

Abstract
Low-temperature NMR studies with a 4-C-methyl-4-O-benzoyl galactopyranosyl donor enable the observation and characterization of a bridged bicyclic dioxacarbenium ion arising from participation by a distal ester. Variable temperature NMR studies reveal this bridged ion to decompose at temperatures above ~−30 °C. In the absence of the methyl group, the formation of a bicyclic ion is not observed. It is concluded that participation by typical secondary distal esters in glycosylation reactions is disfavored in the ground state conformation of the ester from which it is stereoelectronically impossible. Methylation converts the secondary ester to a conformationally more labile tertiary ester, removes this barrier, and renders participation more favorable. Nevertheless, the minor changes in selectivity in model glycosylation reactions on going from the secondary to the tertiary esters at both low and room temperature argue against distal group participation being a major stereodirecting factor even for the tertiary system.
Keywords: Dioxacarbenium ion, distal group participation, glycosylation, variable temperature NMR
Graphical Abstract

Simple methylation at the 4-position of a 4-O-benzoyl galactopyranosyl donor enables direct observation by low temperature NMR spectroscopy of the previously elusive bridging dioxacarbenium ion, supporting the hypothesis that distal group participation by secondary esters is disfavored by the conformation of the ester.
Introduction
Stereodirecting neighboring group participation (NGP) by esters and other groups at the 2-position of glycosyl donors, as first discussed by Frush and Isbell,[1] has been a mainstay of preparative carbohydrate chemistry since the introduction of the Koenigs-Knorr reaction in the very early days of the field.[2] The intervention of fused bicyclic 5-membered 1,3-dioxacarbenium ions, exemplified by 1 (Figure 1), in NGP-directed glycosylation reactions is supported by considerable evidence,[3] including their isolation in the form of crystalline salts,[4] and NMR spectroscopic characterization in organic and superacid media.[5] The parallel concept of stereodirecting participation by more remote or distal esters through bridged bicyclic six or seven-membered dioxacarbenium ions (distal group participation, DGP) in glycosylation is a more nebulous concept. DGP has been frequently invoked,[3, 6] since first advanced in the 1970’s,[7] but the evidence rarely extends beyond frequently small changes in stereoselectivity on replacing an ether by an ester at a distal position in a glycosyl donor. Arguably the best support for bridged ions such as 2 (Figure 1) in DGP was provided by contemporaneous studies from the Boons and Nifantiev laboratories that revealed increased selectivity for the 1,4-trans-glycosides in glycosylations employing p-substituted 4-O-benzoyl galacto- and fucopyranosyl donors with increased electron density on the benzoate,[8] but these observations can also be accommodated by alternative mechanisms in the absence of DGP.[3, 9]
Figure 1.

Five-Membered Fused and Seven-Membered Bridged Bicyclic Dioxacarbenium Ions 1 and 2.
We advanced the concept of tert-butyl carbonate esters (Boc esters) as probes for DGP by esters, with collapse to cyclic carbonates accompanied by formal extrusion of the tert-butyl cation, following cyclic dioxacarbenium ion formation.[10] Based on our inability to isolate bridging carbonates in a number of systems, we concluded that DGP is unlikely to be an important phenomenon, and supported this conclusion with evidence from two further probes for the specific case of esters at the 4-position of galactopyranosyl systems. Exceptions to this conclusion were limited to axial esters at the 3-position of pyranosyl type donors when a six-membered cyclic carbonate could be isolated.[10] Related probes subsequently revealed that participation through fused six-membered dioxacarbenium ions appended to the 2-position is also unlikely.[11] Numerous studies have resulted in the isolation of bridged bicyclic species from DGP by remote trichloroacetimidates, but such groups are more nucleophilic than carboxylate esters, and the conditions employed typically more forcing than standard glycosylation reactions.[3] The recent characterization of a macrocyclic acetal stemming from DGP by the ketone moiety in a remote levulinate ester, in the presence of multiple esters capable of NGP and DGP, is also noteworthy.[12] Multiple computation studies indicate that bridged bicyclic dioxacarbenium ions such as 2 are more stable than the corresponding 4-O-carboxyl glycosyl oxocarbenium ions,[13] and such bridging ions have recently been characterized by IR spectroscopy at liquid helium temperatures when generated in a mass spectrometer lending credence to the DGP concept.[13c, 13d] We have argued, however, that comparison of the relative stabilities of cyclic dioxacarbenium ions from NGP or DGP with those of the corresponding ester substituted oxocarbenium ions is not relevant to the solution phase where the glycosyl oxocarbenium ions are stabilized in the form of glycosyl triflates or other covalent intermediates.[3, 9] We have also argued[9] that DGP is less favored than NGP because i) distal esters are less destabilizing (disarming)[14] toward glycosyl oxocarbenium ion intermediates than neighboring esters, and ii) five-membered cyclic dioxacarbenium ions are more stable than their six and seven-membered cyclic counterparts.[15] Turning to kinetics, we suggest that DGP is slow because it concatenates several unfavorable factors: i) formation of the glycosyl oxocarbenium ion from the activated covalent donor; ii) a change in conformation of the glycosyl oxocarbenium ion; iii) adoption of a high energy ester conformation; iv) and the greater entropic cost of forming the rigid bicyclic system (Figure 2).[3, 9]
Figure 2.

Unfavorable Equilibria Retarding the Formation of a Bridged Dioxocarbenium Ion from 4-O-Acyl Galactopyranosyl Donors
We now show that destabilization of the ground state conformation of the distal ester facilitates formation of a bridged dioxocarbenium ion. Indeed the inclusion of a simple methyl group, to convert a secondary ester to a tertiary ester, enables readily formation and characterization by standard NMR spectroscopic methods in deuteriodichloromethane solution, of a bridged ion at low temperatures under typical solution phase glycosylation conditions.
Results and Discussion
It has long been known that the preferred conformation of esters of secondary alcohols is that in which the carbonyl oxygen is close to eclipsing the α-C-H in the alkyl moiety (Figure 3).[3, 16] It has been estimated that the rotamer of the ester in which the C-H bond is antiperiplanar to the ester C-O bond is some 4–12 kcal.mol−1 higher in energy,[3, 17] thereby providing a considerable barrier to DGP.
Figure 3.

Resting and High Energy Conformations of Esters
We reasoned that destabilization of the low energy conformation of the distal ester, would result in increased population of the conformation required for DGP, and would correspondingly enhance the likelihood of observing a bridged bicyclic dioxacarbenium ion under standard glycosylation conditions. We further reasoned that destabilization of the low energy conformation could be achieved by the simple expedient of introducing a methyl group, i.e., by converting the esterified secondary alcohol to a tertiary one. Accordingly, as described in the Supporting Information, we synthesized the tertiary alcohol 3, and converted it to the corresponding Boc ester 4 and benzoate ester 5, the latter 95% enriched in 13C at the carbonyl position to facilitate NMR studies. Also prepared, as described previously,[8a, 10] were the corresponding secondary derivatives 6-7 for the purposes of comparison. To facilitate low temperature NMR studies thioglycosides 5, and 8 were converted to the corresponding sulfoxides 9, and 10, respectively, each as an unassigned mixture of diastereomers, with mCPBA at −40 °C (Figure 4). Extrapolating from the work of Jensen and Bols,[18] we reason that the presence of the methyl group in 4, 5, and 9 will have only a minor impact on their reactivity in comparison to that of 6-8, and 10, other than by its anticipated influence on the conformation of the Boc and benzoate esters. The methyl group in 3-5 and 9 does not cause any change in the pyranose ring conformation, as noted by Jensen and Bols for an analogous methyl glycoside,[18] and as apparent from the 3J1,2 and 3J2,3 coupling constants (see supporting information) in the 1H NMR spectra of these compounds.
Figure 4.

Compounds 3-10
Inspection of the 1H NMR spectra of 3-8 (Table 1, SI Figures 1a-f) reveals that the installation of the methyl group at C4 in each of the 4-OH, 4-O-Boc, and 4-O-Bz series causes a consistent minor upfield shift in H’s 3 and 5, with both of which it shares a cis-vicinal relationship. The impact of methylation on the chemical shift of H’s 1, 2 and 6a and 6b is less consistent. Small downfield shifts are observed for the alcohol and the Boc ester, but larger downfield shifts are seen for H3 and one of the two H6s in the case of the benzoate ester, suggesting a larger impact of methylation on the conformation of the benzoate ester than on the Boc ester. Inspection of the 3J coupling constants between H5 and the two side chain protons (H’s 6a and 6b) points to a consistent change in the average side chain conformation on methylation, whatever the axial substituent at C4 (OH, OBoc or OBz). Although, the proR and proS protons in the side chain (H’s 6a and 6b) could not be assigned unambiguously,[19] we interpret this change in coupling constants in terms of an increased population of the gauche,trans (gt) rotamer at the expense of the trans,gauche (tg) one.[20],[21]
Table 1.
1H Chemical Shift and Coupling Constant Changes on Methylation at C4.
| C4 subs | δH1 | δH2 | δH3 | δH4 | δH5 | δH6a,b | 3J5a,6a,3J5,6b | |
|---|---|---|---|---|---|---|---|---|
| 3 | Me, OH | 4.69 | 3.85 | 3.30 | - | 3.44 | 3.93, 3.82 | 5.5, 2.9 |
| 6 | H, OH | 4.58 | 3.71 | 3.57 | 4.09 | 3.57 | 3.81, 3.76 | 5.7, 5.7 |
| Δδ | +0.11 | +0.14 | −0.27 | - | −0.13 | - | ||
| 4 | Me, Boc | 4.67 | 3.86 | 3.29 | 3.49 | 3.98, 3.67 | 7.6, 2.5 | |
| 7 | H, OBoc | 4.69 | 3.79 | 3.70 | 5.48 | 3.76 | 3.74, 3.68 | nda |
| Δδ | −0.02 | +0.07 | −0.41 | −0.27 | ||||
| 5 | Me, OBz | 4.85 | 4.03 | 3.52 | - | 3.76 | 4.26, 3.89 | 7.5, 2.5 |
| 8 | H, OBz | 4.64 | 3.67 | 3.75 | 5.89 | 3.87 | 3.69, 3.58 | 6.9, 5.9 |
| Δδ | +0.21 | +0.36 | −0.23 | - | −0.11 |
Not determined because of insufficient resolution
Secure in the knowledge that methylation changes the average conformation of at least the benzoate ester group, we turned to its influence on reactivity. Activation of the methylated Boc ester 4 with diphenyl sulfoxide and triflic anhydride[22] in dichloromethane solution at −78 °C followed by stirring for 3 h and then quenching with aqueous sodium bicarbonate before warming to room temperature and chromatographic isolation afforded the 1,4-bridged bicyclic carbonate 12 in 56% yield, indicative of the formation of the bridged ion 11 on activation, with subsequent loss of the tert-butyl group (Scheme 1). The cyclic carbonate 12 was characterized inter alia by an IR carbonyl stretch at γ 1748 cm−1, and the HMBC correlation of H1 and the carbonyl carbon. In the 1H NMR spectrum H1 had a chemical shift of δ 5.5 and took the form of a multiplet, while H2 was a doublet with 3J1,2 = 2.5 Hz. Additionally, the 1H-1H COSY spectrum shows a four bond W correlation of H1 and H3, whereas only a very weak correlation exists between H2 and H3, all of which is indicative of an approximate 1,4B conformation of the pyranose ring (Figure 5a). When the experiment was repeated with the secondary Boc ester 7 a complex reaction mixture resulted in which we were unable to identify any bicyclic products and from which we were only able to isolate the Freidel-Crafts product 13 in 14% yield. The absence of participation by the 4-O-Boc group in 7 is consistent with our earlier observations with an analogous galactopyranosyl donor under glycosylation conditions,[10] and cyclic ethers such as 13 are typical decomposition products found on warming of ether-protected glycosyl triflates above their decomposition temperature.[23] Clearly the addition of a methyl group at the 4-position in 4-O-Boc protected galactopyranosyl donors significantly impacts the pattern of reactivity by enabling the otherwise disfavored transannular participation.
Scheme 1.
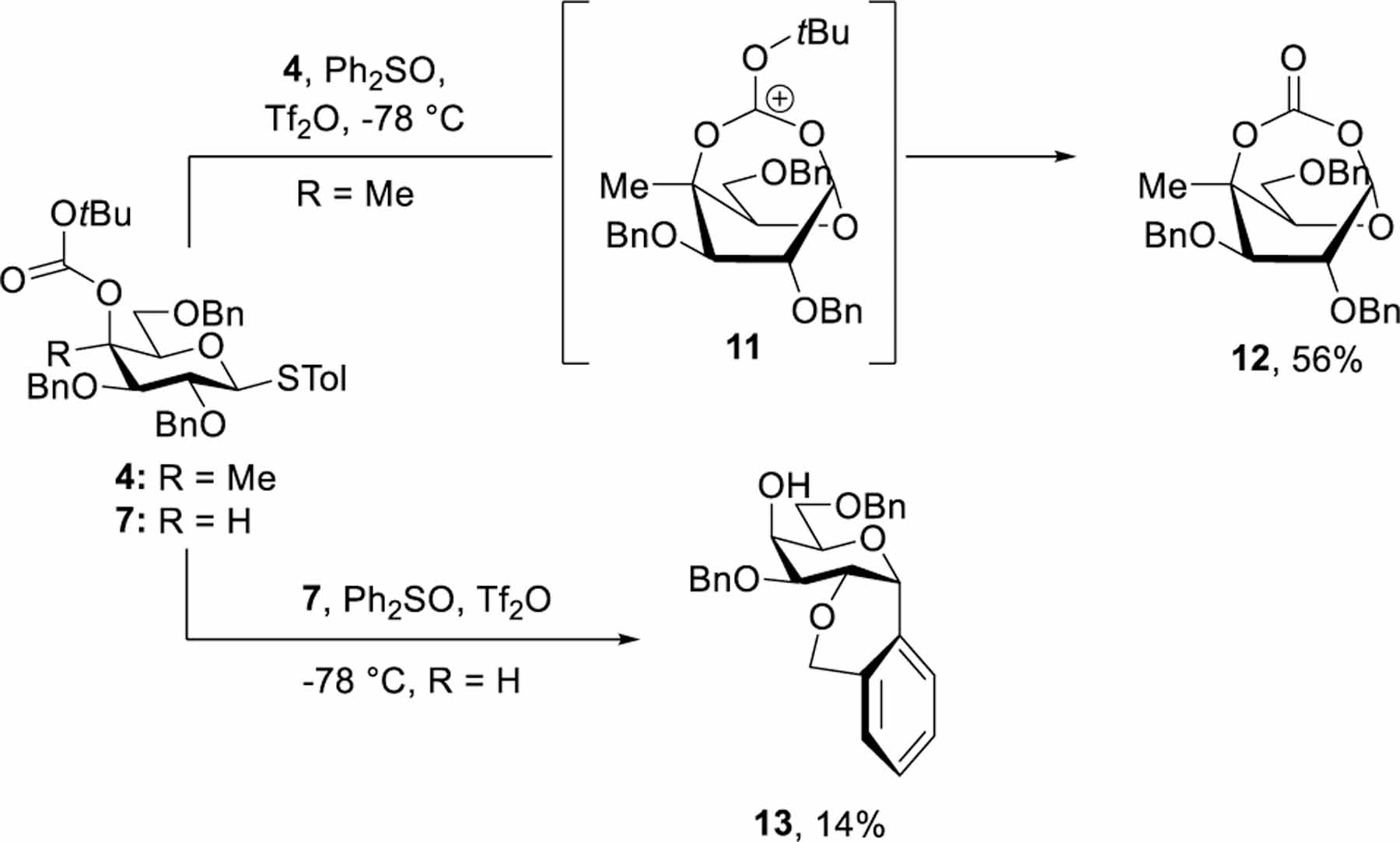
Activation of the Boc Esters 4 and 7 and Influence of the C4 Methyl Group on Reactivity
Figure 5.

Key 1H-1H COSY and HMBC correlations, and coupling constants for a) compound 12 (CDCl3 at 25 oC) and b) 14 (CD2Cl2 at −80 oC)
Turning to the benzoate esters, sulfoxide 9 was dissolved in CD2Cl2 in the presence of the hindered non-nucleophilic base TTBP, cooled to −80 °C in the probe of the spectrometer and 13C (Figure 6a) and the 1H NMR spectra recorded. The tube was then ejected from the probe, rapidly injected with Tf2O, shaken, and reinserted into the cold NMR probe. An immediate 13C NMR spectrum (Figure 6b) indicated ~ 80±10% conversion, depending on the run, of the carbonyl signal at δ165.5 to a new signal at δ177.3 consistent with the clean formation of the bridged bicyclic ion 14 (Scheme 2). In the 1H NMR spectrum at −80 °C (Figure 6c) ion 14 was characterized by a resonance at δ6.85 in the form of a broad doublet of doublet of doublet (3JC,H = 5.59, 3J1,2 ~ 2.95 and 4J1,3 ~ 1.86 Hz) assigned to H1. The structure of ion 14, evident from the heteronuclear coupling of H1 to the downfield-shifted 13C-enriched carbon, was confirmed by the HMBC correlation of H1 to the enriched 13C resonance at δ177.3, the C1 chemical shift of δ110.5, the small 3J coupling of H1 with H2, the 4J W coupling of H1 with H3, and the small unresolved 3J2,3 coupling, all of which again point to a 1,4B conformation of the pyranose ring (Figure 5b). Warming of the NMR sample in 10 °C increments with monitoring by 13C NMR spectroscopy revealed ion 14 to be stable up to ≥−40 °C, but to have decomposed by −30 °C. Further warming to room temperature and purification lead to the characterization of the 1,6-anhydro derivative 15 as the main decomposition product. Some decomposition product 15, accompanied by benzyl triflate (Figure 6c),[24] is present immediately after activation but this arises from partial warming when the tube is ejected from the cold probe for activation, and not decomposition in the probe at −80 °C. When the experiment was repeated with benzoate 10 lacking the methyl group, no evidence was found for formation of a comparable bridged bicyclic ion, with the carbonyl carbon resonance remaining constant at ~ δ164.8–165.6 throughout the course of the experiment; the formation of a new C1 resonance at δ107.9 was consistent with the formation of an α-glycosyl triflate 16,[25] while the 3J1,2 and 3J2,3 coupling constants indicated the retention of the 4C1 conformation of the pyranose ring (Figure 7a-c). No cyclization was observed on warming in 10 °C increments below the decomposition temperature of triflate 16 (~ −20 °C): the intramolecular Friedel-Crafts product 17 and the 1,6-anhydro derivative 18 were the major decomposition products and were isolated in 27 and 29% yield, respectively. Evidently, the incorporation of the methyl group at C4 impacts the reactivity of a 4-O-benzoate ester by facilitating DGP and enabling NMR spectroscopic characterization of a seven-membered bridged bicyclic dioxacarbenium ion for the first time.
Figure 6a-c.

Partial 13C NMR spectra before and after activation of 9, and partial 1H spectrum of activated 9 at −80 °C. The flagged signals belong to 15 formed during the activation process.
Scheme 2.

Activation of the Benzoate Esters 9 and 10 and Influence of the C4 Methyl Group on Reactivity
Figure 7a-c.

Partial 13C NMR spectrum before and after activation of 10, and partial 1H spectrum of activated 10 at −80 °C.
In full agreement with our earlier conclusions,[10] it is clear from these studies that the formation of bridged bicyclic ions arising from DGP by secondary esters at the 4-position of galactopyranosyl donors is not a favored reaction pathway in the solution phase. This situation is reversed by the incorporation of a simple methyl group at the 4-position, when DGP becomes the predominant pathway. We suggest that this drastic change in reactivity on methylation is simply due to the lowering of the barrier to rotation about the ester bond. It is equally apparent from the decomposition of ion 14 below −30 °C that when such ions do form, they are notably less stable than the fused bicyclic 1,3-dioxalenium ion intermediates from NGP-directed glycosylations, which can be observed by NMR spectroscopy up to at least room temperature in organic solution[5a, 5c, 5d] and in some cases isolated as stable crystalline solids.[4]
We next turned our attention to the influence of the C4 methyl group on stereoselectivity in glycosylation reactions. We selected two sets of conditions, the first using activation of a −78 °C dichloromethane solution of the thioglycosides with the diphenyl sulfoxide/triflic anhydride combination,[22] in common use in our laboratories and related to our benzenesulfinyl piperidine/triflic anhydride conditions,[26] and the second using activation by N-iodosuccinimide (NIS) and trimethylsilyl trifluoromethanesulfonate (TMSOTf) of a 3:1 1,4-dioxane:toluene solution of the donor at room temperature, namely the optimal conditions in the Boons DGP study.[8a] 1,2:3,4-Diacetone-α-D-galactopyranose 19 and 1,2:5,6-diacetone-α-D-glucofuranose 20 were selected as model acceptors for direct comparison with the Boons study.[8a] In the event, under the low temperature conditions in dichloromethane the 4-methyl donor 5 gave a 48% of disaccharide 21 on coupling to the primary alcohol 19 in the form of a 1.3:1 α:β mixture, whereas the simple galactosyl donor 8 gave a 43% yield of an 0.67:1 α:β mixture of disaccharide 22 on coupling to the same alcohol (Table 2, entries 1 and 2). Both 5 and 8 gave moderate yields of the respective disaccharides 23 and 24 as a single α-anomers on activation under the same conditions in the presence of acceptor 20 (Table 2, entries 3 and 4). Under the room temperature conditions in 3:1 1,4-dioxane:toluene the 4-methyl donor 5 gave an 80% yield of disaccharide 21 as a 26:1 α:β mixture on coupling to 19, whereas the simple donor 8 gave 44% of a 14.5:1 α:β mixture of 22 (Table 3, entries 1 and 2). As in the low temperature experiments, both 5 and 8 gave exclusively the α-anomers of 23 and 24 on coupling to 20 under the room temperature conditions (Table 3, entries 3 and 4). The 14.5:1 α:β mixture of anomers of 22 observed on coupling of 8 to 19 at room temperature parallels the 17:1 ratio observed by the Boons lab under closely related conditions (albeit concentrations and stoichiometry were not specified), as does the very high α-selectivity observed on coupling of 8 to 20. When 10 mol% of TMSOTf was used for the activation, the coupling reaction of donor 5 with acceptor 19 provided exclusively the α-glycoside 21, whereas donor 8 afforded 44% of a 9.6:1 α:β mixture of 22 (Table 3, entries 5 and 6).
Table 2.
Low Temperature Glycosylations in Dichloromethanea

| |||||
|---|---|---|---|---|---|
|
| |||||
| Entry | Acceptor | Donor | Product | Yield (%)b | α:βc |
| 1 |

|
5 |
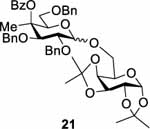
|
48 | 1.3:1 |
| 2 | 19 | 8 |

|
43 | 0.67:1 |
| 3 |

|
5 |

|
58 | α-only |
| 4 | 20 | 8 |

|
56 | α-only |
Conditions: Donor (1.2 eq), Acceptor (1.0 eq), Ph2SO (2.0 eq), TTBP (2.0 eq), Tf2O (1.5 eq), 0.1 M CH2Cl2, 4 Å MS, −78 °C.
Isolated yield.
Anomeric ratios were determined by integration of the 1H NMR spectra of the crude reaction mixtures.
Table 3.
Room Temperature Glycosylations in 3:1 Dioxane:Toluenea

| |||||
|---|---|---|---|---|---|
|
| |||||
| Entry | Acceptor | Donor | Product | Yield (%)b | α:βc |
| 1 | 19 | 5 | 21 | 80 | 26:1 |
| 2 | 19 | 8 | 22 | 44 | 14.5:1 |
| 3 | 20 | 5 | 23 | 64 | α-only |
| 4 | 20 | 8 | 24 | 42 | α-only |
| 5d | 19 | 5 | 21 | 87 | α-only |
| 6d | 19 | 8 | 22 | 44 | 9.6:1 |
Conditions: Donor (1.2 eq), Acceptor (1.0 eq), NIS (2.0 eq), TMSOTf (1.0 eq), 0.1 M 1,4-dioxane:toluene 3:1, 4 Å AWMS, room temp.
Isolated yield.
Anomeric ratios were determined by integration of the 1H NMR spectra of the crude reaction mixtures.
10 mol% of TMSOTf was used.
Overall, the inclusion of the methyl group in donor 5 causes an approximate doubling of α-selectivity on coupling to the primary acceptor 19, when compared to the simple donor 8, whether under the low temperature conditions in dichloromethane or the room temperature conditions in dioxane:toluene. On the other hand it is not possible to discern any methyl group effect on the coupling of 5 and 8 to the less reactive[27] secondary acceptor 20, as very high α-selectivity was observed in all such couplings. As no bridging ion arising from DGP with donor 8 is observed in low temperature NMR experiments, we argue that the high α-selectivity observed on coupling of 8 with acceptor 20 is unlikely to arise from DGP but is reflective of the inherent stereoselectivity of 8 with less reactive acceptors, which may be best understood in terms of α-selective attack on a loose, perhaps solvent-separated ion pair.[28] The reduced selectivity on coupling of 5 and 8 to the more reactive acceptor 19 can then be attributed to competition between the ion pair mechanism and direct β-selective displacement of an intermediate α-glycosyl triflate by the more reactive primary acceptor. The greater selectivity observed on coupling of 5 and 8 to 19 at the higher temperature in dioxane:toluene is unlikely to be the result of DGP in view of the absence of a bridged ion in the low temperature NMR study of 8, and the −30 °C decomposition temperature of the bridging ion 14 derived from 5. Rather the enhanced α-selectivity observed under the second set of conditions must arise from either a greater population of the ion pairs with respect to the covalent triflates owing to the greater polarity of the solvent, or to participation by 1,4-dioxane in the form of a β-glycosyl dioxonium ion. Such β-glycosyl dioxonium ions have yet to be observed spectroscopically, but their decomposition products (β-haloalkyl glycosides) have been isolated on multiple occasions.[29] The approximately two-fold increase in α-selectivity with acceptor 19 on going from the simple donor 8 to its 4-methyl analog 5 (Tables 2 and 3) presumably arises from the observed change in conformation of the side chain on methylation, a phenomenon that is known to affect selectivity,[30] or greater steric shielding of the β-face by the conformationally more mobile tertiary ester in 5.
Conclusion
Incorporation of a methyl group at the 4-position of a galactopyranosyl donor enables for the first time the NMR spectroscopic characterization of a seven-membered bridged bicyclic dioxacarbenium ion arising from DGP at temperatures below −30 °C. In the absence of the methyl group the spectra are consistent with the formation of a classical galactosyl triflate rather than of any bridged ion. These observations are consistent with the ground state conformation of the remote secondary ester presenting a substantial barrier to DGP such that the galactosyl triflate is the more stable species. With the conformationally more labile tertiary benzoate DGP is facilitated and the cyclic ion is readily observed. In model glycosylation reactions, only minor differences in selectivity are observed in going from the secondary to the tertiary systems, at both low and room temperature, suggesting that DGP is not on the main reaction pathway for glycosylation even with the tertiary system. The observation of bridged bicyclic ions from DGP in low temperature gas phase studies in the absence of counter ions does not bear on the mechanism of glycosylation in the condensed phase.
Supplementary Material
Acknowledgements
We thank the NIH (GM62160) for partial support of this work, and Dr Michael G Pirrone, UGA, for help with the low temperature NMR spectra
Footnotes
Supporting information for this article is given via a link at the end of the document
Conflict of Interest
The authors declare no conflict of interest
Institute and/or researcher Twitter usfoernames: @kaps007up; @UGAPharmacy
Contributor Information
Kapil Upadhyaya, Department of Pharmaceutical and Biomedical Sciences, University of Georgia, 250 West Green Street, Athens, GA 30602, USA.
Yagya P. Subedi, Department of Pharmaceutical and Biomedical Sciences, University of Georgia, 250 West Green Street, Athens, GA 30602, USA
David Crich, Department of Pharmaceutical and Biomedical Sciences, University of Georgia, 250 West Green Street, Athens, GA 30602, USA; Complex Carbohydrate Research Center, University of Georgia, 315 Riverbend Road, Athens, GA 30602, USA; Department of Chemistry, University of Georgia, 140 Cedar Street, Athens, GA 30602, USA.
References
- [1].Frush HL, Isbell HS, J. Research Natl. Bur. Standards 1941, 27, 413–428. [Google Scholar]
- [2].Koenigs W, Knorr E, Sitzungsber. Bayr. Akad. Wiss 1900, 103–105. [Google Scholar]
- [3].Hettikankanamalage A, Lassfolk R, Ekholm F, Leino R, Crich D, Chem. Rev 2020, 120, 7104–7151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Paulsen H, Herold C-P, Chem. Ber 1970, 103, 2450–2462. [Google Scholar]
- [5].a) Crich D, Dai Z, Gastaldi S, J. Org. Chem 1999, 64, 5224–5229; [DOI] [PubMed] [Google Scholar]; b) Martin A, Arda A, Désiré J, Martin-Mingot A, Probst N, Sinaÿ P, Jiménez-Barbero J, Thibaudeau S, Blériot Y, Nat. Chem, 2016, 8, 186–191; [DOI] [PubMed] [Google Scholar]; c) Zeng Y, Wang Z, Whitfield D, Huang X, J. Org. Chem 2008, 73, 7952–7962; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Williams RJ, McGill NW, White JM, Williams SJ, J. Carbohydr. Chem 2010, 29, 236–263. [Google Scholar]
- [6].a) Komarova BS, Ustyuzhanina NE, Tsvetkov YE, Nifantiev NE, in Modern Synthetic Methods in Carbohydrate Chemistry; From Monosaccharides to Complex Glycoconjugates (Eds.: Werz DB, Vidal S), Wiley, Weinheim, 2014, pp. 125–160; [Google Scholar]; b) Komarova BS, Tsvetkov YE, Nifantiev NE, Chemical Record 2016, 16, 488–506. [DOI] [PubMed] [Google Scholar]
- [7].Dejter-Juszynski M, Flowers HM, Carbohydr. Res 1972, 23, 41–45. [DOI] [PubMed] [Google Scholar]
- [8].a) Demchenko AV, Rousson E, Boons G-J, Tetrahedron Lett 1999, 40, 6523–6536; [Google Scholar]; b) Gerbst AG, Ustuzhanina NE, Grachev AA, Tsvetkov DE, Khatuntseva EA, Nifant’ev NE, Mendeleev Commun 1999, 9, 114–116. [Google Scholar]
- [9].Crich D, J. Am. Chem. Soc 2021, 143, 17–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Crich D, Hu T, Cai F, J. Org. Chem 2008, 73, 8942–8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Wen P, Crich D, J. Org. Chem 2015, 80, 12300–12310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yang W, Zhang J, Yang C-W, Ramadan S, Staples R, Huang X, Org. Lett 2021, 23, 1153–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].a) Gerbst AG, Ustuzhanina NE, Grachev AA, Khatuntseva EA, Tsvetkov DE, Whitfield DM, Berces A, Nifantiev NE, J. Carbohydr. Chem 2001, 20, 821–831; [Google Scholar]; b) Li Z, Carbohydr. Res 2010, 345, 1952–1957; [DOI] [PubMed] [Google Scholar]; c) Elferink H, Severijnen ME, Martens J, Mensink RA, Berden G, Oomens J, Rutjes FPJT, Rijs AM, Boltje TJ, J. Am. Chem. Soc 2018, 140, 6034–6038; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Matrianski M, Mucha E, Greis K, Moon S, Pardo A, Kirschbaum C, Thomas DA, Meijer G, von Helden G, Gilmore K, Seeberger PH, Pagel K, Angew. Chem. Int. Ed 2020, 59, 6166–6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Zhang Z, Ollmann IR, Ye X-S, Wischnat R, Baasov T, Wong C-H, J. Am. Chem. Soc 1999, 121, 734–753. [Google Scholar]
- [15].a) Paulsen H, Meyborg H, Behre H, Angew. Chem., Int. Ed. Engl 1969, 8, 888–888; [Google Scholar]; b) Larsen JW, Ewing S, J. Am. Chem. Soc 1971, 93, 5107–5111. [Google Scholar]
- [16].a) Mathieson AM, Tetrahedron Lett 1965, 6, 4137–4144; [Google Scholar]; b) Schweizer WB, Dunitz JD, Helv. Chim. Acta 1982, 65, 1547–1554; [Google Scholar]; c) González-Outeiriño J, Nasser R, Anderson JE, J. Org. Chem 2005, 70, 2486–2493. [DOI] [PubMed] [Google Scholar]
- [17].Wiberg KB, Laidig KE, J. Am. Chem. Soc 1987, 109, 5935–5943. [Google Scholar]
- [18].Jensen HH, Bols M, Org. Lett 2003, 5, 3419–3421. [DOI] [PubMed] [Google Scholar]
- [19].Bock K, Duus JO, J. Carbohydr. Chem 1994, 13, 513–543. [Google Scholar]
- [20].The data do not support an increase in population of the gauche,gauche (gg) conformer, which would lead to similar values for the two coupling constants [Google Scholar]
- [21].Dharuman S, Amarasekara H, Crich D, J. Org. Chem 2018, 83, 10334–10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Codée JDC, van den Bos LJ, Litjens REJN, Overkleeft HS, van Boeckel CAA, van Boom JH, van der Marel GA, Tetrahedron 2004, 60, 1057–1064. [Google Scholar]
- [23].a) Crich D, Cai W, Dai Z, J. Org. Chem 2000, 65, 1291–1297; [DOI] [PubMed] [Google Scholar]; b) Crich D, Wu B, Org. Lett 2006, 8, 4879–4882; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Huang M, Retailleau P, Bohé L, Crich D, J. Am. Chem. Soc 2012, 134, 14746–14749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Elferink H, Mensink RA, White PB, Boltje TJ, Angew. Chem. Int. Ed 2016, 55, 11217–11220. [DOI] [PubMed] [Google Scholar]
- [25].a) Crich D, Sun S, J. Am. Chem. Soc 1997, 119, 11217–11223; [Google Scholar]; b) Frihed TG, Bols M, Pedersen CM, Chem. Rev 2015, 115, 4963–5013. [DOI] [PubMed] [Google Scholar]
- [26].Crich D, Smith M, J. Am. Chem. Soc 2001, 123, 9015–9020. [DOI] [PubMed] [Google Scholar]
- [27].Chang C-W, Lin M-H, Chan C-K, Su K-Y, Wu C-H, Lo W-C, Lam S, Cheng Y-T, Liao P-H, Wong C-H, Wang C-C, Angew. Chem. Int. Ed 2021, 60, 12413–12423. [DOI] [PubMed] [Google Scholar]
- [28].a) van der Vorm S, Hansen T, Overkleeft HS, van der Marel GA, Codée JDC, Chem. Sci 2017, 8, 1867–1875; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) van der Vorm S, Overkleeft HS, van der Marel GA, Codée JDC, J. Org. Chem 2017, 82, 4793–4811; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Krumper JR, Salamant WA, Woerpel KA, Org. Lett 2008, 10, 4907–4910; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Adero PO, Amarasekara H, Wen P, Bohé L, Crich D, Chem. Rev 2018, 118, 8242–8284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a) Helferich B, Zirner J, Chem. Ber 1963, 96, 374; [Google Scholar]; b) Wulff G, Schmidt W, Carbohydr. Res 1977, 53, 33–46; [Google Scholar]; c) Dabideen DR, Gervay-Hague J, Org. Lett 2004, 6, 973–975. [DOI] [PubMed] [Google Scholar]
- [30].Upadhyaya K, Bagul R, Crich D, J. Org. Chem 2021, 86, 12199–12225. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.