

Abstract
Purpose
Reliable detection and fitting of macromolecules (MM) are crucial for accurate quantification of brain short‐echo time (TE) 1H‐MR spectra. An experimentally acquired single MM spectrum is commonly used. Higher spectral resolution at ultra‐high field (UHF) led to increased interest in using a parametrized MM spectrum together with flexible spline baselines to address unpredicted spectroscopic components. Herein, we aimed to: (1) implement an advanced methodological approach for post‐processing, fitting, and parametrization of 9.4T rat brain MM spectra; (2) assess the concomitant impact of the LCModel baseline and MM model (ie, single vs parametrized); and (3) estimate the apparent T2 relaxation times for seven MM components.
Methods
A single inversion recovery sequence combined with advanced AMARES prior knowledge was used to eliminate the metabolite residuals, fit, and parametrize 10 MM components directly from 9.4T rat brain in vivo 1H‐MR spectra at different TEs. Monte Carlo simulations were also used to assess the concomitant influence of parametrized MM and DKNTMN parameter in LCModel.
Results
A very stiff baseline (DKNTMN ≥ 1 ppm) in combination with a single MM spectrum led to deviations in metabolite concentrations. For some metabolites the parametrized MM showed deviations from the ground truth for all DKNTMN values. Adding prior knowledge on parametrized MM improved MM and metabolite quantification. The apparent T2 ranged between 12 and 24 ms for seven MM peaks.
Conclusion
Moderate flexibility in the spline baseline was required for reliable quantification of real/experimental spectra based on in vivo and Monte Carlo data. Prior knowledge on parametrized MM improved MM and metabolite quantification.
Keywords: 1H‐MRS, baseline, fitting, macromolecules, parametrization, rat brain, relaxation times, UHF
1. INTRODUCTION
Short echo time (TE) 1H‐MR spectra contain contributions from mobile macromolecules (MM). These are broader resonances characterized by shorter T1 and T2, underlying the narrower peaks of metabolites. 1 , 2 , 3 In healthy brain, the majority of MM signals arise from protons of amino acids that make up the cytosolic proteins. 4 , 5 , 6 , 7 , 8
The MM spectra can be measured in vivo using single and double inversion recovery (IR) techniques, both of which provide adequate suppression of metabolite signals. 3 The availability of ultra‐high magnetic fields (UHF ≥ 7T) leads to better resolved MM, thus more sophisticated approaches need to be used for post‐processing 9 , 10 (eg, elimination of residual metabolites) or for further parametrization of the acquired MM spectrum into individual components. 11 Reliable MM detection and fitting is crucial for quantifying short TE 1H‐MR brain spectra, a topic that has received increased attention in the research community in the recent years. In addition, for accurate assessment, identification and fitting of MM, knowledge of their individual T1 and T2 values is necessary. 12 , 13
Generally, an in vivo acquired and post‐processed single MM spectrum is included in the basis‐set for spectral fitting. 2 , 3 , 14 Given the potential regional or disease 10 , 15 , 16 , 17 , 18 , 19 , 20 , 21 ‐dependent variability in MM spectra, the incorporation of a subject/region‐specific MM spectrum into the fitting analysis may be preferable to avoid bias in metabolite quantification. However, this approach has been of limited use due to the increased scan time needed to acquire a separate spectrum. Recent methodological advances combined with UHF ensure an increased spectral resolution facilitating the separation of the MM spectrum into individual components/peaks. 1 , 11 This parametrization of MM spectra into components and their inclusion in the metabolite basis‐set has already been done for different B0 field strengths. Usually, separately fitted MM peaks were grouped before inclusion in the metabolite basis‐set to reduce the number of independent components and, thus, the risk of over‐fitting by the quantification algorithm. 1 , 13 , 21 , 22 , 23 , 24
Recently, 7T clinical studies showed that the parametrization of MM signals with appropriate prior knowledge (PK) (ie, soft constraints on amplitudes, etc.) is feasible and may facilitate the detection of individual MM components. 11 Considering that an in vivo acquired MM spectrum is preferable to its purely mathematical estimate, typically pre‐acquired representative metabolite‐nulled spectra are used for MM parametrization, 3 bringing an improved MM model for metabolite quantification, simultaneously providing information about the content of individual MM. 11 Parametrization is an accepted method for estimating individual MM peaks, but the increased number of fitted parameters without constraints may lead to overfitting. 3 Therefore, this method needs further evaluation.
In addition, a relatively unconstrained spline baseline is often used during the fitting process to address unpredicted spectroscopic components 25 , 26 (smoothly varying components and spurious signals arising through imperfections during data acquisition). Finding the optimal degree of baseline flexibility is mandatory for reliable metabolite concentration estimates, yet few studies have investigated this topic in detail. 27 , 28 , 29 , 30 , 31 In LCModel, the stiffness of the spline baseline is controlled by the parameter DKNTMN (minimum allowed spacing between spline knots). 25 , 31 The default value is set to 0.15 ppm (low stiffness), and all values equal to or higher than 1 ppm result in a high baseline stiffness. 31 A study performed at 9.4T in humans has reported several changes in metabolite concentrations when quantifying with different DKNTMN values, but no conclusion on the best value was drawn 31 , 32 due to lack of ground truth. To draw such a conclusion, an extensive Monte Carlo (MC) simulation study is necessary.
There are few studies assessing the T2 values of MMs, 13 , 27 , 33 with only two recent studies reporting T2 for individual MM peaks in the full ppm range at 9.4T and 3T in human brain. 9 , 34 , 35 Otherwise, T2 values have only been reported for the entire MM spectrum, 27 grouped MM signals 13 or for the peaks up to 1.7 ppm. Estimating T2 for individual MM signals is not straightforward due to the overlapping metabolites and requires advanced approaches.
In this study, we aimed to: (1) implement an advanced methodological approach for post‐processing, fitting, and parametrization of rat brain MM spectra acquired at 9.4T; (2) assess the concomitant impact of the LCModel baseline stiffness and MM model (ie, single vs parametrized MM in the basis‐set) on metabolite and MM quantification using in vivo and MC simulated spectra; and (3) estimate the apparent T2 relaxation times (J evolution not considered, ) for 7 MM components.
2. METHODS
Wistar male adult rats (n = 13 Charles River Laboratories, L’Arbresle, France) under 1.5‐2.5% isoflurane anesthesia were used for the 1H‐MRS experiments. The body temperature of the animals was kept at 37.5 ± 1.0 °C by circulating warm water. All experiments were approved by The Committee on Animal Experimentation for the Canton de Vaud, Switzerland.
In vivo 1H‐MRS measurements were performed using a horizontal actively shielded 9.4T magnet (Magnex Scientific, Yarnton, UK) interfaced to an Agilent/Varian Direct Drive console (Palo Alto, CA, USA). A home built 1H‐quadrature surface coil was used as the transceiver. First‐ and second‐order shims were adjusted using FASTMAP 36 (water linewidths of 11‐12 Hz in the 3 × 3 × 3 mm3 volume of interest (VOI) for MM spectra; 9‐11 Hz in the 2.0 × 2.8 × 2.0 mm3 VOI for metabolite spectra).
2.1. Acquisition methods
2.1.1. Acquisition of MM spectra
To measure the in vivo MM spectra, the SPECIAL 37 sequence was extended with a 2 ms nonselective hyperbolic secant inversion pulse, 38 applied at TI of 750 ms before the first radiofrequency (RF) pulse of the SPECIAL sequence (ie, the slice selective adiabatic inversion pulse). 39 The MM spectra were acquired with a short repetition time (TR) (TR = 2.5 s), and TE = 2.8 ms, if not stated otherwise. This TI was chosen to minimize the metabolites signals using a series of IR spectra acquired with several TIs (ie, 420, 600, 700, 725, 750, 800, and 1000 ms) and an IR spectrum (TI = 750 ms) acquired with a longer TE (TE = 40 ms). All MM spectra were acquired from the VOI = 3 × 3 × 3 mm3 centered on the rat hippocampus. This VOI was selected to increase the signal‐to‐noise ratio (SNR), as it is well accepted that MM do not substantially change between brain regions in rodents. 3 , 40 , 41 The MM spectra were acquired from n = 6 rats at 13 TEs (TE = 2.8, 4, 6, 8, 10, 12, 16, 20, 40, 60, 100, 120, and 150 ms, TI = 750 ms). To minimize the anesthesia effects due to very long acquisition time (1024 averages per TE) two acquisitions (ie, two rats) for each TE were performed. An additional data set (three rats) was used for MM parametrization at TE = 2.8 ms.
2.1.2. Acquisition of metabolite spectra
Metabolite 1H‐MR spectra were acquired using the SPECIAL sequence (TE = 2.8 ms, TR = 4 s) from a VOI located in the rat hippocampus (2.0 × 2.8 × 2.0 mm3, n = 7, 160 averages). The water signal was suppressed by VAPOR 42 interleaved with outer volume suppression.
2.1.3. MC simulation
To assess the reliability of the estimated concentrations, artificial rat brain 1H‐MR spectra were created (Matlab, MathWorks, Natick, MA, USA) to mimic optimal experimental conditions (metabolites with MM only = “Optimal MC”) and real experimental conditions (metabolites with MM including a baseline = “Real MC”). More details on the MC study can be found in the Supporting Information, which is available online.
2.2. Data processing
2.2.1. Post‐processing: elimination of metabolite residuals from MM spectra at different TEs
The MM spectra were phased individually in jMRUI (http://www.mrui.uab.es/mrui/) and 2 Hz Lorentzian line broadening was applied for visualization. Metabolite residuals present in the acquired MM spectra at all TEs were identified using the procedure highlighted in Figure 1A. 3
FIGURE 1.
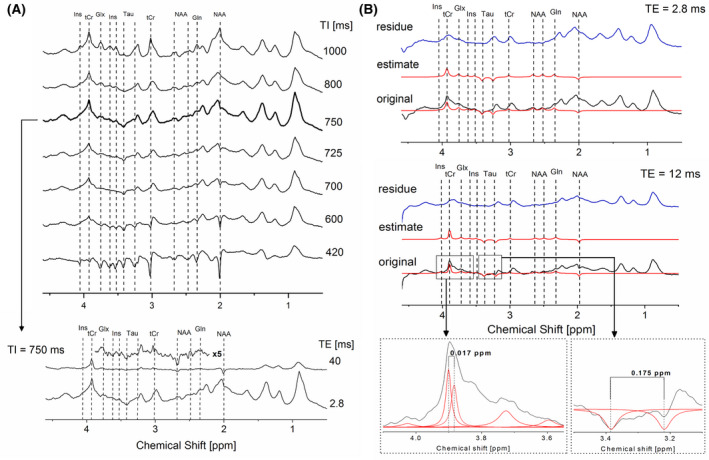
A, (upper panel) In vivo rat brain series of IR spectra with TI ranging from 420 to 1000 ms revealing the evolution of metabolite intensities over a series of different TIs to identify the optimal TI and metabolite residuals (acquisition parameters TE/TR = 2.8/2500 ms); (lower panel) Spectra acquired with a selected TI = 750 ms and TE = 2.8 ms as well as with TE = 40 ms (5× magnified) to confirm the presence of residual metabolite signals. B, (upper panel) Original spectra acquired at TI = 750 ms and TE = 2.8 ms (shown in black), estimated fits of the residual metabolites using AMARES (shown in red) and the residue obtained after subtraction of the estimated metabolites signals remaining from the original spectrum (shown in blue); (lower panel) original spectra acquired at TI = 750 ms and TE = 12 ms (shown in black), estimated fits (shown in red) and obtained residue (shown in blue) with two inserts: (1) an expansion on the fit of tCr at TE = 12 ms with two peaks with 0.017 ppm shift (on the left); (2) an expansion on the fit of two Tau peaks with same amplitude, linwidth, and a chemical shift of 0.175 (on the right)
Using the AMARES algorithm 43 a user‐built set of PK with the constraints on peak frequency, phase, linewidth (full width half maximum [FWHM]), and amplitude was used to fit the metabolite residuals (inositol‐Ins, total creatine‐tCr, taurine‐Tau, N‐acetylaspartate‐NAA) and the sum of glutamine and glutamate (Gln + Glu ‐ Glx) and their contribution was removed from the MM spectra (Figure 1B). To construct such PK, each residual metabolite peak was individually analyzed at a given TI and TE (Table 1) and fitted as a singlet (Lorentzian lineshape). J‐coupled metabolites (eg, Glx and Tau) were fitted with larger linewidths to account for the J‐splitting appearance. This approach is an accepted approximation for the removal of the residual metabolites at short TE and UHF, 10 , 11 , 44 with the main improvement that in this manuscript more metabolite residuals were reliably identified 11 , 23 , 45 (ie, NAA ‐ 2.49 ppm, Tau ‐ 3.42 ppm, Glx ‐ 3.75 ppm, Ins ‐ 4.05 ppm; see Table 1). The identified metabolite residuals were removed from all MM spectra acquired with TE = 2.8 ‐ 40 ms. For the MM spectra acquired with TE ≥ 60 ms (section 2.2.5.) only M0.94, M1.22, and M1.43 components were adequately distinguished and fitted. In addition, M3.21 was still detectable and fitted at TE = 60 ms, having minimal overlap with the Tau resonance. The subsequent iterations were followed to build up the PK for the removal of metabolite residuals: (1) every metabolite was individually removed from the MM spectrum using a flexible, metabolite specific PK; (2) in the second step, the results obtained from the fitting of the individual metabolite residual peaks were combined to form a rigorous PK (leaving some freedom on amplitude for the peaks to adjust to different spectra (no more than 10%)). Finally, the metabolite‐free MM (ie, the residual after AMARES post‐processing) was saved separately.
TABLE 1.
Information on the individual residual metabolite peaks used to build the AMARES PK: This table was created based on previously published data 56 , 57
| Metabolites | Chemical shift (ppm)‐fixed | Multiplicity | Phase ‐fixed | No. of peaks | Linewidth‐soft constraints | Notes |
|---|---|---|---|---|---|---|
| tCr | 3.91 | s | 0 | 1 | 15‐20 | Starting from the TE = 12 ms it is fitted with two peaks with a shift between them of 0.017 ppm (because of different T2 of Cr and PCr) |
| Cr & PCr | 0 & 0 | 2 | 7‐10 | |||
| tCr | 3.027 | s | 0 | 1 | 12‐15 | T1 is longer than for the peak at 3.91 ppm, thus this peak is smaller |
| Glx | 3.75 | dd & t | 0 | 1 | 22‐25 | |
| Ins | 3.61 & 3.52 | dd & t | 0 & 0 | 1 | 20 & 20 | Similar peaks, both disappear at TE = 20 ms |
| Ins | 4.05 | t | 0 | 1 | 20 | |
| Tau | 3.42 & 3.246 | t & t | 180 & 180 | 1 | 20 & 20 | Both peaks must have the same amplitude and lw with a shift between them ≈ 0.175 ppm |
| NAA | 2.67 & 2.49 | dd & dd | 0 & 0 | 1 | 18‐22 & 18‐22 | Both peaks have similar amplitude and lw, they disappear at TE = 20 ms |
| NAA | 2.01 | s | 180 | 1 | 10‐18 | Very well visible even at longer TE |
| Glu | 2.34 | m | 0 | 1 | 20‐21 |
s, singlet; dd, doublet of doublets; t, triplet; m, multiplet. Soft constraint on the chemical shift of 0.01 ppm was added if necessary. PK and starting values example files are provided in the supporting information.
2.2.2. Fitting of the individual MM components
We chose to divide the metabolite‐free MM spectra into 10 components (Table 2). Each MM component was quantified by AMARES using several Lorentzian lines (Table 2) to obtain the best possible match with the original spectra (the number of lines was chosen based on the spectral appearance), without accounting for J‐evolution of these MM peaks. After each fit, the spectra were manually inspected. For longer TE values, soft constraints on the amplitudes of the peaks were additionally imposed to avoid over or underestimation (ie, negative or positive residuals, respectively). To assess the goodness of fit (possible over or under fitting) a “fit quality number” (FQN) was calculated (as a ratio of the variance in the fit residual divided by pure spectral noise) using a Matlab code written in‐house. 14 , 46
TABLE 2.
PK given to AMARES for the MM fitting: constraints in frequency, number of peaks, linewidth, and line shape. Note that if one MM was fitted with multiple peaks, they all had the same constraints on frequency and linewidth. T2 estimates obtained from the exponential fits of MM decay over TEs and its SDs for 7 out of 10 MM. Signal intensity ratios presented as mean values (n = 3 MM spectra from 3 different rats) and their SDs (used for CHRATO parameter in LCModel). PK and starting values example files are provided in the supporting information
| AMARES‐PK for MM fitting | T2 estimates | Signal intensity ratios | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Frequency soft constraints (ppm) | Phase | No. of peaks | Linewidth soft constraints | Shape | T2 [ms] | SD of the fit | Ratios: | Mean | SD | |
| M0.94 | 0.61‐1.05 | 0 | 8 | 0‐15 | lorentzian | 24 | 2 | M1.22/M0.94 | 0.28 | 0.01 |
| M1.22 | 1.10‐1.26 | 0 | 3 | 0‐25 | lorentzian | 24 | 3 | M1.43/M0.94 | 0.59 | 0.08 |
| M1.43 | 1.27‐1.50 | 0 | 3 | 0‐25 | lorentzian | 22 | 2 | M1.70/M0.94 | 0.45 | 0.12 |
| M1.70 | 1.53‐1.76 | 0 | 3 | 0‐35 | lorentzian | 13 | 1 | M2.05/M0.94 | 1.14 | 0.20 |
| M2.05 | 1.77‐2.17 | 0 | 7 | 0‐30 | lorentzian | 15 | 1 | M2.27‐2.26/M0.94 | 0.35 | 0.07 |
| M2.27‐2.36 | 2.17‐2.36 | 0 | 3 | 0‐25 | lorentzian | − | − | M3.00/M0.94 | 0.24 | 0.08 |
| M3.00 | 2.86‐3.05 | 0 | 3 | 0‐30 | lorentzian | 22 | 3 | M3.21/M0.94 | 0.30 | 0.17 |
| M3.21 | 3.10‐3.30 | 0 | 3 | 0‐30 | lorentzian | 12 | 2 | M3.71‐3.97/M0.94 | 0.68 | 0.09 |
| M3.71‐3.97 | 3.70‐4.10 | 0 | 5 | 0‐40 | lorentzian | − | − | M4.20/M0.94 | 0.21 | 0.11 |
| M4.20 | 4.20‐4.40 | 0 | 2 | 0‐35 | lorentzian | − | − | Derived from n = 3 spectra | ||
2.2.3. Parametrization of individual MM components
The 10 individual MM components fitted by AMARES from the spectrum at TE = 2.8 ms and TI = 750 ms were used to create the parametrized MM model (Figure 2). To implement the PK and soft constraints, each MM component was first quantified from three spectra (TE = 2.8 ms) measured from three different rats. Signal intensity ratios of Mxx/M0.94 were then calculated for every spectrum and averaged over the three acquisitions to obtain a mean value and SD for each ratio (Table 2). These values were included in the LCModel control files using the parameter CHRATO as previously described. 11
FIGURE 2.
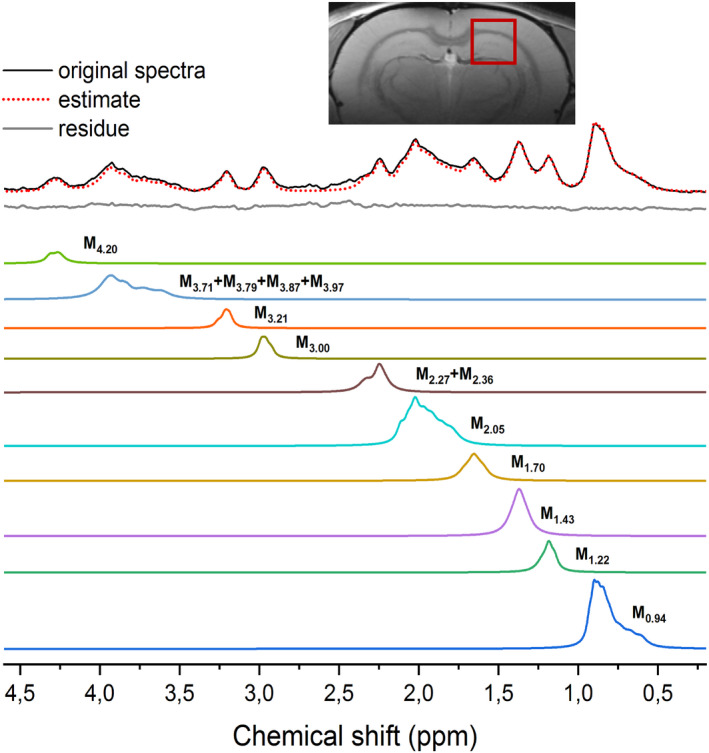
MM spectrum parametrized into 10 individual components using AMARES. The original spectrum was fitted using AMARES and the fits of individual components were saved separately to form a parametrized basis‐set (in color). The insert image shows the VOI = 3 × 3 × 3 mm3 centered on the rat hippocampus (all MM spectra were acquired from VOI positioned in this location)
2.2.4. Quantification of brain metabolites using single vs parametrized MM spectra with varying DKNTMN values
The MC spectra and each in vivo rat brain spectrum (TE = 2.8 ms, n = 7) were quantified with LCModel using the single MM spectrum (standard) and parametrized MM components included in the basis‐set. In addition, different DKNTMN values (0.1, 0.25, 0.4, 0.5, 1, and 5 ppm) were used for fitting. The quantification sets and terminology used are explained in Table 3.
TABLE 3.
Terminology and data sets used for LCModel quantifications with varying DKNTMN values (0.1, 0.25, 0.4, 0.5, 1, 5 ppm)
| QUANTIFICATION SETS → | In vivo + single MM | In vivo + parametrized MM | In vivo + parametrized MM + PK | Optimal MC + single MM | Optimal MC + parametrized MM | Optimal MC + parametrized MM + PK | Real MC + single MM | Real MC + parametrized MM | Real MC + parametrized MM + PK | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Data types | In vivo | In vivo acquired data | ✓ | ✓ | ✓ | ||||||
| Optimal MC | MC simulated data without baseline (metabolites + MM) | ✓ | ✓ | ✓ | |||||||
| Real MC | MC simulated data + the baseline (metabolites, MM and baseline) | ✓ | ✓ | ✓ | |||||||
| MM in the basis set | Single MM | single MM spectrum in the basis set | ✓ | ✓ | ✓ | ||||||
| Parametrized MM | parametrized MM spectrum in the basis set | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||||
| PK | soft constraints on individual MM components ‐ in the LCModel control file | ✓ | ✓ | ✓ | |||||||
2.2.5. Measurement and fitting of MM T2
To measure MM T2, TE was varied from 2.8 to 150 ms (TE = 2.8, 4, 6, 8, 10, 12, 16, 20, 40, 60, 100, 120, and 150 ms, TI = 750 ms). These spectra were individually post‐processed: metabolite residuals were eliminated and individual components were fitted as described in sections 2.2.1 and 2.2.2, respectively. To allow pooling data from all six animals in a joint T2 fitting, inter‐animal scaling of peak amplitudes was done. The amplitude of the M0.94 component was used as a reference because this MM component does not overlap with metabolite resonances and, therefore, is easy to quantify. A scaling factor was calculated for each animal using the ratios between M0.94 components (TE = 2.8 ms). For each MM peak, normalized amplitudes from all rats were fitted to a single exponential decay across the TE series to estimate its T2.
2.2.6. Statistics
Data are presented as mean ± SD. Metabolite concentrations were compared for all the DKNTMN values within groups (single MM and parametrized‐MM groups) using one‐way analysis of variance (ANOVA) with respect to each metabolite in the neurochemical profile followed by Bonferroni’s multi‐comparisons post‐test (DKNTMN value). The significance level in one‐way ANOVA was attributed as follows: *P < .05, **P < .01, ***P < .001, and ****P < .0001. Comparison between groups (single and parametrized MM) was done using two‐way ANOVA with respect to each metabolite in the neurochemical profile followed by Bonferroni’s multi‐comparisons post‐test (with MM type and DKNTMN as factors) (# P < .05, ## P < .01, ### P < .001, #### P < .0001). All tests were two‐tailed. To improve the readability of the text, percentage changes in metabolite concentrations were calculated comparing results for DKNTMN value of 5 vs 0.25 ppm within the same MM model and at DKNTMN of 0.25 ppm when comparing single vs parametrized MM model (values shown in both figures and text).
3. RESULTS
3.1. Post‐processing, fitting, and parametrization into 10 individual MM components
The quality of the experimentally acquired MM spectra is shown in Figure 1. At TE = 2.8 ms, the SNR was 19.1 ± 0.5, calculated using the ratio between the highest peak (M0.94) and the SD of the spectral noise (jMRUI). The proposed post‐processing method using AMARES and advanced PK based on different TI values and TE = 40 ms was efficient and robust in removing all residual metabolites providing clean MM spectra for MM fitting, metabolite and MM quantification (Figure 1). It was also successfully applied to spectra with longer TE values (up to TE = 40 ms). Our approach allowed to reliably identify 12 residuals originating from Ins, tCr, Glx, Tau, and NAA (Table 1).
MM spectra were fitted and parametrized as described in sections 2.2.2. and 2.2.3 (Table 2). The mean FQN value of all fitted spectra was 1.2 ± 0.2 indicating that the fit agrees with the data within the precision allowed by the noise. The final residual spectrum (Figure 2, in gray) was flat and clean of any major MM residual contribution, providing an excellent approximation and parametrization of the in vivo measured MM spectrum. Reliable ratios for each MM component (mean ± SD, Table 2) were obtained and used as PK for the LCModel analysis.
3.2. Quantification of brain metabolites
3.2.1. In vivo and Real MC data quantified using single MM: impact of the DKNTMN parameter
When using the single MM spectrum for the in vivo data quantifications, increasing the stiffness in the spline baseline led to a significant decrease of Gln (−16%,*), Glu (−7%,**), and gamma‐aminobutyric acid (GABA, −30%,***) concentrations. Additional changes were observed for total‐choline (tCho, +23%), aspartate (Asp, −18%), ascorbate (Asc, +37%), alanine (Ala, +18%), lactate (Lac, +15%), and glutathione (GSH, −7%) without reaching significance (Figure 3, black plots).
FIGURE 3.
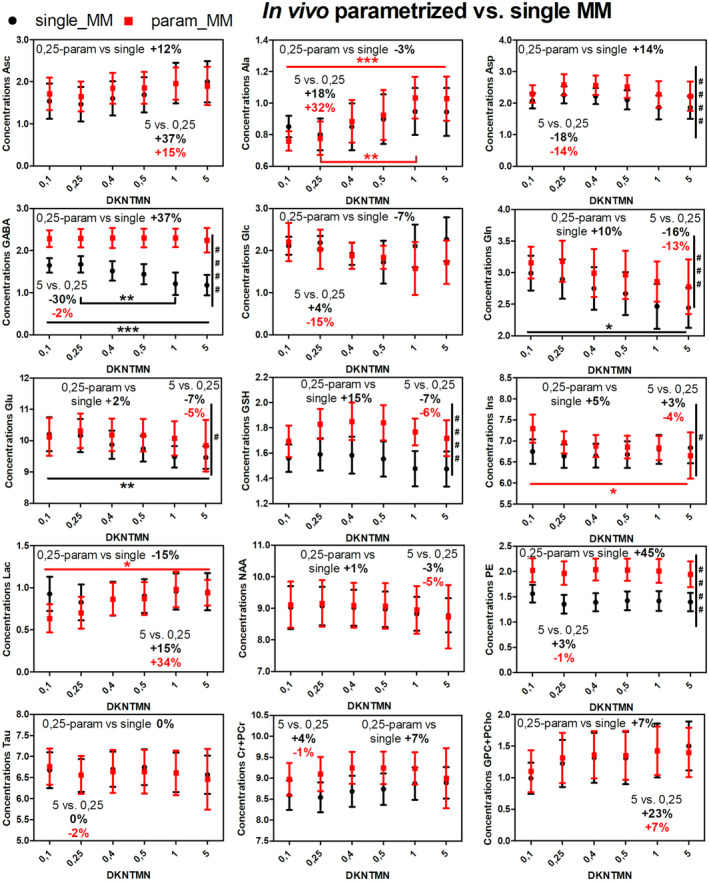
In vivo metabolite changes using two different quantification approaches in LCModel: single MM spectra (black) and parametrized MM spectra (red) over different DKNTMN values (one‐way ANOVA, Bonferroni correction *P < .05, **P < .01, ***P < .001, ****P < .0001). The percentage change between metabolite concentrations when quantified with DKNTMN = 0.25 ppm and DKNTM = 5 ppm are shown in the insert (always in red for the parameterized MM with PK and black for the single MM). The comparison between groups single vs parametrized MM was calculated using two‐way ANOVA (# P < .05, ## P < .01, ### P < .001, #### P < .0001) and is shown on the right in each plot if significant
To validate the changes observed in vivo when using the single MM spectrum with different DKNTMN values, two different MC studies were used. As expected, the Optimal MC study showed a negligible impact of DKNTMN on metabolite concentrations (Supporting Information Figure S2). The Real MC study revealed similar changes in metabolite concentrations to those in vivo for Asp (−52%), GABA (−42%), glucose (Glc, −41%), Gln (−20%), Glu (−10%), and GSH (−9%) when increasing DKNTMN. Additional metabolites, such as Ala, Lac, phosphatidylethanolamine (PE), Tau, tCr, and tCho displayed an increase of 45%, 59%, 16%, 15%, 6%, and 10%, respectively (Figure 4, black plots).
FIGURE 4.
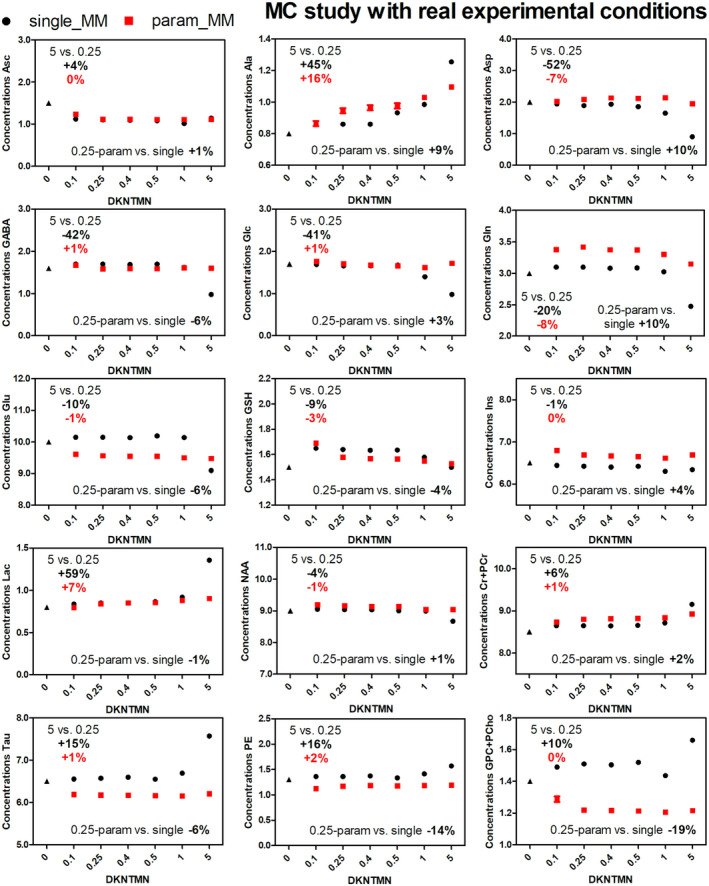
Metabolite concentrations obtained when quantifying MC spectra mimicking real experimental conditions (with baseline) with different DKNTMN values. The quantifications were done using two approaches single MM (black) and parametrized MM with PK (red) in LCModel. The true concentration of each metabolite is represented with a triangle symbol at DKNTMN = 0
The Real MC combined with the single MM spectrum and DKNMTN of 5 ppm showed stronger deviations from the ground truth for most metabolites, suggesting that a too flat baseline should not be used (Supporting Information Table S1). These changes were also supported by the difference between the original baseline used for the simulation of the Real MC spectra and the one resulting from the LCModel analysis (Supporting Information Figures S3, S4). In addition, there was an SNR decrease (Supporting Information Quality of in vivo 1H‐MR spectra) and a mismatch between the raw data and the LCModel fit when increasing DKNTMN (Figure 5A), supporting our quantitative results. Due to the high number of MC realizations and consistent changes in metabolite concentrations, all the variations for the MC studies were statistically significant. In this context, we choose to report only those that were higher than 5% and, thus, could be biologically relevant.
FIGURE 5.
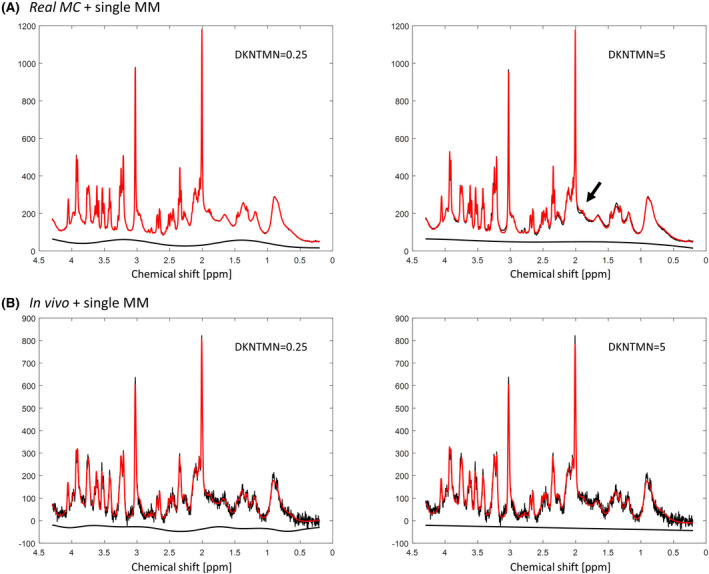
A, Spectra obtained with MC simulation of real experimental conditions (with baseline) showing a mismatch between the raw data and the LCModel fit (arrow) when quantifying with DKNTMN = 5 ppm. B, In vivo acquired spectra from the same rat using DKNTMN = 0.25 ppm (left) and DKNTMN = 5 ppm (right)
3.2.2. In vivo and Real MC data quantified using parametrized MM+PK: impact of the DKNTMN parameter
When using the parametrized MM with PK, the overall in vivo metabolite changes over the DKNTMN values became smaller than for the single MM, except for Ala (+32%, P = .0003(***)), Ins (−4%, P = .0364(*)), and Lac (+34%, P = .0118 (*)) (Figure 3, red plots). Additionally, some overlapping and low concentration metabolites showed noticeable but non‐significant changes (eg, Asc (+15%), Asp (−14%), Gln (−13%), and tCho (+7%)). The smaller impact of DKNTMN when using a parametrized MM spectrum can be explained by the additional flexibility of the fitting model to account for a small mismatch between the experimental spectrum and the spectra in the basis‐set. These findings were also supported by the results obtained using the Real MC with parametrized MM+PK (Figure 4, red plots, Supporting Information Table S1) where metabolite changes followed the same trend as observed for the in vivo MR spectra (ranging from −7% to +16%).
3.2.3. Impact of the MM model (parametrized vs single MM) on the metabolite quantification
When comparing the parametrized MM+PK vs the single MM model, the in vivo spectra revealed an overall increase in metabolite concentrations for parametrized MM+PK, which was significant for Asp (+14%,####), GABA (+37%,####), Gln (+10%,###), Glu (+2%,#), GSH (+15%,####), Ins (+5%,#), PE (+45%,####), and tCr (+7%,####) (Figure 3, of note the changes for Glu and Ins are small in %). When using MC studies, these changes were better assigned due to the known ground truth and were overall in agreement with the in vivo changes with some minor exceptions for low concentrated and/or overlapping metabolites (ie, GABA, GSH, Glu, PE, tCr, Tau, and tCho). For parametrized MM+PK, some metabolites displayed an over‐/under‐estimation compared to single MM and the ground truth for both Optimal and Real MC studies, which was independent of DKNTMN, that is, Gln (+10 to +15%), Glu (−5 to −6%), Ins (+4 to +6%), Tau (−6%), PE (−14 to −20%), and tCho (−7 to −19%) (Supporting Information Table S2).
3.2.4. Impact of PK on MM and metabolite quantification
We have also performed quantifications without using PK on the MM components to evaluate the impact of the added PK. For the in vivo spectra, the lack of PK led to an overall increase in the MM content ranging from 19% to ~250% (Supporting Information Figure S5) and to an overall decrease in metabolite concentrations ranging from −7% to −28% (with significant changes for Ala, Asp, GABA, Glu, GSH, PE, Tau, tCr, and tCho; Supporting Information Figure S6) when compared to parametrized MM+PK. For the Real MC study, no changes were observed when comparing PK with no PK, which can be justified by the fact that the MC study contained the same MM like those included in the basis‐set (Supporting Information Figures S7, S8).
To assess further the impact of PK on metabolite concentrations, the in vivo results obtained with parametrized MM, parametrized MM+PK and single MM were compared at DKNTMN = 0.25 and 5 ppm (Supporting Information Figure S9). For most metabolites, PK led to a better agreement with concentrations obtained when using the single MM spectrum, except for GABA, GSH, and PE where the resulting concentrations for parametrized MM+PK were significantly higher (37%, 15%, and 45%, at DKNTMN = 0.25 ppm, respectively).
3.3. Apparent T2
To determine the MM , all the 10 components were fitted (at different TEs). Acceptable fits of exponentially decaying MM signal intensities were found for 7 MM components (SD of the fitted value was lower than 20%), leading to reliable estimations of (Figure 6). The values were within a narrow range 22‐24 ms for the MM components at 0.94, 1.22, 1.43, and 3.00 ppm and within 12‐15 ms for the MM components at 1.70, 2.05, and 3.21 ppm.
FIGURE 6.
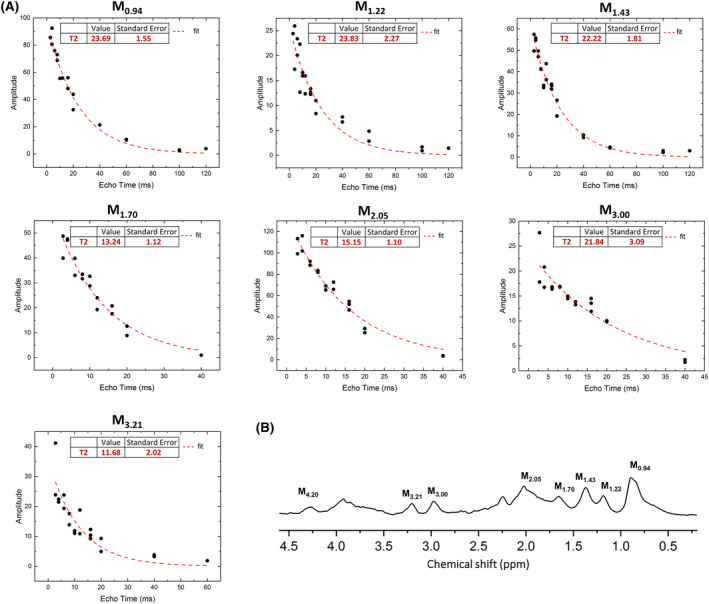
A, Exponential fits which provide T2 relaxation estimates. B, The MM spectra with marked components for which the relaxation times were estimated
4. DISCUSSION
This study reports, for the first time, the of seven individual MM components and the concomitant impact of the LCModel baseline stiffness and MM model (ie, single vs parametrized MM with and without PK) on metabolite and MM quantification using MC simulated spectra and in vivo rat brain spectra acquired at 9.4T. In addition, an advanced methodological approach for post‐processing, fitting, and parametrization of rat brain MM spectra at several TEs using multiple Lorentzian lines per MM component is proposed. Our results showed that a degree of flexibility in the spline baseline is required, while adding PK on the parametrized MM spectrum improved MM and metabolite quantification.
4.1. Post‐processing of individual MM spectra: removal of metabolite residuals
In the present work, AMARES was used with advanced PK for removal of metabolite residuals from MM spectra at TE = 2.8 ms and extended to MM spectra acquired at different TEs (up to 40 ms). Detailed steps for constructing reliable PK are also provided. Furthermore, additional metabolite residuals were identified and reliably removed from MM spectra compared with previous preclinical 10 , 45 (Ins‐4.05 ppm) and clinical (tCr‐3.0 ppm, Ins‐3.6 ppm, and Tau‐3.25 ppm) 9 , 11 , 23 , 34 studies (Table 1).
4.2. Quantification of brain metabolites: impact of varying DKNTMN value
Accurate and reliable quantification of metabolites depends strongly on the MM and baseline estimation. Few studies have assessed the impact of parameters affecting baseline properties (stiffness vs flexibility) on metabolite quantification. These studies have been conducted in humans at B0 ≥ 7T using LCModel and changing the DKNTMN parameter 31 , 32 and have reported an overall tendency of increase in metabolite concentrations with increasing baseline stiffness. 31 These previous studies have suggested that the default LCModel baseline flexibility may not be optimal in some cases but a conclusion on “good” values could not be drawn due to the lack of ground truth when using only in vivo spectra.
In this study, we analyzed a total of six DKNTMN values ranging from 0.1 ppm (high flexibility) to 5 ppm (high stiffness), which were applied to nine quantification sets. Metabolite concentrations from our in vivo data (single MM) mainly displayed a decrease with increasing the DKNTMN value (eg, Gln, Glu, GABA, Figure 3) also confirmed by the Real MC study. When analyzing the Real MC study (single MM) at DKNTMN of 1 or 5 ppm most metabolite concentrations (Ala, Asp, GABA, Glc, Gln, Glu, Lac, PE, Tau, tCr, and tCho) displayed a change that deviated from their ground truth, suggesting that a moderate DKNTMN value might be preferable, that is, below 1 ppm (Figure 4). This result was further supported by the qualitative LCModel output baseline analysis (Supporting Information Figure S3). A recent study presenting a newly developed algorithm for baseline smoothness has reported that the main sources of baseline‐fitting errors are bias from an inflexible baseline (underfitting) and an increased variance from an overly flexible baseline (overfitting), 47 suggesting that a reasonable compromise in baseline flexibility needs to be found. 47 This recommendation agrees well with our results, which suggested that a moderate DKNTMN value might be a better solution than a high one. In our study, metabolite concentrations were more stable over DKNTMN values when using the parametrized MM for all quantification sets, which is due to the additional flexibility of the individual MM components. However, when using the parametrized MM along with a low DKNTMN value the baseline displayed stronger features and distortions with a drop at 0.9 ppm. This is explainable since the MM component at 0.9 ppm was used as a reference for the other components. The drop and other residual features became smaller already at a DKNTMN of 0.25 ppm being comparable to those with single MM (Figure 7).
FIGURE 7.
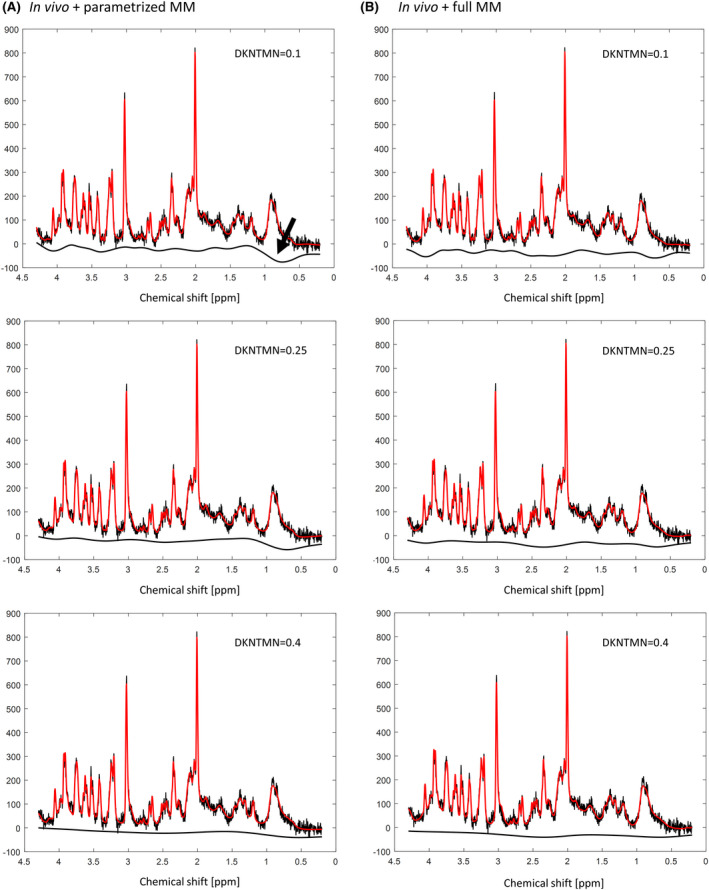
A, LCModel quantification of in vivo acquired spectra with parametrized MM + PK showing a baseline drop at around 0.9 ppm (arrow) when varying the DKNTMN. B, LCModel quantification of in vivo acquired spectra with single MM
4.3. Quantification of brain metabolites: single vs parametrized MM
The standard approach for handling the MM signals during metabolite quantification consists of including a single MM spectrum in the basis‐set or subtracting this MM spectrum from the in vivo acquired spectrum before quantification. Since the MM individual components might change due to disease (eg, brain tumors, multiple sclerosis, and stroke 3 , 21 ), this approach can lead to substantial errors in the quantification when assessing metabolite or MM changes in pathologies. The use of a parametrized MM has been proven helpful for quantification of spectra in pathologically altered cases in humans at 1.5T. 21 Parametrization of MM into individual components is difficult since the signal is a result of many chemical entities whose number and nature are predominantly unknown. To date, the MM spectrum has been parametrized into numerous Lorentzian, Gaussian, or Voigt lines to interpret distinguishable signal entities that were then grouped together to avoid over parametrization and included in the basis‐set for quantification. 3 , 11 , 13 , 21 , 22 , 23 , 24 , 35 , 48 , 49 At UHF, 9 to 24 MM peaks can be distinguished based on the nomenclature used, 3 , 9 , 11 with some peaks needing further investigations. These peaks have been previously fitted using mainly singlets, 9 , 11 , 34 , 45 while some studies used several peaks to fit individual MM components without specific PK, which were then grouped in a limited number of components to avoid over‐parametrization. 13 , 48 , 49 Even though MM signals can be modeled using singlets, they have complex spectral patterns. There are few studies evaluating the impact of parametrized MM with PK on metabolite concentrations, together with the assessment of the best soft constraints while still keeping enough flexibility to estimate MM changes. 11 , 45 , 50
In this study, we fitted and parametrized the experimentally acquired MM spectra into 10 components using AMARES, previously recommended 11 for MM parametrization. We aimed to fit the maximum number of individual MM components by choosing 10 components with 2 of them containing several MM peaks (M3.71‐3.97, M2.27). The MM peaks at 2.56 and 2.70 ppm, previously reported in humans at 9.4T, 9 were not fitted in this study. These two peaks had a relatively small contribution in our rat brain spectra and based on the IR curve they were well modeled by the residual NAA resonances, in agreement with a study at 3T. 34 However, a small MM contribution cannot be excluded. In contrast to previous studies, 11 , 45 , 50 each of the 10 MM components was parametrized by fitting with several Lorentzian functions to consider various unknown constituents that might be contained in one broad resonance. This approach reduced the overlap between MM resonances that might arise when broad functions are used. This parametrization was the result of an iterative process, which led to an efficient fitting procedure giving artifact‐free residuals. Moreover, we used three MM acquisitions that were individually parametrized. The results of signal intensity ratios of Mxx/M0.94 were averaged to obtain mean values and SDs that were then used as PK and soft constraints in the LCModel control files. The PK and soft constraints used herein were in agreement with a previous study in the human brain at 7T, 50 while another study in the rat brain at 7T 45 has reported higher overall MM amplitudes/ratios.
The comparison between the single and parametrized MM model (in vivo) revealed a significant increase in the concentration of several metabolites (Asp, GABA, Gln, Glu, GSH, Ins, PE, and tCr) when the parametrized MM model was used. These findings were confirmed by our MC studies and agreed with previous studies. 11 Importantly, our study evaluated the changes in MM content in addition to metabolite concentrations. We observed an important impact on MM content when no PK was used, emphasizing the need for PK as previously suggested in humans at 7T. 11 Herein, we provided reliable soft constraints for individual estimation of 10 MM components in healthy rat brain. These soft constraints can be extended to pathological conditions by measuring the MM in one to two patients and then parametrizing the in vivo MM spectrum as performed in this study, or using the soft constraints provided. Soft constraints can then be adapted based on visual appearance of the spectra (eg, fits, residuals, and baseline). For instance, if it is known or visible in the 1H‐MR spectrum that a specific MM moiety is changing, then more freedom can be provided for this MM moiety through soft constraints on the PK or a separately simulated MM or lipid component can be added to the basis‐set. 20 Furthermore, the described approach fully characterized the MM spectra at different TIs and TEs; thus, it can provide a comprehensive set of information necessary in a MM dictionary for MR fingerprinting. 35
4.4. Apparent T2
In this study, the of seven individual MM peaks has also been reported. High‐quality spectra and fits were obtained at all TEs. The choice of TEs is important for achieving reliable estimates of T2, but the optimal choice remains an open question and is sometimes dictated by the achievable SNR. Nine to 13 TEs were used in this study to calculate of MM, the longest being 150 ms for M0.94, M1.22, and M1.43. The chosen approach was adequate for modeling the T2 of MM peaks as evidenced by appropriate modeling of the observed signal decay and the low T2 SD. Despite the smaller number of acquisitions per TE, more than nine TEs were used for each fit, resulting in a good confidence curve fit for the seven T2 values. Although previous studies assessing the T2‐s of MM in the rat brain mainly reported results for grouped or full MM rather than individual components, the present results agree well with these published values. 13 , 27 , 33 Our results predominantly fall in the range of recently reported for the human brain at 9.4T for 14 individual MM peaks 9 and at 3T for 10 individual MM peaks assessed in different brain regions. 34
4.5. Limitations and perspectives
This study has a few limitations. The first limitation concerns the usage of only high‐quality data (in vivo and MC studies without and with a small baseline contribution) to evaluate how the inclusion of a single or parametrized MM spectrum together with changes in spline baseline stiffness affect the metabolite quantification. Overall, only high‐quality data should be used for metabolite quantification. Moreover, our aim was to assess the “real impact” of MM in the spectral fitting and quantification process; thus, we did not use lower quality data (eg, low SNR, B0 shimming effects on linewidths, and outer volume contamination). In this context, note that the baseline influence on metabolite concentrations can be slightly different when low‐quality data are used (eg, baseline can become almost flat when processing noisy data or can become critical for spectra with low spectral resolution due to bad quality shimming). An additional limitation of the study concerns the lack of data with outer volume contamination or spectra with important changes in MM content due to different pathological conditions. For spectra with lipid contamination that typically appears at around 1.5 ppm, it would be beneficial to include in the basis‐set one additional broad peak at 1.5 ppm with full flexibility in phase and moderate flexibility in chemical shift and linewidth. Finally, in this study, the purely mathematical estimation of MM was not considered since the smooth approximation of mathematical fitting for MM does not completely reproduce the in vivo spectral pattern at UHF, and as such, experimentally measured MM are recommended for all B0. 51 , 52 , 53 , 54 , 55
5. CONCLUSIONS
This study proposed an advanced methodological approach allowing reliable post‐processing, fitting, and parametrization of MM spectra in the rat brain at 9.4T. Moreover, we performed an extensive assessment on how the inclusion of either a single or parametrized MM spectrum with or without PK in the basis‐set concomitant with changes in the spline baseline stiffness affect the metabolite quantification. The described method also provided an efficient tool for the parametrization of individual MM and estimation of the of seven individual MM components. Using rat brain in vivo MRS data and MC studies, we showed that a degree of flexibility in the spline baseline is required for reliable quantification. A highly stiff baseline led to considerable metabolite changes when using the single MM spectrum for in vivo rat brain spectra, while for the parametrized MM model this effect was less pronounced for stiff baselines, but an overall deviation from the ground truth was measured using MC studies. As such, a generally valid value for DKNTMN cannot be predicted, and it needs to be adapted to the experimental and fitting conditions by keeping a balance between flexibility and stiffness. Finally, our results showed some metabolite changes when including a parametrized MM in the basis‐set vs a single MM. Adding PK on the parametrized MM spectrum improved MM and metabolite quantification, supporting the need of PK when using a parametrized MM spectrum.
Supporting information
FIGURE S1 Spectra simulated to mimic optimal experimental conditions—metabolites with MM only (shown in black); baseline mimicking minor outer volume contamination and insufficient water suppression (shown in red)—contains three resonances at 1.4, 3.2, and 4.7 ppm (simulated in jMRUI); spectra simulated to mimic real experimental conditions ‐ metabolites with MM and baseline (shown in blue)
FIGURE S2 Baseline extracted from the LCModel quantification of one in vivo acquired spectra quantified using different DKNTMN values and single (top panel) or parametrized (bottom panel) MM in the basis‐set
FIGURE S3 Baseline extracted from the LCModel quantification of one real MC simulated spectra quantified using different DKNTMN values and single (top panel) or parametrized (bottom panel) MM in the basis‐set. The original baseline used in MC simulation to create the real MC spectra is shown in bold red
FIGURE S4 Metabolite concentrations obtained when quantifying Monte Carlo spectra mimicking optimal experimental conditions (without baseline) with different DKNTMN values. Quantifications were done using two approaches single MM (black) and parametrized MM with PK (red) in LCModel. True concentration of each metabolite is represented with a triangle symbol at DKNTMN = 0
FIGURE S5 In vivo metabolite changes obtained by the quantification in LCModel using parametrized MM with constraints (with prior knowledge—PK) in form of signal intensity ratios and their standard deviations (included in the control file)—shown in red, and without constraints (no prior knowledge—NoPK)—shown in purple. The comparison between groups NoPK vs PK was calculated using two‐way ANOVA (# P < .05, ## P < .01, ### P < .001, #### P < .0001) and is shown on the right in each plot if significant
FIGURE S6 In vivo macromolecule changes obtained by the quantification in LCModel using parametrized MM with constraints (with prior knowledge—PK) in form of signal intensity ratios and their standard deviations (included in the control file)—shown in red, and without constraints (no prior knowledge—NoPK)—shown in purple. The comparison between groups NoPK vs PK was calculated using two‐way ANOVA (# P < .05, ## P < .01, ### P < .001, #### P < .0001) and is shown on the right in each plot if significant
FIGURE S7 Metabolite concentrations obtained when quantifying (in LCModel) Monte Carlo spectra mimicking real experimental conditions using parametrized MM with constraints (with prior knowledge—PK) in form of signal intensity ratios and their standard deviations (included in the control file)—shown in red, and without constraints (no prior knowledge—NoPK)—shown in purple
FIGURE S8 Macromolecule concentrations obtained when quantifying (in LCModel) Monte Carlo spectra mimicking real experimental conditions using parametrized MM with constraints (with prior knowledge—PK) in form of signal intensity ratios and their standard deviations (included in the control file)—shown in red, and without constraints (no prior knowledge—NoPK)—shown in purple
FIGURE S9 In vivo metabolite changes obtained from LCModel quantifications using single MM spectra‐shown in black, parametrized MM without constraints (no prior knowledge—NoPK)—shown in purple and with constraints (with prior knowledge—PK) in form of signal intensity ratios and their standard deviations (included in the control file)—shown in red at DKNTMN = 0.25 and 5 ppm. One‐way ANOVA, Bonferroni correction *P < .05, **P < .01, ***P < .001, ****P < .0001
TABLE S1 The exact values of metabolite concentrations obtained when quantifying Monte Carlo spectra mimicking real experimental conditions (with baseline) with DKNTMN = 0.25, 1, and 5 ppm. The corresponding percentage change compared to DKNTMN = 0.25 ppm is also reported
TABLE S2 The exact values of metabolite concentrations obtained when quantifying Monte Carlo spectra mimicking real experimental conditions (with baseline) with DKNTMN = 0.25 ppm compared with their true value. The metabolites, which have a bigger deviation from the true value when using parametrized MM, are shown in red
Supinfo 2
ACKNOWLEDGMENTS
Financial support was provided by the Swiss National Science Foundation (project no 310030_173222; DS, VR, CC), National Institutes of Health grants (P41 EB027061 and P30 NS076408; IT), Horizon 2020/ CDS‐QUAMRI Grant number: 634541 (TB), the European Regional Development Fund (MEYS CZ.02.1.01/0.0/0.0/16_013/0001775; ZS, JS). The authors acknowledge access to the facilities and expertise of the CIBM Center for Biomedical Imaging, a Swiss research center of excellence founded and supported by Lausanne University Hospital (CHUV), University of Lausanne (UNIL), Ecole Polytechnique Fédérale de Lausanne (EPFL), University of Geneva (UNIGE), and Geneva University Hospitals (HUG). The authors would like to thank Professors S. R. Williams and Anke Henning for constructive discussions.
Simicic D, Rackayova V, Xin L, et al. In vivo macromolecule signals in rat brain 1H‐MR spectra at 9.4T: Parametrization, spline baseline estimation, and T2 relaxation times. Magn Reson Med. 2021;86:2384‐2401. 10.1002/mrm.28910
Correction added after online publication 10 August 2021. The authors have corrected the images for Figures 3 and 4 which were swapped in the prior version.
DATA AVAILABILITY STATEMENT
The Matlab codes and data that support the findings of this study are openly available in MRShub at https://mrshub.org/datasets/ ; https://doi.org/10.5281/zenodo.3904443.
REFERENCES
- 1. Hofmann L, Slotboom J, Boesch C, Kreis R. Characterization of the macromolecule baseline in localized 1H‐MR spectra of human brain. Magn Reson Med. 2001;46:855‐863. [DOI] [PubMed] [Google Scholar]
- 2. Cudalbu C, Mlynárik V, Gruetter R. Handling macromolecule signals in the quantification of the neurochemical profile. J Alzheimer's Dis. 2012;31:S101‐S115. [DOI] [PubMed] [Google Scholar]
- 3. Cudalbu C, Behar KL, Bhattacharyya PK, et al. Contribution of macromolecules to brain 1H MR spectra: experts’ consensus recommendations. NMR Biomed. 2021;34:e4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Behar KL, Ogino T. Characterization of macromolecule resonances in the 1H NMR spectrum of rat brain. Magn Reson Med. 1993;30:38‐44. [DOI] [PubMed] [Google Scholar]
- 5. Behar KL, Ogino T. Assignment of resonances in the 1H spectrum of rat brain by two‐dimensional shift correlated and J‐resolved NMR spectroscopy. Magn Reson Med. 1991;17:285‐303. [DOI] [PubMed] [Google Scholar]
- 6. Behar KL, Rothman DL, Spencer DD, Petroff OAC. Analysis of macromolecule resonances in 1H NMR spectra of human brain. Magn Reson Med. 1994;32:294‐302. [DOI] [PubMed] [Google Scholar]
- 7. Kauppinen RA, Niskanen T, Hakumäki J, Williams SR. Quantitative analysis of 1H NMR detected proteins in the rat cerebral cortex in vivo and in vitro. NMR Biomed. 1993;6:242‐247. [DOI] [PubMed] [Google Scholar]
- 8. Kauppinen RA, Kokko H, Williams SR. Detection of mobile proteins by proton nuclear magnetic resonance spectroscopy in the Guinea pig brain ex vivo and their partial purification. J Neurochem. 1992;58:967‐974. 10.1111/j.1471-4159.1992.tb09350.x. [DOI] [PubMed] [Google Scholar]
- 9. Murali‐Manohar S, Borbath T, Wright AM, Soher B, Mekle R, Henning A. T2 relaxation times of macromolecules and metabolites in the human brain at 9.4 T. Magn Reson Med. 2020;84:542‐558. [DOI] [PubMed] [Google Scholar]
- 10. Craveiro M, Clément‐Schatlo V, Marino D, Gruetter R, Cudalbu C. In vivo brain macromolecule signals in healthy and glioblastoma mouse models: 1H magnetic resonance spectroscopy, post‐processing and metabolite quantification at 14.1 T. J Neurochem. 2014;129:806‐815. [DOI] [PubMed] [Google Scholar]
- 11. Považan M, Strasser B, Hangel G, et al. Simultaneous mapping of metabolites and individual macromolecular components via ultra‐short acquisition delay 1H MRSI in the brain at 7T. Magn Reson Med. 2018;79:1231‐1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kunz N, Cudalbu C, Mlynarik V, Hüppi PS, Sizonenko SV, Gruetter R. Diffusion‐weighted spectroscopy: a novel approach to determine macromolecule resonances in short‐echo time 1H‐MRS. Magn Reson Med. 2010;64:939‐946. [DOI] [PubMed] [Google Scholar]
- 13. Lopez‐Kolkovsky AL, Mériaux S, Boumezbeur F. Metabolite and macromolecule T1 and T2 relaxation times in the rat brain in vivo at 17.2T. Magn Reson Med. 2016;75:503‐514. [DOI] [PubMed] [Google Scholar]
- 14. Near J, Harris AD, Juchem C, et al. Preprocessing, analysis and quantification in single‐voxel magnetic resonance spectroscopy: experts’ consensus recommendations. NMR Biomed. 2021;34:e4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mader I. Proton MR spectroscopy with metabolite‐nulling reveals elevated macromolecules in acute multiple sclerosis. Brain. 2001;124:953‐961. [DOI] [PubMed] [Google Scholar]
- 16. Pedrosa de Barros N, Meier R, Pletscher M, et al. On the relation between MR spectroscopy features and the distance to MRI‐visible solid tumor in GBM patients. Magn Reson Med. 2018;80:2339‐2355. [DOI] [PubMed] [Google Scholar]
- 17. Howe FA, Barton SJ, Cudlip SA, et al. Metabolic profiles of human brain tumors using quantitative in vivo 1H magnetic resonance spectroscopy. Magn Reson Med. 2003;49:223‐232. [DOI] [PubMed] [Google Scholar]
- 18. Opstad KS, Griffiths JR, Bell BA, Howe FA. Apparent T2 relaxation times of lipid and macromolecules: a study of high‐grade tumor spectra. J Magn Reson Imaging. 2008;27:178‐184. [DOI] [PubMed] [Google Scholar]
- 19. Opstad KS, Wright AJ, Bell BA, Griffiths JR, Howe FA. Correlations between in vivo 1H MRS and ex vivo 1H HRMAS metabolite measurements in adult human gliomas. J Magn Reson Imaging. 2010;31:289‐297. [DOI] [PubMed] [Google Scholar]
- 20. Oz G, Tkac I, Charnas LR, et al. Assessment of adrenoleukodystrophy lesions by high field MRS in non‐sedated pediatric patients. Neurology. 2005;64:434‐441. [DOI] [PubMed] [Google Scholar]
- 21. Seeger U, Klose U, Mader I, Grodd W, Nägele T. Parameterized evaluation of macromolecules and lipids in proton MR spectroscopy of brain diseases. Magn Reson Med. 2003;49:19‐28. [DOI] [PubMed] [Google Scholar]
- 22. Pfeuffer J, Juchem C, Merkle H, Nauerth A, Logothetis NK. High‐field localized 1H NMR spectroscopy in the anesthetized and in the awake monkey. Magn Reson Imaging. 2004;22:1361‐1372. [DOI] [PubMed] [Google Scholar]
- 23. Hong S‐T, Balla DZ, Shajan G, Choi C, Uğurbil K, Pohmann R. Enhanced neurochemical profile of the rat brain using in vivo 1H NMR spectroscopy at 16.4 T. Magn Reson Med. 2011;65:28‐34. [DOI] [PubMed] [Google Scholar]
- 24. Lee HH, Kim H. Parameterization of spectral baseline directly from short echo time full spectra in 1H‐MRS. Magn Reson Med. 2017;78:836‐847. [DOI] [PubMed] [Google Scholar]
- 25. Provencher S. LCModel Manual. Stephen Provencher; 2019. [Google Scholar]
- 26. Provencher SW. Automatic quantitation of localized in vivo 1H spectra with LCModel. NMR Biomed. 2001;14:260‐264. [DOI] [PubMed] [Google Scholar]
- 27. Pfeuffer J, Tkáč I, Provencher SW, Gruetter R. Toward an in vivo neurochemical profile: quantification of 18 metabolites in short‐echo‐time 1H NMR spectra of the rat brain. J Magn Reson. 1999;141:104‐120. [DOI] [PubMed] [Google Scholar]
- 28. Deelchand DK, Marjańska M, Hodges JS, Terpstra M. Sensitivity and specificity of human brain glutathione concentrations measured using short‐TE 1H MRS at 7 T. NMR Biomed. 2016;29:600‐606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Terpstra M, Ugurbil K, Tkac I. Noninvasive quantification of human brain ascorbate concentration using1H NMR spectroscopy at 7 T. NMR Biomed. 2010;23:227‐232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Near J, Andersson J, Maron E, et al. Unedited in vivo detection and quantification of γ‐aminobutyric acid in the occipital cortex using short‐TE MRS at 3T. NMR Biomed. 2013;26:1353‐1362. [DOI] [PubMed] [Google Scholar]
- 31. Giapitzakis IA, Borbath T, Murali‐Manohar S, Avdievich N, Henning A. Investigation of the influence of macromolecules and spline baseline in the fitting model of human brain spectra at 9.4T. Magn Reson Med. 2019;81:746‐758. [DOI] [PubMed] [Google Scholar]
- 32. Marjańska M, Terpstra M. Influence of fitting approaches in LCModel on MRS quantification focusing on age‐specific macromolecules and the spline baseline. NMR Biomed. 2021;34:e4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. De Graaf RA, Brown PB, McIntyre S, Nixon TW, Behar KL, Rothman DL. High magnetic field water and metabolite proton T1 and T2 relaxation in rat brain in vivo. Magn Reson Med. 2006;56:386‐394. [DOI] [PubMed] [Google Scholar]
- 34. Landheer K, Gajdošík M, Treacy M, Juchem C. Concentration and effective T2 relaxation times of macromolecules at 3T. Magn Reson Med. 2020;84:2327‐2337. [DOI] [PubMed] [Google Scholar]
- 35. Hoefemann M, Bolliger CS, Chong DGQ, Veen JW, Kreis R. Parameterization of metabolite and macromolecule contributions in interrelated MR spectra of human brain using multidimensional modeling. NMR Biomed. 2020;33:e4328. [DOI] [PubMed] [Google Scholar]
- 36. Gruetter R, Tkáč I. Field mapping without reference scan using asymmetric echo‐planar techniques. Magn Reson Med. 2000;43:319‐323. [DOI] [PubMed] [Google Scholar]
- 37. Mlynárik V, Gambarota G, Frenkel H, Gruetter R. Localized short‐echo‐time proton MR spectroscopy with full signal‐intensity acquisition. Magn Reson Med. 2006;56:965‐970. [DOI] [PubMed] [Google Scholar]
- 38. Cudalbu C, Mlynrik V, Xin L, Gruetter R. Quantification of in vivo short echo‐time proton magnetic resonance spectra at 14.1 T using two different approaches of modelling the macromolecule spectrum. Meas Sci Technol. 2009;20:104034 (7pp). [Google Scholar]
- 39. Mlynárik V, Cudalbu C, Xin L, Gruetter R. 1H NMR spectroscopy of rat brain in vivo at 14.1 Tesla: improvements in quantification of the neurochemical profile. J Magn Reson. 2008;194:163‐168. [DOI] [PubMed] [Google Scholar]
- 40. Craveiro M, Cudalbu C, Gruetter R. Regional alterations of the brain macromolecule resonances investigated in the mouse brain using an improved method for the pre‐processing of the macromolecular signal. Proceedings of the 20th Annual Meeting ISMRM, Melbourne, Australia, 2012. Abstract 1748.
- 41. Xin L, Mlynarik V, Lei H, Gruetter R. Influence of regional macromolecule baseline on the quantification of neurochemical profile in rat brain. In Proceedings of the 18th Annual Meeting of ISMRM, Stockholm, Sweden, 2010. Abstract 321.
- 42. Tkáč I, Starčuk Z, Choi IY, Gruetter R. In vivo 1H NMR spectroscopy of rat brain at 1 ms echo time. Magn Reson Med. 1999;41:649‐656. [DOI] [PubMed] [Google Scholar]
- 43. Vanhamme L, Van Den Boogaart A, Van Huffel S. Improved method for accurate and efficient quantification of MRS data with use of prior knowledge. J Magn Reson. 1997;129:35‐43. [DOI] [PubMed] [Google Scholar]
- 44. Lee HH, Kim H. Parameterization of spectral baseline directly from short echo time full spectra in 1H‐MRS. Magn Reson Med. 2017;78:836‐847. [DOI] [PubMed] [Google Scholar]
- 45. Fowler CF, Madularu D, Dehghani M, Devenyi GA, Near J. Longitudinal quantification of metabolites and macromolecules reveals age‐ and sex‐related changes in the healthy Fischer 344 rat brain. Neurobiol Aging. 2021;101:109‐122. [DOI] [PubMed] [Google Scholar]
- 46. Kreis R, Boer V, Choi I‐Y, et al. Terminology and concepts for the characterization of in vivo MR spectroscopy methods and MR spectra: Background and experts’ consensus recommendations. NMR Biomed. 2021;34:e4347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wilson M. Adaptive baseline fitting for MR spectroscopy analysis. Magn Reson Med. 2021;85:13‐29. [DOI] [PubMed] [Google Scholar]
- 48. Snoussi K, Gillen JS, Horska A, et al. Comparison of brain gray and white matter macromolecule resonances at 3 and 7 Tesla. Magn Reson Med. 2015;74:607‐613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Otazo R, Mueller B, Ugurbil K, Wald L, Posse S. Signal‐to‐noise ratio and spectral linewidth improvements between 1.5 and 7 Tesla in proton echo‐planar spectroscopic imaging. Magn Reson Med. 2006;56:1200‐1210. [DOI] [PubMed] [Google Scholar]
- 50. Heckova E, Považan M, Strasser B, et al. Effects of different macromolecular models on reproducibility of FID‐MRSI at 7T. Magn Reson Med. 2020;83:12‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chong DGQ, Kreis R, Bolliger CS, Boesch C, Slotboom J. Two‐dimensional linear‐combination model fitting of magnetic resonance spectra to define the macromolecule baseline using FiTAID, a fitting tool for arrays of interrelated datasets. Magn Reson Mater Phys, Biol Med. 2011;24:147‐164. [DOI] [PubMed] [Google Scholar]
- 52. Marjańska M, Deelchand DK, Hodges JS, et al. Altered macromolecular pattern and content in the aging human brain. NMR Biomed. 2018;31:e3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Coenradie Y, De Beer R, Van Ormondt D, Lyon B. Background‐signal parametrization in In Vivo MR Spectroscopy. ProRISC, IEEE Benelux. 2002;248‐254. [Google Scholar]
- 54. Cudalbu C, Beuf O, Cavassila S. In vivo short echo time localized 1H MRS of the rat brain at 7 T: influence of two strategies of background‐accommodation on the metabolite concentration estimation using QUEST. J Signal Process Syst. 2009;55:25‐34. [Google Scholar]
- 55. O’Gorman RL, Michels L, Edden RA, Murdoch JB, Martin E. In vivo detection of GABA and glutamate with MEGA‐PRESS: reproducibility and gender effects. J Magn Reson Imaging. 2011;33:1262‐1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Govindaraju V, Young K, Maudsley AA. Proton NMR chemical shifts and coupling constants for brain metabolites. NMR Biomed. 2000;13:129‐153. [DOI] [PubMed] [Google Scholar]
- 57. Xin L, Gambarota G, Cudalbu C, Mlynárik V, Gruetter R. Single spin‐echo T 2 relaxation times of cerebral metabolites at 14.1 T in the in vivo rat brain. Magn Reson Mater Phys, Biol Med. 2013;26:549‐554. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Spectra simulated to mimic optimal experimental conditions—metabolites with MM only (shown in black); baseline mimicking minor outer volume contamination and insufficient water suppression (shown in red)—contains three resonances at 1.4, 3.2, and 4.7 ppm (simulated in jMRUI); spectra simulated to mimic real experimental conditions ‐ metabolites with MM and baseline (shown in blue)
FIGURE S2 Baseline extracted from the LCModel quantification of one in vivo acquired spectra quantified using different DKNTMN values and single (top panel) or parametrized (bottom panel) MM in the basis‐set
FIGURE S3 Baseline extracted from the LCModel quantification of one real MC simulated spectra quantified using different DKNTMN values and single (top panel) or parametrized (bottom panel) MM in the basis‐set. The original baseline used in MC simulation to create the real MC spectra is shown in bold red
FIGURE S4 Metabolite concentrations obtained when quantifying Monte Carlo spectra mimicking optimal experimental conditions (without baseline) with different DKNTMN values. Quantifications were done using two approaches single MM (black) and parametrized MM with PK (red) in LCModel. True concentration of each metabolite is represented with a triangle symbol at DKNTMN = 0
FIGURE S5 In vivo metabolite changes obtained by the quantification in LCModel using parametrized MM with constraints (with prior knowledge—PK) in form of signal intensity ratios and their standard deviations (included in the control file)—shown in red, and without constraints (no prior knowledge—NoPK)—shown in purple. The comparison between groups NoPK vs PK was calculated using two‐way ANOVA (# P < .05, ## P < .01, ### P < .001, #### P < .0001) and is shown on the right in each plot if significant
FIGURE S6 In vivo macromolecule changes obtained by the quantification in LCModel using parametrized MM with constraints (with prior knowledge—PK) in form of signal intensity ratios and their standard deviations (included in the control file)—shown in red, and without constraints (no prior knowledge—NoPK)—shown in purple. The comparison between groups NoPK vs PK was calculated using two‐way ANOVA (# P < .05, ## P < .01, ### P < .001, #### P < .0001) and is shown on the right in each plot if significant
FIGURE S7 Metabolite concentrations obtained when quantifying (in LCModel) Monte Carlo spectra mimicking real experimental conditions using parametrized MM with constraints (with prior knowledge—PK) in form of signal intensity ratios and their standard deviations (included in the control file)—shown in red, and without constraints (no prior knowledge—NoPK)—shown in purple
FIGURE S8 Macromolecule concentrations obtained when quantifying (in LCModel) Monte Carlo spectra mimicking real experimental conditions using parametrized MM with constraints (with prior knowledge—PK) in form of signal intensity ratios and their standard deviations (included in the control file)—shown in red, and without constraints (no prior knowledge—NoPK)—shown in purple
FIGURE S9 In vivo metabolite changes obtained from LCModel quantifications using single MM spectra‐shown in black, parametrized MM without constraints (no prior knowledge—NoPK)—shown in purple and with constraints (with prior knowledge—PK) in form of signal intensity ratios and their standard deviations (included in the control file)—shown in red at DKNTMN = 0.25 and 5 ppm. One‐way ANOVA, Bonferroni correction *P < .05, **P < .01, ***P < .001, ****P < .0001
TABLE S1 The exact values of metabolite concentrations obtained when quantifying Monte Carlo spectra mimicking real experimental conditions (with baseline) with DKNTMN = 0.25, 1, and 5 ppm. The corresponding percentage change compared to DKNTMN = 0.25 ppm is also reported
TABLE S2 The exact values of metabolite concentrations obtained when quantifying Monte Carlo spectra mimicking real experimental conditions (with baseline) with DKNTMN = 0.25 ppm compared with their true value. The metabolites, which have a bigger deviation from the true value when using parametrized MM, are shown in red
Supinfo 2
Data Availability Statement
The Matlab codes and data that support the findings of this study are openly available in MRShub at https://mrshub.org/datasets/ ; https://doi.org/10.5281/zenodo.3904443.