

Abstract
Background.
Filamin C truncating variants (FLNCtv) cause a form of arrhythmogenic cardiomyopathy (ACM): the mode of presentation, natural history and risk stratification of FLNCtv remain incompletely explored. We sought to develop a risk profile for refractory heart failure and life-threatening arrhythmias in a multicenter cohort of FLNCtv carriers.
Methods.
FLNCtv carriers were identified from ten tertiary care centers for genetic cardiomyopathies. Clinical and outcome data were compiled. Composite outcomes were all-cause mortality/heart transplantation/left ventricle assist device (D/HT/LVAD), non-arrhythmic death/HT/LVAD and SCD/major ventricular arrhythmias (SCD/MVA). Previously established cohorts of 46 patients with LMNA and 60 with DSP-related ACM were used for prognostic comparison.
Results.
Eighty-five patients carrying FLNCtv were included (42±15 years, 53% males, 45% probands). Phenotypes were heterogeneous at presentation: 49% dilated cardiomyopathy, 25% arrhythmogenic left dominant cardiomyopathy, 3% arrhythmogenic right ventricular cardiomyopathy. Left ventricular ejection fraction (LVEF) was <50% in 64% of carriers and 34% had right ventricular fractional area changes (RVFAC=(right ventricular end-diastolic area - right ventricular end-systolic area)/right ventricular end-diastolic area) <35%. During follow-up (median time 61 months), 19 (22%) carriers experienced D/HT/LVAD, 13 (15%) non-arrhythmic death/HT/LVAD and 23 (27%) SCD/MVA. The SCD/MVA incidence of FLNCtv carriers did not significantly differ from LMNA carriers and DSP carriers. In FLNCtv carriers, LVEF was associated with the risk of D/HT/LVAD and non-arrhythmic death/HT/LVAD.
Conclusions.
Among patients referred to tertiary referral centers, FLNCtv ACM is phenotypically heterogeneous and characterized by high risk of life-threatening arrhythmias, which does not seem to be associated with the severity of LV dysfunction.
Keywords: FLNC gene, genotype-phenotype correlation, prognosis, arrhythmogenic cardiomyopathy, sudden cardiac death, heart failure
Introduction
Arrhythmogenic cardiomyopathy (ACM) is a genetic disorder characterized by high risks of life-threatening ventricular arrhythmias, sudden cardiac death (SCD), and progressive heart failure (HF)1, 2. The expression of ACM encompasses a wide phenotypic spectrum ranging from classical dilated cardiomyopathy (DCM) to typical arrhythmogenic right ventricular cardiomyopathy (ARVC) with frequently overlapping features2–5. Recently, truncating variants in the filamin C gene (FLNCtv) have been found to cause ACM with high penetrance, variable phenotypic presentation and prominent ventricular arrhythmias6–9. FLNC encodes a striated muscle protein that cross-links actin and anchors cell membrane proteins to the cytoskeleton, sarcolemma and sarcomere Z-disk10, 11. The clinical spectrum of FLNCtv ACM remains incompletely defined and the natural history and prognosis are largely unexplored. In this study we analyzed longitudinal clinical data from a large multicenter cohort of FLNCtv carriers to define the clinical features of FLNCtv-related cardiomyopathy and the correlations between genotype and phenotype. Factors associated with adverse outcomes were determined to inform on the risk stratification of FLNCtv carriers.
Methods
International FLNCtv Registry
Patients with FLNCtv were collected from ten international tertiary care centers with expertise in the management of inherited cardiomyopathies: University Hospital of Trieste, University of Colorado Cardiovascular Institute, Victor Chang Cardiac Research Institute, Utrecht and Amsterdam University Medical Centers, Brigham and Women’s Hospital, University Hospital of Udine, Johns Hopkins University, Stanford Center for Inherited Cardiovascular Disease, and National Center for Cardiovascular Diseases in Beijing. Data were anonymized and stored in a shared database to create an International Registry of patients with FLNCtv. Institutional review boards approved the study and informed consent was obtained under the institutional review board policies of the hospital administration. Demographic, clinical, imaging and genetic data were retrospectively analyzed by each participating center. In order to minimize the possibility of unintentionally sharing information that can be used to re-identify private information and considering the different institutional review boards policies of the participating centers, the datasets generated for this study will not be made available to other researchers for purposes of reproducing the results or replicating the procedure. The authors declare that all supporting methods are available within the article (and online supplemental material).
Molecular genetics and definition of genetic variants
Genetic testing was done through the participating sites by Next Generation and Sanger sequencing in a clinical or research laboratory. Variants were classified as “pathogenic” or “likely pathogenic” according to the American College of Medical Genetics criteria12 (Table I in the Supplement). To maintain a conservative approach, only ‘pathogenic’ and ‘likely pathogenic’ truncating variants were considered, while missense variants and ‘variants of uncertain significance’ were excluded from the analysis. Probands and available family members carriers of ‘pathogenic’ or ‘likely pathogenic’ FLNCtv were included in the registry. Additional variants of interest inherent to other cardiomyopathies related genes were also reported and patients considered as carriers of multiple mutations.
Phenotypic characterization
Demographic, clinical and therapeutic information were collected at study entry (baseline) and detailed information on family history of cardiomyopathies and SCD were recorded. Clinical data included HF symptoms (New York Heart Association functional class), history of unexplained syncope at enrollment (likely secondary to arrhythmic cause), presence of ventricular arrhythmias (SCD, resuscitated cardiac arrest, sustained ventricular tachycardia [VT] and non-sustained ventricular tachycardia [NSVT]), and atrial fibrillation/atrial flutter. Age of onset was defined based on clinical diagnosis. Data from 12-lead electrocardiograms (ECG), Holter ECG monitoring and signal-averaged electrocardiograms (SAECG) were recorded. Late potentials were determined by SAECG as currently recommended by the 2010 revised Task Force Criteria13.
Echocardiographic biventricular dimensions and systolic function were assessed at transthoracic echocardiography as currently recommended by international guidelines14. Left ventricle (LV) and right ventricle (RV) systolic dysfunction were defined by LV ejection fraction (LVEF) <50% and RV fractional area change (RVFAC=(right ventricular end-diastolic area - right ventricular end-systolic area)/right ventricular end-diastolic area)) <35%, respectively. For the subgroup of patients with available cardiac magnetic resonance (CMR) images, we analyzed the presence, localization (LV, RV or biventricular), distribution (septal, inferoposterolateral or both) and pattern (midwall, subepicardial or both) of late gadolinium enhancement (LGE).
Based on their presenting phenotypes, patients with FLNCtv were classified into six mutually exclusive phenotypic categories based on current international consensus guidance2: DCM, ARVC (definite, borderline or possible), arrhythmogenic left dominant cardiomyopathy (ALVC), biventricular ARVC, ‘minor phenotype’ and ‘unaffected’. The DCM phenotype was defined by a LVEF <50% in the absence of any known possible cause of LV dysfunction15; ARVC phenotype was defined according to the 2010 Task Force Criteria (TFC)13; ALVC phenotype was defined as DCM presentation not fulfilling TFC for ARVC and with ≥1 of the following: SCD/major ventricular arrhythmias (MVA) (resuscitated cardiac arrest, sustained VT, appropriate ICD interventions), unexplained syncope, SVT, ≥1000 premature ventricular contractions (PVCs)/24 hours, ≥50 couplets/24 hours2, 3; biventricular ARVC was defined as “definite” ARVC plus LVEF <50%2. Patients with isolated or multiple pathological findings insufficient to fulfill the criteria for any of the phenotypes defined above were classified as ‘minor phenotype’. Finally, patients without any cardiac pathological finding were considered unaffected.
Study outcomes
The study outcomes were: 1) all-cause mortality/heart transplantation/left ventricular assist device implantation (D/HT/LVAD); 2) non-arrhythmic death (including HF death and death not due to SCD)/HT/LVAD; 3) SCD/major ventricular arrhythmias (MVA). MVA included ventricular fibrillation, sustained VT (lasting >30 s or with hemodynamic instability) and appropriate ICD interventions (shock or anti-tachycardia pacing on ventricular fibrillation or sustained VT) (SCD/MVA). SCD was defined as witnessed SCD with or without documented ventricular fibrillation, death within 1 h of acute symptoms, or nocturnal death with no antecedent history of immediate worsening symptoms. In addition, incident bradyarrhythmias (i.e. sick sinus syndrome, atrio-ventricular blocks, permanent pacemaker/ICD implantation for pacing indications) were recorded. The follow-up date for analysis ended at the date of the first outcome or at the last available contact with the patient. A cohort of 46 carriers with “pathogenic” or “likely pathogenic” LMNA variants and a cohort of 60 carriers with “pathogenic” or “likely pathogenic” DSP variants from three participating Centers (University Hospital of Trieste, University of Colorado Cardiovascular Institute, Brigham and Women’s Hospital), representing arrhythmia-prone populations, were used as comparisons.
Statistical analysis.
Variables were expressed as mean±SD, median and interquartile range (IQR), or counts and percentage, as appropriate. Comparisons between groups were made by the analysis of variance (ANOVA) test on continuous variables using the Brown-Forsythe statistic when the assumption of equal variances did not hold or the nonparametric Mann-Whitney test; the chi-square test or the Fisher’s exact test were calculated for discrete variables. Two independent linear regression models were used to quantify changes of LVEF at follow-up in the overall cohort and in patients with baseline LVEF≥50% and <50%, respectively. Kaplan Meier curves for D/HT/LVAD and Cumulative Incidence Function (CIF) for non-arrhythmic death/HT/LVAD and SCD/MVA were estimated for FLNCtv carriers, LMNA variants carriers and DSP variants carriers and compared by the log rank test. As some patients were grouped as families, in all survival analyses we reported p-values derived from Cox regression models with the family code as a cluster indicator i.e. use a robust sandwich estimator for the standard error16, 17.
The univariate Cox model (D/HT/LVAD) and the cause-specific Cox model (non-arrhythmic death/HT/LVAD and SCD/MVA) were used to assess the association between LVEF and the outcomes. A restricted cubic spline transform (termplot function from the R survival package) was used when the association between LVEF and the outcome was non-linear. To avoid bias due to baseline characteristics missing not at random, multiple imputation (n=5) was performed using predictive mean matching (mice package). A p-value <0.05 was considered statistically significant. Statistical analyses were performed in R version 3.6.2 (R Foundation for Statistical Computing, Vienna, Austria).
Results
Characterization and mapping of FLNC truncating variants
The study included 85 carriers of FLNCtv from 49 families (see Table I in the Supplement for the complete list of variants and cases contribution for each participating center), of whom 38 (45%) were probands. FLNCtv variants included nonsense variants, splice site variants, and insertion/deletion (indel) variants that resulted in downstream premature stop codons. The percentage of variants distribution is reported in Table 1. As shown in Figure 1, variants were distributed throughout the protein, with an apparent cluster in the Z-disk interacting region (Ig-like 19–21) of the Rod Domain 2. Two patients (2.4%) had additional pathogenetic/likely pathogenetic variants on different genes than FLNC (TTN c.76115_76116insA, p.Asn25372Lysfs*5, and PPA2 c.514G>A, p.(Glu172Lys), see Table I in the Supplement).
Table 1.
Baseline clinical characteristics of the overall cohort of FLNC truncating mutations carriers and divided according to proband status.
| total n=85 |
probands n=38 (45%) |
non-probands n=47 (55%) |
p-value | |
|---|---|---|---|---|
| Age (years) | 42±15 | 40±14 | 44±16 | 0.163 |
| Male %(n.) | 53 (45) | 63 (24) | 45 (21) | 0.090 |
| Caucasian %(n.) | 99 (84) | 97 (37) | 100 (47) | 0.263 |
| Dimerization | 4 (3) | 3 (1) | 4 (2) | |
| Family history cardiomyopathy %(n.) | 81 (69) | 61 (23) | 100 (46) | <0.001 |
| Family history SCD %(n.) | 52 (44) | 34 (13) | 66 (31) | 0.004 |
| Unaffected | 11 (9) | 0 (0) | 19 (9) | |
| CPK (U/l) | 80 (55–121) | 75 (55–139) | 94 (50–119) | 0.713 |
| HR (bpm) | 72±21 | 73±21 | 71±21 | 0.567 |
| AF %(n.) | 4 (3) | 5 (2) | 2 (1) | 0.219 |
| LBBB %(n.) | 8 (7) | 8 (3) | 9 (4) | 0.871 |
| NYHA 3–4 %(n.) | 18 (15) | 22 (8) | 15 (7) | 0.424 |
| NYHA 1 %(n.) | 68 (57) | 62 (23) | 72 (34) | 0.321 |
| Negative anterior T waves %(n.) | 21 (18) | 39 (15) | 7 (3) | <0.001 |
| PVCs/24h | 3194±5443 | 4581±6913 | 1806±2994 | 0.108 |
| PVCs>1000/24h %(n.) | 45 (18) | 55 (11) | 35 (7) | 0.204 |
| Positive Late Potentials %(n.) | 8 (7) | 5 (2) | 11 (5) | 0.370 |
| NSVT %(n.) | 48 (28) | 61 (19) | 33 (9) | 0.034 |
| Echo LVEF (%)* | 43±15 | 33±14 | 51±12 | <0.001 |
| Echo LVEF <50 %(n.) | 64 (52) | 89 (33) | 43 (19) | <0.001 |
| Echo LVEF ≦35 %(n.) | 31 (25) | 56 (21) | 9 (4) | 0.046 |
| Echo LVEDD (mm) | 57±10 | 62±8 | 52±9 | <0.001 |
| Echo MWT (mm) | 9±2 | 10±1 | 9±2 | 0.446 |
| Echo LVH (%) | 11 | 6 | 14 | 0.270 |
| Echo RVFAC (%)* | 38±11 | 35±12 | 40±9 | 0.083 |
| Echo RVFAC<35 %(n.) | 34 (17) | 46 (11) | 23 (6) | 0.090 |
| Echo RV WMA %(n.) | 8 (4) | 16 (4) | 0 (0) | 0.031 |
| Echo MR %(n.) | 44 (33) | 58 (19) | 33 (14) | 0.036 |
| CMR LGE %(n.) ** | 53 (23) | 61 (8) | 56 (14) | 0.738 |
| Beta-blockers %(n.) | 64 (52) | 89 (31) | 46 (21) | <0.001 |
| ACEi/ARB/ARNI %(n.) | 57 (46) | 80 (28) | 39 (18) | <0.001 |
| ICD %(n.) | 14 (11) | 20 (7) | 9 (4) | 0.152 |
| ICD at follow-up %(n.) | 48 (39) | 57 (21) | 41 (18) | 0.155 |
ABD=active binding domain, SCD=sudden cardiac death, DCM=dilated cardiomyopathy, ARVC=arrhythmogenic right ventricular cardiomyopathy, ALVC=arrhythmogenic left-dominant cardiomyopathy, CPK=creatine phosphokinase, HR=heart rate, AF=atrial fibrillation, LBBB=left bundle branch block, NYHA=New York heart association, PVC=premature ventricular complex, NSVT=non-sustained ventricular tachycardia, LVEF=left ventricular ejection fraction, LVEDD=left ventricular end-diastolic volume, MWT=maximum wall thickness, LVH=left ventricular hypertrophy, RVFAC=right ventricle fractional area change, WMA=wall motion abnormalities, MR=mitral regurgitation, CMR=cardiac magnetic resonance imaging, LGE=late gadolinium enhancement, ACEi/ARB/ARNI=ACE-inhibitors/angiotensin receptor blockers/angiotensin receptor neprylisin inhibitors, ICD=implantable cardioverter defibrillator.
Values are reported as mean±standard deviation, median and interquartile range or percentage as appropriate.
missing data for LVEF=4 (5%), missing data for RVFAC=35 (41%).
among the 43 pts with available CMR.
Figure 1. Mapping of FLNCtv variants.
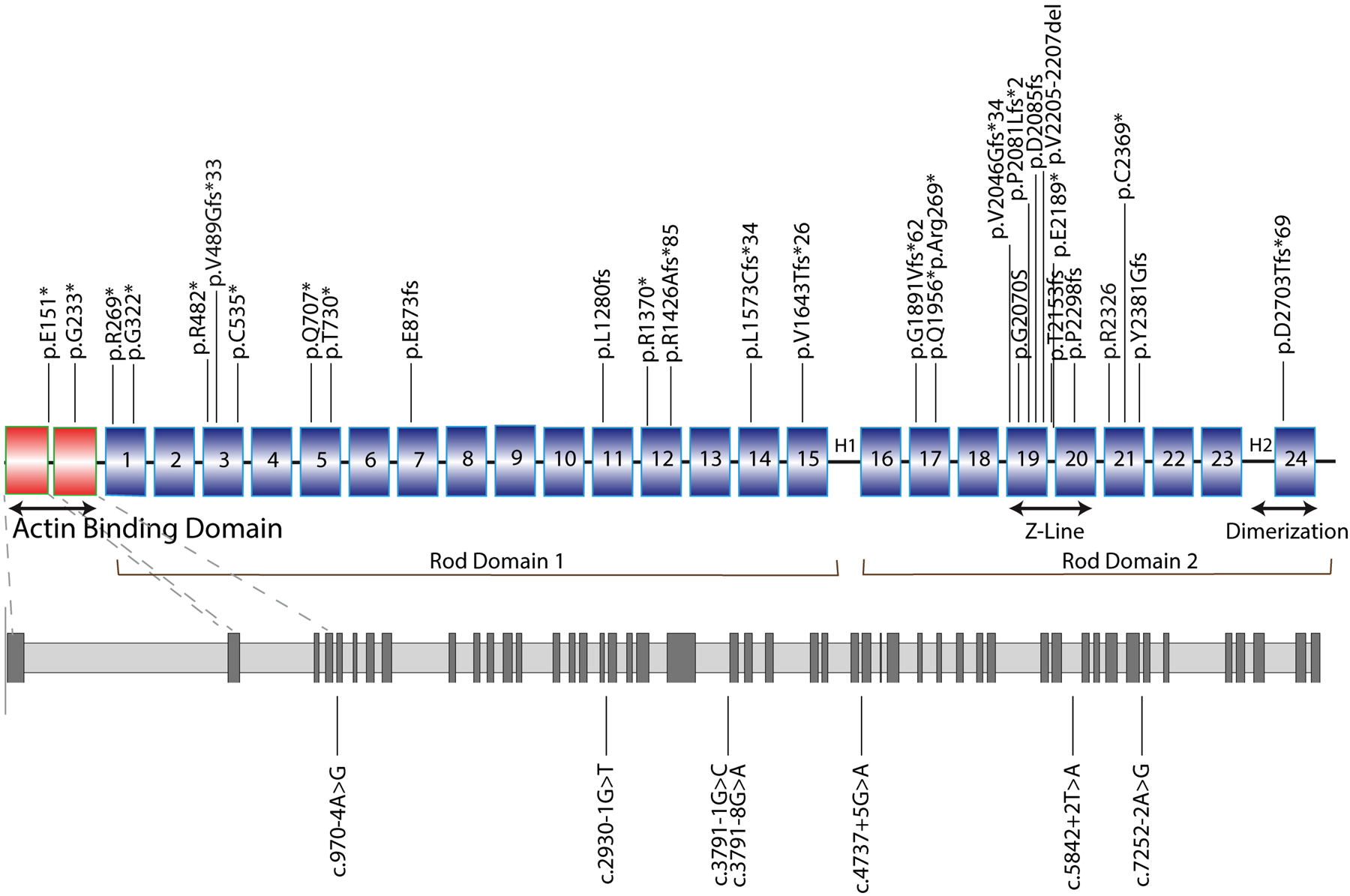
Diagrams representing the structure of FLNC and the distribution of FLNCtv variants. FLNCtv were distributed across all gene domains. However, clusters were noted in the Actin Binding Domain (ABD domains 1 and 2) and in the Z-disk region (Ig-like 19–21) of the Rod Domain 2. Nonsense mutations, and insertion/deletion (indel) variants are indicated in the upper scheme, splice site mutations in the lower (gray) scheme. 1 to 24, Ig-like domains; H1 and H2, hinge domains.
Spectrum of phenotypes in FLNCtv cardiomyopathy
Table 1 shows the baseline characteristics of probands and relatives. Mean age at enrollment was 42±15 years (3 patients were <18 years old: 12 months, 14 months and 14 years, respectively), 53% were male, 99% were European ancestry. The most frequent phenotype at presentation was DCM (42 carriers, 49%) followed by ALVC (21 carriers, 25%), and ARVC (3 carriers, 3%). Nine FLNCtv carriers from 5 families (mean age 45±17 years, 67% males) were unaffected (11%). There were no differences in age (43±15 vs 42±16 years, p=0.854) and gender (male sex 51% vs 70%, p=0.250) between affected and unaffected individuals. Thirty-two percent of subjects had HF-related symptoms (NYHA class >1), 22% had a history of syncope. Negative anterior T-waves (V1-V4) were found in 21% of carriers, 48% had NSVT. Atrial fibrillation/atrial flutter at baseline was present in 4% of the study cohort. No skeletal muscle involvement was reported.
Mean LVEF and RVFAC were 43±15% and 38±11%, respectively; 64% of carriers had LVEF <50% and 34% had RVFAC <35%. Four carriers were missing data for LVEF (5%) and 35 were missing data for RVFAC (41%). Figure 2 shows the distribution of RV and LV dysfunction at enrollment among the 50 patients with both LVEF and RVFAC data available. Eleven (22%) subjects presented with biventricular dysfunction, six (12%) with isolated RV dysfunction, 19 (38%) with isolated LV dysfunction and 14 subjects (28%) with preserved biventricular function. Mild LV hypertrophy was found in 11% of cases. Baseline therapy included 64% of carriers on betablockers, 57% on ACE-inhibitors/angiotensin receptor blockers. In 14% of the cohort, ICDs were already implanted at the time of entry into the study, whereas a total of 48% had ICDs by the end of follow-up.
Figure 2. Distribution of right and left ventricular function in FLNCtv carriers.
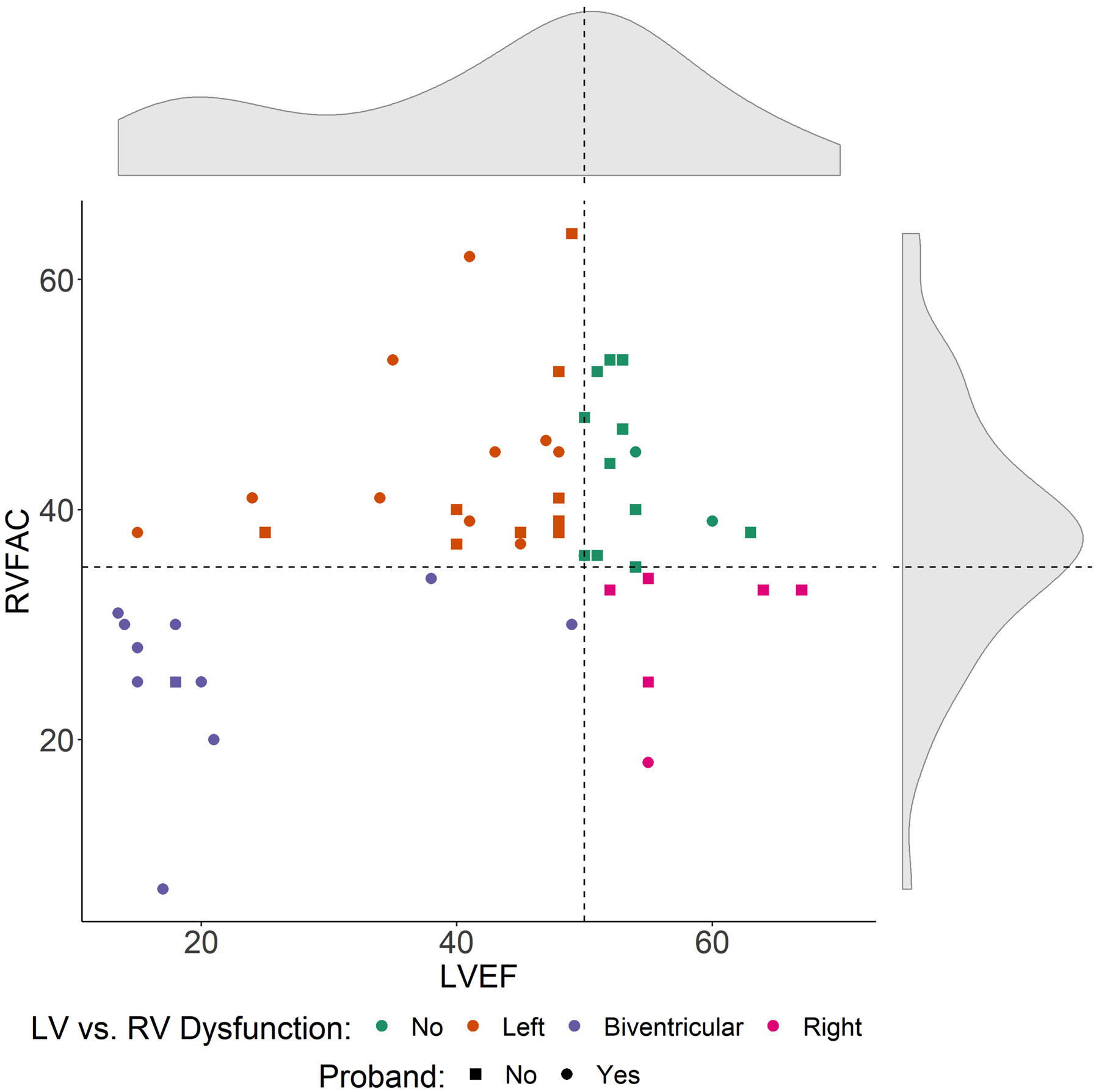
The FLNCtv population showed a wide distribution of RV and LV dysfunction. Dashed lines mark the cut-offs defining LV dysfunction (LVEF<50%) and RV dysfunction (RVFAC<35%). LVEF=left ventricular ejection fraction, RVFAC=right ventricle fractional area change, RV= right ventricular.
As shown in Table 1, probands compared to relatives presented significantly more negative anterior T-waves, NSVT and pathological echocardiographic findings. They were also more likely to be treated with antineurohormonal drugs. Finally, phenotype presentation did not significantly differ according to FLNCtv variant location (Table II in the Supplement).
Cardiac MRI patterns of FLNCtv
There were 43 (51%) FLNCtv carriers with available CMR studies, of which 23 (53%) had evidence of LGE with no differences between probands and family members (44% vs 56%, p=0.738). Figure 3A shows the CMR features of a FLNCtv carrier (proband, male, 48 years old) in early stage of disease (CMR LVEF=54%), with subepicardial “ring-like” LGE indicated by the arrows. Figure 3B reports the characteristics of LGE location, distribution and pattern in the CMR subgroup. All except one case had LV involvement, with the most frequent area of LGE signal distribution being the inferoposterolateral wall (12/23, 52% of LGE positive) and the most frequent LGE pattern was subepicardial (11/23, 48% of LGE positive).
Figure 3. Characteristics of LGE in carriers of FLNCtv.
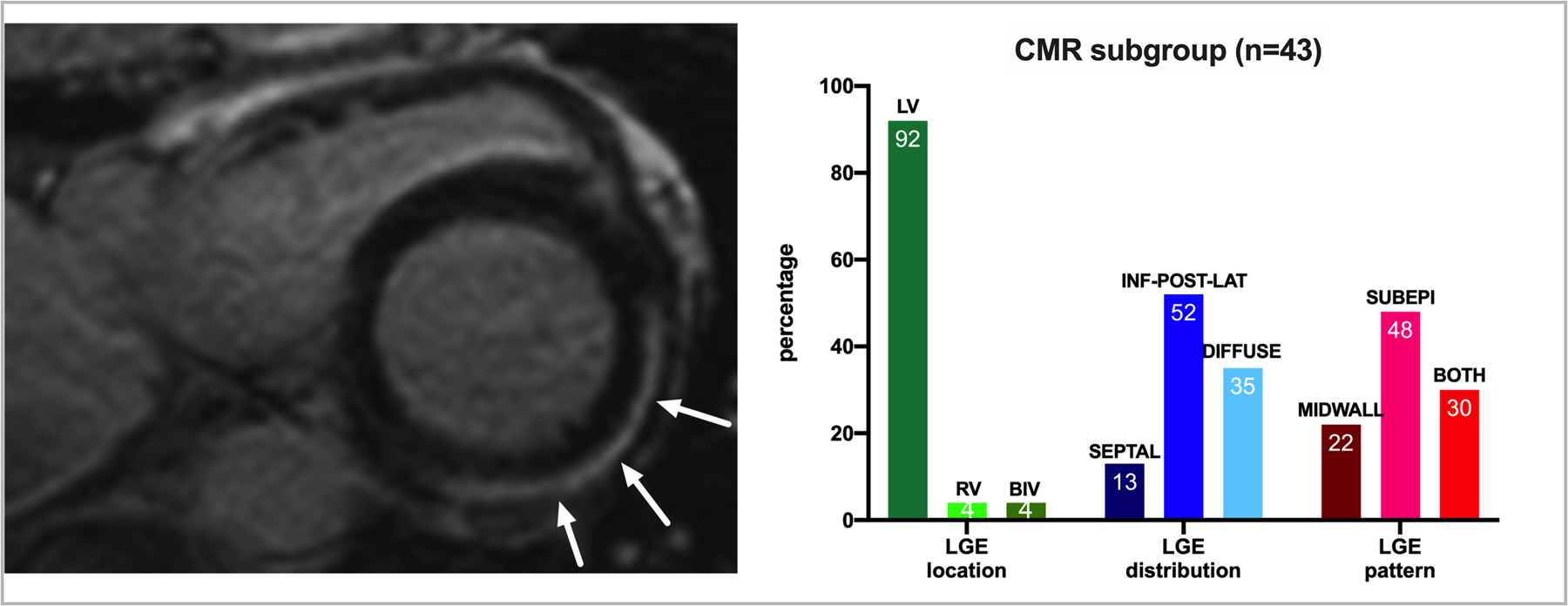
Left panel shows the typical subepicardial “ring-like” distribution of LGE in a carrier of FLNCtv (male, 48 years old, LVEF 54%), involving the inferior, posterior and lateral wall. Right panel summarizes the distribution of LGE in the 43 FLNCtv carriers with available cardiac magnetic resonance (CMR). Percentages are reported within bars.
Variable longitudinal echocardiographic trends at follow-up
Echocardiographic measures at follow-up (median 66 months IQR 19–126) were available in 68 carriers (80%) and are summarized in Table 2. At last follow-up, mean LVEF was 44±14% and mean RVFAC was 41±9%. As shown in Figure I in the Supplement, in both the groups of patients with LVEF ≥50% and LVEF <50% at baseline, no significant mean change was observed during follow-up (coefficient of monthly mean change: −0.01, 95%CI: −0.06–0.03, p=0.5 and −0.04, 95%CI: −0.11–0.02, p=0.2, respectively).
Table 2.
Main echocardiographic measures at follow-up (median time 66 months, IQR 19–126) in the overall subset of patients with available follow-up reassessment and divided according to proband status.
| total n=68 |
probands n=28 (57%) |
non-probands n=21 (43%) |
p-value | |
|---|---|---|---|---|
| Echo LVEF (%) | 44±14 | 38±14 | 49±12 | 0.001 |
| Echo LVEF <50 (%) | 57 | 78 | 39 | 0.001 |
| Echo LVEF ≦35 (%) | 25 | 37 | 14 | 0.025 |
| Echo LVEDD (mm) | 60±10 | 63±9 | 55±10 | 0.008 |
| Echo RVFAC (%) | 41±9 | 41±10 | 40±8 | 0.710 |
| Echo RVFAC<35 (%) | 14 | 17 | 11 | 0.505 |
| Echo MR (%) | 64 | 71 | 55 | 0.277 |
For abbreviations see Table 1
Study endpoints and prognostic stratification
Over a median follow-up of 61 months (IQR 10–139), 19 carriers (15 probands, 4 non-probands, 22% of the total cohort) experienced D/HT/LVAD (11 deaths, 3 HT, 5 LVAD; median age 54, range 47–66), 13 (10 probands, 3 non-probands, 15% of the total cohort) non-arrhythmic death/HT/LVAD (5 deaths, 3 HT, 5 LVAD; median age 51, range 48–61) and 23 (15 probands, 8 non-probands, 27% of the total cohort) SCD/MVA (6 SCD, 6 sustained VT/ventricular fibrillation, 11 ICD interventions; median age 48, range 44–60).
Table 3 reports the main baseline characteristics of the study patients according to outcomes. Compared to patients with no outcome, patients experiencing D/HT/LVAD and patients experiencing non-arrhythmic death/HT/LVAD were more likely probands, reported less frequently a familial history of cardiomyopathy, had more severe symptoms, lower LVEF and larger LV end-diastolic diameter, left bundle-branch block, NSVT (for D/HT/LVAD only), and mitral regurgitation were more prevalent. In patients experiencing SCD/MVA the only differences compared to patients not suffering the arrhythmic outcome concerned the higher rate of probands (65% vs 37%, p=0.021) and of NSVT (82% vs 34%, p=0.001). Of note, no differences were observed in the distribution of FLNCtv and, limited to the 43 patients with available CMR, in the presence or absence of LGE for any of the explored outcomes.
Table 3.
Baseline clinical characteristics of FLNC truncating mutations carriers divided according to study outcomes.
| D/HT/LVAD | Non-arrhythmic death/HT/LVAD | SCD/MVA | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Outcome Yes |
Outcome No |
p-value | Outcome Yes |
Outcome No |
p-value | Outcome Yes |
Outcome No |
p-value | |
| Age (years) | 45±18 | 42±14 | 0.485 | 43±20 | 42±14 | 0.943 | 45±11 | 42±16 | 0.424 |
| Male % | 53 | 53 | 0.976 | 46 | 54 | 0.594 | 56 | 62 | 0.687 |
| Caucasian % | 95 | 100 | 0.061 | 92 | 100 | 0.018 | 100 | 98 | 0.540 |
| Proband (%) | 79 | 35 | 0.001 | 77 | 39 | 0.011 | 65 | 37 | 0.021 |
| Dimerization | 0 | 4 | 0 | 4 | 9 | 2 | |||
| Family history cardiomyopathy % | 63 | 86 | 0.023 | 54 | 86 | 0.006 | 74 | 84 | 0.297 |
| Family history SCD % | 32 | 58 | 0.046 | 31 | 56 | 0.100 | 52 | 52 | 0.963 |
| Unaffected | 0 | 14 | 0 | 12 | 0 | 15 | |||
| HR (bpm) | 74±23 | 71±20 | 0.668 | 74±26 | 71±20 | 0.718 | 70±17 | 72±22 | 0.615 |
| AF % | 32 | 14 | 0.076 | 32 | 15 | 0.186 | 30 | 13 | 0.065 |
| LBBB % | 22 | 5 | 0.017 | 25 | 6 | 0.026 | 4 | 10 | 0.407 |
| NYHA 3–4 % | 42 | 10 | 0.002 | 46 | 13 | 0.004 | 13 | 20 | 0.479 |
| NYHA 1 % | 37 | 77 | 0.001 | 23 | 76 | <0.001 | 74 | 66 | 0.466 |
| Negative anterior T waves % | 32 | 19 | 0.220 | 23 | 21 | 0.875 | 26 | 20 | 0.523 |
| PVCs/24h | 3987±8473 | 3054±4899 | 0.704 | 652±364 | 3476±5673 | 0.331 | 4642±6958 | 2773±4979 | 0.371 |
| Positive Late Potentials % | 5 | 9 | 0.593 | 8 | 8 | 0.938 | 4 | 10 | 0.427 |
| NSVT % | 75 | 41 | 0.038 | 67 | 45 | 0.230 | 82 | 34 | 0.001 |
| Echo LVEF (%) | 30±12 | 46±15 | <0.001 | 27±12 | 45±15 | <0.001 | 40±14 | 43±16 | 0.360 |
| Echo LVEF <50 % | 100 | 55 | 0.001 | 100 | 58 | 0.005 | 73 | 61 | 0.328 |
| Echo LVEF ≦35 % | 65 | 22 | 0.001 | 67 | 25 | 0.004 | 36 | 29 | 0.513 |
| Echo LVEDD (mm) | 67±6 | 54±9 | <0.001 | 67±5 | 55±9 | <0.001 | 59±11 | 56±9 | 0.279 |
| Echo MWT (mm) | 9±2 | 9±2 | 0.966 | 9±2 | 9±2 | 0.973 | 10±1 | 9±2 | 0.170 |
| Echo LVH (%) | 8 | 11 | 0.863 | 10 | 11 | 0.929 | 16 | 9 | 0.358 |
| Echo RVFAC (%) | 34±13 | 39±10 | 0.231 | 35±14 | 38±10 | 0.504 | 34±9 | 39±11 | 0.114 |
| Echo RVFAC<35 % | 62 | 29 | 0.063 | 67 | 30 | 0.072 | 42 | 32 | 0.520 |
| Echo RV WMA % | 20 | 6 | 0.277 | 33 | 6 | 0.086 | 15 | 5 | 0.229 |
| Echo MR % | 69 | 39 | 0.044 | 78 | 39 | 0.030 | 50 | 42 | 0.556 |
| CMR LGE % ** | 50 | 59 | 0.729 | 33 | 60 | 0.367 | 63 | 57 | 0.782 |
D/HT/LVAD=death heart transplant/left ventricular assist device, non-SCD/HT/LVAD=non sudden cardiac death/heart transplant/left ventricular assist device, SCD/MVA=sudden cardiac death/major ventricular arrhythmias, ABD=active binding domain, SCD=sudden cardiac death, DCM=dilated cardiomyopathy, ARVC=arrhythmogenic right ventricular cardiomyopathy, ALVC=arrhythmogenic left-dominant cardiomyopathy, CPK=creatine phosphokinase, HR=heart rate, AF=atrial fibrillation, LBBB=left bundle branch block, NYHA=New York heart association, PVC=premature ventricular complex, NSVT=non-sustained ventricular tachycardia, LVEF=left ventricular ejection fraction, LVEDD=left ventricular end-diastolic volume, MWT=maximum wall thickness, LVH=left ventricular hypertrophy, RVFAC=right ventricle fractional area change, WMA=wall motion abnormalities, MR=mitral regurgitation, CMR=cardiac magnetic resonance imaging, LGE=late gadolinium enhancement.Values are reported as mean±standard deviation, median and interquartile range or percentage as appropriate.
among the 43 pts with available CMR.
As shown in Figure 4, we found an increasing risk of D/HT/VAD and non-arrhythmic death/HT/VAD events with decreasing values of LVEF at baseline. In contrast, there was no significant association between LVEF and the risk of SCD/MVA (Figure 5). Of note, among patients experiencing SCD/MVA, 6 (23%, 5 aborted SCD/sustained VT and one appropriate ICD shock) had LVEF >50%. Their mean age at inclusion was 45±5 years, two were males, one a proband. All were considered affected (2 ARVC, 1 ALVC, 3 minor phenotype).
Figure 4. Association between LVEF and the study outcomes in the FLNCtv cohort.
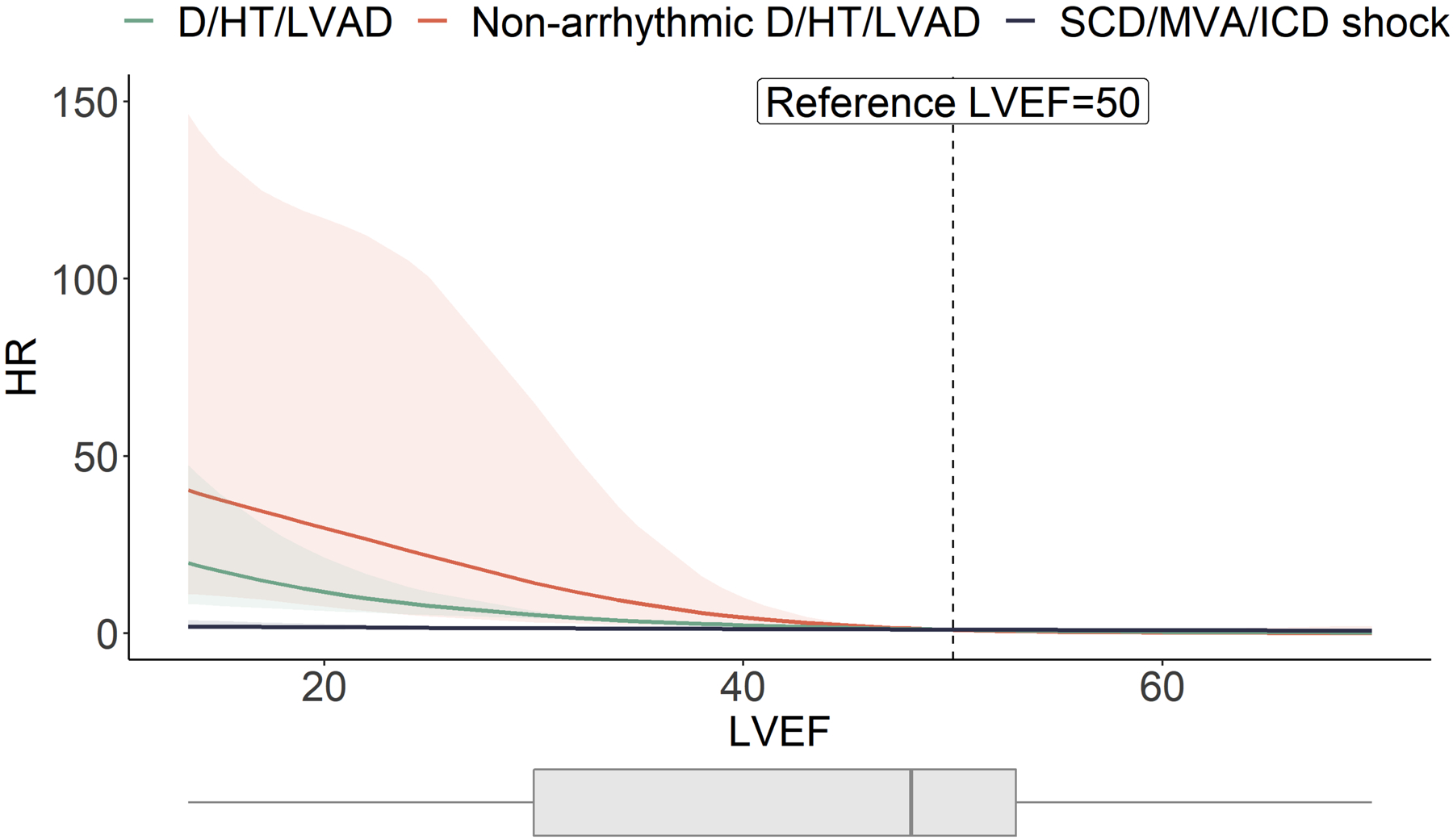
In this high-risk population the burden of D/HT/LVAD and SCD/MVA was 22% and 27%, respectively, over a median ~5 years follow-up. The association of LVEF with the study outcomes varies according to the type of outcome. As LVEF decreases, the HR for the outcomes D/HT/LVAD (green line) and non-arrhythmic death/HT/LVAD (orange line) get progressively higher, whereas no variation in risk was observed for the outcome SCD/MVA (blue line). Light painted areas indicate 95 % confidence intervals.
D/HT/LVAD=all-cause mortality/heart transplantation/left ventricular assist device; HR=hazard ratio, LVEF=left ventricular ejection fraction; SCD/MVA=sudden cardiac death/major ventricular arrhythmias.
Figure 5. Comparison of outcome between the study population of FLNCtv carriers (n=85), LMNA mutation carriers (n=46) and DSP mutation carriers (n=60).
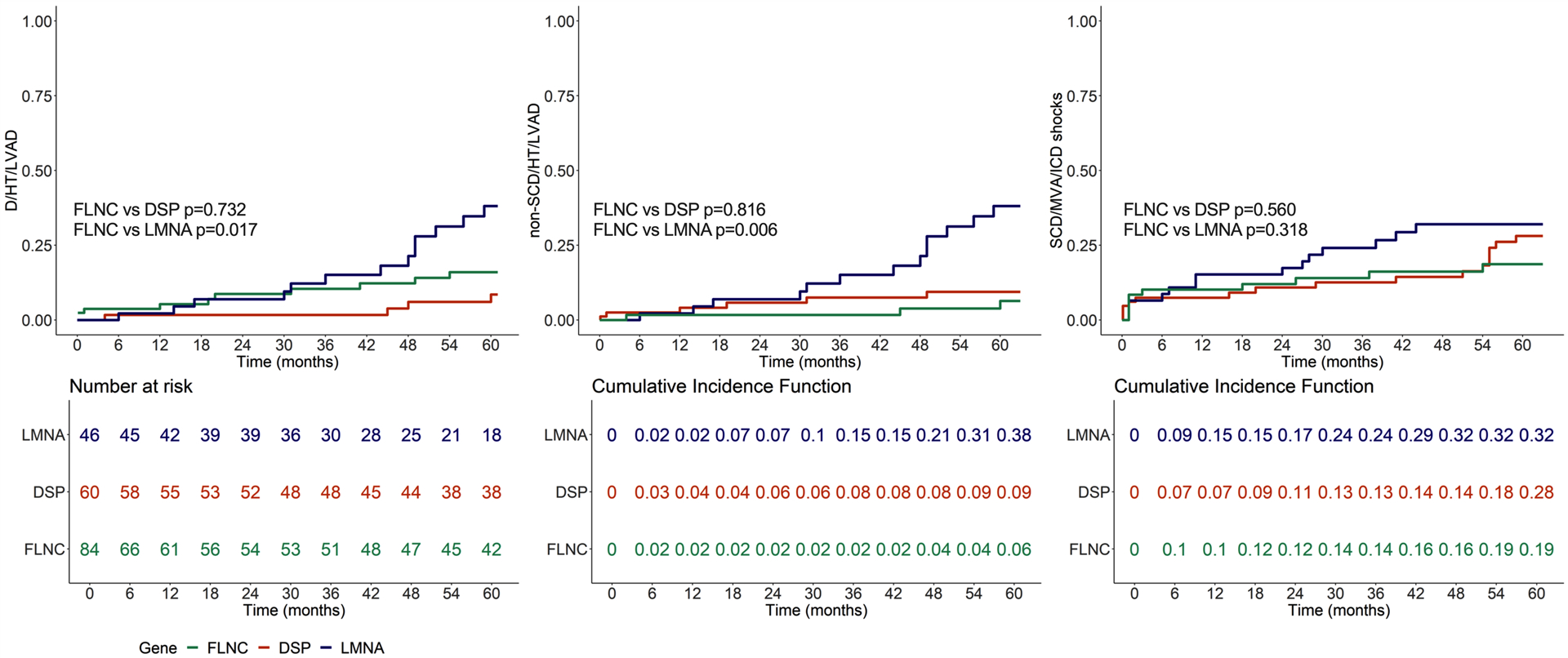
Left panel: all-cause mortality/heart transplantation/left ventricular assist device (D/HT/LVAD) in FLNCtv (green lines) vs LMNA (blue lines) vs DSP carriers (red line). Central panel: Cumulative Incidence Function (CIF) of non-arrhythmic death/HT/LVAD in FLNCtv vs LMNA vs DSP carriers. Right panel: CIF of sudden cardiac death/major ventricular arrhythmias (SCD/MVA) in FLNCtv vs LMNA vs DSP carriers. LMNA patients showed a higher risk of D/HT/LVAD (p=0.017) and non-SCD/HT/LVAD (p=0.006), whereas the risk of SCD/MVA was comparable across the three groups.
We then compared FLNCtv carriers to two separate populations of LMNA and DSP variant carriers as established models of ACM. At baseline, there was no difference in age of onset (LMNA carriers mean age 42±11, vs. FLNCtv p=0.936; DSP carriers mean age 38±14, vs FLNCtv p=0.055). LMNA carriers had a lower LVEF (LMNA carriers mean LVEF 34±13%, vs. FLNCtv, p=0.024; DSP carriers mean LVEF 43±15, vs FLNCtv p=0.779). As shown in Figure 5, compared to LMNA carriers, FLNCtv carriers experienced a lower risk of D/HT/LVAD (p=0.017) and non-arrhythmic death/HT/LVAD (p=0.006), but the risk of SCD/MVA was not significantly different (p=0.318), whereas compared to DSP carriers, FLNCtv carriers experienced a similar risk of D/HT/LVAD (p=0.732), non-arrhythmic death/HT/LVAD (p=0.816) and SCD/MVA (p=0.560). The same results were obtained if only affected carriers (n=76 FLNCtv, n=43 LMNA, n=60 DSP) were considered for the outcome analysis (Figure II in the Supplement). Finally, during follow-up, 2 incident bradyarrhythmias (both first degree atrioventricular blocks) were reported in the FLNCtv carriers, compared to 6 incident bradyarrhythmias (3 first degree atrioventricular blocks, 2 sick sinus syndromes, including 1 requiring pacemaker/ICD implantation) in the LMNA carriers.
Discussion
In the present study, we report the comprehensive characterization of variants, phenotypes and outcomes of FLNCtv cardiomyopathy in an international cohort of FLNCtv carriers from tertiary care centers. Key findings of our study are: 1) FLNCtv cardiomyopathy appears to be a disease with heterogeneous phenotypic presentation ranging from typical DCM to ARVC and with frequently overlapping forms (e.g. biventricular, ALVC); 2) LVEF is associated with the risk of D/HT/LVAD and non-arrhythmic death/HT/LVAD but not with the risk of SCD/MVA, highlighting the need for alternative strategies of stratification of the arrhythmic risk in FLNCtv related cardiomyopathy; 3) FLNCtv cardiomyopathy is associated with a high risk of ventricular arrhythmias, with frequencies of life-threatening ventricular arrhythmias not significantly different from LMNA- and DSP-cardiomyopathy.
Regional distribution of variants across the gene structure
FLNCtvs are an important cause of DCM, accounting for 2% to 4% of DCM patients, and 6% of those with an arrhythmogenic phenotype5, 6. The FLNC protein is composed of an ABD domain, which is critical for actin crosslinking, a series of 24 Ig-like domains divided into ROD1 and ROD2 subdomains and a dimerization domain (Figure 1). In our study, FLNCtvs were distributed across the whole gene, as reported by other investigators18, and we did not find an association between variant position and phenotype or prognosis, as was also observed in one small series19. These findings support the hypothesis that in FLNCtv cardiomyopathy, the disease is the product of haploinsufficiency and that FLNCtv location does not modify the severity of the phenotype. It is noteworthy to mention that similar findings were recently reported by Helms et al. in hypertrophic cardiomyopathy caused by MYBPC3 mutations20 where MYBPC3tvs were homogeneously distributed throughout the gene, in contrast to non-truncating MYBPC3 pathogenic variants that instead, clustered in specific protein domains. Similarly, the severity of the phenotype and outcome were independent of location of the MYBPC3 truncating variants.
The heterogeneous phenotypic presentation of FLNCtv
The causal relation between FLNC variants and cardiomyopathy was initially found by family cosegregation studies, animal models8 and in a large dataset of patients with inherited cardiovascular disease6. Clinical presentation was more frequently DCM, but ALVC was also reported. Moreover, in the study of Ortiz-Genga et al., only half of the relatives harboring the mutation presented with reduced LVEF6. Finally, two studies found overlap between DCM and ARVC in FLNCtv carriers7, 9. Regardless of the subphenotypes and structural abnormalities at presentation, arrhythmogenicity is a constant feature of FLNCtv cardiomyopathies, and indeed, the 2019 Heart Rhythm Society consensus guidelines on ACM included FLNC among the genes responsible for arrhythmogenic cardiomyopathy and suggested they be considered high-risk markers for SCD2.
In our large cohort of FLNCtv carriers, only 11% of carriers showed no sign of myocardial involvement. The clinical-echocardiographic phenotype at presentation was heterogeneous, with left, right or bi-ventricular systolic dysfunction (Figure 2). It has been reported that missense FLNC variants are associated with hypertrophy besides typical DCM18. Interestingly, in our cohort presence of a mild LV hypertrophy was found in 11% of cases. However, more importantly, hallmark of the phenotype were ventricular arrhythmias, which were independent of the degree of LV dysfunction as indicated by the lack of correlation with LVEF. Therefore, the combined presence of some ACM features, such as frequent premature ventricular complex, NSVT or negative anterior T waves should raise the diagnostic suspicion of an underlying FLNCtv. A recent publication also noted the presence of a low voltage electrocardiogram as a potential diagnostic feature21. Our observations support the emerging concept that specific genes (i.e. desmosomal genes, FLNC, PLN, RBM20) demonstrate high variability in phenotypic expression and share a high risk of ventricular arrhythmias2, 5–8, 22, 23.
In our series, echocardiographic follow-up was available in 68 patients. Despite the 5.5 years median interval from baseline to revaluation, LVEF remained stable in the majority of patients, with no significant variations overtime.
In the subgroup with available CMR data, we did not find an association between the presence of LGE and outcome, which could be explained by the limited number of observations and the proportional high overall rate of positive LGE. Notably, CMR features were consistent with the data recently reported by Augusto et al., showing a subepicardial pattern and inferoposterolateral distribution as the more frequent characteristics of LGE in FLNCtv and DSP ACM24. The typical subepicardial “ring-like” distribution of LGE seen in Figure 3 is another marker reported as a defining characteristic that leads one to consider the presence of FLNCtv and DSP cardiomyopathies24.
Survival and arrhythmogenicity
The 27% incidence of SCD/MVA observed in our population was comparable to previous series6–8. The >20% incidence of D/HT/LVAD that we observed was also remarkable. Interestingly, in the study by Ortiz-Genga et al. of a heterogeneous cohort of ~3,000 patients with different inherited cardiovascular diseases, carriers of FLNCtv experiencing SCD had reduced LVEF and the recent study by Akthar et al. suggested that higher LVEF values than those currently recommended for primary prevention ICD were associated with increased arrhythmic risk25. The most recent guidelines on ACM include LVEF as a criterion for the eligibility for primary prevention ICD2, 26, 27. In our cohort, known risk factors such as LVEF and NYHA class were, respectively, lower and more severe in patients experiencing D/HT/LVAD and non-arrhythmic death/HT/LVAD. As summarized in Figure 4, LVEF was associated with the risk of D/HT/LVAD and of non-arrhythmic death/HT/LVAD but not with the risk of SCD/MVA, suggesting that SCD-prevention for FLNCtv patients might not rely exclusively on reduced LVEF, similar to observations in others ACM5, 28. Alternative variables deserve to be explored in order to improve the arrhythmic risk stratification process. In our cohort, for instance, the prevalence of NSVT was significantly higher in patients experiencing SCD/MVA. Finally, we compared the outcome of FLNCtv to LMNA and DSP, two established models of ACM with high risk of arrhythmic and non-arrhythmic events and, particularly for DSP, similarities with FLNC in phenotypic presentation5, 22, 24, 29. FLNCtv carriers showed a similar risk of non-arrhythmic related outcomes compared to DSP, such as irreversible HF, need of HT or LV assist devices, but lower compared to LMNA. On the contrary, in FLNCtv, the risk of life-threatening ventricular arrhythmias (SCD/MVA) was not significantly different from LMNA and DSP, further emphasizing the dominant arrhythmogenic phenotype of FLNCtv. Limiting the analysis to affected carriers did not modify these findings (Figure II in the Supplement). Since LMNA mutations are associated with AV nodal disease, we also compared the incidence of bradyarrhythmias in the FLNC and LMNA populations: unlike LMNA carriers, we observed a lower rate of incident bradyarrhythmias in the FLNCtv carriers, with no severe cases requiring permanent pacing. Although the size of the population and the observational nature of the data do not allow us to support causality, our observation can be considered as hypotheses-generating and, if further confirmed in larger multicenter studies and in validation cohorts, could aid in a more accurate and more precise arrhythmic risk stratification of FLNCtv carriers.
Study Limitations
Despite the multicenter design to overcome limited experience of individual centers, the rarity of the disease leads to a small sample size that limits the power of our observations requiring validation in other cohorts. The enrollment in tertiary care centers for genetic cardiomyopathies allowed us to analyze the largest available cohort of FLNCtv carriers, but also represents potential selection and ascertainment biases that have to be considered in the interpretation of results. The enrolled population was nearly exclusively of Caucasian ancestry which limits the generalizability of our results to non-White populations and highlights the need to conduct similar studies in other less selected populations to comprehensively understand the FLNC genotype-phenotype relationships. The retrospective nature of the study means that some clinical data were not available in all the patients. Finally, imaging studies performed in each center were not reviewed by an independent core-laboratory and the inter-center reproducibility was not tested.
Conclusions.
FLNCtv are variants leading to a heterogeneous ACM clinical presentation with overlapping phenotypic aspects of DCM and ARVC. The frequent ventricular arrhythmias and the significant risk of arrhythmic-related major outcomes support the systematic screening of FLNC in clinical genetic cardiomyopathy panels. FLNC was recently introduced in the new 2019 Heart Rhythm Society expert consensus statement on ACM with specific recommendations for primary prevention of SCD and ICD based exclusively on LVEF < 45% (Class of Recommendation IIa)2. Our findings confirm the high risk phenotype of FLNCtv carriers. However, we show that in these patients, an ICD might be considered regardless of the LVEF. The identification of alternative factors associated with increased risk of SCD/MVA, such as ventricular arrhythmias (NSVT), in larger dedicated studies will foster a precision medicine approach to the risk stratification of patients with FLNCtv.
Supplementary Material
Clinical Perspective.
What is new?
Filamin C truncating variants (FLNCtv) cause a high-risk arrhythmogenic cardiomyopathy with heterogeneous phenotypic presentations
Outcome exposure is mainly characterized by life-threatening arrhythmias.
Left ventricle ejection fraction does not seem to be associated with the risk of life-threatening arrhythmias in carriers of FLNCtv
What are the clinical implications?
Prognostic implications of FLNCtv (i.e. high risk of life-threatening arrhythmias) support the use of genetic testing particularly in patients with features that are suspicious of an underlying FLNCtv
Besides left ventricular ejection fraction, alternative cumulative risk factors may aid the risk stratification of FLNCtv carriers and the adoption of strategies for the prevention of sudden cardiac death.
Acknowledgments
Giulia Barbati PhD, Associate Professor of Biostatistics, University of Trieste, is gratefully acknowledged for her advice and supervision of data analysis. The authors are grateful to the patients and family members for their participation in these studies.
Sources of funding
This study was supported by the following: NIH R01HL69071, R01HL116906, R01HL147064 and AHA 17GRNT33670495 to LM; NIH 1K23HI067915; NIH 2UM1HG006542 and R01HL109209 to MRGT; CRTrieste Foundation and Cassa di Risparmio of Gorizia Foundation to GS. WJM was a UK NIHR Senior Investigator. This work is also supported in part by a Trans-Atlantic Network of Excellence grant from the Fondation Leducq (14-CVD03), NIH/NCATS Colorado CTSA Grant Number UL1 TR002535 and UL1 TR001082, by the Netherlands Cardiovascular Research Initiative, an initiative with support of the Dutch Heart Foundation (grant number CVON2015-12/2018-30 eDETECT/PREDICT2). Dr Te Riele is supported by the Dutch Heart Foundation (grant number 2015T058) and the UMC Utrecht Fellowship Clinical Research Talent. CAJ, HC, and CT are supported by the Leonie-Wild Foundation, the Francis P. Chiaramonte Private Foundation, the Leyla Erkan Family Fund for ARVD Research, the Dr. Satish, Rupal, and Robin Shah ARVD Fund at Johns Hopkins, the Bogle Foundation, the Healing Hearts Foundation, the Campanella family, the Patrick J. Harrison Family, the Peter French Memorial Foundation and the Wilmerding Endowments. VNP is supported by the John Taylor Babbitt Foundation, The Sarnoff Cardiovascular Research Foundation, NIH K08 HL143185. EAA is supported by the NIH Common Fund U01HG007708, and NIH 1U24EB023674. DF is supported by the Victor Chang Cardiac Research Institute, National Health and Medical Research Council, Estate of the Late RT Hall. NKL is supported by the O’Hare Family Foundation for research on cardiolaminopathy.
Disclosures:
D.P.J. has received payments as a consultant from 4D Molecular Therapeutics, ADRx Pharma, MyoKardia, and Pfizer, and has received research funding from Array Biopharma and Eidos Therapeutics, all of which are outside of the scope of this work. NKL has received payments as a consultation from MyoKardia, Pfizer, Tenaya and Array Biopharma, outside the scope of this work.
HC is a consultant for Medtronic Inc. and Abbott and receives research support from Boston Scientific Corp. CT and CJ receive salary support from this grant. BM is a consultant for MyGeneCounsel. CR is a consultant for MyGeneCounsel. The remaining authors have nothing to disclose.
ABBREVIATIONS:
- ACM
Arrhythmogenic Cardiomyopathy
- ARVC
Arrhythmogenic Right Ventricular Cardiomyopathy
- D/HT/LVAD
All-Cause Mortality/Heart Transplantation/Left Ventricular Assist Device
- DCM
Dilated Cardiomyopathy
- HF
Heart Failure
- ICD
Implantable Cardioverter Defibrillator
- LV
Left Ventricle
- LVEF
Left Ventricular Ejection Fraction
- RV
Right Ventricle
- SCD/MVA
Sudden Cardiac Death/Major Ventricular Arrhythmias
Footnotes
References
- 1.Mestroni L and Sbaizero O. Arrhythmogenic Cardiomyopathy: Mechanotransduction Going Wrong. Circulation. 2018;137:1611–1613. doi: 10.1161/CIRCULATIONAHA.118.033558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Towbin JA, McKenna WJ, Abrams DJ, Ackerman MJ, Calkins H, Darrieux FCC, Daubert JP, de Chillou C, DePasquale EC, Desai MY, et al. 2019 HRS expert consensus statement on evaluation, risk stratification, and management of arrhythmogenic cardiomyopathy. Heart Rhythm. 2019;16:e301–e372. doi: 10.1016/j.hrthm.2019.05.007 [DOI] [PubMed] [Google Scholar]
- 3.Spezzacatene A, Sinagra G, Merlo M, Barbati G, Graw SL, Brun F, Slavov D, Di Lenarda A, Salcedo EE, Towbin JA, et al. Arrhythmogenic Phenotype in Dilated Cardiomyopathy: Natural History and Predictors of Life-Threatening Arrhythmias. J Am Heart Assoc. 2015;4:e002149. doi: 10.1161/JAHA.115.002149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Protonotarios A and Elliott PM. Arrhythmogenic cardiomyopathies (ACs): diagnosis, risk stratification and management. Heart. 2019;105:1117–1128. doi: 10.1136/heartjnl-2017-311160 [DOI] [PubMed] [Google Scholar]
- 5.Gigli M, Merlo M, Graw SL, Barbati G, Rowland TJ, Slavov DB, Stolfo D, Haywood ME, Dal Ferro M, Altinier A, et al. Genetic Risk of Arrhythmic Phenotypes in Patients With Dilated Cardiomyopathy. J Am Coll Cardiol. 2019;74:1480–1490. doi: 10.1016/j.jacc.2019.06.072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ortiz-Genga MF, Cuenca S, Dal Ferro M, Zorio E, Salgado-Aranda R, Climent V, Padron-Barthe L, Duro-Aguado I, Jimenez-Jaimez J, Hidalgo-Olivares VM, et al. Truncating FLNC Mutations Are Associated With High-Risk Dilated and Arrhythmogenic Cardiomyopathies. J Am Coll Cardiol. 2016;68:2440–2451. doi: 10.1016/j.jacc.2016.09.927 [DOI] [PubMed] [Google Scholar]
- 7.Begay RL, Graw SL, Sinagra G, Asimaki A, Rowland TJ, Slavov DB, Gowan K, Jones KL, Brun F, Merlo M, et al. Filamin C Truncation Mutations Are Associated With Arrhythmogenic Dilated Cardiomyopathy and Changes in the Cell–Cell Adhesion Structures. JACC Clin Electrophysiol. 2018;4:504–514. doi: 10.1016/j.jacep.2017.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Begay RL, Tharp CA, Martin A, Graw SL, Sinagra G, Miani D, Sweet ME, Slavov DB, Stafford N, Zeller MJ, et al. FLNC Gene Splice Mutations Cause Dilated Cardiomyopathy. JACC Basic Transl Sci. 2016;1:344–359. doi: 10.1016/j.jacbts.2016.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brun F, Gigli M, Graw SL, Judge DP, Merlo M, Murray B, Calkins H, Sinagra G, Taylor MR, Mestroni L, et al. FLNC truncations cause arrhythmogenic right ventricular cardiomyopathy. J Med Genet. 2020;57:254–257. doi: 10.1136/jmedgenet-2019-106394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Furst DO, Goldfarb LG, Kley RA, Vorgerd M, Olive M and van der Ven PF. Filamin C-related myopathies: pathology and mechanisms. Acta Neuropathol. 2013;125:33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van der Flier A and Sonnenberg A. Structural and functional aspects of filamins. Biochim Biophys Acta. 2001;1538:99–117. doi: 10.1007/s00401-012-1054-9 [DOI] [PubMed] [Google Scholar]
- 12.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405–424. doi: 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marcus FI, McKenna WJ, Sherrill D, Basso C, Bauce B, Bluemke DA, Calkins H, Corrado D, Cox MG, Daubert JP, et al. Diagnosis of arrhythmogenic right ventricular cardiomyopathy/dysplasia: proposed modification of the task force criteria. Circulation. 2010;121:1533–1541. doi: 10.1161/CIRCULATIONAHA.108.840827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lang RM, Badano LP, Mor-Avi V, Afilalo J, Armstrong A, Ernande L, Flachskampf FA, Foster E, Goldstein SA, Kuznetsova T, et al. Recommendations for cardiac chamber quantification by echocardiography in adults: an update from the American Society of Echocardiography and the European Association of Cardiovascular Imaging. J Am Soc Echocardiogr. 2015;28:1–39 e14. doi: 10.1016/j.echo.2014.10.003 [DOI] [PubMed] [Google Scholar]
- 15.Elliott P, Andersson B, Arbustini E, Bilinska Z, Cecchi F, Charron P, Dubourg O, Kuhl U, Maisch B, McKenna WJ, et al. Classification of the cardiomyopathies: a position statement from the European Society Of Cardiology Working Group on Myocardial and Pericardial Diseases. Eur Heart J. 2008;29:270–276. doi: 10.1093/eurheartj/ehm342 [DOI] [PubMed] [Google Scholar]
- 16.Therneau TMG, P. M. Modeling Survival Data: Extending the Cox Model. In: Springer NY, ed.; 2000: 170. doi: 10.1007/978-1-4757-3294-8 [DOI] [Google Scholar]
- 17.Klein JPM, M. L. Survival analysis: Techniques for censored and truncated data. In: Springer NY, ed. Survival analysis: Techniques for censored and truncated data; 2003: 436. doi: 10.1007/b97377 [DOI] [Google Scholar]
- 18.Verdonschot JAJ, Vanhoutte EK, Claes GRF, Helderman-van den Enden A, Hoeijmakers JGJ, Hellebrekers D, de Haan A, Christiaans I, Lekanne Deprez RH, Boen HM, et al. A mutation update for the FLNC gene in myopathies and cardiomyopathies. Hum Mutat. 2020;41:1091–1111. doi: 10.1002/humu.24004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ader F, De Groote P, Reant P, Rooryck-Thambo C, Dupin-Deguine D, Rambaud C, Khraiche D, Perret C, Pruny JF, Mathieu-Dramard M, et al. FLNC pathogenic variants in patients with cardiomyopathies: Prevalence and genotype-phenotype correlations. Clin Genet. 2019;96:317–329. doi: 10.1111/cge.13594 [DOI] [PubMed] [Google Scholar]
- 20.Helms AS, Thompson AD, Glazier AA, Hafeez N, Kabani S, Rodriguez J, Yob JM, Woolcock H, Mazzarotto F, Lakdawala NK, et al. Spatial and Functional Distribution of MYBPC3 Pathogenic Variants and Clinical Outcomes in Patients with Hypertrophic Cardiomyopathy. Circ Genom Precis Med. 2020; 13:396–405. doi: 10.1161/CIRCGEN.120.002929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hall CL, Akhtar MM, Sabater-Molina M, Futema M, Asimaki A, Protonotarios A, Dalageorgou C, Pittman AM, Suarez MP, Aguilera B, et al. Filamin C variants are associated with a distinctive clinical and immunohistochemical arrhythmogenic cardiomyopathy phenotype. Int J Cardiol. 2020;307:101–108. doi: 10.1016/j.ijcard.2019.09.048 [DOI] [PubMed] [Google Scholar]
- 22.Smith ED, Lakdawala NK, Papoutsidakis N, Aubert G, Mazzanti A, McCanta AC, Agarwal PP, Arscott P, Dellefave-Castillo LM, Vorovich EE, et al. Desmoplakin Cardiomyopathy, a Fibrotic and Inflammatory Form of Cardiomyopathy Distinct From Typical Dilated or Arrhythmogenic Right Ventricular Cardiomyopathy. Circulation. 2020;141:1872–1884. doi: 10.1161/CIRCULATIONAHA.119.044934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parikh VN, Caleshu C, Reuter C, Lazzeroni LC, Ingles J, Garcia J, McCaleb K, Adesiyun T, Sedaghat-Hamedani F, Kumar S, et al. Regional Variation in RBM20 Causes a Highly Penetrant Arrhythmogenic Cardiomyopathy. Circ Heart Fail. 2019;12:e005371. doi: 10.1161/CIRCHEARTFAILURE.118.005371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Augusto JB, Eiros R, Nakou E, Moura-Ferreira S, Treibel TA, Captur G, Akhtar MM, Protonotarios A, Gossios TD, Savvatis K, et al. Dilated cardiomyopathy and arrhythmogenic left ventricular cardiomyopathy: a comprehensive genotype-imaging phenotype study. Eur Heart J Cardiovasc Imaging. 2020;21:326–336. doi: 10.1093/ehjci/jez188 [DOI] [PubMed] [Google Scholar]
- 25.Akhtar MM, Lorenzini M, Pavlou M, Ochoa JP, O’Mahony C, Restrepo-Cordoba MA, Segura-Rodriguez D, Bermudez-Jimenez F, Molina P, Cuenca S, et al. Association of Left Ventricular Systolic Dysfunction Among Carriers of Truncating Variants in Filamin C With Frequent Ventricular Arrhythmia and End-stage Heart Failure. JAMA Cardiol. 2021;6:891–901. doi: 10.1001/jamacardio.2021.1106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Al-Khatib SM, Stevenson WG, Ackerman MJ, Bryant WJ, Callans DJ, Curtis AB, Deal BJ, Dickfeld T, Field ME, Fonarow GC, et al. 2017 AHA/ACC/HRS Guideline for Management of Patients With Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death: Executive Summary: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines and the Heart Rhythm Society. J Am Coll Cardiol. 2018;72:1677–1749. doi: 10.1016/j.jacc.2017.10.053 [DOI] [PubMed] [Google Scholar]
- 27.Priori SG, Blomstrom-Lundqvist C, Mazzanti A, Blom N, Borggrefe M, Camm J, Elliott PM, Fitzsimons D, Hatala R, Hindricks G, et al. 2015 ESC Guidelines for the management of patients with ventricular arrhythmias and the prevention of sudden cardiac death: The Task Force for the Management of Patients with Ventricular Arrhythmias and the Prevention of Sudden Cardiac Death of the European Society of Cardiology (ESC). Endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC). Eur Heart J. 2015;36:2793–2867. doi: 10.1093/eurheartj/ehv316 [DOI] [PubMed] [Google Scholar]
- 28.Cadrin-Tourigny J, Bosman LP, Wang W, Tadros R, Bhonsale A, Bourfiss M, Lie OH, Saguner AM, Svensson A, Andorin A, et al. Sudden Cardiac Death Prediction in Arrhythmogenic Right Ventricular Cardiomyopathy: A Multinational Collaboration. Circ Arrhythm Electrophysiol. 2021;14:e008509. doi: 10.1161/CIRCEP.120.008509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wahbi K, Ben Yaou R, Gandjbakhch E, Anselme F, Gossios T, Lakdawala NK, Stalens C, Sacher F, Babuty D, Trochu JN, et al. Development and Validation of a New Risk Prediction Score for Life-Threatening Ventricular Tachyarrhythmias in Laminopathies. Circulation. 2019;140:293–302. doi: 10.1161/CIRCULATIONAHA.118.039410 [DOI] [PubMed] [Google Scholar]
- 30.Minoche AE, Horvat C, Johnson R, Gayevskiy V, Morton SU, Drew AP, Woo K, Statham AL, Lundie B, Bagnall RD, et al. Genome sequencing as a first-line genetic test in familial dilated cardiomyopathy. Genet Med. March;21(3):650–662. doi: 10.1038/s41436-018-0084-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.