

Abstract
MicroRNA-155 (miR-155) is overexpressed in various types of lymphomas and leukemias, suggesting that targeting miR-155 could be a potential platform for the development of precision medicine. Here we tested the anti-cancer activity of novel, chemically modified, triplex peptide nucleic acid (PNA)-based antimiRs compared to the current state-of-the-art conventional full length antimiRs. Next-generation modified PNAs that bound miR-155 by Watson-Crick and Hoogsteen domains possessed superior therapeutic efficacy in vivo and ex vivo compared to conventional full-length anti-miR-155. The efficacy of anti-miR-155 targeting in multiple lymphoma cell lines was comprehensively corroborated by gene expression, western blot analysis, and cell viability-based functional studies. Finally, preclinical testing in vivo in xenograft mouse models containing lymphoma cell lines demonstrated that treatment with the miR-155-targeting next generation antimiR resulted in a significant decrease in miR-155 expression followed by reduced tumor growth. These findings support the effective therapeutic application of chemically modified triplex PNAs to target miR-155 to treat lymphoma. Overall, the present proof-of-concept study further implicates the potential for next-generation triplex gamma PNAs to target other miRNAs for treating cancer.
Keywords: microRNA, peptide nucleic acids, Gamma PNA, cancer
INTRODUCTION
MicroRNA (miRNA or miR) is a class of non-coding RNAs that control gene expression at the post-transcription level (1). miRNAs play key roles in maintaining physiological processes by controlling gene expression through regulating messenger RNA (mRNA) stability and translation. Hence, aberrant expression of miRNAs causes several devastating diseases. In cancer, atypical miRNA levels lead to altered processes, including differentiation, proliferation, and apoptosis (2). Hence, miRNAs have been explored as promising molecular targets for the development of precision medicine in cancer (3). Synthetic nucleic acid-based antimiRs have been evaluated in conjunction with delivery systems to repress miRNAs upregulated in multiple tumors (also called oncomiRs) for potential cancer therapeutics (4).
Several antimiRs have been developed to specifically target full-length miRNAs by Watson-Crick recognition to prevent their interaction with target mRNAs. In particular, peptide nucleic acids (PNAs) have gained attention as potential antimiR agents. PNAs are synthetic nucleic acid analogs that possess a neutral backbone and are resistant to enzymatic degradation (5). It is well-known that PNAs can target the full-length miRNAs by Watson-Crick base pairing and thus control gene expression (6,7). Targeting full-length miRNAs provides numerous advantages, especially the sequence-specific targeting of preferred miRNA sites minimizes the off-target toxicity (8). A few studies reported using shorter antimiRs targeting the miRNA seed region to inhibit its function (9,10). Still, clinical translation of targeting seed region-based strategies could be hampered due to off-target effects because of non-specific binding with other coding and non-coding RNAs. Though promising results have been shown, increasing the binding affinity of antimiRs without compromising their specificity remains a continuous goal.
Herein, we demonstrated that compared to full-length PNA, another novel class of PNAs called tail-clamp PNA (tcPNA) possesses superior binding properties to target miRNAs and inhibit their activity. In tcPNA, one strand binds via Watson-Crick base pairing, and the other strand binds via Hoogsteen base pairing (11). Hence, tcPNAs possess a higher binding affinity compared to single-stranded PNAs. tcPNAs have been designed to target double-strand DNA containing homopurine stretches (12). A few biophysical studies have shown that tcPNAs containing regular PNA units can bind to short double-stranded RNA targets (13). However, most of these studies, as mentioned above, were focused only on biophysical studies and not much progress has been made in evaluating their biological activity in cell culture as well as in vivo.
To our knowledge, this is the first study centered on the evaluation of chemically modified tcPNAs for targeting biologically relevant miRNAs for therapeutic application. To boost the binding affinity, we employed gamma (γ) PNA-containing tcPNA based antimiRs. Gamma PNAs are next-generation PNAs that possess superior binding properties due to their preorganized locked structure (14). Due to their high binding affinity, gamma PNAs have been employed in gene editing (15–17) and gene barcoding applications (18).
We established that anti-miR-155 gamma tcPNAs show superior miR-155 inhibition activity compared to conventional single-stranded anti-miR-155 design both in cell culture and in vivo. miR-155 has been upregulated in various cancers, especially in lymphoma and leukemia (19,20). Recent studies have demonstrated that miR-155 is a clinically relevant target for treatment of various lymphomas (6). The drug cobomarsen (MRG-106) is in a phase 2 clinical trial targeting miR-155 for lymphoma therapy (21). Here, we performed comprehensive studies where anti-miR-155 gamma tcPNAs inhibit miR-155 in a lymphoma cell line, and its downstream targets both in the cell culture and in in vivo studies. In a parallel comparison, we demonstrate that anti-miR-155 gamma tcPNAs effectively decrease tumor growth in a lymphoma cell line derived xenograft mouse model compared to the regular PNA. Overall, herein we established that gamma tcPNA based synthetic oncomiR inhibitors could be explored for the next-generation miRNA therapeutics.
MATERIAL AND METHODS
Synthesis of PNA oligomers
Boc-protected regular monomers (for PNA-155) and serine gamma monomers used for gamma tcPNA-155, gamma PNA-155 and Scr-gamma tcPNA-155 synthesis were purchased from ASM Research Chemicals GmbH (Hannover, Germany). The monomers were vacuum dried prior to start of solid-phase synthesis. Around 100 mg arginine-loaded resin was soaked in dichloromethane (DCM) for 5 hours in a reaction vessel. The DCM was drained and the resin was deprotected using trifluoroacetic acid- m-cresol (95:5) mixture for 5 mins. This deprotection step was repeated two additional times followed by washing the resin with DCM and N,N-Dimethylformamide (DMF). The monomer was dissolved in a coupling solution comprising of a mixture of 0.2M N-Methyl pyrrolidone (NMP), 0.52M Di-isopropylethylamine (DIEA), and 0.39M O-Benzotriazole-N,N,N’,N’-tetramethyl-uronium-hexafluoro-phosphate (HBTU). The coupling solution was added to the reaction vessel and rocked for 2 hrs. The resin was capped using a capping solution (a mixture of NMP, Pyridine, and acetic anhydride). The resin was washed with DCM (8X). The entire process was repeated until the last monomer was added. 5-Carboxytetramethylrhodamine (TAMRA) was conjugated to N terminus of gamma tcPNA-155. The PNA was cleaved from the resin using a cleavage cocktail (thioanisole, m-cresol, TMFSA, TFA(1:1:2:6), and the vessel was rocked for 1.5 hrs. The PNA was collected and precipitated using diethyl ether and centrifuged at 3500 rpm for 5 mins. The PNA was washed with ether twice and vacuum dried. The PNA was purified by RP-HPLC and absorbance of the PNA was measured using Nanodrop One (Thermofisher Scientific, MA). The extinction coefficient of the individual monomers used for calculating PNA concentration (6,600 M−1cm−1 (C), 13,700 M−1cm−1 (A), 8,600 M−1cm−1 (T), 11,700 M−1cm−1 (G)).
Gel shift assay
Different concentration of PNA-155 or gamma tcPNA-155 were made in physiological buffer (2mM MgCl2, 150 mM KCl, 10 mM NaPi) and incubated overnight at 37°C with 1 μM DNA-155 target. The samples were run through 8% Polyacrylamide gel electrophoresis (PAGE) gel in IX TBE buffer at 120 volts for 35 mins. The gel was stained with SyBr Gold for two mins and visualized using Gel Doc EZ Imager (Bio-rad).
Cellular Uptake
U2932 are suspended celllines and were purchased from Leibniz Institute (DSMZ, Germany). The cells were regularly tested for mycoplasma contamination using MycoAlert mycoplasma detection kit (Lonza). The authenticity of the cell lines was confirmed by Human cell STR profiling service by ATCC. All the cells used in the experiment were passaged less than 8 times. 50,000 U2932 cells were seeded in twenty four well plate (37°C and 5% CO2). The cells were treated with gamma tcPNA-155 TAMRA (500 nM concentration). After 48 hrs, the cells were washed twice with PBS and then fixed using 4% paraformaldehyde (PFA) for 10 mins at room temperature. The cells were washed with PBS and then permeablized using 0.1% triton X for 10 mins at room temperature. The cells were washed with PBS and the cells were finally resuspended in 50 μL PBS. A drop of mounting media with DAPI (Life technologies) was placed on the slide. 10 μL of cells were mixed with the drop of DAPI on the slide and coverslip was placed on the slide. The slide was allowed to dry overnight and imaged using Keyence digital microscope.
For evaluating cellular uptake by flow cytometry — 400,000 U2932 cells were collected in a 12 well plate (37°C, 5% CO2). Three wells were untreated and three other cells were treated with 500 nM gamma tcPNA-155 TAMRA. After 48 hrs, the cells were washed twice with PBS and then fixed using 4% paraformaldehyde (PFA) for 10 mins at room temperature. The cells were passed through the FACS tube. Further, analysis was performed using LSR Fortessa X-20 Cell analyzer (BD Bioscience). The FACS data was plotted using FlowJo software.
Gene expression by RT-PCR
400,000 U2932 cells were seeded in a 12 well plate. The cells were treated with 500 nM PNA-155, gamma tcPNA-155, gamma PNA-155, Scr gamma tcPNA-155 or were PBS treated (control) for 48 hrs in an incubator (37 °C and 5% CO2). The cells were centrifuged at 2000 rpm for 4 mins at 4 °C. The total RNA from the cell pellet was extracted using RNeasy mini kit (Qiagen). The cDNA was prepared in thermal cycler (Bio-rad) using reverse transcriptase, RNase inhibitor, dNTPs, nuclease-free water and RT primers specific for miR-155 and U6. Random primers were used for the preparation of cDNA for downstream targets. The cDNA was amplified using miR-155 assay, U6 assay, or specific downstream target assays in CFX Connect Real-time PCR detection system (Bio-rad). The samples were subjected to polymerase activation (95°C for 10 mins), followed by 40 cycles of denaturation (95°C for 15 sec) and annealing (60°C for 1 min). The 2 - ΔΔCT method was used to calculate the fold change in target genes.
Similarly, 400,000 SUDHL-2 cells were seeded in a 12 well plate and treated with 500 nM Scr gamma tcPNA-155, PNA-155 and gamma tcPNA-155 for 48 hrs in an incubator (37°C and 5% CO2). The RNA was extracted and the samples for gene expression were prepared in the same manner as described above.
Cell viability by trypan blue assay
Diffused large B cell lymphoma (DLBCL) cell lines like SUDHL-2 (ATCC® CRL-2956™) and SUDHL-5 (ATCC® CRL-2958™) cells were purchased from ATCC (Virginia, USA). The cells were regularly tested for mycoplasma contamination using MycoAlert mycoplasma detection kit (Lonza). All the cells used in the experiment were passaged less than 8 times. 10,000 U2932, SUDHL-5, or SUDHL-2 cells were plated in a 96 well plate. The cells were treated with different doses (500 nM, 1000 nM, 2000 nM and 4000 nM) of PNA-155, gamma tcPNA-155 or Scr-gamma tcPNA-155 for 48 hrs in an incubator (37 °C and 5% CO2). The dead cells were marked with trypan blue. Further counting was performed using an automated cell counter (Bio-rad).
Apoptosis Assay
400,000 U2932 cells were seeded in a 12 well plate. The cells were treated with 500 nM PNA-155, gamma PNA-155, gamma tcPNA-155, Scr-gamma tcPNA-155 or were PBS treated (control) for 48 hrs in an incubator (37 °C, 5% CO2). The cells were washed with PBS twice. The cells were centrifuged at 2000 rpm for 4 mins at 4 °C. The cell pellet was suspended in IX Annexin V binding buffer. The cells were then counted and 100 μL of cell suspension (containing 2.5x105 cells) was passed through the FACS tube. The cells were stained with 12.5 μL Phycoerythrin (PE) Annexin dye and 12.5 μL 7-Amino-Actinomycin (7AAD) and kept in dark for 15 mins. 400 μL of IX Annexin V binding buffer was added to the cells and the cells were then analyzed using LSR Fortessa X-20 Cell analyzer as indicated above.
For Annexin V FITC stained fluorescent imaging method- 10,000 U2932 cells were seeded in 96 well plate (37°C and 5% CO2). The cells were treated with Scr-γtcPNA-155, PNA-155, γPNA-155, γtcPNA-155 (500 nM concentration) for 48 hours. Annexin V FITC diluted 1:10 in 1X Annexin binding buffer was supplemented to each well. The plate was kept at room temperature for 15 mins. The cells were imaged using 10x lens on Keyence digital microscope.
Western blot
400,000 U2932 cells were collected in a 12 well plate and treated with 500 nM PNA-155, gamma PNA-155, gamma tcPNA-155 or Scr-gamma tcPNA-155 for 48 hrs in an incubator. The cell pellet was collected by centrifuging at 2000 rpm for 4 mins at 4 °C. 1X RIPA buffer and 1X protease inhibitor were added to the cell pellet and subjected to intermittent vortexing after 10 mins (3X) to extract the proteins from the cell pellet. The protein was collected after centrifuging the tube at 10,000 rpm for 10 mins at 4 °C. The protein concentration was measured by Lowry protein assay. About 25 μg protein was loaded on SDS PAGE gel (4–20% MP TGX stain-free gels, Bio-rad) and separated at 101 Volts for 90 mins. The proteins were transferred from the gel to a PVDF membrane at 110 Volts for 90 mins. The PVDF membrane was blocked using 5 % milk in 1X tris buffered saline for 1 hr. The western blotting was performed using the following antibodies: Mcl-1 (39224), CASP3 (9622S), Vinculin (13901), antirabbit IgG HRP linked antibody (7074) (Cell Signaling Technology). The blots were imaged using ChemiDoc™ Imaging System (Bio-rad). Protein expression intensities were determined using Image J software and normalized relative to loading control and treatment control.
Safety assessment by trypan blue assay
Primary blood mononuclear cells (PBMC) (ATCC® PCS-800–011™) were purchased from ATCC (Virginia, USA). The cells were regularly tested for mycoplasma contamination using MycoAlert mycoplasma detection kit (Lonza). All the cells used in the experiment were passaged less than 2 times. 10,000 PBMC cells were seeded in a 96 well plate. The cells were treated with 500 nM, 1000 nM, 2000 nM and 4000 nM PNA-155 and γtcPNA-155 for 48 hrs in an incubator. The dead cells were examined with trypan blue and further counted using an automated cell counter (Bio-rad).
Study approval
All the animals’ experiments were performed at the University of Connecticut, Storrs campus, in compliance and approved by the Institutional Animal Care and Use Committee (IACUC). The authorization number for approved IACUC protocol is A21–041.
Mouse tumor xenograft
Female NSG mice (NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ, strain 005557) weighing 20–23g were procured from Jackson laboratories. 1x107 U2932 cells suspended in RPMI-1640 medium were injected subcutaneously in the right and left flank of 6-week-old NSG mice. The mice developed a small bump in 2 weeks. When the tumor volume reached 100–200mm3, the mice were randomly assigned to the treatment group (n=6 per group).
For SUDHL-2 xenografts, 1x107 SUDHL-2 cells suspended in RPMI-1640 medium were injected subcutaneously in the right and left flank of 6-week-old NSG mice. The mice developed a small bump in 2–3 weeks. When the tumor volume reached 100–200mm3, the mice were randomly allocated to the treatment group (n=5 per group).
Biodistribution
The γtcPNA-155 TAMRA PNA was intra-tumorally injected in the NSG mice (n=3) bearing 200 mm3 tumors at the dose of 3mg kg-1 dose at an interval of 1 week. When the tumor volume reached 2000 mm3, the mice were euthanized by CO2 inhalation. The tumors were harvested and imaged using IVIS Spectrum system. The tumors were embedded in OCT compound. The tumors were cryosectioned at 10mm thickness using Leica cryostat. The tumor sections were fixed using 4% formaldehyde for 10 mins, followed by washing with PBS for 10mins. The tumor sections were then permeabilized using 0.2% triton X for 20 mins followed by washing with PBS. A drop of mounting media with DAPI (Life technologies) was placed on the tumor section and a coverslip was placed on it. The tumor sections were imaged using Keyence digital microscope.
RNA and Protein extraction from tumor samples
The mice were injected intra-tumorally with 1mg kg-1 dose of PNA-155, gamma tcPNA-155, gamma PNA-155, Scr gamma tcPNA-155 or were untreated. The injections were repeated two additional times after 1 week each. The length, breadth, and depth of the tumors were measured daily using a vernier caliper. The mice were euthanized when the tumor volumes reached 2000 mm3. The resected tumor sections were finely minced using a sterile blade and suspended in dissociation media (4 ml) comprising of RPMI-1640, 1.2 mg/ml dispase, and 0.5mg/ml collagenase for 90 mins at 500 rpm at 37 °C. The dissociated tumors were washed with buffer saline at 2500 rpm (4 mins) at 4 °C and then suspended in 0.25% trypsin for 4 mins at room temperature. RPMI 1640 media was added to the trypsinized tumor mass and the cells were passed through a 70 μm filter. The cells were centrifuged at 2500 rpm for 4 mins at 4 °C. The cell pellet was then suspended in 1X RBC lysis buffer (Sigma) and incubated on ice for 10 mins. PBS was added to the cells and the cells were passed through a 40 μm filter. The cells were centrifuged at 2500 rpm for 4 mins at 4 °C. The cell pellet was resuspended in 0.5% BSA in PBS. The mouse cells were removed from the tumor cells using a mouse cell depletion kit (Miltenyi Biotec) according to the manufacturer’s protocol. The enriched tumor cells from each tumor sample were divided into two fractions. RNA for gene expression analysis was extracted from one tumor fraction by the same procedure as mentioned earlier. The protein for western blot analysis was extracted from the second fraction using the method described above.
Histopathology and immunohistochemistry
The mice were sacrificed by CO2 inhalation when tumor volume reached 2000 mm3. The tumor and vital organs (e.g., liver, kidney, spleen, lungs, heart) were carefully isolated, weighed, and fixed in the 10% NBF solution. The sections (5 µM) of formalin-fixed paraffin-embedded liver and kidney were stained by hematoxylin and eosin for the histological analysis. The sections of 5 µM of the formalin-fixed paraffin-embedded tumor were heated (95oC, 20 min) in citrate buffer (10 mM) for antigen recovery. Further incubation was performed with primary antibodies. The concentrations of rabbit anti-Ki-67 (D2H10) and rabbit anti-Caspase-3 (9962) were 1:100. The antigen-primary antibody complexes were examined by fluorescent tagged secondary antibodies. Images were taken using a Zeiss confocal microscope (LSM 510).
Statistical analysis
Graphpad Prism 9 software (Version 9.2) was used for all statistical analyses. The data are reported as means SEM of triplicates, and the numbers and replicates are included in the Figure captions. Unpaired two-tailed and multiple student t-test was performed for experiments. For in vivo studies, animals were randomly assigned in groups to minimize the bias, and based on prior experience with animal models, sample size was selected(9,22). The statistical comparisons are significant when *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
RESULTS
Design and Synthesis of anti-miR-155 gamma tcPNA
We designed and synthesized anti-miR-155 gamma tcPNAs (γtcPNA) and control PNA oligomers to test in a series of lymphoma cell lines and a xenograft mouse model (Fig 1A). It has been established that tail clamp PNAs can only bind to target RNA sequences containing homopurine stretches (12). Homopurine stretches are present at the 5’ end of the miR-155 sequence (Fig 1B). Hence, we designed an anti-miR-155 gamma tcPNA containing Watson-Crick and Hoogsteen domain joined by flexible trioxo (-OOO-) linker to target the miR-155 sequence (Fig 1B & C, γtcPNA-155). In prior studies, it has been noted that gamma-modified PNAs exhibit superior binding and biocompatibility features compared to the conventional PNAs due to their right-handed helical pre-organization properties (23). γtcPNA-155 contains pseudoisocytosine (J) units on the Hoogsteen end as J can form hydrogen bonding at physiological pH (24). We also conjugated a TAMRA (5-Carboxytetramethylrhodamine) fluorophore to the 5’ terminal of γtcPNA-155 to study cellular uptake in cell lines and intra-tumoral biodistribution in the xenograft mice (Fig 1C, γtcPNA-155-Tam). For comparison, we synthesized full-length regular PNA (23mer long) as it has been demonstrated to inhibit miR-155 in prior studies (Fig 1C, PNA-155) (6). We synthesized single-stranded full-length serine gamma PNAs that can bind to target miR-155 by Watson-Crick base pairing (Fig 1C, γPNA-155) and a scrambled γtcPNA-155 (Fig 1C, Scr-γtcPNA-155) as control. For cell permeability, we appended two arginine residues to the C- and N-terminus of each PNA (25). We selected four arginine residues to minimize the cytotoxicity associated with cationic domains (9). The PNAs, γPNAs and γtcPNAs were synthesized by established solid-phase synthesis based protocols (26), and quality control analyses were performed by reverse-phase high-performance liquid chromatography (RP-HPLC) (Fig S1).
Figure 1. Design of PNA and gamma PNA-155 oligomers and gel shift binding assay.
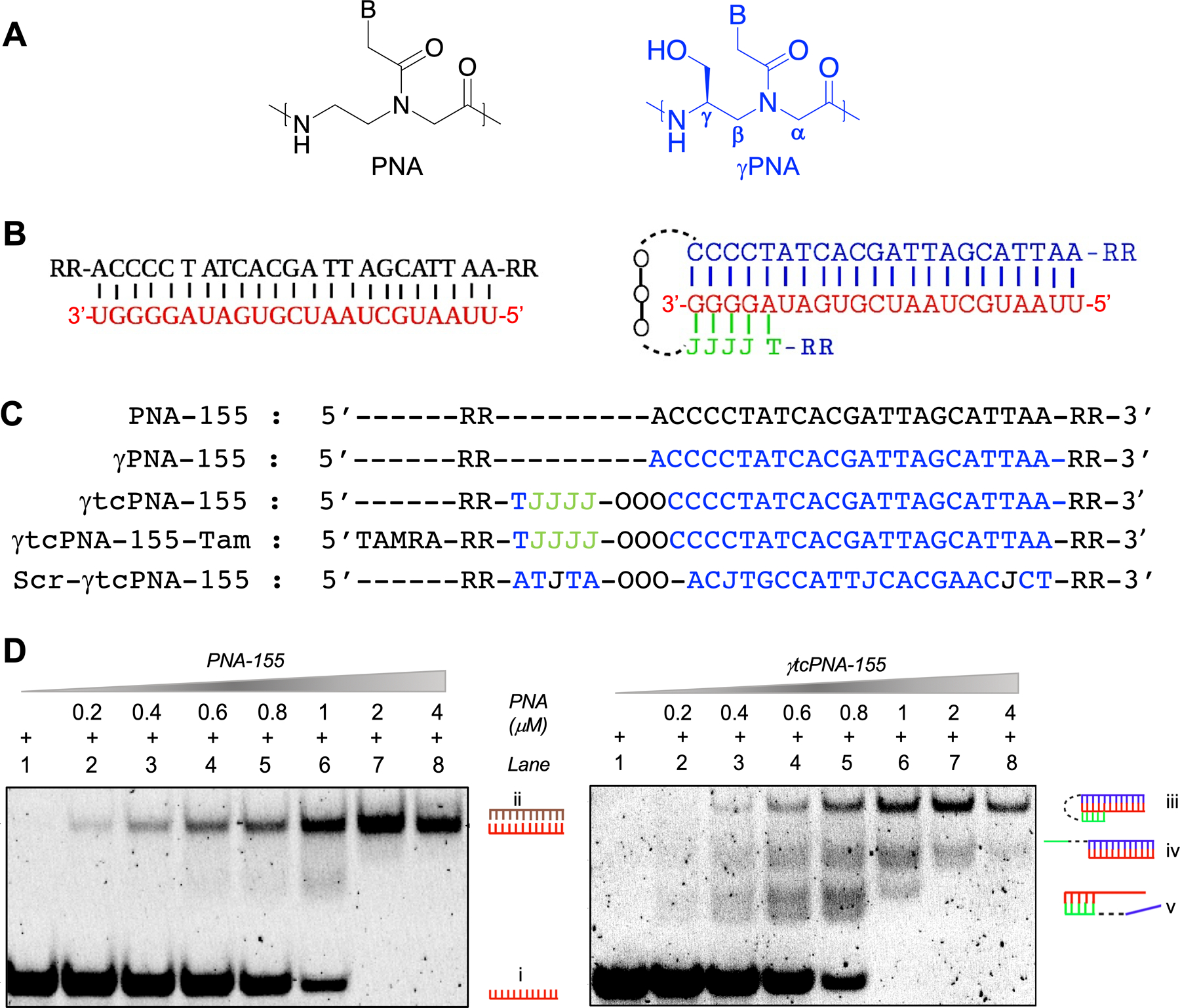
(A) Chemical structure of PNA and serine-γPNA units. B signifies nucleobases Adenine (A), Guanine (G), Cytosine (C) and Thymine (T). (B) Schematic of conventional full length PNA-155 and γtcPNA-155 binding with the target miR-155. (Left) PNA-155 binds by Watson-Crick base pairing whereas (Right) γtcPNA-155 binds by Watson-Crick and Hoogsteen binding domain. J signifies pseudoisocytosine nucleobase. Linker (11-Amino-3,6,9-Trioxaundecanoic Acid, DCHA) is represented as -OOO- (C) The oligomer sequences of PNA-155, γPNA-155 and γtcPNA-155 are designed to bind to the full length of target miR-155. Scramble PNA (Scr-γtcPNA-155) was synthesized as a control. TAMRA (5-Carboxytetramethylrhodamine) is appended to γtcPNA-155. The five PNAs have two arginine (R) residues on each N- and C- terminus ends. Blue are gamma residues. (D) Dose-dependent gel shift binding assay of target miR-155 (1μM) with PNA-155 and γtcPNA-155 at indicated concentrations. The samples were prepared in the physiological buffer (2mM MgCl2, 150 mM KCl, 10 mM NaPi) and incubated for 24 hours at physiological temperature (37°C) followed by PAGE separation and visualization of bands by SyBr Gold staining. Inset number shows different mode of binding (i) unbound miR-155 target (ii) PNA-155 binding with the target miR-155 by Watson-Crick domain. (iii) γtcPNA-155 binding with the target miR-155 by Watson Crick and Hoogsteen base pairing. (iv) γtcPNA-155 binding with the miR-155 by Watson-Crick base pairing. (v) Clamp segment of γtcPNA-155 binding with the target miR-155 by Hoogsteen base pairing.
In vitro binding studies
Next, we evaluated the binding affinity of γtcPNA-155 and PNA-155 with the miR-155 target by polyacrylamide gel electrophoresis (PAGE) based protocols (Fig 1D). We incubated PNA-155 and γtcPNA-155 with miR-155 at indicated concentrations in physiological buffer and temperature and assessed the binding by PAGE followed by SYBR gold staining. As expected, we noticed an increase in the formation of a retarded band (PNA-155-miR-155 bound fraction) with the increase in concentration of PNA-155. We also noted the complete disappearance of the unbound miR-155 target at a miR-155:PNA-155 stoichiometry ratio of 1:2.0 (Fig 1D, left panel). Whereas in the case of γtcPNA-155, we noticed the complete disappearance of unbound miR-155 target at miR-155:γtcPNA-155 stoichiometry ratio of 1:1.0 (Fig 1D, right panel). These results confirmed that γtcPNA-155 possesses a stronger binding affinity compared to regular PNA-155. We also observed three distinct retarded band patterns in the case of γtcPNA-155-incubated with target miR-155. Three separate retarded bands are consistent with tcPNA binding modes with the complementary target strands. The top retarded band corresponds to the presence of γtcPNA-155 Watson-Crick, and the Hoogsteen domain bonded to miR-155 target. The other two retarded bands indicate binding with either Watson-Crick or Hoogsteen domain of γtcPNA-155 with the miR-155 target.
Cellular uptake and efficacy studies in lymphoma cell lines
Various studies established that miR-155 is upregulated in B-cell malignancies and is recognized as a therapeutically bona fide molecular target for treating lymphoma (6). Hence, we performed cellular uptake studies of γtcPNA-155-Tam in lymphoma cell lines (U2932 and SUDHL-2) by flow cytometry analysis. Significant cellular uptake of the TAMRA was observed in the U2932 (Fig 2A) and SUDHL-2 cells (Fig S2A) 24 hours’ post-incubation with γtcPNA-155-TAMRA without using any transfection agent. Further, we also confirmed the cellular uptake of γtcPNA-155-TAMRA in U2932 cells by confocal microscopy (Fig S2B).
Figure 2. Cell culture-based functional assay in U2932 lymphoma cells.
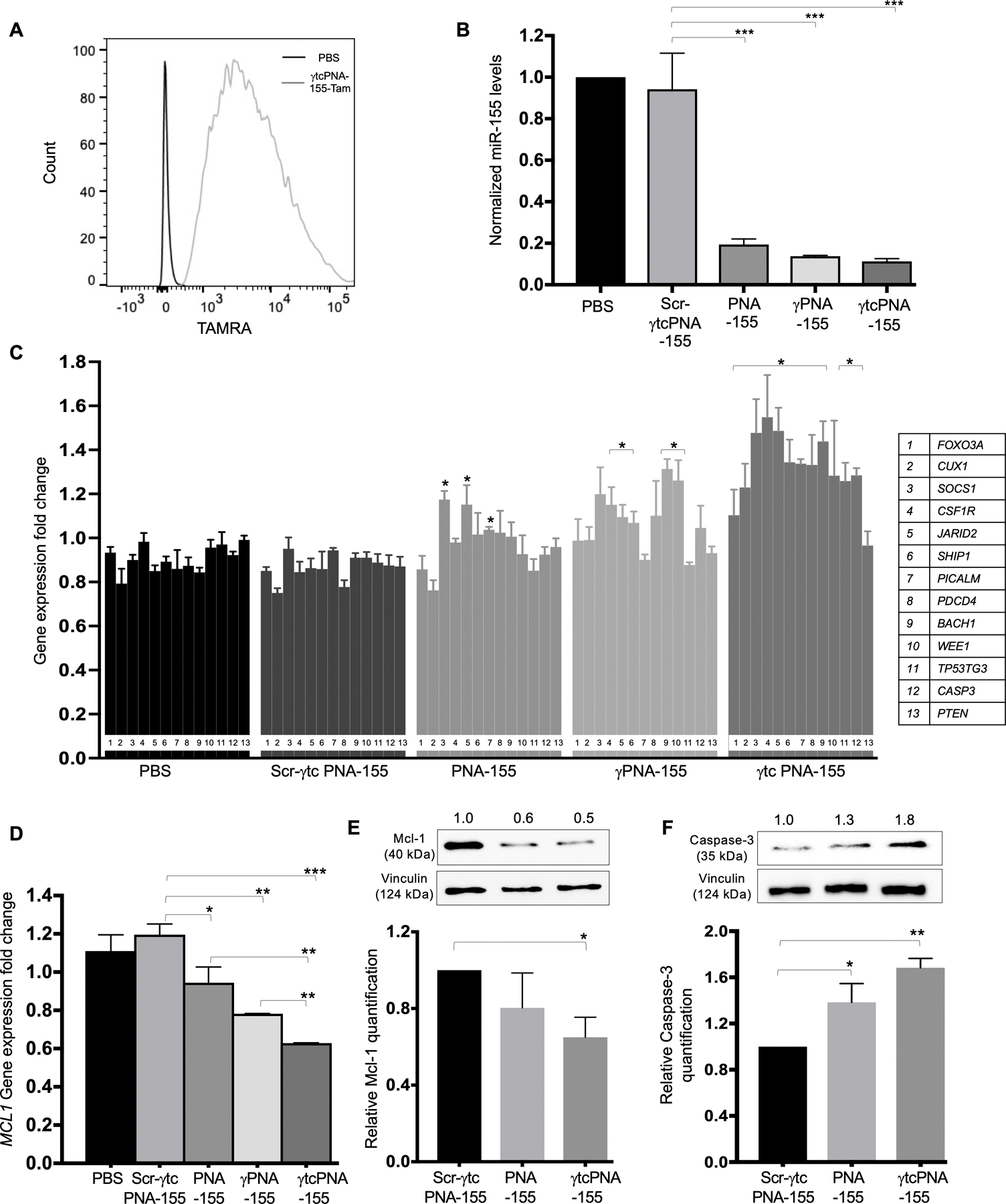
(A) γtcPNA-155-TAMRA uptake in the U2932 lymphoma cell line: Representative flow cytometry traces of TAMRA fluorescence in U2932 cells after treatment with 500 nM dose of γtcPNA-155-TAMRA for 48 hours. The data was analyzed by FlowJo software (B) Normalized miR-155 gene expression levels in U2932 after treatment with phosphate buffer saline (PBS) as a control and with 500 nM dose of Scr-γtcPNA-155, PNA-155, γPNA-155 and γtcPNA-155 for 48 hours compared to average control U6 (n=3), data represented as mean ± standard error mean (SEM), Statistical analysis was performed using Unpaired two-tailed t-test. Further, statistical analysis was performed relative to Scr-γtcPNA-155 treated cells. ***p<0.001. (C) Gene expression level of miR-155 downstream genes, tumor suppressor proteins (FOXO3A, CUX1, SOCS1, CSF1R, JARID2, SHIP1, PICALM, PDCD4, BACH1, WEE1, TP53TG3, CASP3, PTEN) in U2932 cell line after treatment with PBS as a control and with after treatment with 500 nM of Scr-γtcPNA-155, PNA-155, γPNA-155 and γtcPNA-155 for 48 hours. Data is normalized with average GAPDH control (n=3), and represented as mean ± SEM, *p<0.05. Unpaired two-tailed t-test was used for statistical analysis and analysis was performed relative to Scr-γtcPNA-155 treated cells. (D) Gene expression level of miR-155 downstream genes MCL1 in U2932 cell line after treatment with PBS as a control and 500 nM of Scr-γtcPNA-155, PNA-155, γPNA-155 and γtcPNA-155 for 48 hours. Data is normalized with average GAPDH control (n=3), and represented as mean ± SEM, *p<0.05, **p<0.01 and ***p<0.001. Statistical analysis was performed using Unpaired two-tailed t-test. Analysis was performed relative to Scr-γtcPNA-155 treated cells. (E) Representative western blot of Mcl-1 and its quantification (n=3 technical replicate) and (F) Caspase-3 protein and its quantification (n=3 technical replicate) in U2932 cell line after treatment with 500 nM of Scr-γtcPNA-155, PNA-155 and γtcPNA-155 for 48 hours. Data is represented as mean ± SEM and unpaired two-tailed t-test was used for statistical analysis. * p<0.05 **p <0.01. Numbers above western blot panels represent relative quantification of the respective bands using ImageJ software, and normalized relative to loading control and treatment control.
Next, we assessed the miR-155 inhibitory activity of γtcPNA-155, γPNA-155 and PNA-155 in the U2932 cell line. U2932 cells were treated with a 500nM dose of Scr-γtcPNA-155, PNA-155, γPNA-155 and γtcPNA-155 for 48 hours. Our RT-PCR results confirmed ~80%, 83% and 90% miR-155 inhibition with PNA-155, γPNA-155 and γtcPNA-155 treatments, respectively relative to scrambled control (Fig 2B).
To verify miR-155 inhibition by the antimiRs, we evaluated the gene expression of known miR-155 downstream genes by RT-PCR-based gene expression analysis. High miR-155 gene expression results in repression of tumor suppressor genes via activation of the PI3-AKT signaling pathway, thereby promoting tumor cell survival, proliferation, and progression (27). Therefore, downregulating miR-155 will result in the de-repression of tumor suppressor genes. We examined the gene expression of panel of known miR-155 associated tumor suppressor genes by RT-PCR analysis after treatment with γtcPNA-155, γPNA-155 and PNA-155 (Fig 2C) (22). We noted significant de-repression of miR-155 target tumor suppressor genes in the U2932 cells treated with γPNA-155 and γtcPNA-155. In particular, CSF1R, CUX1, and SHIP1 negatively regulate PI3-AKT signaling and are a direct target of miR-155 (28–30). We noted a 1.8-fold increase in CSF1R levels, a 1.6-fold increase in CUX1 levels and a 1.4-fold increase in the SHIP1 levels in γtcPNA-155 treated groups in comparison to PNA-155. Overall, we noted that γtcPNA-155 followed by γPNA-155 shows optimal de-repression of tumor suppressor genes in comparison to PNA-155. One plausible explanation of these findings is that γtcPNA-155 has a higher binding affinity with the miR-155 target.
In addition to the tumor suppressors, miR-155 is also known to impact expression of oncogenes. MCL1 is a known miR-155 downstream oncogene (20,31). Our gene expression results confirmed a 40% reduction in MCL1 gene expression levels after pre-treatment with γtcPNA-155 (Fig 2D). γPNA-155 and PNA-155 pre-treatment results in 19% and 10% decline in MCL1 gene expression, respectively. We confirmed the effect of γtcPNA-155 on the validated miR-155 downstream genes MCL1 and Caspase-3 (CASP3) by western blot analysis. Caspase-3 has also been identified as a one of the direct targets of miR-155 (32). Our results indicated that U2932 pretreatment with γtcPNA-155 at 500nM concentration led to significant downregulation of Mcl-1 (50%) (Fig 2E and S3A) and upregulation of Caspase-3 (80%) (Fig 2F and S3B).
Next, we assessed the efficacy of γtcPNA-155 in SUDHL-2 cell lines that exhibit overexpression of miR-155. Consistent with our aforementioned findings, RT-PCR results show that pre-treatment of SUDHL-2 cell lines with γtcPNA-155 results in a 63% decrease in miR-155 gene expression (Fig S4). We also investigated that the pre-treatment of SUDHL-2 cell lines with γtcPNA-155 results in significant upregulation of miR-155 associated tumor suppressor genes in comparison to Scr-γtcPNA-155 and PNA-155 (Fig S5).
Reduction in tumor cell viability after antimiR treatment
Prior studies indicated that miR-155 inhibition results in a decrease in cell proliferation and viability (22). Therefore, we assessed the dose-dependent cell viability in multiple lymphoma cell lines, U2932, SUDHL-5, and SUDHL-2 by trypan blue assay. As expected, we noticed a dose-dependent reduction in cell viability in U2932, SUDHL-2, and SUDHL-5 cell lines. We observed ~80% decrease in cell viability in three cell lines at a 4μM concentration (Fig 3). We also noted that γtcPNA-155 pretreatment causes a significant reduction in cell viability in U2932, SUDHL-5 and SUDHL-2 cell lines at a lesser concentration of 500nM followed by γPNA-155 compared to the PNA-155 treated group (Fig 3 and Fig S6). These results are consistent with prior results signifying that γtcPNA-155 has a superior binding affinity followed by γPNA-155, and thus anti-miR-155 activity compared to regular full-length PNA-155. We did not notice any effect on cell viability with Scr-γtcPNA-155 in comparison to PBS treated control cells (Fig S7).
Figure 3. Dose-dependent cell viability in U2932, SUDHL-2, and SUDHL-5 cells after treatment with Scr-γtcPNA-155, PNA-155 and γtcPNA-155 for 48 hours.
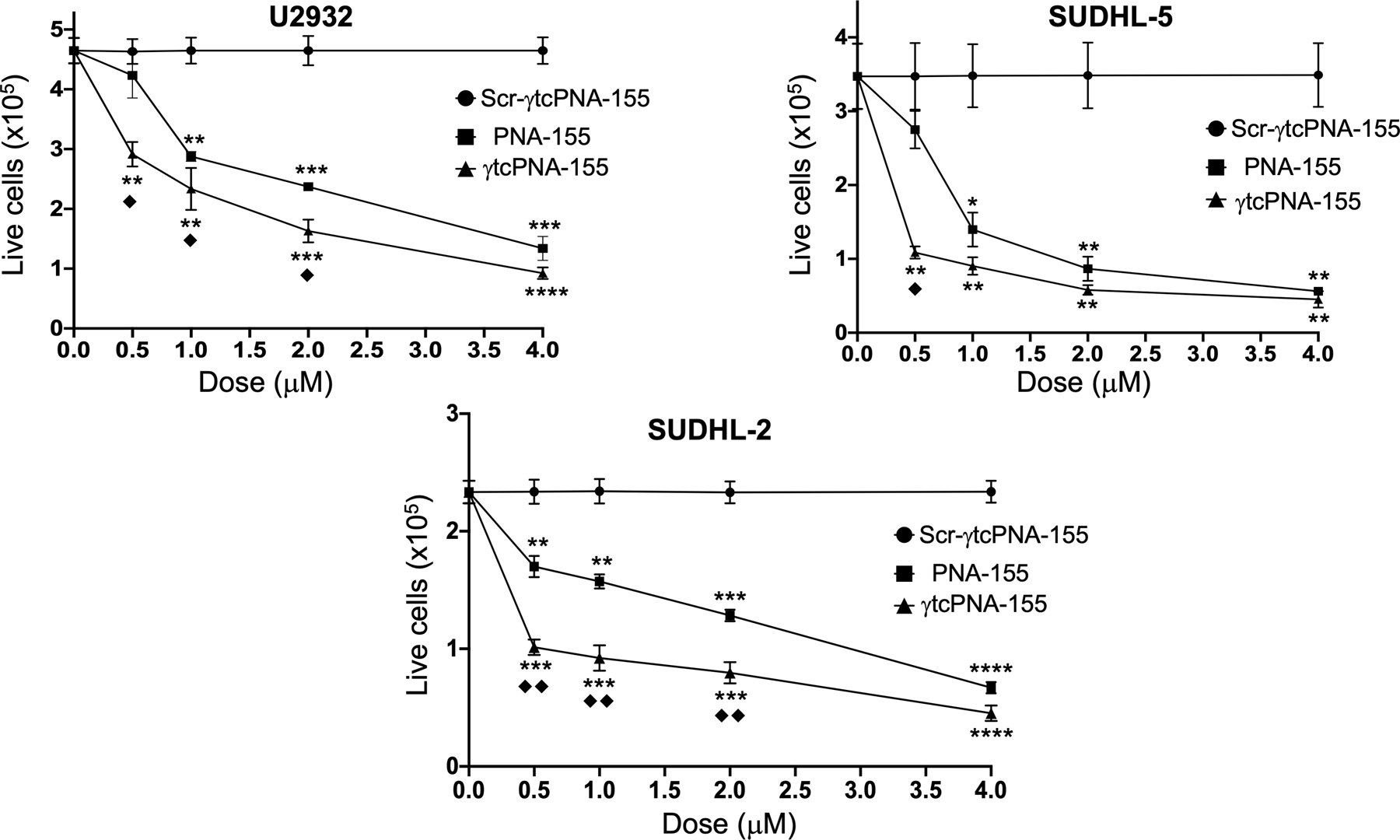
Cell viability was performed using trypan blue-based assay (n=3 technical triplicate), data represented as mean ± (SEM), * represents the statistical analysis were performed relative to Scr-γtcPNA-155 and ◆ represent analysis performed relative to PNA-155. *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001, ◆ p<0.05, ◆◆ p<0.01, unpaired two-tailed t-test was used for statistical analysis. The experiment was repeated three times and in triplicate.
Furthermore, to test whether the decrease in cell viability is due to apoptosis, we treated the U2932 cells with the same amount of γtcPNA-155 and PNA-155 and performed an annexin V-based apoptosis assay. Our results indicated that treatment with γtcPNA-155 results in increased apoptosis in U2932 cells than the PNA-155 and γPNA-155 treated group (Fig 4). We also performed confocal imaging on U2932 cells treated with indicated PNAs and assessed the apoptosis by Annexin-V FITC stained methods. Consistent with our flow cytometry-based results, we noted higher apoptosis in the γtcPNA-155 treated cells (Fig S8).
Figure 4. Quantification of apoptosis by Annexin-based assay.
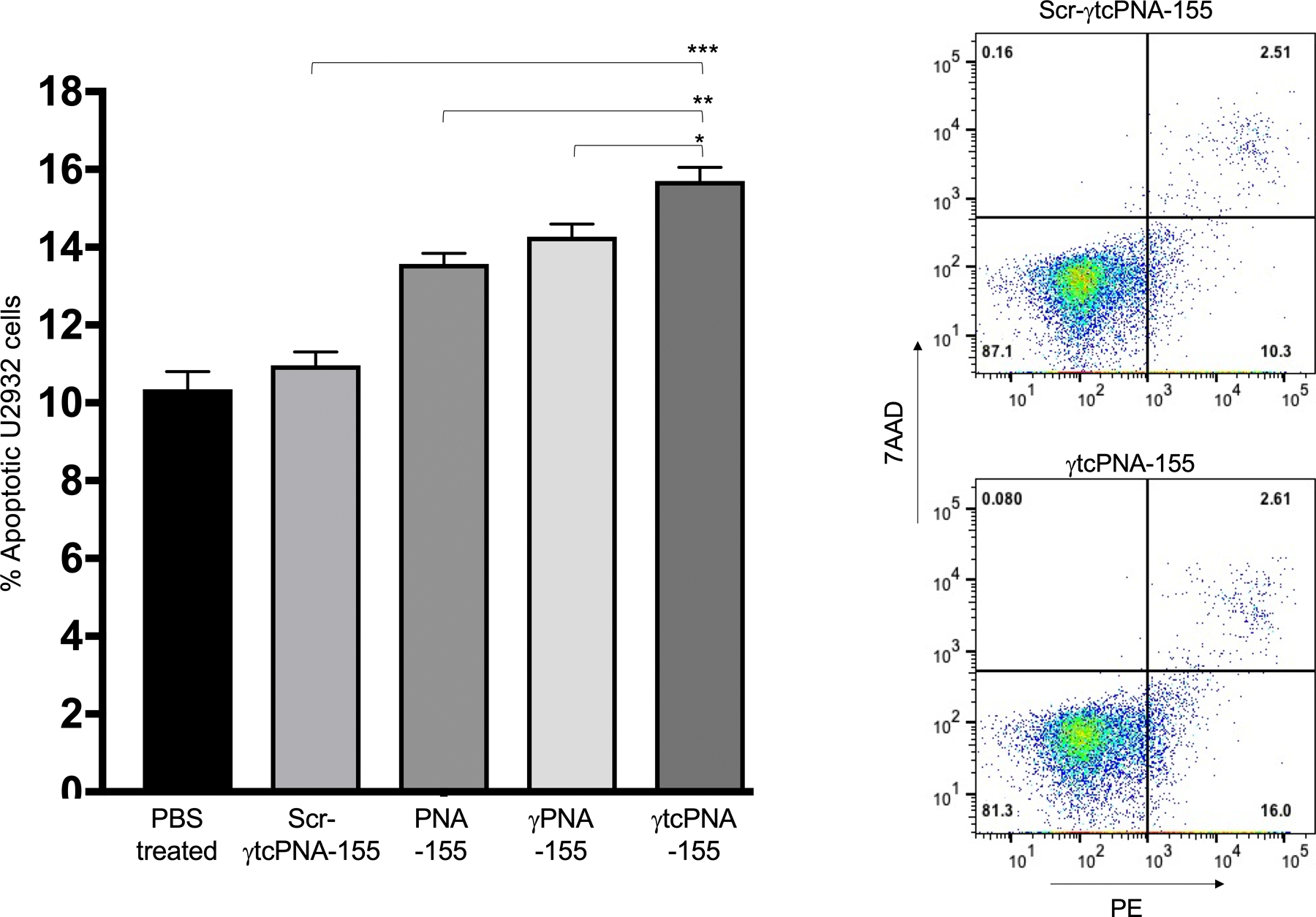
Quantification of apoptotic cells by flow cytometry after treating U2932 cells with PBS as a control and 500nM Scr-γtcPNA-155, PNA-155, γPNA-155 and γtcPNA-155 for 48 hours. The apoptotic cells and necrotic were stained using Phycoerythrin (PE) Annexin V and 7-Amino-Actinomycin (7-AAD) respectively. The % apoptotic cells after treatment was compared to the PBS treated cells, by setting the same threshold. Bar graph of % apoptotic cells (n=3 technical triplicate), data represented as mean ± standard error mean (SEM), *p<0.05, **p<0.01, ***p<0.001, unpaired two-tailed t-test was used for statistical analysis. Representative dot plots of Scr-γtcPNA-155 and γtcPNA-155 treated U2932 cells. The experiment was repeated three times and in triplicate. One out of three representative experiments are shown.
Prior studies successfully confirmed that pre-treatment of cobomarsen at a dose of 10μM results in 4-fold increase in apoptosis in the U2932 lymphoma cell line compared to control (22). In a parallel comparison, we also performed an apoptosis assay after pre-treatment of U2932 cells with γtcPNA-155 at a dose of 10μM. Our results indicated that the γtcPNA-155 result in a 4.5-fold increase in apoptosis in comparison to control (Fig S9). Hence, we suggest that γtcPNA-155 efficacy is comparable to cobomarsen in a cell culture-based analysis.
Overall, these results demonstrate that γtcPNA-155, followed by γPNA-155, led to robust inhibition of miR-155, decreased cell viability, and increased apoptosis in U2932 cell lines. We also evaluated the safety of PNA-155, and γtcPNA-155 in PBMC cell lines in a dose-dependent manner using trypan blue assay. We did not notice a significant reduction in viability of PBMC cell lines indicating that the γtcPNA-155 and Scr-γtcPNA-155 do not cause any non-specific effects in the primary cells (Fig S10).
γtcPNAs suppresses tumor growth in vivo
To evaluate if γtcPNA-155 can inhibit tumor growth more effectively than regular PNA-155 in vivo, we performed studies in xenografts derived from DLBCL cells. DLBCL U2932 cells were selected for implants as they show maximum miR-155 gene expression levels (33). Hence, they provide a robust xenograft model to study miR-155 inhibitory effects of γtcPNA-155. For our in vivo study, we assessed the anti-miR-155 impact of γtcPNA-155 and γPNA-155 compared to PNA-155 via intra-tumoral delivery. Cobomarsen, an investigative drug to inhibit miR-155 has also received orphan drug designation to treat lymphoma by intra-tumoral delivery. Since we selected the intra-tumoral route of delivery, we injected the U2932 cells in the right and left flank of mice (Fig S11). After 10–14 days, when the tumor volume reached 100–200 mm3, the mice were divided into the five treatment groups. We also tested scrambled PNA (Fig 1C, Scr-γtcPNA-155) as a control for in vivo study. The mice were randomized into groups based on the tumor volumes for uniform distribution of tumor volumes in each group. Before the efficacy study, we performed a bio-distribution analysis of TAMRA-conjugated γtcPNA-155 (Fig 1C, γtcPNA-155-Tam) at a dose of 3 mgkg-1 intra-tumoral in the xenograft. We noticed a significant biodistribution in tumor after intra-tumoral injection by IVIS imaging (Fig S12). We confirmed the biodistribution by confocal imaging of cryosections from the TAMRA treated group and control mice. Significant biodistribution of TAMRA fluorescence was noted in the tumor as compared to the control group (Fig 5A).
Figure 5. In vivo studies in U2932 derived xenograft model.
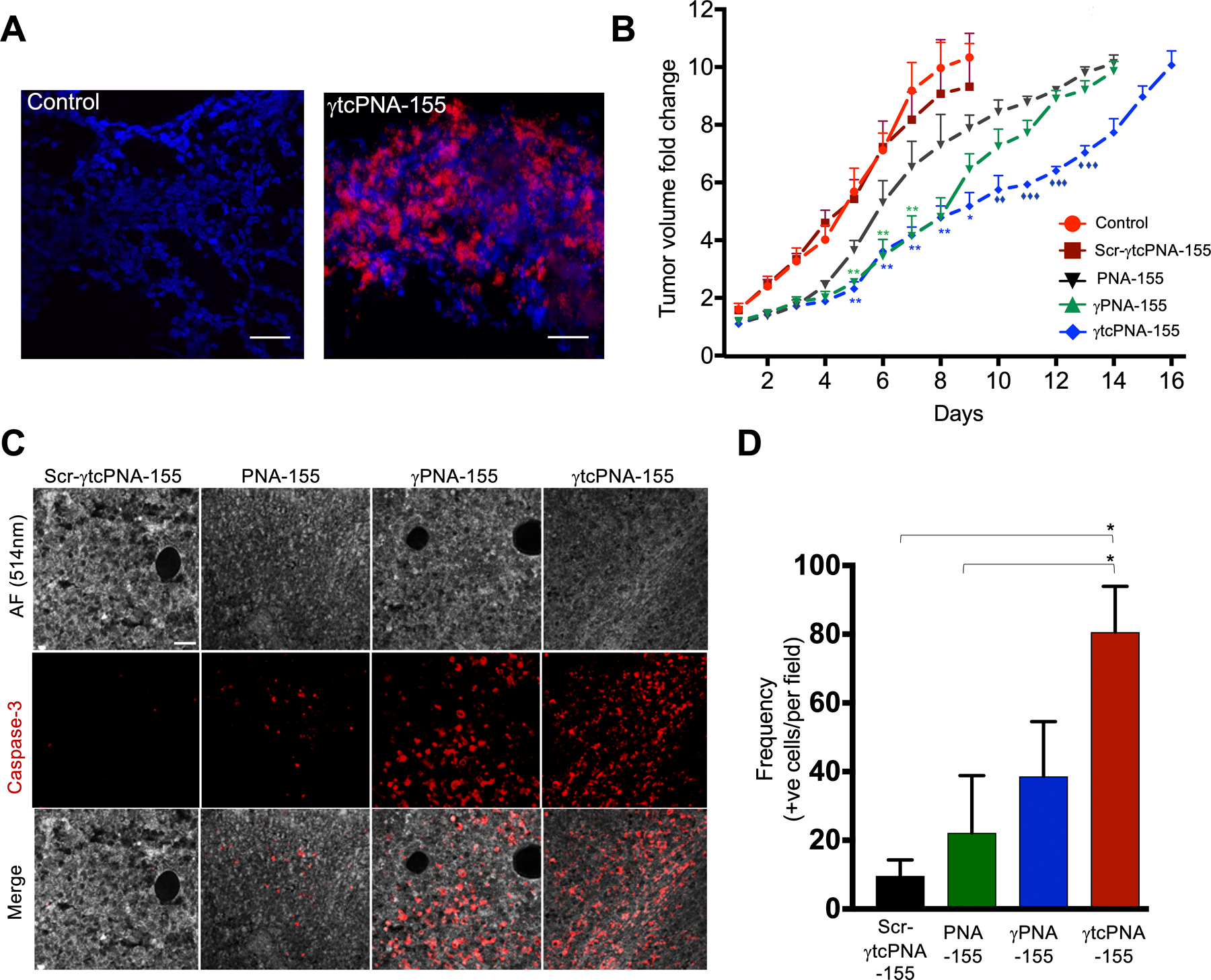
(A) Biodistribution of γtcPNA-155 TAMRA in tumor sections (B) Tumor volume fold change (n=6, data represented as mean ± standard error mean (SEM), *p<0.05, **p<0.01, multiple t-tests one per row was used for statistical analysis). Asterisk denotes the analysis was performed relative to Scr-γtcPNA-155. Diamond symbol denotes the analysis was performed relative to PNA-155. (C) Representative images showing the effects of Scr-γtcPNA-155, PNA-155, γPNA-155 and γtcPNA-155 on Caspase-3 immunostaining of tumor (magnification; ×20). Autofluorescence (AF) at 514 nm was used to show the tumor. Scale bar = 50μm. (D) Caspase-3 immunostaining quantification, *p<0.05. n=15. The n indicates the number of images.
For the efficacy study, each mouse received three intra-tumoral injections of either γtcPNA-155, γPNA-155 or PNA-155 on days 0, 7, 14. We noticed that the control and scrambled treated tumors reached 2000 mm3 much faster during our study, so we could not inject a third time in these mice. We did not observe: a significant decrease in the body weight (Fig S13); histological damage to the kidney and liver (Fig S14); general behavioral change in mice treated with indicated PNAs. Intra-tumoral administration reduced the tumor growth in the γtcPNA-155 treated group, followed by γPNA-155 and PNA-155 (Fig 5B and S15). In contrast, as stated earlier, there was no effect on the control and scrambled PNA control on the tumor volume.
To ascertain the efficacy of γtcPNA-155, the histological sections of tumors isolated from γtcPNA-155 treated mice were immunostained for apoptosis marker Caspase-3 (Fig 5C and 5D) and cell proliferation marker Ki67 (Fig S16A and S16B). We noted a significant decrease in the Ki67 positive cells and an increase in the Caspase-3 positive cells in the tumor sections isolated from γtcPNA-155 treated groups, which further corroborates the anti-tumor efficacy of γtcPNA-155.
We next assessed the levels of miR-155 and its downstream genes in the tumors of xenograft mice to assess the mechanism of tumor reduction. After tumor harvest, mouse and DLBCL cells were separated by a bead separation method to remove the false background from mice tissues. Subsequently, we investigated the level of miR-155 gene expression. Our results indicated that the γtcPNA-155 treated group shows a 90% decrease in miR-155 gene expression, whereas PNA-155 and γPNA-155 treated tumors indicated an 75% and 80% decline in the miR-155 expression respectively (Fig 6A). As anticipated, we did not observe a change in the miR-155 expression level in PBS treated cells and scrambled groups.
Figure 6. In vivo gene expression and protein level analysis.
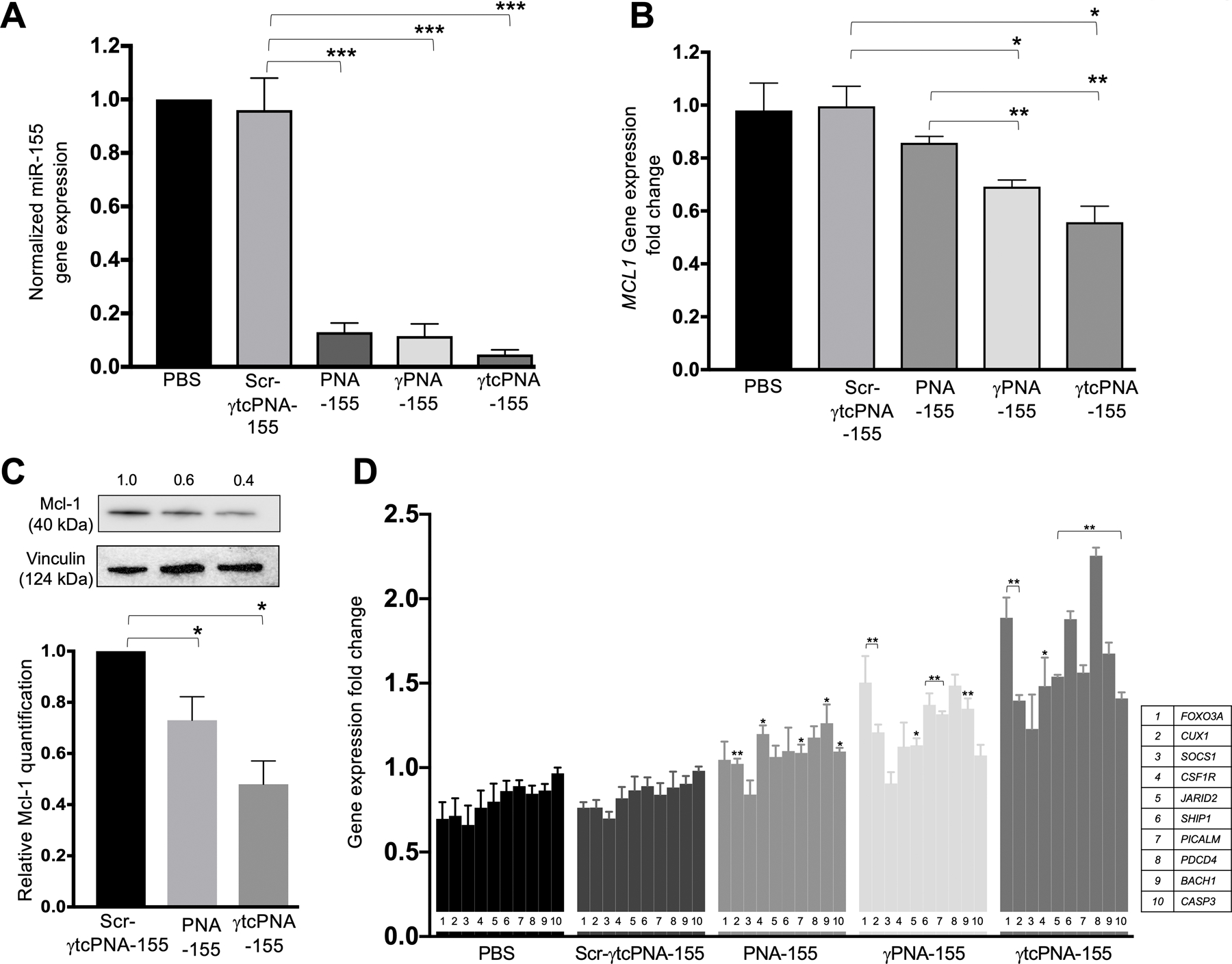
(A) miR-155 gene expression levels in U2932 tumor samples after in vivo treatment with indicated PNAs. Data was normalized with average control U6 (n=4, data represented as mean ± standard error mean). ***p<0.001 (B) Gene expression level of downstream target genes of miR-155: MCL1 in U2932 tumor samples after treatment with total 3mg kg-1 dose of Scr-γtcPNA-155, PNA-155, γPNA-155 and γtcPNA-155 and normalized with average control GAPDH (n=3, data represented as mean ± SEM), unpaired two-tailed t-test was performed for analysis). *p<0.05, **p<0.01(C) Representative western blot of Mcl-1protein and its quantification (n=3 technical replicate) in U2932 tumor samples treated with total 3mg kg-1 dose of Scr-γtcPNA-155, PNA-155 and γtcPNA-155. Data is represented as mean ± SEM and unpaired two-tailed t-test was performed for analysis. * p <0.05. The relative protein levels were determined from the band intensity using ImageJ software, and normalized relative to loading control and treatment control. (D) Gene expression level of tumor suppressor proteins (FOXO3A, CUX1, SOCS1, CSF1R, JARID2, SHIP1, PICALM, PDCD4, BACH1, CASP3) in U2932 tumor samples after treatment with total 3mg kg-1 dose of Scr-γtcPNA-155, PNA-155, γPNA-155 and γtcPNA-155 and normalized with average control GAPDH (n=3, data represented as mean ± SEM, unpaired two-tailed t-test was performed for statistical analysis). *p<0.05, **p<0.01.
Further, we measured the expression of miR-155 downstream genes, both tumor suppressor and oncogenes, as described in the in vitro result section. Upon γtcPNA-155 treatment, there was a significant reduction in the oncogenes and de-repression of tumor suppressor genes compared to the PNA-155 treated group. Compared to PNA-155 treated group, we observed about a 50% decrease in MCL1 mRNA levels in the γtcPNA-155 treated group (Fig 6B). Further, we confirmed these results by measuring reduction in the protein levels of Mcl-1 in γtcPNA-155 treated tumors (Fig 6C and Fig S17). Consistent with tumor growth studies, we did not notice any alteration in the gene expression and Mcl-1 protein level for the Scr-γtcPNA-155 treated groups (Fig S18). We also examined the in vivo de-repression of tumor suppressor genes as mentioned in the cell culture studies. Consistent with our cell culture-based results, we noted significant upregulation of tumor suppressor genes in the tumors of mice that received γtcPNA-155 intratumorally (Fig 6D). We also measured Caspase-3 protein levels in the tumors and observed a 23% increase in the in vivo γtcPNA-155-treated tumors than Scr-γtcPNA-155-treated tumors (Fig S19). These results collectively indicated that γtcPNA-155 exhibits a superior anti-miR-155 effect, followed by γPNA-155, decreasing tumor growth compared to its scrambled control and classical PNA-155.
Further, we corroborated our results in the SUDHL-2 derived xenograft mouse model. We injected the SUDHL-2 cells in the right and left flank of mice. After 14–21 days, when the tumor volume reached 100–200 mm3, the mice were randomized into three treatment groups based on uniform tumor volume distribution in each group. Consistent with prior in vivo results, we noted that γtcPNA-155 reduced the tumor growth as compared to the Scr-γtcPNA-155 and PNA-155 control (Fig S20). We further confirmed that PNA-155 significantly reduces miR-155 expression level in the SUDHL-2 cell line-based xenograft after in vivo treatment. (Fig. S21)
DISCUSSION
The antisense field has seen a rapid surge of FDA approvals of various nucleic acid-based drugs; Nusinersen, Onpattro, Gilvari, Milasen to name a few, targeting coding mRNA for diverse therapeutic applications (34). However, targeting non-coding RNAs like miRNAs for clinical applications lags behind and still needs further optimization as a broader platform. miRNAs have been established for their key roles in cancer progression and transformation (35). Especially, miR-155 is considered an important biomarker and highly expressed in B-cell lymphoma and DLBCL (36). Hence, in recent years, interest and research in this area has grown. miRagen Therapeutics (now Viridian Therapeutics) has made strides in developing cobomarsen as a drug candidate to target miR-155 for cutaneous T-cell lymphoma treatment. Cobomarsen has shown promise in decreasing miR-155 levels followed by reduced tumor burden. Recent studies also indicated that systemic delivery of cobomarsen shows favorable outcomes in patients that developed resistance to CHOP and CAR-T-cell therapy (22). Overall, these studies underpin the significance of miR-155 as an important molecular target for developing precision medicine for lymphoma therapy.
However, practicing miRNA therapeutics has been challenging due to delivery, plasma stability, and moderate efficacy of nucleic acid analogs-based antimiRs (37). Novel chemical modifications have been performed to increase the efficacy and enzymatic stability of next-generation antimiRs with minimal off-target effects. Though numerous synthetic nucleic acid analogs have been developed as potential antimiRs, LNA and PNA have gained enormous attention due to their enzymatic stability and superior physico-biochemical properties (38,39). Both full-length and seed-targeting LNAs have been explored as potential anti-miR-155 based therapeutics (10,40). Though LNA-based cobomarsen has shown promise, the field still needs to increase the antimiR-based repertoire that can be utilized for broader applications and targeting of diverse miRNAs with increased efficacy.
PNA has been widely used as antimiR for targeting full-length miRNA. Apart from its binding properties, PNA-based technology has been amenable to several delivery platforms like nanoparticles (41), liposomes (42), and peptide conjugations (43), to target the tumor microenvironment and inhibit the target miRNA selectively. Conventional regular PNA targets full-length miRNAs and inhibits their target mRNA interaction by steric hindrance (44). Herein, we tested novel gamma modified tcPNA antimiRs that can bind with both Watson-Crick and Hoogsteen base pairing with a miR-155 target containing a homopurine stretch and inhibit its activity. In prior studies, it has been well-established that gamma tcPNAs can induce a higher percentage of gene editing than the regular PNA due to their high binding affinity (15,17,45). We report that gamma tcPNAs can inhibit the miR-155 at a higher level than conventional full-length PNAs.
Herein, we also noted that full-length serine-gamma PNA causes increased miR-155 inhibition in both cell culture and in vivo analysis compared to conventional PNA-155. Prior studies indicated that poly-lactic-co-glycolic acid (PLGA) nanoparticles loaded with diethylene-glycol units containing gamma anti-miR-210 PNA results in significant miR-210 inhibition in a HeLa cell line derived xenograft (23). Diethylene-glycol containing gamma PNAs have enhanced loading and release in the PLGA NPs as compared to the regular PNA-210, resulting in increased antimiR efficacy. To the best of our knowledge, our study here is the first where a parallel comparison of the antimiR efficacy of cationic serine gamma PNAs and conventional PNAs based on their binding affinity has been performed. In addition, serine gamma PNAs have not been explored before for in vitro and in vivo antimiR efficacy. Though we noted that gamma tcPNAs show superior inhibition due to the gamma substitutions and the presence of Hoogsteen binding domains, it would also be interesting to study the full-length gamma PNAs for targeting mixed sequence containing target miRNAs.
We performed an extensive analysis of gamma tcPNA’s anti-miR-155 activity in both cell culture and in vivo analysis. We validated our anti-miR-155 results by gene expression and protein level analysis of direct and indirect miR-155 targets in U2932 lymphoma cell lines. We demonstrated that repressing miR-155 levels decreases cell viability in multiple lymphoma cell lines SUDHL-2, SUDHL-5, and U2932. We also found that gamma tcPNA-155 induced apoptosis in vitro. Our in vivo results in the xenograft mice indicated that gamma tcPNA design could inhibit miR-155 and further effect the miR-155 downstream targets in vivo more efficiently compared to regular PNA-155. Further, a decrease in cellular proliferation and an increase in apoptosis markers mechanistically supports the retardation of tumor growth in mice receiving intra-tumoral injections of gamma tcPNA-155. Prior studies successfully demonstrated that pre-treatment of cobomarsen at a dose of 10µM results in 4-fold increase in apoptosis in the U2932 lymphoma cell line compared to control. Our results indicate that treatment of U2932 cell lines with gamma tcPNAs at a dose of 10μM causes a 4.5-fold increase in apoptosis compared to control. Hence, we examined that gamma tcPNA results are comparable to cobomarsen based on in-vitro studies. Various important miR-155 targets are involved in lymphoma cell proliferation. In a prior study, it has been noted that CUX1 and WEE1 are established miR-155 predicted targets and play an essential role in tumor proliferation (22,46). In both in vitro and in vivo analysis, we noted that gene expression of multiple miR-155 targets, including CUX1 and WEE1, increased after treatment with gamma tcPNA-155.
We have presented a novel antimiR strategy that could target multiple miRNA-based molecular targets for diverse therapeutic applications. One possible limitation of gamma tcPNA design is that it can only target miRNA-containing homopurine stretches. Still, numerous miRNAs containing homopurine stretches play a crucial role in various cancers and other metabolic diseases. It would be noteworthy to target other miRNAs that contain more than five homopurine stretches. It has been demonstrated that targeting longer homopurine stretches results in stronger binding affinity and increases the PNA-mediated gene-editing efficacy (47). Hence, we would anticipate that preorganized features of gamma PNAs and targeting the extended Hoogsteen domain will enhance antimiR efficacy. In addition, it would be interesting to study the direct systemic delivery of gamma tcPNAs in conjunction with nanoparticles or pH low insertion peptide (pHLIP)-based technology that directly targets these novel antimiRs in the acidic tumor microenvironment.
In conclusion, an efficient antimiR-based repertoire is required to target the miRNAs that are involved in disease pathogenesis. Our results demonstrate that gamma-modified tail-clamp- PNA-based anti-miR-155 not only improves its affinity toward the target miR-155 but also increases its efficacy in retarding tumor growth. Further, gamma-modified tail-clamp-PNA-based antimiRs appeared safe in our mice throughout treatment. Hence, gamma tail clamp PNA-based molecules hold the potential to be used in combination with other drugs like R-CHOP and radiation therapy as adjuvant therapy to improve the drug response of DLBCL patients. Lastly, the gamma tail-clamp PNA technology can also be employed to target mRNAs and other non-coding RNAs involved in the development of other health disorders.
Supplementary Material
SIGNIFICANCE.
This study demonstrates the utility of novel oncomiR inhibitors as cancer therapeutics, providing a new approach for targeting microRNAs and other noncoding RNAs.
ACKNOWLEDGEMENTS
This work was supported by Charles H. Hood Foundation Grant (to R.B) and National Institutes of Health (CA241194 to R.B. and F.J.S).
Footnotes
AUTHORS’ DISCLOSURES
The authors declare no competing interests.
REFERENCES
- 1.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 2004;116:281–97 [DOI] [PubMed] [Google Scholar]
- 2.Hatziapostolou M, Polytarchou C, Iliopoulos D. miRNAs link metabolic reprogramming to oncogenesis. Trends Endocrinol Metab 2013;24:361–73 [DOI] [PubMed] [Google Scholar]
- 3.Peng Y, Croce CM. The role of MicroRNAs in human cancer. Signal Transduct Target Ther 2016;1:15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Baumann V, Winkler J. miRNA-based therapies: strategies and delivery platforms for oligonucleotide and non-oligonucleotide agents. Future Med Chem 2014;6:1967–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pellestor F, Paulasova P. The peptide nucleic acids (PNAs), powerful tools for molecular genetics and cytogenetics. Eur J Hum Genet 2004;12:694–700 [DOI] [PubMed] [Google Scholar]
- 6.Babar IA, Cheng CJ, Booth CJ, Liang X, Weidhaas JB, Saltzman WM, et al. Nanoparticle-based therapy in an in vivo microRNA-155 (miR-155)-dependent mouse model of lymphoma. Proc Natl Acad Sci U S A 2012;109:E1695–704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cheng CJ, Bahal R, Babar IA, Pincus Z, Barrera F, Liu C, et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature 2015;518:107–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Broderick JA, Zamore PD. MicroRNA therapeutics. Gene Ther 2011;18:1104–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Malik S, Lim J, Slack FJ, Braddock DT, Bahal R. Next generation miRNA inhibition using short anti-seed PNAs encapsulated in PLGA nanoparticles. J Control Release 2020;327:406–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Obad S, dos Santos CO, Petri A, Heidenblad M, Broom O, Ruse C, et al. Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet 2011;43:371–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Economos NG, Oyaghire S, Quijano E, Ricciardi AS, Saltzman WM, Glazer PM. Peptide Nucleic Acids and Gene Editing: Perspectives on Structure and Repair. Molecules 2020;25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ricciardi AS, Quijano E, Putman R, Saltzman WM, Glazer PM. Peptide Nucleic Acids as a Tool for Site-Specific Gene Editing. Molecules 2018;23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Quijano E, Bahal R, Ricciardi A, Saltzman WM, Glazer PM. Therapeutic Peptide Nucleic Acids: Principles, Limitations, and Opportunities. Yale J Biol Med 2017;90:583–98 [PMC free article] [PubMed] [Google Scholar]
- 14.Sahu B, Sacui I, Rapireddy S, Zanotti KJ, Bahal R, Armitage BA, et al. Synthesis and characterization of conformationally preorganized, (R)-diethylene glycol-containing gamma-peptide nucleic acids with superior hybridization properties and water solubility. J Org Chem 2011;76:5614–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bahal R, Ali McNeer N, Quijano E, Liu Y, Sulkowski P, Turchick A, et al. In vivo correction of anaemia in beta-thalassemic mice by gammaPNA-mediated gene editing with nanoparticle delivery. Nat Commun 2016;7:13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McNeer NA, Anandalingam K, Fields RJ, Caputo C, Kopic S, Gupta A, et al. Nanoparticles that deliver triplex-forming peptide nucleic acid molecules correct F508del CFTR in airway epithelium. Nat Commun 2015;6:6952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ricciardi AS, Bahal R, Farrelly JS, Quijano E, Bianchi AH, Luks VL, et al. In utero nanoparticle delivery for site-specific genome editing. Nat Commun 2018;9:2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Singer A, Rapireddy S, Ly DH, Meller A. Electronic barcoding of a viral gene at the single-molecule level. Nano Lett 2012;12:1722–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cui B, Chen L, Zhang S, Mraz M, Fecteau JF, Yu J, et al. MicroRNA-155 influences B-cell receptor signaling and associates with aggressive disease in chronic lymphocytic leukemia. Blood 2014;124:546–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eis PS, Tam W, Sun L, Chadburn A, Li Z, Gomez MF, et al. Accumulation of miR-155 and BIC RNA in human B cell lymphomas. Proc Natl Acad Sci U S A 2005;102:3627–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seto AG, Beatty X, Lynch JM, Hermreck M, Tetzlaff M, Duvic M, et al. Cobomarsen, an oligonucleotide inhibitor of miR-155, co-ordinately regulates multiple survival pathways to reduce cellular proliferation and survival in cutaneous T-cell lymphoma. Br J Haematol 2018;183:428–44 [DOI] [PubMed] [Google Scholar]
- 22.Anastasiadou E, Seto AG, Beatty X, Hermreck M, Gilles ME, Stroopinsky D, et al. Cobomarsen, an Oligonucleotide Inhibitor of miR-155, Slows DLBCL Tumor Cell Growth In Vitro and In Vivo. Clin Cancer Res 2021;27:1139–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gupta A, Quijano E, Liu Y, Bahal R, Scanlon SE, Song E, et al. Anti-tumor Activity of miniPEG-gamma-Modified PNAs to Inhibit MicroRNA-210 for Cancer Therapy. Mol Ther Nucleic Acids 2017;9:111–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Egholm M, Christensen L, Dueholm KL, Buchardt O, Coull J, Nielsen PE. Efficient pH-independent sequence-specific DNA binding by pseudoisocytosine-containing bis-PNA. Nucleic Acids Res 1995;23:217–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ghavami M, Shiraishi T, Nielsen PE. Cooperative Cellular Uptake and Activity of Octaarginine Antisense Peptide Nucleic acid (PNA) Conjugates. Biomolecules 2019;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Manna A, Rapireddy S, Bahal R, Ly DH. MiniPEG-gammaPNA. Methods Mol Biol 2014;1050:1–12 [DOI] [PubMed] [Google Scholar]
- 27.Fu X, Wen H, Jing L, Yang Y, Wang W, Liang X, et al. MicroRNA-155–5p promotes hepatocellular carcinoma progression by suppressing PTEN through the PI3K/Akt pathway. Cancer Sci 2017;108:620–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Faraoni I, Antonetti FR, Cardone J, Bonmassar E. miR-155 gene: a typical multifunctional microRNA. Biochim Biophys Acta 2009;1792:497–505 [DOI] [PubMed] [Google Scholar]
- 29.Ji WG, Zhang XD, Sun XD, Wang XQ, Chang BP, Zhang MZ. miRNA-155 modulates the malignant biological characteristics of NK/T-cell lymphoma cells by targeting FOXO3a gene. J Huazhong Univ Sci Technolog Med Sci 2014;34:882–8 [DOI] [PubMed] [Google Scholar]
- 30.Pedersen IM, Otero D, Kao E, Miletic AV, Hother C, Ralfkiaer E, et al. Onco-miR-155 targets SHIP1 to promote TNFalpha-dependent growth of B cell lymphomas. EMBO Mol Med 2009;1:288–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim JH, Kim WS, Park C. Epstein-Barr virus latent membrane protein-1 protects B-cell lymphoma from rituximab-induced apoptosis through miR-155-mediated Akt activation and up-regulation of Mcl-1. Leuk Lymphoma 2012;53:1586–91 [DOI] [PubMed] [Google Scholar]
- 32.De Santis R, Liepelt A, Mossanen JC, Dueck A, Simons N, Mohs A, et al. miR-155 targets Caspase-3 mRNA in activated macrophages. RNA Biol 2016;13:43–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Higgs G, Slack F. The multiple roles of microRNA-155 in oncogenesis. J Clin Bioinforma 2013;3:17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dhuri K, Bechtold C, Quijano E, Pham H, Gupta A, Vikram A, et al. Antisense Oligonucleotides: An Emerging Area in Drug Discovery and Development. J Clin Med 2020;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov 2017;16:203–22 [DOI] [PubMed] [Google Scholar]
- 36.Sandhu SK, Volinia S, Costinean S, Galasso M, Neinast R, Santhanam R, et al. miR-155 targets histone deacetylase 4 (HDAC4) and impairs transcriptional activity of B-cell lymphoma 6 (BCL6) in the Emu-miR-155 transgenic mouse model. Proc Natl Acad Sci U S A 2012;109:20047–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cheng CJ, Saltzman WM, Slack FJ. Canonical and non-canonical barriers facing antimiR cancer therapeutics. Curr Med Chem 2013;20:3582–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Demidov VV, Potaman VN, Frank-Kamenetskii MD, Egholm M, Buchard O, Sonnichsen SH, et al. Stability of peptide nucleic acids in human serum and cellular extracts. Biochem Pharmacol 1994;48:1310–3 [DOI] [PubMed] [Google Scholar]
- 39.Khvorova A, Watts JK. The chemical evolution of oligonucleotide therapies of clinical utility. Nat Biotechnol 2017;35:238–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Y, Roccaro AM, Rombaoa C, Flores L, Obad S, Fernandes SM, et al. LNA-mediated anti-miR-155 silencing in low-grade B-cell lymphomas. Blood 2012;120:1678–86 [DOI] [PubMed] [Google Scholar]
- 41.Gupta A, Bahal R, Gupta M, Glazer PM, Saltzman WM. Nanotechnology for delivery of peptide nucleic acids (PNAs). J Control Release 2016;240:302–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Avitabile C, Accardo A, Ringhieri P, Morelli G, Saviano M, Montagner G, et al. Incorporation of Naked Peptide Nucleic Acids into Liposomes Leads to Fast and Efficient Delivery. Bioconjug Chem 2015;26:1533–41 [DOI] [PubMed] [Google Scholar]
- 43.Turner JJ, Ivanova GD, Verbeure B, Williams D, Arzumanov AA, Abes S, et al. Cell-penetrating peptide conjugates of peptide nucleic acids (PNA) as inhibitors of HIV-1 Tat-dependent trans-activation in cells. Nucleic Acids Res 2005;33:6837–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dean DA. Peptide nucleic acids: versatile tools for gene therapy strategies. Adv Drug Deliv Rev 2000;44:81–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Malik S, Oyaghire S, Bahal R. Applications of PNA-laden nanoparticles for hematological disorders. Cell Mol Life Sci 2019;76:1057–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tili E, Michaille JJ, Wernicke D, Alder H, Costinean S, Volinia S, et al. Mutator activity induced by microRNA-155 (miR-155) links inflammation and cancer. Proc Natl Acad Sci U S A 2011;108:4908–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perera JDR, Carufe KEW, Glazer PM. Peptide nucleic acids and their role in gene regulation and editing. Biopolymers 2021:e23460. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.