

Abstract
As a privileged structural motif, tetrahydroquinoline skeletons widely exist in biologically active natural products and pharmaceuticals. In this protocol, a highly diastereoselective [4 + 2] annulation of ortho-tosylaminophenyl-substituted p-QMs and cyanoalkenes to construct tetrahydroquinoline derivatives has been successfully achieved. This strategy proceeds efficiently under mild condition, offering straightforward route to a variety of 4-aryl-substituted tetrahydroquinolines with high yields, excellent diastereoselectivities, broad functional group tolerance as well as gram-scale capacity. Moreover, a one-pot reaction sequence utilizing in situ generated p-QMs under the similar condition to build tetrahydroquinoline framework is smoothly conducted with good reaction performance as well as step and atom economy.
Keywords: tetrahydroquinolines, p-quinone methides, multi-substituted alkenes, Aza-Michael, 1, 6-conjugate addition
Introduction
As privileged structural motifs, nitrogen-containing heterocycles widely exist in biologically active natural products and pharmaceuticals (Noolvi et al., 2011; Solomon and Lee, 2011; Afzal et al., 2015; Murlykina et al., 2018; Harikandei et al., 2019; Staskiewicz et al., 2021). Among them, 4-phenyl-substituted tetrahydroquinolines are of great importance owing to their wide range of applications in medicinal chemistry, exhibiting antitumor and antibacterial properties (Figure 1). For example, 4-phenylquinolin-2 (1H)-one I shows potential as a specific allosteric inhibitor of Akt (Huang et al., 2017). Compound II has been designed as novel anticancer agents that induce apoptosis with cell cycle arrest at G2/M phase (Chen et al., 2013). As a rationally developed antitumoral agent, compound III displays excellent bioactivity to kill parasite 14DM (Kraus et al., 2009). Known as a novel synthetic molecule, compound IV exhibits antitumoral and antiplasmodial activities (Vladimir et al., 2010). The well-designed compound V owns good antibacterial activity against microorganisms of Escherichia coli (Ramesh et al., 2009). Peniprequinolone VI, isolated from Penicillium sp. FKI-2140, demonstrates impressive insecticidal activity (Uchida et al., 2006). Thus, considering the significant research value of tetrahydroquinolines derivatives in medicinal chemistry, the development of efficient and facile methods to build these valuable skeletons is highly demanded.
FIGURE 1.

Selected natural products and synthetic compounds containing di- or tetra-hydroquinoline frameworks.
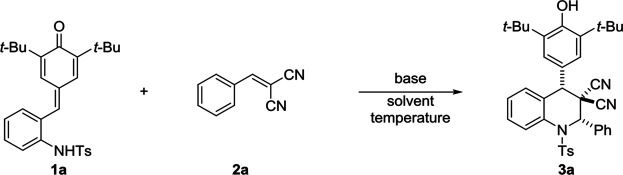
Owing to the remarkable chemical reactivity comprising reactive carbonyl and olefinic moieties, p-quinone methides (p-QMs) have been considered versatile building blocks in organic synthesis (Toteva and Richard, 2011; Parra and Tortosa, 2015; Chauhan et al., 2017; Lima et al., 2020; Wang J.-Y. et al., 2020). Inspired by the seminal works of Fan and Jorgensen (Chu et al., 2013; Caruana et al., 2014), numerous of methods utilizing p-QMs as vinylogous Michael acceptors have been successfully developed through 1,6-conjugate addition and annulation reactions (Lou et al., 2015; Wang et al., 2015; Deng et al., 2016; Dong et al., 2016; He et al., 2016; Li et al., 2016; Ma et al., 2016; Zhang et al., 2016; Roiser and Waser, 2017; Zhang et al., 2017). Recently, Enders and co-workers first demonstrated the potential of ortho-hydroxyphenyl-substituted p-QMs in [4 + 2] cyclization reaction (Zhao et al., 2016), which subsequently enables the extensively investigation of [4 + 1] (Chen et al., 2018; Liu L. et al., 2018; Xiong et al., 2018; Zhi et al., 2018; Zhou et al., 2018; Lu et al., 2019; Tan et al., 2019) [4 + 2] (Jiang X. L. et al., 2018; Mei et al., 2018; Zhang et al., 2018; Yang et al., 2019; Zhang et al., 2019; Huang et al., 2020; Roy et al., 2020; Tan et al., 2020; You et al., 2020) and [4 + 3] (Jiang F. et al., 2018; Li et al., 2018; Liu Q. et al., 2018; Chen et al., 2019) annulations by various research groups. Although great progress has been witnessed in this field, the employment of p-QMs substrates in the construction of heterocyclic frameworks, especially nitrogen-containing heterocyclic frameworks, still remains underdeveloped (Scheme 1A). Very recently, the group of Hu and Zhao pioneered the design of in situ generated ortho-tosylaminophenyl-substituted p-QMs and transformations of this class of substrates in [4 + 1] (Wang et al., 2019; Wang et al., 2020a) and [4 + 2] (Wang et al., 2018; Si et al., 2020) annulation reactions, providing a straightforward access to construct valuable tetrahydroquinoline and 2,3-dihydroindole derivatives, respectively (Scheme 1B). Those transformations avoid the utilization of presynthesized p-QMs, which greatly enhance the step and atom economy of this strategy. To date, currently limited reports almost concentrate on applications between ortho-tosylaminophenyl-substituted p-QMs and disubstituted alkenes. However, rare explorations regarding multifunctional alkenes like tri- or tetra-substituted alkenes, which contain large steric hindrance or relatively poor reactivity, have been successfully achieved so far (Wang et al., 2020b). Hence, to address the aforementioned challenges, we herein reported a highly diastereoselective [4 + 2] annulation of ortho-tosylaminophenyl-substituted p-QMs and cyanoalkenes for the efficient synthesis of 4-aryl-substituted tetrahydroquinolines under mild conditions. This reaction features high yield (up to 96% yield), excellent diastereoselectiveties (>20:1 dr), broad functional group tolerance as well as gram-scale capacity (Scheme 1C).
Scheme 1.

Advancements based on functional p-QMs (A, B) and our strategy for synthesis of multi-substituted tetrahydroquinolines (C).
Results and Discussion
To verify the feasibility of our protocol, a serial of reaction parameters was investigated to explore the best condition for the [4 + 2] annulation. Initially, we established the model reaction using ortho-tosylaminophenyl-p-QMs 1a and α,α-dicyanoalkenes 2a as substrates (Table 1). To our delight, with the help of Cs2CO3 as base, the desired product 3a was successfully obtained in DCM at room temperature in 80% yield with excellent diastereoselectivity of >20:1 (Table 1, entry 1). Encouraged by this promising result, different bases were systematically evaluated, and we found that Na2CO3, pyrolidine, and triethylamine (TEA) were inefficient for this reaction compared to Cs2CO3 (entries 2–4). Delightfully, the organic base 1,8-diazabicyclo [5.4.0]undec-7-ene (DBU) was beneficial to this transformation, generating product 3a in 89% yield (entry 5). Then, we switched attention to solvents screening to further improve reaction efficiency (entries 6–10), and the toluene serving as reaction mediate performed best with up to 96% yield (entry 8). After the confirmation of the optimized base and solvent, the factors of temperature and substrate ratio in this base-mediated catalyst-free protocol was subsequently investigated, and it was found that none of improvement of reaction efficiency was observed when applying other temperature or substrate ratio (entries 11–14). Furthermore, we found that reducing the DBU loading to 0.01 mmol and raising the reaction temperature led to slight decrease in yield with prolonged reaction time (entries 15–16).
TABLE 1.
Optimization of [4 + 2] annulation conditions.
a
| Entry | Base | Solvent | Yield b (%) |
|---|---|---|---|
| 1 | Cs2CO3 | DCM | 80 |
| 2 | Na2CO3 | DCM | <10 |
| 3 | Pyrrolidine | DCM | 59 |
| 4 | TEA | DCM | 74 |
| 5 | DBU | DCM | 89 |
| 6 | DBU | MeCN | 80 |
| 7 | DBU | THF | 82 |
| 8 | DBU | Toluene | 96 |
| 9 | DBU | Ethyl Acetate | 91 |
| 10 | DBU | 1,4-Dioxane | 84 |
| 11 c | DBU | Toluene | 96 |
| 12 d | DBU | Toluene | 93 |
| 13 e | DBU | Toluene | 96 |
| 14 f | DBU | Toluene | 90 |
| 15 g | DBU | Toluene | 89 |
| 16 h | DBU | Toluene | 86 |
Unless other noted, all reactions were conducted with 1a (0.1 mmol), 2a (0.12 mmol) and base (0.02 mmol) in anhydrous solvent (2 ml) at room temperature for 1 h.
Determined by 1H NMR using bromoform as an internal standard; dr > 20:1.
The reaction was performed at 0°C for 3 h;
The reaction was performed at 40°C for 30 min.
0.15 mmol 2a was added.
0.15 mmol 1a was added.
0.01 mmol base was added.
0.01 mmol base was added at the temperature of 80°C.
With the optimal reaction conditions in hand (Table 1, entry 8), we started to explore the substrate scope of this [4 + 2] annulation reaction (Scheme 2). Firstly, the scope of the α, a-dicyanoalkenes part was examined, and we were pleased to find that this protocol tolerated a wide range of α, a-dicyanoalkenes 2, which could readily react with 1a to afford 3a–3u in 55–96% yields. In detail, the α, a-dicyanoalkenes 2 bearing electron-withdrawing (F, Cl, Br, I, ethynyl) and electron-donating (Me, MeO) groups at the phenyl ring could be efficiently converted into the desired products 3a–3n in 62–96% yields (Scheme 2, line 1). The relative configuration of 3a (CCDC 2100705) was determined by X-ray crystallographic analysis, and the relative configurations of other products three were tentatively assigned by analogy. Besides, the α, a-dicyanoalkenes 2 containing challenging disubstituted groups.
Scheme 2.

Investigation of the substrate scope of the [4 + 2] annulation. a,b . a Unless other noted, the reaction was carried out with 1a (46.36 mg, 0.1 mmol), 2a (18.5 mg, 0.12 mmol) and base (0.02 mmol) in anhydrous solvent (2 ml) at room temperature for 1 h b Yields are those of isolated products 3 after column chromatography; the diastereomeric ratio was determined by 1H NMR, dr > 20:1. c Gram scale reaction of 1a (1.02 g, 2.2 mmol) in toluene (5 ml) was conducted under the standard condition.
Were also available, giving the corresponding products 3o–3r in consistent high yields (Scheme 2, line 1). Moreover, aromatic series (naphthyl, furyl, and thienyl) could also participate in this [4 + 2] annulation sequence to provide expected products 3s, 3t and 3u in 79, 55 and 65% yields, respectively (Scheme 2, line 2). Subsequently, several ortho-tosylaminophenyl-p-QMs 1 were investigated to further verify the generality of this method. The results indicated that electronic-withdrawing substituents on the phenyl ring of substrate 1 showed rare affection on efficiency, delivering target products 3v and 3w in high yields (Scheme 2, line 2). Changing the type of protecting group still resulted in desire compound 3x with excellent reaction performance (Scheme 2, line 2). However, the unavailability of dimethyl substituted substrate failed to provide the expected product 3y. To evaluate the general utility and robustness of this protocol, we also conducted the gram-scale reaction under the standard condition, and the target product 3a could be smoothly isolated in 93% yield.
To enrich the diversification of this protocol towards functionalized tetrahydroquinoline derivatives, we established the verification with represented substrates 2 (Scheme 3). Fixed different electronic properties groups (such as nitro, benzoxyl and COOEt groups) on the position of R 2 and/or R3, all reaction could move forward the production of compounds 3z-3bb under optimal condition in 42, 90 and 92% yields, respectively (Scheme 3A). However, substrate containing two ester groups could not offer the target product 3cc under this condition. The relative configuration of 3aa (CCDC 2100706) was determined by X-ray crystallographic analysis, and the relative configurations of 3z–3bb were tentatively assigned by analogy. Moreover, we attempted to construct valuable spirocyclic frameworks employing tetra-substituted substrates four and six within the established condition, and successfully obtained desirable products five and seven in 90 and 62% yields, respectively (Scheme 3B). Regretfully, we failed to accurately assign the relative configuration of compound 5 and 7 with limited information (for details please see ESI). In order to further explore the robustness of this methodology, a preliminary attempt of one-pot synthesis of functionalized tetrahydroquinoline compound starting from precursor 1a was successfully conducted, producing target molecule 3a in 63% yield (Scheme 3C, Wang et al., 2018).
Scheme 3.

Diversification of multifunctional (A) and spirocyclic (B) tetrahydroquinoline derivatives, and one-pot synthesis of compound 3a (C). All the reactions were conducted under the standard condition, dr > 20:1.
Meanwhile, the successful transformation of reducing cyan group into primary amine delivered the valuable product 8 with good reaction performance and undefined-relative configur–ation (for details please see ESI), which may show the potential application in medicinal chemistry (Scheme 4, right column). However, the removal of para-toluene sulfonamide and di-tertiary butyl groups failed to provide the expected products 9 and 10 (Scheme 4, left column).
Scheme 4.

Transformation of multifunctional tetrahydroquinoline derivatives.
Conclusion
In conclusion, we have developed a DBU-mediated catalyst-free [4 + 2] annulation between ortho-tosylaminophenyl-substituted p-QMs and cyanoalkenes for synthesis of valuable tetrahydroquinoline derivatives through an aza-Michael/1,6-conjugate addition sequence. This protocol features broad of tolerance and diversification on substrates, offering straightforward route to various of 4-aryl-substituted tetrahydroquinolines with high yields, excellent diastereoselectivities as well as gram-scale capacity. Moreover, a one-pot reaction sequence utilizing in situ generated p-QMs under the similar condition to build tetrahydroquinoline framework is smoothly conducted with good reaction performance as well as step and atom economy. Further studies on the bioactivity of those promising tetrahydroquinolines will be reported in due course.
Experiment
General Information
NMR data were obtained for 1H at 400 MHz and for 13C at 100 MHz, or for 1H at 600 MHz and for 13C at 150 MHz. Chemical shifts were reported in parts per million (ppm) using tetramethyl silane as internal standard with solvent resonance in CDCl3. UV detection was performed at 254 nm. ESI-HRMS spectra were measured with a Q-TOF instrument. Column chromatography was performed on a silica gel (200–300 mesh) using an eluent of ethyl acetate and petroleum ether. TLC was performed on glass-backed silica plates; products were visualized using UV light. Melting points were determined on a Mel-Temp apparatus. All reagents and solvents were obtained from commercial sources and used without further purification. Substrates 1 and precursor 1a were prepared according to the literature procedures (Wang et al., 2018; Wang J.-Y. et al., 2020). Substrates 2 (Ghosh et al., 2021) and other cyanoalkenes (Fioravanti et al., 2012; Donckele et al., 2015; Zhu et al., 2017; Chen et al., 2021) were prepared through the Knoevenagel reactions, and all substrates can be stored at 4°C for 25 days without decomposition.
General procedure for the synthesis of 3: The reaction was carried out with 1 (0.1 mmol), 2 (0.12 mmol) and DBU (0.02 mmol) in anhydrous toluene (2 ml) at room temperature for 1 h. Upon the consumption of substrate 1 (monitored by TLC), the reside was directly purified by flash column chromatography (PE/EA = 20:1 to 10:1) to provide the desired product 3. The one-pot synthesis of product 3a was conducted in similar manner and MnO2 (5.0 eq.) was added as oxidant in the reaction.
4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-phenyl-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3a). Compound 3a was synthesized in a manner of the general procedure for the synthesis of 3. Yield 96%, white powder solid, >20:1 dr, m. p. 215–216°C. 1H NMR (600 MHz, CDCl3) δ ppm 7.94 (dd, J = 7.8, 1.2 Hz, 1H), 7.92–7.87 (m, 1H), 7.63–7.57 (m, 2H), 7.57–7.52 (m, 2H), 7.49–7.37 (m, 5H), 7.32–7.28 (m, 2H), 6.84 (d, J = 7.8 Hz, 1H), 6.34 (s, 1H), 5.79 (s, 1H), 5.34 (s, 1H), 3.17 (s, 1H), 2.45 (s, 3H), 1.39 (s, 18H). 13C NMR (150 MHz, CDCl3) δ ppm 158.9, 153.5, 143.8, 135.6, 134.4, 133.61, 133.59, 132.4, 129.7, 128.9, 128.7, 128.6, 128.5, 128.0, 127.4, 126.9, 126.5, 126.4, 126.2, 121.2, 112.7, 111.4, 66.3, 49.9, 49.8, 30.6, 29.1, 20.7. HRMS (ESI): m/z [M + Na]+ calcd for C38H39N3NaO3S+: 640.2610; found 640.2614.
2-(2-chlorophenyl)-4-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3b). Compound 3b was synthesized in a manner of the general procedure for the synthesis of 3. Yield 91%, white powder solid, >20:1 dr, m. p. 254–255 °C. 1H NMR (400 MHz, CDCl3) δ ppm 7.88 (d, J = 8.0 Hz, 1H), 7.59–7.54 (m, 2H), 7.53–7.46 (m, 3H), 7.42 (s, 1H), 7.35–7.24 (m, 5H), 6.89 (d, J = 7.6 Hz, 1H), 6.36 (s, 2H), 5.34 (s, 1H), 3.10 (s, 1H), 2.46 (s, 3H), 1.40 (s, 18H). 13C NMR (100 MHz, CDCl3) δ ppm 154.6, 145.1, 136.7, 135.6, 134.7, 134.2, 133.05, 133.02, 130.6, 130.3, 130.1, 129.9, 129.8, 128.5, 127.9, 127.6, 127.4, 122.1, 112.8, 112.2, 61.7, 51.6, 49.3, 34.3, 30.2, 21.8. HRMS (ESI): m/z [M + Na]+ calcd for C38H38ClN3NaO3S+: 674.2220; found 674.2215.
2-(2-bromophenyl)-4-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3c). Compound 3c was synthesized in a manner of the general procedure for the synthesis of 3. Yield 71%, white powder solid, >20:1 dr, m. p. 241–242 °C. 1H NMR (400 MHz, CDCl3) δ ppm 7.87 (d, J = 8.4 Hz, 1H), 7.66 (dd, J = 8.0, 1.2 Hz, 1H), 7.57–7.53 (m, 2H), 7.51–7.49 (m, 2H), 7.42–7.37 (m, 2H), 7.34–7.30 (m, 3H), 7.26 (td, J = 8.0, 2.0 Hz, 1H), 6.89 (d, J = 7.6 Hz, 1H), 6.34 (s, 1H), 6.33 (s, 2H), 5.34 (s, 1H), 3.08 (s, 1H), 2.47 (s, 3H), 1.40 (s, 18H). 13C NMR (100 MHz, CDCl3) δ ppm 154.6, 145.0, 136.7, 136.2, 135.6, 134.2, 133.3, 133.1, 130.9, 130.7, 130.1, 129.8, 128.5, 128.0, 127.9, 127.60, 127.5, 123.4, 122.1, 112.8, 112.1, 63.8, 51.7, 49.3, 34.3, 30.2, 21.8. HRMS (ESI): m/z [M + Na]+ calcd for C38H38BrN3NaO3S+: 718.1715; found 718.1721.
4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-(2-iodophenyl)-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3d). Compound 3d was synthesized in a manner of the general procedure for the synthesis of 3. Yield 77%, white powder solid, >20:1 dr, m. p. 247–248°C. 1H NMR (400 MHz, CDCl3) δ ppm 7.95 (dd, J = 8.0, 1.2 Hz, 1H), 7.87 (dd, J = 8.1, 1.2 Hz, 1H), 7.67–7.48 (m, 4H), 7.46–7.38 (m, 2H), 7.34 (d, J = 8.0 Hz, 3H), 7.09 (td, J = 7.6, 1.6 Hz, 1H), 6.88 (d, J = 8.0 Hz, 1H), 6.33 (s, 1H), 6.15 (s, 1H), 5.33 (s, 1H), 3.06 (s, 1H), 2.47 (s, 3H), 1.40 (s, 18H). 13C NMR (100 MHz, CDCl3) δ ppm 154.6, 145.0, 140.3, 139.2, 136.7, 135.7, 134.2, 133.2, 131.0, 130.4, 130.0, 129.8, 128.8, 128.5, 127.9, 127.7, 127.5, 122.1, 112.8, 112.1, 99.4, 67.5, 51.6, 49.4, 34.3, 30.2, 21.8. HRMS (ESI): m/z [M + Na]+ calcd for C38H38IN3NaO3S+: 766.1576; found 766.1585.
4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-(3-fluorophenyl)-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3e). Compound 3e was synthesized in a manner of the general procedure for the synthesis of 3. Yield 89%, white powder solid, >20:1 dr, m. p. 215–216 °C. 1H NMR (400 MHz, CDCl3) δ ppm 7.96 (d, J = 8.0 Hz, 1H), 7.55 (td, J = 7.6, 1.2 Hz, 1H), 7.47–7.38 (m, 5H), 7.34–7.30 (m, 4H), 7.18–7.05 (m, 1H), 6.86 (d, J = 8.0 Hz, 1H), 6.34 (s, 1H), 5.79 (s, 1H), 5.35 (s, 1H), 3.13 (s, 1H), 2.45 (s, 3H), 1.40 (s, 18H). 13C NMR (100 MHz, CDCl3) δ ppm 164.1 (d, J CF = 246.7 Hz), 154.6, 145.1, 139.2 (d, J CF = 6.8 Hz), 136.8, 135.1, 134.4, 133.2, 130.8 (d, J CF = 8.1 Hz), 130.0, 129.9, 128.4, 128.1, 127.6, 127.4, 123.0, 122.9 (d, J CF = 3.1 Hz), 116.8 (d, J CF = 21.1 Hz), 114.6 (d, J CF = 23.1 Hz), 113.6, 112.2, 66.7, 50.9, 50.5, 34.3, 30.2, 21.8. HRMS (ESI): m/z [M + Na]+ calcd for C38H38FN3NaO3S+: 658.2516; found 658.2512.
2-(3-bromophenyl)-4-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3f). Compound 3f was synthesized in a manner of the general procedure for the synthesis of 3. Yield 93%, white powder solid, >20:1 dr, m. p. 223–224°C. 1H NMR (400 MHz, CDCl3) δ ppm 7.94 (dd, J = 8.0, 1.2 Hz, 1H), 7.70 (d, J = 2.0 Hz, 1H), 7.64–7.47 (m, 3H), 7.42 (dd, J = 6.4, 1.6 Hz, 3H), 7.37–7.28 (m, 4H), 6.85 (d, J = 7.6 Hz, 1H), 6.34 (s, 1H), 5.72 (s, 1H), 5.35 (s, 1H), 3.13 (s, 1H), 2.45 (s, 3H), 1.40 (s, 18H). 13C NMR (100 MHz, CDCl3) δ ppm 154.6, 145.1, 138.8, 136.8, 135.1, 134.3, 133.1, 132.8, 130.6, 130.3, 130.0, 129.9, 128.4, 128.1, 127.6, 127.4, 126.0, 123.1, 122.1, 113.5, 112.2, 66.6, 50.9, 50.6, 34.7, 30.2, 21.8. HRMS (ESI): m/z [M + Na]+ calcd for C38H38BrN3NaO3S+: 718.1715; found 718.1720.
2-(4-chlorophenyl)-4-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3g). Compound 3g was synthesized in a manner of the general procedure for the synthesis of 3. Yield 95%, white powder solid, >20:1 dr, m. p. 217–218°C. 1H NMR (400 MHz, CDCl3) δ ppm 7.93 (d, J = 8.0 Hz, 1H), 7.63–7.51 (m, 3H), 7.42 (dd, J = 8.4, 3.6 Hz, 5H), 7.30 (t, J = 7.6 Hz, 3H), 6.84 (d, J = 7.6 Hz, 1H), 6.34 (s, 1H), 5.74 (s, 1H), 5.35 (s, 1H), 3.13 (s, 1H), 2.45 (s, 3H), 1.40 (s, 18H). 13C NMR (100 MHz, CDCl3) δ ppm 171.2, 154.6, 145.1, 136.8, 135.6, 135.3, 135.2, 134.3, 133.2, 130.0, 129.9, 129.3, 128.7, 128.3, 128.1, 127.6, 127.4, 123.9, 122.1, 113.5, 112.3, 66.8, 60.4, 50.6, 34.5, 30.2, 21.7. HRMS (ESI): m/z [M + Na]+ calcd for C38H38ClN3NaO3S+: 674.2220; found 674.2223.
2-(4-bromophenyl)-4-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3h). Compound 3h was synthesized in a manner of the general procedure for the synthesis of 3. Yield 95%, white powder solid, >20:1 dr, m. p. 235–236°C. 1H NMR (400 MHz, CDCl3) δ ppm 7.93 (d, J = 8.0 Hz, 1H), 7.59 (d, J = 8.0 Hz, 2H), 7.54 (t, J = 7.6 Hz, 1H), 7.48 (d, J = 8.4 Hz, 2H), 7.42 (d, J = 8.0 Hz, 3H), 7.30 (t, J = 7.6 Hz, 3H), 6.84 (d, J = 7.6 Hz, 1H), 6.34 (s, 1H), 5.72 (s, 1H), 5.35 (s, 1H), 3.13 (s, 1H), 2.45 (s, 3H), 1.40 (s, 18H). 13C NMR (100 MHz, CDCl3) δ ppm 154.6, 145.1, 136.8, 135.8, 135.1, 134.3, 133.2, 132.3, 130.0, 129.9, 129.0, 128.3, 128.1, 127.6, 127.4, 123.9, 122.1, 113.5, 112.3, 66.9, 50.9, 50.5, 34.5, 30.2, 21.8. HRMS (ESI): m/z [M + Na]+ calcd for C38H38BrN3NaO3S+: 718.1715; found 718.1709.
4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-(4-ethynylphenyl)-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3i). Compound 3i was synthesized in a manner of the general procedure for the synthesis of 3. Yield 92%, white powder solid, >20:1 dr, m. p. 248–249°C. 1H NMR (400 MHz, CDCl3) δ ppm 7.94 (d, J = 7.2 Hz, 1H), 7.62–7.50 (m, 5H), 7.43–7.41 (m, 3H), 7.35–7.28 (m, 3H), 6.85 (d, J = 7.6 Hz, 1H), 6.35 (s, 1H), 5.77 (s, 1H), 5.35 (s, 1H), 3.15 (s, 1H), 3.12 (s, 1H), 2.45 (s, 3H), 1.40 (s, 18H). 13C NMR (100 MHz, CDCl3) δ ppm 154.6, 145.1, 137.2, 136.8, 135.2, 134.4, 133.3, 132.8, 130.0, 129.8, 128.4, 128.7, 127.6, 127.4, 127.3, 123.5, 122.1, 113.5, 112.3, 82.9, 78.4, 67.0, 51.0, 50.5, 34.5, 30.2, 21.8. HRMS (ESI): m/z [M + Na]+ calcd for C40H39N3NaO3S+: 664.2610; found 664.2614.
4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-(o-tolyl)-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3j). Compound 3j was synthesized in a manner of the general procedure for the synthesis of 3. Yield 96%, white powder solid, >20:1 dr, m. p. 233–234°C. 1H NMR (400 MHz, CDCl3) δ ppm 7.81 (dd, J = 8.0, 1.2 Hz, 1H), 7.52 (td, J = 8.0, 1.6 Hz, 1H), 7.47–7.36 (m, 3H), 7.36–7.20 (m, 7H), 6.92 (dd, J = 7.6, 1.2 Hz, 1H), 6.41 (s, 1H), 6.13 (s, 1H), 5.34 (s, 1H), 3.25 (s, 1H), 2.75 (s, 3H), 2.45 (s, 3H), 1.41 (s, 18H). 13C NMR (100 MHz, CDCl3) δ ppm 154.6, 144.8, 136.6, 136.0, 135.6, 135.2, 134.9, 133.3, 130.9, 129.9, 129.7, 129.3, 129.0, 128.1, 127.8, 127.5, 127.3, 126.5, 122.2, 113.3, 112.9, 77.3, 77.0, 76.7, 62.1, 51.6, 50.0, 34.7, 30.2, 21.8, 21.7, 19.5. HRMS (ESI): m/z [M + Na]+ calcd for C39H41N3NaO3S+: 654.2766; found 654.2762.
4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-(2-methoxyphenyl)-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3k). Compound 3k was synthesized in a manner of the general procedure for the synthesis of 3. Yield 94%, white powder solid, >20:1 dr, m. p. 198–199°C. 1H NMR (400 MHz, CDCl3) δ ppm 7.97 (d, J = 8.0 Hz, 1H), 7.67–7.41 (m, 5H), 7.36–7.27 (m, 4H), 6.99 (t, J = 8.0 Hz, 2H), 6.94 (d, J = 8.4 Hz, 1H), 6.46 (s, 1H), 6.36 (s, 1H), 5.32 (s, 1H), 3.93 (s, 3H), 3.17 (s, 1H), 2.45 (s, 3H), 1.40 (s, 18H). 13C NMR (100 MHz, CDCl3) δ ppm 155.9, 154.5, 144.7, 136.3, 136.0, 135.5, 132.8, 130.4, 130.0, 129.5, 128.9, 128.0, 127.9, 127.6, 127.4, 126.2, 122.1, 121.0, 114.3, 113.2, 110.5, 60.6, 55.0, 51.1, 50.1, 34.6, 30.2, 21.8. HRMS (ESI): m/z [M + Na]+ calcd for C39H41N3NaO4S+: 670.2715; found 670.2718.
4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-(p-tolyl)-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3l). Compound 3l was synthesized in a manner of the general procedure for the synthesis of 3. Yield 93%, white powder solid, >20:1 dr, m. p. 223–224°C. 1H NMR (400 MHz, CDCl3) δ ppm 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 8.0 Hz, 1H), 7.45 (t, J = 7.6 Hz, 1H), 7.40–7.34 (m, 5H), 7.24–7.19 (m, 3H), 7.17 (d, J = 8.2 Hz, 2H), 6.75 (d, J = 7.6 Hz, 1H), 6.26 (s, 1H), 5.67 (s, 1H), 5.25 (s, 1H), 3.07 (s, 1H), 2.37 (s, 3H), 2.29 (s, 3H), 1.32 (s, 18H). 13C NMR (100 MHz, CDCl3) δ ppm 154.5, 144.8, 139.4, 136.7, 135.5, 134.7, 133.8, 133.4, 129.9, 129.7, 128.4, 127.9, 127.5, 127.4, 127.1, 124.0, 122.4, 113.7, 112.6, 67.3, 50.95, 50.92, 34.4, 30.2, 21.7, 21.3. HRMS (ESI): m/z [M + Na]+ calcd for C39H41N3NaO3S+: 654.2766; found 654.2773.
4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-(4-methoxyphenyl)-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3m). Compound 3m was synthesized in a manner of the general procedure for the synthesis of 3. Yield 88%, white powder solid, >20:1 dr, m. p. 210–211°C. 1H NMR (400 MHz, CDCl3) δ ppm 7.92 (dd, J = 8.0, 1.2 Hz, 1H), 7.54–7.50 (m, 3H), 7.43 (d, J = 8.0 Hz, 3H), 7.32–7.27 (m, 3H), 6.96 (d, J = 8.8 Hz, 2H), 6.83 (dt, J = 8.0, 1.6 Hz, 1H), 6.35 (s, 1H), 5.75 (s, 1H), 5.34 (s, 1H), 3.82 (s, 3H), 3.16 (s, 1H), 2.45 (s, 3H), 1.40 (s, 18H).13C NMR (100 MHz, CDCl3) δ ppm 160.4, 154.5, 144.8, 136.7, 135.5, 134.7, 133.5, 129.9, 129.7, 128.8, 128.6, 128.4, 127.9, 127.5, 127.4, 122.4, 114.4, 113.8, 112.6, 67.1, 55.3, 51.1, 50.9, 34.9, 30.2, 21.8. HRMS (ESI): m/z [M + Na]+ calcd for C39H41N3NaO4S+: 670.2715; found 670.2721.
4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-(3,4-dimethoxyphenyl)-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3n). Compound 3n was synthesized in a manner of the general procedure for the synthesis of 3. Yield 62%, white powder solid, >20:1 dr, m. p. 202–203°C. 1H NMR (400 MHz, CDCl3) δ ppm 7.92 (t, J = 1.6 Hz, 1H), 7.52 (td, J = 7.6, 1.6 Hz, 1H), 7.43 (d, J = 8.0 Hz, 3H), 7.30 (t, J = 7.2 Hz, 3H), 7.18 (dd, J = 8.0, 2.0 Hz, 1H), 7.08 (s, 1H), 6.92 (d, J = 8.4 Hz, 1H), 6.85 (d, J = 7.6 Hz, 1H), 6.36 (s, 1H), 5.76 (s, 1H), 5.34 (s, 1H), 3.89 (s, 3H), 3.87 (s, 3H), 3.17 (s, 1H), 2.45 (s, 3H), 1.40 (s, 18H). 13C NMR (100 MHz, CDCl3) δ 154.5, 149.9, 149.2, 144.9, 136.7, 135.5, 134.7, 133.4, 129.9, 129.7, 129.1, 128.1, 127.9, 127.6, 127.4, 122.3, 119.9, 113.9, 112.6, 111.3, 110.1, 67.2, 55.9, 55.8, 51.2, 50.9, 34.5, 30.2, 21.7. HRMS (ESI): m/z [M + Na]+ calcd for C40H43N3NaO5S+: 700.2821; found 700.2828.
4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-(2,4-dichlorophenyl)-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3o). Compound 3o was synthesized in a manner of the general procedure for the synthesis of 3. Yield 91%, white powder solid, >20:1 dr, m. p. 250–251°C. 1H NMR (400 MHz, Chloroform-d) δ ppm 7.91 (dd, J = 8.0, 1.2 Hz, 1H), 7.57 (td, J = 7.6, 1.6 Hz, 1H), 7.52 (d, J = 2.8 Hz, 1H), 7.46 (d, J = 8.4 Hz, 2H), 7.42 (d, J = 8.8 Hz, 2H), 7.39–7.30 (m, 4H), 6.90 (d, J = 7.6 Hz, 1H), 6.34 (s, 1H), 6.27 (s, 1H), 5.35 (s, 1H), 3.04 (s, 1H), 2.47 (s, 3H), 1.40 (s, 18H). 13C NMR (100 MHz, CDCl3) δ ppm 154.7, 145.2, 136.3, 135.3, 133.9, 133.4, 132.7, 131.3, 131.0, 130.8, 130.3, 130.1, 130.0, 128.5, 128.1, 127.6, 127.5, 121.9, 112.5, 111.9, 61.4, 51.6, 49.0, 34.7, 30.2, 21.8. HRMS (ESI): m/z [M + Na]+ calcd for C38H37Cl2N3NaO3S+: 708.1830; found 708.1841.
4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-(2,6-dichlorophenyl)-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3p). Compound 3p was synthesized in a manner of the general procedure for the synthesis of 3. Yield 89%, white powder solid, >20:1 dr, m. p. 303–304°C. 1H NMR (400 MHz, CDCl3) δ 7.85 (d, J = 8.0 Hz, 1H), 7.45 (t, J = 7.8 Hz, 1H), 7.43–7.24 (m, 5H), 7.22–7.20 (m, 3H), 7.17 (d, J = 8.4 Hz, 2H), 6.75 (d, J = 7.7 Hz, 1H), 6.26 (s, 1H), 5.67 (s, 1H), 5.25 (s, 1H), 3.07 (s, 1H), 2.37 (s, 3H), 2.29 (s, 3H), 1.32 (s, 18H).13C NMR (100 MHz, CDCl3) δ ppm 154.7, 145.1, 136.6, 136.55, 136.47, 136.0, 133.5, 131.8, 131.5, 131.0, 130.1, 130.0, 129.9, 129.4, 129.3, 128.3, 127.7, 127.4, 127.2, 124.3, 122.1, 113.0, 112.1, 62.5, 52.0, 47.1, 34.7, 34.2, 30.3, 30.1, 21.8. HRMS (ESI): m/z [M + Na]+ calcd for C38H37Cl2N3NaO3S+: 708.1830; found 708.1841.
2-(2-chloro-5-fluorophenyl)-4-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-tosyl-1,4-dihydro-quinoline-3,3(2H)-dicarbonitrile (3q). Compound 3q was synthesized in a manner of the general procedure for the synthesis of 3. Yield 93%, white powder solid, >20:1 dr, m. p. 232–233°C. 1H NMR (400 MHz, CDCl3) δ ppm 7.91 (d, J = 8.0 Hz, 1H), 7.57 (t, J = 8.0 Hz, 1H), 7.51–7.42 (m, 3H), 7.41–7.32 (m, 4H), 7.29 (dt, J = 9.2, 2.4 Hz, 1H), 7.08 (td, J = 8.0, 3.2 Hz, 1H), 6.90 (d, J = 7.6 Hz, 1H), 6.34 (s, 1H), 6.29 (s, 1H), 5.36 (s, 1H), 3.06 (s, 1H), 2.47 (s, 3H), 1.40 (s, 18H). 13C NMR (100 MHz, CDCl3) δ ppm 162.5 (d, J CF = 249.5 Hz), 154.7, 145.2, 136.6 (d, J CF = 7.0 Hz), 135.3, 133.9, 132.8, 131.3 (d, J CF = 7.7 Hz), 130.1, 130.0, 128.5, 128.1, 128.0 (d, J CF = 3.5 Hz), 127.6, 127.5, 124.0, 121.9, 118.2 (d, J CF = 23.3 Hz), 117.4 (d, J CF = 24.4 Hz), 112.5, 112.0, 61.6, 51.6, 49.0, 34.6, 30.2, 21.8. HRMS (ESI): m/z [M + Na]+ calcd for C38H37ClFN3NaO3S+: 692.2126; found 692.2131.
2-(5-bromo-2-fluorophenyl)-4-(3,5-di-tert-butyl-4-hydroxyphenyl)-1-tosyl-1,4-dihydro-quinoline-3,3(2H)-dicarbonitrile (3r). Compound 3r was synthesized in a manner of the general procedure for the synthesis of 3. Yield 91%, white powder solid, >20:1 dr, m. p. 206–207°C. 1H NMR (400 MHz, CDCl3) δ ppm 7.92 (d, J = 8.0 Hz, 1H), 7.65 (dd, J = 6.0, 2.4 Hz, 1H), 7.57 (t, J = 8.0 Hz, 1H), 7.42 (d, J = 8.0 Hz, 3H), 7.32 (d, J = 8.0 Hz, 3H), 7.07 (t, J = 8.8 Hz, 1H), 6.89 (d, J = 7.6 Hz, 1H), 6.34 (s, 1H), 6.05 (s, 1H), 5.35 (s, 1H), 3.06 (s, 1H), 2.46 (s, 3H), 1.40 (s, 18H). 13C NMR (150 MHz, CDCl3) δ ppm 154.8, 145.4, 136.8 (d, J CF = 12.9 Hz), 135.2, 134.4 (d, J CF = 7.5 Hz), 134.1, 132.8, 132.4, 130.2, 130.1, 128.6, 128.2, 128.1, 127.7, 127.6, 124.1, 122.0, 117.8, 117.4, 112.5, 59.3, 51.3, 49.6, 34.7, 34.3, 30.3, 30.2, 21.9. HRMS (ESI): m/z [M + Na]+ calcd for C38H37BrFN3NaO3S+: 736.1621; found 736.1615.
4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-(naphthalen-1-yl)-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3s). Compound 3s was synthesized in a manner of the general procedure for the synthesis of 3. Yield 79%, white powder solid, >20:1 dr, m. p. 255–256°C. 1H NMR (400 MHz, CDCl3) δ ppm 8.43 (d, J = 8.4 Hz, 1H), 8.00–7.83 (m, 3H), 7.74 (d, J = 7.2 Hz, 1H), 7.67 (td, J = 8.8, 1.6 Hz, 1H), 7.60–7.51 (m, 3H), 7.48 (t, J = 8.0 Hz 3H), 7.35 (td, J = 7.6, 1.2 Hz, 1H), 7.28 (d, J = 8.0 Hz, 2H), 6.98 (d, J = 7.8 Hz, 1H), 6.81 (s, 1H), 6.50 (s, 1H), 5.34 (s, 1H), 3.45 (s, 1H), 2.44 (s, 3H), 1.40 (s, 18H). 13C NMR (100 MHz, CDCl3) δ ppm 153.6, 143.8, 135.6, 135.1, 133.8, 132.7, 132.2, 132.1, 129.0, 128.9, 128.7, 128.1, 127.2, 126.8, 126.6, 126.4, 126.4, 126.0, 125.1, 124.2, 121.2, 121.1, 112.3, 111.7, 60.0, 50.8, 49.5, 33.4, 29.2, 20.7. HRMS (ESI): m/z [M + Na]+ calcd for C42H41N3NaO3S+: 690.2766; found 690.2769.
4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-(furan-2-yl)-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3t). Compound 3t was synthesized in a manner of the general procedure for the synthesis of 3. Yield 55%, white powder solid, >20:1 dr, m. p. 179–180°C. 1H NMR (400 MHz, CDCl3) δ ppm 7.67 (dd, J = 8.1, 1.3 Hz, 1H), 7.39–7.36 (m, 2H), 7.31 (td, J = 7.8, 1.6 Hz, 1H), 7.22 (d, J = 8.1 Hz, 2H), 7.15 (td, J = 7.6, 1.2 Hz, 1H), 6.60 (s, 2H), 6.50 (d, J = 7.8 Hz, 1H), 5.22 (s, 2H), 5.16 (s, 1H), 4.12 (q, J = 7.2 Hz, 1H), 2.97 (s, 1H), 2.46 (s, 3H), 2.04 (s, 1H), 1.37 (s, 18H). 13C NMR (100 MHz, CDCl3) δ ppm 153.2, 152.1, 145.5, 137.6, 135.9, 134.4, 133.5, 129.84, 129.77, 127.9, 127.4, 127.3, 126.9, 126.7, 125.7, 118.4, 60.4, 50.1, 41.8, 34.3, 30.2, 21.7. HRMS (ESI): m/z [M + Na]+ calcd for C36H37N3NaO4S+: 630.2402; found 630.2409.
4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-(thiophen-2-yl)-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3u). Compound 3u was synthesized in a manner of the general procedure for the synthesis of 3. Yield 65%, white powder solid, >20:1 dr, m. p. 238–239°C. 1H NMR (400 MHz, CDCl3) δ ppm 7.89 (d, J = 8.0 Hz, 1H), 7.51 (td, J = 7.6, 1.6 Hz, 1H), 7.44–7.42 (m, 3H), 7.40–7.34 (m, 2H), 7.33–7.28 (m, 3H), 7.07 (dd, J = 5.2, 3.6 Hz, 1H), 6.84 (d, J = 7.6 Hz, 1H), 6.32 (s, 1H), 6.13 (s, 1H), 5.34 (s, 1H), 3.12 (s, 1H), 2.45 (s, 3H), 1.40 (s, 18H). 13C NMR (100 MHz, CDCl3) δ ppm 154.6, 145.1, 140.4, 136.7, 134.6, 134.5, 133.3, 130.0, 129.7, 128.8, 128.2, 127.7, 127.6, 127.4, 126.5, 126.3, 122.1, 113.7, 112.1, 63.8, 51.3, 50.6, 34.6, 30.2, 21.6. HRMS (ESI): m/z [M + Na]+ calcd for C36H37N3NaO3S2 +: 646.2174; found 646.2171.
7-bromo-4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-phenyl-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3v). Compound 3v was synthesized in a manner of the general procedure for the synthesis of 3. Yield 93%, white powder solid, >20:1 dr, m. p. 225–226°C. 1H NMR (400 MHz, CDCl3) δ ppm 7.74 (d, J = 8.5 Hz, 1H), 7.59 (dd, J = 8.2, 2.4 Hz, 1H), 7.51–7.45 (m, 2H), 7.41–7.29 (m, 6H), 7.26 (d, J = 8.0 Hz, 2H), 6.96 (s, 1H), 6.28 (s, 1H), 5.69 (s, 1H), 5.31 (s, 1H), 3.04 (s, 1H), 2.39 (s, 3H), 1.33 (s, 18H). 13C NMR (100 MHz, CDCl3) δ ppm 154.9, 145.2, 136.9, 136.3, 135.1, 134.6, 134.4, 132.9, 130.8, 130.1, 129.9, 129.7, 129.1, 127.4, 127.2, 121.8, 121.4, 113.4, 112.2, 67.2, 50.9, 50.7, 34.3, 30.2, 21.8. HRMS (ESI): m/z [M + Na]+ calcd for C38H38BrN3NaO3S+: 718.1715; found 718.1721.
6-chloro-4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-phenyl-1-tosyl-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3w). Compound 3w was synthesized in a manner of the general procedure for the synthesis of 3. Yield 87%, white powder solid, >20:1 dr, m. p. 250–251°C. 1H NMR (400 MHz, CDCl3) δ ppm 7.88 (d, J = 8.8 Hz, 1H), 7.62–7.53 (m, 2H), 7.51 (dd, J = 8.4, 2.4 Hz, 1H), 7.50–7.36 (m, 6H), 7.34–7.32 (m, 2H), 6.87 (d, J = 2.4 Hz, 1H),6.34 (s, 1H), 5.77 (s, 1H), 5.38 (s, 1H), 3.12 (s, 1H), 2.47 (s, 3H), 1.41 (s, 18H). 13C NMR (100 MHz, CDCl3) δ ppm 154.8, 145.2, 136.9, 136.3, 135.0, 134.4, 134.1, 133.9, 130.1, 129.9, 129.7, 129.6, 129.1, 127.9, 127.4, 127.2, 121.5, 113.4, 112.2, 67.2, 60.4, 50.8, 34.4, 30.2, 21.8. HRMS (ESI): m/z [M + Na]+ calcd for C38H38ClN3NaO3S+: 674.2220; found 674.2223.
4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-phenyl-1-(phenylsulfonyl)-1,4-dihydroquinoline-3,3(2H)-dicarbonitrile (3y). Compound 3y was synthesized in a manner of the general procedure for the synthesis of 3. Yield 90%, white powder solid, >20:1 dr, m. p. 211–212°C. 1H NMR (400 MHz, CDCl3) δ ppm 7.95 (d, J = 8.0 Hz, 1H), 7.70 (tt, J = 6.8, 2.0 Hz, 1H), 7.63–7.58 (m, 2H), 7.57–7.49 (m, 5H), 7.48–7.37 (m, 4H), 7.32 (t, J = 7.6 Hz, 1H), 6.82 (d, J = 8.0 Hz, 1H), 6.23 (s, 1H), 5.79 (s, 1H), 5.33 (s, 1H), 2.96 (s, 1H), 1.39 (s, 18H). 13C NMR (100 MHz, CDCl3) δ ppm 154.5, 137.4, 136.7, 136.6, 135.3, 133.8, 133.6, 129.8, 129.6, 129.4, 129.1, 128.5, 128.1, 127.5, 127.3, 127.2, 123.9, 122.1, 113.7, 112.4, 67.4, 50.9, 50.7, 34.5, 30.2. HRMS (ESI): m/z [M + Na]+ calcd for C37H37N3NaO3S+: 626.2453; found 626.2457.
3-benzoyl-4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-phenyl-1-tosyl-1,2,3,4-tetrahydroquinoline-3-carbonitrile (3z). Compound 3z was synthesized in a manner of the general procedure for the synthesis of 3. Yield 42%, white powder solid, >20:1 dr, m. p. 269–270°C. 1H NMR (400 MHz, CDCl3) δ ppm 8.04 (dd, J = 8.0, 1.2 Hz, 1H), 7.60–7.54 (m, 2H), 7.50 (dd, J = 8.4, 7.6 Hz, 1H), 7.46–7.30 (m, 7H), 7.28–7.26 (m, 1H), 7.25–7.20 (m, 1H), 7.16 (d, J = 2.4 Hz, 1H), 7.04–7.00 (m, 2H), 6.81 (dd, J = 6.4, 1.6 Hz, 1H), 6.69–6.55 (m, 2H), 6.27 (s, 1H), 6.13 (d, J = 2.4 Hz, 1H), 5.11 (s, 1H), 3.18 (s, 1H), 2.45 (s, 3H), 1.41 (s, 9H), 1.01 (s, 9H). 13C NMR (150 MHz, CDCl3) δ ppm 196.0, 153.8, 144.3, 138.9, 137.5, 136.4, 136.0, 136.0, 135.2, 134.8, 132.1, 129.6, 129.0, 128.8, 128.7, 128.6, 128.4, 127.9, 127.6, 127.3, 127.1, 125.2, 122.7, 118.6, 67.9, 67.7, 53.3, 34.2, 34.2, 30.2, 30.0, 21.8. HRMS (ESI): m/z [M + Na]+ calcd for C44H44N2NaO4S+: 719.2919; found 719.2925.
Ethyl-3-cyano-4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-phenyl-1-tosyl-1,2,3,4-tetrahydro-quinoline-3-carboxylate (3aa). Compound 3aa was synthesized in a manner of the general procedure for the synthesis of 3. Yield 90%, white powder solid, >20:1 dr, m. p. 255–256°C. 1H NMR (400 MHz, CDCl3) δ ppm 7.99 (d, J = 8.0 Hz, 1H), 7.58–7.43 (m, 4H), 7.36–7.35 (m, 4H), 7.34–7.28 (m, 1H), 7.28–7.24 (m, 3H), 6.89 (d, J = 8.0 Hz, 1H), 6.13 (s, 1H), 6.10 (s, 1H), 5.23 (s, 1H), 3.91–3.71 (m, 2H), 3.02 (s, 1H), 2.42 (s, 3H), 1.36 (s, 18H), 0.73 (t, J = 7.6 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ ppm 166.2, 154.0, 144.2, 138.6, 136.1, 135.9, 135.8, 134.8, 134.7, 129.5, 128.79, 128.77, 128.6, 128.5, 127.9, 127.7, 127.3, 127.0, 126.7, 125.2, 122.9, 116.0, 66.1, 64.7, 63.2, 50.9, 34.5, 34.1, 30.3, 30.1, 21.7, 13.3. HRMS (ESI): m/z [M + Na]+ calcd for C40H44N2NaO5S+: 687.2869; found 687.2873.
Ethyl-4-(3,5-di-tert-butyl-4-hydroxyphenyl)-3-nitro-2-phenyl-1-tosyl-1,2,3,4-tetrahydro-quinoline-3-carboxylate (3bb). Compound 3bb was synthesized in a manner of the general procedure for the synthesis of 3. Yield 92%, white powder solid, >20:1 dr, m. p. 166–167°C. 1H NMR (600 MHz, CDCl3) δ ppm 7.92 (d, J = 7.8 Hz, 1H), 7.56 (d, J = 8.4 Hz, 2H), 7.45 (t, J = 7.8 Hz, 1H), 7.31–7.20 (m, 8H), 7.15 (s, 1H), 6.95 (d, J = 8.4 Hz, 1H), 6.56 (s, 2H), 5.20 (s, 1H), 3.35 (dd, J = 10.8, 7.2 Hz, 1H), 3.28 (s, 1H), 3.18 (dd, J = 10.8, 7.2 Hz, 1H), 2.39 (s, 3H), 1.34 (s, 18H), 0.56 (t, J = 7.2 Hz, 3H). 13C NMR (150 MHz, CDCl3) δ ppm 163.7, 153.9, 144.3, 139.4, 136.1, 135.9, 135.6, 134.6, 129.8, 128.4, 128.4, 128.1, 127.7, 127.5, 127.4, 126.9, 123.2, 106.0, 65.4, 62.2, 52.8, 34.3, 30.3, 21.9, 12.9. HRMS (ESI): m/z [M + Na]+ calcd for C39H44N2NaO7S+: 707.2767; found 707.2772.
4'-(3,5-di-tert-butyl-4-hydroxyphenyl)-1,3-dioxo-1′-tosyl-1,1′,3,4′-tetrahydro-3′H-spiro[indene-2,2′-quinoline]-3′,3′-dicarbonitrile (5). Compound 5 was synthesized in a similar manner of the general procedure for the synthesis of 3. Yield 90%, white powder solid, >20:1 dr, m. p. 239–240°C. 1H NMR (400 MHz, CDCl3) δ ppm 8.17–7.13 (m, 2H), 8.06–7.91 (m, 4H), 7.68 (d, J = 8.0 Hz, 1H), 7.35–7.32 (m, 3H), 7.20 (td, J = 8.4, 1.6 Hz, 1H), 7.10 (s, 1H), 7.01 (t, J = 7.6 Hz, 1H), 6.94 (d, J = 7.6 Hz, 1H), 5.38 (s, 1H), 5.14 (s, 1H), 2.40 (s, 3H), 1.45 (s, 9H), 1.38 (s, 9H). 13C NMR (100 MHz, CDCl3) δ ppm 195.4, 191.1, 154.9, 145.4, 139.0, 138.9, 137.4, 137.2, 136.9, 136.4, 135.8, 130.1, 129.3, 129.0, 128.4, 124.9, 124.7, 124.3, 123.9, 123.3, 119.5, 110.5, 110.3, 68.7, 47.4, 45.6, 34.7, 34.4, 30.3, 21.7. HRMS (ESI): m/z [M + Na]+ calcd for C40H37N3NaO5S+: 694.2352; found 694.2359.
4'-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-oxo-1′-tosyl-1′,4′-dihydro-3′H-spiro[indoline-3,2′-quinoline]-3′,3′-dicarbonitrile (7). Compound 7 was synthesized in a similar manner of the general procedure for the synthesis of 3. Yield 62%, red powder solid, >20:1 dr, m. p. 201–202°C. 1H NMR (600 MHz, CDCl3) δ ppm 8.11 (d, J = 7.8 Hz, 1H), 7.63 (d, J = 8.4 Hz, 2H), 7.47 (dd, J = 7.8, 1.8 Hz, 1H), 7.36–7.30 (m, 1H), 7.30–7.26 (m, 1H), 7.19 (d, J = 8.4 Hz, 2H), 7.16 (dd, J = 7.8, 1.8 Hz, 1H), 7.13 (dd, J = 7.8, 1.2 Hz, 1H), 7.06 (td, J = 7.8, 1.2 Hz, 1H), 6.82 (m, 2H), 6.76 (s, 1H), 6.52 (s, 1H), 6.37 (d, J = 8.4 Hz, 1H), 5.32 (s, 1H), 2.36 (s, 3H), 1.33 (s, 18H). 13C NMR (150 MHz, CDCl3) δ ppm 163.2, 154.1, 148.7, 146.0, 144.2, 137.1, 136.9, 136.7, 135.0, 131.4, 129.9, 129.6, 128.9, 127.1, 126.8, 126.6, 126.5, 125.0, 124.5, 123.8, 118.6, 113.0, 112.4, 110.7, 82.9, 55.7, 34.5, 30.2, 21.7. HRMS (ESI): m/z [M + Na]+ calcd for C39H38N7NaO4S+: 681.2511; found 681.2514.
Procedure for the synthesis of 8: The reaction was carried out with 1a (0.1 mmol), NaBH4 (10.0 eq.) in anhydrous MeOH (2 ml) at room temperature for several hours. Upon the consumption of substrate 1 (monitored by TLC), the reaction was quenched with aqueous NaHCO3, and extraction was carried out with CH2Cl2. The organic layer was dried over Na2SO4 and concentrated. Then, the reside was directly purified by flash column chromatography (PE/EA = 10:1 to 8:1) to provide the desired product 8.
3-(aminomethyl)-4-(3,5-di-tert-butyl-4-hydroxyphenyl)-2-phenyl-1-tosyl-1,2,3,4-tetrahydro-quinoline-3-carbonitrile (8). Yield 72%, white powder solid, 10:1 dr, m. p. 235–236°C. 1H NMR (600 MHz, CDCl3) δ ppm 7.82 (dd, J = 8.4, 1.0 Hz, 1H), 7.38 (d, J = 8.4 Hz, 3H), 7.35 (d, J = 7.2 Hz, 3H), 7.28 (t, J = 7.8 Hz, 2H), 7.26–7.19 (m, 2H), 7.13 (d, J = 8.4 Hz, 3H), 6.65 (d, J = 7.8 Hz, 1H), 5.80 (s, 1H), 5.19 (s, 1H), 2.82 (s, 1H), 2.58 (dd, J = 28.8 Hz, 13.8 Hz, 2H), 2.33 (s, 3H), 1.31 (s, 18H), 0.78 (s, 2H).13C NMR (150 MHz, CDCl3) δ ppm 153.8, 143.9, 139.5, 137.1, 136.7, 136.1, 135.7, 129.3, 128.4, 128.4, 128.3, 128.2, 127.9, 127.8, 127.2, 126.8, 124.4, 120.2, 62.9, 56.6, 47.0, 43.6, 34.5, 30.4, 21.8. HRMS (ESI): m/z [M + H]+ calcd for C38H44N3O3S+: 622.3103; found 622.3107.
Procedure for the synthesis of 9: The reaction was carried out with 1a (0.1 mmol), AlCl3 (6.0 eq.) in anhydrous Toluene (2 ml) at room temperature. Upon the end of the reaction, the reaction mixture was quenched with aqueous NaHCO3, and extraction was carried out with CH2Cl2. The organic layer was dried over Na2SO4 and concentrated. Then, the reside was directly purified by flash column chromatography. This condition led to a mess reaction and could not offer target product 9.
Procedure for the synthesis of 10: condition A: the reaction was carried out with 1a (0.1 mmol), HCl (1N) in anhydrous MeOH (2 ml) at room temperature for several hours; condition B: the reaction was carried out with 1a (0.1 mmol), KOH (1N) in anhydrous EtOH (2 ml) at room temperature for several hours; condition C: the reaction was carried out with 1a (0.1 mmol), Mg (1 eq.) and NH4Cl (1.2 eq.) in anhydrous MeOH (2 ml) at room temperature for several hours. The condition A and C led to no reaction, and condition B led to a mess reaction.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
TD and YQ contributed to the conception and design of the study. The synthetic work and data collection were carried out by TD, ML, and FG. The first draft of the article was done by TD and YQ. YQ and PW contributed to the article revision. All authors read and approved the submitted version.
Funding and Acknowledgements
We thank for financial support provided by the National Natural Science Foundation of China, Grant/Award Number: 82104412; Shaanxi Natural Science Basic Research Program, Grant/Award Number: 2020JQ-865; Scientifific Research Projects of Shaanxi Provincial Department of Education, Grant/Award Number: 20JK0597; Subject Innovation Team of Shaanxi University of Chinese Medicine, Grant/Award Number: 2019-QN02.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fchem.2021.764866/full#supplementary-material.
References
- Afzal O., Kumar S., Haider M. R., Ali M. R., Kumar R., Jaggi M., et al. (2015). A Review on Anticancer Potential of Bioactive Heterocycle Quinoline. Eur. J. Med. Chem. 97, 871–910. 10.1016/j.ejmech.2014.07.044 [DOI] [PubMed] [Google Scholar]
- Babanezhad Harikandei K., Salehi P., Ebrahimi S. N., Bararjanian M., Kaiser M., Khavasi H. R., et al. (2019). N-substituted Noscapine Derivatives as New Antiprotozoal Agents: Synthesis, Antiparasitic Activity and Molecular Docking Study. Bioorg. Chem. 91, 103116. 10.1016/j.bioorg.2019.103116 [DOI] [PubMed] [Google Scholar]
- Caruana L., Kniep F., Johansen T. K., Poulsen P. H., Jørgensen K. A. (2014). A New Organocatalytic Concept for Asymmetric α-Alkylation of Aldehydes. J. Am. Chem. Soc. 136, 15929–15932. 10.1021/ja510475n [DOI] [PubMed] [Google Scholar]
- Chauhan P., Kaya U., Enders D. (2017). Advances in Organocatalytic 1,6-Addition Reactions: Enantioselective Construction of Remote Stereogenic Centers. Adv. Synth. Catal. 359, 888–912. 10.1002/adsc.201601342 [DOI] [Google Scholar]
- Chen C., Janoszka N., Wong C. K., Gramse C., Weberskirch R., Gröschel A. H. (2021). Scalable and Recyclable All‐Organic Colloidal Cascade Catalysts. Angew. Chem. Int. Ed. 60, 237–241. 10.1002/anie.202008104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K., Hao W.-J., Tu S.-J., Jiang B. (2019). Metal-free Synthesis of Triarylated (Z)-nitrones via H2O-Mediated 1,3-dipolar Transfer under Aerobic Conditions. Green. Chem. 21, 675–683. 10.1039/C8GC03593H [DOI] [Google Scholar]
- Chen X.-M., Xie K.-X., Yue D.-F., Zhang X.-M., Xu X.-Y., Yuan W.-C. (2018). Catalyst-free Synthesis of 2,3-dihydrobenzofurans through [4+1] Cycloaddition of Ortho -hydroxyphenylsubstituted Para- Quinone Methides and Sulfur Ylides. Tetrahedron 74, 600–605. 10.1016/j.tet.2017.12.038 [DOI] [Google Scholar]
- Chen Y.-F., Lin Y.-C., Huang P.-K., Chan H.-C., Kuo S.-C., Lee K.-H., et al. (2013). Design and Synthesis of 6,7-Methylenedioxy-4-Substituted Phenylquinolin-2(1h)-One Derivatives as Novel Anticancer Agents that Induce Apoptosis with Cell Cycle Arrest at G2/M Phase. Bioorg. Med. Chem. 21, 5064–5075. 10.1016/j.bmc.2013.06.046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu W.-D., Zhang L.-F., Bao X., Zhao X.-H., Zeng C., Du J.-Y., et al. (2013). Asymmetric Catalytic 1,6-Conjugate Addition/Aromatization Ofpara-Quinone Methides: Enantioselective Introduction of Functionalized Diarylmethine Stereogenic Centers. Angew. Chem. Int. Ed. 52, 9229–9233. 10.1002/anie.201303928 [DOI] [PubMed] [Google Scholar]
- Deng Y.-H., Zhang X.-Z., Yu K.-Y., Yan X., Du J.-Y., Huang H., et al. (2016). Bifunctional Tertiary Amine-Squaramide Catalyzed Asymmetric Catalytic 1,6-conjugate Addition/aromatization of Para-Quinone Methides with Oxindoles. Chem. Commun. 52, 4183–4186. 10.1039/C5CC10502A [DOI] [PubMed] [Google Scholar]
- Donckele E. J., Finke A. D., Ruhlmann L., Boudon C., Trapp N., Diederich F. (2015). The [2 + 2] Cycloaddition-Retroelectrocyclization and [4 + 2] Hetero-Diels-Alder Reactions of 2-(Dicyanomethylene)indan-1,3-Dione with Electron-Rich Alkynes: Influence of Lewis Acids on Reactivity. Org. Lett. 17, 3506–3509. 10.1021/acs.orglett.5b01598 [DOI] [PubMed] [Google Scholar]
- Dong N., Zhang Z.-P., Xue X.-S., Li X., Cheng J.-P. (2016). Phosphoric Acid Catalyzed Asymmetric 1,6-Conjugate Addition of Thioacetic Acid to para -Quinone Methides. Angew. Chem. Int. Ed. 55, 1460–1464. 10.1002/anie.201509110 [DOI] [PubMed] [Google Scholar]
- Fioravanti S., Pellacani L., Vergari M. C. (2012). Domino Reactions for the Synthesis of Various α-substituted nitro Alkenes. Org. Biomol. Chem. 10, 524–528. 10.1039/C1OB06260C [DOI] [PubMed] [Google Scholar]
- Ghosh K. G., Das D., Chandu P., Sureshkumar D. (2021). Visible‐Light Driven Organo‐photocatalyzed Multicomponent Reaction for C( Sp 3 )−H Alkylation of Phosphoramides with In Situ Generated Michael Acceptors. Eur. J. Org. Chem. 2021, 4293–4298. 10.1002/ejoc.202100561 [DOI] [Google Scholar]
- He F.-S., Jin J.-H., Yang Z.-T., Yu X., Fossey J. S., Deng W.-P. (2016). Direct Asymmetric Synthesis of β-Bis-Aryl-α-Amino Acid Esters via Enantioselective Copper-Catalyzed Addition of P-Quinone Methides. ACS Catal. 6, 652–656. 10.1021/acscatal.5b02619 [DOI] [Google Scholar]
- Huang B. X., Newcomer K., Kevala K., Barnaeva E., Zheng W., Hu X., et al. (2017). Identification of 4-Phenylquinolin-2(1h)-One as a Specific Allosteric Inhibitor of Akt. Sci. Rep. 7, 11673. 10.1038/s41598-017-11870-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H.-M., Wu X.-Y., Leng B.-R., Zhu Y.-L., Meng X.-C., Hong Y., et al. (2020). Cu(ii)-Catalyzed Formal [4 + 2] Cycloaddition between Quinone Methides (QMs) and Electron-Poor 3-vinylindoles. Org. Chem. Front. 7, 414–419. 10.1039/C9QO01343A [DOI] [Google Scholar]
- Jiang F., Yuan F.-R., Jin L.-W., Mei G.-J., Shi F. (2018a). Metal-Catalyzed (4 + 3) Cyclization of Vinyl Aziridines with Para-Quinone Methide Derivatives. ACS Catal. 8, 10234–10240. 10.1021/acscatal.8b03410 [DOI] [Google Scholar]
- Jiang X. L., Wu S. F., Wang J. R., Mei G. J., Shi F. (2018b). Catalytic Asymmetric [4+2] Cyclization of para ‐Quinone Methide Derivatives with 3‐Alkyl‐2‐vinylindoles. Adv. Synth. Catal. 360, 4225–4235. 10.1002/adsc.201800829 [DOI] [Google Scholar]
- Kraus J. M., Verlinde C. L. M. J., Karimi M., Lepesheva G. I., Gelb M. H., Buckner F. S. (2009). Rational Modification of a Candidate Cancer Drug for Use against Chagas Disease. J. Med. Chem. 52, 1639–1647. 10.1021/jm801313t [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Yuan H., Liu Z., Zhang Z., Cheng Y., Li P. (2018). NHC-catalyzed Enantioselective [4+3] Cycloaddition of Ortho -Hydroxyphenyl Substituted Para -Quinone Methides with Isatin-Derived Enals. Adv. Synth. Catal. 360, 2460–2464. 10.1002/adsc.201800337 [DOI] [Google Scholar]
- Li X., Xu X., Wei W., Lin A., Yao H. (2016). Organocatalyzed Asymmetric 1,6-Conjugate Addition of Para-Quinone Methides with Dicyanoolefins. Org. Lett. 18, 428–431. 10.1021/acs.orglett.5b03471 [DOI] [PubMed] [Google Scholar]
- Lima C. G. S., Pauli F. P., Costa D. C. S., de Souza A. S., Forezi L. S. M., Ferreira V. F., et al. (2020). para -Quinone Methides as Acceptors in 1,6-Nucleophilic Conjugate Addition Reactions for the Synthesis of Structurally Diverse Molecules. Eur. J. Org. Chem. 2020, 2650–2692. 10.1002/ejoc.201901796 [DOI] [Google Scholar]
- Liu L., Yuan Z., Pan R., Zeng Y., Lin A., Yao H., et al. (2018a). 1,6-Conjugated Addition-Mediated [4 + 1] Annulation: an Approach to 2,3-dihydrobenzofurans. Org. Chem. Front. 5, 623–628. 10.1039/C7QO00846E [DOI] [Google Scholar]
- Liu Q., Li S., Chen X.-Y., Rissanen K., Enders D. (2018b). Asymmetric Synthesis of Spiro-oxindole-ε-lactones through N-Heterocyclic Carbene Catalysis. Org. Lett. 20, 3622–3626. 10.1021/acs.orglett.8b01400 [DOI] [PubMed] [Google Scholar]
- Lou Y., Cao P., Jia T., Zhang Y., Wang M., Liao J. (2015). Copper-Catalyzed Enantioselective 1,6-Boration Ofpara-Quinone Methides and Efficient Transformation Ofgem-Diarylmethine Boronates to Triarylmethanes. Angew. Chem. Int. Ed. 54, 12134–12138. 10.1002/anie.201505926 [DOI] [PubMed] [Google Scholar]
- Lu H., Zhang H.-X., Tan C.-Y., Liu J.-Y., Wei H., Xu P.-F. (2019). One-Pot Asymmetric Synthesis of Spiropyrazolone-Linked Cyclopropanes and Benzofurans through a General Michael Addition/Chlorination/Nucleophilic Substitution Sequence. J. Org. Chem. 84, 10292–10305. 10.1021/acs.joc.9b01454 [DOI] [PubMed] [Google Scholar]
- Ma C., Huang Y., Zhao Y. (2016). Stereoselective 1,6-Conjugate Addition/Annulation of Para-Quinone Methides with Vinyl Epoxides/Cyclopropanes. ACS Catal. 6, 6408–6412. 10.1021/acscatal.6b01845 [DOI] [Google Scholar]
- Mei G.-J., Xu S.-L., Zheng W.-Q., Bian C.-Y., Shi F. (2018). [4 + 2] Cyclization of Para-Quinone Methide Derivatives with Alkynes. J. Org. Chem. 83, 1414–1421. 10.1021/acs.joc.7b02942 [DOI] [PubMed] [Google Scholar]
- Murlykina M. V., Morozova A. D., Zviagin I. M., Sakhno Y. I., Desenko S. M., Chebanov V. A. (2018). Aminoazole-Based Diversity-Oriented Synthesis of Heterocycles. Front. Chem. 6. 10.3389/fchem.2018.00527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noolvi M. N., Patel H. M., Bhardwaj V., Chauhan A. (2011). Synthesis and In Vitro Antitumor Activity of Substituted Quinazoline and Quinoxaline Derivatives: Search for Anticancer Agent. Eur. J. Med. Chem. 46, 2327–2346. 10.1016/j.ejmech.2011.03.015 [DOI] [PubMed] [Google Scholar]
- Parra A., Tortosa M. (2015). para-Quinone Methide: a New Player in Asymmetric Catalysis. ChemCatChem 7, 1524–1526. 10.1002/cctc.201500176 [DOI] [Google Scholar]
- Ramesh E., Manian R. D. R. S., Raghunathan R., Sainath S., Raghunathan M. (2009). Synthesis and Antibacterial Property of Quinolines with Potent DNA Gyrase Activity. Bioorg. Med. Chem. 17, 660–666. 10.1016/j.bmc.2008.11.058 [DOI] [PubMed] [Google Scholar]
- Roiser L., Waser M. (2017). Enantioselective Spirocyclopropanation of Para-Quinone Methides Using Ammonium Ylides. Org. Lett. 19, 2338–2341. 10.1021/acs.orglett.7b00869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S., Pradhan S., Kumar K., Chatterjee I. (2020). Asymmetric Organocatalytic Double 1,6-addition: Rapid Access to Chiral Chromans with Molecular Complexity. Org. Chem. Front. 7, 1388–1394. 10.1039/D0QO00354A [DOI] [Google Scholar]
- R. Solomon V., Lee H. (2011). Quinoline as a Privileged Scaffold in Cancer Drug Discovery. Cmc 18, 1488–1508. 10.2174/092986711795328382 [DOI] [PubMed] [Google Scholar]
- Si W., Xu F., Liu Z., Song R., Lv J. (2020). High Diastereoselective [4 + 2] Annulation of β,γ-unsaturated α-keto Esters and P-Quinone Methides: Approach to Polysubstituted 4-aryl Chromans and Tetrahydroquinolines. Tetrahedron Lett. 61, 152171. 10.1016/j.tetlet.2020.152171 [DOI] [Google Scholar]
- Staśkiewicz A., Ledwoń P., Rovero P., Papini A. M., Latajka R. (2021). Triazole-Modified Peptidomimetics: An Opportunity for Drug Discovery and Development. Front. Chem. 9. 10.3389/fchem.2021.674705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J.-P., Yu P., Wu J.-H., Chen Y., Pan J., Jiang C., et al. (2019). Bifunctional Phosphonium Salt Directed Enantioselective Formal [4 + 1] Annulation of Hydroxyl-Substituted Para-Quinone Methides with α-Halogenated Ketones. Org. Lett. 21, 7298–7302. 10.1021/acs.orglett.9b02560 [DOI] [PubMed] [Google Scholar]
- Tan J. P., Zhang H., Jiang Z., Chen Y., Ren X., Jiang C., et al. (2020). Enantioselective Construction of Spiro[chroman‐thiazolones]: Bifunctional Phosphonium Salt‐Catalyzed [2+4] Annulation between 5‐Alkenyl Thiazolones and Ortho ‐Hydroxyphenyl‐Substituted Para‐ Quinone Methides. Adv. Synth. Catal. 362, 1058–1063. 10.1002/adsc.201901413 [DOI] [Google Scholar]
- Toteva M. M., Richard J. P. (2011). “The Generation and Reactions of Quinone Methides,” in Advances in Physical Organic Chemistry. Editor Richard J. P. (Academic Press; ), 39–91. 10.1016/b978-0-12-386047-7.00002-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida R., Imasato R., Tomoda H., Ōmura S. (2006). Yaequinolones, New Insecticidal Antibiotics Produced by Penicillium Sp. FKI-2140. J. Antibiot. 59, 652–658. 10.1038/ja.2006.87 [DOI] [PubMed] [Google Scholar]
- V. Kouznetsov V., R. Merchan Arenas D., Arvelo F., S. Bello Forero J., Sojo F., Munoz A. (2010). 4-Hydroxy-3-methoxyphenyl Substituted 3-Methyl-Tetrahydroquinoline Derivatives Obtained through Imino Diels-Alder Reactions as Potential Antitumoral Agents. Lddd 7, 632–639. 10.2174/157018010792929577 [DOI] [Google Scholar]
- Wang J.-Y., Hao W.-J., Tu S.-J., Jiang B. (2020c). Recent Developments in 1,6-addition Reactions of Para-Quinone Methides (P-QMs). Org. Chem. Front. 7, 1743–1778. 10.1039/d0qo00387e [DOI] [Google Scholar]
- Wang J., Pan X., Liu J., Zhao L., Zhi Y., Zhao K., et al. (2018). Diastereoselective Synthesis of Tetrahydroquinolines via [4 + 2] Annulation between In Situ Generated P-Quinone Methides and Nitroalkenes. Org. Lett. 20, 5995–5998. 10.1021/acs.orglett.8b02127 [DOI] [PubMed] [Google Scholar]
- Wang J., Pan X., Rong Q., Zhao L., Zhao L., Dai W., et al. (2020a). One-pot Synthesis of Indoles and Quinolinones from Ortho-Tosylaminophenyl-Substituted Para-Quinone Methides. RSC Adv. 10, 33455–33460. 10.1039/D0RA05497F [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Pan X., Zhao L., Zhao L., Liu J., Zhi Y., et al. (2019). Diastereoselective Construction of 3-Aryl-Substituted Indolines via Annulation of In Situ Generated P-Quinone Methides. Org. Biomol. Chem. 17, 10158–10162. 10.1039/C9OB02351H [DOI] [PubMed] [Google Scholar]
- Wang J., Zhao L., Zhao L., Pan X., Lv C., Zhi Y., et al. (2020b). Diastereoselective Synthesis of Tetrahydroquinolines Bearing Oxindole Scaffolds via Annulation of In Situ Generated P ‐Quinone Methides. Adv. Synth. Catal. 362, 2755–2759. 10.1002/adsc.202000436 [DOI] [Google Scholar]
- Wang Z., Wong Y. F., Sun J. (2015). Catalytic Asymmetric 1,6-Conjugate Addition Ofpara-Quinone Methides: Formation of All-Carbon Quaternary Stereocenters. Angew. Chem. Int. Ed. 54, 13711–13714. 10.1002/anie.201506701 [DOI] [PubMed] [Google Scholar]
- Xiong Y.-J., Shi S.-Q., Hao W.-J., Tu S.-J., Jiang B. (2018). A New Dehydrogenative [4 + 1] Annulation between Para-Quinone Methides (P-QMs) and Iodonium Ylides for the Synthesis of 2,3-dihydrobenzofurans. Org. Chem. Front. 5, 3483–3487. 10.1039/C8QO00950C [DOI] [Google Scholar]
- Yang G.-H., Zhao Q., Zhang Z.-P., Zheng H.-L., Chen L., Li X. (2019). Asymmetric Cycloaddition of Ortho-Hydroxyphenyl-Substituted Para-Quinone Methides and Enamides Catalyzed by Chiral Phosphoric Acid. J. Org. Chem. 84, 7883–7893. 10.1021/acs.joc.9b00749 [DOI] [PubMed] [Google Scholar]
- You Y., Li T.-T., Yuan S.-P., Xie K.-X., Wang Z.-H., Zhao J.-Q., et al. (2020). Catalytic Asymmetric [4+2] Cycloaddition of 1-((2-Aryl)vinyl)naphthalen-2-Ols with In Situ Generated Ortho-Quinone Methides for the Synthesis of Polysubstituted Chromanes. Chem. Commun. 56, 439–442. 10.1039/C9CC08316B [DOI] [PubMed] [Google Scholar]
- Zhang J. R., Jin H. S., Wang R. B., Zhao L. M. (2019). Synthesis of 2,4‐Diaryl‐1,3‐benzoxazines via FeCl 3 ‐Catalyzed Annulation of ortho ‐Hydroxyphenyl‐Substituted para ‐Quinone Methides with Imidates. Adv. Synth. Catal. 361, 4811–4816. 10.1002/adsc.201900842 [DOI] [Google Scholar]
- Zhang X.-Z., Deng Y.-H., Gan K.-J., Yan X., Yu K.-Y., Wang F.-X., et al. (2017). Tandem Spirocyclopropanation/Rearrangement Reaction of Vinyl P-Quinone Methides with Sulfonium Salts: Synthesis of Spirocyclopentenyl P-Dienones. Org. Lett. 19, 1752–1755. 10.1021/acs.orglett.7b00516 [DOI] [PubMed] [Google Scholar]
- Zhang X.-Z., Du J.-Y., Deng Y.-H., Chu W.-D., Yan X., Yu K.-Y., et al. (2016). Spirocyclopropanation Reaction of Para-Quinone Methides with Sulfonium Salts: The Synthesis of Spirocyclopropanyl Para-Dienones. J. Org. Chem. 81, 2598–2606. 10.1021/acs.joc.5b02725 [DOI] [PubMed] [Google Scholar]
- Zhang Z.-P., Chen L., Li X., Cheng J.-P. (2018). Organocatalytic Asymmetric Sequential 1,6-Addition/Acetalization of 1-Oxotetralin-2-Carbaldehyde to Ortho-Hydroxyphenyl-Substituted Para-Quinone Methides for Synthesis of Spiro-3,4-dihydrocoumarins. J. Org. Chem. 83, 2714–2724. 10.1021/acs.joc.7b03177 [DOI] [PubMed] [Google Scholar]
- Zhao K., Zhi Y., Shu T., Valkonen A., Rissanen K., Enders D. (2016). Organocatalytic Domino Oxa-Michael/1,6-Addition Reactions: Asymmetric Synthesis of Chromans Bearing Oxindole Scaffolds. Angew. Chem. Int. Ed. 55, 12104–12108. 10.1002/anie.201606947 [DOI] [PubMed] [Google Scholar]
- Zhi Y., Zhao K., von Essen C., Rissanen K., Enders D. (2018). Synthesis of Trans-disubstituted-2,3-dihydrobenzofurans by a Formal [4 + 1] Annulation between Para-Quinone Methides and Sulfonium Salts. Org. Chem. Front. 5, 1348–1351. 10.1039/C8QO00008E [DOI] [Google Scholar]
- Zhou J., Liang G., Hu X., Zhou L., Zhou H. (2018). Facile Synthesis of 3-aryl 2,3-dihydrobenzofurans via Novel Domino 1,6-addition/O-Alkylation Reactions of Para-Quinone Methides. Tetrahedron 74, 1492–1496. 10.1016/j.tet.2018.02.008 [DOI] [Google Scholar]
- Zhu L., Yan P., Zhang L., Chen Z., Zeng X., Zhong G. (2017). TiCl4/DMAP Mediated Z-Selective Knovenagel Condensation of Isatins with Nitroacetates and Related Compounds. RSC Adv. 7, 51352–51358. 10.1039/C7RA09951G [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.