

Abstract
Aims
Epidemiologic evidence links ischemic stroke to age, yet the mechanisms that underlie the specific and independent effects of age on stroke remain elusive, impeding the development of targeted treatments. This study tested the hypothesis that age directly aggravates stroke outcomes and proposes inflamm‐aging as a mediator and potential therapeutic target.
Methods
3 months‐ (young) and 18‐20 months‐old (old) mice underwent transient middle cerebral artery occlusion (tMCAO) for 30 minutes followed by 48 hours of reperfusion. Old animals received weekly treatment with the TNF‐α neutralizing antibody adalimumab over 4 weeks before tMCAO in a separate set of experiments. Plasma levels of TNF‐ α were assessed in patients with ischemic stroke and correlated with age and outcome.
Results
Old mice displayed larger stroke size than young ones with increased neuromotor deficit. Immunohistochemical analysis revealed impairment of the blood‐brain barrier in old mice, i.e. increased post‐stroke degradation of endothelial tight junctions and expression of tight junctions‐digesting and neurotoxic matrix metalloproteinases. At baseline, old animals showed a broad modulation of several circulating inflammatory mediators. TNF‐α displayed the highest increase in old animals and its inhibition restored the volume of stroke, neuromotor performance, and survival rates of old mice to the levels observed in young ones. Patients with ischemic stroke showed increased TNF‐α plasma levels which correlated with worsened short‐term neurological outcome as well as with age.
Conclusions
This study identifies TNF‐α as a causative contributor to the deleterious effect of aging on stroke and points to inflamm‐aging as a mechanism of age‐related worsening of stroke outcomes and potential therapeutic target in this context. Thus, this work provides a basis for tailoring novel stroke therapies for the particularly vulnerable elderly population.
Keywords: ageing, inflamm‐ageing, inflammation, ischaemic stroke, matrix metalloproteinases, TNF‐α
1. INTRODUCTION
Age associates with the incidence of ischaemic stroke in both men and women. 1 Given the ongoing demographic shift, stroke will likely pose an ever‐greater threat to individual's independence and quality of life with ageing, and strain on healthcare systems in the coming decades. 2 This challenge stands in stark contrast to the relatively limited treatment options for this disease which aim at early reperfusion of the ischaemic territory. Limitations of reperfusion therapies include a narrow temporal window and numerous contraindications particularly prevalent in the elderly. 3 Furthermore, despite the strong epidemiological link with cerebrovascular accidents, 4 , 5 the specific molecular mechanisms that explain the effect of ageing on stroke outcome remain obscure. The lack of effective age‐tailored therapies reflects this knowledge gap. Studying ageing and its effects in humans is complex as it always occurs in parallel to other risk factors for stroke. 6 From this point of view, mice offer an experimental advantage since as they age, they do not spontaneously develop cardiovascular risk factors such as hypertension, hyperlipidemia or diabetes and thus permit isolation of effects of ageing. In rodents, ageing ‘per se’ associates with a decline in cardiac and vascular function suggesting that a genetically determined biological clock governs lifespan and does so by promoting adverse changes causing organ dysfunction. 7
Considered as ‘immune‐privileged’ under physiological conditions, the central nervous system shows deep interactions with the immune system during pathological states. In this regard, inflammation has recently emerged as an important determinant of stroke progression. 8 The innate immune system with its cellular and humoral factors responds first. Indeed, inflammatory cytokines, secreted by circulating and resident immune cells, as well as dying neurons, regulate important processes related to the response to ischaemic brain injury. Levels of these mediators portend adverse outcome in stroke patients. 9 , 10 Recent experimental evidence also support their role as promising therapeutic targets in cardiovascular (CV) accidents. 11 With age, the innate immune system becomes chronically activated as reflected by persistently increased levels of pro‐inflammatory cytokines, while the adaptive immune response wanes. 12 Hallmarks of this dysregulation include increased systemic levels of interleukin (IL)‐1β, IL‐6, tumour necrosis factor (TNF)‐α and of the biomarker C‐reactive protein (CRP). 13 This state of sterile persistent chronic low‐grade inflammation, known as ‘inflamm‐ageing’, likely contributes influentially to the pathophysiology of different age‐dependent conditions, including cerebrovascular diseases. 14 , 15
Hence, we hypothesize that age, independently of concomitant risk factors, not only pre‐disposes to strokes, but also directly adversely affects outcome. This study therefore investigated whether inflamm‐ageing represents a pathophysiological link between age and adverse stroke outcome and if intervention by direct targeting of specific pro‐inflammatory cytokines ameliorates stroke outcome in old animals. To test the translational relevance of our findings and investigate whether the elderly exhibit a dysregulated inflammatory response in response to sterile insults, plasma levels of TNF‐α were assessed in ischaemic stroke patients and correlated with age and outcome.
2. METHODS
2.1. Animals
Male C57BL/6 WT mice from the ageing colony established in our facility at the University of Zurich, Zurich, Switzerland, we used for all experiments. This study used 3‐month‐old mice for the young arm and 18‐ to 20‐month‐old mice for the old arm. All rodents were kept in a temperature‐controlled animal facility under normal light/dark cycle with free access to food and water. All procedures were approved by the local Ethical Committee for animal research and the Cantonal Veterinary Authority. Animal experiments conformed to the Directive 2010/63/EU of the European Parliament and of the Council of 22 September 2010 on the protection of animals used for scientific purposes.
2.2. Transient middle cerebral artery occlusion
Transient middle cerebral artery occlusion (tMCAO) induced ischaemia/reperfusion (I/R) brain injury, as previously described. 16 , 17 Briefly, mice were anaesthetized using isoflurane 5% and 1.5% for induction and maintenance, respectively, while body temperature was tightly maintained at 37°C For analgesia, buprenorphine HCl was infiltrated at the incision side (0.1 mg/Kg). Ischaemia was induced by inserting a 6‐0 silicone‐coated filament (Doccol Corporation) into the common carotid artery until the origin of the left MCA after the dissection of common, internal and external carotid arteries. After 30 minutes of ischaemia, the filament was retracted, and reperfusion allowed for 48 hours before animal euthanasia with carbon dioxide. During this time, animals were carefully observed and received analgesia with buprenorphine‐HCL at a dose of 0.1 mg/kg s.c. every 6 hours. A score sheet approved by the Cantonal Veterinary Office of the Canton of Zurich monitored the well‐being of mice during the experimental period.
2.3. Adalimumab treatment
In a set of experiments, old mice were randomly assigned to either weekly intraperitoneal injections of adalimumab (Humira®, AbbVie) at the dose of 10 mg/kg bodyweight for 4 weeks (old and adalimumab) or an equivalent regimen of vehicle (ie distilled water; old). Young animals received the same vehicle treatment (Young). Such animals underwent tMCAO one week after the latest injection. Adalimumab was acquired from the Zurich cantonal pharmacy and previously showed to neutralize both human and murine TNF‐α. 18
2.4. Cerebral infarct volume
After euthanasia, mice were perfused with 10 mL of phosphate‐buffered saline (PBS) and relevant organs were excised. Murine brains were cut into 5 equally spaced (2 mm) coronal sections and immersed in a 2% solution of 2,3,5‐triphenyltetrazolium chloride (TTC) (Sigma‐Aldrich, Chemie GmbH) at 37°C for 20 minutes. 19 To measure infarct size, ipsilateral and contralateral hemispheres were quantified using ImageJ software (Image J, NIH). To correct infarct size measurement for cerebral oedema and consequent overestimation, we applied the following formula as previously described 20 : Corrected infarct volume =contralateral hemisphere volume ‐ (ipsilateral hemisphere volume ‐ infarct volume). Infarct size was expressed as volume in mm3.
2.5. Neurological assessment
Baseline and post‐infarction neurological status were assessed by a four‐point scale neurological score according to Bederson et al and the Rotarod test as previously described. 21 Both composite sensory‐motor tests evaluate motor functions, proprioception, spatial orientation and balance. The neurological score test according to Bederson was performed at baseline, 2, 24 and 48 hours after reperfusion according to the following scores: grade 0, normal neurological function; grade 1, forelimb and torso flexion on and towards the contralateral side upon lifting of the animal by the tail to 1 m above the work surface; grade 2, circling to the contralateral side; grade 3, leaning to the contralateral side at rest; grade 4, no spontaneous motor activity. The Rotarod test was performed at baseline, 24 and 48 hours after reperfusion. Mice were placed on a rotating rod at increasing speed (4‐44 rot/min), and the time to fall was measured in seconds. Three consecutive measurements were performed at each time point for each animal, and the best score was used.
2.6. Plasma and brain sampling for cytokine assessment
A dedicated set of mice not undergoing tMCAO was employed for baseline plasma and brain cytokine sampling. Blood was collected via intracardiac puncture and immediately mixed with EDTA. The EDTA‐blood solution was then centrifuged for 15 minutes at 3000 g as previously described. 22 Plasma was collected and snap‐frozen in liquid nitrogen. The Proteome Profiler™ Antibody Array (R&D Systems) was employed for the semi‐quantitative determination of several mediators of inflammation in murine pooled plasma (3 mice).
Brains were collected from after euthanasia and immediately snap‐frozen in liquid nitrogen. Later, they were homogenized in the lysis buffer (Tris 50 mM, NaCl 150 mM, EDTA 1 mmol/L, NaF 1 mmol/L, DTT 1 mmol/L, aprotinin 10 mg/mL, leupeptin 10 mg/mL, Na3VO4 0.1 mmol/L, phenylmethylsulfonyl fluoride (PMSF) 1 mmol/L and NP‐40 0.5%) and total protein concentration was determined by the Bradford protein assay according to the manufacturer's recommendations (VWR Life Science AMRESCO).
Colorimetric enzyme‐linked immunosorbent assays (ELISA) quantitatively assessed IL‐1β and TNF‐α levels in murine EDTA‐plasma and brain samples following the manufacturer instruction (R&D Systems). Mean intra‐ and inter‐assay coefficients of variation were <10%, and lower level of detection was 0.8 pg/mL for both. For brain lysates, protein concentration as detected by ELISA was then normalized according to the total protein content of the sample and expressed as pg per mg of total protein.
2.7. Immunohistologic analyses
Immunohistochemical staining was performed as previously described. 16 Briefly, after 48 h from tMCAO mice were euthanized and perfused with PBS (Sigma‐Aldrich, Chemie GmbH). The brains were removed and consecutively incubated overnight in 4.0% paraformaldehyde (PFA; Sigma‐Aldrich, Chemie GmbH) at 4°C and afterwards transferred to 30% sucrose in PBS for 36 hours. Cryoprotected brains were cut into 100‐µm thick free‐floating sections using a microtome (Leica Jung HN40), pre‐treated with proteinase K or 1 M HCl for antigen retrieval and immune‐blocked with 10% donkey serum. After these steps, they were incubated with primary antibodies at the following dilutions: Iba‐1 (1:500; Wako Chemicals), Occludin (1:200; Santa Cruz Biotechnology), the endothelial marker CD31 (1:50; BD Pharmingen), claudin‐5 (1:200; Abcam), MMP‐3 (1:100; Abcam) and MMP‐9 (1:500; Abcam) at 4°C overnight, respectively. Secondary antibodies were added at a dilution of 1:750 (Jackson Immunoresearch) for 24 hours at 4°C. Images were acquired using a confocal microscope (Leica SP8; Leica). Cells positively stained for the microglial and activated macrophage marker Iba‐1 were counted in specified ipsilateral area (the CA1 hippocampal area) using ImageJ software. Stained areas of claudin‐5 and occludin were measured in the penumbra area of the stroke using ImageJ software and co‐localized to the area positively stained with the endothelial marker CD31. The area stained for both proteins of interest and endothelial marker is expressed as a percentage of the total endothelial surface area. Stained areas of MMP‐9 and MMP‐3 were assessed in the penumbra area of the stroke using ImageJ and normalized to the total endothelial cell surface area assessed by CD31 staining.
BBB permeability was assessed by quantifying endogenous immunoglobulin G (IgG) extravasation. Sections were incubated with Alexa 647‐conjugated donkey anti‐mouse IgG for 24 hours (1:600; Jackson Immunoresearch). IgG‐stained area was expressed as a percentage of the contralateral hemisphere.
2.8. Acute ischaemic stroke patients
Twenty‐three patients admitted to the emergency room of San Raffaele Hospital (OSR, Milan, Italy) with a diagnosis of acute ischaemic stroke who presented within 6 hours from symptom onset were enrolled. Five patients had wake‐up stroke and were recruited within 6 hours from awakening. The initial diagnosis was based on clinical history, neurological examination (conducted by certified neurologists) and head computed tomography (CT). Eighteen sex‐ and age‐matched healthy volunteers (either relatives or visitors of in‐hospital patients), with a negative history of cardio‐ and cerebrovascular diseases, were included as controls. Patients diagnosed with diabetes, systemic inflammatory diseases, acute infections and malignancy were excluded. Blood was withdrawn from the antecubital vein at 6 and 24 hours after initial stroke symptoms (for stroke patients), whereas control subjects donated blood once.
Ischaemic strokes were clinically classified according to the Oxford Community Stroke Project classification (also known as the Bamford or OXFORD classification). Stroke aetiology was classified according to the Trial of ORG 10172 in Acute Stroke Treatment criteria. Stroke severity was assessed using NIHSS on hospital admission and at discharge. Furthermore, ΔNIHSS% was calculated as the difference between the NIHSS presented at discharge and the NIHSS presented at admission relativized on initial NIHSS [ΔNIHSS% = (NIHSS discharge −NIHSS admission)/NIHSS admission * 100]; thereby, positive values indicate short‐term neurologic worsening while negative values indicate neurological improvement. The study was approved by the local Ethics Committee at San Raffaele Scientific Institute, Milan, Italy and was performed conform to the declaration of Helsinki. All participants (or their representative relatives) provided signed informed consent.
Circulating levels of TNF‐α were assessed in EDTA‐plasma samples by mean of high sensitivity ELISA kit following the manufacturer instructions (R&D system, HSTA00E). Lower level of detection was 0.2 pg/mL.
2.9. Statistical analysis
Data are expressed as mean ± SEM. All statistical analyses were performed using GraphPad Prism 6 software (GraphPad Software, Inc). Data were analysed by one‐way analysis of variance (ANOVA) with Tukey's post hoc test for multiple comparisons or unpaired two‐tailed Student's t test, as appropriate. For repeated measurements, two‐way ANOVA with Tukey's post hoc test was used. Statistical analysis for survival studies was performed using log‐rank (Mantel‐Cox) test. A probability value (p) below 0.05 was considered as statistically significant.
3. RESULTS
3.1. Ageing ‘per se’ causes increased infarct size and worsened post‐stroke neurological deficit
To assess the specific effect of ageing on ischaemic stroke outcome, 3‐month‐old (young) and 18‐ to 20‐month‐old (old) mice underwent tMCAO for 30 minutes followed by 48 h of reperfusion (Figure 1A). Young mice displayed a stroke size 2 times smaller than that of old animals, as assessed by TTC staining (Figure 1B). Furthermore, 24 and 48 h after stroke the neurological performance as assessed by Bederson scale was less impaired in young mice compared to old ones (Figure 1C). In line with the above, old animals also had a worse neuromotor function than young ones, falling markedly earlier from the rotating rod as assessed by Rotarod test (Figure 1D).
FIGURE 1.
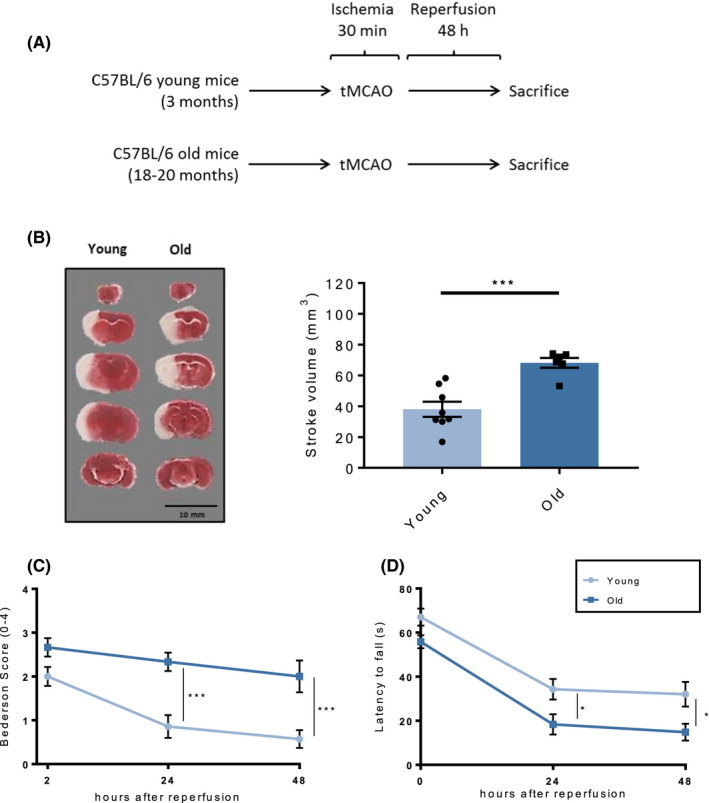
Morphological and functional ischaemic stroke outcome in young and old mice. A, Schematic of the experimental study design. B, 48 h after tMCAO, old mice showed larger cerebral infarcted areas as assessed on TTC stained coronal sections in which the white colour distinguishes the stroke area from viable tissue (representative staining on the left, quantification on the right). Accordingly, old animals also showed worsened post‐stroke neuromotor function as assessed by C, RotaRod test or D, Bederson‐based neurological score, as compared to young mice. n = 6‐8 different mice per group. B: unpaired two‐tailed Student's t test, C‐D: two‐way analysis of variance (ANOVA) with Tukey's post hoc test. *P < .05, ***P < .001. tMCAO, transient middle cerebral artery occlusion; TTC = 2,3,5‐triphenyltetrazolium chloride
3.2. Ageing associates with increased I/R‐induced blood‐brain barrier (BBB) disruption and tight junction protein (TJP) degradation
Brain I/R‐injury affects many cerebral components, including the BBB. BBB damage is a major determinant of stroke outcome as it contributes to extravasation of large circulating molecules (such as IgG), vasogenic oedema and haemorrhagic transformation. As expected, immunohistochemical analysis demonstrated IgG extravasation into ischaemic hemispheres of all experimental groups with increased leakage in old mice indicating aggravated BBB disruption upon I/R as compared with young animals (P < .01, Figure 2A).
FIGURE 2.
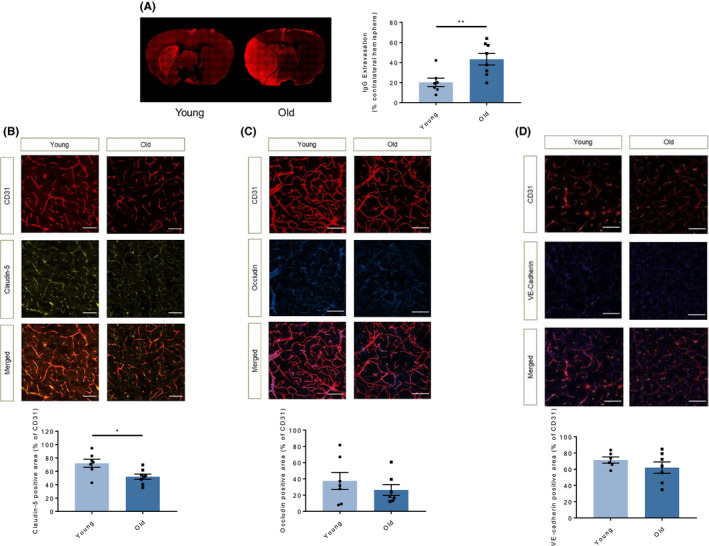
Ageing associates with increased blood‐brain barrier permeability through decreased tight junction protein levels. A, Representative images showing endogenous IgG extravasation (red) into the brain parenchyma 48 h after tMCAO in the two study groups. BBB permeability of old mice was increased as compared to that observed in young animals. B, Immunostaining for claudin‐5 (light yellow) and the endothelial marker CD31 (red) in ipsilateral hemispheres 48 h after tMCAO showed significantly reduced endothelial expression of this molecule in old animals as compared to young ones. C, The expression of occludin (blue) showed a similar (although not significant) trends in endothelial cells (CD31, red) of the penumbra area of the two study groups. D, The endothelial expression (CD31, red) of another protein involved in regulation of BBB function VE‐cadherin (dark yellow) did not show any difference between young and old mice. n = 6‐8 different mice per group. A‐D: unpaired two‐tailed Student's t test. *P <.05, **P < .01, (white bar = 50μm for all). IgG, immunoglobulin G; tMCAO, transient middle cerebral artery occlusion; TNF, tumour necrosis factor; VE‐cadherin, vascular endothelium cadherin
Tight and adherens‐junctional proteins (TJP and AJP, respectively) regulate the integrity of the endothelial component of the BBB. During stroke, the concentration of these proteins declines and the paracellular permeability of the BBB increases. Concordant with the IgG extravasation data, immunohistochemical analysis of endothelial claudin‐5—one of the TJP deeply implicated in stroke pathophysiology—showed a significant reduction of this protein in the penumbra area of old animals as compared to young ones (P < .05, Figure 2B). Occludin and VE‐cadherin showed a similar trend towards decreased expression in old animals, although in these cases it did not reach statistical significance (Figure 2C‐D). The representative images depict a specified area in the ipsilateral hemisphere, where the vasculature appears in red in the top panel (for the endothelial marker CD31) and the respective AJP and TJP in light yellow (claudin‐5), blue (occludin) or dark yellow (VE‐cadherin) in the middle panel. The bottom panel shows an overlay of the junctional protein staining with the total endothelial surface (Figure 2B‐D).
3.3. Age increases I/R‐induced metalloproteinase expression in the penumbra
Following ischaemia, activation of resident immune cells of the brain (ie microglia) and infiltration of monocytes from the circulating pool contributes importantly to brain tissue damage. To assess the contribution of activated microglia and invading macrophages to the observed alterations in stroke outcomes and BBB function, we measured their marker Iba‐1 in the ipsilateral hemisphere of animals after tMCAO. Immunohistochemical counting of Iba1+ cells did not show any difference in terms of activated macrophages in the penumbra area of the different study groups (Figure 3A).
FIGURE 3.
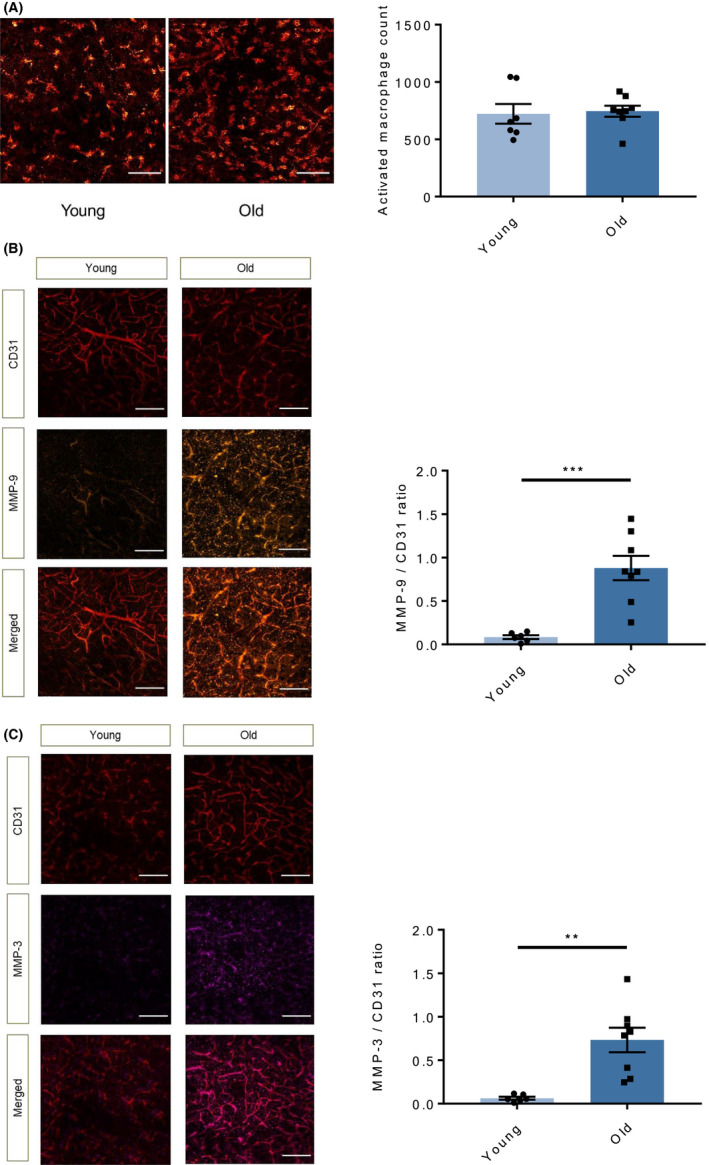
Ageing increases MMP content in the penumbra area of stroke without affecting mononuclear phagocyte activation. A, Immunostaining for the activated microglia/macrophage marker Iba1 (red) in the penumbra area of brain 48 h after tMCAO showed similar counts in the two study groups. B, Immunostaining quantification of MMP‐9 levels (orange) normalized to total endothelial surface (CD31 positive area, red) showed significantly increased levels of this mediator in the penumbra area of old animals as compared with young ones. C, Staining for MMP‐3 yielded similar findings (violet). n = 6‐8 different mice per group. A: B‐C: unpaired two‐tailed Student's t test. **P < .01, ***P < .001, (white bar =100μm for A, white bar =50μm for B and C). MMP, matrix metalloproteinases; tMCAO, transient middle cerebral artery occlusion; TNF, tumour necrosis factor
Among inflammatory mediators, MMPs may exacerbate cerebral parenchymal damage through different mechanisms ranging from enzymatic degradation of TJP and AJP to direct neurotoxicity. 48 hours after tMCAO, immunoreactive MMP‐9 was higher in the penumbra area of old mice as compared to that of younger animals (P < .001, Figure 3B). MMP‐3 expression was also higher in aged mice compared to young ones (P < .01, Figure 3C). The representative images depict a specified area in the ipsilateral hemisphere, where the vasculature appears in red in the top panel (for the endothelial marker CD31) and the respective MMPs in orange (MMP‐9) or magenta (MMP‐3) in the middle panel. The bottom panel shows an overlay of MMP immunoreactivity with the total endothelial surface, demonstrating increase of said enzymes in the old animals.
3.4. Plasma levels of soluble mediators of inflammation, TNF‐α in particular, increase with age
Increased levels of MMPs point towards inflammation as a mechanism underlying the observed age‐dependent differences. Indeed, a state of chronic low‐grade inflammation accompanies ageing that is termed inflamm‐ageing. To characterize the age‐associated changes in circulating levels of soluble inflammatory mediators, we analysed the pooled plasma from different animals (young vs old, three for each group, not undergoing tMCAO) by using a multiplex antibody array assessing 111 different mouse cytokines simultaneously. As hypothesized, ageing associates with a broad modulation of cytokines and chemokines with some of them being increased and others showing a reduction (Figure 4A; relative values above or below 1, respectively). Among the most modulated cytokines, TNF‐α levels increased over 10‐fold in plasma of aged mice compared to young mice. This observation was confirmed quantitatively by using a commercially available ELISA kit (P < .05, Figure 4B). In contrast, the increase in IL‐1β plasma levels observed by multiplex analysis (Figure 4A) was not confirmed by using the specific quantitative ELISA kit (Figure 4C). Interestingly, when measured locally in brain tissue extracts, neither TNF‐α nor IL‐1β differed significantly in either age groups (Figure 4D‐E).
FIGURE 4.
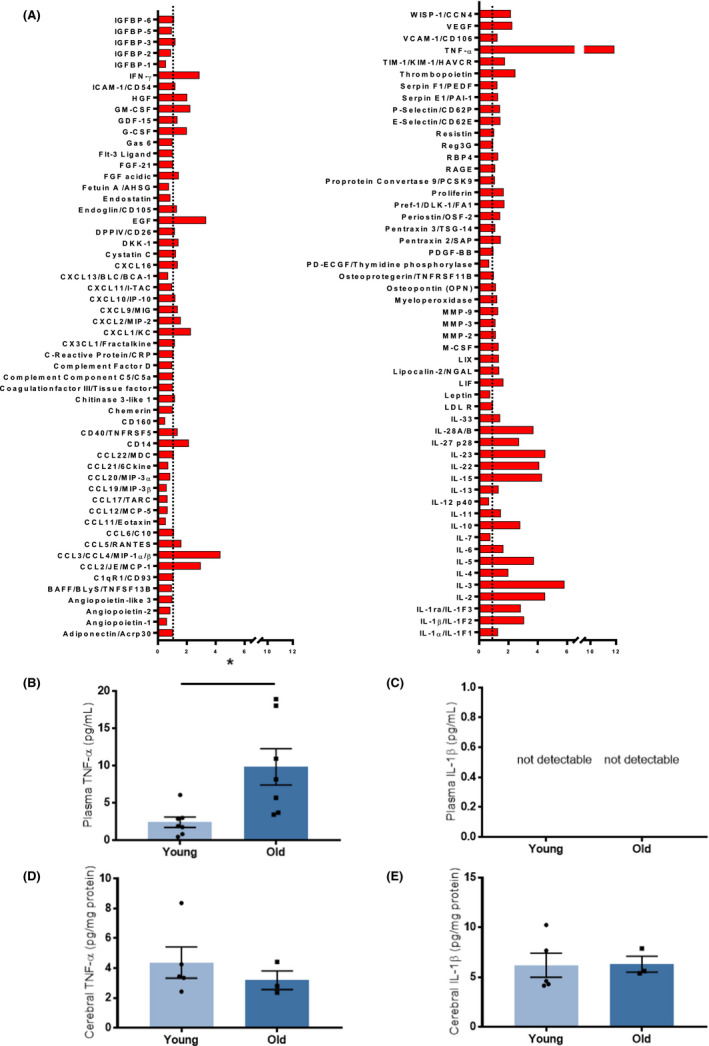
Systemic and cerebral mediators of inflammation in young and old mice. A, Multiplex antibody array assessing 111 different mouse chemokines, cytokines and growth factors simultaneously in pooled plasma from three different animals (young and old, not undergoing tMCAO). Relative expression of mediators of inflammation in old animals as compared with young ones, expression levels were normalized against those from young plasma. Ageing associates with a broad modulation of cytokines and chemokines with some of them being increased and others showing a reduction. B, Circulating levels of TNF‐α increase in plasma samples from old animals as compared to young ones, as assed by specific ELISA. C, Circulating levels of IL‐1β remained below the detection limit of the ELISA kit in both young and old animals. Cerebral levels of TNF‐α D and IL‐1β E, were similar in brain lysates from young and old animals. n = 3‐7 different mice per group. B, D‐E: unpaired two‐tailed Student's t test. *P < .05. IL, interleukin; tMCAO, transient middle cerebral artery occlusion; TNF, tumour necrosis factor
3.5. TNF‐α inhibition rescues the effect of ageing on infarct size, post‐stroke neurological deficit and survival
To assess the implications on stroke outcome of the increased systemic TNF‐α levels observed in old animals, a set of WT mice received the clinically approved TNF‐α‐inhibiting antibody adalimumab (Figure 5A). Weekly treatment with adalimumab over 4 weeks before tMCAO (30 minutes ischaemia/ 48 hours reperfusion), restored stroke volumes in old animals to the levels observed in young ones (P < .001, Figure 5B). Adalimumab treatment also reversed the effect of ageing on post‐stroke neurological function thereby restoring the Bederson performances observed after tMCAO in young mice (Figure 5C). Specifically, adalimumab‐treated old animals showed reduced Bederson scores already 2 hours after the ischaemic events, a finding consolidating over time being highly significant after 24 and 48 hours of reperfusion (P < .05 and P < .01, respectively; Figure 5C). Similarly, old animals show a tendency towards reduced post‐stroke deficit with adalimumab treatment shown by the RotaRod test already 24 h after tMCAO that reached the statistical significance at 48 hours (P < .01, Figure 5D). Moreover, TNF‐α inhibition also improved the survival rate of old animals after stroke as showed by Kaplan‐Meyer survival curves (P < .05, Figure 5E).
FIGURE 5.
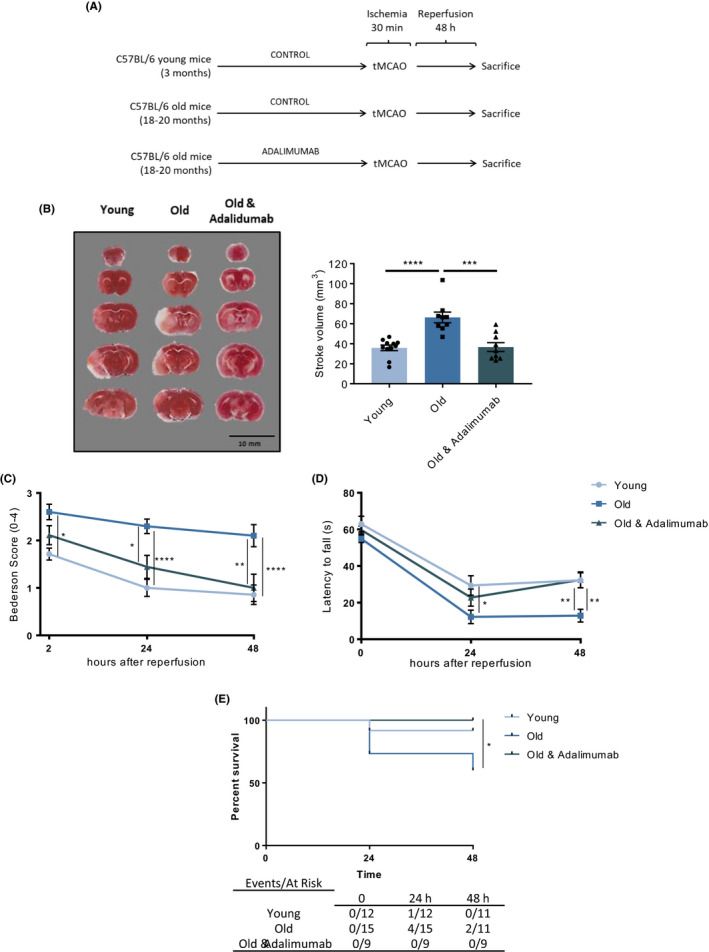
Treatment with adalimumab reverts the effect of ageing on stroke size, functional outcome and survival. A, Schematic of the experimental study design. B, When weekly treated with adalimumab for 4 weeks before tMCAO, old animals showed a significant reduction of the stroke volumes as assessed on TTC‐stained coronal sections in which the white colour distinguishes the stroke area from viable tissue (representative staining on the left, quantification on the right). Accordingly, treatment with the TNF‐α neutralizing antibody improved post‐stroke neurological function as assessed by C, Bederson‐based neurological score or D, Rotarod test, as compared to vehicle‐treated animals. E, Treatment with Adalimumab significantly increased survival rate of old animals at 48 hours after stroke. n = 9‐12 different mice per group. B: one‐way analysis of variance (ANOVA) with Tukey's post hoc test, C‐D: two‐way analysis of variance (ANOVA) with Tukey's post hoc test, E: Log‐rank (Mantel‐Cox test) with Bonferroni's post hoc test. *P <.05, **P <.01, ***P <.001, ****P <.0001. tMCAO, transient middle cerebral artery occlusion; TTC, 2,3,5‐triphenyltetrazolium chloride; TNF, tumour necrosis factor
3.6. Plasma TNF‐α concentrations increase in patients with ischaemic stroke and correlate with age and short‐term outcome
To substantiate the translational relevance of our data, we analysed TNF‐α levels in plasma of 23 ischaemic stroke patients and in 18 age‐ and sex‐matched healthy subjects. Clinical characteristics of both groups did not statistically differ (Table 1). TNF‐α plasma levels increased significantly 6 hours after initial stroke symptoms as compared to healthy controls (P < .05, Figure 6A). A similar trend was observed in plasma isolated 24 hours after the stroke onset although it did not reach the statistical significance (Figure 6A). Circulating TNF‐α levels 24 hours after the stroke onset positively correlated with ΔNIHSS% as a measure of short‐term neurological outcome (r = .532, P < .05, Figure 6B). Levels of TNF‐α did not correlate with the age of the subject in the healthy group (Figure 6C). On the other hand, a positive correlation was found among the age of the patients and plasma levels of the cytokine at 6 hours (r = .446, P < .05, Figure 6D) and 24 hours (r = .611, P < .05, Figure 6E) after the ischaemic stroke onset.
TABLE 1.
Characteristics of the study population
| Controls (n = 18) | Stroke patients (n = 23) | P‐value | |
|---|---|---|---|
| Demographic, risk factors and comorbidities | |||
| Age, years (range) | 72.7 (63‐83) | 74.2 (52‐90) | NS |
| Female, n (%) | 11 (61.1%) | 12 (52.2%) | NS |
| Smoking, n (%) | 1 (5.6%) | 6 (26.1%) | NS |
| Hypertension, n (%) | 8 (44.4%) | 12 (52.2%) | NS |
| Dyslipidaemia, n (%) | 1 (5.6%) | 4 (17.4%) | NS |
| Coronary artery disease, n (%) | 1 (5.6%) | 5 (21.7%) | NS |
| Previous TIA/stroke, n (%) | 0 (‐) | 3 (13%) | NS |
| Peripheral artery disease, n (%) | 0 (‐) | 3 (13%) | NS |
| Medication History | |||
| No antithrombotics, n (%) | 15 (83%) | 11 (48%) | NS |
| Antiplatelets, n (%) | 3 (18%) | 11 (48%) | |
| Anticoagulants, n (%) | 0 (‐) | 1 (4.3%) | |
| Acute phase treatment | |||
| Thrombolysis, n (%) | ‐ | 11 (47.8) | ‐ |
| Loading dose of antiplatelets, n (%) | ‐ | 10 (43%) | ‐ |
| Initiation of anticoagulants, n (%) | ‐ | 1 (4.3%) | ‐ |
| TOAST classification | |||
| Large vessel atherosclerosis, n (%) | ‐ | 5 (21.7%) | ‐ |
| Cardioembolism, n (%) | ‐ | 11 (47.8%) | ‐ |
| Small vessel disease, n (%) | ‐ | 1 (4.3%) | ‐ |
| Undetermined cause, n (%) | ‐ | 3 (13.0%) | ‐ |
| Other cause, n (%) | ‐ | 3 (13.0%) | ‐ |
| Stroke severity assessment | |||
| NIHSS admission, mean (SD) | ‐ | 11.3 (6.0) | ‐ |
| NIHSS discharge, mean (SD) | ‐ | 8.7 (10.2) | ‐ |
| Early complications | |||
| Haemorrhagic transformation, n (%) | ‐ | 2 (8.7%) | ‐ |
| Cerebral oedema, n (%) | ‐ | 3 (13.0%) | ‐ |
Abbreviations: NIHSS, National Institute of Health Stroke Scale; NS, Not Significant; TIA, transient ischaemic attack.
FIGURE 6.
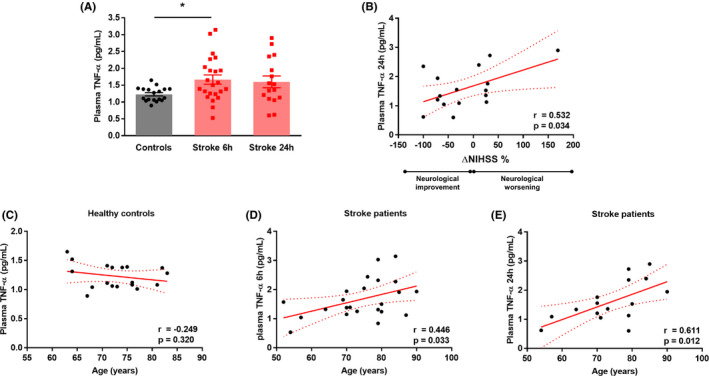
Circulating TNF‐α levels increase in stroke patients and correlate with clinical outcome. A, Circulating levels of TNF‐α 6 h after stroke onset increase significantly in stroke patients compared to healthy age‐ and sex‐matched controls. A similar trend although not significant is observed for levels of this cytokine 24 h after the stroke (n = 18‐23 patients). B, Ischaemic stroke patients show a linear correlation between TNF‐α levels and short‐term neurological outcome as assessed by the National Institute of Health Stroke Scale (NIHSS) (n = 16 patients). C, Healthy controls show no correlation between plasma TNF‐α and age. In contrast, TNF‐α levels at 6 D and 24 h E, after symptom onset correlate with age in patients with ischaemic stroke. A: one‐way analysis of variance (ANOVA) with Tukey's post hoc test, B‐E: Pearson (r) correlation. *P <.05. NIHSS, National Institutes of Health Stroke Scale; TNF, tumour necrosis factor
4. DISCUSSION
Age is a major risk factor for the development of CV disease. Thus, investigating the molecular mechanisms that underlie the effects of age on the vasculature and target organs merits scientific priority, especially in view of the ageing of the global population. 6 This study identifies TNF‐α as a causative mediator of the deleterious effect of ageing on stroke and highlights inflamm‐ageing as a potential therapeutic target in the context of ischaemic stroke of the elderly. Specifically, we demonstrated that old mice, devoid of other traditional CV risk factors, experience larger strokes, increased neuromotor deficit and reduced survival compared to young ones. This effect accompanies aggravated BBB damage as confirmed by reduced levels of the TJP claudin‐5 and increased levels of MMP‐3 and MMP‐9. Furthermore, we systematically characterized humoral mediators of inflammation and show that aged animals have heightened systemic inflammation as reflected by increased levels of cytokines among which TNF‐α showed the largest modulation. In line with a causal effect of TNF‐α on stroke size, the TNF‐α inhibitory antibody adalimumab reversed the deleterious effects of ageing and restored neurological outcomes to that observed in young animals. Lastly, a clinical proof‐of‐principle study demonstrated that plasma levels of TNF‐α indeed increase in patients with acute ischaemic stroke and correlate positively with age and worse short‐term neurological outcome.
The finding that old animals in the absence of any other CV risk factors (a condition rarely found in elderly humans) show bigger ischaemic lesions, worsened post‐stroke neurological performance, decreased survival and increased BBB damage as compared with younger mice, provides novel evidence to underscore the importance of ageing in brain physiology and pathology. 23 Thus, age not only pre‐disposes to stroke incidence, 24 but also unfavourably affects its outcome. As such, mechanisms involved in ageing hold potential as therapeutic targets for age‐tailored management of ischaemic cerebral events. 25 Over the last years, we and others showed that ageing, vascular dysfunction and age‐dependent cardio‐ and cerebrovascular diseases involve similar pathophysiological processes including the production of reactive oxygen species and the surge of inflammation. 16 , 19 , 20 , 26 , 27 , 28 Indeed, age associates with an augmented chronic smouldering innate immune response, known as inflamm‐ageing, that occurs in the absence of appropriate inflammatory stimuli. This inflammatory state may have several causes including immune‐senescence, telomere shortening and defective protein catabolism, autophagy and mitophagy. 29
Given the prominent role of inflammation in determining stroke outcome and the readily availability of clinically applied biologicals that target specifically inflammatory mediators, we sought to investigate soluble inflammation molecules that rise with inflamm‐ageing as mediators and potential target to blunt the deleterious effect of ageing on stroke outcome. We found broad derangement of circulating levels of inflammatory mediators in aged mice in the absence of any other noxious stimulus. Previous experimental and clinical observations reported deranged inflammatory mediators with age 30 , 31 , 32 ; however, the broad approach employed here allowed for the detection of alterations of a wide range of inflammatory mediators showing the highest degree of modulation. The pro‐inflammatory cytokine TNF‐α rose the most among cytokines studied in plasma of aged animals. This result agrees with a previous report 30 and seems to involve circulating pool selectively as indicated by the unaltered levels we observed in cerebral lysates of aged mice. Similarly, in healthy subjects circulating levels of TNF‐α rise with age, 13 they associate with frailty and atherosclerosis 33 , 34 and predict mortality in the elderly. 35 , 36 Consistent with a causal link, employing adalimumab, a monoclonal TNF‐α antibody widely used clinically in a wide range of conditions including bowel inflammatory diseases and different rheumatological diseases, rescued the effects of ageing on stroke size, post‐stroke neurological deficit and mortality. 37
TNF‐α is a cell signalling protein involved in the acute phase reaction and produced by white blood cells (mainly macrophages), endothelial and smooth muscle cells as well as neurons. 38 , 39 By binding to TNF receptor‐1 and 2, TNF‐α activates different intracellular pathways including NF‐κB, mitogen‐activated protein kinases (MAPK) and the apoptotic cascade through caspase‐8, eventually leading to inflammation, fever and cachexia. 39 In addition to its fundamental physiological roles in host defence, tissue repair and inhibition of tumorigenesis, TNF‐α contributes to the development of age‐dependent pathological conditions such as cardio‐ and cerebrovascular diseases. Both experimental and clinical evidence demonstrated a deleterious role for this cytokine in primary and secondary ischaemic stroke. 40 , 41 , 42 , 43 Yet, previous studies have not elucidated fully its function as a mediator of inflamm‐ageing and its specific effects on the stroke of aged animals.
Post‐ischaemic BBB function influences ischaemic stroke outcomes. Indeed, BBB disruption (as assessed by magnetic resonance imaging and single photon emission computed tomography studies) correlates with haemorrhagic transformation, worsened Rankin scores and recovery at time of discharge. 44 Here we show that age associates with increased post‐ischaemic BBB permeability through reduced levels of the junctional protein claudin‐5. Several changes that occur in the aged brain may underlie the higher susceptibility of BBB to the ischaemic damage including arterial remodelling, glial cell activation and increased apoptosis, 45 , 46 , 47 , 48 with previous reports already pointing towards TJP and AJP disassembly as an important culprit mechanism. 49 , 50 In this regard, TNF‐α dose‐dependently decreases the expression of junctional proteins in vitro and play a role in the determination of stroke outcome in experimental inflammatory disease. 41
MMPs are important mediators of inflammation rising in the ischaemic and penumbra area of stroke brains and participate in the degradation of TJP and AJP as well as having direct neurotoxic effects. 51 Among the MMPs, MMP‐9 and MMP‐3—proteinases deeply involved in ischaemic stroke pathophysiology—rose in the penumbra of old animals as compared to young ones. 52 , 53 Microglia and invading macrophages are major sources of MMPs at the site of ischaemic stroke and TNF‐α rapidly induces MMP transcription in those cells contributing to BBB dysfunction and junctional protein degradation in the early phase of stroke. 52 Accordingly, despite similar activated macrophage counts in the experimental groups, old animals showed increased MMP penumbra levels as compared to young ones.
Post‐ischaemic inflammation contributes importantly to determining the outcome after an ischaemic stroke. The complex cytokine network is tightly regulated at different levels and often redundant, showing negative and positive feedback that can further fuel inflammation but can also foster its resolution. Specifically, TNF‐α can induce the transcription of other cytokines including IL‐1 and IL‐6. 54 Our previous work demonstrated specific detrimental effects of different pro‐inflammatory cytokines in this context. Cerebral levels of IL‐1α and β rise after ischaemia/reperfusion and their post‐ischaemic neutralization by inhibitory antibodies reduces infarct size and blunts post‐stroke neurological deficit in mice. 17 , 55 Of note, both IL‐1α and IL‐1β inhibition exert their beneficial effects through preservation of BBB integrity. Nonetheless, whether IL‐1β blockade reduced post‐ischaemic cerebral neutrophil infiltration and MMP‐2 thereby preserving VE‐cadherin expression, 55 inhibition of IL‐1α did not modulate the endothelial expression of such regulators of paracellular BBB permeability. Rather, blocking IL‐1α modulated endothelial activation in the penumbra area of the stroke, thereby blunting monocyte/macrophage recruitment, activation and release of neurotoxic mediators such as MMP‐9. 17 Similarly, we previously reported that TNF‐α mediates the worsened stroke outcome observed in a mouse model of rheumatoid arthritis through increased BBB disruption, post‐ischaemic inflammation and oxidative stress. 56 Ischaemic stroke induces local and systemic levels of IL‐6. Yet, the role of IL‐6 in ischaemic cerebrovascular diseases remains to be fully elucidated. Distinct from IL‐1, IL‐6 can exert pro‐ or anti‐inflammatory functions, depending on activation of the classic or the trans‐signalling pathway, a dichotomy that may contribute to apparently conflicting results in the literature. 57 Indeed, although IL‐6 prompts inflammation in the early phase after stroke, 58 it may also exert neurotrophic protective functions at later time points. 59
In some but not all human studies, circulating and cerebrospinal fluid TNF‐α levels increase after ischaemic stroke and correlate with its severity. 60 , 61 , 62 To probe the translational relevance of our findings, we assessed plasma levels of this cytokine at 6 and 24 hours after symptom onset in ischaemic stroke patients and in sex‐ and age‐matched controls. In keeping with previous reports, we show increased circulating TNF‐α levels already 6 hours after the stroke that seems maintained even after one day. In our cohort, higher plasma levels associated with worsened short‐term neurological outcome as assessed by NIHSS during hospitalization. The low number of patients enrolled in our cohort did not permit reliable correlation between age and TNF‐α levels in healthy subjects in contrast with previous reports. 63 Yet, stroke patients showed a strong relationship between these two variables, suggesting a dysregulated inflammatory response of the elderly towards sterile insults—a characteristic of inflamm‐ageing. 14 Whether the increase of systemic TNF‐alpha levels observed in the elderly exacerbate post‐ischaemic BBB damage, and thus, stroke outcome in humans requires further study.
Some limitations pertain to the interpretation of these results. First, the present study focuses only up to 48 hours after stroke which is considered as an early time point when inflammation plays major deleterious effects in the penumbra area. 17 , 55 Further studies including later time points will be needed to address the long‐term effects of TNF‐α antagonization in old animals. Finally, to support our conclusions with respect to possible clinical applications, additional studies including larger animal models should be conducted.
In conclusion, this study demonstrates a direct adverse effect of age ‘per se’ on stroke outcome and mortality in a mouse model of disease, avoiding many confounding factors. Elevated systemic TNF‐α plasma levels in old mice underlie ‘inflamm‐ageing’ and may act via increased MMPs production, junctional protein degradation and impaired BBB function. The beneficial effects observed with adalimumab which rescued the deleterious effects of ageing on stroke outcome and survival support causality of TNF‐α in the pathogenesis of cerebral ischaemic injury. These findings could help identify treatments tailored to the particularly vulnerable and growing elderly population.
CONFLICTS OF INTEREST
LL, PL and GGC are coinventors on the International Patent WO/2020/226993 filed in April 2020. The patent relates to the use of antibodies which specifically bind IL‐1α to reduce various sequelae of ischaemia‐reperfusion injury to the central nervous system. GGC is a consultant to Sovida solutions limited. PL is an unpaid consultant to, or involved in clinical trials for Amgen, AstraZeneca, Baim Institute, Beren Therapeutics, Esperion, Therapeutics, Genentech, Kancera, Kowa Pharmaceuticals, Medimmune, Merck, Norvo Nordisk, Merck, Novartis, Pfizer, Sanofi‐Regeneron. PL is a member of scientific advisory board for Amgen, Corvidia Therapeutics, DalCor Pharmaceuticals, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis, XBiotech, Inc PL’s laboratory has received research funding in the last 2 years from Novartis. Dr Libby is on the Board of Directors of XBiotech, Inc PL has a financial interest in XBiotech, a company developing therapeutic human antibodies. PL’s interests were reviewed and are managed by Brigham and Women's Hospital and Partners HealthCare in accordance with their conflict of interest policies. FR has not received personal payments by pharmaceutical companies or device manufacturers in the last three years (remuneration for the time spent in activities, such as participation in steering committee member of clinical trials, were made directly to the University of Zurich). The Department of Cardiology (University Hospital of Zurich/University of Zurich) reports research, educational and/or travel grants from Abbott, Amgen, Astra Zeneca, Bayer, B. Braun, Biosense Webster, Biosensors Europe AG, Biotronik, BMS, Boehringer Ingelheim, Boston Scientific, Bracco, Cardinal Health Switzerland, Daiichi, Diatools AG, Edwards Lifesciences, Guidant Europe NV (BS), Hamilton Health Sciences, Kaneka Corporation, Labormedizinisches Zentrum, Medtronic, MSD, Mundipharma Medical Company, Novartis, Novo Nordisk, Orion, Pfizer, Quintiles Switzerland Sarl, Sanofi, Sarstedt AG, Servier, SIS Medical, SSS International Clinical Research, Terumo Deutschland, V‐ Wave, Vascular Medical, Vifor, Wissens Plus, ZOLL. The research and educational grants do not impact on FR`s personal remuneration. The other authors report no conflict of interest. TFL reports educational and research grants and speaker fees outside this work from Abbott, Amgen, Ablative Solutions, AstraZeneca, Bayer HealthCare, Boehringer‐Ingelheim, Daichi‐Sankyo, Novartis, Servier, Vifor. LL reports speaker fees outside of this work from Daichi‐Sankyo.
AUTHOR CONTRIBUTIONS
LL, NRB, FM, FR, TFL, PL, JHB and GGC conceived and planned the study. LL, NRB, YMP, AV, CD‐C and SK performed experiments. AA and MM provided critical technical support. AS, GG and MS enrolled the clinical cohort. LL, NRB and GGC analysed the data and drafted the manuscript. All authors revised the article for important intellectual content, reviewed the data and their analyses and approved this article.
ACKNOWLEDGEMENTS
None.
Liberale L, Bonetti NR, Puspitasari YM, et al. TNF‐α antagonism rescues the effect of ageing on stroke: Perspectives for targeting inflamm‐ageing. Eur J Clin Invest. 2021;51:e13600. 10.1111/eci.13600
Luca Liberale and Nicole R. Bonetti equally contributed as first author.
Funding information
The present work was supported by the Swiss National Science Foundation (to GGC [310030_197510]), the Swiss Heart Foundation to GGC and LL, the Alfred and Annemarie von Sick Grants for Translational and Clinical Research Cardiology and Oncology to GGC and the Foundation for Cardiovascular Research–Zurich Heart House. GGC is recipient of a Sheikh Khalifa's Foundation Assistant Professorship at the Faculty of Medicine, University of Zurich. PL receives funding from the US National Heart, Lung and Blood Institute (R01HL080472 and 1R01HL134892), the American Heart Association (18CSA34080399), and the RRM and Simard Charitable Funds
REFERENCES
- 1. Liberale L, Carbone F, Montecucco F, et al. Ischemic stroke across sexes: What is the status quo? Front Neuroendocrinol. 2018;50:3‐17. [DOI] [PubMed] [Google Scholar]
- 2. Ovbiagele B, Goldstein LB, Higashida RT, et al. Forecasting the future of stroke in the United States: a policy statement from the American Heart Association and American Stroke Association. Stroke. 2013;44:2361‐2375. [DOI] [PubMed] [Google Scholar]
- 3. Fugate JE, Rabinstein AA. Absolute and relative contraindications to IV rt‐PA for acute ischemic stroke. Neurohospitalist. 2015;5:110‐121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Steensig K, Olesen KKW, Thim T, et al. Predicting stroke in patients without atrial fibrillation. Eur J Clin Invest. 2019;49(6):e13103. [DOI] [PubMed] [Google Scholar]
- 5. Potpara TS, Simovic S, Pavlovic N, et al. Stroke prevention in elderly patients with non‐valvular atrial fibrillation in the BALKAN‐AF survey. Eur J Clin Invest. 2020;50:e13200. [DOI] [PubMed] [Google Scholar]
- 6. Camici GG, Liberale L. Aging: the next cardiovascular disease? Eur Heart J. 2017;38:1621‐1623. [DOI] [PubMed] [Google Scholar]
- 7. Stampfli SF, Akhmedov A, Gebhard C, et al. Aging induces endothelial dysfunction while sparing arterial thrombosis. Arterioscler Thromb Vasc Biol. 2010;30:1960‐1967. [DOI] [PubMed] [Google Scholar]
- 8. Steinman L. Elaborate interactions between the immune and nervous systems. Nat Immunol. 2004;5:575‐581. [DOI] [PubMed] [Google Scholar]
- 9. Vila N, Castillo J, Davalos A, Chamorro A. Proinflammatory cytokines and early neurological worsening in ischemic stroke. Stroke. 2000;31:2325‐2329. [DOI] [PubMed] [Google Scholar]
- 10. Liberale L, Montecucco F, Bonaventura A, et al. Monocyte count at onset predicts poststroke outcomes during a 90‐day follow‐up. Eur J Clin Invest. 2017;47:702‐710. [DOI] [PubMed] [Google Scholar]
- 11. Ministrini S, Carbone F, Montecucco F. Updating concepts on atherosclerotic inflammation: From pathophysiology to treatment. Eur J Clin Invest. 2021;51:e13467. [DOI] [PubMed] [Google Scholar]
- 12. Fulop T, Larbi A, Dupuis G, et al. Immunosenescence and Inflamm‐Aging As Two Sides of the Same Coin: Friends or Foes? Front Immunol. 2017;8:1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alvarez‐Rodriguez L, Lopez‐Hoyos M, Munoz‐Cacho P, Martinez‐Taboada VM. Aging is associated with circulating cytokine dysregulation. Cell Immunol. 2012;273:124‐132. [DOI] [PubMed] [Google Scholar]
- 14. Liberale L, Montecucco F, Tardif JC, Libby P, Camici GG. Inflamm‐ageing: the role of inflammation in age‐dependent cardiovascular disease. Eur Heart J. 2020;41(31):2974‐2982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Liberale L, Montecucco F, Schwarz L, Luscher TF, Camici GG. Inflammation and cardiovascular diseases: lessons from seminal clinical trials. Cardiovasc Res. 2020; 117(2):411‐422. [DOI] [PubMed] [Google Scholar]
- 16. Diaz‐Canestro C, Merlini M, Bonetti NR, et al. Sirtuin 5 as a novel target to blunt blood‐brain barrier damage induced by cerebral ischemia/reperfusion injury. Int J Cardiol. 2018;260:148‐155. [DOI] [PubMed] [Google Scholar]
- 17. Liberale L, Bonetti NR, Puspitasari YM, et al. Postischemic administration of IL‐1alpha neutralizing antibody reduces brain damage and neurological deficit in experimental stroke. Circulation. 2020;142:187‐189. [DOI] [PubMed] [Google Scholar]
- 18. Ubah OC, Steven J, Porter AJ, Barelle CJ. An Anti‐hTNF‐alpha variable new antigen receptor format demonstrates superior in vivo preclinical efficacy to humira(R) in a transgenic mouse autoimmune polyarthritis disease model. Front Immunol. 2019;10:526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Spescha RD, Klohs J, Semerano A, et al. Post‐ischaemic silencing of p66Shc reduces ischaemia/reperfusion brain injury and its expression correlates to clinical outcome in stroke. Eur Heart J. 2015;36:1590‐1600. [DOI] [PubMed] [Google Scholar]
- 20. Liberale L, Gaul DS, Akhmedov A, et al. Endothelial SIRT6 blunts stroke size and neurological deficit by preserving blood‐brain barrier integrity: a translational study. Eur Heart J. 2020;41:1575‐1587. [DOI] [PubMed] [Google Scholar]
- 21. Akhmedov A, Bonetti NR, Reiner MF, et al. Deleterious role of endothelial lectin‐like oxidized low‐density lipoprotein receptor‐1 in ischaemia/reperfusion cerebral injury. J Cereb Blood Flow Metab. 2019;39:2233‐2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liberale L, Holy EW, Akhmedov A, et al. Interleukin‐1beta mediates arterial thrombus formation via NET‐associated tissue factor. J Clin Med. 2019;8(12):2072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pluvinage JV, Wyss‐Coray T. Systemic factors as mediators of brain homeostasis, ageing and neurodegeneration. Nat Rev Neurosci. 2020;21:93‐102. [DOI] [PubMed] [Google Scholar]
- 24. Virani SS, Alonso A, Aparicio HJ, et al. Heart disease and stroke statistics‐2021 update: a report from the American Heart Association. Circulation. 2021:CIR0000000000000950. [DOI] [PubMed] [Google Scholar]
- 25. Liberale L, Kraler S, Camici GG, Luscher TF. Ageing and longevity genes in cardiovascular diseases. Basic Clin Pharmacol Toxicol. 2020;127:120‐131. [DOI] [PubMed] [Google Scholar]
- 26. Spescha RD, Shi Y, Wegener S, et al. Deletion of the ageing gene p66(Shc) reduces early stroke size following ischaemia/reperfusion brain injury. Eur Heart J. 2013;34:96‐103. [DOI] [PubMed] [Google Scholar]
- 27. Camici GG, Savarese G, Akhmedov A, Luscher TF. Molecular mechanism of endothelial and vascular aging: implications for cardiovascular disease. Eur Heart J. 2015;36:3392‐3403. [DOI] [PubMed] [Google Scholar]
- 28. Diaz‐Canestro C, Reiner MF, Bonetti NR, et al. AP‐1 (Activated protein‐1) transcription factor JunD regulates ischemia/reperfusion brain damage via IL‐1beta (Interleukin‐1beta). Stroke. 2019;50:469‐477. [DOI] [PubMed] [Google Scholar]
- 29. Ferrucci L, Fabbri E. Inflammageing: chronic inflammation in ageing, cardiovascular disease, and frailty. Nat Rev Cardiol. 2018;15:505‐522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bauernfeind F, Niepmann S, Knolle PA, Hornung V. Aging‐associated TNF production primes inflammasome activation and NLRP3‐related metabolic disturbances. J Immunol. 2016;197:2900‐2908. [DOI] [PubMed] [Google Scholar]
- 31. Koelman L, Pivovarova‐Ramich O, Pfeiffer AFH, Grune T, Aleksandrova K. Cytokines for evaluation of chronic inflammatory status in ageing research: reliability and phenotypic characterisation. Immun Ageing. 2019;16:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Puzianowska‐Kuznicka M, Owczarz M, Wieczorowska‐Tobis K, et al. Interleukin‐6 and C‐reactive protein, successful aging, and mortality: the PolSenior study. Immun Ageing. 2016;13:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Marcos‐Perez D, Sanchez‐Flores M, Maseda A, et al. Frailty in older adults is associated with plasma concentrations of inflammatory mediators but not with lymphocyte subpopulations. Front Immunol. 2018;9:1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bruunsgaard H, Skinhoj P, Pedersen AN, Schroll M, Pedersen BK. Ageing, tumour necrosis factor‐alpha (TNF‐alpha) and atherosclerosis. Clin Exp Immunol. 2000;121:255‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bruunsgaard H, Ladelund S, Pedersen AN, Schroll M, Jorgensen T, Pedersen BK. Predicting death from tumour necrosis factor‐alpha and interleukin‐6 in 80‐year‐old people. Clin Exp Immunol. 2003;132:24‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bruunsgaard H, Andersen‐Ranberg K, Hjelmborg J, Pedersen BK, Jeune B. Elevated levels of tumor necrosis factor alpha and mortality in centenarians. Am J Med. 2003;115:278‐283. [DOI] [PubMed] [Google Scholar]
- 37. Monaco C, Nanchahal J, Taylor P, Feldmann M. Anti‐TNF therapy: past, present and future. Int Immunol. 2015;27:55‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Warner SJ, Libby P. Human vascular smooth muscle cells. Target for and source of tumor necrosis factor. J Immunol. 1989;142:100‐109. [PubMed] [Google Scholar]
- 39. Palladino MA, Bahjat FR, Theodorakis EA, Moldawer LL. Anti‐TNF‐alpha therapies: the next generation. Nat Rev Drug Discov. 2003;2:736‐746. [DOI] [PubMed] [Google Scholar]
- 40. Barone FC, Arvin B, White RF, et al. Tumor necrosis factor‐alpha. A mediator of focal ischemic brain injury. Stroke. 1997;28:1233‐1244. [DOI] [PubMed] [Google Scholar]
- 41. Bonetti NR, Diaz‐Canestro C, Liberale L, et al. Tumour necrosis factor‐alpha inhibition improves stroke outcome in a mouse model of rheumatoid arthritis. Sci Rep. 2019;9:2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tuttolomondo A, Di Raimondo D, di Sciacca R, Pinto A, Licata G. Inflammatory cytokines in acute ischemic stroke. Curr Pharm Des. 2008;14:3574‐3589. [DOI] [PubMed] [Google Scholar]
- 43. Domac FM, Somay G, Misirli H, Erenoglu NY. Tumor necrosis factor alpha serum levels and inflammatory response in acute ischemic stroke. Neurosciences (Riyadh). 2007;12:25‐30. [PubMed] [Google Scholar]
- 44. Yang Y, Rosenberg GA. Blood‐brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke. 2011;42:3323‐3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Montagne A, Barnes SR, Sweeney MD, et al. Blood‐brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85:296‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Diaz‐Otero JM, Garver H, Fink GD, Jackson WF, Dorrance AM. Aging is associated with changes to the biomechanical properties of the posterior cerebral artery and parenchymal arterioles. Am J Physiol Heart Circ Physiol. 2016;310:H365‐H375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roussel BD, Macrez R, Jullienne A, et al. Age and albumin D site‐binding protein control tissue plasminogen activator levels: neurotoxic impact. Brain. 2009;132:2219‐2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Bors L, Toth K, Toth EZ, et al. Age‐dependent changes at the blood‐brain barrier. A Comparative structural and functional study in young adult and middle aged rats. Brain Res Bull. 2018;139:269‐277. [DOI] [PubMed] [Google Scholar]
- 49. DiNapoli VA, Huber JD, Houser K, Li X, Rosen CL. Early disruptions of the blood‐brain barrier may contribute to exacerbated neuronal damage and prolonged functional recovery following stroke in aged rats. Neurobiol Aging. 2008;29:753‐764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kaur J, Tuor UI, Zhao Z, Barber PA. Quantitative MRI reveals the elderly ischemic brain is susceptible to increased early blood‐brain barrier permeability following tissue plasminogen activator related to claudin 5 and occludin disassembly. J Cereb Blood Flow Metab. 2011;31:1874‐1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kauppinen TM, Swanson RA. Poly(ADP‐ribose) polymerase‐1 promotes microglial activation, proliferation, and matrix metalloproteinase‐9‐mediated neuron death. J Immunol. 2005;174:2288‐2296. [DOI] [PubMed] [Google Scholar]
- 52. Yong VW, Power C, Forsyth P, Edwards DR. Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci. 2001;2:502‐511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase‐mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007;27:697‐709. [DOI] [PubMed] [Google Scholar]
- 54. Loppnow H, Libby P. Proliferating or interleukin 1‐activated human vascular smooth muscle cells secrete copious interleukin 6. J Clin Invest. 1990;85:731‐738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Liberale L, Diaz‐Canestro C, Bonetti NR, et al. Post‐ischaemic administration of the murine Canakinumab‐surrogate antibody improves outcome in experimental stroke. Eur Heart J. 2018;39(38):3511‐3517. [DOI] [PubMed] [Google Scholar]
- 56. Bonetti N, Diaz‐Canestro C, Liberale L, et al. Tumour necrosis factor‐alpha ihibition improves stroke outcome in a mouse model of rheumatoid arthritis. Sci Rep. 2019;9(1):2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Scheller J, Chalaris A, Schmidt‐Arras D, Rose‐John S. The pro‐ and anti‐inflammatory properties of the cytokine interleukin‐6. Biochim Biophys Acta. 2011;1813:878‐888. [DOI] [PubMed] [Google Scholar]
- 58. Clark WM, Rinker LG, Lessov NS, et al. Lack of interleukin‐6 expression is not protective against focal central nervous system ischemia. Stroke. 2000;31:1715‐1720. [DOI] [PubMed] [Google Scholar]
- 59. Gertz K, Kronenberg G, Kalin RE, et al. Essential role of interleukin‐6 in post‐stroke angiogenesis. Brain. 2012;135:1964‐1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zaremba J, Losy J. Early TNF‐alpha levels correlate with ischaemic stroke severity. Acta Neurol Scand. 2001;104:288‐295. [DOI] [PubMed] [Google Scholar]
- 61. Kim JW, Park MS, Kim JT, et al. The Impact of Tumor Necrosis Factor‐alpha and Interleukin‐1beta levels and polymorphisms on long‐term stroke outcomes. Eur Neurol. 2018;79:38‐44. [DOI] [PubMed] [Google Scholar]
- 62. Oto J, Suzue A, Inui D, et al. Plasma proinflammatory and anti‐inflammatory cytokine and catecholamine concentrations as predictors of neurological outcome in acute stroke patients. J Anesth. 2008;22:207‐212. [DOI] [PubMed] [Google Scholar]
- 63. de Gonzalo‐Calvo D, Neitzert K, Fernandez M, et al. Differential inflammatory responses in aging and disease: TNF‐alpha and IL‐6 as possible biomarkers. Free Radic Biol Med. 2010;49:733‐737. [DOI] [PubMed] [Google Scholar]