

Abstract
Background:
Endothelial cells depend on glycolysis for much of their energy production. Impaired endothelial glycolysis has been associated with various vascular pathobiologies, including impaired angiogenesis and atherogenesis. Interferon-gamma (IFN-γ)-producing CD4+ and CD8+ T-lymphocytes have been identified as the predominant pathologic cell subsets in human atherosclerotic plaques. While the immunological consequences of these cells have been extensively evaluated, their IFN-γ-mediated metabolic effects on endothelial cells remain unknown. The purpose of this study was to determine the metabolic consequences of the T-lymphocyte cytokine, IFN-γ, on human coronary artery endothelial cells (HCAEC).
Methods:
The metabolic effects of IFN-γ on primary HCAEC were assessed by unbiased transcriptomic and metabolomic analyses combined with real-time extracellular flux analyses and molecular mechanistic studies. Cellular phenotypic correlations were made by measuring altered endothelial intracellular cyclic guanosine monophosphate (cGMP) content, wound healing capacity, and adhesion molecule expression.
Results:
IFN-γ exposure inhibited basal glycolysis of quiescent primary HCAEC by 20 % through the global transcriptional suppression of glycolytic enzymes resulting from decreased basal hypoxia inducible factor 1α (HIF1α) nuclear availability in normoxia. The decrease in HIF1α activity was a consequence of IFN-γ-induced tryptophan catabolism resulting in ARNT (aryl hydrocarbon receptor nuclear translocator)/HIF1β sequestration by the kynurenine-activated aryl hydrocarbon receptor (AHR). Additionally, IFN-γ resulted in a 23% depletion of intracellular NAD+ in HCAEC. This altered glucose metabolism was met with concomitant activation of fatty acid oxidation, which augmented its contribution to intracellular ATP balance by over 20%. These metabolic derangements were associated with adverse endothelial phenotypic changes, including decreased basal intracellular cGMP, impaired endothelial migration, and a switch to a pro-inflammatory state.
Conclusions:
IFN-γ impairs endothelial glucose metabolism via altered tryptophan catabolism destabilizing HIF1, depletes NAD+, and results in a metabolic shift toward increased fatty acid oxidation. This work suggests a novel mechanistic basis for pathologic T-lymphocyte-endothelial interactions in atherosclerosis mediated by IFN-γ, linking endothelial glucose, tryptophan, and fatty acid metabolism with NAD(H) and ATP generation, and their adverse endothelial functional consequences.
Keywords: coronary artery, endothelium, metabolism, atherosclerosis, T-lymphocytes, interferon-gamma, glycolysis, fatty acid oxidation, tryptophan, HIF
INTRODUCTION
Glucose metabolism is fundamental to endothelial cell survival and function, serving as an essential source for energy generation, biomass synthesis, and redox hemostasis (reviewed in1). Endothelial cells depend on glycolysis for much of their adenosine triphosphate (ATP) production, up to 80% even in the presence of abundant oxygen2, 3, and on the pentose phosphate pathway for redox balance and nucleotide synthesis. Dysregulated endothelial glycolysis has been linked to various vascular pathologies, including atherogenesis4, pulmonary hypertension5, and impaired angiogenesis6.
The interface between the immunological and endothelial systems forms a shared mechanistic basis for a broad spectrum of diseases, including atherosclerosis. Interferon-gamma (IFN-γ)-producing CD8+ and CD4+ T helper type 1 cells have been identified as the most abundant and prominent pathological immune cell subsets in human carotid atherosclerotic plaques correlating with worse clinical outcomes7. IFN-γ has been characterized as proatherogenic in mice,8, 9 and has been linked to neointima formation in in-stent stenosis10 and allograft vasculopathy11 in humans. While their contributions as immune effector cells to atheroma development and progression have been extensively studied, the metabolic consequences of their actions, and of IFN-γ in particular, on endothelial cells remain unknown.
Here, we study the metabolic effects of IFN-γ on human coronary artery endothelial cells (HCAEC) with unbiased transcriptomic and metabolomic analyses. Given that most systemic vascular beds affected by atherosclerosis are non-proliferating in the absence of significant hypoxic stress, our study focuses on how basal metabolism of quiescent endothelial cells is altered by IFN-γ exposure in normoxia. We demonstrate that IFN-γ exposure results in significant impairment of basal endothelial glucose metabolism via altered tryptophan metabolism with downstream inhibition of HIF1 activity resulting from AHR activation by the tryptophan catabolite, kynurenine. These effects are accompanied by concomitant intracellular NAD+ depletion and heightened endothelial oxygen consumption from accelerated fatty acid oxidation, and are associated with endothelial dysfunction. These metabolic derangements and their associated pathologic phenotypic changes present a novel mechanistic link between the endothelial cell and T-cell-dependent, IFN-γ-mediated cardiovascular pathobiology.
METHODS
An expanded methods section is provided in the Supplemental Material.
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
No animal experiments were included in this study. While we used human cells, they were purchased from a vendor, Lonza; were anonymous with respect to donor; and for which IRB review was not required.
Cell culture and treatments
Primary human coronary artery endothelial cells (HCAEC) (Lonza) were cultured in Endothelial Cell Growth Basal Medium-2 supplemented with EGM-2 Microvascular Endothelial Cell Growth Medium-2 BulletKit without antibiotics unless otherwise specified at 37°C with 5 % CO2. Glucose concentration of this medium was 1.0 g/L (5.5 mM) unless otherwise indicated. Low and high glucose media were prepared using the basal medium containing no glucose (Cell Biologics) with the above BulletKit supplemented with glucose (Boston Bioproducts) at 2.5 mM and 25 mM, respectively. For studying the effects of tryptophan depletion, confluent HCAECs were incubated with the Dulbecco’s Modified Eagle Medium (DMEM) containing 1.0 g/L of glucose and deficient in tryptophan (Thermo Fisher Scientific) with or without exogenous tryptophan supplementation (16.0 mg/L; Sigma) equivalent to the concentration in the standard DMEM formulation. The cells had been prepared commercially from deceased adult male and female donors ranging in age from 46 – 55 yr without diabetes or known cardiac diseases whose causes of deaths were non-cardiac. All experiments were performed with cells grown to confluence between passages five and seven before being treated with human recombinant IFN-γ (R&D Systems; 1–50 ng/mL) in vitro for the durations specified. For the real-time extracellular flux assays and high performance liquid chromatography (HPLC) measurements of ATP and ADP, only passage six cells were used. For indoleamine-2,3, -dioxygenase 1 (IDO1) inhibition, cells were co-treated with 1-methyltryptophan (1-MT) (Sigma-Aldrich; 1, 3, 5 mM) and IFN-γ at 50 ng/mL for 24 h. For poly (ADP-ribose) polymerase inhibition, cells were pretreated with 3-aminobenzamide (3-AB) (Sigma-Aldrich; 5 mM) for 1 h prior to 24 h of IFN-γ treatment. For NAD precursor supplementation, nicotinamide mononucleotide (NMN) was added at 0.5 mM to cells pre-treated with IFN-γ for 24 h for an additional 24 h before measuring intracellular NAD(H). For fatty oxidation (FAO) inhibition, 2[6(4-chlorophenoxy)hexyl]oxirane-2-carboxylate (etomoxir) (Cayman; 10 μM) was added during the last hour of 24 h of IFN-γ treatment at 50 ng/mL. For hypoxia studies, cells were treated for 24 h with 1 % O2, 5 % CO2, with N2 balance, at 37°C in a hypoxia chamber glove box with or without IFN-γ at 50 ng/mL.
Statistical Analyses
Statistical analyses were carried out with GraphPad Prism 8. Data represent mean ± standard error of the mean (SEM) from at least 3 independent experiments as specified. Normal distribution was assessed by the Shapiro-Wilk test. Homogeneity of variances was assessed by the F-test or Brown-Forsythe test. For normally distributed data, two-tailed Student’s t-test or Welch’s t-test (for unequal variances) was used for 2-group analyses; and one- or two-way ANOVA with post hoc Bonferroni’s, Tukey’s, or Sidak’s multiple comparisons test or Welch’s ANOVA test (for unequal variances) with post hoc Dunnett’s T3 multiple comparisons test was used for multi-group analyses, as specified. For data that deviate from a normal distribution, the Mann-Whitney U test or Wilcoxon matched-pairs signed-rank test was used for 2-group analyses; and the Kruskal-Wallis test with post hoc Dunn’s multiple comparisons test was used for multi-group analyses. One-sample t-test was used to analyze the fold changes in normalized protein densitometric analysis data or intracellular cGMP as specified. p < 0.05 was used to define statistical significance.
RESULTS
Interferon-γ impairs basal glycolysis in human coronary artery endothelial cells (HCAEC)
IFN-γ exposure to confluent, resting HCAEC for 24 h in vitro in culture medium containing physiological glucose concentration (5.5 mM) resulted in a 20 % decrease in basal extracellular acidification rate (ECAR) (p = 0.006) measured by the real-time extracellular flux assay, indicating decreased endothelial glycolytic activity (Figure 1A). Consistent with this finding, a 27% decrease in intracellular lactate was observed (p = 0.010) (Figure 1B).
Figure 1. IFN-γ impairs basal glucose metabolism in HCAEC via global transcriptional suppression of glycolytic enzymes.
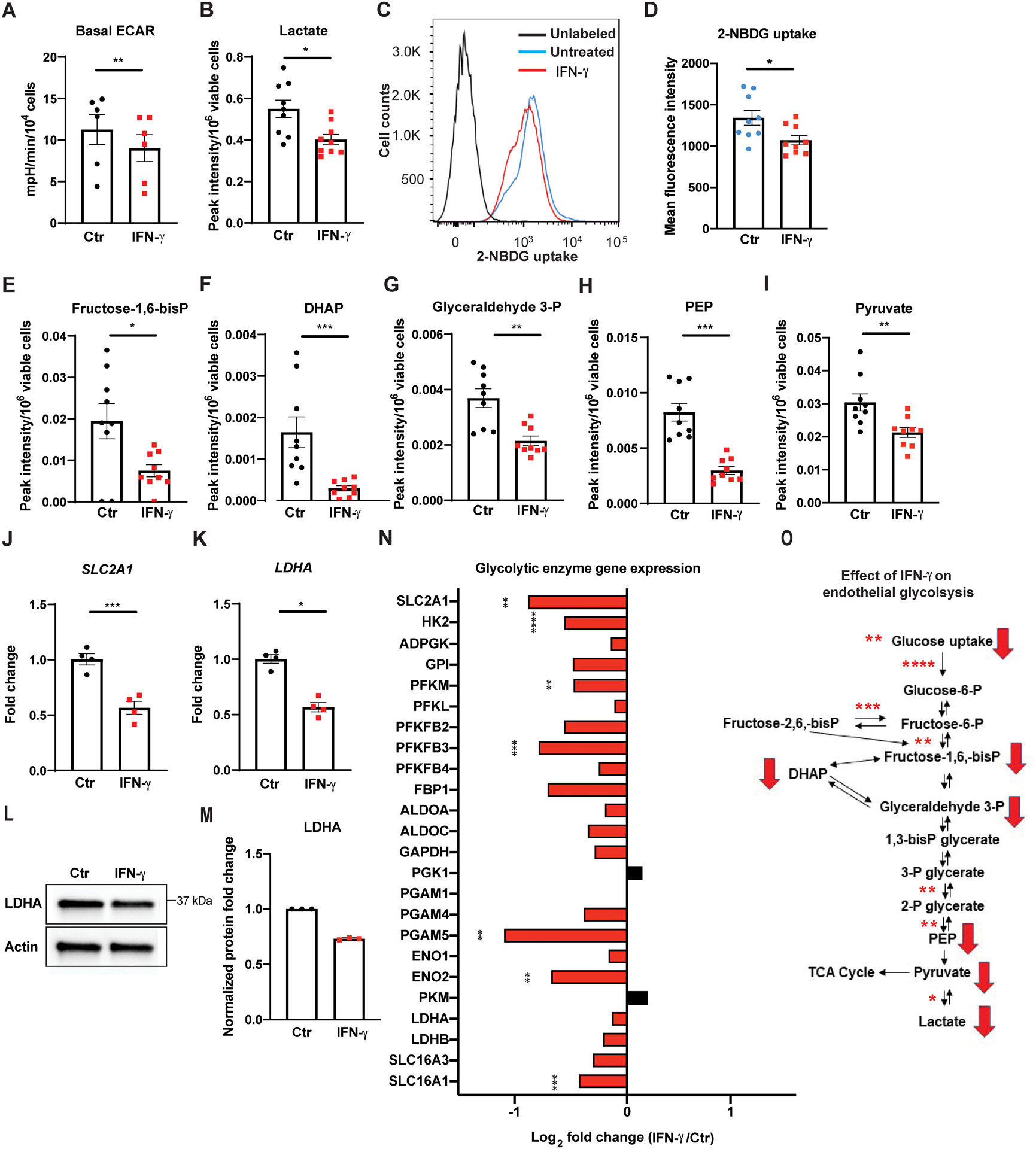
A, Real time extracellular flux analysis of extracellular acidification rate (ECAR) of HCAEC following 24 h of IFN-γ treatment at 50 ng/ml. Data represent mean ± SEM from 6 independent experiments.
B, Intracellular lactate levels in HCAEC measured by LC-MS following the same IFN-γ treatment. Mean ± SEM from 9 biological replicates from 3 independent experiments.
C-D, Cellular uptake of the glucose analogue 2-NBDG by HCAEC assessed by flow cytometry following 48 h of IFN-γ treatment. Mean ± SEM from 9 biological replicates from 3 independent experiments.
E-I, Intracellular metabolites of the glycolytic pathway in HCAEC measured by LC-MS following 24 h of IFN-γ treatment. Mean ± SEM from 9 biological replicates from 3 independent experiments.
J-K, Gene expression changes of SLC2A1 (J) and LDHA (K) measured by real-time PCR in HCAEC following the same IFN-γ treatment. Mean ± SEM from 4 independent experiments. Statistical significance was assessed using paired (A) or unpaired (D) two-tailed Student’s t-tests, unpaired Welch’s t-tests (B, E, I-J), and Mann-Whitney U tests (F-H, K).
L-M, Protein expression changes of LDHA by western blot in HCAEC following 24–48 h of IFN-γ (L) and densitometric quantification of 3 independent experiments (M).
N, IFN-γ induced gene expression changes involving the glycolytic enzymes by RNASeq analysis of HCAEC obtained in biological triplicates. Statistical significance was determined based on the p-values adjusted for multiple test hypotheses by Benjamini-Hochberg procedure with a false discovery rate threshold of < 0.05.
O, Summary of IFN-γ impairment of HCAEC glycolysis at the transcriptional (asterisks) and metabolite (red arrows) levels. Red arrows denote the significantly decreased intracellular metabolites measured by LC-MS while asterisks indicate the significantly decreased gene expressions of the enzymes involved in the reactions and the extents of the statistical significance.
* p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001
ADPGK: ADP dependent glucokinase, ALDO: Aldolase, DHAP: Dihydroxyacetone phosphate, ENO: Enolase, FBP1: Fructose-bisphosphatase 1, Fructose-1,6-bisP: Fructose-1,6,-bisphospate, Fructose-2,6-bisP: Fructose-2,6,-bisphospate, Fructose-6-P: Fructose-6-phosphate, GAPDH: Glyceraldehyde 3-phosphate dehydrogenase, GPI: Glucose-6-phosphate isomerase, Glucose-6-P: glucose-6-phosphate, Glyceraldehyde 3-P: glyceraldehyde 3-phosphate, HK: Hexokinase, IFN: Interferon, PFK: Phosphofructokinase, SLC2A1: Solute carrier family 2 Member 1, PEP: Phosphoenolpyruvate, PFKFB: 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase, PGAM: Phosphoglycerate mutase 1, PGK: Phosphoglycerate kinase, PKM: Pyruvate kinase M, LDH: Lactate dehydrogenase, SLC16A1: Solute carrier family 16A member 1 (encodes monocarboxylate transporter 1), and SLC16A3: Solute carrier family 16A member 3 (encodes monocarboxylate transporter 4), TCA: tricarboxylic acid, 1, 3-bisP glycerate: 1,3-bisphosphoglycerate, 2-NBDG: 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose, 2-P glycerate: 2-phosphoglycerate, 3-P glycerate: 3-phosphoglycerate.
HCAEC exposed to IFN-γ had significantly reduced intracellular glucose transport in vitro as demonstrated by a 20% decrease in the fluorescent glucose analogue 2-NBDG uptake (Figure 1C–D). Similarly, many of the intracellular intermediates in glycolytic pathways measured by LC-MS, including fructose-1,6-bisphosphate, dihydroxyacetone phosphate, glyceraldehyde 3-phosphate, phosphoenolpyruvate, and pyruvate, were significantly decreased after 24 h of IFN-γ exposure throughout (Figure 1E–I). Taken together, these findings indicate that IFN-γ impairs endothelial glycolysis (Figure 1O). Similarly, impaired glycolysis was observed in the absence of serum or growth factor supplementation to the culture medium and, in addition, in low–glucose medium (2.5 mM) (Figure I in the Supplement).
IFN-γ induces global suppression of endothelial glycolytic enzyme gene expression
Given the notable suppression of basal glycolysis, including glucose uptake and pyruvate-to-lactate conversion, we hypothesized that IFN-γ impairs glycolysis via inhibition of multiple glycolytic enzymes at the transcriptional level. Indeed, expression of both solute carrier family 2 member 1 (SLC2A1) and lactate dehydrogenase A (LDHA), the genes encoding the enzymes regulating glucose uptake and pyruvate-to-lactate conversion, respectively, was suppressed after 24 h of IFN-γ exposure in vitro, each by over 40% (p = 0.0008 and 0.0286, respectively) (Figure 1J–K). LDHA protein expression was similarly decreased (Figure 1L–M). Additionally, RNA sequencing analysis of HCAEC following 24 h of IFN-γ exposure was notable for suppression of the expression of the majority of the glycolytic enzymes (Figure 1N), further supporting the conclusion that IFN-γ interferes with endothelial glucose metabolism at the level of transcription (Figure 1O).
IFN-γ accelerates tryptophan catabolism resulting in an intracellular kynurenine surge in HCAEC
Given the significant alterations in endothelial glucose metabolism induced by IFN-γ, we next sought to obtain a more global assessment of the landscape of different cellular metabolic pathways by targeted LC-MS analysis of 144 common intracellular metabolites extracted from HCAEC treated with IFN-γ for 24 h in vitro (Figure 2A). We observed a decrease in intracellular tryptophan by up to 78% (p < 0.0001) (Figure 2B) following 24 h of IFN-γ exposure with robust antiparallel accumulation of kynurenine (the first major stable metabolite of tryptophan catabolism) by 720% compared with untreated control (p < 0.0001) (Figure 2C). We also observed concomitant increases in mRNA and protein expression of indoleamine-2, 3, -dioxygenase 1 (IDO1), an IFN-γ-activated enzyme catalyzing tryptophan degradation to kynurenine (Figure 2D–E). Increasing IDO activity over time was suggested by a time-dependent increase in the intracellular kynurenine-to-tryptophan ratio (Figure 2F). These findings are consistent with prior reports of tryptophan depletion in non-coronary artery endothelial cells upon IFN-γ exposure12–14.
Figure 2. IFN-γ accelerates tryptophan degradation to kynurenine in HCAEC.
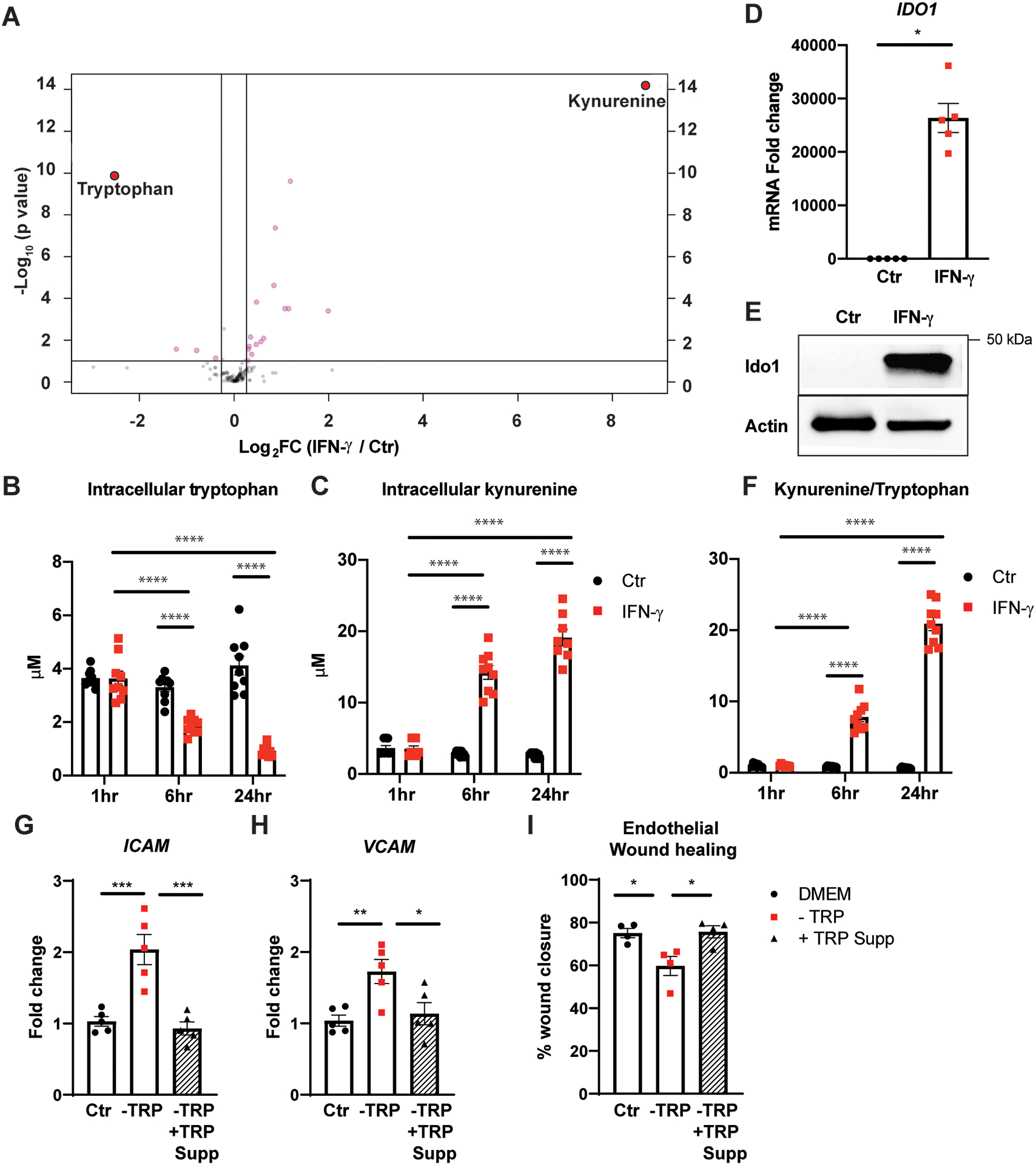
A, Targeted LC-MS analysis of 144 common intracellular metabolites. Log2 of fold changes in intracellular metabolites extracted from HCAEC following 24 h of IFN-γ at 50 ng/mL are depicted on the x-axis with their p values on the y-axis. Mean from 3 independent experiments.
B-C, Intracellular tryptophan (B) and kynurenine (C) in HCAEC measured by LC-MS following 1, 6, 24 h of IFN-γ treatment. Mean ± SEM from 9 biological replicates from 3 independent experiments.
D, IDO1 mRNA expression fold change in HCAEC following the same IFN-γ treatment. Mean ± SEM from 3 independent experiments.
E, IDO1 protein expression changes in HCAEC following the same IFN-γ treatment. Representative Western blot.
F, Intracellular kynurenine/tryptophan ratio over time indicating increased indoleamine 2,3-dioxygenase 1 (IDO1) activity. Mean ± SEM from 9 biological replicates from 3 independent experiments.
G-H, Adhesion molecules, ICAM (G) and VCAM (H), mRNA expression fold changes in HCAEC measured by real-time PCR following 24 h incubation in control Dulbecco’s Modified Eagle Medium (DMEM) (Ctrl) or DMEM deficient in tryptophan (TRP) (−TRP) with (black triangles with hatched bars) or without (red squares) TRP supplementation (−TRP + TRP Supp). Mean ± SEM from 3 independent experiments.
I, Percentages of wound closure of monolayer re-endothelialization following 24 h incubation in the above medium calculated by the following formula = [(open area at 0 h) - (open area at 48 h)]/(open area at 0 h) × 100. Mean ± SEM from 4 independent experiments.
Statistical significance was determined by two-way ANOVA with post hoc Tukey’s multiple comparisons test (B-C, F), Welch’s t-test (D), or one-way ANOVA with post hoc Bonferroni’s pairwise comparisons test (G-I).
* p < 0.05, ** p < 0.01, *** < 0.001, **** p < 0.0001
ICAM: intercellular adhesion molecule; VCAM: vascular cell adhesion molecule.
To understand better the functional consequences of tryptophan depletion on endothelial cells, confluent HCAECs were incubated in DMEM deficient in tryptophan with or without exogenous tryptophan supplementation for 24 h followed by assessment of changes in endothelial adhesion molecule expression and migratory capacity. Exposure to the tryptophan-deficient medium resulted in a pro-inflammatory phenotype with robust upregulation of ICAM (p = 0.0005) and VCAM (p = 0.0083) gene expression (Figure 2G–H) and a significant impairment in HCAEC migratory capacity following mechanical (scratch) injury to the endothelial monolayer (Figure 2I; Figure II in the Supplement).
IFN-γ activates AHR and promotes its dimerization with ARNT/ HIF1β with a concomitant decrease in nuclear HIF1α
Kynurenine is an endogenous ligand for the intracellular transcription factor, aryl hydrocarbon receptor (AHR)15. Upon ligand- and non-ligand-specific activation, AHR translocates to the nucleus, dimerizes with aryl hydrocarbon receptor nuclear translocator (ARNT)/ hypoxia-inducible factor 1 beta (HIF1β) (henceforth referred to as ARNT), and binds to the xenobiotic response element [XRE; also known as the dioxin response element (DRE)] to regulate downstream target gene transcription16. Following IFN-γ treatment and the subsequent intracellular kynurenine surge in HCAEC, we detected an increase in AHR protein expression in endothelial nuclei and nuclear extracts (Figure 3A–C). This increase in nuclear AHR was accompanied by downstream transcriptional activation of its canonical target genes, such as cytochrome P450 family 1 subfamily B member 1 (CYP1B1) with an over 27-fold change in its mRNA expression (Figure 3D). Given that ARNT is a shared binding partner for AHR and HIF1α and necessary for their stabilization and activity as transcription factors17, 18, we hypothesized that AHR activation and nuclear translocation by the IFN-γ-induced kynurenine increase leads to sequestration of the limited nuclear ARNT and, as a consequence, destabilizes residual HIF1α in normoxia (Figure 3H). Such competition between HIF1α and AHR (activated by an exogenous agonist) has been demonstrated in other endothelial cell types in hypoxia19. We further hypothesized this mechanism likely would explain IFN-γ-induced transcriptional suppression of SLC2A1 and LDHA expression— both well-characterized HIF1 target genes—as well as the decreases in the expression of other glycolytic enzymes. We found that IFN-γ treatment of HCAEC for 38 h in normoxia resulted in significant HIF1α destabilization with a 29 % decrease in total nuclear protein expression (p = 0.012), while total nuclear ARNT remained unchanged (Figure 3C). A similar and significant decrease in nuclear HIF1α was noted with IFN-γ treatment in hypoxia (1% O2), although the extent of HIF1α destabilization was somewhat less (17 %) (data not shown). Co-immunoprecipitation analysis (Figure 3E–G) comparing the relative quantities of ARNT-bound HIF1α and ARNT-bound AHR in nuclear extracts following the same IFN-γ exposure showed a 30% decrease in ARNT-bound HIF1α/ARNT-bound AHR when compared to untreated controls (p = 0.036). Consistent with the decreased nuclear HIF1α availability by IFN-γ, IFN-γ abrogated cobalt chloride (CoCl2)-induced HRE-luciferase reporter activity in HCAECs (Figure 3I) and suppressed another canonical HIF1 target gene expression, VEGFA, by more than 50 % (Figure 3J).
Figure 3. IFN-γ results in AHR activation, nuclear translocation, dimerization with ARNT, and transcriptional activation of its target gene and concomitantly decreases total nuclear HIF1α in HCAEC.
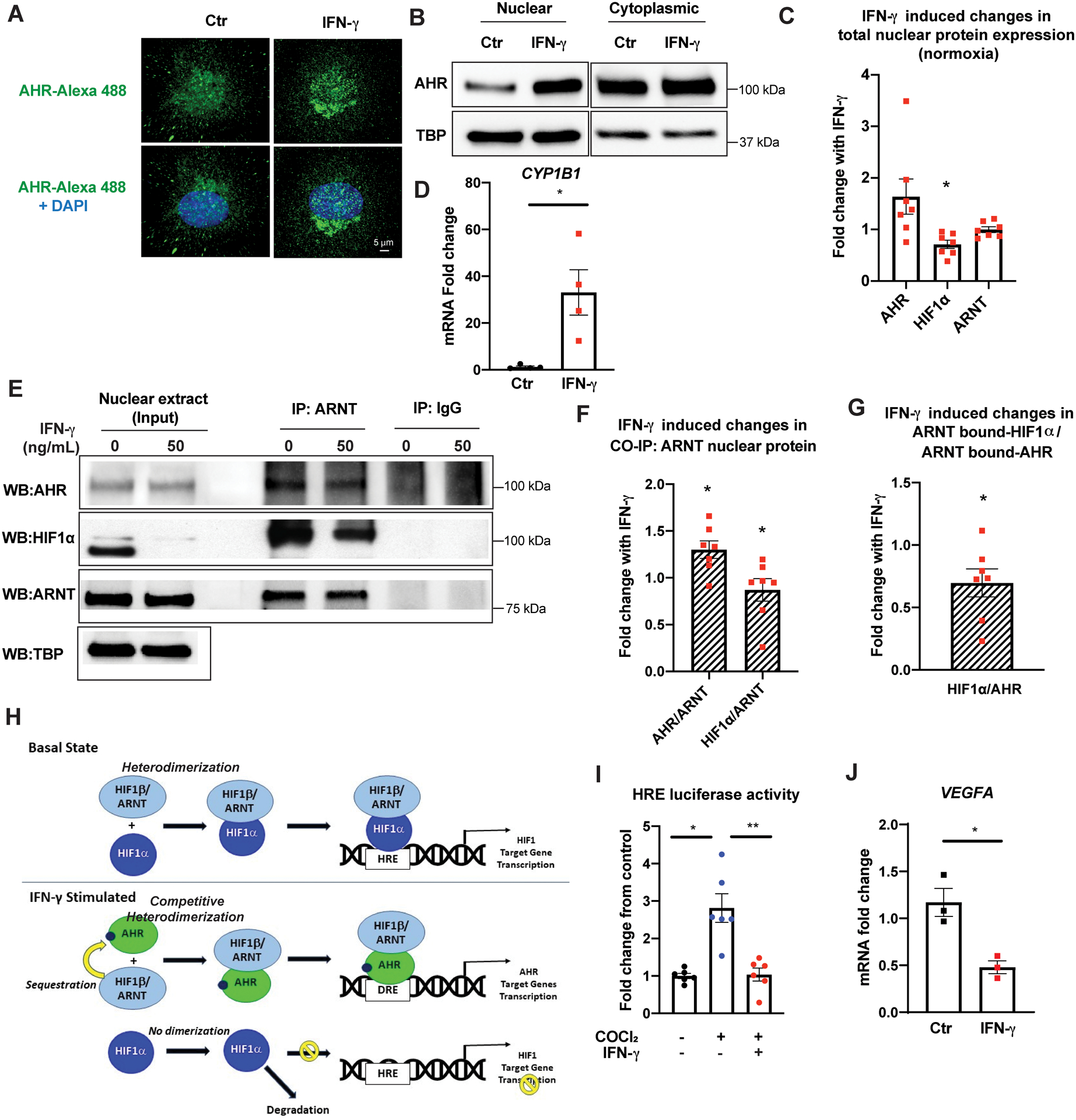
A, Confocal microscopy analysis of intracellular AHR (green: Alexa Flour 488) superimposed with DAPI nuclei as staining (blue) following 24 h of IFN-γ treatment at 50 ng/mL. Representative images.
B, Protein expression of AHR in the nuclear and cytoplasmic extracts following IFN-γ treatment. Representative Western blots.
C, IFN-γ-induced fold changes in total nuclear protein expression levels of AHR, HIF1α, and ARNT in HCAEC following 38 h of IFN-γ treatment in normoxia. Densitometric quantification of each protein level was normalized by loading control protein, TATA binding protein (TBP), before the IFN-γ-treated group was compared to the untreated control within each experiment. Mean ± SEM from 7 independent experiments. AHR: fold change (FC) 1.64; p= 0.11. HIF1α: FC 0.71; p= 0.012. ARNT: FC 1.0; p= 0.97.
D, Transcriptional activation of a canonical AHR target gene CYP1B1 by qPCR by IFN-γ. Mean ± SEM from 3 independent experiments.
E, Nuclear protein extracts of HCAEC were co-immunoprecipitated (CO-IP) with ARNT following IFN-γ treatment and probed with antibodies against AHR, HIF1α, and ARNT. The left panel represents protein detection in the 8% of the total unprocessed nuclear lysates (input) used for each CO-IP experiment. Representative Western blot.
F, IFN-γ-induced fold changes in nuclear AHR and HIF1α co-immunoprecipitated with ARNT in normoxia. Densitometric quantification of each protein was normalized by the co-immunoprecipitated ARNT level before the IFN-γ-treated group was compared to the untreated control within each experiment. Mean ± SEM from 7 independent experiments. IP ARNT: AHR FC 1.30; p = 0.018. IP ARNT: HIF1α: FC 0.86; p = 0.036.
G, Fold changes in the relative abundance of ARNT-bound nuclear HIF1α over ARNT-bound AHR in HCAEC following IFN-γ treatment over untreated control, p = 0.036. Mean ± SEM from 7 independent experiments.
H, Schematic summary of the proposed mechanism.
I, HRE luciferase activity after 48 h cobalt chloride (CoCl2) treatment with or without IFN-γ. HRE firefly luciferase activity was normalized by the cotransfected Renilla luciferase activity in each sample before quantifying the fold changes in the reporter activity from the untreated control group. Mean ± SEM from 3 independent experiments.
J, Transcriptional suppression of a canonical HIF1 target gene VEGFA by qPCR by IFN-γ. Mean ± SEM from 3 independent experiments.
Statistical significance was determined by one-sample t-test (C, F, G), Welch’s t-test (D), Welch’s ANOVA with post hoc Dunnett’s T3 multiple comparisons test (I), and unpaired two-tailed student’s t-test (J).
AHR: Aryl hydrocarbon receptor, ARNT: Aryl hydrocarbon receptor nuclear translocator, HIF1α: Hypoxia inducible factor 1 alpha, CYP1B1: Cytochrome P450 family 1 subfamily B Member 1, TBP: Tata binding protein, DRE: digoxin responsive element, HRE: hypoxia responsive element, VEGFA: vascular endothelial growth factor A.
* p <0.05, ** p < 0.01
IFN-γ exposure results in widespread suppression of HIF1 target gene expression in HCAEC
Downstream of IFN-γ-induced HIF1α destabilization in quiescent HCAEC in normoxia, we found global suppression of HIF1 target gene expression (Figure III in the Supplement; Table I in the Supplement). In addition to the significant decreases in the expression of genes encoding almost all of the glycolytic enzymes, other markedly suppressed genes involve broad aspects of endothelial cell survival and function, including cell growth and viability, with a 51% decrease in insulin-like growth factor (IGF2) and its binding protein (IGFBP) isotypes, as well as a 22% decrease in cyclin-dependent kinase inhibitor (CDKN1). Additionally, there were decreases in the expression of genes involved in vascular remodeling [transforming growth factor beta (TGFB)], vasomotor tone [adrenomedullin (ADM), adrenoceptor alpha 1B (ADRA1B)], thrombosis [serpin family E member 1 (SERPINE1)], and heme [(heme oxygenase 1 (HMOX1)] and iron metabolism [transferrin receptor (TFRC)].
IFN-γ-induced suppression of endothelial glycolytic genes is mediated by IDO1-AHR-HIF1 pathway interactions
In order to link further IFN-γ-induced activation of IDO1-kyneurenine-AHR pathways and concomitant HIF1α destabilization with IFN-γ-induced suppression of glycolytic enzyme gene expression, we carried out a series of targeted gene silencing and/or pharmacological inhibition studies specific to each pathway. Pharmacological inhibition of IDO1 by 1-methyltryptophan (1-MT) abrogated IFN-γ-induced suppression of SLC2A1 gene expression (Figure 4A) and partially attenuated suppression of LDHA (Figure 4B) in HCAEC. Similarly, gene silencing of AHR attenuated IFN-γ-induced suppression of LDHA gene expression (Figure 4C–D; Figure IV in the Supplement). Lastly, silencing of the von Hippel-Lindau tumor suppressor gene (VHL)20 successfully stabilized nuclear HIF1α protein expression in normoxia (Figure 4E) beyond that of the control siRNA-treated sample, and abrogated the IFN-γ-induced suppression of LDHA gene expression (Figure 4F–G; Figure IV in the Supplement; Table II–III in the Supplement).
Figure 4. IFN-γ-induced suppression of glycolytic gene expression is mediated by the interactions between the IDO1-AHR-HIF1 transcriptional regulatory pathways.
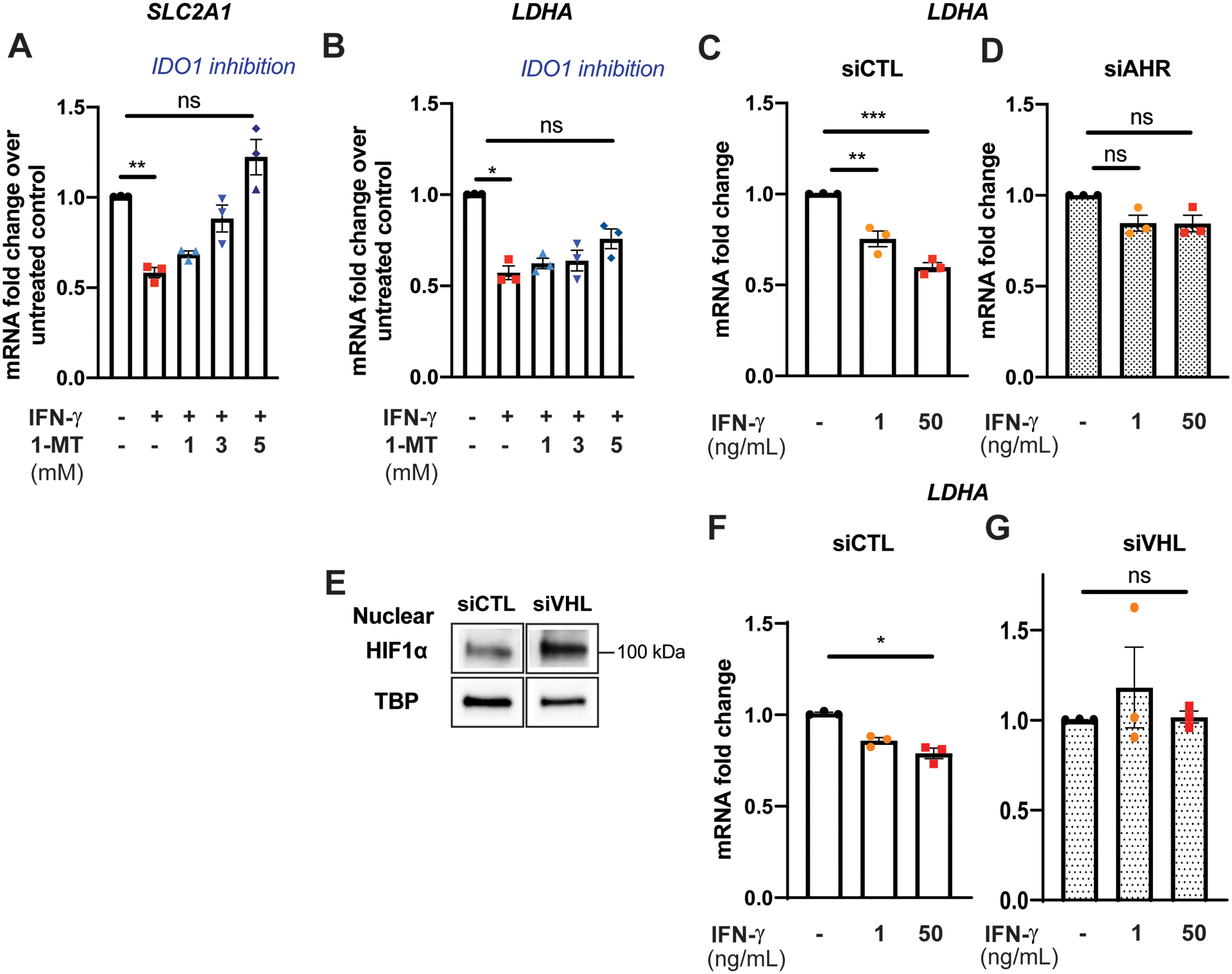
A-B, Pharmacological inhibition of IDO1 by 1-methyltryptophan (1-MT) abrogates IFN-γ induced suppression of SLC2A1 gene expression (A) and partially attenuates suppression of LDHA (B) in HCEAC. Data represent mean ± SEM from 3 independent experiments.
C-D, Gene silencing of AHR attenuates IFN-γ induced suppression of LDHA gene expression in HCEAC. Data represent mean mRNA fold changes with IFN-γ treatment compared to those not exposed to IFN-γ within each siRNA treatment group ± SEM from 3 independent experiments.
E, Gene silencing of von Hippel-Lindau tumor suppressor (VHL) stabilizes nuclear HIF1α protein expression in normoxia. TATA binding protein (TBP) was used as a loading control. Representative Western blot.
F-G, HIF1α stabilization in normoxia by gene silencing of VHL abrogates IFN-γ induced suppression of LDHA gene expression in HCAEC. Data represent mean mRNA fold changes with IFN-γ treatment compared to those not exposed to IFN-γ within each siRNA treatment group ± SEM from 3 independent experiments.
Statistical significance was determined by one-way ANOVA with post hoc Tukey’s multiple comparisons test (A-D) or Kruskal-Wallis test with post hoc Dunn’s multiple comparisons test (F-G).
*p < 0.05, ** p < 0.01, *** < 0.001
IFN-γ depletes basal intracellular NAD(H) in HCAEC
Nicotinamide adenine dinucleotide (NAD+) and its reduced form (NADH) are essential elements of cellular bioenergetics and redox homeostasis, serving not only as cofactors for glycolysis but also as important regulators of glycolysis and intracellular signaling [reviewed in21]. IFN-γ exposure resulted in a significant dose-dependent depletion of intracellular NAD(H) by 20 % (p = 0.0078) (without a significant change in the NAD+/NADH ratio) (Figure 5A–B). These findings were contrary to our initial expectation that the IFN-γ-induced intracellular kynurenine surge would promote de novo NAD+ synthesis and increase total intracellular NAD(H). Pretreatment of HCAEC with 3-aminobenzamide (3-AB), an inhibitor of poly (ADP-ribose) polymerase (PARP), completely prevented IFN-γ-induced depletion of intracellular NAD(H) (Figure 5A), suggesting such NAD+ depletion is largely driven by PARP activation. Of note, no significant differences in cell viability were noted between the IFN-γ-treated cells and untreated controls. Supplementation with the NAD+ precursor, nicotinamide mononucleotide (NMN), also effectively restored the intracellular NAD(H) level following IFN-γ exposure (Figure 5C).
Figure 5. IFN-γ exposure depletes basal intracellular NAD(H) via increased ADP-ribosylation in HCAEC.
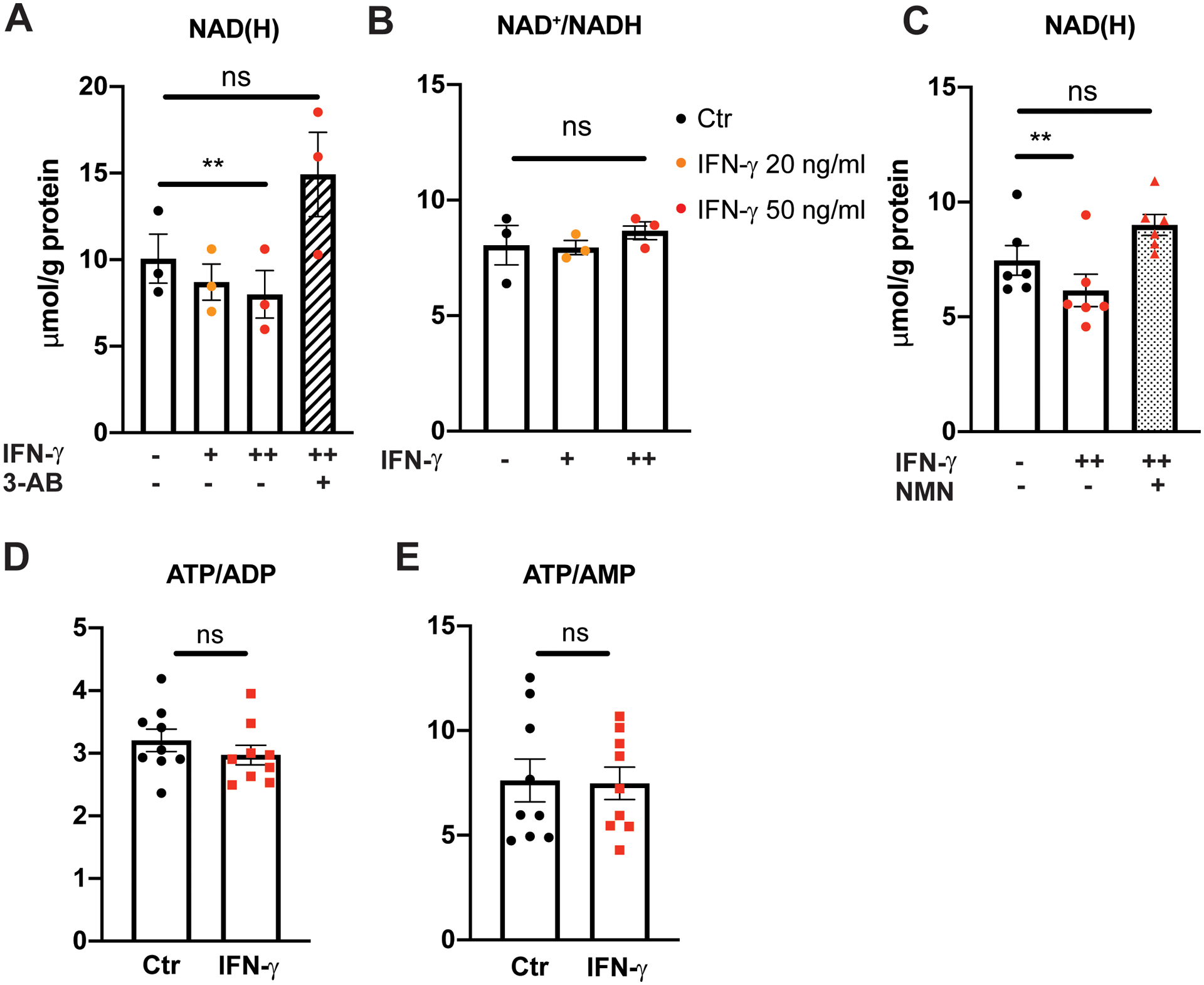
A, Intracellular NAD(H) (total NAD+ and NADH) in HCAEC following IFN-γ treatment for 24 h at 20 or 50 ng/mL detected by bioluminescent assay with or without 1 h pretreatment with a poly (ADP-ribose) polymerase inhibitor, 3 aminobenzamide (3-AB).
B, Intracellular NAD+/NADH ratio following the same IFN-γ treatments.
C, Intracellular NAD(H) in HCAEC following IFN-γ treatment for 24 h at 50 ng/mL with or without subsequent nicotinamide mononucleotide (NMN) supplementation at 0.5 mM for an additional 24 h. Data represent mean ± SEM from 3 independent experiments.
D-E, Intracellular ATP/ADP (D) and ATP/AMP (E) ratios in HCAEC following 24 h of IFN-γ treatment measured by HPLC. Data represent mean ± SEM from 9 independent experiments.
Statistical significance was assessed using one-way ANOVA with post hoc Tukey’s multiple comparisons (A-C) or an unpaired two-tailed Student’s t-test (D-E).
ADP: Adenosine diphosphate, ATP: Adenosine triphosphate, HPLC: High performance liquid chromatography, NAD(H): Nicotinamide adenine dinucleotide.
** p < 0.01
With marked impairment of glycolysis and NAD(H) depletion, we next hypothesized that total intracellular energy balance would be impaired. Interestingly, ATP/ADP and ATP/AMP ratios were unchanged with IFN-γ (Figure 5D–E), raising a question as to the nature of alternative metabolic pathways and energy source(s) that compensate for impaired glycolysis.
IFN-γ results in endothelial metabolic reprogramming to augment fatty acid oxidation
Indeed, despite the inhibition of glycolysis by IFN-γ resulting in a marked depletion of intracellular pyruvate (Figure 1I), a 23% increase in basal mitochondrial oxygen consumption rate (OCR) or oxidative phosphorylation (OXPHOS (defined as [basal OCR] – [OCR after antimycin A and rotenone injection]) was noted with IFN-γ (p = 0.0032) (Figure 6A). This finding was independent of the presence of serum or growth factor supplementation in the basal medium (Figure IB–C in the Supplement). Furthermore, there was a 30% increase in mitochondrial respiration associated with ATP production (ATP-linked OCR; defined as [basal OCR]-[OCR following oligomycin injection]) (p = 0.0021) (Figure 6B). Overall, there was a significant shift away from glycolysis to oxidative phosphorylation in basal endothelial metabolism following IFN-γ exposure as demonstrated by an increased basal mitochondrial OCR-to-ECAR ratio (Figure 6C) and a left- and upward-shift in the representative ECAR-OCR plot (Figure 6D). This finding raised the important issue of the identity of the alternative fuel source(s) that supplies the mitochondrial electron transport chain in the face of impaired glucose metabolism.
Figure 6. IFN-γ activates fatty acid oxidation in HCAEC.
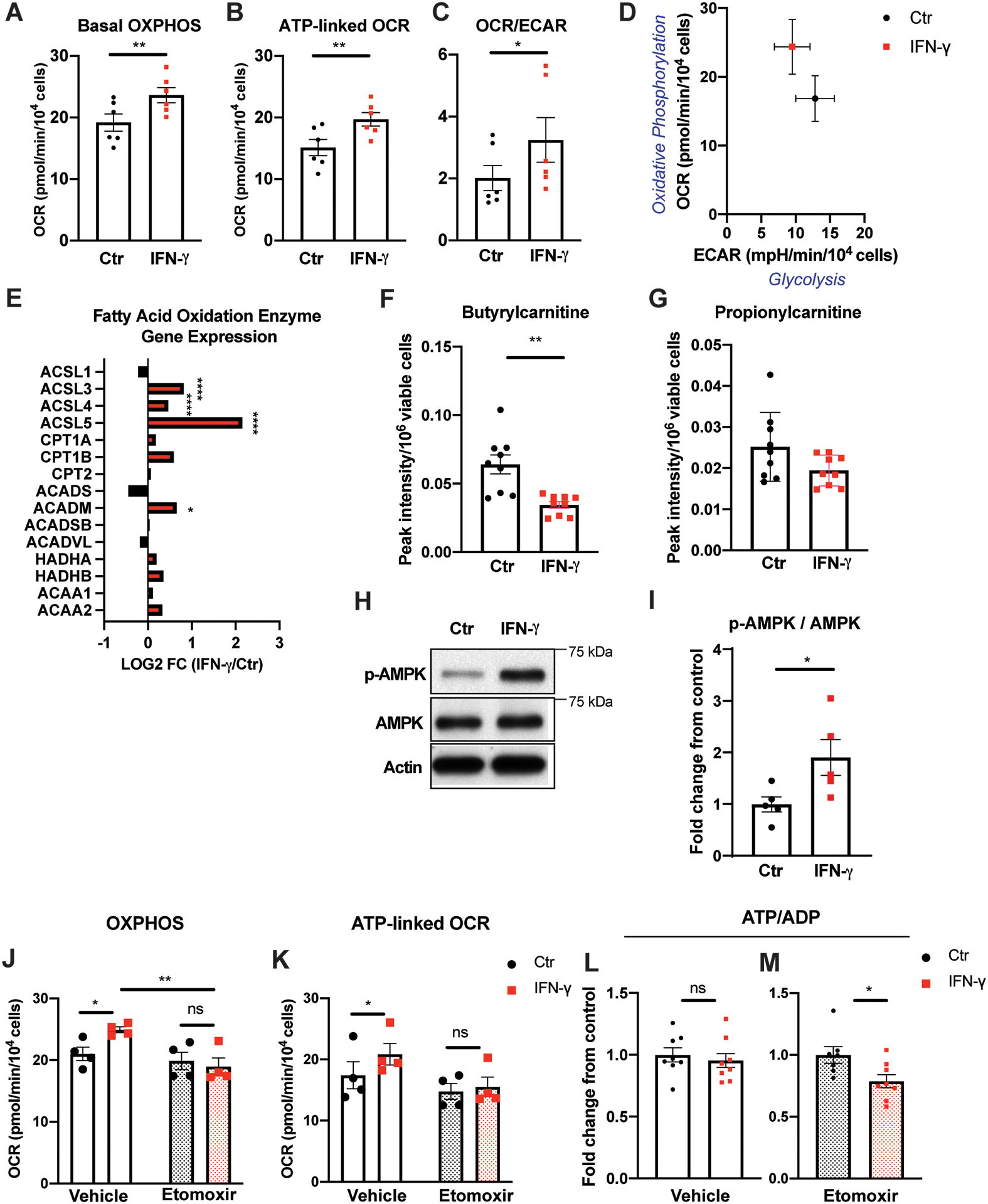
A, Real time extracellular flux analysis of mitochondrial oxygen consumption rate reflecting oxidative phosphorylation (OXPHOS) of HCAEC following IFN-γ treatment at 50 ng/ml for 24 h.
B, Mitochondrial oxygen consumption associated with ATP production from the same experiments.
C, IFN-γ-induced changes in the ratio between mitochondrial OCR and ECAR representing decreased glycolysis and increased OXPHOS in HCAEC. Mean ± SEM from 6 independent experiments (A-C). Statistical significance was assessed using paired two-tailed Student’s t-tests (A-B) or Wilcoxon matched-pairs signed rank test (C).
D, Representative plot demonstrating altered basal glycolysis and oxidative phosphorylation activities in HCAEC following the same IFN-γ treatment in a single experiment. Mean ± SEM from 9–10 biological replicates.
E, IFN-γ induces gene expression changes in the fatty acid oxidation enzymes by RNASeq analysis of HCAEC obtained in biological triplicates. Statistical significance was determined based on the p-values adjusted for multiple test hypotheses by Benjamini-Hochberg procedure.
F-G, Intracellular acylcarnitines (butyrylcarnitine (F) and propionylcarnitine (G)) in HCAEC measured by LC-MS following the same IFN-γ treatment. Mean ± SEM of 9 biological replicates from 3 independent experiments. Statistical significance was assessed using Welch’s t-test.
H-I, Phosphorylation of AMP-activated protein kinase (AMPK) at Thr 172 following 7 h of IFN-γ treatment at 50 ng/mL. Representative Western blot (H) and densitometric analysis (I). Mean ± SEM from 3 independent experiments analyzed by two-tailed Student’s t-test.
J, Real time extracellular flux analysis of basal OXPHOS of HCAEC following the same IFN-γ treatment with and without 1 h 10 μM etomoxir incubation just prior to analysis.
K, Mitochondrial oxygen consumption associated with ATP production from the same experiments. Mean ± SEM from 4 independent experiments (J-K). Statistical significance was assessed by 2-way ANOVA with Sidak’s multiple comparison tests.
L-M, Intracellular ATP/ADP ratios detected in HCAEC following the IFN-γ treatment with (M) and without (L) etomoxir. Data represent mean fold changes with IFN-γ treatment compared to those not exposed to IFN-γ within each inhibitor treatment group ± SEM from 7–9 independent experiments. Statistical significance was assessed using a two-tailed Student’s t-test.
*p < 0.05, ** p < 0.01, **** p < 0.0001
ACAA: Acetyl-CoA acyltransferase, ACADM: Acyl-CoA dehydrogenase medium chain, ACADS: Acyl-CoA dehydrogenase short chain, ACADSB: Acyl-CoA dehydrogenase short/branched chain, ACADVL: Acyl-CoA dehydrogenase very long chain, ACSL: Acyl-CoA synthetase long chain family member, CPT: Carnitine palmitoyltransferase, HADH: Hydroxyacyl-CoA dehydrogenase trifunctional multienzyme complex.
In contrast to the global transcriptional suppression of glycolytic enzymes by IFN-γ (Figure 1N), the gene expression levels of multiple key enzymes regulating fatty acid oxidation (FAO) were significantly elevated in HCAEC treated with IFN-γ (Figure 6E). These included up to a 450% increase in acyl-CoA synthetase long chain family members (ACSL) expression as well as a 57% increase in acyl-CoA dehydrogenase medium chain (ACADM) expression. In order for an activated cytosolic long chain fatty acid to cross the mitochondrial inner membrane for oxidation, transfer of its acyl moiety to carnitine (acyl-carnitine) by carnitine palmitoyl transferase I (CPT1) is required. Once in the mitochondrial matrix, the acyl group of the fatty acid is transferred to CoA (fatty acyl-CoA), which is subsequently oxidized to generate ATP. There was a 47% decrease in intracellular butyrylcarnitine, an acyl-carnitine, in IFN-γ-treated HCAEC as well as a similarly decreasing trend in propionylcarnitine (Figure 6F–G). Together with the transcriptional activation of multiple FAO enzymes, such a decrease in precursors to fatty acyl-CoAs was consistent with increased FAO in IFN-γ-treated HCAEC. This metabolic shift from glycolysis to FAO was accompanied by earlier activation of AMP-activated protein kinase (AMPK) by IFN-γ (Figure 6H–I).
Upon pharmacologic inhibition of CPT1 (etomoxir), the rate limiting FAO enzyme, IFN-γ-treated HCAEC could no longer augment basal OXPHOS (Figure 6J) or ATP-linked mitochondrial OCR (Figure 6K). Similarly, gene silencing of CPT1A attenuated the IFN-γ-induced increase in OXPHOS (Figure VA–C in the Supplement).
Furthermore, inhibition of FAO disrupted intracellular energy balance in IFN-γ-treated cells as noted by over 20% reduction in the ATP/ADP ratio (Figure 6L–M), supporting the novel role of FAO in maintaining endothelial intracellular energy balance in the face of IFN-γ-induced impaired glucose metabolism. Of note, no transcriptional activation of the enzymes involved in glutaminolysis or significant changes in intracellular glutamine or glutamate levels were observed with IFN-γ treatment (Figure VD–F in the Supplement), eliminating this amino acid pathway from consideration as an alternate energy source.
IFN-γ exposure depletes basal endothelial cyclic guanosine 5ʹ-monophosphate (cGMP) and impairs endothelial proliferative and migratory capacity
Metabolic derangements induced by IFN-γ treatment were accompanied by unfavorable phenotypic changes in HCAEC. Endothelial dysfunction in association with cGMP-nitric oxide (NO) insufficiency has been well established as an important correlate of heightened cardiovascular risk22 and deemed a precursor to coronary artery atherogenesis23. Twenty-four h of IFN-γ exposure at 50 ng/mL resulted in a 29 % reduction in basal intracellular cGMP in HCAEC (p = 0.0247) (Figure 7A–B). This reduction was observed despite the increased phosphorylation of the serine 1177 residue of endothelial nitric oxide synthetase (eNOS) (Figure VIA–D in the Supplement) consistent with recent examples of discordance between eNOS phosphorylation in human umbilical vein endothelial cells (HUVEC) and nitric oxide formation24. Total eNOS protein expression did not change (Figure VIE–F in the Supplement).
Figure 7. IFN-γ exposure depletes endothelial basal intracellular cGMP and impairs proliferation and migratory capacity.
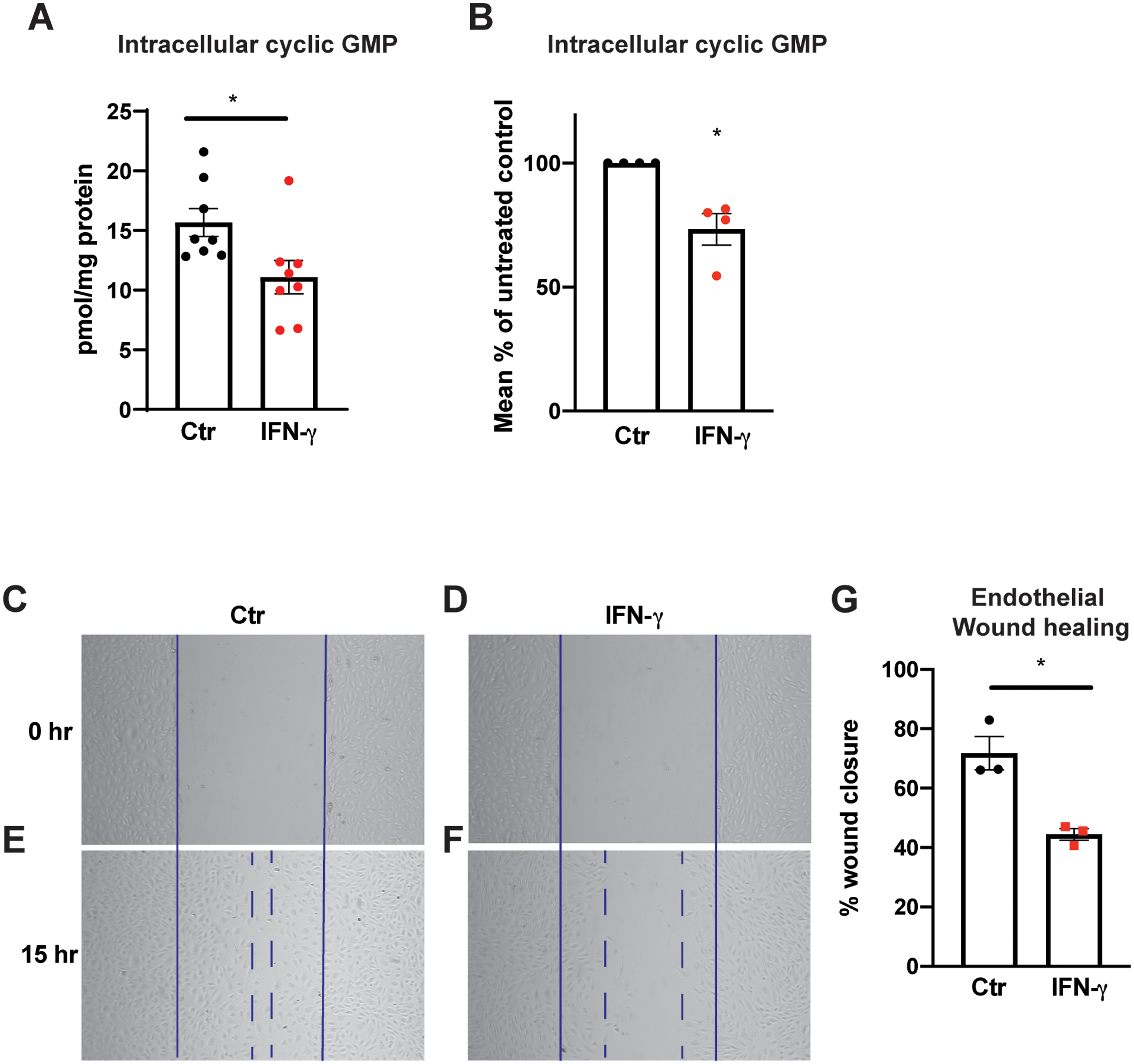
A-B, Intracellular cGMP levels from HCAEC quantified using ELISA following 25 h of IFN-γ treatment at 50 ng/mL. Absolute intracellular cGMP measurements normalized to total protein/plate (pmol/mg protein) (A) and relative intracellular cGMP levels expressed as the percentages of those of untreated control groups (B). Mean ± SEM from 8 biological replicates from 4 independent experiments.
C-F, HCAEC proliferation and migration capacities were assessed by scratch assay after IFN-γ exposure for 30 h. Representative bright field images are shown at 0 h (C-D) and 15 h the scratch (E-F).
G, Percentages of wound closure or monolayer re-endothelialization following IFN-γ treatment were calculated by the following formula = [(open area at 0 h) - (open area at 15 h)]/(open area at 0 h) × 100. Mean ± SEM from 3 independent experiments.
Statistical significance was assessed using a two-tailed Student’s t-test (A,G) or one-sample t-test (B).
* p < 0.05
Similar to the effect of tryptophan depletion (Figure 2I; Figure II in the Supplement), 15 h of IFN-γ exposure resulted in impaired healing of an endothelial monolayer following mechanical (scratch) injury, a process dependent on endothelial proliferative and migratory function (p = 0.0098) (Figure 7C–G), and consistent with prior reports of the anti-angiogenic effect of IFN-γ25.
DISCUSSION
In the present study, IFN-γ impaired coronary artery endothelial glucose metabolism by glycolysis inhibition--via accelerated tryptophan catabolism--and NAD(H) depletion while activating fatty acid oxidation. Without FAO upregulation, IFN-γ-exposed-HCAEC suffered a significant intracellular energy deficit, which highlights the novel role of endothelial FAO in preserving overall intracellular energy balance in the face of T-cell-mediated endothelial metabolic derangements induced by this cytokine. These metabolic changes were associated with pathological endothelial phenotypic changes, including intracellular cyclic GMP depletion and impaired migration and proliferation. To the best of our knowledge, this is the first report of the metabolic shift from glucose utilization to FAO in any cell type induced by IFN-γ, a major cytokine in human atheroma as well as in anti-viral, anti-tumor, and autoimmune milieux.
Studies have established glycolysis as essential for endothelial survival, proliferation, and angiogenesis6, 26. While studies linking impaired glycolysis with vascular pathologies centered on the inhibition or insufficiency of specific glycolytic regulators, especially PFKFB35, 6, IFN-γ treatment of quiescent HCAEC in normoxia in the present study resulted in a far more extensive (global) suppression of nearly all glycolytic enzymes at the transcriptional level with a correlative decline in associated intracellular metabolites. Suppression of glycolysis by IFN-γ was specific to endothelial cells, in striking contrast to their neighboring macrophages in atheroma in which IFN-γ promotes glycolysis and a switch to a pro-inflammatory phenotype27. To the best of our knowledge, the only other biological context wherein IFN-γ resulted in a similar directional change in glycolysis as in endothelial cells involves Chlamydia trachomatis-infected epithelial cell lines28.
The role of HIF as a major regulator of endothelial function and metabolism has been established first in hypoxia29–31 with now growing appreciation of its role in normoxia32–34. The biological consequences of IFN-γ-induced loss of tonic HIF activity in diseased and non-diseased endothelial cells are likely system-wide—well beyond its implications in atherobiology35–37--either as a direct sequela of disrupted glucose metabolism or via impaired transcriptional activation of the multitude of genes governing important aspects of endothelial function (Figure III in the Supplement). It is also important to note that endothelial glucose metabolism is regulated by closely intertwined networks of numerous transcription factors and signaling pathways in addition to HIF, including, but not limited to, MYC, mammalian targets of rapamycin (mTOR), AMPK, Kruppel-like factors (KLF), and their interactions1.
Maintenance of intracellular NAD(H) balance is tightly regulated and indispensable for cellular bioenergetics. Despite robust activation of the rate-limiting step for de novo NAD+ synthesis, IFN-γ resulted in a net 20 % depletion in total intracellular NAD(H) driven largely by PARP activation by IFN-γ similar to that observed in cancer cell lines38 and peripheral blood mononuclear cells39. While IFN-γ-induced cellular apoptosis has been reported in various biological contexts, the exact mechanism by which IFN-γ induces PARP activation remains incompletely understood. The consequences of NAD+ depletion in relation to glucose metabolism are several-fold. In addition to creating cofactor deficiency for multiple glycolytic enzymes, NAD+ depletion alone can result in PARP activation40, which, in turn, can inhibit glycolysis further independent of NAD+ via PARPylation of HK141, 42. In the present study, pretreatment of HCAECs with a PARP inhibitor blocked IFN-γ-mediated NAD+ depletion, suggesting that the increased PARP activity was driven by IFN-γ first and not a mere consequence of the NAD+ depletion that ensued. The current study highlights a PARP-mediated degradation mechanism, but it is worth noting that total intracellular NAD(H) reflects the summation and complex cross-regulation of major NAD+ biosynthesis pathways and degradation pathways [reviewed in21] as demonstrated by our ability to restore intracellular NAD(H) by NMN following IFN-γ exposure.
Unexpectedly, conserved intracellular ATP/ADP balance was found in this study despite a substantial impairment of glycolysis and NAD(H) loss following IFN-γ-exposure. Further investigation of compensatory metabolic pathways led to the identification of novel effects of IFN-γ on endothelial FAO and a role for fatty acids in fueling ATP-coupled mitochondrial respiration. While IFN-γ had been noted to promote adipocyte lipolysis43 and modulate fatty acid incorporation in monocyte and macrophage membrane phospholipids44, its role in promoting FAO as an alternative fuel source has not been previously described.
Endothelial FAO had been known mostly for non-exergonic roles, such as cell fate45, proliferation and vessel sprouting46–48, vascular permeability48, nucleotide synthesis47, and redox homeostasis49. Undiminished intracellular ATP/ADP despite FAO inhibition in basal quiescent HCAEC in the present study (in the absence of IFN-γ) is consistent with the current understanding that FAO provides minimal contribution to basal endothelial energy balance49. More than 20% loss of the intracellular ATP/ADP resulting from acute CPT1 inhibition in IFN-γ-exposed HCAEC, however, signifies that IFN-γ-augmented endothelial FAO’s contributes to overall cellular bioenergetics in the face of disrupted glucose utilization. Complete50 or partial51 extracellular glucose deprivation in culture media had been shown to increase FAO in HUVEC or bovine aortic ECs, respectively. Our observation is especially intriguing because the endothelial metabolic shift from glycolysis to FAO occurred without a glucose deficit, being purely driven by IFN-γ. We propose IFN-γ creates a relative hypoglycemic or starvation state in the intracellular compartment in HCAEC via inhibition of glucose uptake and glycolysis, requiring that the cell resort to an alternative fuel source even with abundant extracellular glucose available. The AMPK activation we observed in these cells likely contributes to facilitating this metabolic shift. The extent of intracellular ATP/ADP (21%) balance contributed by FAO in IFN-γ-treated HCAECs closely matched the 20% decrease in glycolytic activity resulting from IFN-γ. This finding illustrates the tightly regulated, yet highly fluid, crosstalk between multiple metabolic pathways coexisting within a single cell type where perturbation in one pathway has widespread impact on others (Figure 8). The complex interplay between glycolysis and FAO has been well described in other tissues and cell types (e.g., the Randle cycle)52, but remains incompletely characterized in general and not previously studied in endothelial cells in particular.
Figure 8.
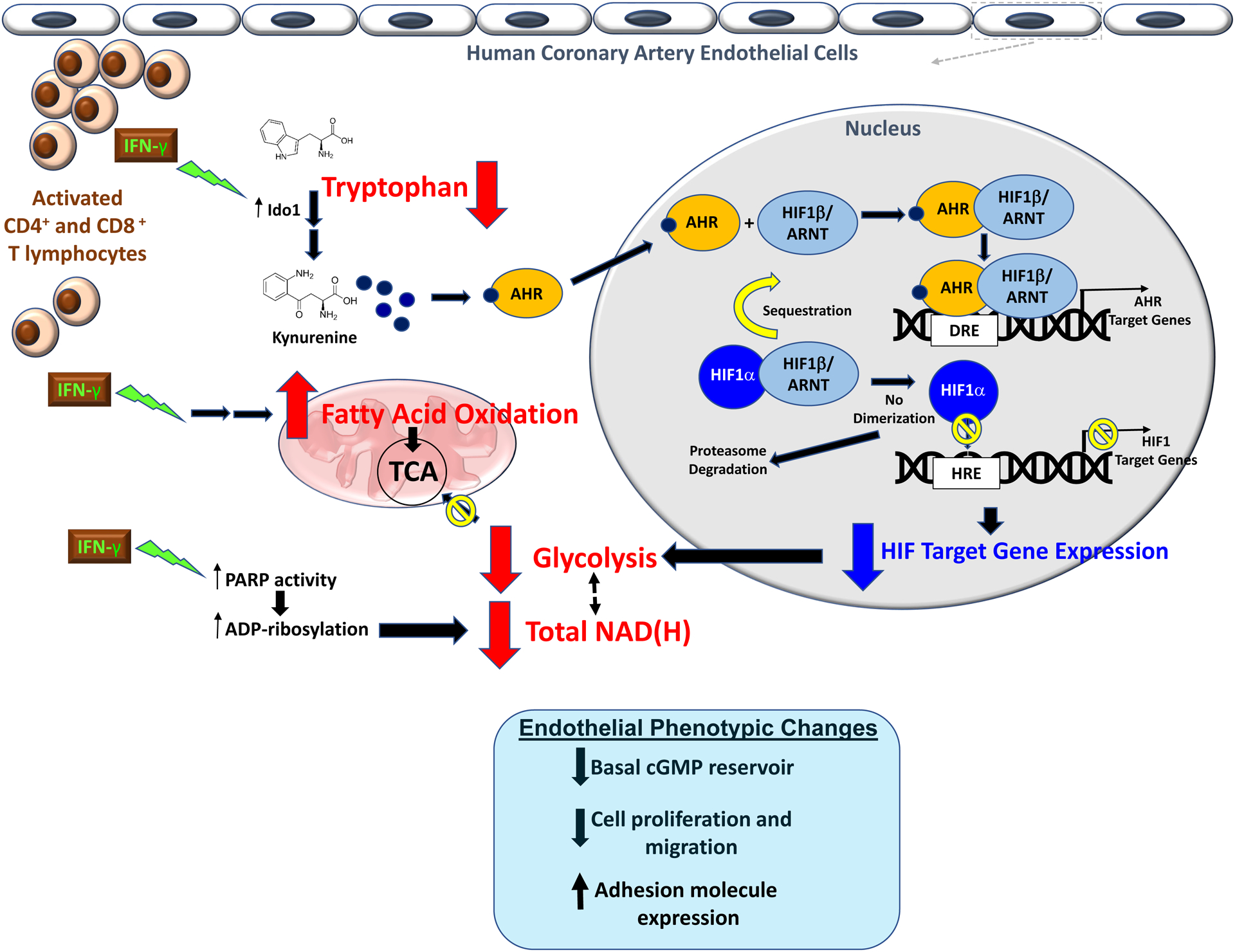
Proposed mechanisms for the widespread IFN-γ-induced metabolic derangements in human coronary artery endothelial cells.
The potential clinical implications of endothelial metabolic derangements from chronic IFN-γ exposure in atheroma rich in activated T-lymphocytes in ischemic heart disease are paramount. NAD+ depletion in other cell types has been demonstrated to result in necrotic cell death53. Impaired glycolysis and suppression of growth factor signaling likely inhibit collateral vessel formation as demonstrated by the significantly impaired endothelial wound healing capacity with IFN-γ. With local ischemia in the setting of hemodynamically significant coronary artery luminal stenosis, impaired HIF stabilization by IFN-γ-mediated AHR activation would compromise essential cellular adaptive responses to hypoxia, while chronically increased endothelial oxygen consumption in the intima may exacerbate myocardial ischemia by oxygen “steal” during the delivery of an already limited oxygen supply. In addition to the adverse phenotypic consequences of the above metabolic pathways, we report for the first time that tryptophan depletion similarly impairs the endothelial migratory capacity in vitro while switching HCAEC to a proinflammatory phenotype with upregulation of adhesion molecule expression as demonstrated in aortic endothelial cells54.
Two prior studies involving different human venous endothelial cell sources (HUVEC) have reported contrasting directional changes in intracellular cGMP levels in response to IFN-γ55, 56. By contrast, our results in arterial endothelial cells (HCAEC) showing that IFN-γ induces a reduction in basal cGMP corroborates and provides mechanistic insight into the over three-decades-old seminal observation linking decreased cGMP bioavailability to atheroma-laden coronary arteries57. More importantly, this significant loss of basal endothelial cGMP likely results in a system-wide disruption of key aspects of vascular homeostasis. When combined with the sequelae of the concomitant loss of tonic HIF activity and deranged metabolic balance demonstrated in this study, IFN-γ-induced endothelial cGMP depletion likely has important clinical consequences especially in the context of atherogenesis and its thrombotic and ischemic complications. Chronically decreased NO-cGMP bioavailability has been clearly associated with an increased risk for atherothrombotic diseases and adverse clinical outcomes22, 58.
In summary, IFN-γ impairs basal glycolysis of HCAEC via activation of the IDO-kynurenine-AHR pathway resulting in decreased HIF1 activity and depletes intracellular NAD+ while augmenting fatty acid oxidation as an alternative fuel source (Figure 8). We propose such metabolic and phenotypic derangements resulting from pathologic T-lymphocyte-endothelial interaction as a fundamental mechanism linking adaptive immunity and vascular pathobiology.
Supplementary Material
CLINICAL PERSPECTIVE.
What is new?
Interferon-gamma (IFN-γ), a major cytokine in human atheroma secreted by CD8+ and CD4+ T-lymphocytes, impairs glucose metabolism of primary human coronary artery endothelial cells by inhibiting glycolysis and depleting nicotinamide adenine dinucleotide (NAD(H)).
Glycolysis inhibition by IFN-γ was mediated by hypoxia inducible factor 1α (HIF1α) destabilization resulting from tryptophan catabolism to kynurenine with downstream sequestration of HIF1β by aryl hydrocarbon receptor and was offset by increased fatty acid oxidation to preserve cellular energy balance.
IFN-γ-induced metabolic derangements were accompanied by adverse endothelial phenotypic changes, including intracellular cyclic GMP depletion, impaired migratory capacity, and increased adhesion molecule expression.
What are the clinical implications?
Endothelial cells depend on glycolysis for energy, angiogenesis, and survival, and its impairment by IFN-γ in atheroma-laden coronary artery endothelium likely accelerates atherosclerosis and impairs collateral vessel formation.
Our metabolomic and transcriptomic analyses demonstrate IFN-γ induces widespread perturbations across a network of metabolic pathways (glycolysis, NAD(H), tryptophan, and fatty acid metabolism) and regulatory and signaling pathways (HIF1, AMPK, NO-cGMP) essential for vascular homeostasis and function, highlighting the fluid crosstalk between, and cross-regulation of, multiple endothelial metabolic pathways in CAD.
This study provides a novel pathobiological mechanism linking T-cell activation and CAD through endothelial metabolic derangements by IFN-γ.
ACKNOWLEDGEMENTS
We thank Stephanie C. Tribuna for expert technical assistance with the manuscript preparation. We thank Drs. Jane Leopold and Wusheng Xiao for sharing their expertise in endothelial biology and Dr. Andriy Samokhin for sharing his expertise in confocal microscopy. We thank Dr. Andrew Lichtman for the valuable discussions of T cell biology. We also thank Dr. Zachery Herbert at the Molecular Biology Core Facility, Dana-Farber Cancer Institute, and Dr. Grigoriy Losyev at the BWH Flow Cytometry Core Facility.
L. Lee designed the study, performed all experiments, analyzed all data, and wrote the manuscript. W. Oldham provided expertise in metabolomics and performed LC-MS measurements of metabolites. H. He provided expertise in metabolomics and performed HPLC measurements of metabolites. R. Wang analyzed the RNA sequencing study data and provided expert statistical analysis. R. Mulhern assisted with experiments. D. Handy provided critical advice during study design and experiments. J. Loscalzo designed and supervised the overall study and manuscript preparation. All authors contributed to final approval of the manuscript.
SOURCES OF FUNDING
This study was supported, in part by the National Institutes of Health (NIH) grant T32 HL760434 to LYL; NIH grant HL128802 to WMO; and NIH grants HG007690, HL108630, HL119145, HL155096, and HL155107, and American Heart Association grants D700382 and CV-19 to JL.
Non-standard Abbreviations and Acronyms
- 3-AB
3-aminobenzamide
- ACADM
acyl-CoA dehydrogenase medium chain
- ACSL
acyl-CoA synthetase long chain family members
- ADM
adrenomedullin
- ADP
adenosine diphosphate
- ADRA1B
adrenoceptor alpha 1B
- AMPK
adenosine monophosphate-activated protein kinase
- ATP
adenosine triphosphate
- AHR
aryl hydrocarbon receptor
- ARNT
aryl hydrocarbon receptor nuclear translocator
- CDKN1
cyclin-dependent kinase inhibitor
- cGMP
cyclic guanosine 5ʹ-monophosphate
- CPT1
carnitine palmitoyl transferase I
- CYP1B1
cytochrome P450 family 1 subfamily B member 1
- DMEM
Dulbecco’s Modified Eagle Medium
- DRE
dioxin response element
- ECAR
extracellular acidification rate
- eNOS
endothelial nitric oxide synthetase
- FAO
fatty acid oxidation
- FLT1
fms-related receptor tyrosine kinase
- HCAEC
human coronary artery endothelial cell
- HIF1
hypoxia inducible factor 1
- HMOX1
heme oxygenase 1
- HUVEC
human umbilical vein endothelial cell
- ICAM
intercellular adhesion molecule
- IDO1
indoleamine-2, 3, -dioxygenase 1
- IFN-γ
Interferon-gamma
- IGF2
insulin-like growth factor 1
- IGFBP
insulin-like growth factor binding protein
- KLF
Kruppel like factors
- LC-MS
liquid chromatography mass spectrometry
- LDHA
lactate dehydrogenase A
- 1-MT
1-methyltryptophan
- MYC
MYC Proto-Oncogene, BHLH Transcription Factor
- mTOR
mammalian targets of rapamycin
- NAD(H)
nicotinamide adenine dinucleotide
- 2-NBDG
2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose
- NMN
nicotinamide mononucleotide
- NO
nitric oxide
- OCR
oxygen consumption rate
- OXPHOS
oxidative phosphorylation
- PARP
poly (ADP-ribose) polymerase
- PFKFB
gene encoding 6-Phosphofructo-2-Kinase/Fructose-2,6-Biphosphatase
- SERPINE1
serpin family E member 1
- SLC2A1
solute carrier family 2 member 1
- TBP
TATA-box binding protein
- TCA
tricarboxylic acid cycle
- TFRC
transferrin receptor
- TGFB
transforming growth factor beta
- VCAM
vascular cell adhesion molecule
- VHL
von Hippel-Lindau tumor suppressor
- XRE
xenobiotic response element
Footnotes
DISCLOSURES
There is nothing to disclose.
SUPPLEMENTAL MATERIAL
REFERENCES
- 1.Li X, Sun X and Carmeliet P. Hallmarks of Endothelial Cell Metabolism in Health and Disease. Cell Metabolism. 2019;30:414–433. [DOI] [PubMed] [Google Scholar]
- 2.Krützfeldt A, Spahr R, Mertens S, Siegmund B and Piper HM. Metabolism of exogenous substrates by coronary endothelial cells in culture. J Mol Cell Cardiol. 1990;22:1393–404. [DOI] [PubMed] [Google Scholar]
- 3.Culic O, Gruwel ML and Schrader J. Energy turnover of vascular endothelial cells. Am J Physiol. 1997;273:C205–13. [DOI] [PubMed] [Google Scholar]
- 4.Yang Q, Xu J, Ma Q, Liu Z, Sudhahar V, Cao Y, Wang L, Zeng X, Zhou Y, Zhang M, et al. PRKAA1/AMPKα1-driven glycolysis in endothelial cells exposed to disturbed flow protects against atherosclerosis. Nature Communications. 2018;9:4667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cao Y, Zhang X, Wang L, Yang Q, Ma Q, Xu J, Wang J, Kovacs L, Ayon RJ, Liu Z, et al. PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc Natl Acad Sci U S A. 2019;116:13394–13403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Bock K, Georgiadou M, Schoors S, Kuchnio A, Wong Brian W, Cantelmo Anna R, Quaegebeur A, Ghesquière B, Cauwenberghs S, Eelen G, et al. Role of PFKFB3-Driven Glycolysis in Vessel Sprouting. Cell. 2013;154:651–663. [DOI] [PubMed] [Google Scholar]
- 7.Fernandez DM, Rahman AH, Fernandez NF, Chudnovskiy A, Amir E-aD, Amadori L, Khan NS, Wong CK, Shamailova R, Hill CA, et al. Single-cell immune landscape of human atherosclerotic plaques. Nature Medicine. 2019;25:1576–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tellides G, Tereb DA, Kirkiles-Smith NC, Kim RW, Wilson JH, Schechner JS, Lorber MI and Pober JS. Interferon-gamma elicits arteriosclerosis in the absence of leukocytes. Nature. 2000;403:207–11. [DOI] [PubMed] [Google Scholar]
- 9.Buono C, Come CE, Stavrakis G, Maguire GF, Connelly PW and Lichtman AH. Influence of interferon-gamma on the extent and phenotype of diet-induced atherosclerosis in the LDLR-deficient mouse. Arterioscler Thromb Vasc Biol. 2003;23:454–60. [DOI] [PubMed] [Google Scholar]
- 10.Zohlnhofer D, Richter T, Neumann F, Nuhrenberg T, Wessely R, Brandl R, Murr A, Klein CA and Baeuerle PA. Transcriptome analysis reveals a role of interferon-gamma in human neointima formation. Mol Cell. 2001;7:1059–69. [DOI] [PubMed] [Google Scholar]
- 11.Tellides G and Pober JS. Interferon-gamma; Axis in Graft Arteriosclerosis. Circulation Research. 2007;100:622–632. [DOI] [PubMed] [Google Scholar]
- 12.Sakash JB, Byrne GI, Lichtman A and Libby P. Cytokines induce indoleamine 2,3-dioxygenase expression in human atheroma-asociated cells: implications for persistent Chlamydophila pneumoniae infection. Infect Immun. 2002;70:3959–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lahdou I, Engler C, Mehrle S, Daniel V, Sadeghi M, Opelz G and Terness P. Role of Human Corneal Endothelial Cells in T-Cell–Mediated Alloimmune Attack In Vitro. Investigative Ophthalmology & Visual Science. 2014;55:1213–1221. [DOI] [PubMed] [Google Scholar]
- 14.Liu R, Merola J, Manes TD, Qin L, Tietjen GT, Lopez-Giraldez F, Broecker V, Fang C, Xie C, Chen PM, et al. Interferon-gamma converts human microvascular pericytes into negative regulators of alloimmunity through induction of indoleamine 2,3-dioxygenase 1. JCI Insight. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller M, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203. [DOI] [PubMed] [Google Scholar]
- 16.Hankinson O The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol. 1995;35:307–40. [DOI] [PubMed] [Google Scholar]
- 17.Wang GL and Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–7. [DOI] [PubMed] [Google Scholar]
- 18.Wood SM, Gleadle JM, Pugh CW, Hankinson O and Ratcliffe PJ. The role of the aryl hydrocarbon receptor nuclear translocator (ARNT) in hypoxic induction of gene expression. Studies in ARNT-deficient cells. J Biol Chem. 1996;271:15117–23. [DOI] [PubMed] [Google Scholar]
- 19.Jacob A, Potin S, Saubaméa B, Crete D, Scherrmann JM, Curis E, Peyssonnaux C and Declèves X. Hypoxia interferes with aryl hydrocarbon receptor pathway in hCMEC/D3 human cerebral microvascular endothelial cells. J Neurochem. 2015;132:373–83. [DOI] [PubMed] [Google Scholar]
- 20.Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V and Kaelin WG. Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol. 2000;2:423–7. [DOI] [PubMed] [Google Scholar]
- 21.Xiao W, Wang RS, Handy DE and Loscalzo J. NAD(H) and NADP(H) Redox Couples and Cellular Energy Metabolism. Antioxid Redox Signal. 2018;28:251–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vita JA, Treasure CB, Nabel EG, McLenachan JM, Fish RD, Yeung AC, Vekshtein VI, Selwyn AP and Ganz P. Coronary vasomotor response to acetylcholine relates to risk factors for coronary artery disease. Circulation. 1990;81:491–7. [DOI] [PubMed] [Google Scholar]
- 23.Loscalzo J Nitric Oxide Signaling and Atherothrombosis Redux: Evidence From Experiments of Nature and Implications for Therapy. Circulation. 2018;137:233–236. [DOI] [PubMed] [Google Scholar]
- 24.Eroglu E, Saravi SSS, Sorrentino A, Steinhorn B and Michel T. Discordance between eNOS phosphorylation and activation revealed by multispectral imaging and chemogenetic methods. Proceedings of the National Academy of Sciences. 2019;116:20210–20217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friesel R, Komoriya A and Maciag T. Inhibition of endothelial cell proliferation by gamma-interferon. J Cell Biol. 1987;104:689–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Merchan JR, Kovacs K, Railsback JW, Kurtoglu M, Jing Y, Pina Y, Gao N, Murray TG, Lehrman MA and Lampidis TJ. Antiangiogenic activity of 2-deoxy-D-glucose. PLoS One. 2010;5:e13699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang F, Zhang S, Jeon R, Vuckovic I, Jiang X, Lerman A, Folmes CD, Dzeja PD and Herrmann J. Interferon Gamma Induces Reversible Metabolic Reprogramming of M1 Macrophages to Sustain Cell Viability and Pro-Inflammatory Activity. EBioMedicine. 2018;30:303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shima K, Kaeding N, Ogunsulire IM, Kaufhold I, Klinger M and Rupp J. Interferon-γ interferes with host cell metabolism during intracellular Chlamydia trachomatis infection. Cytokine. 2018;112:95–101. [DOI] [PubMed] [Google Scholar]
- 29.Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, et al. Role of HIF-1alpha in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–90. [DOI] [PubMed] [Google Scholar]
- 30.Manalo DJ, Rowan A, Lavoie T, Natarajan L, Kelly BD, Ye SQ, Garcia JG and Semenza GL. Transcriptional regulation of vascular endothelial cell responses to hypoxia by HIF-1. Blood. 2005;105:659–69. [DOI] [PubMed] [Google Scholar]
- 31.Tang N, Wang L, Esko J, Giordano FJ, Huang Y, Gerber H-P, Ferrara N and Johnson RS. Loss of HIF-1α in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell. 2004;6:485–495. [DOI] [PubMed] [Google Scholar]
- 32.Wei H, Bedja D, Koitabashi N, Xing D, Chen J, Fox-Talbot K, Rouf R, Chen S, Steenbergen C, Harmon JW, et al. Endothelial expression of hypoxia-inducible factor 1 protects the murine heart and aorta from pressure overload by suppression of TGF-β signaling. Proceedings of the National Academy of Sciences. 2012;109:E841–E850. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 33.Huang Y, Lei L, Liu D, Jovin I, Russell R, Johnson RS, Di Lorenzo A and Giordano FJ. Normal glucose uptake in the brain and heart requires an endothelial cell-specific HIF-1α–dependent function. Proceedings of the National Academy of Sciences. 2012;109:17478–17483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang Y-Z, Li Y, Wang K, Dai C-F, Huang S-A, Chen D-B and Zheng J. Distinct roles of HIF1A in endothelial adaptations to physiological and ambient oxygen. Molecular and cellular endocrinology. 2014;391:60–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akhtar S, Hartmann P, Karshovska E, Rinderknecht FA, Subramanian P, Gremse F, Grommes J, Jacobs M, Kiessling F, Weber C, et al. Endothelial Hypoxia-Inducible Factor-1α Promotes Atherosclerosis and Monocyte Recruitment by Upregulating MicroRNA-19a. Hypertension. 2015;66:1220–6. [DOI] [PubMed] [Google Scholar]
- 36.Feng S, Bowden N, Fragiadaki M, Souilhol C, Hsiao S, Mahmoud M, Allen S, Pirri D, Ayllon BT, Akhtar S, et al. Mechanical Activation of Hypoxia-Inducible Factor 1α Drives Endothelial Dysfunction at Atheroprone Sites. Arteriosclerosis, thrombosis, and vascular biology. 2017;37:2087–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wu D, Huang RT, Hamanaka RB, Krause M, Oh MJ, Kuo CH, Nigdelioglu R, Meliton AY, Witt L, Dai G, et al. HIF-1α is required for disturbed flow-induced metabolic reprogramming in human and porcine vascular endothelium. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aune TM and Pogue SL. Inhibition of tumor cell growth by interferon-gamma is mediated by two distinct mechanisms dependent upon oxygen tension: induction of tryptophan degradation and depletion of intracellular nicotinamide adenine dinucleotide. J Clin Invest. 1989;84:863–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Grant RS. Indoleamine 2,3-Dioxygenase Activity Increases NAD+ Production in IFN-gamma-Stimulated Human Primary Mononuclear Cells. Int J Tryptophan Res. 2018;11:1178646917751636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM and Swanson RA. NAD+ depletion is necessary and sufficient for poly(ADP-ribose) polymerase-1-mediated neuronal death. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2010;30:2967–2978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andrabi SA, Umanah GK, Chang C, Stevens DA, Karuppagounder SS, Gagne JP, Poirier GG, Dawson VL and Dawson TM. Poly(ADP-ribose) polymerase-dependent energy depletion occurs through inhibition of glycolysis. Proc Natl Acad Sci U S A. 2014;111:10209–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fouquerel E, Goellner EM, Yu Z, Gagne JP, Barbi de Moura M, Feinstein T, Wheeler D, Redpath P, Li J, Romero G, et al. ARTD1/PARP1 negatively regulates glycolysis by inhibiting hexokinase 1 independent of NAD+ depletion. Cell Rep. 2014;8:1819–1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patton JS, Shepard HM, Wilking H, Lewis G, Aggarwal BB, Eessalu TE, Gavin LA and Grunfeld C. Interferons and tumor necrosis factors have similar catabolic effects on 3T3 L1 cells. Proc Natl Acad Sci U S A. 1986;83:8313–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jackson SK, Darmani H, Stark JM and Harwood JL. Interferon-gamma increases macrophage phospholipid polyunsaturation: a possible mechanism of endotoxin sensitivity. Int J Exp Pathol. 1992;73:783–91. [PMC free article] [PubMed] [Google Scholar]
- 45.Xiong J, Kawagishi H, Yan Y, Liu J, Wells QS, Edmunds LR, Fergusson MM, Yu ZX, Rovira II, Brittain EL, et al. A Metabolic Basis for Endothelial-to-Mesenchymal Transition. Mol Cell. 2018;69:689–698 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Faulkner A, Lynam E, Purcell R, Jones C, Lopez C, Board M, Wagner KD, Wagner N, Carr C and Wheeler-Jones C. Context-dependent regulation of endothelial cell metabolism: differential effects of the PPARβ/δ agonist GW0742 and VEGF-A. Sci Rep. 2020;10:7849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Schoors S, Bruning U, Missiaen R, Queiroz KC, Borgers G, Elia I, Zecchin A, Cantelmo AR, Christen S, Goveia J, et al. Fatty acid carbon is essential for dNTP synthesis in endothelial cells. Nature. 2015;520:192–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Patella F, Schug ZT, Persi E, Neilson LJ, Erami Z, Avanzato D, Maione F, Hernandez-Fernaud JR, Mackay G, Zheng L, et al. Proteomics-based metabolic modeling reveals that fatty acid oxidation (FAO) controls endothelial cell (EC) permeability. Mol Cell Proteomics. 2015;14:621–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kalucka J, Ghesquière B, Fendt SM and Carmeliet P. Analysis of Endothelial Fatty Acid Metabolism Using Tracer Metabolomics. Methods Mol Biol. 2019;1978:259–268. [DOI] [PubMed] [Google Scholar]
- 50.Dagher Z, Ruderman N, Tornheim K and Ido Y. Acute regulation of fatty acid oxidation and amp-activated protein kinase in human umbilical vein endothelial cells. Circ Res. 2001;88:1276–82. [DOI] [PubMed] [Google Scholar]
- 51.Kajihara N, Kukidome D, Sada K, Motoshima H, Furukawa N, Matsumura T, Nishikawa T and Araki E. Low glucose induces mitochondrial reactive oxygen species via fatty acid oxidation in bovine aortic endothelial cells. J Diabetes Investig. 2017;8:750–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hue L and Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab. 2009;297:E578–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Berger NA. Poly(ADP-ribose) in the cellular response to DNA damage. Radiat Res. 1985;101:4–15. [PubMed] [Google Scholar]
- 54.Eleftheriadis T, Pissas G, Sounidaki M, Antoniadi G, Rountas C, Liakopoulos V and Stefanidis I. Correction to: Tryptophan depletion under conditions that imitate insulin resistance enhances fatty acid oxidation and induces endothelial dysfunction through reactive oxygen species-dependent and independent pathways. Mol Cell Biochem. 2018;440:221. [DOI] [PubMed] [Google Scholar]
- 55.Werner-Felmayer G, Werner ER, Fuchs D, Hausen A, Reibnegger G, Schmidt K, Weiss G and Wachter H. Pteridine biosynthesis in human endothelial cells. Impact on nitric oxide-mediated formation of cyclic GMP. J Biol Chem. 1993;268:1842–6. [PubMed] [Google Scholar]
- 56.Ng CT, Fong LY, Low YY, Ban J, Hakim MN and Ahmad Z. Nitric oxide participates in IFN-gamma-induced HUVECs hyperpermeability. Physiol Res. 2016;65:1053–1058. [DOI] [PubMed] [Google Scholar]
- 57.Bossaller C, Habib GB, Yamamoto H, Williams C, Wells S and Henry PD. Impaired muscarinic endothelium-dependent relaxation and cyclic guanosine 5’-monophosphate formation in atherosclerotic human coronary artery and rabbit aorta. J Clin Invest. 1987;79:170–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Casas JP, Bautista LE, Humphries SE and Hingorani AD. Endothelial nitric oxide synthase genotype and ischemic heart disease: meta-analysis of 26 studies involving 23028 subjects. Circulation. 2004;109:1359–65. [DOI] [PubMed] [Google Scholar]
- 59.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M and Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Anders S, Pyl PT and Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Robinson MD, McCarthy DJ and Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Samokhin AO, Stephens T, Wertheim BM, Wang RS, Vargas SO, Yung LM, Cao M, Brown M, Arons E, Dieffenbach PB, et al. NEDD9 targets COL3A1 to promote endothelial fibrosis and pulmonary arterial hypertension. Sci Transl Med. 2018;10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lazzarino G, Amorini AM, Fazzina G, Vagnozzi R, Signoretti S, Donzelli S, Di Stasio E, Giardina B and Tavazzi B. Single-sample preparation for simultaneous cellular redox and energy state determination. Anal Biochem. 2003;322:51–9. [DOI] [PubMed] [Google Scholar]
- 64.Leopold JA, Zhang YY, Scribner AW, Stanton RC and Loscalzo J. Glucose-6-phosphate dehydrogenase overexpression decreases endothelial cell oxidant stress and increases bioavailable nitric oxide. Arterioscler Thromb Vasc Biol. 2003;23:411–7. [DOI] [PubMed] [Google Scholar]
- 65.Semenza GL. Hypoxia-inducible factor 1: oxygen homeostasis and disease pathophysiology. Trends Mol Med. 2001;7:345–50. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
No animal experiments were included in this study. While we used human cells, they were purchased from a vendor, Lonza; were anonymous with respect to donor; and for which IRB review was not required.