

Abstract
Recognition of distinct phenotypic features is an important component of genetic diagnosis. Although CHARGE syndrome, Kabuki syndrome, and a recently delineated KMT2D Ex 38/39 allelic disorder exhibit significant overlap, differences on neuroimaging may help distinguish these conditions and guide genetic testing and variant interpretation. We present an infant clinically diagnosed with CHARGE syndrome but subsequently found to have a de novo missense variant in exon 38 of KMT2D, the gene implicated in both Kabuki syndrome and a distinct KMT2D allelic disorder. We compare her brain and inner ear morphology to a retrospective cohort of 21 patients with classic Kabuki syndrome and to typical CHARGE syndrome findings described in the literature. Thirteen of the 21 Kabuki syndrome patients had temporal bone imaging (5/13 CT, 12/13 MRI) and/or brain MRI (12/13) which revealed findings distinct from both CHARGE syndrome and the KMT2D allelic disorder. Our findings further elucidate the spectrum of inner ear dysmorphology distinguishing Kabuki syndrome and the KMT2D allelic disorder from CHARGE syndrome, suggesting that these three disorders may be differentiated at least in part by their inner ear anomalies.
Keywords: KMT2D, Kabuki syndrome, CHARGE syndrome, temporal bone
INTRODUCTION:
As many genetic disorders share overlapping features, phenotype delineation is crucial to guiding genetic testing and interpretation of results. For example, CHARGE syndrome, Kabuki syndrome, and a KMT2D allelic disorder distinct from Kabuki syndrome and described in two recent publications (Baldridge et al., 2020; Cuvertino et al., 2020) exhibit significant clinical overlap (Butcher et al., 2017; Verhagen et al., 2014), and distinguishing between them may be difficult, particularly since Kabuki syndrome and the KMT2D Ex 38/39 disorder are both caused by variants in KMT2D. However, the KMT2D Ex 38/39 disorder is caused by missense variants in a specific, highly conserved region spanning exons 38 and 39 and leads to features distinct from Kabuki syndrome. Since a number of variants in this region have been associated with a similar phenotype, there appears to be allelic heterogeneity and thus going forward we will refer to this disorder as the KMT2D Ex 38/39 allelic disorder.
CHARGE syndrome is well characterized by its mnemonic CHARGE (C – coloboma, H – heart disease, A – atresia choanae, R – retarded growth and development and/or CNS anomalies, G – genital hypoplasia, and E – ear anomalies and/or deafness) representing the unifying features (Abruzzo & Erickson, 1977; Pagon et al., 1981). Disease-causing variants in CHD7 have been found in approximately 70–90% of clinically diagnosed cases of CHARGE syndrome (Hale et al., 2016). As such, CHD7 sequence analysis can confirm a diagnosis suspected on the basis of clinical features in the majority of cases. Kabuki syndrome is characterized by classic clinical findings of distinct facial features, skeletal anomalies, persistence of fetal fingertip pads, intellectual disability, and postnatal growth deficiency (Niikawa et al., 1981). In 2010 it was reported to be associated with pathogenic variants in KMT2D/MLL2 and KDM6A (Lederer et al., 2012; Ng et al., 2010) which have been found to be present in approximately 55–80% and 9–14%, respectively, of patients meeting clinical diagnostic criteria (Dentici et al., 2015).
Overlapping features of CHARGE syndrome and Kabuki syndrome include growth retardation, cleft lip/palate, hearing loss, congenital heart defects, genitourinary malformations, and developmental delay. Even features traditionally thought to be specific to CHARGE syndrome, such as ocular coloboma and choanal atresia, have also been described in Kabuki syndrome, and there are a number of reports of patients with overlapping features who exhibited features of CHARGE syndrome but were found to have a molecular diagnosis of Kabuki syndrome (Patel & Alkuraya, 2015; Schulz et al., 2014; Verhagen et al., 2014). However, it was recently discovered that some patients with overlapping phenotypic features but a known variant in KMT2D who were therefore thought to have a molecular diagnosis of Kabuki syndrome actually have a different disorder altogether. Unlike KMT2D variants leading to Kabuki syndrome, which are typically nonsense variants that subsequently lead to protein truncation and resultant haploinsufficiency (Faundes et al., 2019), this cluster of heterozygous KMT2D variants in a highly conserved key region of exons 38–39 were all missense and led to a distinct methylation pattern and therefore an epigenetically distinct disorder (Baldridge et al., 2020; Cuvertino et al., 2020). These patients’ unique clinical features were most notable for choanal atresia, hypoplastic nipples, branchial apparatus abnormalities, neck pits, lacrimal duct anomalies, hearing loss, external ear malformations, and thyroid abnormalities. One group also noted the presence of interstitial lung disease in 2 out of their 3 patients, which had not been previously reported (Baldridge et al., 2020). This constellation of features overlaps with both CHARGE syndrome and Kabuki syndrome but is distinct from both.
One particularly interesting aspect of the clinical overlap between CHARGE syndrome, Kabuki syndrome, and the KMT2D Ex 38/39 allelic disorder that has not yet been well described is the presence of inner ear anomalies. Although inner ear anomalies have been well described in CHARGE syndrome and include hypoplasia of the vestibule, semicircular canal (SCC) hypoplasia/aplasia, incomplete partition type II deformity of the cochlea, other cochlear malformations with stenosis or atresia of the cochlear aperture, cochlear nerve hypoplasia/aplasia, and a small internal auditory canal (D’Arco et al., 2020; Legendre et al., 2017; Morimoto et al., 2006; A. C. Vesseur et al., 2016; Wineland et al., 2017), unifying findings in Kabuki syndrome and the KMT2D Ex 38/39 allelic disorder have not been thoroughly described in a unifying manner despite some case reports of abnormalities including dysplastic cochlea with an enlarged vestibule, absence of the cochlea and semicircular canals, a unilateral enlarged vestibule, an enlarged vestibular aqueduct, and aural atresia with fusion of the incus and malleus (Hempel et al., 2005; Igawa et al., 2000; Tekin et al., 2006; Toutain et al., 1997; Zarate et al., 2012). We present a female infant who met clinical criteria for CHARGE syndrome but was later found to have a missense variant in KMT2D. This patient was initially diagnosed with Kabuki syndrome, but the diagnosis was ultimately more consistent with the newly described KMT2D Ex 38/39 MV allelic disorder. In particular, we focus on her inner ear findings, which were initially misinterpreted as supportive of a CHARGE syndrome diagnosis, and contrast this to neuroimaging findings observed in Kabuki syndrome.
MATERIALS AND METHODS:
The index patient and her parents were enrolled in an Institutional Review Board-approved research study at the Manton Center for Orphan Disease Research at Boston Children’s Hospital. Exome sequencing (ES) and data processing were performed by the Genomics Platform at the Broad Institute of MIT and Harvard. Libraries from DNA samples (>250 ng of DNA, at >2 ng/ul) were created with an Illumina exome capture (38 Mb target) and sequenced (150 bp paired reads) to cover >90% of targets at 20x and a mean target coverage of >100x. Sample identity quality assurance checks were performed on each sample. The ES data was de-multiplexed and each sample’s sequence data were aggregated into a single Picard BAM file. ES data were processed through a pipeline based on Picard, using base quality score recalibration and local realignment at known indels. The BWA aligner was used for mapping reads to the human genome build 37 (hg19). Single Nucleotide Polymorphism (SNPs) and insertions/deletions (indels) were jointly called across all samples using Genome Analysis Toolkit (GATK) HaplotypeCaller package version 3.4. Default filters were applied to SNP and indel calls using the GATK Variant Quality Score Recalibration (VQSR) approach. Annotation was performed using Variant Effect Predictor (VEP).
We subsequently performed a search of our electronic medical records database from 1998–2018 to identify patients with a molecular diagnosis of Kabuki Syndrome (pathogenic or likely pathogenic variant in KMT2D or KDM6A, or a variant of unknown significance in which the treating clinical geneticist determined that the variant was explanatory for the phenotype). We reviewed the otologic phenotype, including audiograms, of these patients. Neuroimaging studies were reviewed by a pediatric neuroradiologist with over 25 years of experience with subspecialty expertise in temporal bone imaging. This retrospective study was also approved by the Institutional Review Board at Boston Children’s Hospital with a waiver of informed consent due to the nature of the study.
Helical temporal bone CT exams were acquired on 1 of 3 different CT units with parameters as follows. Axial images were reconstructed in a plane parallel to that of the horizontal semicircular canals and coronal images were reconstructed perpendicular to the plane of the horizontal semicircular canals. General Electric (GE) Lightspeed Pro16 CT scanner: effective mAs: 2–5; mA: 60 – 105; rotation time: 698–1081 ms; pitch: 0.6–0.94; kVp: 100–120; and slice thickness: 0.625 mm. Siemens Sensation 40 and 64: effective mAs: 100 – 141; mA: 55–78; rotation time: 1000 ms; pitch: 0.55; kVp120–140; slice thickness 0.6 mm.
Temporal bone MRI exams were performed on 1 of 3 different MR units with parameters as follows. GE 1.5 Tesla (T) Signa Excite using a 8 channel head coil and the axial constructive interference in steady state (CISS) sequence: repetition time (TR): 4.97; echo train length (ETL): 1; Echo time (TE): 1.66; slice thickness: 0.8 mm/0 mm gap; flip angle (FA): 65; number of excitations (NEX): 3; acquisition matrix (AQM): 256×320. Siemens 3T Tim Trio or 3T Skyra using a 32 channel head coil and axial and reconstructed or directly acquired sagittal oblique T2 spatial and chemical-shift encoded excitation (SPACE) sequences: TR: 700 −100, ETL: 46–65; TE: 126 – 133; thickness: 0.4–0.53 mm/0 mm gap; flip angle: 120 – 140; NEX: 2–4; AQM: 256–326×320. Conventional brain sequences were also obtained including sagittal T1 magnetization prepared rapid gradient echo (MPRAGE), axial turbo spin echo T2, axial diffusion weighted imaging and either axial turbo T1 (less than 1 month of age) or axial fluid attenuated inversion recovery (FLAIR) pulse sequences.
RESULTS:
Case Description
A female infant was born at 33 weeks and 6 days to a 30 year old G2P1→2 woman following a pregnancy complicated by insulin-dependent gestational diabetes mellitus, intrauterine growth restriction, and oligohydramnios. Prenatal ultrasounds were notable for a moderate membranous ventricular septal defect (VSD), mild tricuspid regurgitation, a small pericardial effusion, dilated small bowel, echogenic bowel, and small/immature kidneys with duplicated collecting systems bilaterally. There was no family history of birth defects or genetic disorders. The infant was delivered via scheduled Cesarean section due to worsening oligohydramnios and decreased fetal movements. After delivery, the infant was intubated due to respiratory distress, and she was subsequently transferred to our institution for further management. Postnatal echocardiogram demonstrated pulmonary hypertension and a small muscular VSD. A renal ultrasound demonstrated small, echogenic kidneys with small left renal cysts. Studies for genetic causes of surfactant dysfunction (SP-B, SP-C, ABCA3) and alveolar capillary dysplasia (FOXF1) were negative, and her respiratory failure was felt to be due to pulmonary hypoplasia secondary to oligohydramnios.
The newborn screen showed undetectable T-cell receptor excision circles (T-RECs) concerning for severe combined immunodeficiency (SCID), with follow-up laboratory investigation revealing severe T-cell and B-cell lymphopenia with decreased levels of naïve CD4 T-cells. Mitogen proliferation testing was consistent with severely depressed T-cell function. Immunoglobulin levels were also low, and she was treated with intravenous immunoglobulin. However, repeat evaluations throughout her admission demonstrated improvement in lymphocyte subsets and mitogen proliferation studies which were not consistent with a SCID diagnosis. In addition, a gene sequencing panel evaluating 18 genes associated with SCID was unrevealing (GeneDx, Gaithersburg, MD). The infant’s newborn screen was also notable for hypothyroidism which was confirmed on serum studies and she was subsequently started on levothyroxine.
Brain magnetic resonance imaging (MRI) at 1 month of age revealed multiple anomalies (Fig. 1), including mild hypoplasia of the pons which appeared narrowed in transverse and shortened in craniocaudad dimensions. There was a 10 × 5 mm cystic structure in the expected location of the anterior pituitary gland (possible Rathke cleft cyst) effacing the posterior pituitary gland. The right olfactory bulb was absent with an absent right olfactory sulcus. The left olfactory sulcus appeared shallow and the bulb appeared prominent. Both the MR and facial CT showed bilateral part osseous, part membranous choanal atresia (Figs. 1D and 2A). Multiple bilateral temporal bone anomalies were also seen and affected external, middle and inner ear structures (Figs. 1C and 2B–G). The right external auditory canal appeared stenotic. The malleus and incus were malformed and abnormally oriented, with deficiency of the lenticular process of the incus and absent incudostapedial joints bilaterally. Small stapedial remnants were present which appeared severely malformed and angulated, in association with oval window stenosis. The facial nerve canals could not be accurately assessed due to the low dose CT technique that was optimized for assessment of the nasal cavity. The cochlear middle turns appeared misshapen, hypoplastic and offset, with small, flattened apical turns. This appeared most consistent with cochlear hypoplasia type IV according to the revised Sennaroglu classification (Sennaroğlu & Bajin, 2017). The cochlear modioli were malformed and the cochlear apertures were stenotic, measuring less than 0.4 mm transverse. The vestibules appeared globular with small protrusions at the expected junctions with the posterior SCCs which were absent. The lateral and superior semicircular canals were fully formed with mild luminal narrowing. The basiocciput appeared normal.
Figure 1:
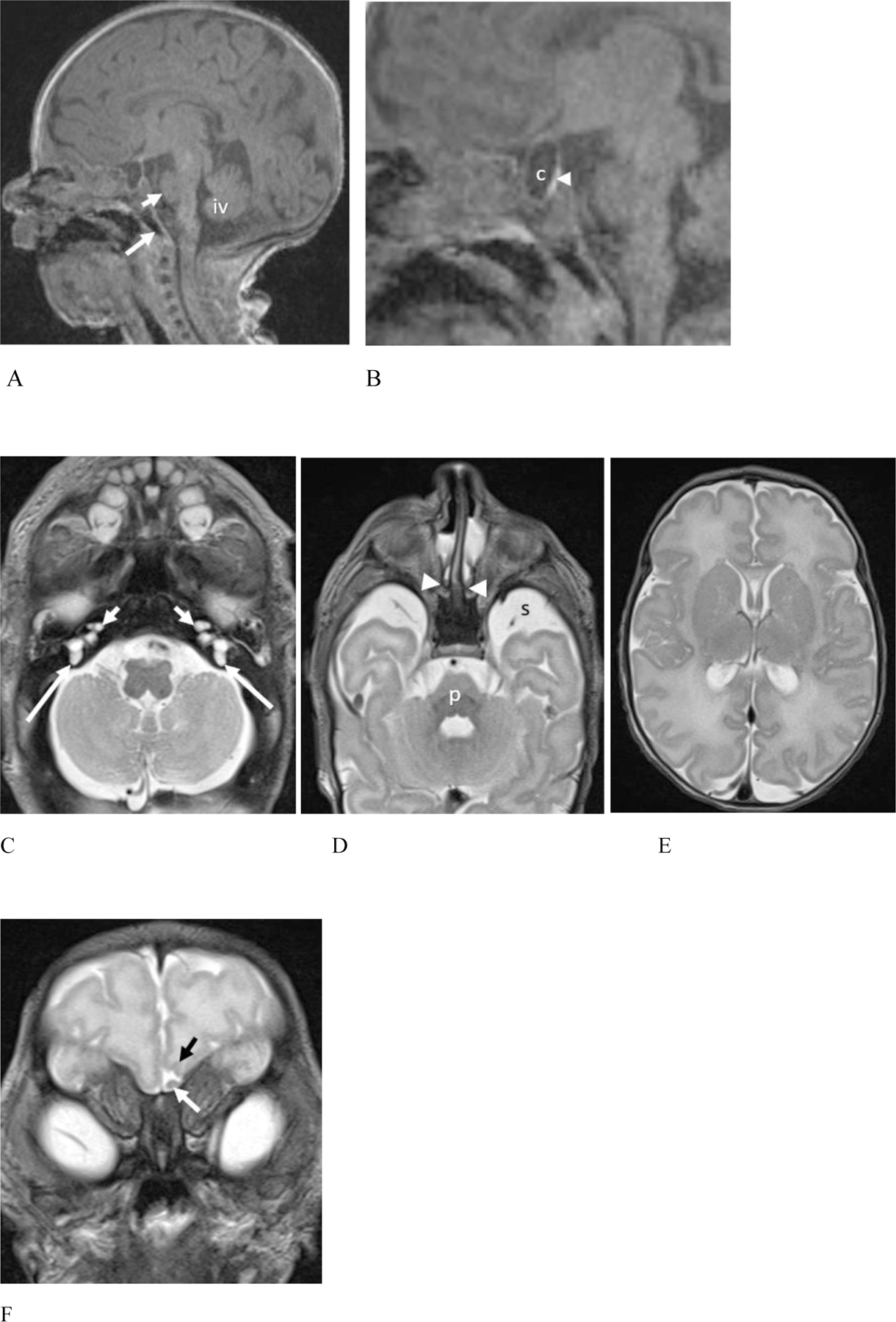
(Patient #22) 1 month old female (feed and swaddle MR examination performed without sedation). (A – B) 3T Sagittal T1 magnetization prepared rapid gradient echo (MPRAGE) MR images. (A) There is hypoplasia of the pons which is shortened in height (short white arrow). The inferior vermis (iv) is mildly uplifted but normal size. Note the normal morphology of the basiocciput (long white arrow). (B) A magnified sagittal image reveals a cyst (c) in the expected location of the anterior pituitary gland with partial effacement of the posterior pituitary gland (arrowhead).
(C – E). Axial T2 weighted MR images. (C) The cochleae appear misshapen with hypoplastic, anteriorly offset middle turns and markedly hypoplastic apical turns (short arrows). The cochlear modiolus is misshapen on the right and absent on the left. Unlike CHARGE syndrome, the vestibules are misshapen with slight protrusions at the expected take off of the posterior semicircular canals (SCCs) which are essentially absent (arrows). The lateral and superior SCC were present (not shown). (D) The pons (p) is small. At this level, the extraaxial spaces (s) ventral to the anterior temporal lobes appear prominent. Note high signal intensity fluid pooling in the nasal cavities ventral to choanal atresia (arrowheads). (E) There is a somewhat simplified gyral pattern with relatively shallow sulci, particularly in the frontal lobes, for the given corrected postnatal age of 38 weeks.
(F). Coronal T2 weighted MR image shows absence of the right olfactory bulb and sulcus. Note the mildly prominent left olfactory bulb (white arrow) and shallow left olfactory sulcus (black arrow).
Figure 2.
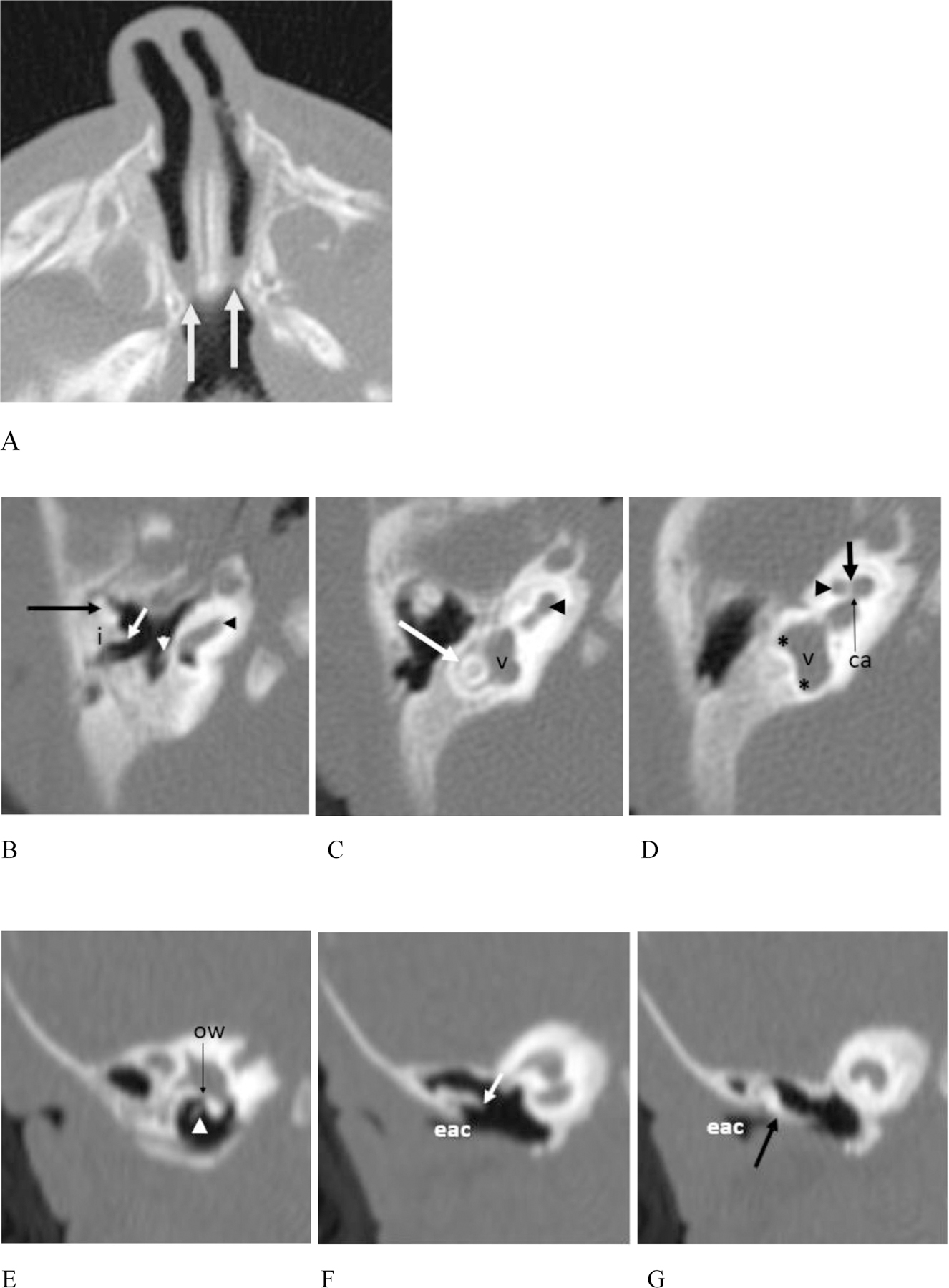
(Patient #22) (A – G). CT of the face at 1 month of age. (A) There is bilateral choanal atresia that is part osseous (medial deviation of the lateral nasal cavity walls) and part membranous (arrows).
(B) – (D). Images from the facial CT exam also demonstrate multiple temporal bone anomalies. (B) The head of the malleus is flattened (long black arrow). The short process of the incus (i) is abnormally broad and appears laterally ankylosed to the epitympanum. The long process (short white arrow) is abnormally oriented and rotated into the same plane as the short process. The lenticular process of the incus and incudostapedial articulation are absent. A short, thick, malformed and angulated stapedial remnant is seen (white arrowhead). The basal turn of the cochlea appears normal (black arrowhead). (C & D) More cephalad images show malformation, hypoplasia and anterior offset of the middle cochlear turn (black arrowheads) with deficiency of the modiolus (short black arrow). The apical turn is markedly hypoplastic and the interscalar septum is not seen. The cochlear aperture is narrowed (thin black arrow ca). The vestibule (v) is enlarged and globular. The horizontal (long white arrow) and superior SCCs (not shown) are fully formed with luminal narrowing. The posterior SCC is absent with slight thickening of the vestibule at the expected connections with the SCC (black asterisks), as seen on the MR in (C).
(E) – (G). Reformatted coronal CT images show stenosis of the external auditory canal (eac). (E) This image demonstrates the stapedial remnant (arrowhead) with oval window atresia. (F) The incus is abnormally oriented, and malformed, with deficiency of the distal long and lenticular process (short white arrow). There is osseous fixation of the incus to the lateral wall of the attic. (G) There is relatively low placement of the tegmen tympani. The malleus (black arrow) is abnormally approximated and likely ankylosed to the scutum.
Due to the critical nature of her illness, she did not have a newborn hearing screen. She underwent successful surgical repair of the choanal atresia. Her ears were small, low-set, and dysplastic with skin tags on the helices bilaterally. She was also noted to have bilateral creases beneath her eyes, downslanting palpebral fissures, and a broad nasal root. Given her clinical features of dysmorphic ears, immunologic dysfunction, dysplastic kidneys, and the CT findings of choanal atresia, temporal bone anomalies, and the unilaterally absent olfactory bulb, CHARGE syndrome was initially felt to be a likely unifying explanation. However, CHD7 sequencing and deletion/duplication analysis were normal. The search for an underlying explanation for her presentation also included a chromosomal microarray and GATA6 sequencing and deletion/duplication analysis, which were also unrevealing.
Ultimately, the infant’s respiratory failure worsened and she continued to require maximal ventilatory support. With the support of the medical team her parents made the decision to redirect to comfort care at three months of age and she passed away following terminal extubation. Autopsy determined the underlying cause of death to be disseminated CMV infection, likely related to her immunodeficiency, that had not been previously detected.
Sequencing Results
The index patient was found to have a de novo novel variant, c.10624C>G (p.Leu3542Val), located on exon 38 of KMT2D (RefSeq NM_003482.4). This variant is absent from the Exome Aggregation Consortium and Genome Aggregation Database, overlaps with an indel variant classified as pathogenic in ClinVar, occurs at a highly-conserved residue, and is predicted to be “probably damaging” by Polyphen, “damaging” by SIFT, and “disease causing” by Mutation Taster (Karczewski et al., 2020; Lek et al., 2016).
Neuroimaging in Kabuki Syndrome
We identified 21 patients with a confirmed molecular diagnosis of Kabuki syndrome. 20 had presumed causative variants in KMT2D and one in KDM6A (Table S1). None had missense variants in KMT2D exons 38–39. Fifteen patients had hearing loss (3 sensorineural, 5 conductive, 6 mixed, 1 unknown type), with 13/15 managed using ear tubes and 10/15 using hearing aids. Two had unknown hearing status.
Imaging studies were available in 13 patients. These exams consisted of temporal bone CT only (one patient), temporal bone CT and brain MRI (four patients) with additional temporal bone sequences in two of the brain MRIs. Brain MRI only was performed in eight patients with additional temporal bone sequences in one patient. In total six patients had detailed temporal bone imaging (5 CT, 3 MRI). The imaging findings are outlined in Table 1 and illustrated in Figs. 3–5. The pons appeared shortened in height (8/13) or mildly misshapen (obtuse angle between the pons and medulla) (5/13) in all patients (Fig. 3). The inferior vermis was uplifted and mildly to moderately hypoplastic in 8/13 patients and uplifted but normally sized in another 2/13 (Figs. 3 & 4). Malformation of the cerebellar hemispheres was seen in one patient (Fig. 4C). Craniosynostosis occurred in three patients, affecting the metopic suture in two and a single coronal suture in one patient (Fig. 7). Lobar holoprosencephaly and hypogenesis of the corpus callosum occurred in one patient (Figs. 4A & B). Temporal bone findings (Fig. 5) included low tegmen tympani (3/5 temporal bone CTs), ossicular anomalies (4/5), and oval window atresia (3/5). Cochlear anomalies included thickened, dysmorphic modioli in 5/6 patients with cochlear aperture stenosis in two of these (Fig. 6A). Horizontal SCC anomalies were seen in only two patients (Figs. 6B–D) consisting of a mildly widened anterior limb of the horizontal SCCs in one patient and globular anterior limb of the horizontal SCCs which were deficient posteriorly with absent horizontal SCC bone islands in the other patient. Flared morphology of the internal auditory canals occurred in 4/6 patients (Fig. 6B).
Table 1.
Neuroradiology findings of patients with available imaging.
| Study ID | EAC | Ossicles | MEC & Mastoid | Round & oval windows | Cochlea | Cochlear aperture | Vestibule & SCC | IAC | Pons | Other |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | - | - | - | - | - | - | - | - | Misshapen | Slight IVH |
| 3 | N | Thickened stapedial crura Malleus fixation Malformed incus |
Under-pneumatized Lt mastoid air cells Low tegmen tympani |
Stenotic Lt oval & round windows | N | N | Mildly dilated horizontal SCC & small bone island | Flared | Slightly short | Metopic suture synostosis |
| 4 | N | N | N | N | Thick right modiolus | Stenotic | N | N | Slightly short | Slight IVH |
| 5 | - | - | - | - | - | - | - | - | Slightly short | Slight IVH |
| 6 | N | Thick Lt stapedial posterior crus Possible ossicular fixation Malformed incus |
Under-pneumatized mastoid air cells Low tegmen tympani |
N | N | N | N | N | Misshapen | Slight IVH |
| 7 | - | - | - | - | - | - | - | - | Misshapen | Slight IVH Malformed cerebellum Delayed myelination |
| 8 | - | - | - | - | Malformed modioli & apical turns | N | N | Flared | Slightly short | Slight IVH |
| 9 | N | Thickened stapedial crura Slightly malformed malleus & incus Malleus ankylosed |
Slightly under-pneumatized mastoid air cells | Stenotic Lt oval & round windows | Malformed modioli | N | Small bone island right horizontal SCC | Flared | Misshapen | |
| 12 | - | - | - | - | - | - | - | - | Short | Slightly uplifted vermis Metopic suture synostosis |
| 14 | N | Thickened stapedial crura Lt malleus ankylosed Malformed incus |
Low tegmen tympani | Slightly stenotic oval windows | Dysmorphic modioli | N | N | Flared | Short | Slightly uplifted vermis Unilateral coronal suture synostosis |
| 16 | - | - | - | - | - | - | - | - | Slightly short | |
| 17 | - | - | - | - | - | - | Malformed horizontal SCCs & absent bone islands | N | Misshapen | IVH |
| 20 | - | - | - | - | - | - | - | - | Slightly short | Slight IVH Hypoplasia CC Lobar HPE Absent right olfactory bulb |
| 22† | Rt stenotic | Rt tilted small monopod stapes Lt short thick crura absent IS joint Malleus ankylosed Incus malformed |
Low tegmen tympani | Stenotic oval windows & Lt round window | Tapered, small middle turns absent apical turns Malformed modioli |
Stenotic | Absent posterior SCCs Partial absence anterior limb superior SCCs |
N | Slightly short |
Proband
Abbreviations: N: Normal; EAC: External auditory canal; MEC: Middle ear cavity; SCC: Semicircular canal; IAC: Internal auditory canal; IVH: inferior vermian hypoplasia; Lt: left, Rt: right; CC: corpus callosum; HPE: holoprosencephaly
Figure 3.
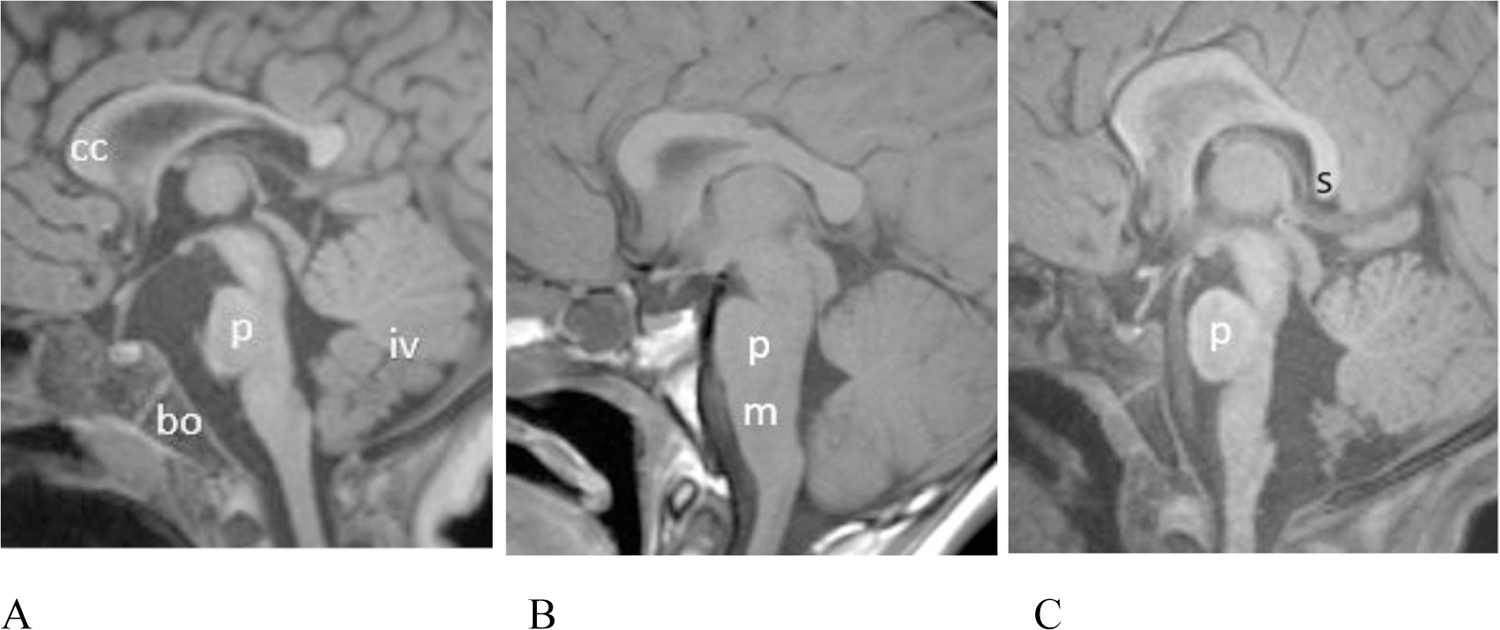
(A–C) Midline sagittal T1 weighted MR images showing variable pontine dysmorphism. (A) Patient # 17, 15 month old female. T1 MPPRAGE image shows a relative short pons (p) that is mildly narrowed in AP diameter, especially superiorly. There is hypoplasia of the inferior vermis (iv). The corpus callosum (cc) is mildly thinned. Note the normal basiocciput (bo). (B) Patient #6, 11 year old male. Sagittal spin echo T1 weighted image shows that the pons (p) is slightly shortened in height and there is an unusually obtuse angle between the ventral aspect of the pons and medulla (m). (C) Patient #14 (22 month old female). T1 MPRAGE image shows that the pons is mildly shortened in height. The vermis is uplifted. The posterior body of the corpus callosum is mildly thinned and the splenium has a vertical orientation.
Figure 5.
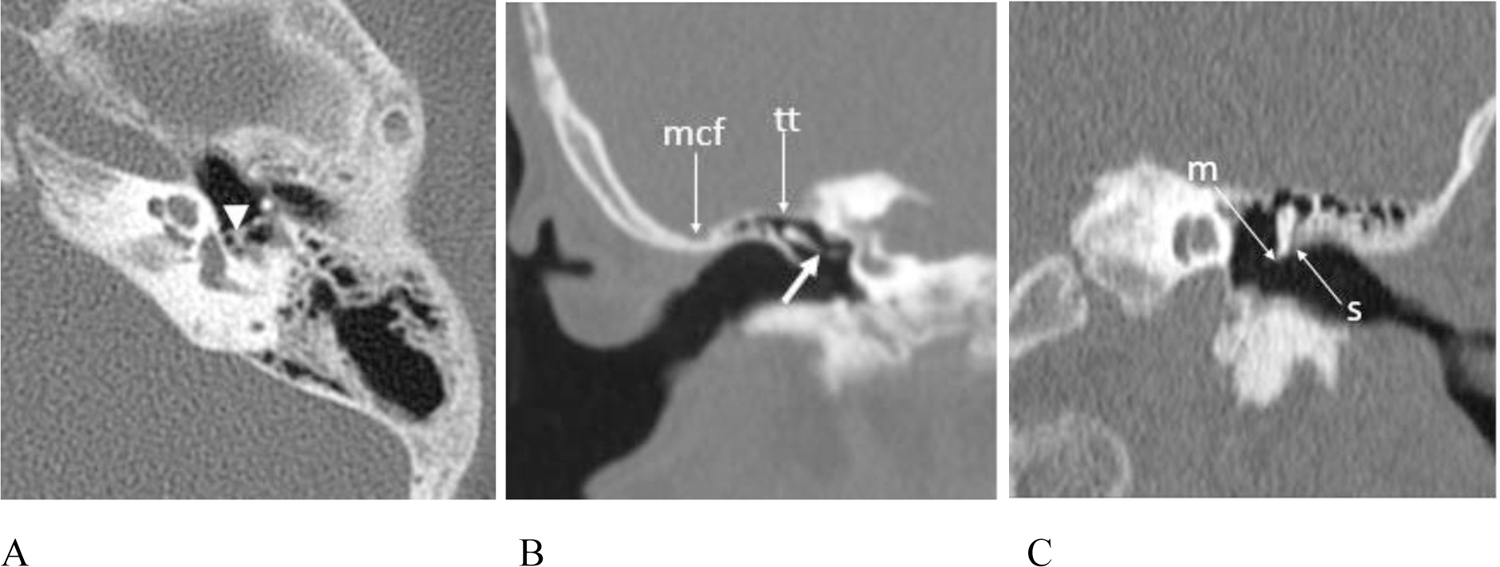
(A – C) CT exams showing ossicular anomalies (A) Patient #9, 11 year old male, axial image left temporal bone. The stapedial crura are short with abnormal osseous thickening at the junction with the capitulum (arrowhead). (B) Patient #3, 8 year old female, reformatted coronal image right temporal bone. The incus is malformed and abnormally oriented with an obtuse incudostapedial angle and deficiency of the distal lenticular process (short white arrow). The epitympanum is shallow with low placement of the tegmen tympani (arrow tt) and the middle cranial fossa (arrow mcf) is in close proximity to the normal appearing external auditory canal. (C) Patient # 9, 11 year old male, reformatted coronal image left temporal bone. The malleus is misshapen with a hypoplastic manubrium (arrow m). It is abnormally oriented and closely approximated to the scutum (arrows).
Figure 4.
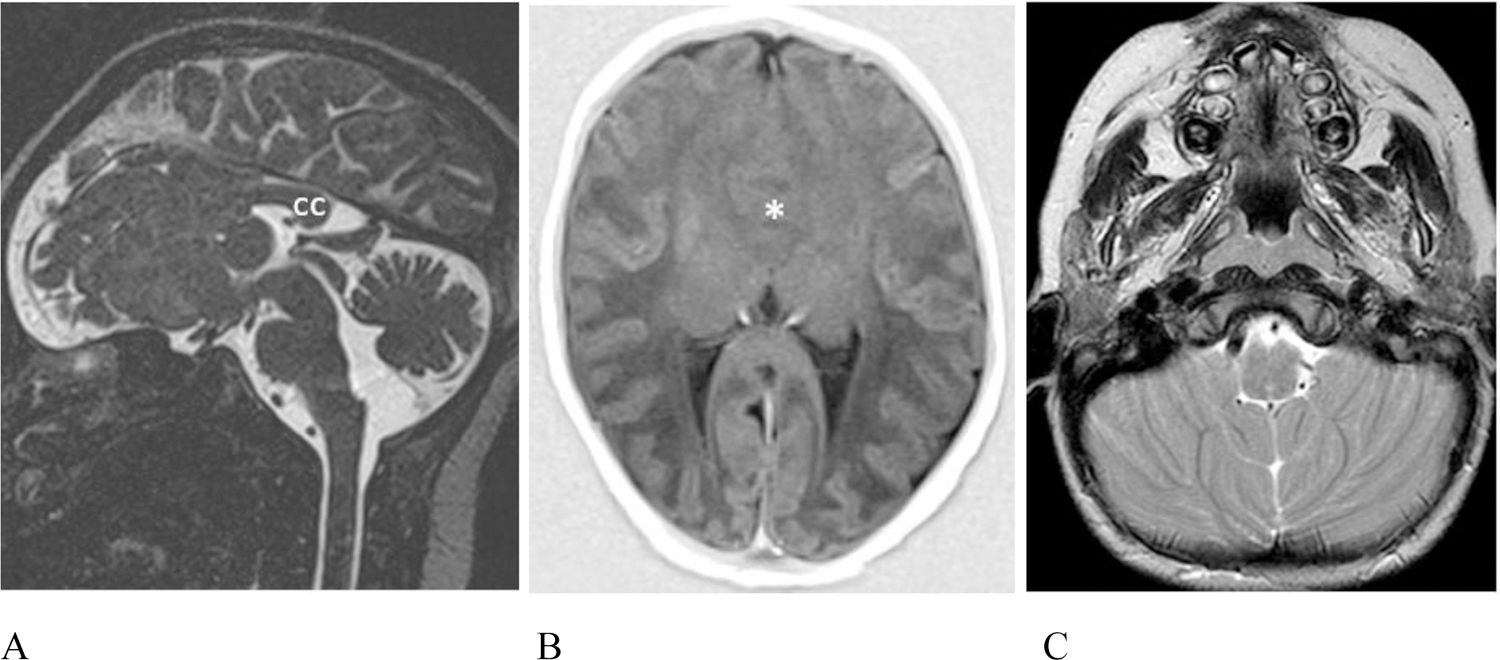
Additional intracranial anomalies. (A – B) Patient #20, 5 day old female. (A) Sagittal T2 weighted sampling perfection with application-optimized contrasts using different flip angle evolution (SPACE) image reveals absence of the anterior 2/3 of the corpus callosum (cc). The vermis is uplifted and the pons is mildly misshapen and slightly short. (B) Axial T1 inversion recovery MR image shows lobar holoprosencephaly with lack of cleavage of the frontal lobes (asterisk) and dysmorphic lateral ventricles with non-visualization of the frontal horns. The right olfactory bulb was absent (not shown). (C) Patient #7, 4 year old male. Axial T2 weighted MR image shows mildly malformed cerebellar hemispheres which are coapted in the midline and the inferior vermis which is hypoplastic is not seen as expected.
Figure 7.
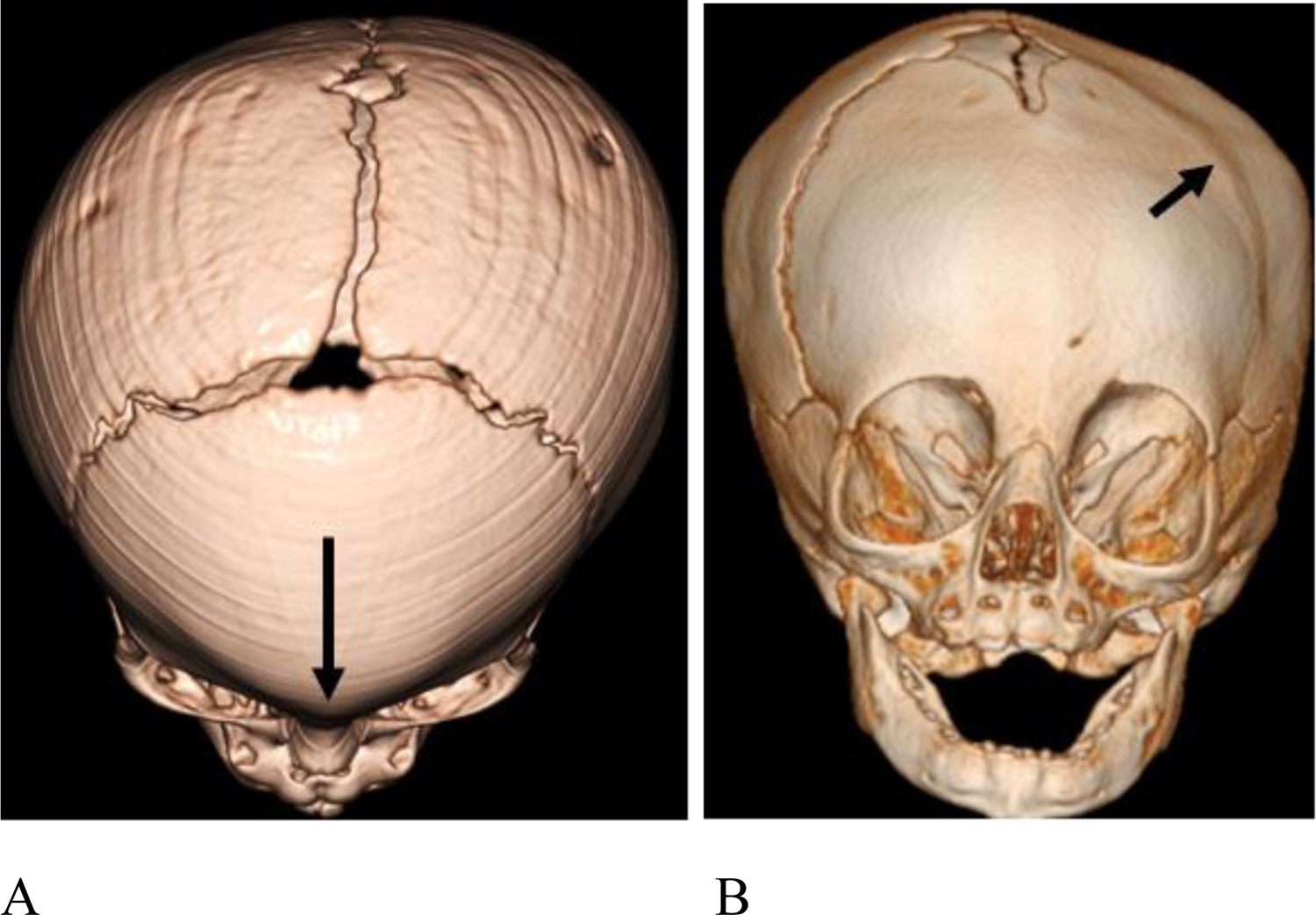
3D CT images demonstrating craniosynostosis. (A) Patient #3, 7 month old female. There is trigonocephaly with a pointed appearance of the frontal bones in the midline due to premature fusion of the metopic suture (long arrow). There is associated hypotelorism (not shown). (B) Patient #14, 6 month old female with left frontal plagiocephaly due to premature fusion of the left coronal suture (short arrow).
Figure 6.
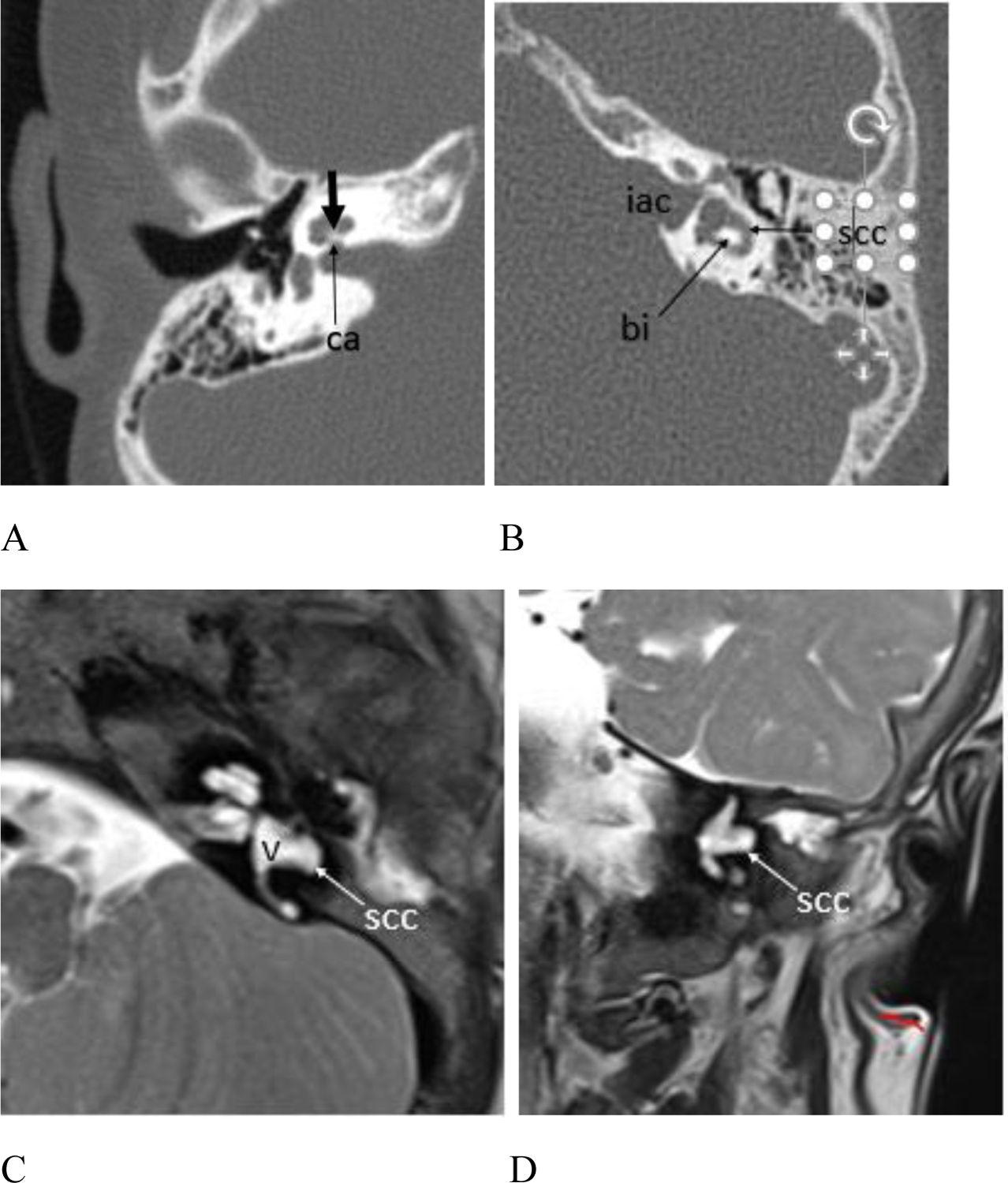
(A – B) CT and (C – D) MR exams showing inner ear anomalies. (A) Patient #4, 6 year old female, axial CT image right temporal bone. The cochlear modiolus is thickened (short arrow) and there is mild stenosis of the cochlear aperture (arrow ca). (B) Patient #3, 8 year old female, axial CT image left temporal bone. The horizontal SCC lumen is mildly widened anteriorly and laterally (arrow scc), and the bone island (arrow bi) between the SCC and the vestibule is misshapen. Note the widened and somewhat bulbous appearance of the internal auditory canal (iac). Figure 6. cont. Patient #17, 15 month old female. (C) Axial and (D) coronal T2 weighted MR images show malformation of the horizontal SCC (arrow scc) which appears globular anteriorly and deficient posteriorly with absence of the bone island between the vestibule (v) and the horizontal SCC.
DISCUSSION:
We describe a neonatal presentation of the recently delineated KMT2D Ex 38/39 allelic syndrome diagnosed by ES that had been previously clinically diagnosed as CHARGE syndrome and molecularly diagnosed as Kabuki syndrome. Although these three disorders share phenotypic overlap, their distinct temporal bone imaging findings may be helpful in phenotypically distinguishing them. In addition, the typical skull base findings of CHARGE syndrome (constricted or hypoplastic basiocciput) were not seen in our index patient. Our review of the temporal bone imaging of this patient and of a series of Kabuki syndrome patients at our institution illustrates that the inner ear findings in patients with Kabuki syndrome and the KMT2D Ex 38/39 allelic disorder are distinct both from each other and from those seen in CHARGE syndrome.
The infant’s ultimate diagnosis of an allelic disorder caused by KMT2D MVs in a region spanning exons 38 and 39 was realized after two reports were published on separate series of patients with this newly described disorder (Baldridge et al., 2020; Cuvertino et al., 2020). In addition to the 12 new patients reported on by the combination of these reports, our review of previously published case reports of patients with molecularly diagnosed Kabuki syndrome and clinical features reminiscent of CHARGE syndrome demonstrated that at least 4 of these patients actually had the Ex 38/39 KMT2D allelic disorder rather than classic Kabuki syndrome (Badalato et al., 2017; Sakata et al., 2017).
Table 2 summarizes the clinical features of all presently known Ex 38/39 KMT2D MV cases based on our literature review. Though there is phenotypic overlap between CHARGE syndrome, Kabuki syndrome, and the KMT2D Ex 38/39 allelic disorder, each is distinct (Table 3). External ear malformations are seen in all three disorders but with differences – external ears are small and variably dysplastic in KMT2D Ex 38/39 allelic disorder patients, large and prominent in Kabuki syndrome, and small with a classic appearance in CHARGE syndrome.
Table 2.
Clinical features of described Ex 38/39 KMT2D MV patients.
| Index patient (1) | Cuvertino (9) | Baldridge (3) | Badalato (3) | Sakata (1) | All Ex 38/39 KMT2D MVs (n = 17)† | |
|---|---|---|---|---|---|---|
| Variant in KMT2D | c.10624C>G (p.Leu3542Val) | Variety of Ex 38/39 MVs | Variety of Ex 38/39 MVs | c.10725G>C (p.Gln3575His) | c.10690C>G (p.Leu3564Val) | Variety of Ex 38/39 MVs |
| Physical feature | ||||||
| Athelia/hypoplastic nipples | No | 6/9 | 3/3 | 0/3 | Yes | 10/17 |
| Brain malformation | Yes | 1/3 | 0/3 | No MRI | No | 2/5 |
| Branchial sinus/neck pits | No | 7/9 | 2/3 | 0/3 | No | 9/17 |
| Choanal atresia | Yes | 7/9 | 3/3 | 3/3 | Yes | 15/17 |
| Cleft lip/palate | No | 0/9 | 0/3 | 0/3 | Yes | 1/17 |
| Congenital heart defect | Yes | 3/9 | 0/3 | 1/3 | No | 5/17 |
| Dental abnormalities | N/A | 3/8 | 3/3 | 3/3 | Yes | 10/15 |
| Ear abnormalities (external) | Yes | 6/9 | 3/3 | 0/3 | Yes | 11/17 |
| Ear abnormalities (internal) | Yes | 1/2 | 2/2 (1 without imaging) | 1/1 (2 without imaging) | Yes | 6/7 |
| Eye abnormalities | No | 2/9 | 0/3 | 0/3 | No | 2/17 |
| Feeding difficulties | N/A | 5/9 | 3/3 | 0/3 | No | 8/16 |
| Hearing loss | Unknown | 8/9 | 3/3 | 3/3 | Yes | 15/16 |
| Immunodeficiency | Yes | 4/9 | 0/3 | 0/3 | No | 5/17 |
| Lacrimal duct abnormalities | No | 7/9 | 2/3 | 0/3 | No | 9/17 |
| Renal abnormalities | Yes | 0/9 | 1/3 | 0/3 | No | 2/17 |
| Short stature | N/A | 5/9 | 3/3 | 1/3 | Yes | 10/16 |
| Thyroid abnormalities | Yes | 6/9 | 3/3 | 0/3 | Yes | 11/17 |
Patients from Cuvertino et al. (9), Baldridge et al. (3), Badalato et al. (3), Sakata et al. (1) and proband (1)
Table 3.
Clinical features of Ex 38/39 KMT2D MV disorder, Kabuki syndrome, and CHARGE syndrome. Note: Listed brain malformations, external ear abnormalities, and internal ear abnormalities are not observed in all patients and are not exhaustive but rather are a summary of features that have been classically/commonly described.
| Physical feature | Ex 38/39 KMT2D MVs (n = 17)† | Kabuki syndrome | CHARGE syndrome |
|---|---|---|---|
| Athelia/hypoplastic nipples | Common | Rare | Rare |
| Brain malformation | Common | ‡Common | Common |
| - Hypoplasia of pons | - Shortened or misshapen pons - Uplifted and hypoplastic inferior vermis |
- Hypoplasia and/or clefting of basiocciput - Hypoplasia of vermis - Hypo/aplasia of olfactory bulbs/sulci |
|
| Branchial sinus/neck pits | Common | Rare | Rare |
| Choanal atresia | Common | Rare | Common |
| Cleft lip/palate | Rare | Common | Common |
| Congenital heart disease | Common | Common | Common |
| Dental abnormalities | Common | Common | Common |
| Ear abnormalities (external) | Common | Common | Common |
| - Small - Variably dysplastic +/− tags or pits |
- Large, prominent, cupped | - Short, small ear with minimal lobe - Snipped off helix and prominent anti-helix |
|
| Ear abnormalities (internal) | Common | ‡Common | Common |
| - Absent posterior SCCs - Misshapen vestibules - Cochlear hypoplasia - Cochlear aperture stenosis - Ossicular anomalies |
- Cochlear anomalies (thickened, dysmorphic modioli with variable cochlear aperture stenosis) - Ossicular anomalies - Flared morphology of the IACs - Low tegmen typani - Oval window atresia - SCC abnormalities less common |
- Absence or hypoplasia of all SCCs - Cochlear and vestibular hypoplasia - Cochlear aperture stenosis - Ossicular anomalies |
|
| Eye abnormalities | Rare | Rare | Common |
| Feeding difficulties | Common | Common | Common |
| Hearing loss | Common | Common | Common |
| Immunodeficiency | Common | Common | Common |
| Lacrimal duct abnormalities | Common | Rare | Rare |
| Renal abnormalities | Rare | Common | Common |
| Short stature | Common | Common | Common |
| Thyroid abnormalities | Common | Rare | Rare |
Patients from Cuvertino et al. (9), Baldridge et al. (3), Badalato et al. (3), Sakata et al. (1) and proband (1)
Not previously reported as common but retrospective review of our patients suggests otherwise
Inner ear abnormalities have been well recognized as one of, if not the most, distinctive features of CHARGE syndrome, with semicircular canal aplasia or hypoplasia being the classic finding (Legendre et al., 2017; Wineland et al., 2017). Although inner ear anatomy is not yet widely described in the KMT2D Ex 38/39 allelic disorder, most previously reported patients did not have imaging. Of those that did, features described included absence of posterior SCCs and other unspecified vestibular and cochlear abnormalities. One patient had a number of temporal bone abnormalities that were well described, most notably bilateral atresia of the external auditory meati, hypoplasia of the right vestibular aqueduct, bilateral hypoplasia of the long process of the incus, and bilateral agenesis of the stapes and posterior SCC (Sakata et al., 2017).
Careful review of the temporal bone imaging in our index patient demonstrated a distinctive anomaly of the vestibules and SCCs that appeared quite unlike the characteristic hypoplasia/aplasia of the vestibules and all SCCs characteristic of CHARGE syndrome. The vestibules appeared misshapen with slight protrusions at the expected take off of the posterior SCCs. The posterior SCCs were essentially absent and there was partial absence of the anterior limbs of the superior SCCs. The cochlear anomalies with deficient modioli, hypoplastic apical turns and stenotic cochlear apertures were similar to findings seen in CHARGE, however, unlike CHARGE, the middle and apical turns appeared offset anteriorly, more reminiscent of branchio-oto-renal syndrome. Similarities with CHARGE were seen with respect to ossicular anomalies, oval window atresia, dysmorphism of the pons and mild uplifting of the cerebellar vermis. The typical central skull base anomalies (hypoplasia and/or clefting of the basiocciput) seen in CHARGE were not seen in our patient. More information is needed in order to determine the typical morphology associated with the Ex 38/39 KMT2D MV disorder, but there are now multiple lines of evidence suggesting that inner ear abnormalities may be one of the hallmark features of this new disorder.
Kabuki syndrome, while frequently associated with hearing loss, has been less widely associated with inner ear or temporal abnormalities. There have been several case reports of inner ear abnormalities in select children with Kabuki syndrome, particularly involving the cochleae and SCC. These include dysplastic cochlea with an enlarged vestibule, absence of the cochlea and semicircular canals, a unilateral enlarged vestibule, an enlarged vestibular aqueduct, and aural atresia with fusion of the incus and malleus (Igawa et al., 2000; Tekin et al., 2006; Toutain et al., 1997; Zarate et al., 2012). However, this is the first systematic case series reviewing the temporal bone imaging of Kabuki syndrome patients. Our review of brain and temporal bone imaging of patients with classic Kabuki syndrome who received neuroimaging revealed a number of interesting findings. For example, all patients were noted to have an abnormal pons and most (8/13) had an uplifted and hypoplastic vermis. Although rare cases of structural brain malformations have been previously described in Kabuki syndrome, these particular anomalies were surprisingly consistent throughout our series of patients. In addition, temporal bone findings were common among the patients who had dedicated temporal bone imaging, with cochlear anomalies, ossicular anomalies, and flared morphology of the IACs noted. Other temporal bone findings included low tegmen tympani and oval window atresia. SCC anomalies, which are the hallmark of inner ear anomalies in CHARGE syndrome, were not invariably present, being seen in only 2 patients of the 6 with detailed temporal bone imaging. One limitation of our retrospective review is that since patients with Kabuki syndrome do not routinely receive temporal bone imaging, abnormalities are likely overrepresented in those for whom there was enough clinical suspicion to obtain imaging. Furthermore, there are published case reports of patients with Kabuki syndrome who did have temporal bone imaging that was unremarkable (Lu & Cao, 2014; Pepe et al., 2020). Even so, the high prevalence of abnormalities seen in our cohort suggests that patients with Kabuki syndrome, and especially those with hearing loss, should routinely undergo temporal bone imaging.
Hearing loss, which is reported in approximately 40% of patients with Kabuki syndrome (Tekin et al, 2006), was present in 15 of the 19 patients in our Kabuki cohort with known hearing status. The nature of the hearing loss seen in Kabuki syndrome has not been well-defined and historically has been attributed largely to frequent otitis media (Barozzi et al., 2009). However, the underlying cause is not always found, and it is unclear how many inner ear anomalies may have been missed due to lack of imaging. It is possible that the hearing loss so commonly seen in Kabuki syndrome is more often attributable to inner ear and ossicular anomalies than previously thought. Identification of anatomic inner ear abnormalities may have implications for treatment of the hearing loss. For example, one case study demonstrated the efficacy of cochlear implantation in a patient with Kabuki syndrome and semicircular canal malformation (A. Vesseur et al., 2016).
In addition to clinical similarities, molecular links between CHARGE, Kabuki, and the KMT2D Ex 38/39 syndromes have also been discovered. It is known that CHD7, KMT2D, and KDM6A are all involved in chromatin remodeling, and a molecular link between CHD7 and KMT2D based upon their shared interaction with members of the WAR complex has been postulated (Aref-Eshghi et al., 2018). KMT2D encodes a methyltransferase that is critical for transcriptional regulation. However, the methyltransferase activity is only active in the context of a multi-subunit complex, one important aspect of which is the WAR complex. It has been previously demonstrated that CHD7 also interacts with the WAR complex, which infers a possible functional connection between the two genes (Schulz et al., 2014). Other groups subsequently strengthened the notion that CHD7 and KMT2D regulate some of the same genes, such as HOXA5 and SLITRK5, involved in embryonic development of numerous cell types and tissues (Aref-Eshghi et al., 2018; Butcher et al., 2017). Future work may clarify the molecular relationship with these particular features of clinical overlap.
In summary, we provide further evidence for a newly described KMT2D Ex 38/39 allelic disorder and emphasize the clinical similarities but also key differences between this disorder, CHARGE syndrome, and Kabuki syndrome with a particular focus on inner ear abnormalities. Though subtle, there are key differences that may help with clinical differentiation. We propose that inner ear and ossicular abnormalities may be more common in Kabuki syndrome than previously thought and may be a distinguishing feature of the KMT2D Ex 38/39 allelic disorder, though further research is needed to fully understand the spectrum of inner ear anomalies in Kabuki syndrome and related KMT2D disorders. Nonetheless, temporal imaging should be widely considered in cases of suspected or confirmed Kabuki syndrome or the KMT2D Ex 38/39 allelic disorder.
Supplementary Material
Acknowledgements:
The Broad Institute of MIT and Harvard Center for Mendelian Genomics (Broad CMG) is funded by the National Human Genome Research Institute, the National Eye Institute, and the National Heart, Lung and Blood Institute grant UM1 HG008900 and in part by National Human Genome Research Institute grant R01 HG009141. M.H.W. is supported by K23 HD102589.
Footnotes
Conflicts of Interest:
The authors have no conflicts of interest to disclose.
Data Availability:
The data that supports the findings of this study are available in the supplementary material of this article.
References:
- Abruzzo MA, & Erickson RP (1977). A new syndrome of cleft palate associated with coloboma, hypospadias, deafness, short stature, and radial synostosis. J Med Genet, 14(1), 76–80. 10.1136/jmg.14.1.76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aref-Eshghi E, Rodenhiser DI, Schenkel LC, Lin H, Skinner C, Ainsworth P, Paré G, Hood RL, Bulman DE, Kernohan KD, Boycott KM, Campeau PM, Schwartz C, Sadikovic B, & Consortium CRC (2018). Genomic DNA Methylation Signatures Enable Concurrent Diagnosis and Clinical Genetic Variant Classification in Neurodevelopmental Syndromes. Am J Hum Genet, 102(1), 156–174. 10.1016/j.ajhg.2017.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badalato L, Farhan SM, Dilliott AA, Bulman DE, Hegele RA, Goobie SL, & Consortium CRC (2017). KMT2D p.Gln3575His segregating in a family with autosomal dominant choanal atresia strengthens the Kabuki/CHARGE connection. Am J Med Genet A, 173(1), 183–189. 10.1002/ajmg.a.38010 [DOI] [PubMed] [Google Scholar]
- Baldridge D, Spillmann RC, Wegner DJ, Wambach JA, White FV, Sisco K, Toler TL, Dickson PI, Cole FS, Shashi V, & Grange DK (2020). Phenotypic expansion of KMT2D-related disorder: Beyond Kabuki syndrome. Am J Med Genet A, 182(5), 1053–1065. 10.1002/ajmg.a.61518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barozzi S, Di Berardino F, Atzeri F, Filipponi E, Cerutti M, Selicorni A, & Cesarani A (2009). Audiological and vestibular findings in the Kabuki syndrome. Am J Med Genet A, 149A(2), 171–176. 10.1002/ajmg.a.32610 [DOI] [PubMed] [Google Scholar]
- Butcher DT, Cytrynbaum C, Turinsky AL, Siu MT, Inbar-Feigenberg M, Mendoza-Londono R, Chitayat D, Walker S, Machado J, Caluseriu O, Dupuis L, Grafodatskaya D, Reardon W, Gilbert-Dussardier B, Verloes A, Bilan F, Milunsky JM, Basran R, Papsin B, Stockley TL, Scherer SW, Choufani S, Brudno M, & Weksberg R (2017). CHARGE and Kabuki Syndromes: Gene-Specific DNA Methylation Signatures Identify Epigenetic Mechanisms Linking These Clinically Overlapping Conditions. Am J Hum Genet, 100(5), 773–788. 10.1016/j.ajhg.2017.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuvertino S, Hartill V, Colyer A, Garner T, Nair N, Al-Gazali L, Canham N, Faundes V, Flinter F, Hertecant J, Holder-Espinasse M, Jackson B, Lynch SA, Nadat F, Narasimhan VM, Peckham M, Sellers R, Seri M, Montanari F, Southgate L, Squeo GM, Trembath R, van Heel D, Venuto S, Weisberg D, Stals K, Ellard S, Barton A, Kimber SJ, Sheridan E, Merla G, Stevens A, Johnson CA, Banka S, Research GE, & Consortium. (2020). A restricted spectrum of missense KMT2D variants cause a multiple malformations disorder distinct from Kabuki syndrome. Genet Med, 22(5), 867–877. 10.1038/s41436-019-0743-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Arco F, Youssef A, Ioannidou E, Bisdas S, Pinelli L, Caro-Dominguez P, Nash R, Siddiqui A, & Talenti G (2020). Temporal bone and intracranial abnormalities in syndromic causes of hearing loss: an updated guide. Eur J Radiol, 123, 108803. 10.1016/j.ejrad.2019.108803 [DOI] [PubMed] [Google Scholar]
- Dentici ML, Di Pede A, Lepri FR, Gnazzo M, Lombardi MH, Auriti C, Petrocchi S, Pisaneschi E, Bellacchio E, Capolino R, Braguglia A, Angioni A, Dotta A, Digilio MC, & Dallapiccola B (2015). Kabuki syndrome: clinical and molecular diagnosis in the first year of life. Arch Dis Child, 100(2), 158–164. 10.1136/archdischild-2013-305858 [DOI] [PubMed] [Google Scholar]
- Faundes V, Malone G, Newman WG, & Banka S (2019). A comparative analysis of KMT2D missense variants in Kabuki syndrome, cancers and the general population. J Hum Genet, 64(2), 161–170. 10.1038/s10038-018-0536-6 [DOI] [PubMed] [Google Scholar]
- Hale CL, Niederriter AN, Green GE, & Martin DM (2016). Atypical phenotypes associated with pathogenic CHD7 variants and a proposal for broadening CHARGE syndrome clinical diagnostic criteria. Am J Med Genet A, 170A(2), 344–354. 10.1002/ajmg.a.37435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hempel JM, Jäger L, Naumann A, & Schorn K (2005). [Niikawa-Kuroki syndrome. Which characteristics must the HNO doctor consider in its diagnosis]. HNO, 53(3), 253–256. 10.1007/s00106-004-1071-7 [DOI] [PubMed] [Google Scholar]
- Igawa HH, Nishizawa N, Sugihara T, & Inuyama Y (2000). Inner ear abnormalities in Kabuki make-up syndrome: report of three cases. Am J Med Genet, 92(2), 87–89. [DOI] [PubMed] [Google Scholar]
- Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, Collins RL, Laricchia KM, Ganna A, Birnbaum DP, Gauthier LD, Brand H, Solomonson M, Watts NA, Rhodes D, Singer-Berk M, England EM, Seaby EG, Kosmicki JA, Walters RK, Tashman K, Farjoun Y, Banks E, Poterba T, Wang A, Seed C, Whiffin N, Chong JX, Samocha KE, Pierce-Hoffman E, Zappala Z, O’Donnell-Luria AH, Minikel EV, Weisburd B, Lek M, Ware JS, Vittal C, Armean IM, Bergelson L, Cibulskis K, Connolly KM, Covarrubias M, Donnelly S, Ferriera S, Gabriel S, Gentry J, Gupta N, Jeandet T, Kaplan D, Llanwarne C, Munshi R, Novod S, Petrillo N, Roazen D, Ruano-Rubio V, Saltzman A, Schleicher M, Soto J, Tibbetts K, Tolonen C, Wade G, Talkowski ME, Neale BM, Daly MJ, MacArthur DG, & Consortium G. A. D. (2020). The mutational constraint spectrum quantified from variation in 141,456 humans. Nature, 581(7809), 434–443. 10.1038/s41586-020-2308-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederer D, Grisart B, Digilio MC, Benoit V, Crespin M, Ghariani SC, Maystadt I, Dallapiccola B, & Verellen-Dumoulin C (2012). Deletion of KDM6A, a histone demethylase interacting with MLL2, in three patients with Kabuki syndrome. Am J Hum Genet, 90(1), 119–124. 10.1016/j.ajhg.2011.11.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legendre M, Abadie V, Attié-Bitach T, Philip N, Busa T, Bonneau D, Colin E, Dollfus H, Lacombe D, Toutain A, Blesson S, Julia S, Martin-Coignard D, Geneviève D, Leheup B, Odent S, Jouk PS, Mercier S, Faivre L, Vincent-Delorme C, Francannet C, Naudion S, Mathieu-Dramard M, Delrue MA, Goldenberg A, Héron D, Parent P, Touraine R, Layet V, Sanlaville D, Quélin C, Moutton S, Fradin M, Jacquette A, Sigaudy S, Pinson L, Sarda P, Guerrot AM, Rossi M, Masurel-Paulet A, El Chehadeh S, Piguel X, Rodriguez-Ballesteros M, Ragot S, Lyonnet S, Bilan F, & Gilbert-Dussardier B (2017). Phenotype and genotype analysis of a French cohort of 119 patients with CHARGE syndrome. Am J Med Genet C Semin Med Genet, 175(4), 417–430. 10.1002/ajmg.c.31591 [DOI] [PubMed] [Google Scholar]
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, Tukiainen T, Birnbaum DP, Kosmicki JA, Duncan LE, Estrada K, Zhao F, Zou J, Pierce-Hoffman E, Berghout J, Cooper DN, Deflaux N, DePristo M, Do R, Flannick J, Fromer M, Gauthier L, Goldstein J, Gupta N, Howrigan D, Kiezun A, Kurki MI, Moonshine AL, Natarajan P, Orozco L, Peloso GM, Poplin R, Rivas MA, Ruano-Rubio V, Rose SA, Ruderfer DM, Shakir K, Stenson PD, Stevens C, Thomas BP, Tiao G, Tusie-Luna MT, Weisburd B, Won HH, Yu D, Altshuler DM, Ardissino D, Boehnke M, Danesh J, Donnelly S, Elosua R, Florez JC, Gabriel SB, Getz G, Glatt SJ, Hultman CM, Kathiresan S, Laakso M, McCarroll S, McCarthy MI, McGovern D, McPherson R, Neale BM, Palotie A, Purcell SM, Saleheen D, Scharf JM, Sklar P, Sullivan PF, Tuomilehto J, Tsuang MT, Watkins HC, Wilson JG, Daly MJ, MacArthur DG, & Consortium EA (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, & Cao K (2014). A case report: Hearing disorder in Kabuki make-up (Niikawa-Kuroki) syndrome in China. In Journal of Otology (Vol. 9, pp. 136–140). [Google Scholar]
- Morimoto AK, Wiggins RH, Hudgins PA, Hedlund GL, Hamilton B, Mukherji SK, Telian SA, & Harnsberger HR (2006). Absent semicircular canals in CHARGE syndrome: radiologic spectrum of findings. AJNR Am J Neuroradiol, 27(8), 1663–1671. [PMC free article] [PubMed] [Google Scholar]
- Ng SB, Bigham AW, Buckingham KJ, Hannibal MC, McMillin MJ, Gildersleeve HI, Beck AE, Tabor HK, Cooper GM, Mefford HC, Lee C, Turner EH, Smith JD, Rieder MJ, Yoshiura K, Matsumoto N, Ohta T, Niikawa N, Nickerson DA, Bamshad MJ, & Shendure J (2010). Exome sequencing identifies MLL2 mutations as a cause of Kabuki syndrome. Nat Genet, 42(9), 790–793. 10.1038/ng.646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niikawa N, Matsuura N, Fukushima Y, Ohsawa T, & Kajii T (1981). Kabuki make-up syndrome: a syndrome of mental retardation, unusual facies, large and protruding ears, and postnatal growth deficiency. J Pediatr, 99(4), 565–569. 10.1016/s0022-3476(81)80255-7 [DOI] [PubMed] [Google Scholar]
- Pagon RA, Graham JM, Zonana J, & Yong SL (1981). Coloboma, congenital heart disease, and choanal atresia with multiple anomalies: CHARGE association. J Pediatr, 99(2), 223–227. 10.1016/s0022-3476(81)80454-4 [DOI] [PubMed] [Google Scholar]
- Patel N, & Alkuraya FS (2015). Overlap between CHARGE and Kabuki syndromes: more than an interesting clinical observation? Am J Med Genet A, 167A(1), 259–260. 10.1002/ajmg.a.36804 [DOI] [PubMed] [Google Scholar]
- Pepe G, Negri M, Falcioni M, Di Lella F, & Vincenti V (2020). Bonebridge implantation for mixed hearing loss in a patient with Kabuki syndrome. Acta Biomed, 91(3), e2020079. 10.23750/abm.v91i3.8257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata S, Okada S, Aoyama K, Hara K, Tani C, Kagawa R, Utsunomiya-Nakamura A, Miyagawa S, Ogata T, Mizuno H, & Kobayashi M (2017). Individual Clinically Diagnosed with CHARGE Syndrome but with a Mutation in. Front Genet, 8, 210. 10.3389/fgene.2017.00210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz Y, Freese L, Mänz J, Zoll B, Völter C, Brockmann K, Bögershausen N, Becker J, Wollnik B, & Pauli S (2014). CHARGE and Kabuki syndromes: a phenotypic and molecular link. Hum Mol Genet, 23(16), 4396–4405. 10.1093/hmg/ddu156 [DOI] [PubMed] [Google Scholar]
- Sennaroğlu L, & Bajin MD (2017). Classification and Current Management of Inner Ear Malformations. Balkan Med J, 34(5), 397–411. 10.4274/balkanmedj.2017.0367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tekin M, Fitoz S, Arici S, Cetinkaya E, & Incesulu A (2006). Niikawa-Kuroki (Kabuki) syndrome with congenital sensorineural deafness: evidence for a wide spectrum of inner ear abnormalities. Int J Pediatr Otorhinolaryngol, 70(5), 885–889. 10.1016/j.ijporl.2005.09.025 [DOI] [PubMed] [Google Scholar]
- Toutain A, Plée Y, Ployet MJ, Benoit S, Perrot A, Sembely C, Barthez MA, & Moraine C (1997). Deafness and Mondini dysplasia in Kabuki (Niikawa-Kuroki) syndrome. Report of a case and review of the literature. Genet Couns, 8(2), 99–105. [PubMed] [Google Scholar]
- Verhagen JM, Oostdijk W, Terwisscha van Scheltinga CE, Schalij-Delfos NE, & van Bever Y (2014). An unusual presentation of Kabuki syndrome: clinical overlap with CHARGE syndrome. Eur J Med Genet, 57(9), 510–512. 10.1016/j.ejmg.2014.05.005 [DOI] [PubMed] [Google Scholar]
- Vesseur A, Cillessen E, & Mylanus E (2016). Cochlear Implantation in a Patient with Kabuki Syndrome. J Int Adv Otol, 12(1), 129–131. 10.5152/iao.2016.2004 [DOI] [PubMed] [Google Scholar]
- Vesseur AC, Verbist BM, Westerlaan HE, Kloostra FJJ, Admiraal RJC, van Ravenswaaij-Arts CMA, Free RH, & Mylanus EAM (2016). CT findings of the temporal bone in CHARGE syndrome: aspects of importance in cochlear implant surgery. Eur Arch Otorhinolaryngol, 273(12), 4225–4240. 10.1007/s00405-016-4141-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wineland A, Menezes MD, Shimony JS, Shinawi MS, Hullar TE, & Hirose K (2017). Prevalence of Semicircular Canal Hypoplasia in Patients With CHARGE Syndrome: 3C Syndrome. JAMA Otolaryngol Head Neck Surg, 143(2), 168–177. 10.1001/jamaoto.2016.3175 [DOI] [PubMed] [Google Scholar]
- Zarate YA, Zhan H, & Jones JR (2012). Infrequent Manifestations of Kabuki Syndrome in a Patient with Novel MLL2 Mutation. Mol Syndromol, 3(4), 180–184. 10.1159/000342253 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that supports the findings of this study are available in the supplementary material of this article.