using a colorimetric Bradford assay (Bio-Rad Protein Assay Dye Reagent Concentrate). 20 μg protein/well were loaded on 4%–20% Mini-PROTEAN TGX Stain-Free Protein Gels (Bio-Rad) or 4%–15% Mini-PROTEAN TGX Stain-Free Protein Gels for SDS-PAGE, using the following running buffer (190 mM glycine, 25 mM Tris base, 0.1% [v/v] SDS). Proteins were then blotted onto a 0.45 μm nitrocellulose membrane (GE Healthcare) using either a wet blot assembly with the following cold transfer buffer (200 mM glycine, 25 mM Tris base, 20% [v/v] ethanol) or the standard mixed molecular weight protocol on the Trans-Blot Turbo (Bio-Rad). Membranes were blocked in 1% BSA in TBT (150 mM sodium chloride, 6 mM Tris base, 19 mM Tris HCl, 0.1% [v/v] Tween20) for 40 min at RT and incubated with primary antibody overnight at 4°C (shaking). The antibodies used: are specified in the Key resources table. Membranes were washed 3x with TBT and incubated for 1 h at RT with horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit secondary antibodies (1:10,000, Abliance). Blots were washed again 3x in TBT and revealed using Amersham ECL Prime (Thermo Fisher Scientific) or Clarity Western ECL substrate (Bio-Rad). Imaging and quantification of the blots were performed with a ChemiDoc XRS+ system (Bio-Rad) and the Image Lab software (Bio-Rad).
Differential staining of Transferrin or c-Met receptors
In order to monitor endocytosis and recycling of the Transferrin and c-Met receptors at the plasma membrane, we performed differential staining according to Connor et al. (2018). HCT116 cells were seeded 48 h before the assay in black 96-well plates (Greiner Bio One) at a density of 1 × 104 cells/well (six wells per condition) or on coverslips in 24-well plates (TPP) at a density of 6 × 104 cells/well. Cells previously maintained in D-glucose- or D-galactose-containing media were starved for 30 min in medium without D-glucose/galactose or FBS; control samples were incubated in the same medium supplemented with 20 μM dynasore hydrate (Sigma-Aldrich). The cells were further incubated in presence of a rabbit polyclonal anti-Transferrin receptor antibody (ab84036, Abcam) (1:1,000) or a rabbit monoclonal anti-c-Met antibody (ab51067, Abcam) (1:300). Infection for 1 h with GFP-expressing L. monocytogenes EGD (MOI 20) was performed as described above and was started at the same time as the antibody incubation. At the indicated time points, the cells were gently washed 3x with ice-cold acidic stripping buffer (50 mM glycine, 150 mM NaCl, 0.2% [w/v] BSA in Hanks buffered salt solution [HBSS] [pH 4]) to remove surface-bound antibody, leaving only endocytosed antibody-receptor complexes. Cells were then fixed with 2.5% (v/v) PFA/DPBS for 15 min at RT and then incubated in permeabilization and blocking buffer (0.5% [v/v] Tween 20, 3% [w/v] BSA in DPBS) overnight at 4°C. Finally, cells were incubated with secondary anti-rabbit antibody conjugated to Alexa Fluor 488 (1:500;Thermo Fisher Scientific) or Alexa Fluor 594 (Thermo Fisher Scientific, 1:500) and Hoechst 33342 (1:2,000;Thermo Fisher Scientific) in permeabilization and blocking buffer for 1 h. After washing the cells 2x in DPBS, they were stored at 4°C until acquisition with a Cytation 5 microplate reader (Biotek). Fluorescence intensity quantification was performed using Fiji and signal intensity per cell was calculated as follows: (fluorescence signal x number of endosomes)/number of nuclei, reflecting the amount of endocytosed receptor per cell.
Measurement of total cellular ATP levels
Total cellular ATP levels were evaluated through a luminescence-based assay. HCT116 cells were seeded in a white 96-well plate at a density of 4 × 104 cells/well and incubated for 48 h hours. Infection with L. monocytogenes EGDe, MOI 20, 1 h, was performed as described above. As controls, cells were treated with 0.1% Triton X-100 for 30 min. Cells were processed with the ATPlite kit (Perkin-Elmer) according to the manufacturer’s instructions and luminescence was recorded in a Cytation 5 microplate reader (Biotek).
Cell death analysis
For cell death analysis, cells were seeded in 6-well plates two days prior to the assay. Infections were carried out with GFP-expressing L. monocytogenes EGDe as described in the cell infection section. To collect both live and dead cells, the supernatant of the cells (medium) and of the DPBS used for washing were set aside. Adherent cells were detached with EGTA/EDTA/DPBS and pooled with supernatant and washing DPBS. As a positive control for cell death, cells were heat-shocked for 10 min at 60°C. The samples were then centrifuged at 1,500 rpm, 4°C for 5 min, washed with DPBS and centrifuged again. Dead cells were stained by resuspending the cell pellet in the LIVE/DEAD Fixable Red Dead Cell Staining dye (Thermo Fisher Scientific, diluted 1:300 in 0.5% [w/v] bovine serum albumin [BSA]/DPBS) and incubating it for 30 min in the dark, at 4°C. After centrifugation at 2,000 rpm, 4°C for 5 min and washing 2x with 0.5% BSA/DPBS, the cells were then fixed in 4% (v/v) PFA/DPBS and again washed 2x with DBPS. Cells were resuspended in 0.5% BSA/DPBS, filtered through a 35 μm cell strainer (Corning) and if applicable stored at 4°C until FACS analysis (maximal overnight). Samples were run on a CytoFLEX S flow cytometer (Beckman Coulter) and analyzed on FlowJo v10 (BD Biosciences).
Mitochondrial morphology imaging
Cells were seeded in CellCarrier-96well Ultra microplate (Perkin Elmer) at a density of 5 × 104 cells/well and incubated for four days in growth media prior to bacterial infection. Fluorescent labeling of mitochondria was achieved using MitoTracker Deep Red (Thermo Fisher Scientific) at 100 nM for 30 min at 37°C. Nuclei were labeled with NucBlue Live ReadyProbes Reagent (Thermo Fisher Scientific). Depending on the experiments, cells were either (mock) treated with 6 nM recombinant LLO (in serum-free medium) or infected with GFP-expressing L. monocytogenes EGD (MOI 20, 1 h incubation prior to gentamicin addition, described in the “Cell infection” section). After addition of the LLO- or gentamicin-containing medium, images were acquired using the Operetta CLS High-Content Analysis system (Perkin Elmer), with 40x Air/0.6 NA or 63x Water/1.15 NA. GFP-expressing L. monocytogenes (460-490 nm), MitoTracker Deep Red (615-645nm) and NucBlue (355-385 nm) were excited the appropriate LEDs (Operetta CLS). Images were acquired using Harmony 4.9 (Perkin Elmer).
Statistical analyses
Details regarding the specific statistical tests, dispersion and precision measures (e.g., standard deviation, standard error of the mean), and number of replicates (n) are indicated for each experiment in the respective figure legends. Statistical analyses were performed in Prism 8 (GraphPad Software) and statistical significance between groups was determined using the t test or one-way ANOVA with multiple comparisons or nonparametric tests. P values were considered statistically significant at p ≤ 0.05. Significance levels are indicated as follows: ns, not significant p ≥ 0.05; ∗, p ≤ 0.05; ∗∗, p ≤ 0.01; ∗∗∗, p ≤ 0.001. All experiments (excluding 13C isotopologue profiling) were performed at least two to three times.
Acknowledgments
We dedicate this work to the memory of Fabrizia Stavru. We thank Frédéric Bouillaud, Anne Lombès, and Nathalie Sauvonnet for useful advice, discussions, and support with Seahorse and endocytosis assays; Nam Tham for technical support; Werner Goebel for insightful discussions; Pierre-Henri Commere and members of the Cytometry & Biomarkers facility (CB_UTechS) at Institut Pasteur for training and help with flow cytometry and the Seahorse analyzer; and Alessandro Pagliuso and Simonetta Gribaldo for critical reading of the manuscript. Other bacterial species were GFP-expressing Salmonella enterica serovar Typhimurium strain 12023 (BUG2562) (a gift from Stéphane Méresse, Centre d’Immunologie de Marseille-Luminy, France) and Shigella flexneri M09T (BUG 2505) (a gift from Phillipe Sansonetti,Unité de Pathogénie Microbienne Moléculaire, Institut Pasteur, Paris). We thank Dr. Matthew Lawrenz at the University of Louisville for sharing the Rab11bCA construct generated by M.G.C. while in his laboratory. This study was supported by the European Research Council (H2020-ERC-2014-ADG 670823-BacCellEpi to P.C.), the DFG (Project-ID EI 384/16-1 to W.E.), Institut Pasteur, and the Centre National de la Recherche Scientifique (CNRS). A.S. was supported by a BioSPC doctoral fellowship from the Université de Paris. M.G.C. is funded by a Pasteur Foundation Fellowship and his work was funded by ANR JCJC grant “Epibactin.”
Author contributions
Conceived and designed the experiments, A.S., T.S., M.G.C., F.C., W.E., T.W., and F.S.; performed the experiments, A.S., T.S., M.G.C., and F.C.; analyzed data, A.S., T.S., M.G.C., W.E., T.W., P.C., and F.S.; wrote the manuscript, A.S., T.S., and F.S.; all authors read and commented the manuscript; supervised the project, F.S.; provided funding, P.C., W.E., and F.S.
Declaration of interests
The authors declare no competing interests.
Published: November 9, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109989.
Contributor Information
Pascale Cossart, Email: pascale.cossart@pasteur.fr.
Timothy Wai, Email: timothy.wai@pasteur.fr.
Supplemental information
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. GC-MS data have been deposited (doi: 10.11922/sciencedb.997) and are publicly available. The accession number is listed in the Key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. This paper does not report original code.
References
- Aguer C., Gambarotta D., Mailloux R.J., Moffat C., Dent R., McPherson R., Harper M.E. Galactose enhances oxidative metabolism and reveals mitochondrial dysfunction in human primary muscle cells. PLoS One. 2011;6:e28536. doi: 10.1371/journal.pone.0028536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Zubeidi D., Thangarajh M., Pathak S., Cai C., Schlaggar B.L., Storch G.A., Grange D.K., Watson M.E., Jr. Fatal human herpesvirus 6-associated encephalitis in two boys with underlying POLG mitochondrial disorders. Pediatr. Neurol. 2014;51:448–452. doi: 10.1016/j.pediatrneurol.2014.04.006. [DOI] [PubMed] [Google Scholar]
- Alston C.L., Rocha M.C., Lax N.Z., Turnbull D.M., Taylor R.W. The genetics and pathology of mitochondrial disease. J. Pathol. 2017;241:236–250. doi: 10.1002/path.4809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benthani F.A., Herrmann D., Tran P.N., Pangon L., Lucas M.C., Allam A.H., Currey N., Al-Sohaily S., Giry-Laterriere M., Warusavitarne J., et al. “MCC” protein interacts with E-cadherin and β-catenin strengthening cell-cell adhesion of HCT116 colon cancer cells. Oncogene. 2018;37:663–672. doi: 10.1038/onc.2017.362. [DOI] [PubMed] [Google Scholar]
- Bonazzi M., Veiga E., Pizarro-Cerdá J., Cossart P. Successive post-translational modifications of E-cadherin are required for InlA-mediated internalization of Listeria monocytogenes. Cell. Microbiol. 2008;10:2208–2222. doi: 10.1111/j.1462-5822.2008.01200.x. [DOI] [PubMed] [Google Scholar]
- Bradley C.A., Dunne P.D., Bingham V., McQuaid S., Khawaja H., Craig S., James J., Moore W.L., McArt D.G., Lawler M., et al. Transcriptional upregulation of c-MET is associated with invasion and tumor budding in colorectal cancer. Oncotarget. 2016;7:78932–78945. doi: 10.18632/oncotarget.12933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cairns R.A., Harris I.S., Mak T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer. 2011;11:85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- Carvalho F., Spier A., Chaze T., Matondo M., Cossart P., Stavru F. Listeria monocytogenes exploits mitochondrial contact site and cristae organizing system complex subunit Mic10 to promote mitochondrial fragmentation and cellular infection. mBio. 2020;11:e03171-19. doi: 10.1128/mBio.03171-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chico-Calero I., Suárez M., González-Zorn B., Scortti M., Slaghuis J., Goebel W., Vázquez-Boland J.A., European Listeria Genome Consortium Hpt, a bacterial homolog of the microsomal glucose- 6-phosphate translocase, mediates rapid intracellular proliferation in Listeria. Proc. Natl. Acad. Sci. USA. 2002;99:431–436. doi: 10.1073/pnas.012363899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury S.R., Reimer A., Sharan M., Kozjak-Pavlovic V., Eulalio A., Prusty B.K., Fraunholz M., Karunakaran K., Rudel T. Chlamydia preserves the mitochondrial network necessary for replication via microRNA-dependent inhibition of fission. J. Cell Biol. 2017;216:1071–1089. doi: 10.1083/jcb.201608063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor M.G., Pulsifer A.R., Chung D., Rouchka E.C., Ceresa B.K., Lawrenz M.B. Yersinia pestis targets the host endosome recycling pathway during the biogenesis of the Yersinia-containing vacuole to avoid killing by macrophages. mBio. 2018;9:1–19. doi: 10.1128/mBio.01800-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart P., Sansonetti P.J. Bacterial invasion: the paradigms of enteroinvasive pathogens. Science. 2004;304:242–248. doi: 10.1126/science.1090124. [DOI] [PubMed] [Google Scholar]
- Da-Rè C., von Stockum S., Biscontin A., Millino C., Cisotto P., Zordan M.A., Zeviani M., Bernardi P., De Pittà C., Costa R. Leigh syndrome in Drosophila melanogaster: morphological and biochemical characterization of Surf1 post-transcriptional silencing. J. Biol. Chem. 2014;289:29235–29246. doi: 10.1074/jbc.M114.602938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darin N., Oldfors A., Moslemi A.R., Holme E., Tulinius M. The incidence of mitochondrial encephalomyopathies in childhood: clinical features and morphological, biochemical, and DNA abnormalities. Ann. Neurol. 2001;49:377–383. [PubMed] [Google Scholar]
- de Noordhout C.M., Devleesschauwer B., Angulo F.J., Verbeke G., Haagsma J., Kirk M., Havelaar A., Speybroeck N. The global burden of listeriosis: a systematic review and meta-analysis. Lancet Infect. Dis. 2014;14:1073–1082. doi: 10.1016/S1473-3099(14)70870-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell’agnello C., Leo S., Agostino A., Szabadkai G., Tiveron C., Zulian A., Prelle A., Roubertoux P., Rizzuto R., Zeviani M. Increased longevity and refractoriness to Ca(2+)-dependent neurodegeneration in Surf1 knockout mice. Hum. Mol. Genet. 2007;16:431–444. doi: 10.1093/hmg/ddl477. [DOI] [PubMed] [Google Scholar]
- Dowd G.C., Mortuza R., Bhalla M., Van Ngo H., Li Y., Rigano L.A., Ireton K. Listeria monocytogenes exploits host exocytosis to promote cell-to-cell spread. Proc. Natl. Acad. Sci. USA. 2020;117:3789–3796. doi: 10.1073/pnas.1916676117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dramsi S., Biswas I., Maguin E., Braun L., Mastroeni P., Cossart P. Entry of Listeria monocytogenes into hepatocytes requires expression of inIB, a surface protein of the internalin multigene family. Mol. Microbiol. 1995;16:251–261. doi: 10.1111/j.1365-2958.1995.tb02297.x. [DOI] [PubMed] [Google Scholar]
- Dramsi S., Lévi S., Triller A., Cossart P. Entry of Listeria monocytogenes into neurons occurs by cell-to-cell spread: an in vitro study. Infect. Immun. 1998;66:4461–4468. doi: 10.1128/iai.66.9.4461-4468.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagle H., Barban S., Levy M., Schulze H.O. The utilization of carbohydrates by human cell cultures. J. Biol. Chem. 1958;233:551–558. [PubMed] [Google Scholar]
- Edmonds J.L., Jr. Surgical and anesthetic management of patients with mitochondrial dysfunction. Mitochondrion. 2004;4:543–548. doi: 10.1016/j.mito.2004.07.012. [DOI] [PubMed] [Google Scholar]
- Edmonds J.L., Kirse D.J., Kearns D., Deutsch R., Spruijt L., Naviaux R.K. The otolaryngological manifestations of mitochondrial disease and the risk of neurodegeneration with infection. Arch. Otolaryngol. Head Neck Surg. 2002;128:355–362. doi: 10.1001/archotol.128.4.355. [DOI] [PubMed] [Google Scholar]
- Eisenreich W., Heesemann J., Rudel T., Goebel W. Metabolic adaptations of intracellullar bacterial pathogens and their mammalian host cells during infection (“pathometabolism”) Microbiol. Spectr. 2015;3 doi: 10.1128/microbiolspec.MBP-0002-2014. [DOI] [PubMed] [Google Scholar]
- Escoll P., Buchrieser C. Metabolic reprogramming of host cells upon bacterial infection: Why shift to a Warburg-like metabolism? FEBS J. 2018;285:2146–2160. doi: 10.1111/febs.14446. [DOI] [PubMed] [Google Scholar]
- Escoll P., Song O.R., Viana F., Steiner B., Lagache T., Olivo-Marin J.C., Impens F., Brodin P., Hilbi H., Buchrieser C. Legionella pneumophila modulates mitochondrial dynamics to trigger metabolic repurposing of infected macrophages. Cell Host Microbe. 2017;22:302–316.e7. doi: 10.1016/j.chom.2017.07.020. [DOI] [PubMed] [Google Scholar]
- Eylert E., Schär J., Mertins S., Stoll R., Bacher A., Goebel W., Eisenreich W. Carbon metabolism of Listeria monocytogenes growing inside macrophages. Mol. Microbiol. 2008;69:1008–1017. doi: 10.1111/j.1365-2958.2008.06337.x. [DOI] [PubMed] [Google Scholar]
- Ferro E., Bosia C., Campa C.C. RAB11-mediated trafficking and human cancers: an updated review. Biology (Basel) 2021;10:26. doi: 10.3390/biology10010026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francione L., Smith P.K., Accari S.L., Taylor P.E., Bokko P.B., Bozzaro S., Beech P.L., Fisher P.R. Legionella pneumophila multiplication is enhanced by chronic AMPK signalling in mitochondrially diseased Dictyostelium cells. Dis. Model. Mech. 2009;2:479–489. doi: 10.1242/dmm.003319. [DOI] [PubMed] [Google Scholar]
- Frey P.A. The Leloir pathway: a mechanistic imperative for three enzymes to change the stereochemical configuration of a single carbon in galactose. FASEB J. 1996;10:461–470. [PubMed] [Google Scholar]
- Garaude J., Acín-Pérez R., Martínez-Cano S., Enamorado M., Ugolini M., Nistal-Villán E., Hervás-Stubbs S., Pelegrín P., Sander L.E., Enríquez J.A., Sancho D. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat. Immunol. 2016;17:1037–1045. doi: 10.1038/ni.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudó P., Emperador S., Garrido-Pérez N., Ruiz-Pesini E., Yubero D., García-Cazorla A., Artuch R., Montoya J., Bayona-Bafaluy M.P. Infectious stress triggers a POLG-related mitochondrial disease. Neurogenetics. 2020;21:19–27. doi: 10.1007/s10048-019-00593-2. [DOI] [PubMed] [Google Scholar]
- Gillmaier N., Götz A., Schulz A., Eisenreich W., Goebel W. Metabolic responses of primary and transformed cells to intracellular Listeria monocytogenes. PLoS One. 2012;7:e52378. doi: 10.1371/journal.pone.0052378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleeson L.E., Sheedy F.J., Palsson-McDermott E.M., Triglia D., O’Leary S.M., O’Sullivan M.P., O’Neill L.A., Keane J. Cutting edge: Mycobacterium tuberculosis induces aerobic glycolysis in human alveolar macrophages that is required for control of intracellular bacillary replication. J. Immunol. 2016;196:2444–2449. doi: 10.4049/jimmunol.1501612. [DOI] [PubMed] [Google Scholar]
- Gorman G.S., Schaefer A.M., Ng Y., Gomez N., Blakely E.L., Alston C.L., Feeney C., Horvath R., Yu-Wai-Man P., Chinnery P.F., et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease. Ann. Neurol. 2015;77:753–759. doi: 10.1002/ana.24362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorman G.S., Chinnery P.F., DiMauro S., Hirano M., Koga Y., McFarland R., Suomalainen A., Thorburn D.R., Zeviani M., Turnbull D.M. Mitochondrial diseases. Nat. Rev. Dis. Primers. 2016;2:16080. doi: 10.1038/nrdp.2016.80. [DOI] [PubMed] [Google Scholar]
- Grant B.D., Donaldson J.G. Pathways and mechanisms of endocytic recycling. Nat. Rev. Mol. Cell Biol. 2009;10:597–608. doi: 10.1038/nrm2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubmüller S., Schauer K., Goebel W., Fuchs T.M., Eisenreich W. Analysis of carbon substrates used by Listeria monocytogenes during growth in J774A.1 macrophages suggests a bipartite intracellular metabolism. Front. Cell. Infect. Microbiol. 2014;4:156. doi: 10.3389/fcimb.2014.00156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann D.W., Fine M., Linder M.E., Jennings B.C., Lin M.J. Massive endocytosis triggered by surface membrane palmitoylation under mitochondrial control in BHK fibroblasts. eLife. 2013;2:e01293. doi: 10.7554/eLife.01293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong J.-Y., Yim H.S., Ryu J.Y., Lee H.S., Lee J.H., Seen D.S., Kang S.G. One-step sequence- and ligation-independent cloning as a rapid and versatile cloning method for functional genomics studies. Appl. Environ. Microbiol. 2012;78:5440–5443. doi: 10.1128/AEM.00844-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph B., Mertins S., Stoll R., Schär J., Umesha K.R., Luo Q., Müller-Altrock S., Goebel W. Glycerol metabolism and PrfA activity in Listeria monocytogenes. J. Bacteriol. 2008;190:5412–5430. doi: 10.1128/JB.00259-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Käding N., Kaufhold I., Müller C., Szaszák M., Shima K., Weinmaier T., Lomas R., Conesa A., Schmitt-Kopplin P., Rattei T., Rupp J. Growth of Chlamydia pneumoniae is enhanced in cells with impaired mitochondrial function. Front. Cell. Infect. Microbiol. 2017;7:499. doi: 10.3389/fcimb.2017.00499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalghatgi S., Spina C.S., Costello J.C., Liesa M., Morones-Ramirez J.R., Slomovic S., Molina A., Shirihai O.S., Collins J.J. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in Mammalian cells. Sci. Transl. Med. 2013;5:192ra85. doi: 10.1126/scitranslmed.3006055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M., et al. Live imaging reveals Listeria hijacking of E-cadherin recycling as it crosses the intestinal barrier. Curr. Biol. 2020;31:1–11. doi: 10.1016/j.cub.2020.11.041. [DOI] [PubMed] [Google Scholar]
- Kovářová N., Cížková Vrbacká A., Pecina P., Stránecký V., Pronicka E., Kmoch S., Houštěk J. Adaptation of respiratory chain biogenesis to cytochrome c oxidase deficiency caused by SURF1 gene mutations. Biochim. Biophys. Acta. 2012;1822:1114–1124. doi: 10.1016/j.bbadis.2012.03.007. [DOI] [PubMed] [Google Scholar]
- Kowarz E., Löscher D., Marschalek R. Optimized Sleeping Beauty transposons rapidly generate stable transgenic cell lines. Biotechnol. J. 2015;10:647–653. doi: 10.1002/biot.201400821. [DOI] [PubMed] [Google Scholar]
- Kühbacher A., Cossart P., Pizarro-Cerdá J. Listeria monocytogenes. Springer Nature; 2014. Internalization assays for Listeria monocytogenes; pp. 167–178. [DOI] [PubMed] [Google Scholar]
- Kühbacher A., Emmenlauer M., Rämo P., Kafai N., Dehio C., Cossart P., Pizarro-Cerdá J. Genome-wide siRNA screen identifies complementary signaling pathways involved in Listeria infection and reveals different actin nucleation mechanisms during Listeria cell invasion and actin comet tail formation. mBio. 2015;6:e00598-15. doi: 10.1128/mBio.00598-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachmandas E., Beigier-Bompadre M., Cheng S.C., Kumar V., van Laarhoven A., Wang X., Ammerdorffer A., Boutens L., de Jong D., Kanneganti T.D., et al. Rewiring cellular metabolism via the AKT/mTOR pathway contributes to host defence against Mycobacterium tuberculosis in human and murine cells. Eur. J. Immunol. 2016;46:2574–2586. doi: 10.1002/eji.201546259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lake N.J., Compton A.G., Rahman S., Thorburn D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016;79:190–203. doi: 10.1002/ana.24551. [DOI] [PubMed] [Google Scholar]
- Landry M.-C., Champagne C., Boulanger M.C., Jetté A., Fuchs M., Dziengelewski C., Lavoie J.N. A functional interplay between the small GTPase Rab11a and mitochondria-shaping proteins regulates mitochondrial positioning and polarization of the actin cytoskeleton downstream of Src family kinases. J. Biol. Chem. 2014;289:2230–2249. doi: 10.1074/jbc.M113.516351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebreton A., Stavru F., Cossart P. Organelle targeting during bacterial infection: insights from Listeria. Trends Cell Biol. 2015;25:330–338. doi: 10.1016/j.tcb.2015.01.003. [DOI] [PubMed] [Google Scholar]
- Lee W.-N.P., Byerley L.O., Bergner E.A., Edmond J. Mass isotopomer analysis: theoretical and practical considerations. Biol. Mass Spectrom. 1991;20:451–458. doi: 10.1002/bms.1200200804. [DOI] [PubMed] [Google Scholar]
- Lee I.-C., El-Hattab A.W., Wang J., Li F.Y., Weng S.W., Craigen W.J., Wong L.J. SURF1-associated Leigh syndrome: a case series and novel mutations. Hum. Mutat. 2012;33:1192–1200. doi: 10.1002/humu.22095. [DOI] [PubMed] [Google Scholar]
- Mátés L., Chuah M.K., Belay E., Jerchow B., Manoj N., Acosta-Sanchez A., Grzela D.P., Schmitt A., Becker K., Matrai J., et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat. Genet. 2009;41:753–761. doi: 10.1038/ng.343. [DOI] [PubMed] [Google Scholar]
- Mayle K.M., Le A.M., Kamei D.T. The intracellular trafficking pathway of transferrin. Biochim. Biophys. Acta. 2012;1820:264–281. doi: 10.1016/j.bbagen.2011.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee D.H., Cooper P.N., Denning D.W. Invasive aspergillosis in a patient with MELAS syndrome. J. Neurol. Neurosurg. Psychiatry. 2000;68:765–767. doi: 10.1136/jnnp.68.6.765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengaud J., Ohayon H., Gounon P., Mege R-M, Cossart P. E-cadherin is the receptor for internalin, a surface protein required for entry of L. monocytogenes into epithelial cells. Cell. 1996;84:923–932. doi: 10.1016/s0092-8674(00)81070-3. [DOI] [PubMed] [Google Scholar]
- Morgan-Hughes J.A. Mitochondrial diseases. Trends Neurosci. 1986;9:15–19. [Google Scholar]
- Newsholme P., Gordon S., Newsholme E.A. Rates of utilization and fates of glucose, glutamine, pyruvate, fatty acids and ketone bodies by mouse macrophages. Biochem. J. 1987;242:631–636. doi: 10.1042/bj2420631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng Y.S., Bindoff L.A., Gorman G.S., Klopstock T., Kornblum C., Mancuso M., McFarland R., Sue C.M., Suomalainen A., Taylor R.W., Thorburn D.R., Turnbull D.M. Mitochondrial disease in adults: recent advances and future promise. Lancet Neurol. 2021;20:573–584. doi: 10.1016/S1474-4422(21)00098-3. [DOI] [PubMed] [Google Scholar]
- Nichols C.D., Casanova J.E. Salmonella-directed recruitment of new membrane to invasion foci via the host exocyst complex. Curr. Biol. 2010;20:1316–1320. doi: 10.1016/j.cub.2010.05.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J., Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan M.J., Lindsay A.J. The endosomal recycling pathway—at the crossroads of the cell. Int. J. Mol. Sci. 2020;21:6074. doi: 10.3390/ijms21176074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlou S., Wang L., Xu H., Chen M. Higher phagocytic activity of thioglycollate-elicited peritoneal macrophages is related to metabolic status of the cells. J. Inflamm. (Lond.) 2017;14:4. doi: 10.1186/s12950-017-0151-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizarro-Cerdá J., Cossart P. Listeria monocytogenes: cell biology of invasion and intracellular growth. Microbiol. Spectr. 2018;6 doi: 10.1128/microbiolspec.gpp3-0013-2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock S.R., Schinlever A.R., Rohani A., Kashatus J.A., Kashatus D.F. RalA and RalB relocalization to depolarized mitochondria depends on clathrin-mediated endocytosis and facilitates TBK1 activation. PLoS One. 2019;14:e0214764. doi: 10.1371/journal.pone.0214764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rampersad S.N. Multiple applications of Alamar Blue as an indicator of metabolic function and cellular health in cell viability bioassays. Sensors (Basel) 2012;12:12347–12360. doi: 10.3390/s120912347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013;8:2281–2308. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitzer L.J., Wice B.M., Kennell D. Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J. Biol. Chem. 1979;254:2669–2676. [PubMed] [Google Scholar]
- Ren M., Xu G., Zeng J., De Lemos-Chiarandini C., Adesnik M., Sabatini D.D. Hydrolysis of GTP on rab11 is required for the direct delivery of transferrin from the pericentriolar recycling compartment to the cell surface but not from sorting endosomes. Proc. Natl. Acad. Sci. USA. 1998;95:6187–6192. doi: 10.1073/pnas.95.11.6187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson B.H., Petrova-Benedict R., Buncic J.R., Wallace D.C. Nonviability of cells with oxidative defects in galactose medium: a screening test for affected patient fibroblasts. Biochem. Med. Metab. Biol. 1992;48:122–126. doi: 10.1016/0885-4505(92)90056-5. [DOI] [PubMed] [Google Scholar]
- Rossignol R., Gilkerson R., Aggeler R., Yamagata K., Remington S.J., Capaldi R.A. Energy substrate modulates mitochondrial structure and oxidative capacity in cancer cells. Cancer Res. 2004;64:985–993. doi: 10.1158/0008-5472.can-03-1101. [DOI] [PubMed] [Google Scholar]
- Sauer J.D., Herskovits A.A., O’Riordan M.X.D. Metabolism of the Gram-positive bacterial pathogen Listeria monocytogenes. Microbiol. Spectr. 2019;7:139–148. doi: 10.1128/microbiolspec.gpp3-0066-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt-Rohr K. Oxygen is the high-energy molecule powering complex multicellular life: fundamental corrections to traditional bioenergetics. ACS Omega. 2020;5:2221–2233. doi: 10.1021/acsomega.9b03352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y., Naujokas M., Park M., Ireton K. InIB-dependent internalization of Listeria is mediated by the Met receptor tyrosine kinase. Cell. 2000;103:501–510. doi: 10.1016/s0092-8674(00)00141-0. [DOI] [PubMed] [Google Scholar]
- Sousa S., Cabanes D., Bougnères L., Lecuit M., Sansonetti P., Tran-Van-Nhieu G., Cossart P. Src, cortactin and Arp2/3 complex are required for E-cadherin-mediated internalization of Listeria into cells. Cell. Microbiol. 2007;9:2629–2643. doi: 10.1111/j.1462-5822.2007.00984.x. [DOI] [PubMed] [Google Scholar]
- Spier A., Stavru F., Cossart P. Interaction between intracellular bacterial pathogens and host cell mitochondria. Microbiol. Spectr. 2019;7 doi: 10.1128/microbiolspec.BAI-0016-2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavru F., Bouillaud F., Sartori A., Ricquier D., Cossart P. Listeria monocytogenes transiently alters mitochondrial dynamics during infection. Proc. Natl. Acad. Sci. USA. 2011;108:3612–3617. doi: 10.1073/pnas.1100126108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stavru F., Palmer A.E., Wang C., Youle R.J., Cossart P. Atypical mitochondrial fission upon bacterial infection. Proc. Natl. Acad. Sci. USA. 2013;110:16003–16008. doi: 10.1073/pnas.1315784110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiku V., Tan M.-W., Dikic I. Mitochondrial functions in infection and immunity. Trends Cell Biol. 2020;30:263–275. doi: 10.1016/j.tcb.2020.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiranti V., Hoertnagel K., Carrozzo R., Galimberti C., Munaro M., Granatiero M., Zelante L., Gasparini P., Marzella R., Rocchi M., et al. Mutations of SURF-1 in Leigh disease associated with cytochrome c oxidase deficiency. Am. J. Hum. Genet. 1998;63:1609–1621. doi: 10.1086/302150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiranti V., Galimberti C., Nijtmans L., Bovolenta S., Perini M.P., Zeviani M. Characterization of SURF-1 expression and Surf-1p function in normal and disease conditions. Hum. Mol. Genet. 1999;8:2533–2540. doi: 10.1093/hmg/8.13.2533. [DOI] [PubMed] [Google Scholar]
- Ullrich O., Reinsch S., Urbé S., Zerial M., Parton R.G. Rab11 regulates recycling through the pericentriolar recycling endosome. J. Cell Biol. 1996;135:913–924. doi: 10.1083/jcb.135.4.913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Langendonck N., Velge P., Bottreau E. Host cell protein tyrosine kinases are activated during the entry of Listeria monocytogenes. Possible role of pp60c-src family protein kinases. FEMS Microbiol. Lett. 1998;162:169–176. doi: 10.1111/j.1574-6968.1998.tb12995.x. [DOI] [PubMed] [Google Scholar]
- Varghese M., Cafferkey M., O’Regan M., Monavari A.A., Treacy E.P. Should children with inherited metabolic disorders receive varicella vaccination? Arch. Dis. Child. 2011;96:99–100. doi: 10.1136/adc.2008.147934. [DOI] [PubMed] [Google Scholar]
- Veiga E., Cossart P. Listeria hijacks the clathrin-dependent endocytic machinery to invade mammalian cells. Nat. Cell Biol. 2005;7:894–900. doi: 10.1038/ncb1292. [DOI] [PubMed] [Google Scholar]
- Velge P., Bottreau E., Van-Langendonck N., Kaeffer B. Cell proliferation enhances entry of Listeria monocytogenes into intestinal epithelial cells by two proliferation-dependent entry pathways. J. Med. Microbiol. 1997;46:681–692. doi: 10.1099/00222615-46-8-681. [DOI] [PubMed] [Google Scholar]
- Venter G., Oerlemans F.T., Wijers M., Willemse M., Fransen J.A., Wieringa B. Glucose controls morphodynamics of LPS-stimulated macrophages. PLoS One. 2014;9:e96786. doi: 10.1371/journal.pone.0096786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker M.A., Slate N., Alejos A., Volpi S., Iyengar R.S., Sweetser D., Sims K.B., Walter J.E. Predisposition to infection and SIRS in mitochondrial disorders: 8 years’ experience in an academic center. J. Allergy Clin. Immunol. Pract. 2014;2:465–468. doi: 10.1016/j.jaip.2014.02.009. [DOI] [PubMed] [Google Scholar]
- Wedatilake Y., Brown R.M., McFarland R., Yaplito-Lee J., Morris A.A., Champion M., Jardine P.E., Clarke A., Thorburn D.R., Taylor R.W., et al. SURF1 deficiency: a multi-centre natural history study. Orphanet J. Rare Dis. 2013;8:96. doi: 10.1186/1750-1172-8-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiner A., Mellouk N., Lopez-Montero N., Chang Y.Y., Souque C., Schmitt C., Enninga J. Macropinosomes are key players in early Shigella invasion and vacuolar escape in epithelial cells. PLoS Pathog. 2016;12:e1005602. doi: 10.1371/journal.ppat.1005602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z., Yao J., Johns T., Fu K., De Bie I., Macmillan C., Cuthbert A.P., Newbold R.F., Wang J., Chevrette M., et al. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat. Genet. 1998;20:337–343. doi: 10.1038/3804. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. GC-MS data have been deposited (doi: 10.11922/sciencedb.997) and are publicly available. The accession number is listed in the Key resources table. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request. This paper does not report original code.