

Abstract
Cancer is a complex disease and cancer cells typically harbor multiple genetic and epigenetic alterations. Large-scale sequencing of patient-derived cancer samples has identified several druggable driver oncogenes. Many of these oncogenes can be pharmacologically targeted to provide effective therapies for breast cancer, leukemia, lung cancer, melanoma, lymphoma, and other cancer types. Initial responses to these agents can be robust in many cancer types and some cancer patients experience sustained tumor inhibition. However, resistance to these targeted therapeutics frequently emerges, either from intrinsic or acquired mechanisms, posing a major clinical hurdle for effective treatment. Several resistance mechanisms, both cell autonomous and cell non-autonomous, have been identified in different cancer types. Here we describe how alterations of the transcriptome, transcription factors, DNA, and chromatin regulatory proteins confer resistance to targeted therapeutic agents. We also elaborate on how these studies have identified underlying epigenetic factors that drive drug resistance and oncogenic pathways, with direct implications for the prevention and treatment of drug-resistant cancer.
Introduction
It is estimated that more than 19 million men and women worldwide were diagnosed with cancer last year, leading to over 9.6 million cancer-related deaths (1). Anti-cancer therapies have advanced considerably in the last two decades, with targeted therapeutic agents often replacing chemotherapy as front-line therapy for most cancers with oncogenic alterations for which clinical-grade pharmacological inhibitors have been developed (2,3). These targeted therapeutic agents provide more clinical benefits than chemotherapy for some cancer patients, offering reduced therapy-related side effects, increased survival, and even long-term cure in some cases.
However, many cancer patients do not respond to targeted therapeutics, due to either intrinsic drug resistance or the development of acquired resistance after treatment (4–6). Drug resistance renders targeted therapeutic agents ineffective and leads to disease progression. Consequently, drug-resistant tumors are a major clinical challenge in modern oncology practice, and there is dire need for new therapeutic agents to treat these cancers. Several mechanisms of targeted therapy resistance development have been identified, and notably, many of these discoveries have resulted in development of new drugs that target resistance mechanisms and their translation into the clinic (7–10). These “bench-to-bedside” translational successes have provided hope to patients with drug-resistant tumors. Furthermore, ongoing efforts to identify additional resistance mechanisms will likely prove useful for the development of additional treatments for drug-resistant cancer by uncovering novel mechanisms and driver proteins that activate drug resistance pathways (Fig. 1A) (11–15).
Figure 1. Epigenetic mechanisms and regulators that drive resistance to targeted cancer therapeutics.
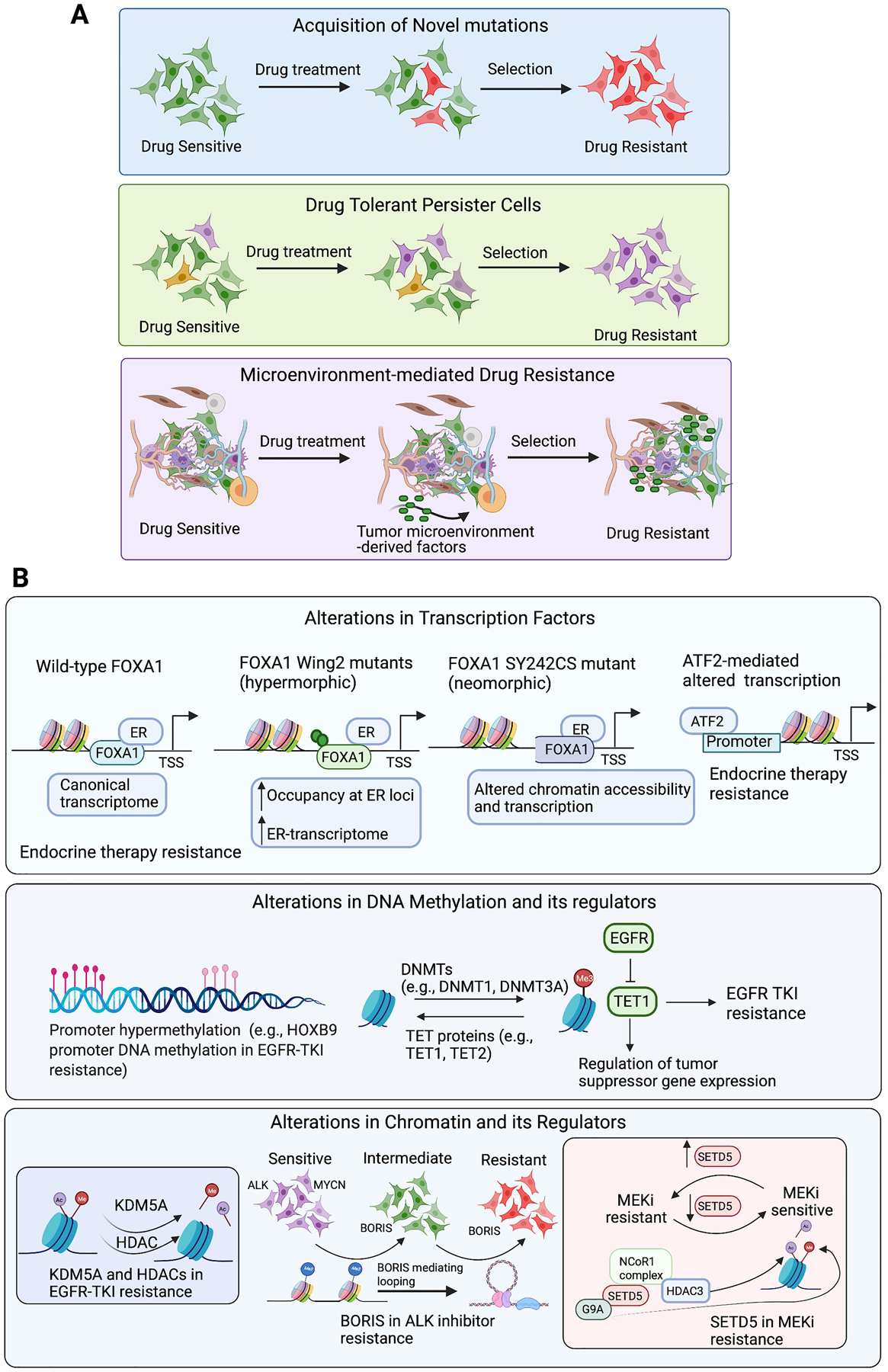
A. Therapeutic agents can activate cell autonomous and/or cell non-autonomous mechanisms to confer drug resistance in cancer. This may involve acquisition of novel mutations, enrichment of drug-tolerant persister cells, and microenvironment-mediated drug resistance. B. The illustration depicts transcription factors, DNA modifiers, and chromatin regulators that contribute to resistance against targeted cancer therapies.
Epigenetic alterations refer to heritable changes that can modulate gene transcription without causing change in DNA sequence. The most common examples of epigenetic alterations are DNA and histone protein modifications, which affect transcription factor binding and function, leading to transcriptomic changes. Epigenetic alterations in DNA and histone proteins are important for cancer initiation and progression (16–18), and they can also drive resistance to targeted therapies (19–22). In this review, we provide an overview of the epigenetic mechanisms associated with cancer-drug resistance and discuss the implications of these findings for treatment of drug-resistant tumors (Supplementary Table S1). For the purpose of this review, we will consider non-genetic alterations of DNA and histone proteins, as well as their impact on transcription factor function and the cancer cell transcriptome, as epigenetic changes. The role of non-coding (nc)RNAs in cancer drug resistance has been previously reviewed by others (23,24) and will not be discussed.
Altered Transcriptional Profiles of Cancer Cells Resistant To Targeted Therapeutic Agents
Cancer types and subtypes can be classified with great certainty using mRNA expression profiles (25–29). Similarly, mRNA profiling approaches have been instrumental for distinguishing drug-sensitive and drug-resistant cancer cells, as well as for facilitating identification of potential therapeutic targets (30–32). As an example, one study performed differential mRNA expression analysis of the epidermal growth factor receptor (EGFR)-mutant lung adenocarcinoma (LUAD) cell line PC9 (PC9GR) that is resistant to EGFR inhibitor (gefitinib) and identified 865 mRNAs with altered expression (396 upregulated, 469 downregulated) in gefitinib-resistant PC9GR cells compared to parental PC9 cells (33). Using the Cancer Cell Line Encyclopedia (CCLE) and Cancer Therapeutics Response Portal (CTRP), the authors identified 17 drugs whose responses were associated with four or more of these upregulated genes. Of these drugs, they tested four—dasatinib, KPT-185, trametinib, and pluripotin—and found that KPT-185 most potently inhibited growth of PC9GR cells.
KPT-185 is an inhibitor of Exportin 1 (XPO1), a protein that mediates nuclear export of proteins and RNAs (34,35), and has documented anti-cancer activity in leukemia (36) and lung cancer (37). KPT-185 was shown to inhibit PC9GR cells as effectively as PC9 parental cells (33), providing an excellent example of transcriptional profiling as an approach for identifying new therapeutics to treat drug-resistant cancers. However, in this study, it was not clear why no differences in KPT-185 sensitivity were observed in gefitinib-sensitive and gefitinib-resistant cells. A likely possibility is that the growth-suppressive effect of KPT-185 results from its ability to target essential cellular functions (e.g., protein and RNA transport), although the precise KPT-185 mechanism-of-action was not identified. Therefore, the toxicity profiles and off-target effects of XPO1 inhibitors should be carefully assessed before they can be considered as possible therapeutics.
Bulk RNA-sequencing (RNA-seq), although useful for identifying important transcriptional features of drug-resistant tumors, fails to capture heterogeneous drug-resistant cell populations and their relative abundance. However, these limitations are overcome through use of single-cell (sc)RNA-seq, which can deconvolute the heterogeneous nature and complex evolution of drug resistance (38–43). An excellent example of using scRNA-seq to better understand the response to therapeutic agents is a study by Xue and colleagues that identified a rapid, non-uniform adaptation to conformation-specific KRAS(G12C) inhibition (44). The authors assessed adaptation of KRAS(G12C)-mutant lung cancer cells to the conformation-specific KRAS(G12C) inhibitor ARS1620, which binds the inactive non-GTP-bound form of KRAS(G12C), but not the GTP-bound form. Results showed that KRAS(G12C)-mutant cells undergo two alternative fates after G12C inhibitor treatment. The first subset of cells shows attenuated growth and inhibited expression of the KRAS(G12C) target gene signature. Conversely, the second subset of cells displays restored proliferation after transient growth inhibition and higher expression of the KRAS(G12C) target gene signature, indicating restoration of the KRAS(G12C) signaling pathway.
Combining the results of scRNA-seq with a CRISPR-based screen, the authors identified heparin-binding EGF-like growth factor (HB-EGF) and Aurora kinase A (AURKA) as mediators of G12C inhibitor drug resistance. Non-uniform drug resistance adaptation occurs, in part, due to newly synthesized KRAS(G12C) that rapidly binds GTP and becomes insensitive to ARS1620. This also promotes cell proliferation in response to signaling upstream of KRAS, in which receptor tyrosine kinases (RTKs), such as EGFR, facilitate nucleotide exchange. AURKA further promotes adaptive reactivation of KRAS-GTP and CRAF-MEK-ERK signaling, which also allows cells to overcome the effect of G12C inhibitor. Notably, consistent with the role of these proteins in ARS1620 adaptation, EGFR and AURKA inhibitors enhance therapeutic response to ARS1620. These findings highlight the importance of profiling at the single-cell transcriptome level to identify adaptive drug responses, as these data can lead to development of combination therapies for preventing drug resistance and extending duration of therapeutic efficacy. Future studies in other cancer types treated with ARS1620, such as KRAS(G12C)-mutant pancreatic ductal adenocarcinoma (PDAC) cells, will reveal if the same or different mechanisms of drug resistance operate in these cancers. Similarly, use of other mutant KRAS inhibitors, such as AMG510, and JDQ443, may uncover distinct inhibitor-specific effects.
Role of Transcription Factors in Targeted Therapy Resistance
The above studies demonstrate that drug-resistant cancer cell transcriptomes are distinct from those of drug-sensitive cancer cells. It can be further rationalized that changes in mRNA abundance in drug-resistant cells are driven, at least in part, by transcription factors, and this is supported by several published studies (45–50). In one example, Ninad et al. identified novel transcription factors that regulate glucocorticoid resistance in acute lymphoblastic leukemia (ALL) using two clones, CEM-C1 (dexamethasone-resistant) and CEM-C7 (dexamethasone-sensitive), derived from a 3-year-old ALL patient (48). Here, the authors analyzed the CEM-C1 and CEM-C7 clones using transposase-accessible chromatin-sequencing (ATAC-seq) and RNA-seq to simultaneously profile their chromatin states and mRNA expression patterns, respectively. DNA sequence recognition motifs for three transcription factor families—RUNX, ETS, and E-protein—were present in more accessible peaks in dexamethasone-sensitive ALL cells. RNA-seq was then used to identify three transcription factors that bind these motifs in dexamethasone-sensitive ALL cells: RUNX2, ETV5, and TCF4, indicating reduced binding for these transcription factors in dexamethasone-resistant ALL cells. Although this finding is intriguing, further studies are required to establish whether RUNX2, ETV5, and/or TCF4 inhibition can promote glucocorticoid resistance. Another limitation of this study is the use of single patient-derived clones; consequently, further validation in clones derived from multiple patients is needed. Nonetheless, similar studies can be conducted in other cancer types and for other drugs to identify key transcription factors involved in conferring drug resistance to cancer therapeutics.
One cancer for which targeted therapeutic agents have provided significant benefits to patients is breast cancer (51). In particular, as estrogen receptor (ER)α drives growth in two-thirds of all breast cancers, endocrine therapy can be effectively used to treat this breast cancer sub-type (52). However, approximately 50% of ER+ breast cancers do not respond or becomes resistant to endocrine therapy (52,53). One previous study investigated this phenomenon and uncovered an important role for activating transcription factor 2 (ATF2), a member of the leucine zipper family of DNA-binding proteins, in endocrine therapy (tamoxifen) resistance in breast cancer (49). Knockdown of ATF2 using small-interfering (si)RNA in tamoxifen-resistant ER+ breast cancer cells was found to inhibit their growth. Intriguingly, however, significant growth inhibition was only noted in tamoxifen-resistant ER+ breast cancer cells, although similar ATF2 protein levels were detected in tamoxifen-sensitive and tamoxifen-resistant cells. Gene expression profiling revealed the emergence of an alternative hormone-independent mechanism in tamoxifen-resistant ER+ breast cancer cells, as shown by decreased estrogen receptor 1 (ESR1) levels and reduced ER-regulatory gene enrichment (Fig. 1B) (49). These tamoxifen-resistant cells shift to an ATF2-enriched network and transforming growth factor (TGF)-β signaling pathway. Notably, however, ATF2 knockdown in tamoxifen-resistant cells reduces MYC transcription factor activity and restores gene expression signatures associated with ER signaling, which partially explains the reappearance of tamoxifen sensitivity in response to ATF2 inhibition. This study provides an example of transcription factor functional modulation that occurs independently of changes in expression levels and drives drug resistance by reprogramming transcription in drug-resistant cells. Importantly, an interesting paradigm to emerge from this study is that drug-resistant cells can “rewire” themselves to rely on alternative survival pathways and thereby acquire autonomy from pathways they were dependent upon before becoming resistant. Further studies are needed, however, to elucidate how ATF2 acquires the ability to differentially regulate transcription in tamoxifen-resistant and tamoxifen-sensitive cells.
Another key study characterized the role of the Forkhead Box A1 (FOXA1) transcription factor in regulating response to aromatase inhibitors in breast cancer cells (50). These inhibitors block the enzyme aromatase, which converts androgen to estrogen (54), and are therefore beneficial for treating ER+ breast cancer patients (50,55). However, resistance to aromatase inhibitors is commonly observed (50,55). In one study, analysis of about 5,000 breast cancer samples from a clinically and genomically curated cohort of patients, combined with a series of breast cancer models, revealed that mutations in FOXA1 promote aromatase inhibitor resistance (50). In particular, the authors identified several hotspot mutations in the Wing2 region of FOXA1, as well as the breast cancer-specific mutation SY242CS, with evidence for two distinct mechanisms of resistance in the Wing2 and SY242CS FOXA1 mutants (Fig. 1B) (50). Notably, Wing2 mutants were found to be more responsive (hypermorphic) to estrogen stimulation, as shown by enhanced mutant binding to ER-responsive loci and increased ER-driven transcription. However, changes in chromatin accessibility were not observed with Wing2 mutations. In contrast, the FOXA1 SY242CS mutation results in a conformational change that promotes neomorphic function. That is, this mutant can open distinct chromatin regions and activate transcription of alternative genes not normally regulated by wild-type FOXA1. Overall, this study demonstrates that transcription factor mutations can alter their function, impacting both chromatin accessibility and the transcriptome in cancer cells, and leading to drug resistance. These findings highlight the importance of considering the potential for transcription factor mutations to alter chromatin accessibility, either directly or possibly indirectly, by affecting function of interacting proteins. Future studies will be critical for resolving this issue. Similarly, studies in other cancer types harboring FOXA1 mutations (56), such as prostate cancer, can determine whether similar mutations cause resistance in these cells in response to other targeted therapies.
In addition to cell intrinsic mechanisms, cell extrinsic mechanisms may also be involved in cancer drug resistance development (57–59). For example, one study reported that interaction between cancer stem cells (CSCs) and tumor-associated macrophages (TAMs) promotes resistance to androgen-deprivation therapy (ADT) in prostate cancer (60). Here, the authors found that autophagy-related gene 7 (ATG7), which facilitates transcription of octamer-binding transcription factor 4 (OCT4) in a β-catenin-dependent manner, stimulates cells with CSC characteristics. These CSCs educate monocytes/macrophages toward TAMs, and in turn, CSC-educated TAMs enhance the stem-like properties of CSCs, as well as prostate cancer progression and ADT resistance by activating interleukin (IL)-6/signal transducer and activator of transcription 3 (STAT3). Interestingly, combined suppression of ATG7/OCT4 and IL6/STAT3 restored ADT sensitivity in a prostate cancer model. Overall, this study highlights the importance of cell extrinsic factors, such as the tumor microenvironment, in promoting drug resistance and established the role of transcription factors OCT4 and STAT3 in this process. Additionally, these findings suggest that drug-resistant tumors can be treated by disrupting transcription networks in the tumor microenvironment. In future studies, it will be important to evaluate cancer cell–microenvironment interactions in the context of other immune and non-immune cell types, such as tumor-associated T-cells and fibroblasts, to identify similar transcriptional networks.
Role of DNA Modification Regulatory Proteins in Targeted Therapy Resistance
DNA modifications can affect the ability of transcription factors to bind DNA, thereby either activating or repressing mRNA expression. Consequently, DNA modification regulatory proteins, including DNA methyltransferases (e.g., DNMT1, DNMT3A, and DNMT3B) and DNA demethylases (e.g., TET1, TET2, and TET3), likely contribute to targeted therapy resistance. Additionally, other enzymes, such as IDH1 and IDH2, can indirectly affect DNA methylation levels by regulating activity of DNA modification enzymes, such as the TET proteins (61–63).
The relationship between global DNA methylation changes and EGFR-tyrosine kinase inhibitor (TKI) response rate is illustrated in a study of 79 tumor samples from patients with advanced LUAD, before and after EGFR-TKI treatment (64). The authors identified 216 CpG sites that were differentially methylated in EGFR-TKI responders and non-responders. Most probes (203/216) displayed higher DNA methylation in poor responders than in patients with a favorable response, indicating that, in general, DNA hypermethylation correlates with poor EGFR-TKI response. Further analysis of publicly available DNA methylation and RNA-seq data for LUAD identified 37 sites as potential transcriptionally repressed sites, with eight of these sites annotated to five transcription factors (IKZF1, HOXB9, SP8, LASS4, and VAX2). However, only HOXB9 methylation was significantly associated with patients experiencing cancer progression after EGFR-TKI treatment (Fig. 1B). Although interesting, this study lacked validation to establish whether HOXB9 is a driver, or just a marker, of EGFR-TKI resistance. Thus, future studies addressing this question will reveal the true impact of HOXB9 epigenetic silencing in driving EGFR-TKI resistance.
Similarly, another study identified a role for TET1 in EGFR-TKI response (65). TET1 is a methyl-cytosine dioxygenase that demethylates methylated-cytosine to promote gene reactivation (66). This protein was repressed by EGFR in LUAD cells, and knockdown of TET1 causes resistance to the EGFR-TKI gefitinib (65). Additionally, analysis of tumors derived from EGFR inhibitor-sensitive and -resistant EGFR-driven mouse models revealed that tumors resistant to the EGFR-TKI erlotinib had lower 5-hydroxymethylcytosine (5hmC) levels. Furthermore, patient-derived EGFR-TKI-resistant samples displayed lower TET1 expression than EGFR-TKI-sensitive samples. However, although this study identified several TET1 targets, none of the candidate genes were confirmed as mediators of TET1 function. Therefore, future studies are needed to address this important question. Nonetheless, this study provides key evidence for the role of TET1, and potentially TET1-regulated DNA methylation levels, in EGFR-TKI resistance.
A third study reported that resistance to multiple poly (ADP-ribose) polymerase (PARP) inhibitors emerges when TET2 expression is decreased in BRAC2-deficient cells (67). Like TET1, TET2 is a member of the TET family of methyl-cytosine dioxygenases. TET2 affects stability of replication forks by modulating 5hmC levels, and loss of TET2 stabilized stalled replication forks in BRCA2-deficient cells, leading to PARP resistance (67). Although the authors did not rule out the possibility that this effect is due to TET2’s ability to regulate transcription, they noted the mechanism is likely driven by a non-transcription-related function, as no significant changes in expression of key regulators of replication fork integrity were observed. This study therefore indicates that DNA modification regulatory proteins may play roles in therapy resistance that are independent of their gene expression regulation activity. Thus, in future studies involving TET proteins, it will be important to assess whether the observed phenotypes are driven through their canonical function (e.g., transcriptional regulation) or via non-canonical activities, such as in the study described above.
Overall, the studies in this section collectively demonstrate that changes in DNA methylation can determine the response and resistance to targeted therapeutic agents in cancer. Thus, proteins and pathways that modulate DNA methylation may represent attractive therapeutic targets for some resistance cancers.
Role of Chromatin Regulators and Chromatin State Deregulation in Driving Resistance to Targeted Therapeutic Agents
Proteins that regulate chromatin are classified by their activity as either writers (e.g., histone acetyl transferases), erasers (e.g., histone deacetylases), or readers (e.g., bromodomain proteins) (68–70), and these are known to be important for conferring drug resistance (71–73). One example of chromatin regulator-driven drug resistance was documented in the context of EGFR-TKI-resistant LUAD and other cancers (71). This study discovered a reversible drug-tolerant chromatin state that is dependent on IGF1R signaling and lysine demethylase 5A (KDM5A) and could be depleted after treatment with HDAC inhibitors (Fig. 1B) (71). This finding is consistent with the observation of increased histone 3 lysine 14 acetylation (H3K14ac) in drug-tolerant cells. The authors further showed that the effect of HDAC inhibitor (Tricostatin A) is partly due to its ability to induce DNA damage-related caspase-independent apoptosis. Additionally, this phenomenon could be generalized to other drugs, including BRAF inhibitor (AZ628) and the chemotherapeutic agent cisplatin. These findings indicate that drug tolerance is often an intermediate state, the disruption of which may prevent emergence of resistance to a variety of anticancer agents. However, it is important to note that some of inhibitors tested, such as Tricostatin A, are broad-spectrum HDAC inhibitors. Therefore, it is not clear if one or more HDACs are involved in this process. Future studies will be needed to reveal the precise contributions of specific HDACs in development of drug-tolerant cells.
In another example, the BORIS protein, a paralogue of CCCTC-binding factor (CTCF), was found to promote chromatin regulatory interactions in anaplastic lymphoma kinase (ALK) inhibitor-resistant neuroblastoma cells (Fig. 1B) (72). This study showed that continuous treatment with TAE684 and other ALK inhibitors causes MYC-amplified ALK+ cells to overexpress BORIS and lose MYC expression due to epigenetic silencing. Survival in these cells also became independent of MYC and dependent upon BORIS. Notably, ectopic BORIS expression was not sufficient to induce ALK inhibitor resistance; this was only achieved by combined MYC inhibition and ectopic BORIS expression during treatment with ALK inhibitor. In drug-resistant cells, BORIS facilitated new long-range interactions by promoting chromatin looping, and in most cases, these interactions match BORIS occupancy sites. Analysis of the super-enhancer landscape revealed enrichment of super-enhancers unique to resistant cells at BORIS-positive regulatory loops. These super-enhancer-associated genes were also enriched in a chromatin state that switched from a closed to neutral to active configuration in drug-resistant cells, explaining their higher expression. Thus, this study showed that cancer cells can acquire an alternative chromatin–transcription state in response to drug treatment that allows their survival. These findings also highlight the importance of identifying factors that mediate such states, to uncover new dependencies in drug-resistant tumors, not responsive to available therapies. Once again, similar to the ATF2 study described above (49), this study reveals how drug-resistant cells become dependent upon alternative oncogenic pathways. Interestingly, the authors also showed that BORIS is upregulated in lapatinib-resistant breast cancer and cisplatin-resistant ovarian cancer. Thus, future functional studies are required to determine whether a mechanism similar to that shown for neuroblastoma/ALK inhibitors also operates in the context of other cancers and therapeutic agents.
Some chromatin modifiers play unexpected non-canonical roles in cancer drug resistance. One report evaluated 95 known and putative human methyltransferases using a short-hairpin (sh)RNA-based screen in the MIAPaCa2 PDAC cell line treated with MEK inhibitor trametinib (73). This screen identified the putative methyltransferase set domain-containing 5 (SETD5) as the most potent sensitizer to trametinib treatment (Fig. 1B). Notably, the authors found that SETD5 does not display histone H3 lysine 36 methyltransferase activity, but rather, functions as part of the Nuclear Receptor Corepressor 1 (NCoR1) complex in combination with G9a (also known as EHMT2) and HDAC3. In this context, SETD5 promoted transcriptional repression via methylation and deacetylation of histone H3. Additionally, although HDAC3 typically deacetylates a broad spectrum of histone acetyl-lysine substrates, its catalytic activity was limited only to acetylated histone H3 lysine 9 and lysine 27 when complexed with SETD5. Consistent with a role for SETD5 in pancreatic cancer cell resistance, treating MEK inhibitor-resistant PDAC cells with a triple combination of G9a inhibitor UNC0638, HDAC3 inhibitor RGFP966, and MEK inhibitor trametinib strongly inhibited tumor growth and significantly increased survival in mouse models of PDAC tumor growth. Thus, this study identified a new role for SETD5 in MEK inhibitor resistance in PDAC cells and demonstrates that chromatin modifier proteins can acquire new molecular functions via interactions with other proteins. Because, in most cases, MEK-targeted therapies have been shown to induce mutations in MEK kinase or its downstream effector ERK (74), it would be interesting to determine whether PDAC cells lacking SETD5 acquire these mutations at some point. This will also provide improved therapeutic modalities for treating PDAC, for example, by combining G9A and HDAC3 inhibitors with ERK inhibitors, if MEK kinases acquire constitutively active mutations.
Overall, these studies demonstrate that chromatin regulatory proteins play a complex and important role in conferring resistance to a variety of targeted cancer therapies. Therefore, these proteins represent attractive new targets for drug-resistant tumor treatment.
Conclusions and Future Directions
Drug resistance to targeted therapies has become inevitable for many patients (7–15), necessitating development of contingency plans for treating drug-resistant tumors. To this end, an improved understanding of the epigenetic drivers of drug resistance can provide new therapeutic approaches. In this review, we present several examples that document how targeting such epigenetic regulators in cancer cells or tumor microenvironments can facilitate development of new treatments for drug-resistant tumors.
One unifying theme that emerges from the studies described above is that, in response to drug treatment, cancer cells can evolve independence from the oncogenic pathway they originally relied upon. Consequently, they acquire new dependencies, such as for tamoxifen-resistant ER+ breast cancer with ATF2 (49) and ALK inhibitor-resistant neuroblastoma with BORIS (72). Importantly, these new driver proteins can then be targeted for treating drug-resistant tumors. However, almost all studies described in this review are in early pre-clinical validation stages or have only been validated with clinical samples. Therefore, to determine the true translational impact of these findings, clinical trials with candidate therapeutic agents that block key epigenetic driver targets are needed. For example, a clinical trial assessing combined administration of G9A, HDAC3, and MEK inhibitors will reveal whether such an approach can benefit patients with KRAS-mutant PDAC.
A few previous clinical trials have reported the successful use of therapeutic agents that block epigenetic targets for treating previously untreated cancers, either alone or in combination with other anti-cancer agents (75,76). These results provide hope that similar epigenetic-targeting therapeutic agents will show efficacy for treating drug-resistant tumors. Consistent with this possibility, one study assessed the use of EZH2 inhibitor tazemetostat for treating patients with relapsed or refractory follicular lymphoma (77). Here, it was found that tazemetostat monotherapy lead to clinically meaningful and durable responses in this cohort of pretreated patients and therefore shows promise for treating drug-resistant tumors (77).
It is also important to remember that drug resistance is a dynamic state, which continually evolves under various selective pressures, making cancer treatment, in many ways, a “moving target”. Therefore, drug-resistant tumors must be continuously evaluated to determine the best therapeutic interventions at any given stage of evolution. A worthy step in this direction is the TRACERx project, which integrates several technologies to study evolution of different cancer types under therapeutic intervention (78–81). These studies are expected to lay the foundation for providing long-term care for cancer patients, even when first-line therapies fail, and drug resistance emerge. Taken together, the studies described in this review suggest that, although resistance to targeted therapies remains a challenge to modern oncology, deeper understanding of drug-resistant tumors and the epigenetic drivers of therapy resistance have the potential to identify new therapeutic opportunities for treating drug-resistant tumors in the near future.
Supplementary Material
Acknowledgements
We gratefully acknowledge the following grants from the National Institutes of Health: R01CA196566, R01CA200919, and R01CA257046 to N.W and R01CA233481 to R.G., as well as the grants W81XWH-19-1-0480 and W81XWH-19-1-0084 from the Department of Defense to N.W. The figure in this review was created using BioRender.Com
Footnotes
Conflict of Interest Statement: Authors have no conflict of interest to declare.
References
- 1.Open Database: Global Cancer Observatory, International Agency for Research on Cancer. World Health Organization. Available from : https://gco.iarc.fr/today/data/factsheets/cancers/39-All-cancers-fact-sheet.pdf.
- 2.Schilsky RL. Personalized medicine in oncology: the future is now. Nat Rev Drug Discov 2010;9:363–6 [DOI] [PubMed] [Google Scholar]
- 3.Salgado R, Moore H, Martens JWM, Lively T, Malik S, McDermott U, et al. Steps forward for cancer precision medicine. Nat Rev Drug Discov 2018;17:1–2 [DOI] [PubMed] [Google Scholar]
- 4.Boumahdi S, de Sauvage FJ. The great escape: tumour cell plasticity in resistance to targeted therapy. Nat Rev Drug Discov 2020;19:39–56 [DOI] [PubMed] [Google Scholar]
- 5.Vander Velde R, Yoon N, Marusyk V, Durmaz A, Dhawan A, Miroshnychenko D, et al. Resistance to targeted therapies as a multifactorial, gradual adaptation to inhibitor specific selective pressures. Nat Commun 2020;11:2393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin JJ, Shaw AT. Resisting Resistance: Targeted Therapies in Lung Cancer. Trends Cancer 2016;2:350–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cortes JE, Kantarjian H, Shah NP, Bixby D, Mauro MJ, Flinn I, et al. Ponatinib in refractory Philadelphia chromosome-positive leukemias. N Engl J Med 2012;367:2075–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mok TS, Wu YL, Ahn MJ, Garassino MC, Kim HR, Ramalingam SS, et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N Engl J Med 2017;376:629–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shaw AT, Friboulet L, Leshchiner I, Gainor JF, Bergqvist S, Brooun A, et al. Resensitization to Crizotinib by the Lorlatinib ALK Resistance Mutation L1198F. N Engl J Med 2016;374:54–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yang X, Wang D, Lin J, Yang X, Zhao H. Atezolizumab plus bevacizumab for unresectable hepatocellular carcinoma. Lancet Oncol 2020;21:e412. [DOI] [PubMed] [Google Scholar]
- 11.Sabnis AJ, Bivona TG. Principles of Resistance to Targeted Cancer Therapy: Lessons from Basic and Translational Cancer Biology. Trends Mol Med 2019;25:185–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bisserier M, Wajapeyee N. Mechanisms of resistance to EZH2 inhibitors in diffuse large B-cell lymphomas. Blood 2018;131:2125–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Janostiak R, Malvi P, Wajapeyee N. Anaplastic Lymphoma Kinase Confers Resistance to BRAF Kinase Inhibitors in Melanoma. iScience 2019;16:453–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Webster MR, Fane ME, Alicea GM, Basu S, Kossenkov AV, Marino GE, et al. Paradoxical Role for Wild-Type p53 in Driving Therapy Resistance in Melanoma. Mol Cell 2020;77:633–44 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Obenauf AC, Zou Y, Ji AL, Vanharanta S, Shu W, Shi H, et al. Therapy-induced tumour secretomes promote resistance and tumour progression. Nature 2015;520:368–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baylin SB, Jones PA. Epigenetic Determinants of Cancer. Cold Spring Harb Perspect Biol 2016;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gupta R, Janostiak R, Wajapeyee N. Transcriptional regulators and alterations that drive melanoma initiation and progression. Oncogene 2020;39:7093–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bradner JE, Hnisz D, Young RA. Transcriptional Addiction in Cancer. Cell 2017;168:629–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shaffer SM, Dunagin MC, Torborg SR, Torre EA, Emert B, Krepler C, et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017;546:431–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Strub T, Ghiraldini FG, Carcamo S, Li M, Wroblewska A, Singh R, et al. SIRT6 haploinsufficiency induces BRAF(V600E) melanoma cell resistance to MAPK inhibitors via IGF signalling. Nat Commun 2018;9:3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gupta R, Bugide S, Wang B, Green MR, Johnson DB, Wajapeyee N. Loss of BOP1 confers resistance to BRAF kinase inhibitors in melanoma by activating MAP kinase pathway. Proc Natl Acad Sci U S A 2019;116:4583–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bugide S, Parajuli KR, Chava S, Pattanayak R, Manna DLD, Shrestha D, et al. Loss of HAT1 expression confers BRAFV600E inhibitor resistance to melanoma cells by activating MAPK signaling via IGF1R. Oncogenesis 2020;9:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu K, Gao L, Ma X, Huang JJ, Chen J, Zeng L, et al. Long non-coding RNAs regulate drug resistance in cancer. Mol Cancer 2020;19:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Allen KE, Weiss GJ. Resistance may not be futile: microRNA biomarkers for chemoresistance and potential therapeutics. Mol Cancer Ther 2010;9:3126–36 [DOI] [PubMed] [Google Scholar]
- 25.Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A 2001;98:10869–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chung CH, Parker JS, Karaca G, Wu J, Funkhouser WK, Moore D, et al. Molecular classification of head and neck squamous cell carcinomas using patterns of gene expression. Cancer Cell 2004;5:489–500 [DOI] [PubMed] [Google Scholar]
- 27.Cieslik M, Chinnaiyan AM. Cancer transcriptome profiling at the juncture of clinical translation. Nat Rev Genet 2018;19:93–109 [DOI] [PubMed] [Google Scholar]
- 28.Golub TR, Slonim DK, Tamayo P, Huard C, Gaasenbeek M, Mesirov JP, et al. Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science 1999;286:531–7 [DOI] [PubMed] [Google Scholar]
- 29.Ayars M, Goggins M. Pancreatic cancer: Classifying pancreatic cancer using gene expression profiling. Nat Rev Gastroenterol Hepatol 2015;12:613–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szakacs G, Annereau JP, Lababidi S, Shankavaram U, Arciello A, Bussey KJ, et al. Predicting drug sensitivity and resistance: profiling ABC transporter genes in cancer cells. Cancer Cell 2004;6:129–37 [DOI] [PubMed] [Google Scholar]
- 31.Kobayashi S, Shimamura T, Monti S, Steidl U, Hetherington CJ, Lowell AM, et al. Transcriptional profiling identifies cyclin D1 as a critical downstream effector of mutant epidermal growth factor receptor signaling. Cancer Res 2006;66:11389–98 [DOI] [PubMed] [Google Scholar]
- 32.Smalley I, Kim E, Li J, Spence P, Wyatt CJ, Eroglu Z, et al. Leveraging transcriptional dynamics to improve BRAF inhibitor responses in melanoma. EBioMedicine 2019;48:178–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei N, Song Y, Zhang F, Sun Z, Zhang X. Transcriptome Profiling of Acquired Gefitinib Resistant Lung Cancer Cells Reveals Dramatically Changed Transcription Programs and New Treatment Targets. Front Oncol 2020;10:1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okamura M, Inose H, Masuda S. RNA Export through the NPC in Eukaryotes. Genes (Basel) 2015;6:124–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nachmias B, Schimmer AD. Targeting nuclear import and export in hematological malignancies. Leukemia 2020;34:2875–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ranganathan P, Yu X, Na C, Santhanam R, Shacham S, Kauffman M, et al. Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood 2012;120:1765–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang S, Han X, Wang J, Yao J, Shi Y. Antitumor effects of a novel chromosome region maintenance 1 (CRM1) inhibitor on non-small cell lung cancer cells in vitro and in mouse tumor xenografts. PLoS One 2014;9:e89848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yeo SK, Zhu X, Okamoto T, Hao M, Wang C, Lu P, et al. Single-cell RNA-sequencing reveals distinct patterns of cell state heterogeneity in mouse models of breast cancer. Elife 2020;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fan J, Lee HO, Lee S, Ryu DE, Lee S, Xue C, et al. Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RNA-seq data. Genome Res 2018;28:1217–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aissa AF, Islam A, Ariss MM, Go CC, Rader AE, Conrardy RD, et al. Single-cell transcriptional changes associated with drug tolerance and response to combination therapies in cancer. Nat Commun 2021;12:1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharma A, Cao EY, Kumar V, Zhang X, Leong HS, Wong AML, et al. Longitudinal single-cell RNA sequencing of patient-derived primary cells reveals drug-induced infidelity in stem cell hierarchy. Nat Commun 2018;9:4931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee MC, Lopez-Diaz FJ, Khan SY, Tariq MA, Dayn Y, Vaske CJ, et al. Single-cell analyses of transcriptional heterogeneity during drug tolerance transition in cancer cells by RNA sequencing. Proc Natl Acad Sci U S A 2014;111:E4726–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ho YJ, Anaparthy N, Molik D, Mathew G, Aicher T, Patel A, et al. Single-cell RNA-seq analysis identifies markers of resistance to targeted BRAF inhibitors in melanoma cell populations. Genome Res 2018;28:1353–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xue JY, Zhao Y, Aronowitz J, Mai TT, Vides A, Qeriqi B, et al. Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition. Nature 2020;577:421–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rothenberg SM, Concannon K, Cullen S, Boulay G, Turke AB, Faber AC, et al. Inhibition of mutant EGFR in lung cancer cells triggers SOX2-FOXO6-dependent survival pathways. Elife 2015;4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramsdale R, Jorissen RN, Li FZ, Al-Obaidi S, Ward T, Sheppard KE, et al. The transcription cofactor c-JUN mediates phenotype switching and BRAF inhibitor resistance in melanoma. Sci Signal 2015;8:ra82. [DOI] [PubMed] [Google Scholar]
- 47.Jeselsohn R, Cornwell M, Pun M, Buchwalter G, Nguyen M, Bango C, et al. Embryonic transcription factor SOX9 drives breast cancer endocrine resistance. Proc Natl Acad Sci U S A 2017;114:E4482–E91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ninad Walavalkar N, MichaelGuertin. Identifying Transcription Factors that Dictate Drug-Resistance in Acute Lymphoblastic Leukemia. Clinical Lymphoma Myeloma and Leukemia 2017;17:S258–S9 [Google Scholar]
- 49.Giannoudis A, Malki MI, Rudraraju B, Mohhamed H, Menon S, Liloglou T, et al. Activating transcription factor-2 (ATF2) is a key determinant of resistance to endocrine treatment in an in vitro model of breast cancer. Breast Cancer Res 2020;22:126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Arruabarrena-Aristorena A, Maag JLV, Kittane S, Cai Y, Karthaus WR, Ladewig E, et al. FOXA1 Mutations Reveal Distinct Chromatin Profiles and Influence Therapeutic Response in Breast Cancer. Cancer Cell 2020;38:534–50 e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Masoud V, Pages G. Targeted therapies in breast cancer: New challenges to fight against resistance. World J Clin Oncol 2017;8:120–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hart CD, Migliaccio I, Malorni L, Guarducci C, Biganzoli L, Di Leo A. Challenges in the management of advanced, ER-positive, HER2-negative breast cancer. Nat Rev Clin Oncol 2015;12:541–52 [DOI] [PubMed] [Google Scholar]
- 53.Hanker AB, Sudhan DR, Arteaga CL. Overcoming Endocrine Resistance in Breast Cancer. Cancer Cell 2020;37:496–513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith IE, Dowsett M. Aromatase inhibitors in breast cancer. N Engl J Med 2003;348:2431–42 [DOI] [PubMed] [Google Scholar]
- 55.Brueggemeier RW, Hackett JC, Diaz-Cruz ES. Aromatase inhibitors in the treatment of breast cancer. Endocr Rev 2005;26:331–45 [DOI] [PubMed] [Google Scholar]
- 56.Shah N, Brown M. The Sly Oncogene: FOXA1 Mutations in Prostate Cancer. Cancer Cell 2019;36:119–21 [DOI] [PubMed] [Google Scholar]
- 57.Corso S, Giordano S. Cell-autonomous and non-cell-autonomous mechanisms of HGF/MET-driven resistance to targeted therapies: from basic research to a clinical perspective. Cancer Discov 2013;3:978–92 [DOI] [PubMed] [Google Scholar]
- 58.Apicella M, Giannoni E, Fiore S, Ferrari KJ, Fernandez-Perez D, Isella C, et al. Increased Lactate Secretion by Cancer Cells Sustains Non-cell-autonomous Adaptive Resistance to MET and EGFR Targeted Therapies. Cell Metab 2018;28:848–65 e6 [DOI] [PubMed] [Google Scholar]
- 59.Blum D, LaBarge S, Reproducibility Project: Cancer B. Registered report: Tumour micro-environment elicits innate resistance to RAF inhibitors through HGF secretion. Elife 2014;3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang H, Wang C, Liu F, Li HZ, Peng G, Gao X, et al. Reciprocal Network between Cancer Stem-Like Cells and Macrophages Facilitates the Progression and Androgen Deprivation Therapy Resistance of Prostate Cancer. Clin Cancer Res 2018;24:4612–26 [DOI] [PubMed] [Google Scholar]
- 61.Raineri S, Mellor J. IDH1: Linking Metabolism and Epigenetics. Front Genet 2018;9:493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Penard-Lacronique V, Bernard OA. IDH1, Histone Methylation, and So Forth. Cancer Cell 2016;30:192–4 [DOI] [PubMed] [Google Scholar]
- 63.Bledea R, Vasudevaraja V, Patel S, Stafford J, Serrano J, Esposito G, et al. Functional and topographic effects on DNA methylation in IDH1/2 mutant cancers. Sci Rep 2019;9:16830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sheng-Fang Su C-HL, Chiou-Ling Cheng, Chao-Chi Ho, Tsung-Ying Yang, Kun-Chieh Chen. Genome-Wide Epigenetic Landscape of Lung Adenocarcinoma Links HOXB9 DNA Methylation to Intrinsic EGFR-TKI Resistance and Heterogeneous Responses JCO Precision Oncology 2021:418431. [DOI] [PMC free article] [PubMed]
- 65.Forloni M, Gupta R, Nagarajan A, Sun LS, Dong Y, Pirazzoli V, et al. Oncogenic EGFR Represses the TET1 DNA Demethylase to Induce Silencing of Tumor Suppressors in Cancer Cells. Cell Rep 2016;16:457–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pastor WA, Aravind L, Rao A. TETonic shift: biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol 2013;14:341–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kharat SS, Ding X, Swaminathan D, Suresh A, Singh M, Sengodan SK, et al. Degradation of 5hmC-marked stalled replication forks by APE1 causes genomic instability. Sci Signal 2020;13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer 2010;10:457–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet 2011;12:7–18 [DOI] [PubMed] [Google Scholar]
- 70.Yun M, Wu J, Workman JL, Li B. Readers of histone modifications. Cell Res 2011;21:564–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010;141:69–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Debruyne DN, Dries R, Sengupta S, Seruggia D, Gao Y, Sharma B, et al. BORIS promotes chromatin regulatory interactions in treatment-resistant cancer cells. Nature 2019;572:676–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang Z, Hausmann S, Lyu R, Li TM, Lofgren SM, Flores NM, et al. SETD5-Coordinated Chromatin Reprogramming Regulates Adaptive Resistance to Targeted Pancreatic Cancer Therapy. Cancer Cell 2020;37:834–49 e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kun E, Tsang YTM, Ng CW, Gershenson DM, Wong KK. MEK inhibitor resistance mechanisms and recent developments in combination trials. Cancer Treat Rev 2021;92:102137. [DOI] [PubMed] [Google Scholar]
- 75.DiNardo CD, Jonas BA, Pullarkat V, Thirman MJ, Garcia JS, Wei AH, et al. Azacitidine and Venetoclax in Previously Untreated Acute Myeloid Leukemia. N Engl J Med 2020;383:617–29 [DOI] [PubMed] [Google Scholar]
- 76.Bates SE. Epigenetic Therapies for Cancer. N Engl J Med 2020;383:650–63 [DOI] [PubMed] [Google Scholar]
- 77.Morschhauser F, Tilly H, Chaidos A, McKay P, Phillips T, Assouline S, et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. Lancet Oncol 2020;21:1433–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Swanton C Take lessons from cancer evolution to the clinic. Nature 2020;581:382–3 [DOI] [PubMed] [Google Scholar]
- 79.Jamal-Hanjani M, Hackshaw A, Ngai Y, Shaw J, Dive C, Quezada S, et al. Tracking genomic cancer evolution for precision medicine: the lung TRACERx study. PLoS Biol 2014;12:e1001906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.consortium TRR. TRACERx Renal: tracking renal cancer evolution through therapy. Nat Rev Urol 2017;14:575–6 [DOI] [PubMed] [Google Scholar]
- 81.Bailey C, Black JRM, Reading JL, Litchfield K, Turajlic S, McGranahan N, et al. Tracking Cancer Evolution through the Disease Course. Cancer Discov 2021;11:916–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.