

Abstract
Introduction.
Protein phosphorylation is a primary mechanism of signal transduction in cellular systems. Isobaric tagging can be used to investigate alterations in phosphorylation events in sample multiplexing experiments where quantification extends across all conditions. As such, innovations in tandem mass tag methods can facilitate the expansion of the depth and breadth of phosphoproteomics analyses.
Areas covered.
This review discusses the current state of tandem mass tag-centric phosphoproteomics and highlights advances in reagent chemistry, instrumentation, data acquisition, and data analysis. We stress that approaches for phosphoproteomic investigations require high-specificity enrichment, sensitive detection, and accurate phosphorylation site localization.
Expert opinion.
Tandem mass tag-centric phosphoproteomics will continue to be an important conduit for our understanding of signal transduction in living organisms. We anticipate that progress in phosphopeptide enrichment methodologies, enhancements in instrumentation and data acquisition technologies, and further refinements in analytical strategies will be key to the discovery of biologically relevant data from phosphoproteomics studies.
Keywords: FAIMS, isobaric tagging, phosphoproteome, real-time search, TMT, TMTpro
1. Introduction
Protein phosphorylation on serine, threonine, and tyrosine amino acids is a highly studied post-translational modification (PTM) that coordinates a wide and diverse array of cellular and biochemical processes. Phosphorylation events in human cells are dynamically regulated by more than 500 kinases and approximately 200 phosphatases [1]. As phosphorylation is a readily and rapidly reversible process, its importance in signal transduction and protein activity modulation cannot be understated. For example, protein phosphorylation and associated cellular machinery (e.g., kinase, phosphatases, and phosphoprotein-binding proteins) are intricately involved in processes, such as apoptosis, cell division, response to extracellular signals and growth factor stimulation, among many other pathways [2]. Recent studies have mapped over 50,000 phosphorylation sites in a human cell line [3]. However, estimates have been made that approximately 1.85 million phosphorylatable residues are available across the entire human proteome [4]. Assuming ~11,000 proteins are expressed in a given human cell roughly suggests ~700,000 potential phosphorylation sites, each contributing to the millions of potential proteoforms that exist at any one time in human cells [5,6]. Resources, such as PhosphoSitePlus [7] and eukaryotic phosphorylation site database (EPSD) [8] are available that extensively collect, curate and annotate phosphorylation sites on eukaryotic proteins.
Sample multiplexing is a frequently used approach that can increase data throughput. Analyzing multiple samples simultaneously limits missing values resulting from stochastic sampling across experiments and improves statistical power by allowing replicates to be analyzed within a single experiment. Isobaric tagging strategies, such as tandem mass tags (TMT) [9] and isobaric tags for relative and absolute quantitation (iTRAQ) [10], are increasingly used for proteomic and phosphoproteomic profiling of complex peptide mixtures [11,12]. These NHS ester-based isobaric labels generally consist of three distinct functional regions: the reporter ion region, the mass balancer region, and the amine reactive region. Each tag differs in the distribution of heavy isotopes across their structure. The amine reactive region modifies the N-terminal amine groups and ε-amine groups of lysine residues thereby conjugating labels to peptides. The mass balancer region ensures that isobaric TMT tags have identical overall mass, while allowing for reporter ions to have different masses and therefore enabling the relative quantification of associated peptides. Using these isobaric labeling reagents allows for the relative quantification of individual (phospho)peptides across all samples.
Sample multiplexing can offer several benefits to phosphoproteomics analysis. For instance, the sensitivity and dynamic range of phosphorylation profiling is limited by the general low abundance of phosphopeptides among all peptides, as well as the low stoichiometry of the phosphorylated peptide verses its unphosphorylated version. Sample multiplexing can essentially boost sensitivity as the intensities of chromatographic peaks, precursor ions, and fragment ions are increased due to the additive effects of a given peptide having identical mass and chromatographic properties across all isobaric tag-labeled samples. Moreover, missing values are reduced when samples are arranged in a single multiplexed experiment, eliminating the caveats of stochastic precursor sampling across multiple, individually analyzed samples. Overcoming the challenges of sensitivity and dynamic range is essential for acquiring comprehensive phosphoproteomics datasets and to extract biologically relevant findings therefrom. As such, progress in phosphopeptide enrichment methodologies, enhancements in instrumentation and data acquisition technologies, and further refinements in data analysis strategies will be key to improved tandem mass tag-centric phosphoproteomics studies.
2. Sample preparation strategies for isobaric tag-labeled phosphopeptides.
Sample preparation techniques originally designed for global, whole proteome profiling can be readily applied to phosphoproteomics profiling (summarized in Figure 1). Such protocols, specifically the streamlined-TMT (“SL-TMT”) method have been used extensively for phosphoproteomics profiling, with a seamless integration of a phosphopeptide enrichment step [13]. For bottom-up phosphoproteomics experiments as highlighted in this review, proteins are extracted from cells, tissue, or fluids and are enzymatically digested (Figure 1A) prior to labeling (Figure 1B) or phosphopeptide enrichment (Figure 1C). Much like endogenous proteases can adversely affect proteome depth, phosphatases can lower the number of phosphorylation events observed. Extra care is often necessary to rapidly lyse cells, typically done so on-plate, with chilled lysis buffer containing phosphatase inhibitors so as to minimize phosphorylation losses [14]. Phosphoproteome datasets may also exhibit a higher number of miscleaved peptides which are more prevalent when a phosphorylated residue is proximal to a tryptic cleavage site [15].
Figure 1: General strategy for phosphoproteome profiling using isobaric labels.
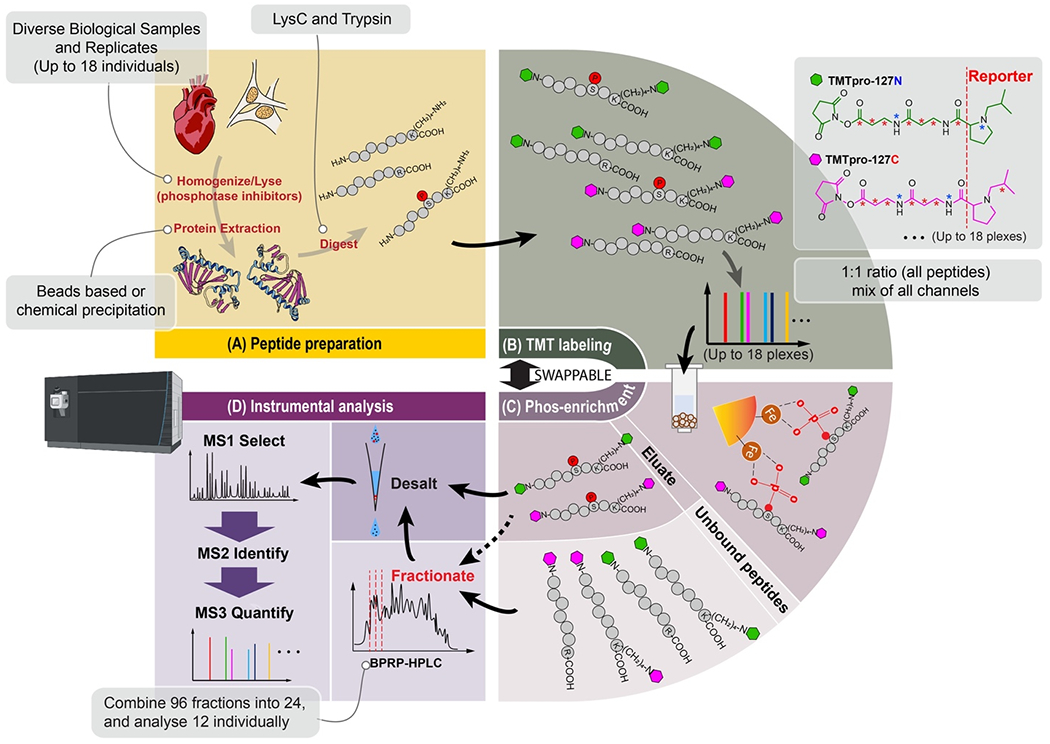
A) Peptides can be prepared by a variety of methods; however, efficient lysis and protein extraction (either chemical or bead-based precipitation) is necessary. Proteins are often digested with trypsin followed by LysC. B) Peptides are then typically labeled with isobaric reagent. C) Phosphopeptide enrichment is performed, and data may also be acquired for unbound (non-phosphorylated) proteins to determine protein-level abundance alterations. Alternatively, peptides can be enriched first and then labeled, which conserves isobaric labeling reagent at the expense of potentially increasing variability due to individual enrichments and desalting steps. D) Samples may be pre-fractionated off-line but must be desalted prior to mass spectrometry analysis. Data may be acquired using a variety of modalities, such as MS3-based methods, as depicted here, or by hrMS2 in which peptide identification and quantification are both elucidated at the MS2 stage.
The peptides may now either be labeled before phosphopeptide enrichment or enriched-prior to labeling (as discussed later). Moreover, once labeled, the peptides can be phosphopeptide-enriched and then fractioned offline or vice versa. For large-scale preparations (>1.5 mg total starting material), we recommend fractionating first and then performing individual phosphopeptide enrichments. Phosphopeptides may then be fractionated off-line (for example, with basic pH reversed phase chromatography) and then on-line (with the mass spectrometer) prior to analysis (Figure 1D). We note that peptides with the same amino acid sequence (with identical modifications) are chromatographically indistinguishable, regardless of the isobaric tag. These peptides not only co-elute but as they have the same mass, they will appear as a single composite peak with an identical mass-to-charge (m/z) value in an MS1 scan. The fragmentation of the labeled peptides during the MS2 or MS3 stage generates reporter ion peaks of different masses, whose intensities are used for the relative quantification across samples and downstream bioinformatics analysis [12]. As such, careful sample preparation, including sufficient fractionation is critical for successful phosphopeptide quantification workflows.
Isobaric labeling.
Proteome profiling using isobaric tags is a frequently used and well-accepted technology for sample multiplexing of complex peptide mixtures [11,12]. Tandem mass tag (TMT) reagents from Thermo Scientific (licensed by Proteome Sciences) are among the most common isobaric tagging reagents. Two distinct TMT reagents are currently available, one with its reporter ion based dimethylpiperidine ring (with an 11-plex limit) and the second based on an isobutyl-proline (with an 18-plex limit, Figure 1B) [9,16–18]. The original TMT reagent set began as an isobaric pair of mass tags, the TMTduplex, with a single isotopic substitution [9]. The addition of four more stable isotopes (for a total of five) between the reporter ion and mass balancer regions of this structure generated a TMTsix-plex, in which the reporter ions are 1 Da apart [19]. Leveraging the 0.0063 Da mass difference of a neutron from 13C and 15N enabled a TMT10/11-plex to be assembled, again with the same backbone structure and five stable isotopes [20]. The feasibility to distribute heavy isotopes across the reporter ion and mass normalization groups limits the multiplexing capability of these isobaric labels. In efforts to increase the degree of multiplexing, the new isobaric labeling set based on an isobutyl-proline-based reagents, named TMTpro (Figure 1B), was developed [17,18]. These reagents use nine stable isotopes which can accommodate up to an 18-plex using the 0.0063 Da elemental isotope mass difference, or a TMTpro10-plex set at unit resolution [16]. We note that whereas all the reagents of the TMTpro16 set are isobaric, a mass shift may be observed for peptides labeled with the 17th and 18th reagents when using very high resolution (>250,000). This phenomenon may be exacerbated if peptides labeled this these two additional reagents are of higher abundance than those conjugated to the remaining reagents. Nonetheless, the limits imposed by the number of potential heavy isotopes has been reached on the current TMTpro structure such that any higher degree of multiplexing will require a new molecule.
In addition to TMT reagent sets, several other isobaric labeling formats are currently available or being developed. Along with TMT, iTRAQ is among the most used isobaric tag systems. Like TMT, iTRAQ has two distinct sets of reagents, a 4-plex [10] and an 8-plex [21] set, which differ in structure, but uses the same NHS-ester chemistry. Moreover, several other existing tag sets enable higher multiplexing capacity. For example, the chemistries underlying DiLeu [22] and Combinatorial Isobaric Mass Tags (CMTs) can reach multiplexing levels of 21 and 28 samples, respectively [23]. Another labeling strategy, the EASI-tag system [24], is based on the TMT complement ion (TMTc/TMTproC) concept [25], which quantifies the relative difference among samples using the peptide fragment bound to the balancer region of the TMT molecule. As such, this system does not rely on reporter ion detection, which is often prone to ratio compression due to interference from co-isolated, co-fragmented, and co-analyzed precursors [26–29]. Future improvements in software for the data analysis may overcome some of the caveats that hinder the broad acceptance of these emerging isobaric tag-based technologies. The wide array of available and developing chemical labeling strategies, all of which can be applied to phosphoproteomics analyses, are a testament to the utility and promise of isobaric labeling for such investigations.
Selective and efficient phosphopeptide enrichment.
Phosphoproteomics is challenging as phosphopeptides must be identified among orders of magnitude more non-phosphorylated peptides. Protocols for whole proteome profiling are readily applicable to phosphorylation profiling. Unfortunately, phosphoproteomic analyses often require greater than 100 times more starting material than whole-proteome profiling as phosphopeptides comprise <2-3% of peptides in a typical tryptic digest [30,31]. The low stoichiometry of phosphopeptides is a major obstacle for their investigation, as phosphoproteins are typically at very low abundance in any given cell. As such, highly specific enrichment strategies are needed to segregate phosphopeptides from the vast background of unmodified peptides.
The low stoichiometry of phosphorylated peptides raises the question of when it is best in the workflow to label the samples. A key point of isobaric tagging is that peptides are analyzed simultaneously across all samples, thereby reducing sample-to-sample variability. The earlier in the protocol that the samples are mixed, the less variability is expected. The selection of the phosphopeptide enrichment strategy may alter the degree of variability introduced into a phosphoproteomics workflow. More specifically, labeling prior to enrichment (Figure 1B and C) allows for earlier sample mixing and only a single desalting and enrichment step is needed, thereby decreasing potential variability. This advantage, however, is at the expense of cost as >95% of the labeled sample is not phosphorylated and thus is destined to be discarded as the flow-through of the enrichment column. This cost may be somewhat recouped if the non-phosphorylated peptides are profiled in terms of a global proteome analysis. Conversely, one may label peptides following enrichment [32]. Such a workflow conserves labeling reagent but requires multiple desalting and enrichment steps which will likely contribute to sample variability. However, greater phosphoproteome depth is often dependent on increased starting material (at times >20 mg of peptide), which becomes cost-prohibitive if labeling is performed before enrichment. As such, a cost verses benefit decision must be made when determining if samples should be labeled before or after enrichment.
As the importance of enrichment cannot be overstated, a wide range of methodologies for phosphopeptide enrichment have been developed. Often enrichment entails ion exchange chromatography, metal oxide affinity chromatography (MOAC), metal ion affinity chromatography (IMAC), and phosphopeptide-specific immunoprecipitation (Figure 2).
Figure 2: Common strategies for phosphopeptide enrichment.

Low abundance phosphopeptides may be enriched from a much larger background of non-phosphorylated peptides through a variety of techniques. The most common techniques include ion exchange chromatography, metal oxide affinity chromatography (MOAC), metal ion affinity chromatography (IMAC), and immunoprecipitation methods (with phosphorylated tyrosine antibody-based enrichment being the most popular). Combining these methods in series is also common, such as SCX (strong cation exchange) enrichment or pY-enrichment followed by MOAC or IMAC, IMAC followed by MOAC, or sequential IMAC.
Strong cation exchange (SCX) is regularly used as a pre-fractionation technique but may also be used to selectively pre-enrich phosphopeptides. In SCX, peptides are retained by their positively charged side chains binding to the negatively charged resin [33]. Subsequently, peptides elute from an SCX column as the salt concentration increases according to isoelectric point. It follows that a phosphorylated peptide has less affinity toward the negatively charged matrix due to the negative charge of the phosphoryl group, and thereby phosphopeptides can be found in the column flowthrough, or early fractions [34]. Most often, SCX is coupled with MOAC or IMAC for enhanced enrichment [35]. In fact, the two most common phosphopeptide enrichment strategies are currently MOAC [36,37] and IMAC [38]. Accordingly, titanium dioxide (TiO2) is one of the most widely used MOAC affinity matrices. Phosphopeptides bind the metal oxide at acidic pH and are eluted at pH > 10 [37,39]. Likewise, Fe3+-NTA has been widely used for IMAC-based enrichment. IMAC in a spin column format offers a balance between recovery, enrichment, throughput, and efficiency [40]. Commercial spin cartridges enable the processing of 50 µg to 5 mg of peptide and can yield up to 150 µg of phosphopeptides [40]. These spin columns can be integrated seamlessly in the SL-TMT-based “mini-phos” protocol [13]. The high yield, enrichment, and recovery from these Fe3+-NTA spin columns makes them a suitable phosphopeptide enrichment strategy for users of all experience levels. IMAC has the advantages of high enrichment efficiency, and reusability, whereas MOAC has a higher tolerance to salts and detergents [41]. As expected, each enrichment strategy has its own advantages and limitations.
In addition, these enrichment strategies can be applied in tandem with other chromatographic techniques or enrichment methods to achieve greater phosphoproteome coverage [14,42]. For instance, the sequential elution from IMAC (SIMAC) on the same sample [43] has shown increased yields, as has SMOAC (Sequential enrichment of Metal Oxide Affinity Chromatography) which uses serial enrichment with TiO2 and Fe3+-NTA columns [44]. IMAC, such as Fe3+-NTA, generally results in the higher detection of multiply phosphorylated peptides, while TiO2 enrichment results in a greater number of identified singly phosphorylated peptides [45]. In addition, chromatographic strategies can improve dynamic range by fractionation, either pre- or post-enrichment. Such techniques include high pH reversed-phase chromatography (as discussed above), SCX [46], hydrophilic interaction chromatography (HILIC) [47], and electrostatic repulsion–hydrophilic interaction chromatography (ERLIC) [48]. Also, other non-chromatographic alternatives also can be used for phosphopeptide enrichment. For example, antibody-based methods have been used for enriching phosphopeptides, including pan-pS/pT antibody enrichment [49], and phosphotyrosine immunoprecipitation to enrich phosphorylated tyrosine residues [50] or another mass spectrometry-based targeted approach, such as SureQuant pTyr [51]. Other enrichment approaches have been developed including Phos-tag [52], polymer-based metal ion affinity capture [53], lanthanide ion-based enrichment [54], and affinity precipitation with SH2 domains, which are also specific for phosphorylated tyrosine residues [55]. Each strategy has its own sets of benefits and challenges which must be assessed on a case-dependent basis for a given experimental set-up.
Studies comparing multiple enrichment protocols have shown limited overlap, which is likely due to the modest phosphoproteome coverage achieved with these methods and the multitude of phosphopeptides available to sample in a given system. Consequently, many of these strategies have shown good orthogonality. A comparison of ERLIC with weak and strong anion exchange (WAX and SAX) showed that ERLIC enriched and identified more than double the number of phosphopeptides as either of the two ion exchange systems, but together their orthogonal phosphopeptide separation can increase phosphoproteomic depth [56]. Likewise, several groups have used HILIC with IMAC enrichment and SCX separation and have shown increased phosphopeptide yields [34,57–60]. Of specific interest is that HILIC and ERLIC can separate singly from multiply phosphorylated peptides with the adjustment of salt concentration, pH, and/or organic solvent compositions in the mobile phase [48]. The selection of the number of enrichment and fractionation methods to be used depends ultimately on the potential yield of phosphopeptides. For example, if only several micrograms of phosphopeptide are expected, a single enrichment step should suffice, whereas if hundreds of micrograms of peptides are expected, a deeper fractionation would most definitely increase phosphopeptide yields.
3. Tandem mass spectrometry of isobaric tag-labeled phosphopeptides
Spectral acquisition modes.
Phosphopeptide profiling may be performed using a variety of strategies, many of which are based on methods developed for global proteomics profiling (Figure 3). However, phosphopeptides are unique as along with limited sample availability, the labile nature of the phosphoester bond can adversely affect phosphopeptide profiling. Specifically, the presence of the phosphate moiety can impair precursor fragmentation behavior when compared to non-modified precursor peptides [61]. In classic ion trap-based collision-induced dissociation (CID), many phosphopeptides undergo a neutral loss of phosphoric acid corresponding to a mass difference of −98 Da at the expense of backbone fragmentation. Often a neutral loss peak dominates the fragmentation spectrum with an intensity that can be orders of magnitude higher than that of backbone fragment ions, and thereby is detrimental to database matching and amino acid sequence assignment. This phenomenon is true for both label-free and TMT-labeled phosphopeptides [62]. Alternative fragmentation strategies have been developed to either overcome or take advantage of this characteristic neutral loss in efforts to improve the profiling depth of phosphopeptides. Some fragmentation techniques that have been evaluated for phosphopeptide profiling include: electron transfer dissociation (ETD) [63], higher-energy collisional dissociation (HCD) [64], and ultraviolet photodissociation (UVPD) [65], as well as combinations thereof (such as electron-transfer/higher-energy collision dissociation (EThcD)) [66], with HCD-type fragmentation providing the greatest performance in typical large-scale phosphoproteomics experiments [67]. Although many of these methodologies have been performed in label-free settings, all are readily applicable to isobaric tag-centric experiments.
Figure 3: Potential analytical workflows.
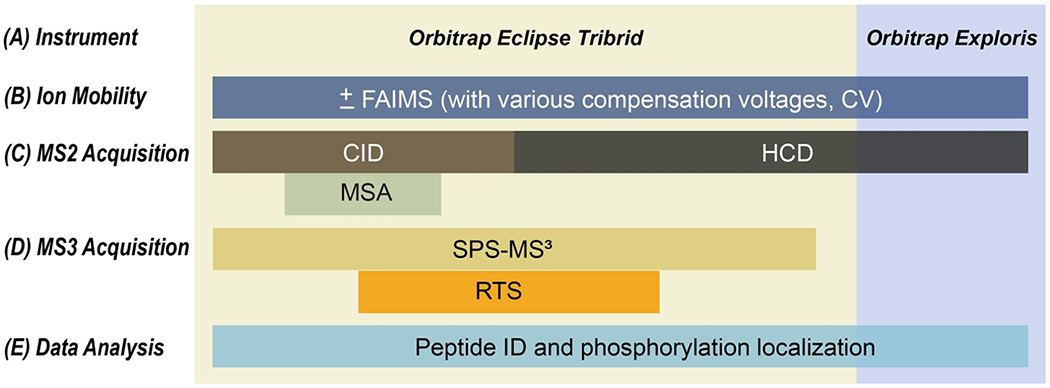
Various modalities are possible for the analysis of enriched phosphopeptides. A) The overall architecture of the instrument may limit the options for data acquisition. Such limitations are specifically with respect to mass analyzers, as high-resolution mass analyzers are required to detect mass differences of 6 mDa between isotopologue pairs of isobaric reagents. B) Ion mobility (such as FAIMS) can add an orthogonal dimension of separation. Alternating the number and value of compensation voltages (CVs) can enhance phosphoproteome coverage. C) Options for MS2 acquisition can include CID, CID-MSA, or HCD fragmentation on Tribrid mass spectrometers - such as the Orbitrap Fusion, Lumos, and Eclipse mass spectrometers), but commercial Orbitrap Exploris and Q-Exactive instruments are limited to only HCD fragmentation. D) Tribrid mass spectrometers also allow for MS3 scan-based quantification. In addition, newer instruments can leverage the advantages of real-time database searching (RTS). E) A variety of data analysis strategies are possible including various search engines and probability-based algorithms to determine phosphorylation site localization.
Ion trap CID is among the most commonly used fragmentation strategies [68] (Figure 3C). In ion trap CID, peptides collide with an inert gas (usually helium) which typically fragments a given peptide ion once at the most labile bond. For phosphopeptides, the most labile bond is the phosphoester bond. Fragmentation at this bond leads to a loss of phosphoric acid (−98 Da) from the precursor that results in a neutral loss peak that typically dominates a phosphopeptide CID tandem MS spectrum, as mentioned above. Methods have been developed to leverage the presence of this neutral loss peak. The incorporation of MS3-based fragmentation [69–75] has been used to improve database matching for phosphopeptides. One commonly-used method is pseudoMS3, or multistage activation (MSA) (Figure 3C, CID), which isolates the neutral loss product ion in the MS/MS stage, fragments it, and then traps and analyzes it along with the original MS/MS fragment ions without an additional isolation cycle [76–82]. The result is a mosaic MS/MS spectrum with the original MS/MS fragments together with those fragments produced via MSA which shows improved search scores [83]. A comparison among MS2-alone, MSA, and MS2/MS3 methodologies has shown better performance with MSA for phosphopeptide identification [84].
Many current high resolution, high mass accuracy, benchtop Orbitrap instruments, such as the Q Exactive HFX [85] and the Orbitrap Exploris [86] are not equipped with an ion trap and must rely on beam-type CID (i.e., higher-energy collisional dissociation, HCD) for fragmentation (Figure 3A and C). These instrument types have widespread use for both whole proteome and phosphoproteome profiling. Here, collisions towards the inert gas, typically nitrogen, are assisted by an electrical potential that accelerates peptide ions. Due to the higher energy collisions, multiple fragmentation events are more likely to occur per precursor. As such, the neutral loss precursor intensity is significantly reduced, and a more complex and richer fragmentation spectrum is generated. We have shown previously that sequential acquisition of CID-MSA and HCD show complimentary phosphopeptide identification for medium scale “mini-phos”-based analyses [76] with similar numbers of phosphopeptide identifications.
Other fragmentation strategies can also be utilized that can preserve the labile phosphoric acid moiety and facilitate phosphopeptide identification, specifically, electron capture dissociation (ECD) [63] and electron transfer dissociation (ETD) [63]. In ECD, multiply charged precursor ions capture a low-energy electron, while in ETD analysis, an electron is transferred from a radical anion with low electron affinity to the peptide precursor cation. Both methods fragment the peptide backbone and retain the phosphoric acid moiety on a fragment ion. Moreover, instruments that can perform ETD and HCD can leverage a combination of the two fragmentation techniques (i.e., EThcD). EThcD can generate MS/MS spectra with both b/y and c/z ion pairs [66,87] thereby yielding higher peptide sequence coverage and thus more confident localization of phosphorylation sites [66]. A caveat for both fragmentation schemes is that they are slightly less sensitive, slower scanning, and are only efficient for peptides with charge states greater than 2+. Methods with charge-dependent decision trees can be constructed such that peptides with a charge state of 2+ can be fragmented with HCD, while higher charge states can be subjected to ETD/ECD fragmentation [88]. Overall, techniques such as EThcD, ETD, and ECD, can complement well the more commonly used CID and HCD fragmentation strategies. Further development of these techniques can only improve their utility, specifically for phosphoproteome profiling.
Ion mobility
Ion mobility is a technique that separates gas phase ions based on their mobility in an inert buffer gas in the presence of an electric field as a function of their charge, shape, and size [89]. Recently, ion mobility has been integrated into routine mass spectrometry applications using High-Field Asymmetric waveform Ion Mobility Spectrometry (FAIMS) through a FAIMSPro interface on Orbitrap mass spectrometers [76,90–93] (Figure 3B). FAIMS separation is based on differences in ion mobility in high and low electric fields as ions transit between an inner and outer electrode in the presence of an asymmetric waveform [94–96]. The drift of an ion towards electrodes can be stopped by the application of a small, tunable DC voltage (‘compensation voltage’ or ‘CV’), which in turn can separate ions by rapidly alternating the CV values [97]. As such, FAIMS separates ions in a manner orthogonal to that of reversed-phased liquid chromatography-based fractionation [98].
Recent studies have used the FAIMSPro interface for a variety of proteomic, as well as phosphoproteomic, applications. For example, the use of FAIMS can improve the sensitivity of phosphopeptide quantification in stable-isotope incorporated samples [99]. Also, a study with label-free phosphopeptide mixtures has similarly shown an increase in peptide identifications and improved quantification when FAIMS is incorporated into their workflow [100]. In addition, several recent studies have used FAIMS to analyze isobaric tag-labeled, enriched phosphopeptide samples [91,93,101]. For TMT-labeled phosphopeptides, we refined further the optimal CVs (i.e., −40, −60, −80V) in a single 2.5 hr method. We also showed that consecutive analyses using MSA-CID and HCD fragmentation at the MS2 stage with FAIMS increased the depth of phosphorylation profiling, without loss of quantitative accuracy [76]. Continued improvement to the FAIMS hardware, the enhancement of associated software, and the integration of FAIMS or other ion mobility modalities into additional mass spectrometry instrument platforms will enhance further the coverage and quantitative accuracy of isobaric tag-labeled phosphopeptide profiling.
Real-time searching.
Real-time database searching (RTS) can be used to improve the depth and accuracy of global proteomics applications. RTS is applied to SPS-MS3-based data acquisition strategies in which peptide identification and quantification are decoupled (Figure 3D). The usage of RTS results in higher accuracy compared to traditional MS2-only methods for isobaric tag-centric quantitative analyses [26–29]. More specifically, fragment ions from the MS2 stage are isolated, fragmented further to release reporter ions, and a time-consuming Orbitrap scan is performed. SPS-MS3 suffers from long acquisition duty cycles that are detrimental to proteome depth, specifically for low abundance samples, such as small-scale (less than 1 mg of starting protein) phosphorylation analyses. RTS offers a potential solution to the lengthy duty cycle. Here, a database search is performed on a single MS2 spectrum (in ~10 ms) and a decision is made as to whether an MS3 scan will be acquired [102]. With the availability of search results in real-time, if an MS2 spectra matches a non-phosphorylated peptide, a time-intensive MS3 scan will not be acquired. However, when a peptide match occurs, the RTS application inserts an MS3 scan using preselected SPS ions thereby ensuring that all reporter ion signal originates from the precursor of interest. As such, RTS-MS3 can decrease phosphopeptide analysis cycle time and increase quantitative accuracy by specifically targeting phosphopeptide precursors and associated SPS ions.
The RTS strategy has been implemented directly as a node in the Thermo Instrument Acquisition Software. A multiplexed proteome can be comprehensively characterized with 50% less data acquisition time when using RTS-MS3 compared to traditional SPS-MS3 [102,103]. Using RTS-MS3 for phosphopeptide quantification may be computationally challenging as an expanded database is necessary, which in turn can slow spectral matching. However, recent work with RTS-MS3 has shown that RTS is capable of rapidly matching phosphopeptides to spectra in real-time [102]. We expect that applying the RTS strategy to multiplexed phosphoproteome profiling will show a strong improvement in data acquisition throughput and quantitative accuracy.
4. Data analysis
Data analysis.
Many aspects of data analysis for phosphoproteomics experiments are similar to those used in global proteome analysis, but several elements are specific to phosphorylation (Figure 3E). As with routine data processing, the acquired spectra are matched against a database of known peptides, and false discovery rates (FDR) are controlled using the target-decoy strategy [104,105]. However, a dynamic modification of +79.9663 Da must be added to every serine, threonine, and tyrosine amino acid, resulting in longer search times due to the larger database search space. Moreover, neutral loss fragment ions may also be included when matching MS/MS spectra. Along with traditional search engines, such as Sequest [106], Mascot [107], and Comet [108], recently developed frameworks that can perform open database searches (with wide precursor tolerances) which may be used to identify phosphorylation and/or multiple modifications on a given peptide [109–113]. Precursor mass deviations can be compared to the masses of hundreds of potential post-translational modifications. MSFragger, in particular, can be easily accessed through a graphical user interface, FragPipe, and is fully compatible with isobaric tagging reagents, such as TMT and TMTpro [114].
Nonetheless, phosphorylation-specific searching, remains more complicated than simply searching for global proteomics profiling. For instance, in the event that more than one phosphorylatable amino acid is present on a given peptide (i.e., a positional isomer), the probability of a phosphorylation event being at a specific amino acid must be determined. Several algorithms have been developed to assign a site localization score, including AScore [115], PTM score [116], the Mascot delta score [117], Phosphinator [118], PhosphoRS [119], PhosphoScore [120], LuciPHOr [121,122], among others. Much like an FDR is calculated at the peptide and protein level, the analogous false localization rate (FLR) should also be determined to avoid the common practice of arbitrary score cut-offs as a surrogate for the FLR [123]. For example, the SLIP method performs searches in which phosphorylations are allowed on non-phosphorylatable glutamate and proline amino acids [124]. Likewise, the LuciPHOr program reports FLR estimates based on smoothed distributions of the localization delta scores [121,122]. We stress that ascertaining the precise localization of a phosphosite along with its associated FLR is required for pinpointing which residues to target for downstream applications. With the availability of numerous search engines and localization algorithms, various strategies may be designed for bottom-up phosphoproteomics analysis. In fact, Locard-Paulet et al have recently compared over 20 phosphoproteomics analysis pipelines highlighting the pros and cons of each [125].
Once phosphorylation sites are quantified, various bioinformatics analysis strategies can be implemented to extract biologically relevant data. For instance, the changes in the phosphorylation state of a given peptide can be normalized back to protein changes as found in a global proteomics experiment to account for protein abundance difference [126]. This calculation can confirm if the observed alterations in phosphorylation coincided with alterations in the expression of a given protein. Moreover, while thousands and even tens of thousands of phosphorylation events can be quantified, identifying the kinases and phosphatases regulating these events is non-trivial. It is noteworthy that the regulatory kinase of only approximately 3% of reported phosphosites are known, leaving the rest to be determined [127]. As such, several bioinformatics approaches have been developed in an attempt to assign kinases to given phosphorylation sites [128–130]. Various tools are freely available to search for kinase motifs among regulated phosphorylation sites, including motif-x [131], NetworKIN [128], and Phospho.ELM [132]. Knowing the kinase associated with a specific phosphorylation site can be used to measure kinase activity in a given system. Specifically, the KAYAK (Kinase ActivitY Assay for Kinome profiling) assay was designed to measure the phosphorylation rates of dozens of peptide substrates simultaneously and directly from cell lysates and has been used in sample multiplexing experiments with isobaric tagging [133,134]. Having a greater number of kinases with known substrates will permit further expansion of this assay. These and many other application-specific bioinformatics tools can be used to extract meaningful and relevant biological insights. It follows that the development of novel sample preparation methodologies and analytical technologies can enable the collection of more robust and more clearly interpretable datasets and thus can enhance phosphopeptide profiling applications.
5. Emerging Applications
New applications of phosphoproteomics have emerged with the expansion of studies focusing on the myriad of proteoforms that exist in a given cellular context. Modern applications of phosphoproteomics and sample multiplexed phosphoproteomics are continuing to define our understanding of proteins, protein complexes, and essential signaling axes. Integration of phosphoproteomics with thermal proteome profiling (TPP) has shown particular promise in terms of uncovering how specific phosphorylation events regulate protein stability. TPP and related methods – e.g., cellular thermal shift assay (CETSA) [135], proteome Integral Solubility Alteration (PISA) [136], Dali [137] – measure protein abundance as a function of exposure to increasing temperatures. The resulting data can then be used to estimate melting temperatures for proteins. Seminal work on TPP successfully uncovered protein-ligand and protein-protein stability changes across a wide range of samples. Recently, three groups integrated phosphoproteomics and TPP to determine how phosphorylation modifies the stability response [137–139]. These studies identified key phosphorylation sites in proteins, including the receptor tyrosine kinase LYN that specifically altered protein stability, and uncovered new roles for phosphorylation in protein regulation.
Protein phosphorylation is a well-known regulator of protein complex formation. Yet, unbiased identification of protein complexes and subunits across the proteome remains challenging. Recent work from Bludau et al. highlight how size-exclusion chromatography (SEC) in combination with high-throughput proteomics can be used to decipher how proteoforms – including those due to phosphorylation events – affect protein complex composition [140]. In a comparison of cell cycle stages (interphase vs. mitosis), Bludau, et al. identified cell cycle regulated phosphoproteins that correlated with significant changes in complex formation. For example, they observed mitosis-specific proteoform associations for the Ataxin-2-like protein (ATXN2L) without prior enrichment. Future studies aiming to improve the sensitivity of phosphopeptide detection in combination with SEC and unbiased phosphoproteomic detection could markedly improve our understanding of how phosphorylation governs protein complexes and proteoforms.
6. Conclusion
Phosphorylation events are extremely dynamic and firmly dependent on a system’s biological and/or physiological context. Here, we have highlighted several proteomics-based methods that can be used to investigate the phosphoproteome. We summarize the major methods for phosphoproteomics and highlight their benefits and caveats, as well as the expected proteome depth in Table S1. A drawback of studying phosphorylation is the low abundance and low stoichiometry of this PTM in a background of a large amount of the unphosphorylated state of the peptide, the proverbial needle in the haystack. Moreover, difficulties arise with respect to determining kinase-substrate relationships and the elucidation of meaningful biological consequences of alterations in phosphorylation states. Isobaric tagging strategies permit multiple samples to be analyzed simultaneously thereby reducing instrument time and costs, while generating fewer missing values within an experiment, so as to allow for better statistical inference. Future improvements in the breadth and depth of phosphoproteome profiling will likely be the result of multiple integrated factors including refinements of sample preparation techniques, reduction in sample complexity due to advances in fractionation, enhancement in the speed and sensitivity of mass spectrometers, incorporation of different technologies (e.g., FAIMS and RTS), and innovations in data analysis strategies. With such improvements, we can best overcome the compromise among phosphoproteome coverage, the number of replicates and/or conditions to be analyzed, and instrument analysis time, which will lead to a more complete elucidation and a better understanding of cellular phosphoproteomes.
7. Expert Opinion
Sample multiplexing can greatly enhance phosphoproteome analysis by producing more comprehensive datasets both in the number of phosphorylation sites identified and the number of samples compared in a single sample multiplexing experiment. Consequently, improvements are needed to overcome the low abundance of phosphopeptides while processing samples in a high throughput format. Even more challenging for high-throughput phosphorylation profiling is that biologically relevant phosphorylation events are generally of low abundance and low stoichiometry. Moreover, the precise localization of a given phosphorylation site on a peptide is often difficult to determine when multiple phosphorylatable residues are present [115,141]. In other words, discerning a biologically meaningful phosphorylation site from biological noise is difficult. As we mentioned previously, the abundance of phosphorylated peptides is, in general, orders of magnitude below that of non-phosphorylated peptides. This low abundance is closely related to the stoichiometry of a phosphorylation site. This stoichiometry - i.e., the percent phosphorylation site occupancy - is essentially the abundance of the phosphopeptide compared to that of the phosphorylated plus unphosphorylated states of a given peptide. We acknowledge that stoichiometry has a major impact on phosphopeptide quantification, but it is also of significant relevance biologically [142]. More specifically, the stoichiometry of a given phosphosite can represent the point at which a phosphorylation event results in a biologically relevant alteration. To this end, several approaches have been developed to enable the global estimation of phosphorylation site occupancy [142,143]. Moreover, data have shown that cellular conditions can alter phosphorylation site stoichiometry across different phases of the cell cycle [144]. However, only with increases both in sensitivity and dynamic range (due to improvements in instrumentation and/or upstream enrichment/fractionation) can we expand our understanding of phosphorylation-related cellular signaling events.
In addition, standardization of sample preparation protocols can enhance reproducibility and improve the elucidation of biologically-relevant findings across different laboratories. The assembly of a comprehensive isobaric tag-labeled phosphoproteome dataset is a multi-step process. Additionally, isobaric tagging allows for the collection of time-series data, different pharmacological doses, and multiple control samples to be analyzed together in a single experiment, which multiplies the number of procedural steps. Even more so, when several replicates are required, the entire process sums to a formidable undertaking. The capability to automate tasks in a high-throughput format will also benefit data reproducibility. Automation assures that samples are processed in parallel and thereby decreases the likelihood of human error, thus improving the quantitative integrity of the data. As such, some progress has been made toward automation to streamline phosphopeptide enrichment workflows. For example, EasyPhos is a high-throughput phosphoproteomics platform that uses a single 96-well plate starting from cell lysis and terminating with protease digestion [145]. EasyPhos has been used to identify over 20,000 distinct phosphopeptides from only 200 μg of protein in less than 24 hrs. The EasyPhos workflow is fully compatible with isobaric labeling. Moreover, automation and robotics, such as the use of liquid handlers can increase throughput and reproducibility, by performing highly repetitive and potentially human error-prone sample preparation steps [146–148]. We, and others, have used precipitation on nanoparticles (single-pot, solid phase sample preparation, “SP3”) as an alternative method to overcome variability-prone chemical-based precipitation methods, such as methanol-chloroform or trichloroacetic acid (TCA) precipitation [149–152]. We have adapted the SL-TMT protocol to use SP3 technology [149–152], now termed SL-SP3-TMT [153], to further streamline the process. Automated liquid handlers, such as the opentrons OT-2 [154], Thermo KingFisher Flex [155], or Hamilton Vantage [156] systems, include magnetic modules which can readily accommodate paramagnetic beads for nanoparticle-based precipitation or phosphopeptide enrichment in a 96-well format. This workflow is amenable to a 96-well format for some or the entire isobaric tag-centric phosphoproteomics procedure [145]. As the degree of multiplexing (and thus, the number of samples) increases, we envision the need for and the broader use of automation as a means to increase throughput.
The application of the methodologies and technologies described herein is not limited to traditional phosphorylation and can be developed further to profile other PTMs. For example, another area requiring further study is the investigation of non-canonical phosphorylation, that is, phosphorylation on residues other than serine, threonine, and tyrosine. For example, several studies have shown the importance of histidine phosphorylation [157–164] in both prokaryotic and eukaryotic organisms. Another study has postulated the overall occurrence of histidine, arginine, lysine, aspartate, glutamate, and cysteine phosphorylation, constitutes up to one-third of total protein phosphorylation [165]. These non-canonical phosphorylations are not only low abundant, but are labile such that current standard enrichment workflows may not be amendable to investigating these modifications [158]. However, we are confident that the general workflows for sample preparation and data analysis described herein can form the basis for isobaric tag-centric analysis of other PTMs.
In conclusion, phosphorylation is an important mechanism of signal transduction. Consequently, sample multiplexing using isobaric tags generates datasets in which all phosphorylation sites are quantified across all samples. Improvements in sample preparation, instrumentation, and data analysis may overcome the hurdles of low abundance and low stoichiometry associated with phosphopeptide profiling. We are confident that future innovations in these areas will enhance our feasibility to elucidate relevant biological insights about phosphorylation-related mechanisms.
Supplementary Material
Article Highlights.
Phosphorylation plays a key role in signal transduction and protein activity modulation.
The abundance of phosphorylated peptides (without enrichment) is orders of magnitude below that of the unphosphorylated peptides.
Overcoming the challenges of sensitivity and dynamic range is essential for acquiring comprehensive phosphoproteomics datasets.
Isobaric tagging allows for multiple control samples to be analyzed together in a single experiment, thereby reducing missing values among (phospho)peptide abundance measurements.
Improvements in sample preparation, instrumentation, and data analysis may overcome the hurdles of low abundance and low stoichiometry associated with phosphopeptide profiling.
High-Field Asymmetric waveform Ion Mobility Spectrometry (FAIMS) can improve the sensitivity of phosphopeptide quantification in stable-isotope incorporated samples.
Real-time database searching (RTS) can improve the depth and accuracy of global proteomics applications.
Determining the precise localization of a phosphosite along with its associated false localization rate is needed for pinpointing which residues to target for downstream validation studies.
Automation assures that samples are processed in parallel and thereby decreases the likelihood of human error when dozens of samples are prepared simultaneously.
Acknowledgement.
We would like to thank the members of the Gygi Lab and the Taplin Mass Spectrometry Facility at Harvard Medical School, in particular Prof. Steven P Gygi, for his invaluable advice and use of his instrumentation.
Funding.
This work was funded in part by NIH/NIGMS grant R01 GM132129 (J.A.P.).
List of Abbreviations.
- CID
collision-induced dissociation
- BPRP
basic pH reversed phase
- ERLIC
electrostatic repulsion–hydrophilic interaction chromatography
- ETD
electron transfer dissociation
- FAIMS
high-Field Asymmetric waveform Ion Mobility Spectrometry
- FDR
false discovery rate
- Fe3+-NTA
ferric nitrilotriacetate
- FLR
false localization rate
- HCD
higher-energy collisional dissociation
- HILIC
hydrophilic interaction chromatography
- IMAC
metal ion affinity chromatography
- iTRAQ
isobaric tag for relative and absolute quantitation
- KAYAK
Kinase ActivitY Assay for Kinome profiling
- MOAC
metal oxide affinity chromatography
- MSA
multistage activation
- NHS
N-Hydroxysuccinimide
- PTM
post translational modification
- RTS
Real-time database searching
- SCX
strong cation exchange chromatography
- SIMAC
sequential elution from IMAC
- SL-TMT
streamlined tandem mass tag
- SMOAC
sequential enrichment of Metal Oxide Affinity Chromatography
- SP3
single-pot, solid phase sample preparation
- TMT
tandem mass tags
- TMTc
TMT complement ion
- UVPD
ultraviolet photodissociation
Footnotes
Declaration of interest. The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Reviewer disclosures. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
Papers of special note have been highlighted as:
* of interest
** of considerable interest
- 1.Manning G, Whyte DB, Martinez R, et al. The protein kinase complement of the human genome. Science. 2002. December 6;298(5600):1912–1934. [DOI] [PubMed] [Google Scholar]
- 2.Janne PA, Gray N, Settleman J. Factors underlying sensitivity of cancers to small-molecule kinase inhibitors. Nat Rev Drug Discov. 2009. September;8(9):709–723. [DOI] [PubMed] [Google Scholar]
- 3.Sharma K, D’Souza RC, Tyanova S, et al. Ultradeep human phosphoproteome reveals a distinct regulatory nature of tyr and ser/thr-based signaling. Cell Rep. 2014. September 11;8(5):1583–1594. [DOI] [PubMed] [Google Scholar]
- 4.von Stechow L, Francavilla C, Olsen JV. Recent findings and technological advances in phosphoproteomics for cells and tissues. Expert Rev Proteomics. 2015;12(5):469–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beltrao P, Bork P, Krogan NJ, et al. Evolution and functional cross-talk of protein post-translational modifications. Mol Syst Biol. 2013;9:714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith LM, Kelleher NL, Consortium for Top Down P. Proteoform: A single term describing protein complexity. Nature methods. 2013;10(3):186–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hornbeck PV, Zhang B, Murray B, et al. Phosphositeplus, 2014: Mutations, ptms and recalibrations. Nucleic Acids Res. 2015. January;43(Database issue):D512–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lin S, Wang C, Zhou J, et al. Epsd: A well-annotated data resource of protein phosphorylation sites in eukaryotes. Brief Bioinform. 2021. January 18;22(1):298–307. [DOI] [PubMed] [Google Scholar]
- 9.Thompson A, Schafer J, Kuhn K, et al. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by ms/ms. Anal Chem. 2003. April 15;75(8):1895–1904. [DOI] [PubMed] [Google Scholar]; ** This paper introduces Tandem Mass Tag reagents.
- 10.Ross PL, Huang YN, Marchese JN, et al. Multiplexed protein quantitation in saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol Cell Proteomics. 2004. December;3(12):1154–1169. [DOI] [PubMed] [Google Scholar]
- 11.Hogrebe A, von Stechow L, Bekker-Jensen DB, et al. Benchmarking common quantification strategies for large-scale phosphoproteomics [Research Support, Non-U.S. Gov’t]. Nature communications. 2018. March 13;9(1):1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rauniyar N, Yates JR 3rd. Isobaric labeling-based relative quantification in shotgun proteomics [Research Support, N.I.H., Extramural Review]. J Proteome Res. 2014. December 5;13(12):5293–5309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Navarrete-Perea J, Yu Q, Gygi SP, et al. Streamlined tandem mass tag (sl-tmt) protocol: An efficient strategy for quantitative (phospho)proteome profiling using tandem mass tag-synchronous precursor selection-ms3. J Proteome Res. 2018. June 1;17(6):2226–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]; **This paper introduces the Streamlined Tandem Mass Tag (SL-TMT) protocol which has become the standard workflow for TMT and TMTpro experiments.
- 14.Fila J, Honys D. Enrichment techniques employed in phosphoproteomics. Amino Acids. 2012. September;43(3):1025–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dickhut C, Feldmann I, Lambert J, et al. Impact of digestion conditions on phosphoproteomics. J Proteome Res. 2014. June 6;13(6):2761–2770. [DOI] [PubMed] [Google Scholar]
- 16.Li J, Cai Z, Bomgarden RD, et al. TMTpro-18plex: The expanded and complete set of tmtpro reagents for sample multiplexing. J Proteome Res. 2021. May 7;20(5):2964–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, Van Vranken JG, Pontano Vaites L, et al. TMTpro reagents: A set of isobaric labeling mass tags enables simultaneous proteome-wide measurements across 16 samples. Nat Methods. 2020. April;17(4):399–404. [DOI] [PMC free article] [PubMed] [Google Scholar]; **This paper introduces TMTpro reagents and highlights an application for global phosphoproteomics.
- 18.Thompson A, Wolmer N, Koncarevic S, et al. TMTpro: Design, synthesis, and initial evaluation of a proline-based isobaric 16-plex tandem mass tag reagent set. Anal Chem. 2019. December 17;91(24):15941–15950. [DOI] [PubMed] [Google Scholar]
- 19.Dayon L, Hainard A, Licker V, et al. Relative quantification of proteins in human cerebrospinal fluids by ms/ms using 6-plex isobaric tags. Anal Chem. 2008. April 15;80(8):2921–2931. [DOI] [PubMed] [Google Scholar]
- 20.McAlister GC, Huttlin EL, Haas W, et al. Increasing the multiplexing capacity of tmts using reporter ion isotopologues with isobaric masses. Anal Chem. 2012. September 4;84(17):7469–7478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pierce A, Unwin RD, Evans CA, et al. Eight-channel itraq enables comparison of the activity of six leukemogenic tyrosine kinases. Mol Cell Proteomics. 2008. May;7(5):853–863. [DOI] [PubMed] [Google Scholar]
- 22.Frost DC, Feng Y, Li L. 21-plex dileu isobaric tags for high-throughput quantitative proteomics. Anal Chem. 2020. June 16;92(12):8228–8234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Braun CR, Bird GH, Wuhr M, et al. Generation of multiple reporter ions from a single isobaric reagent increases multiplexing capacity for quantitative proteomics. Anal Chem. 2015. October 6;87(19):9855–9863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Virreira Winter S, Meier F, Wichmann C, et al. EASI-tag enables accurate multiplexed and interference-free ms2-based proteome quantification. Nat Methods. 2018. July;15(7):527–530. [DOI] [PubMed] [Google Scholar]
- 25.Sonnett M, Yeung E, Wuhr M. Accurate, sensitive, and precise multiplexed proteomics using the complement reporter ion cluster. Analytical Chemistry. 2018. April 17;90(8):5032–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gygi JP, Ramin R, Navarrete-Perea J, et al. A triple knockout isobaric-labeling quality control platform with an integrated online database search. J Am Soc Mass Spectrom. 2020. July 1;31(7):1344–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gygi JP, Yu Q, Navarrete-Perea J, et al. Web-based search tool for visualizing instrument performance using the triple knockout (TKO) proteome standard. J Proteome Res. 2019. February 1;18(2):687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paulo JA, Navarrete-Perea J, Guha Thakurta S, et al. TKO6: A peptide standard to assess interference for unit-resolved isobaric labeling platforms. J Proteome Res. 2019. January 4;18(1):565–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Paulo JA, O’Connell JD, Gygi SP. A triple knockout (TKO) proteomics standard for diagnosing ion interference in isobaric labeling experiments. J Am Soc Mass Spectrom. 2016. October;27(10):1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]; ** This paper highlights interference for isobaric tagging and introduces the TKO quality control standard.
- 30.Manning G, Plowman GD, Hunter T, et al. Evolution of protein kinase signaling from yeast to man. Trends Biochem Sci. 2002. October;27(10):514–520. [DOI] [PubMed] [Google Scholar]
- 31.Moorhead GB, De Wever V, Templeton G, et al. Evolution of protein phosphatases in plants and animals. Biochem J. 2009. January 15;417(2):401–409. [DOI] [PubMed] [Google Scholar]
- 32.Kreuzer J, Edwards A, Haas W. Multiplexed quantitative phosphoproteomics of cell line and tissue samples. Methods Enzymol. 2019;626:41–65. [DOI] [PubMed] [Google Scholar]
- 33.Mohammed S, Heck A Jr. Strong cation exchange (scx) based analytical methods for the targeted analysis of protein post-translational modifications. Curr Opin Biotechnol. 2011. February;22(1):9–16. [DOI] [PubMed] [Google Scholar]
- 34.Villen J, Gygi SP. The scx/imac enrichment approach for global phosphorylation analysis by mass spectrometry. Nat Protoc. 2008;3(10):1630–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhai B, Villen J, Beausoleil SA, et al. Phosphoproteome analysis of drosophila melanogaster embryos. J Proteome Res. 2008. April;7(4):1675–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou H, Ye M, Dong J, et al. Robust phosphoproteome enrichment using monodisperse microsphere-based immobilized titanium (iv) ion affinity chromatography. Nat Protoc. 2013. March;8(3):461–480. [DOI] [PubMed] [Google Scholar]
- 37.Ficarro SB, McCleland ML, Stukenberg PT, et al. Phosphoproteome analysis by mass spectrometry and its application to saccharomyces cerevisiae. Nat Biotechnol. 2002. March;20(3):301–305. [DOI] [PubMed] [Google Scholar]
- 38.Pinkse MW, Uitto PM, Hilhorst MJ, et al. Selective isolation at the femtomole level of phosphopeptides from proteolytic digests using 2d-nanolc-esi-ms/ms and titanium oxide precolumns. Anal Chem. 2004. July 15;76(14):3935–3943. [DOI] [PubMed] [Google Scholar]
- 39.Ndassa YM, Orsi C, Marto JA, et al. Improved immobilized metal affinity chromatography for large-scale phosphoproteomics applications. J Proteome Res. 2006. October;5(10):2789–2799. [DOI] [PubMed] [Google Scholar]
- 40.Paulo JA, Navarrete-Perea J, Erickson AR, et al. An internal standard for assessing phosphopeptide recovery from metal ion/oxide enrichment strategies. J Am Soc Mass Spectrom. 2018. July;29(7):1505–1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Thingholm TE, Larsen MR. The use of titanium dioxide for selective enrichment of phosphorylated peptides. Methods Mol Biol. 2016;1355:135–146. [DOI] [PubMed] [Google Scholar]
- 42.Qiu W, Evans CA, Landels A, et al. Phosphopeptide enrichment for phosphoproteomic analysis-a tutorial and review of novel materials. Anal Chim Acta. 2020. September 8;1129:158–180. [DOI] [PubMed] [Google Scholar]
- 43.Thingholm TE, Jensen ON, Robinson PJ, et al. Simac (sequential elution from imac), a phosphoproteomics strategy for the rapid separation of monophosphorylated from multiply phosphorylated peptides. Mol Cell Proteomics. 2008. April;7(4):661–671. [DOI] [PubMed] [Google Scholar]
- 44.Choi J, Snovida SI, Bomgarden RD, et al. Sequential enrichment from metal oxide affinity chromatography (smoac), a phosphoproteomics strategy for the separation of multiply phosphorylated from monophosphorylated peptides. American Society for Mass Spectrometry Conference. 2017:WP601. [Google Scholar]
- 45.Thingholm TE, Larsen MR. Sequential elution from imac (simac): An efficient method for enrichment and separation of mono- and multi-phosphorylated peptides. Methods Mol Biol. 2016;1355:147–160. [DOI] [PubMed] [Google Scholar]
- 46.Ballif BA, Villen J, Beausoleil SA, et al. Phosphoproteomic analysis of the developing mouse brain [Research Support, U.S. Gov’t, P.H.S.]. Mol Cell Proteomics. 2004. November;3(11):1093–1101. [DOI] [PubMed] [Google Scholar]
- 47.McNulty DE, Annan RS. Hydrophilic interaction chromatography for fractionation and enrichment of the phosphoproteome. Methods Mol Biol. 2009;527:93–105, x. [DOI] [PubMed] [Google Scholar]
- 48.Loroch S, Zahedi RP, Sickmann A. Highly sensitive phosphoproteomics by tailoring solid-phase extraction to electrostatic repulsion-hydrophilic interaction chromatography. Anal Chem. 2015. February 3;87(3):1596–1604. [DOI] [PubMed] [Google Scholar]
- 49.Possemato AP, Paulo JA, Mulhern D, et al. Multiplexed phosphoproteomic profiling using titanium dioxide and immunoaffinity enrichments reveals complementary phosphorylation events. J Proteome Res. 2017. April 07;16(4):1506–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rush J, Moritz A, Lee KA, et al. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol. 2005. January;23(1):94–101. [DOI] [PubMed] [Google Scholar]
- 51.Stopfer LE, Flower CT, Gajadhar AS, et al. High-density, targeted monitoring of tyrosine phosphorylation reveals activated signaling networks in human tumors. Cancer Res. 2021. May 1;81(9):2495–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kinoshita E, Kinoshita-Kikuta E, Koike T. Advances in phos-tag-based methodologies for separation and detection of the phosphoproteome. Biochim Biophys Acta. 2015. June;1854(6):601–608. [DOI] [PubMed] [Google Scholar]
- 53.Iliuk AB, Martin VA, Alicie BM, et al. In-depth analyses of kinase-dependent tyrosine phosphoproteomes based on metal ion-functionalized soluble nanopolymers. Mol Cell Proteomics. 2010. October;9(10):2162–2172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rainer M, Bonn GK. Enrichment of phosphorylated peptides and proteins by selective precipitation methods. Bioanalysis. 2015;7(2):243–252. [DOI] [PubMed] [Google Scholar]
- 55.Machida K, Khenkhar M, Nollau P. Deciphering phosphotyrosine-dependent signaling networks in cancer by sh2 profiling. Genes Cancer. 2012. May;3(5-6):353–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Loroch S, Schommartz T, Brune W, et al. Multidimensional electrostatic repulsion-hydrophilic interaction chromatography (erlic) for quantitative analysis of the proteome and phosphoproteome in clinical and biomedical research. Biochim Biophys Acta. 2015. May;1854(5):460–468. [DOI] [PubMed] [Google Scholar]
- 57.Trinidad JC, Specht CG, Thalhammer A, et al. Comprehensive identification of phosphorylation sites in postsynaptic density preparations. Mol Cell Proteomics. 2006. May;5(5):914–922. [DOI] [PubMed] [Google Scholar]
- 58.Meijer LA, Zhou H, Chan OY, et al. Quantitative global phosphoproteomics of human umbilical vein endothelial cells after activation of the rap signaling pathway. Mol Biosyst. 2013. April 5;9(4):732–749. [DOI] [PubMed] [Google Scholar]
- 59.Doubleday PF, Ballif BA. Developmentally-dynamic murine brain proteomes and phosphoproteomes revealed by quantitative proteomics. Proteomes. 2014. June;2(2):197–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dephoure N, Gygi SP. A solid phase extraction-based platform for rapid phosphoproteomic analysis. Methods. 2011. August;54(4):379–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Boersema PJ, Mohammed S, Heck AJ. Phosphopeptide fragmentation and analysis by mass spectrometry. J Mass Spectrom. 2009. June;44(6):861–878. [DOI] [PubMed] [Google Scholar]
- 62.Everley RA, Huttlin EL, Erickson AR, et al. Neutral loss is a very common occurrence in phosphotyrosine-containing peptides labeled with isobaric tags. J Proteome Res. 2017. February 3;16(2):1069–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Syka JE, Coon JJ, Schroeder MJ, et al. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc Natl Acad Sci U S A. 2004. June 29;101(26):9528–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Olsen JV, Macek B, Lange O, et al. Higher-energy c-trap dissociation for peptide modification analysis. Nat Methods. 2007. September;4(9):709–712. [DOI] [PubMed] [Google Scholar]
- 65.Robinson MR, Taliaferro JM, Dalby KN, et al. 193 nm ultraviolet photodissociation mass spectrometry for phosphopeptide characterization in the positive and negative ion modes. J Proteome Res. 2016. August 5;15(8):2739–2748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Frese CK, Zhou H, Taus T, et al. Unambiguous phosphosite localization using electron-transfer/higher-energy collision dissociation (ethcd). J Proteome Res. 2013. March 1;12(3):1520–1525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Frese CK, Altelaar AF, Hennrich ML, et al. Improved peptide identification by targeted fragmentation using cid, hcd and etd on an ltq-orbitrap velos [Comparative Study Research Support, Non-U.S. Gov’t]. J Proteome Res. 2011. May 6;10(5):2377–2388. [DOI] [PubMed] [Google Scholar]
- 68.Jedrychowski MP, Huttlin EL, Haas W, et al. Evaluation of hcd- and cid-type fragmentation within their respective detection platforms for murine phosphoproteomics. Mol Cell Proteomics. 2011. December;10(12):M111 009910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Palumbo AM, Reid GE. Evaluation of gas-phase rearrangement and competing fragmentation reactions on protein phosphorylation site assignment using collision induced dissociation-ms/ms and ms3. Anal Chem. 2008. December 15;80(24):9735–9747. [DOI] [PubMed] [Google Scholar]
- 70.Villen J, Beausoleil SA, Gygi SP. Evaluation of the utility of neutral-loss-dependent ms3 strategies in large-scale phosphorylation analysis [Evaluation Studies Research Support, N.I.H., Extramural]. Proteomics. 2008. November;8(21):4444–4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wan Y, Cripps D, Thomas S, et al. Phosphoscan: A probability-based method for phosphorylation site prediction using ms2/ms3 pair information. J Proteome Res. 2008. July;7(7):2803–2811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jiang X, Han G, Feng S, et al. Automatic validation of phosphopeptide identifications by the ms2/ms3 target-decoy search strategy. J Proteome Res. 2008. April;7(4):1640–1649. [DOI] [PubMed] [Google Scholar]
- 73.Lee J, Xu Y, Chen Y, et al. Mitochondrial phosphoproteome revealed by an improved imac method and ms/ms/ms. Mol Cell Proteomics. 2007. April;6(4):669–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Amoresano A, Monti G, Cirulli C, et al. Selective detection and identification of phosphopeptides by dansyl ms/ms/ms fragmentation. Rapid Commun Mass Spectrom. 2006;20(9):1400–1404. [DOI] [PubMed] [Google Scholar]
- 75.Wolschin F, Lehmann U, Glinski M, et al. An integrated strategy for identification and relative quantification of site-specific protein phosphorylation using liquid chromatography coupled to ms2/ms3. Rapid Commun Mass Spectrom. 2005;19(24):3626–3632. [DOI] [PubMed] [Google Scholar]
- 76.Schweppe DK, Rusin SF, Gygi SP, et al. Optimized workflow for multiplexed phosphorylation analysis of tmt-labeled peptides using high-field asymmetric waveform ion mobility spectrometry. J Proteome Res. 2020. January 3;19(1):554–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jiang X, Bomgarden R, Brown J, et al. Sensitive and accurate quantitation of phosphopeptides using tmt isobaric labeling technique. J Proteome Res. 2017. November 3;16(11):4244–4252. [DOI] [PubMed] [Google Scholar]
- 78.Fischer J, Dos Santos MDM, Marchini FK, et al. A scoring model for phosphopeptide site localization and its impact on the question of whether to use msa. J Proteomics. 2015. November 3;129:42–50. [DOI] [PubMed] [Google Scholar]
- 79.Wiese H, Kuhlmann K, Wiese S, et al. Comparison of alternative ms/ms and bioinformatics approaches for confident phosphorylation site localization. J Proteome Res. 2014. February 7;13(2):1128–1137. [DOI] [PubMed] [Google Scholar]
- 80.Linke D, Hung CW, Cassidy L, et al. Optimized fragmentation conditions for itraq-labeled phosphopeptides. J Proteome Res. 2013. June 07;12(6):2755–2763. [DOI] [PubMed] [Google Scholar]
- 81.Vandenbogaert M, Hourdel V, Jardin-Mathe O, et al. Automated phosphopeptide identification using multiple ms/ms fragmentation modes. J Proteome Res. 2012. December 7;11(12):5695–5703. [DOI] [PubMed] [Google Scholar]
- 82.Palumbo AM, Smith SA, Kalcic CL, et al. Tandem mass spectrometry strategies for phosphoproteome analysis. Mass Spectrom Rev. 2011. Jul-Aug;30(4):600–625. [DOI] [PubMed] [Google Scholar]
- 83.Schroeder MJ, Shabanowitz J, Schwartz JC, et al. A neutral loss activation method for improved phosphopeptide sequence analysis by quadrupole ion trap mass spectrometry. Anal Chem. 2004. July 1;76(13):3590–3598. [DOI] [PubMed] [Google Scholar]
- 84.Ulintz PJ, Yocum AK, Bodenmiller B, et al. Comparison of ms(2)-only, msa, and ms(2)/ms(3) methodologies for phosphopeptide identification. J Proteome Res. 2009. February;8(2):887–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kelstrup CD, Bekker-Jensen DB, Arrey TN, et al. Performance evaluation of the q exactive hf-x for shotgun proteomics. J Proteome Res. 2018. January 5;17(1):727–738. [DOI] [PubMed] [Google Scholar]
- 86.Bekker-Jensen DB, Martinez-Val A, Steigerwald S, et al. A compact quadrupole-orbitrap mass spectrometer with faims interface improves proteome coverage in short lc gradients. Mol Cell Proteomics. 2020. April;19(4):716–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Liu F, van Breukelen B, Heck AJ. Facilitating protein disulfide mapping by a combination of pepsin digestion, electron transfer higher energy dissociation (ethcd), and a dedicated search algorithm slinks. Mol Cell Proteomics. 2014. October;13(10):2776–2786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yu Q, Shi X, Feng Y, et al. Improving data quality and preserving hcd-generated reporter ions with ethcd for isobaric tag-based quantitative proteomics and proteome-wide ptm studies. Anal Chim Acta. 2017. May 22;968:40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jiang W, Chung NA, May JC, et al. Ion mobility–mass spectrometry. Encyclopedia of analytical chemistry. p. 1–34. [Google Scholar]
- 90.Pfammatter S, Bonneil E, Lanoix J, et al. Extending the comprehensiveness of immunopeptidome analyses using isobaric peptide labeling. Anal Chem. 2020. July 7;92(13):9194–9204. [DOI] [PubMed] [Google Scholar]
- 91.Pfammatter S, Bonneil E, McManus FP, et al. A novel differential ion mobility device expands the depth of proteome coverage and the sensitivity of multiplex proteomic measurements. Mol Cell Proteomics. 2018. October;17(10):2051–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pfammatter S, Bonneil E, Thibault P. Improvement of quantitative measurements in multiplex proteomics using high-field asymmetric waveform spectrometry. J Proteome Res. 2016. December 2;15(12):4653–4665. [DOI] [PubMed] [Google Scholar]; ** This paper shows the advantage of FAIMS for multiplex proteomics.
- 93.Schweppe DK, Prasad S, Belford MW, et al. Characterization and optimization of multiplexed quantitative analyses using high-field asymmetric-waveform ion mobility mass spectrometry. Anal Chem. 2019. March 19;91(6):4010–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hebert AS, Prasad S, Belford MW, et al. Comprehensive single-shot proteomics with faims on a hybrid orbitrap mass spectrometer. Analytical chemistry. 2018. August 7;90(15):9529–9537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Prasad S, Belford MW, Dunyach JJ, et al. On an aerodynamic mechanism to enhance ion transmission and sensitivity of faims for nano-electrospray ionization-mass spectrometry. Journal of the American Society for Mass Spectrometry. 2014. December;25(12):2143–2153. [DOI] [PubMed] [Google Scholar]
- 96.Purves RW, Prasad S, Belford M, et al. Optimization of a new aerodynamic cylindrical faims device for small molecule analysis. Journal of the American Society for Mass Spectrometry. 2017. March;28(3):525–538. [DOI] [PubMed] [Google Scholar]
- 97.Saba J, Bonneil E, Pomies C, et al. Enhanced sensitivity in proteomics experiments using faims coupled with a hybrid linear ion trap/orbitrap mass spectrometer. J Proteome Res. 2009. July;8(7):3355–3366. [DOI] [PubMed] [Google Scholar]
- 98.Kanu AB, Dwivedi P, Tam M, et al. Ion mobility-mass spectrometry. J Mass Spectrom. 2008. January;43(1):1–22. [DOI] [PubMed] [Google Scholar]
- 99.Zhao H, Cunningham DL, Creese AJ, et al. Faims and phosphoproteomics of fibroblast growth factor signaling: Enhanced identification of multiply phosphorylated peptides. J Proteome Res. 2015. December 4;14(12):5077–5087. [DOI] [PubMed] [Google Scholar]
- 100.Bridon G, Bonneil E, Muratore-Schroeder T, et al. Improvement of phosphoproteome analyses using faims and decision tree fragmentation. Application to the insulin signaling pathway in drosophila melanogaster s2 cells. J Proteome Res. 2012. February 3;11(2):927–940. [DOI] [PubMed] [Google Scholar]
- 101.Pfammatter S, Bonneil E, McManus FP, et al. Accurate quantitative proteomic analyses using metabolic labeling and high field asymmetric waveform ion mobility spectrometry (faims). J Proteome Res. 2019. May 3;18(5):2129–2138. [DOI] [PubMed] [Google Scholar]
- 102.Schweppe DK, Eng JK, Yu Q, et al. Full-featured, real-time database searching platform enables fast and accurate multiplexed quantitative proteomics. J Proteome Res. 2020. May 1;19(5):2026–2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Erickson BK, Mintseris J, Schweppe DK, et al. Active instrument engagement combined with a real-time database search for improved performance of sample multiplexing workflows. J Proteome Res. 2019. March 1;18(3):1299–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This paper introduces and applies real-time search for isobaric tagging experiments.
- 104.Elias JE, Gygi SP. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol Biol. 2010;604:55–71. [DOI] [PMC free article] [PubMed] [Google Scholar]; * This paper highlights the target-decoy for false discovery rate determination.
- 105.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007. March;4(3):207–214. [DOI] [PubMed] [Google Scholar]
- 106.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994. November;5(11):976–989. [DOI] [PubMed] [Google Scholar]; * This paper introduces the COMET search engine.
- 107.Perkins DN, Pappin DJ, Creasy DM, et al. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999. December;20(18):3551–3567. [DOI] [PubMed] [Google Scholar]
- 108.Eng JK, Jahan TA, Hoopmann MR. Comet: An open-source ms/ms sequence database search tool. Proteomics. 2013. January;13(1):22–24. [DOI] [PubMed] [Google Scholar]
- 109.Devabhaktuni A, Lin S, Zhang L, et al. Taggraph reveals vast protein modification landscapes from large tandem mass spectrometry datasets. Nat Biotechnol. 2019. April;37(4):469–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Kong AT, Leprevost FV, Avtonomov DM, et al. Msfragger: Ultrafast and comprehensive peptide identification in mass spectrometry-based proteomics. Nat Methods. 2017. May;14(5):513–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li D, Fu Y, Sun R, et al. Pfind: A novel database-searching software system for automated peptide and protein identification via tandem mass spectrometry. Bioinformatics. 2005. July 1;21(13):3049–3050. [DOI] [PubMed] [Google Scholar]
- 112.Solntsev SK, Shortreed MR, Frey BL, et al. Enhanced global post-translational modification discovery with metamorpheus. J Proteome Res. 2018. May 4;17(5):1844–1851. [DOI] [PubMed] [Google Scholar]
- 113.Wang LH, Li DQ, Fu Y, et al. Pfind 2.0: A software package for peptide and protein identification via tandem mass spectrometry. Rapid Commun Mass Spectrom. 2007;21(18):2985–2991. [DOI] [PubMed] [Google Scholar]
- 114.Tarasova IA, Chumakov PM, Moshkovskii SA, et al. Profiling modifications for glioblastoma proteome using ultra-tolerant database search: Are the peptide mass shifts biologically relevant or chemically induced? J Proteomics. 2019. January 16;191:16–21. [DOI] [PubMed] [Google Scholar]
- 115.Beausoleil SA, Villen J, Gerber SA, et al. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006. October;24(10):1285–1292. [DOI] [PubMed] [Google Scholar]; * This paper introduces the AScore as a metric for probability-based approach for phosphorylation site localization.
- 116.Olsen JV, Blagoev B, Gnad F, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006. November 03;127(3):635–648. [DOI] [PubMed] [Google Scholar]
- 117.Savitski MM, Lemeer S, Boesche M, et al. Confident phosphorylation site localization using the mascot delta score. Mol Cell Proteomics. 2011. February;10(2):M110 003830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Swaney DL, Wenger CD, Thomson JA, et al. Human embryonic stem cell phosphoproteome revealed by electron transfer dissociation tandem mass spectrometry. Proc Natl Acad Sci U S A. 2009. January 27;106(4):995–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Taus T, Kocher T, Pichler P, et al. Universal and confident phosphorylation site localization using phosphors. J Proteome Res. 2011. December 2;10(12):5354–5362. [DOI] [PubMed] [Google Scholar]
- 120.Ruttenberg BE, Pisitkun T, Knepper MA, et al. Phosphoscore: An open-source phosphorylation site assignment tool for msn data. J Proteome Res. 2008. July;7(7):3054–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fermin D, Avtonomov D, Choi H, et al. Luciphor2: Site localization of generic post-translational modifications from tandem mass spectrometry data. Bioinformatics. 2015. April 1;31(7):1141–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Fermin D, Walmsley SJ, Gingras AC, et al. Luciphor: Algorithm for phosphorylation site localization with false localization rate estimation using modified target-decoy approach. Mol Cell Proteomics. 2013. November;12(11):3409–3419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Potel CM, Lemeer S, Heck AJR. Phosphopeptide fragmentation and site localization by mass spectrometry: An update. Anal Chem. 2019. January 2;91(1):126–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Baker PR, Trinidad JC, Chalkley RJ. Modification site localization scoring integrated into a search engine. Mol Cell Proteomics. 2011. July;10(7):M111 008078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Locard-Paulet M, Bouyssie D, Froment C, et al. Comparing 22 popular phosphoproteomics pipelines for peptide identification and site localization. J Proteome Res. 2020. March 6;19(3):1338–1345. [DOI] [PubMed] [Google Scholar]
- 126.Li J, Paulo JA, Nusinow DP, et al. Investigation of proteomic and phosphoproteomic responses to signaling network perturbations reveals functional pathway organizations in yeast. Cell Rep. 2019. November 12;29(7):2092–2104 e2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Needham EJ, Parker BL, Burykin T, et al. Illuminating the dark phosphoproteome. Sci Signal. 2019. January 22;12(565). [DOI] [PubMed] [Google Scholar]
- 128.Linding R, Jensen LJ, Pasculescu A, et al. Networkin: A resource for exploring cellular phosphorylation networks. Nucleic Acids Res. 2008. January;36(Database issue):D695–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Munk S, Refsgaard JC, Olsen JV, et al. From phosphosites to kinases. Methods Mol Biol. 2016;1355:307–321. [DOI] [PubMed] [Google Scholar]
- 130.Yaffe MB, Leparc GG, Lai J, et al. A motif-based profile scanning approach for genome-wide prediction of signaling pathways. Nat Biotechnol. 2001. April;19(4):348–353. [DOI] [PubMed] [Google Scholar]
- 131.Schwartz D, Gygi SP. An iterative statistical approach to the identification of protein phosphorylation motifs from large-scale data sets. Nat Biotechnol. 2005. November;23(11):1391–1398. [DOI] [PubMed] [Google Scholar]
- 132.Dinkel H, Chica C, Via A, et al. Phospho.Elm: A database of phosphorylation sites--update 2011. Nucleic Acids Res. 2011. January;39(Database issue):D261–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kunz RC, McAllister FE, Rush J, et al. A high-throughput, multiplexed kinase assay using a benchtop orbitrap mass spectrometer to investigate the effect of kinase inhibitors on kinase signaling pathways. Anal Chem. 2012. July 17;84(14):6233–6239. [DOI] [PubMed] [Google Scholar]
- 134.Kubota K, Anjum R, Yu Y, et al. Sensitive multiplexed analysis of kinase activities and activity-based kinase identification [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov’t]. Nat Biotechnol. 2009. October;27(10):933–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Martinez Molina D, Jafari R, Ignatushchenko M, et al. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013. July 5;341(6141):84–87. [DOI] [PubMed] [Google Scholar]
- 136.Gaetani M, Sabatier P, Saei AA, et al. Proteome integral solubility alteration: A high-throughput proteomics assay for target deconvolution. J Proteome Res. 2019. November 1;18(11):4027–4037. [DOI] [PubMed] [Google Scholar]
- 137.Smith IR, Hess KN, Bakhtina AA, et al. Identification of phosphosites that alter protein thermal stability. Nature Methods. 2021. 2021/06/17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Huang JX, Lee G, Cavanaugh KE, et al. High throughput discovery of functional protein modifications by hotspot thermal profiling. Nature Methods. 2019. 2019/09/01;16(9):894–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Potel CM, Kurzawa N, Becher I, et al. Impact of phosphorylation on thermal stability of proteins. Nature Methods. 2021. 2021/06/17. [DOI] [PubMed] [Google Scholar]
- 140.Bludau I, Frank M, Dörig C, et al. Systematic detection of functional proteoform groups from bottom-up proteomic datasets. Nature Communications. 2021. 2021/06/21;12(1):3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Olsen JV, Mann M. Improved peptide identification in proteomics by two consecutive stages of mass spectrometric fragmentation. Proc Natl Acad Sci U S A. 2004. September 14;101(37):13417–13422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Lim MY, O’Brien J, Paulo JA, et al. Improved method for determining absolute phosphorylation stoichiometry using bayesian statistics and isobaric labeling. J Proteome Res. 2017. November 3;16(11):4217–4226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wu R, Haas W, Dephoure N, et al. A large-scale method to measure absolute protein phosphorylation stoichiometries. Nat Methods. 2011;8(8):677–683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Olsen JV, Vermeulen M, Santamaria A, et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010;3(104):ra3. [DOI] [PubMed] [Google Scholar]
- 145.Humphrey SJ, Karayel O, James DE, et al. High-throughput and high-sensitivity phosphoproteomics with the easyphos platform. Nat Protoc. 2018. September;13(9):1897–1916. [DOI] [PubMed] [Google Scholar]
- 146.Zhao Y, Liu G, Zambito FC, et al. A multiplexed immunocapture liquid chromatography tandem mass spectrometry assay for the simultaneous measurement of myostatin and gdf-11 in rat serum using an automated sample preparation platform. Anal Chim Acta. 2017. August 1;979:36–44. [DOI] [PubMed] [Google Scholar]
- 147.Ruelcke JE, Loo D, Hill MM. Reducing the cost of semi-automated in-gel tryptic digestion and gelc sample preparation for high-throughput proteomics. J Proteomics. 2016. October 21;149:3–6. [DOI] [PubMed] [Google Scholar]
- 148.Ippoliti PJ, Kuhn E, Mani DR, et al. Automated microchromatography enables multiplexing of immunoaffinity enrichment of peptides to greater than 150 for targeted ms-based assays. Anal Chem. 2016. August 2;88(15):7548–7555. [DOI] [PubMed] [Google Scholar]
- 149.Hughes CS, Sorensen PH, Morin GB. A standardized and reproducible proteomics protocol for bottom-up quantitative analysis of protein samples using sp3 and mass spectrometry. Methods Mol Biol. 2019;1959:65–87. [DOI] [PubMed] [Google Scholar]; *This paper introduces the single-pot, solid-phase-enhanced sample preparation (SP3) strategy as an alternative to chemical precipitation methods.
- 150.Hughes CS, Moggridge S, Muller T, et al. Single-pot, solid-phase-enhanced sample preparation for proteomics experiments. Nat Protoc. 2019. January;14(1):68–85. [DOI] [PubMed] [Google Scholar]
- 151.Moggridge S, Sorensen PH, Morin GB, et al. Extending the compatibility of the sp3 paramagnetic bead processing approach for proteomics. J Proteome Res. 2018. April 6;17(4):1730–1740. [DOI] [PubMed] [Google Scholar]
- 152.Hughes CS, Foehr S, Garfield DA, et al. Ultrasensitive proteome analysis using paramagnetic bead technology. Mol Syst Biol. 2014. October 30;10:757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Paulo JA, Navarrete-Perea J, Gygi SP. Multiplexed proteome profiling of carbon source perturbations in two yeast species with sl-sp3-tmt. J Proteomics. 2020. January 6;210:103531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Liu X, Gygi SP, Paulo JA. A semiautomated paramagnetic bead-based platform for isobaric tag sample preparation. J Am Soc Mass Spectrom. 2021. May 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Leutert M, Rodriguez-Mias RA, Fukuda NK, et al. R2-p2 rapid-robotic phosphoproteomics enables multidimensional cell signaling studies. Mol Syst Biol. 2019. December;15(12):e9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Gaun A, Lewis Hardell KN, Olsson N, et al. Automated 16-plex plasma proteomics with real-time search and ion mobility mass spectrometry enables large-scale profiling in naked mole-rats and mice. J Proteome Res. 2021. February 5;20(2):1280–1295. [DOI] [PubMed] [Google Scholar]
- 157.Kalagiri R, Adam K, Hunter T. Empirical evidence of cellular histidine phosphorylation by immunoblotting using phis mabs. Methods Mol Biol. 2020;2077:181–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hardman G, Eyers CE. High-throughput characterization of histidine phosphorylation sites using upax and tandem mass spectrometry. Methods Mol Biol. 2020;2077:225–235. [DOI] [PubMed] [Google Scholar]
- 159.Attwood PV. A quantitative method for the measurement of protein histidine phosphorylation. Methods Mol Biol. 2020;2077:51–61. [DOI] [PubMed] [Google Scholar]
- 160.Potel CM, Lin MH, Heck AJR, et al. Widespread bacterial protein histidine phosphorylation revealed by mass spectrometry-based proteomics. Nat Methods. 2018. March;15(3):187–190. [DOI] [PubMed] [Google Scholar]
- 161.Fuhs SR, Hunter T. Phisphorylation: The emergence of histidine phosphorylation as a reversible regulatory modification. Curr Opin Cell Biol. 2017. April;45:8–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sun Q, Julian RR. Probing sites of histidine phosphorylation with iodination and tandem mass spectrometry. Rapid Commun Mass Spectrom. 2011. August 15;25(15):2240–2246. [DOI] [PubMed] [Google Scholar]
- 163.Zu XL, Besant PG, Imhof A, et al. Mass spectrometric analysis of protein histidine phosphorylation. Amino Acids. 2007;32(3):347–357. [DOI] [PubMed] [Google Scholar]
- 164.Sickmann A, Meyer HE. Histidine phosphorylation site identification by esi-ms. CSH Protoc. 2007. September 1;2007:pdb prot4628. [DOI] [PubMed] [Google Scholar]
- 165.Hardman G, Perkins S, Brownridge PJ, et al. Strong anion exchange-mediated phosphoproteomics reveals extensive human non-canonical phosphorylation. EMBO J. 2019. October 4;38(21):e100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.