

Abstract
Lead‐free double perovskites, A2M+M′3+X6, are considered as promising alternatives to lead‐halide perovskites, in optoelectronics applications. Although iodide (I) and bromide (Br) mixing is a versatile tool for bandgap tuning in lead perovskites, similar mixed I/Br double perovskite films have not been reported in double perovskites, which may be due to the large activation energy for ion migration. In this work, mixed Br/I double perovskites were realized utilizing an anion exchange method starting from Cs2AgBiBr6 solid thin‐films with large grain‐size. The optical and structural properties were studied experimentally and theoretically. Importantly, the halide exchange mechanism was investigated. Hydroiodic acid was the key factor to facilitate the halide exchange reaction, through a dissolution–recrystallization process. In addition, the common organic iodide salts could successfully perform halide‐exchange while retaining high mixed‐halide phase stability and strong light absorption capability.
Keywords: bandgap engineering, density functional calculations, ion exchange, lead-free double perovskites, solar cells
Ion exchange: A halide‐exchange method is introduced to narrow the bandgap of double perovskite Cs2AgBiBr6 solid thin‐films, resulting in over 0.3 eV bandgap change under ambient conditions, with superior stability. Hydroiodic acid is demonstrated to be a key factor to facilitate the anion‐exchange using organic iodide salt, making the proposed method versatile.
Introduction
Lead‐halide perovskites with a general formula APbX3 [A=Cs+, CH3NH3 + (MA); X=Cl−, Br−, I−] have shown great potential in optoelectronic device applications. [1] However, problems such as toxicity of lead and instability of these materials have been noticed. [2] Therefore, research to address these issues by replacing lead (Pb) with low‐toxic elements while keeping superior optoelectronic properties is imperative. Among the materials reported to date, the materials with a general expression of Cs2MM′X6 show great promise in realizing low‐toxic and long‐term stable optoelectronic applications. [3] These kinds of compounds are generally formed by replacing two Pb2+ ions into one monovalent M+ and one trivalent M′3+ ion, leading to an elpasolite (“double perovskite”) structure (Figure 1a). Several combinations of M+ and M′3+ ions are proved to be stable, such as Cu+, Ag+, Na+, Bi3+, Sb3+, In3+. [4]
Figure 1.
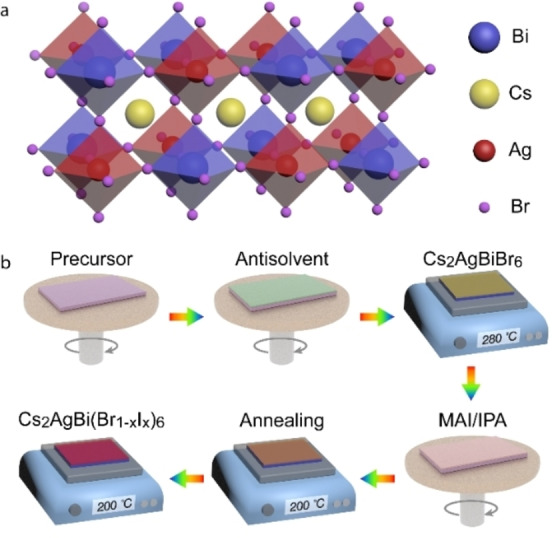
(a) Double perovskite crystal structure of Cs2AgBiBr6. (b) Schematic of the post‐treatment process.
So far, only a few types of double perovskites have been experimentally prepared, such as Cs2AgBiBr6, Cs2AgBiCl6, Cs2AgInCl6, Cs2NaBiCl6, as well as some mixed compositions. [5] Among these compounds, Cs2AgBiBr6 has attracted wide attention for photovoltaic applications, because it can absorb light in the visible range of the solar spectrum and exhibit an impressive heat and moisture stability. [6] Besides, Bi3+, being isoelectronic with Pb2+, provides great possibility of defect‐tolerance. [7] A long photoluminescence (PL) lifetime of microseconds has also been demonstrated, indicating the promising charge transport property. [6a] Photovoltaic devices based on Cs2AgBiBr6 have been studied in recent years, with rising photovoltaic power conversion efficiency over 3 %. [8] However, the relatively large bandgap is still considered as one of the main limiting factors for photovoltaic applications.
Compositional engineering in lead‐halide perovskites, which creates advantageous optical and electrical properties, has been investigated widely, leading to optoelectronic devices with advanced performances. [9] In lead‐free double perovskites, mixed compositions have been explored by trivalent or monovalent metal alloying,[ 4b , 10 ] as well as changing Br/Cl ratios. [11] However, Br/I mixed double perovskites have not previously been obtained. The direct fabrication of Cs2AgBiI6 or Cs2AgBi(Br/I)6 thin‐films from corresponding precursors followed by annealing always failed, resulting in Cs3Bi2I9 product (Figure S1), due to the either negative or slightly positive enthalpies. [12] Due to the low activation energy for ionic migration, the anion exchange method is quite applicable in lead perovskites for both bulk and nanocrystal (NC) forms, treated by halide‐containing solutions or vapors. [13] However, the ionic migration activation energy of double perovskite Cs2AgBiBr6 (348 meV) is nearly three times that of MAPbBr3 (126 meV), making halide less mobile and halide‐exchange more difficult. [3d] Until now, Cs2AgBiI6 only in NC form from direct synthesis, [14] and from Cs2AgBiBr6 NCs via anion exchange by trimethylsilyl iodide (TMSI) has been achieved. [15] TMSX (X=Cl, Br, I) were allowed to conduct anion exchange in double perovskite NCs, while the common reagents caused NC decomposition and formed impurity phases. However, it would be advantageous to obtain the bulk form of mixed‐halide double perovskites, since most of the applications are in the form of solid thin films.
In this work, we demonstrate halide‐exchange on Cs2AgBiBr6 solid thin‐films by post‐treating with methylammonium iodide (MAI) salt. To the best of our knowledge, this is the first successful method to obtain mixed‐halide double perovskite films. The compositions, crystal structures and band gaps were studied to demonstrate the successful incorporation of iodine. Moreover, the anion‐exchange mechanism was studied, where hydro‐iodide was demonstrated to be a key factor. Assisted by hydro‐iodide acid, the common iodide salts are allowed to replace bromine with iodine while keeping the cubic crystal structure, which makes the anion‐exchange method a more robust and versatile tool in double perovskites.
Results and Discussion
The post‐treatment process for double perovskite Cs2AgBiBr6 films is illustrated in Figure 1b. In brief, CsBr (1 mmol), AgBr (0.5 mmol), and BiBr3 (0.5 mmol) were dissolved in DMSO (1 mL) as precursor to fabricate Cs2AgBiBr6 via spin‐coating. Chlorobenzene (CB) was used as antisolvent, followed by 280 °C annealing for 5 min. Then, MAI dissolved in isopropanol (IPA) at certain concentrations was dropped onto the Cs2AgBiBr6 film, followed by spinning to remove the excess solution. Except the wide utilization in lead perovskites, the key advantage of MAI includes its volatility at high temperature, avoiding undesired by‐products. After 3 min annealing, we found that the film changed from yellow to red. The whole process was performed in a nitrogen‐filled dry box, and the details of the fabrication process can be found in the Experimental Section.
To investigate the effect of MAI treatment on the Cs2AgBiBr6 film properties, MAI solution with various concentrations (1, 10, 20, 25, 35 mg mL−1) were employed, named as C1, C2, C3, C4, C5, respectively, while using a Cs2AgBiBr6 film as the reference sample. X‐ray photoelectron spectroscopy (XPS) spectra of I 3d for these six samples clearly confirm the presence of iodine after MAI post‐treatment (Figure 2a). According to the N 1 s XPS spectra (Figure S2a), there were almost no MA+ ions detected, and we therefore conclude that the iodine ions could at least partly stay in films while the MA cations were evaporated during annealing. For perovskites, an appropriate Goldschmidt tolerance factor (between 0.8 and 1) is critical for achieving a stable structure. We illustrate this point in Figure 2b, which shows the calculated tolerance factor (α) of Cs2AgBiBr6‐x I x (x=0–6). This calculation is according to Equation 1:
| (1) |
Figure 2.

(a) XPS spectra of I 3d for Cs2AgBiBr6 film and treated by MAI (at various concentrations C1–C5: 1, 10, 20, 25, 35 mg mL−1). (b) Calculated Goldschmidt tolerance factor of Cs2AgBiBr6‐x I x double perovskite, according to the ion radii in Table S1 in the Supporting Information. (c) The left column represents XRD patterns of the Cs2AgBiBr6 film and the films exposed to MAI post‐treatment at various concentrations, while the right column represents the enlarged view of the (400) peak for all samples in XRD patterns. The peak marked with “&” is from an impure phase.
where R A, R X, and R B are the ion radii for A, B, and X in the ABX3 perovskite. [16] In our case, R B was modified as the average value for Ag+ and Bi3+. The ion radii used during the calculation are listed in Table S1 in the Supporting Information, taken from Ref. [17]. The tolerance factor shows that the values of α (over 0.83) of Cs2AgBiBr6‐x I x (x=0–6) with different iodine contents fall into the range required for stable perovskites, indicating that the fabrication of Cs2AgBiBr6‐x I x is theoretically feasible.
X‐ray diffraction (XRD) patterns of these six samples are shown in Figure 2c. The Cs2AgBiBr6 film showed a typical double‐perovskite structure, matching well with the reference pattern [Crystallography Open Database (COD) CIF 4131244] in Figure S3a. Also, the lattice parameter a of Cs2AgBiBr6 was calculated from the (400) reflection, yielding a=11.26 Å, which is consistent with previous reports. [15] After post‐treatment with MAI, the major diffraction peaks can be observed, but with shifted angles. An extra peak at around 17° appeared, which existed in previously reported Cs2AgBiI6 NCs and was ascribed to an artifact or impurity.[ 14 , 15 ] Apparently, the diffraction peaks shifted to smaller angle with increasing MAI treatment, especially the main peak of (400) (as shown in the enlarged view), which thereby suggests formation of Cs2AgBiBr6‐x I x mixed halide phase. There could be a possibility that ion exchange only occurred close to the film surface. In order to investigate it, XRD patterns with various incident angles were measured for the C5 sample. As shown in Figure S3, the main peaks remain at the same positions when varying the X‐ray incident angle, confirming the assumption of a uniform phase. In addition, the Br 3d and I 3d5/2 XPS elemental mapping for the C5 film also supports the presence and uniform distribution of I (Figure S2b).
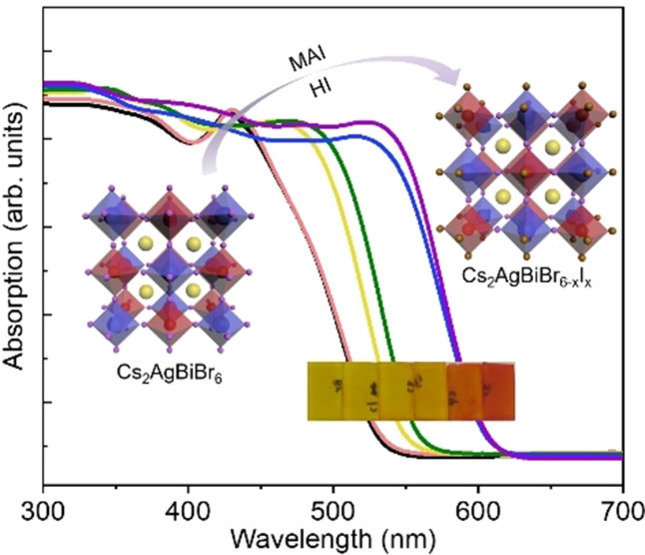
To examine the optical properties of the mixed‐halide double perovskites, UV/Vis reflectance (R) and transmittance (T) spectra for these samples were collected and converted into absorptance (A), via A=1‐R‐T. In Figure 3a, the absorptance of Cs2AgBiBr6 film is shown as the black line, with a peak at 426 nm induced from s‐p transitions in bismuth (Bi), and long band tail ascribed to indirect absorption.[ 8a , 18 ] Post‐treatment with MAI resulted in changes of absorption features, such as the sharper band tail and the wider absorption within visible wavelength range, suggesting a change in band structure after I incorporation. Moreover, Figure 3b shows that the film color is turned from yellow to orange and then red with increasing concentrations of MAI in the post‐treatment, confirming a narrowed bandgap, as expected.
Figure 3.
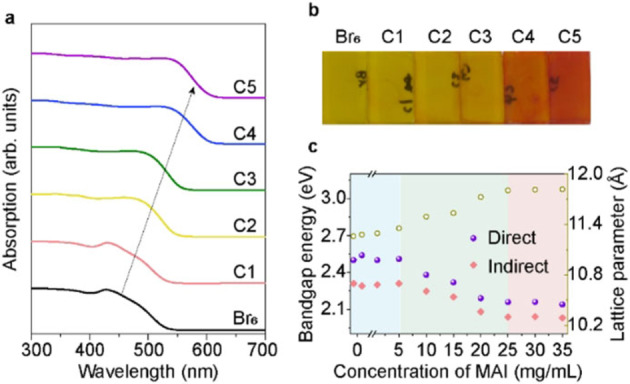
(a) UV/Vis absorption spectra and (b) optical images of six samples treated with different concentrations of MAI, increasing from left to right (Br6 to C5). (c) Change of bandgap energy (direct and indirect bandgap) and lattice parameter of obtained films as function of MAI concentrations in the preparation of the film. The lattice parameters were calculated from the (400) reflection by Gauss fitting to obtain peak position (in Figure S7).
XPS measurements were performed to determine the atomic ratio in the obtained samples. Figure S4 presents the survey XPS and detailed Ag 3d, Cs 3d, Bi 4f, and Br 3d core‐level spectra for samples treated by various MAI concentrations. Table S2 summarizes the atomic ratios for the different samples. The highest I/Br ratio was 4.5 : 1.0, indicating that iodine did not completely replace bromine. XPS results also suggest that the halide content is less than expected from the ideal ratio for Cs/Ag/Bi/(Br+I) of 2 : 1 : 1 : 6, indicating halide deficiency in the samples. Halide deficiency may lead to point defect formation, which could act as carrier traps. According a report by Xiao et al., [19] halide vacancy‐induced defects located 0.03 eV below the conduction band minimum (CBM) may act as shallow donors, affecting the electrical properties of the material. The halide deficiency might result in uncoordinated Bi3+, and then reduction to metallic bismuth (Bi0), indicated by the Bi0 peaks (157 eV at Bi 4f7/2 of Bi metal) in Figure S4d.
In order to understand the halide‐exchange mechanism and gain a higher I/Br ratio, we also investigated in detail the effect of MAI concentrations on the structural and optical properties (in Figures S5–S7). In Figure 3c, we monitor the trend for bandgap energies (calculated from Tauc plots) and lattice parameters as a function of MAI concentration (0–35 mg mL−1), also summarized in Table 1. Three distinct regions are clearly seen: at low concentration (below 5 mg mL−1), halide exchange was hardly observed; at concentrations between 5–25 mg mL−1, bromine was gradually replaced by iodine almost linearly; after that region, the I/Br ratio hardly changed. When the MAI concentration is further increased (higher than 35 mg mL−1), the absorptance (Figure S6a) and XRD patterns (Figure S7) both show that the obtained samples changed to a new phase. Since Cs3Bi2I9 phase is a common impurity in Cs2AgBiX6, we fabricated Cs3Bi2I9 thin film from the stoichiometry precursor solution and measured the XRD and optical absorption, as shown in Figures S5–S7. Apparently, when MAI concentration is higher than 35 mg mL−1, the obtained films is more similar to Cs3Bi2I9 phase. For the double perovskite films treated with lower MAI concentration, the main phase remained as mixed‐halide double perovskite. Thus, the study on mixed‐halide double perovskite in the following discussion will focus on samples treated by MAI concentration lower than 40 mg mL−1. The lattice parameter increases as expected during halide exchange reaction in Figure 3c. According to the lattice parameters of these mixed samples, we estimated the intermediate compositions using Vegard's law. Since it was not possible to fabricate Cs2AgBiI6, the reported lattice parameter of 12.09 Å for the pure iodine phase was used here. [15] Figure S8 and Table 1 show the estimated iodine amount (x ranges from 0 to 6) in final films at various MAI concentrations. The iodine amount x could reach at most 4.02, which is close to the XPS results obtained above. For electronic structure, the direct bandgap decreased from 2.50 to 2.14 eV, while indirect bandgap decreased from 2.31 to 2.03 eV. For Cs2AgBiBr6‐x I x , a nonlinear dependence is observed for the bandgap and x in Figure S8b. The relationship between bandgap (E g) of Cs2AgBiBr6‐x I x and iodine ratio (y=x/6 in our case) can be expressed in Equation 2:[ 13a , 20 ]
| (2) |
Table 1.
Summary of the experimentally determined bandgap energies, lattice parameters and estimated iodine amount as function of MAI concentrations.
|
MAI concentration [mg mL−1] |
Direct bandgap [eV] |
Indirect bandgap [eV] |
Lattice parameter [Å] |
Iodine amount x [0–6] |
|---|---|---|---|---|
|
0 |
2.50 |
2.31 |
11.26 |
0 |
|
0.1 |
2.54 |
2.29 |
11.28 |
0.16 |
|
1 |
2.50 |
2.30 |
11.30 |
0.26 |
|
5 |
2.51 |
2.31 |
11.35 |
0.69 |
|
10 |
2.38 |
2.24 |
11.49 |
1.69 |
|
15 |
2.32 |
2.20 |
11.53 |
2.00 |
|
20 |
2.19 |
2.08 |
11.73 |
3.36 |
|
25 |
2.16 |
2.04 |
11.80 |
3.94 |
|
30 |
2.16 |
2.04 |
11.81 |
3.99 |
|
35 |
2.14 |
2.03 |
11.82 |
4.02 |
The bowing parameter b [0.36 eV for indirect bandgap, almost linear (0.02 eV) for direct bandgap] may reflect the miscibility between I and Br material. A nonlinear dependence for the bandgap versus halide ratio is not unfamiliar in lead halide perovskite films. The Cs2AgBiBr6 bandgap energies obtained here are slightly larger than previous reports,[ 8a , 8b ] which could be due to differences in measurements and the fitting procedures when extracting the bandgap from Tauc plots. The obtained approximately 0.3 eV decrease in bandgap and redshift of the absorption edge for increased iodine content demonstrate that we have achieved halide composition mixing in the double perovskite thin‐films. A redshift of photoluminescence for the mixed‐halide double perovskite shown in Figure S8c is also consistent with the decreased bandgap energy. However, both samples exhibited weak emission at room temperature, due to the indirect bandgap nature and the existence of defect states which can suppress emission intensity. [10a]
In addition, we also performed first‐principles density functional theory (DFT) calculations to study the electronic structure of mixed‐halide double perovskites. The calculations were carried out in the CP2K package, [21] using the Perdew‐Burke‐Ernzerhof (PBE) functional, [22] with DFT‐D3 dispersion interactions, [23] and Godecker‐Teter‐Hutter (GTH) pseudopotentials. [24] The system geometry was generated starting from previous experimental results of Cs2AgBiBr6 and then randomly substituting the halides until the target composition was reached. [10b] The optimized lattice parameters (as shown in Table S3) were in good agreement with the results from XRD, within approximately 1 % difference and the substitution‐induced shift was well reproduced. The projected electronic density of states (PDOS) was calculated in an optimized 2×2×2 supercell representation of the system at the Γ‐point while the electronic band structure calculations were performed using an optimized unit cell with k‐point sampling over a 3×3×3 Monkhorst‐Pack mesh, [25] including spin‐orbit coupling (SOC) effects. In order to consider the SOC, the band structure was calculated in the Quantum ESPRESSO program, [26] at the PBE level employing fully relativistic Rappe‐Rabe‐Kaxiras‐Joannopoulos ultrasoft pseudopotentials. [27] More details about the calculations and the level of theory used as well as additional results including the band structure without SOC effects and more detailed PDOS can be found in the Supporting Information. Figures 4 and S9–S10 show the PDOS and band structures calculated for various halide compositions.
Figure 4.
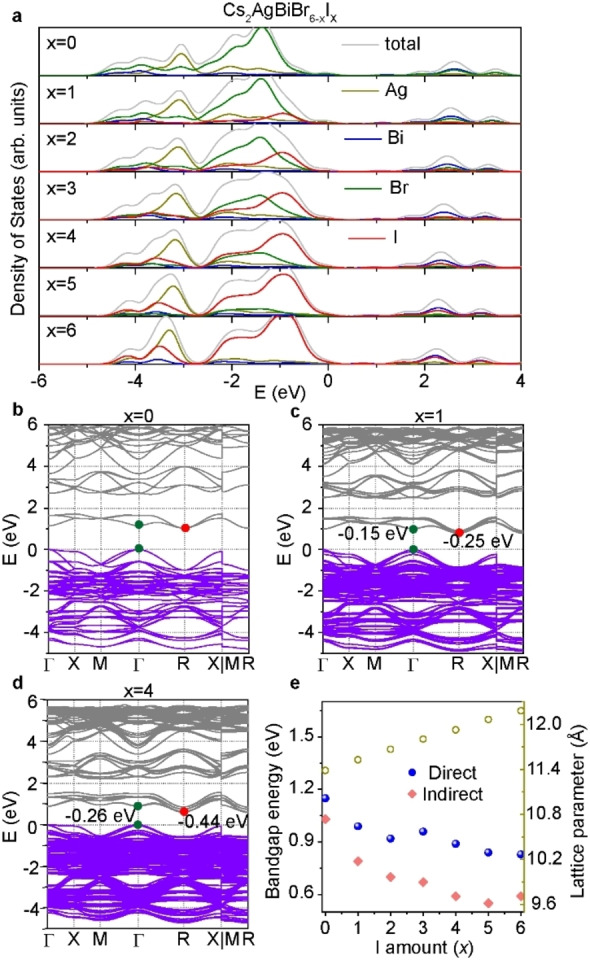
(a) PDOS graph of double perovskite with various halide compositions. DFT calculated band structures of (b) Cs2AgBiBr6, (c) Cs2AgBiBr5I1, and (d) Cs2AgBiBr2I4. VBM of all figures are aligned at 0 eV for easy comparison. The energy values [eV] represent movement of the CBM compared to the Cs2AgBiBr6 phase. (e) Summary of the calculated bandgap energies with SOC and lattice parameters for various compositions.
For our reference sample Cs2AgBiBr6 (Figure S9a), the CBM is mainly made up of Bi p/Br p orbitals with slight contributions from Br p‐ and s‐states, and the valence band maximum (VBM) corresponds to Ag d and Br p orbitals, yielding an indirect bandgap in the Γ‐R direction. In Figure 4a, the substitution of I for Br in Cs2AgBiBr6‐x I x introduces a shift in the CBM (Bi p and halide p orbitals) towards lower energies while causing an only marginal energy increase for the VBM, thereby reducing the bandgap of Cs2AgBiBr6. The bandgap of Cs2AgBiBr6‐x I x remains indirect when more bromine atoms are substituted by iodine atoms, consistent with the above bandgap analysis. The calculated bandgap energies at various compositions are summarized in Table S3 and Figure 4e, engineering from 1.15 eV (Cs2AgBiBr6) to 0.83 eV (Cs2AgBiI6) at direct bandgap. Note that the bandgaps are underestimated from theory due to the poor description of the energy derivative discontinuity of generalized gradient approximation (GGA) level DFT. [28] However, the relative bandgap shift caused by iodine substitution is in good agreement with the experimental results, reproducing both the direction and only slightly overestimating magnitude of the shifts. The kink in both direct and indirect bandgap in Figure 4e is likely related to the distribution of halides within unit cell. Including SOC lowers the overall dispersion and considerably reduces the difference between the direct and indirect bandgap (as shown in Figure S11). In addition to shrinking the bandgap, iodine substitution also causes a split in the lower part of the main valence feature at −3 eV in the PDOS seen in Figure S9b, which for the case of the pure Cs2AgBiI6 becomes an independent peak with a strong Ag d state component, separating the antibonding and bonding Ag and halide orbitals. The states composed of bonding orbitals close to the VBM broadens slightly as the iodide content increases.
Surface morphology properties of these mixed‐halide samples were studied with scanning electron microscopy (SEM), as shown in Figure S12. The Cs2AgBiBr6 film is composed of closely packed grains, with diameters of 200–800 nm (average diameter of 420 nm). When the film was treated by MAI, pin‐holes appeared and the grains were not stacked as closely as in the reference sample. The grain diameter and size distribution also changed with MAI post‐treatment. The average diameter firstly decreased to around 230 nm at small I inclusion, then increased to 450 and 600 nm at larger I inclusion, respectively. According to the Kirkendall effect, [29] film morphology might change after Br/I exchange between perovskites and reagents, due to the different diffusion rates of bromine and iodine. In lead‐halide perovskites, halide‐exchange does not induce large morphology variation due to the robust Pb‐MA framework and the high diffusion ability of the halides. Only long‐time exposure of the sample to reagents made surface rougher. [30] In our case, exposure of double perovskites into MAI reagents lasts within 30 s; thus, there is a possibility that grains may to some degree be dissolved and recrystallized during the halide‐exchange reaction, inducing morphology variation.
To understand the halide exchange process, we monitored the XRD patterns (Figure S13a). After MAI solution cast on the Cs2AgBiBr6 film, but before annealing the films, the XRD diffraction peak turned to be much weaker and wider, indicating partial dissolution of the initial double perovskite crystals. The slight shift at peak position might be ascribed to iodine bonded at the crystal surface, since a control sample with MABr/IPA treatment (instead of MAI/IPA) did not cause such a peak shift (Figure S13b). After annealing, the main diffraction peaks appeared stronger, along with larger shift to smaller angles, suggesting recrystallization to larger grain size and increased iodide substitution in the crystal structure. Previous results indicate that ion migration is more probable at grain boundaries and surfaces, due to the almost half activation energy compared to that in the bulk. [31] Therefore, the partial dissolution of Cs2AgBiBr6 could boost halide migration for desired composition.
It is also interesting to investigate if other organic iodide salts could work for halide exchange. Then, we employed formamidinium iodide (CH5IN2, FAI) and guanidinium iodide (CH6IN3, GAI) with the same iodide molar concentration as the MAI solution (35 mg mL−1) to treat the Cs2AgBiBr6 film, as shown in Figure S14a. Although the XRD diffraction peaks from these post‐treated samples also are shifted to smaller angle, the FAI and GAI‐treated samples exhibited less change, when compared to sample treated by MAI. One main difference between MAI and FAI is that MAI is more prone to decompose, resulting in hydro ionic acid (HI) generation. [32] Thus, we added very little HI into 4 mL of FAI/IPA and GAI/IPA solution, respectively. The post‐treated samples showed obvious change after the addition of HI (Figure 5a and Figure S14b). The XRD diffraction peaks and the light absorption onset exhibited clear shift with 10 μL of HI; however, no further change was observed upon the addition of more HI amount. Finally, the 2θ at the (400) peak shifted from 31.76° (for Cs2AgBiBr6 film) to 30.20° for both samples treated by FAI and GAI, assisted by HI, in contrast to 30.22° for MAI‐treated sample. The light absorption onset for samples treated by FAI and GAI shifted to around 600 nm, which are relatively close to MAI‐treated samples.
Figure 5.
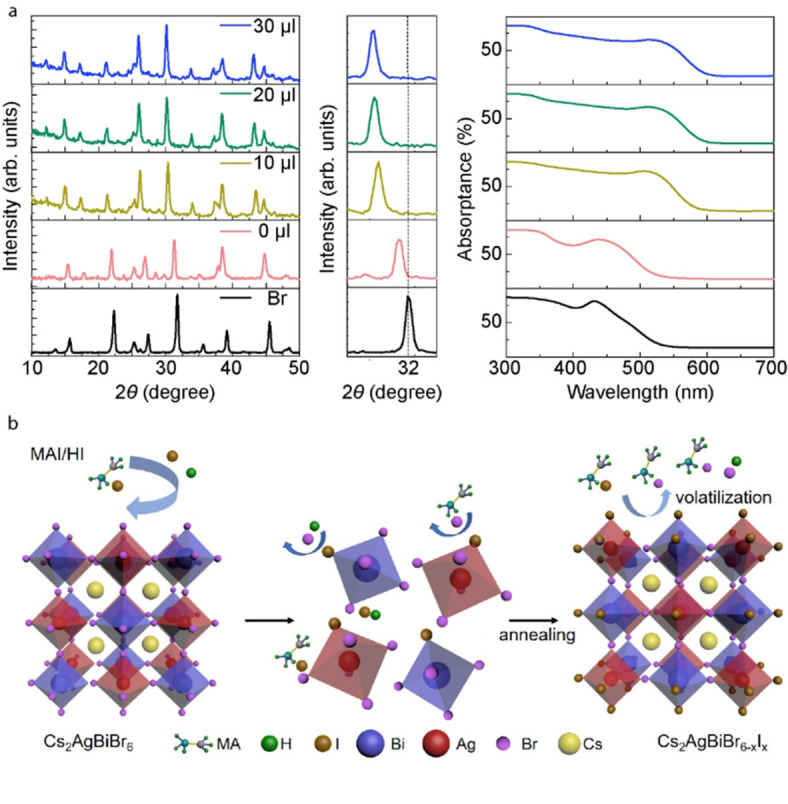
(a) Effect of HI on the halide‐exchange process. Comparison of XRD patterns (left column), enlarged view of (400) peak (middle column), and UV/Vis absorption spectra (right column) when adding different amount of HI into FAI post‐treatment solution. (b) Schematic of the halide‐exchange reaction process to fabricate mixed Br/I double perovskite (as an example of MAI/HI post‐treatment solution).
To further understand the role of the organic salt and HI during the halide reaction, pure HI (in IPA) and FAI (108 mg mL−1 to get more HI decomposition) solutions were used to post‐treat the samples (XRD shown in Figure S15a). Both treatments did not result in a mixed‐halide phase, indicating the important role of the combination of HI and the organic iodide salt. In addition, as shown in Figure S15b, when adding HI to the lower concentration MAI solution, the halide exchange reaction was also promoted, while the reaction was limited at C5 even with HI addition. Thus, we can conclude that with the assistance of HI and certain content of organic iodide salt, the Br/I halide exchange could be promoted in the double perovskite. From the obtained results, we infer that in the anion exchange process (Figure 5b) the Cs2AgBiBr6 film is first partially dissolved (with the HI addition in IPA), while iodine ions mainly bind at the boundaries and surfaces, where ion‐exchange occurs more easily due to decreased activation energy needed. During annealing, the organic bromide salt together with HBr is formed via exchange reaction due to the stronger binding energy. Then iodide and bromide salts would evaporate with excess bromide, [33] and the iodine ions left will enter into the structure to fill the vacant position of bromine. The proposed ion exchange method using a combination of hydroiodic acid and organic iodide salt can be a promising way to tune bandgap energies for the double perovskites.
Stability is also an important factor to consider when evaluating the potential use for optoelectronic applications. Regarding the structural stability of the substituted double perovskite, the samples were stored in a dry box for six months under dark condition, and the XRD patterns for the samples are shown in Figure S16a, b. Neither Cs2AgBiBr6 nor mix‐halide film showed any significant changes. We further tracked the absorbance changes of the mixed‐halide sample under 120 °C annealing or under continuous light illumination in air (Figure S16c, d). Compared to the temperature and light‐induced phase segregation in lead mixed‐halide perovskites, [34] there was no obvious sign of phase separation in the samples studied here, indicating phase stability of the obtained mixed‐halide films. However, decrease in absorbance occurred after 2 h annealing or illumination in air, which was not recovered in dark condition at room temperature. After continuous illumination or annealing, partial decomposition was observed in XRD patterns (Figure S16e).
The slightly broader absorption towards long wavelength at high temperature indicates a thermochromic behavior with a decreased bandgap, which is consistent with measurements of a double perovskite in a previous report. [35]
Conclusion
We report a halide exchange method on solid thin‐films to obtain mixed‐halide double perovskites, Cs2AgBiBr6‐x I x , with tunable bandgap energy as a function of iodide ratio x. The composition, band structure, and bandgap of Cs2AgBiBr6‐x I x were studied using different experimental techniques and density functional theory. Band structure calculations indicate that halide substitutions shift conduction band minimum and valence band maximum positions, inducing bandgap energy change in agreement with the measured samples. Compared to the reference sample of Cs2AgBiBr6, the iodide‐substituted systems exhibit expanded cubic lattice constants, as well as decreased bandgaps. The results suggest a promising way for flexible engineering of the double perovskite bandgap. Importantly, the obtained mixed‐halide double perovskites also show a stable phase, for both long‐time storage and continuous light illumination/thermal annealing. Finally, it was found that hydro‐iodide could be the key factor to facilitate halide‐exchange process using volatile organic iodide salts, making our exchange method more broadly applicable on double perovskites.
Experimental Section
Chemicals
Bismuth bromide (BiBr3, 98 %), cesium bromide (CsBr, 99.9 %), chlorobenzene (CB, 99.8 %), dimethyl sulfoxide (DMSO, anhydrous, 99.8 %), isopropanol (IPA, 99.5 %), and hydroiodic acid (HI, 57 wt% in H2O, 99.95 %) were obtained from Sigma‐Aldrich. Silver bromide (AgBr, 99.998 %) was bought from Alfa‐Aesar. Methylammonium iodide (MAI, >99.99 %), formamidinium iodide (FAI, >99.99 %), and guanidinium iodide (GAI, >99 %) were obtained from Greatcell Solar Materials. All solvents were used without further purification.
Fabrication of Cs2AgBiBr6 double perovskite film
For preparing Cs2AgBiBr6 precursor solution, a mixture of CsBr (1.0 mmol, 212.8 mg), AgBr (0.5 mmol, 93.9 mg) and BiBr3 (0.5 mmol, 224.3 mg) were dissolved in DMSO (1 mL) solvent. After being heated at 75 °C for several hours, a light‐yellow solution was obtained.
The Cs2AgBiBr6 film was deposited via spin‐coating method. Drops of the prepared solution were spun on the mesoporous TiO2 substrate at 3000 rpm for 30 s. 200 μL of CB was dipped onto the film after 10 s of spinning. The obtained film was annealed at 280 °C for 5 min to form the double perovskite phase. The fabrication of TiO2 substrates was according to our previous work. [36]
Halide‐exchange via post‐treatment
MAI/IPA solutions with different concentrations (0.1, 1, 5, 10, 15, 20, 25, 30, 35, 40, 45, 50 mg mL−1) were used to treat Cs2AgBiBr6 film to replace bromide with iodide. For each treatment, 50 μL of MAI solution was dropped on Cs2AgBiBr6 film, followed by spinning at 3000 rpm for 30 s and annealing at 200 °C for 3 min. Then, the mixed‐halide perovskite films were obtained as the film color changed from yellow to orange or red. A control sample with only IPA treatment did not show any changes. In this work, all used MAI solutions were freshly prepared (without overnight storage before use) to avoid the effect of further decomposition on halide exchange.
Characterizations
UV/Vis absorption spectra were obtained from reflectance and transmittance spectra, via A(λ)=1‐R(λ)‐T(λ). Reflectance and transmittance spectra of films were measured using a PerkinElmer LAMBDA 900 spectrophotometer. XRD measurements were carried out via a Siemens D5000 goniometer with CuKα radiation (λ=1.54051 Å). XPS was performed using PHI Quantum 2000 Scanning ESCA Microprobe spectrometer using AlKα (photon energy=1486.6 eV). The composition ratios were analyzed by Multipak software. The steady‐state PL spectra were recorded using a Fluorolog spectrophotometer (HORIBA JOBIN YNON) with an excitation at 365 nm. SEM measurements were performed using a LEO 1550 FEG instrument and a secondary electron detector operating at 3 kV (10 KV for mapping). The photoluminescence quantum yield (PLQY) was measured using FLS 1000 photoluminescence spectrometer from Edinburgh Instruments, attached with a barium sulfate‐coated integrating sphere.
Stability measurement
The absorbance of double perovskite film was measured by in‐situ UV/Vis spectroscopy set‐up with Ocean Optics QE6500 spectrometer and DH‐2000‐BAL light source. For the thermal stress test, the temperature was controlled by TMS‐93 Stage Temp Controller (Linkam), and absorbance data was recorded using the Ocean Optics OceanView software every 5 min for 300 min. For the illumination test, the sample was illuminated under AM 1.5 G (100 mW cm−2) illumination from a solar simulator (Model: 91160), which was calibrated with a standard Silicon solar cell (Fraunhofer ISE). The absorbance spectra were recorded with Ocean Optics OceanView manually, almost every 30 min for 9 h. Both tests were performed in air.
Theoretical section
The geometry optimizations were performed at the PBE level, [22] including DFT‐D3 dispersion interactions, [23] and with GTH pseudopotentials, [24] in the CP2K package using the Gaussian and plane waves (GPW) method.[ 21a , 37 ] The Kohn‐Sham orbitals were described in a local Gaussian TZVP basis set, [21b] and the electron density was expanded in a plane‐wave basis set with a kinetic energy cutoff of 600 Ry. Here, the number of valence electrons in the pseudopotentials were Ag 4d105s1, Bi 5d106s26p3, Cs 5p66s1, I 5s25p5, and Br 4s24p5. The initial system geometry was set up following previous experimental results [10b] and was represented both as a unit cell and as a 2×2×2 super cell. For the unit cell representations, k‐point sampling over a 3×3×3 Monkhorst‐Pack mesh was employed. [25] The correct halide distribution was generated by randomly substituting Br until the wanted composition was achieved and both the atomic coordinates and cell parameter were optimized while enforcing simple cubic symmetry. The PDOS, projected on both the orbital angular momenta and the atom element was obtained from optimized super cells at the Γ‐point. Here, we interpret the occupied Kohn‐Sham orbital energies as the electron binding energies. An ad‐hoc shift was added to set the highest occupied orbital energy to zero for better comparison with the band structure and the result was broadened using a Gaussian convolution with a full width at half maximum (FWHM) of 0.4 eV for easier interpretation and comparison. Note that the same energy shift, derived from the Cs2AgBiBr6, was used for all compositions. Starting from an optimized unit cell representation, band structure calculations were performed by calculating the Kohn‐Sham orbital energies along high symmetric k‐paths in the first Brillouin zone using the previously converged potential. While the pure Cs2AgBiBr6 has a face‐centered cubic crystal symmetry, this symmetry is lost for the mixed systems, and hence we use simple‐cubic symmetry and its corresponding high symmetric k‐paths for the band structure. The band structure calculations were carried out with the inclusion of SOC effects in the Quantum Espresso (QE) program, [26] using fully relativistic Rappe‐Rabe‐Kaxiras‐Joannopoulos ultrasoft pseudopotentials, [27] at the PBE level of theory and with kinetic energy cutoffs of 45 Ry and 455 Ry for the wave functions and electron density, respectively. In this case, the number of explicit electrons included in the calculations were Ag 4d105s1, Bi 5d106s26p3, Cs 5p66s1, I 5s25p5, and Br 3d104s24p5. SOC effects were not included in the cell and geometry optimizations. However, the forces were verified to be small at the SOC level in the unit cell cases. For the geometry optimizations and PDOS calculations, the results for each mixed composition, hence excluding the pure systems as they are unambiguous, were averaged over five different realizations in order to obtain sufficient statistics over the possible halide distributions. For the band structures and bandgaps, only a single realization for each composition was used.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We acknowledge financial support obtained from the Swedish Research Council (V.R.), ÅForsk, Olle Engkvist Foundation, Swedish Energy Agency. The calculations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) partially funded by the Swedish Research Council through grant agreement no. 2018‐05973. The authors acknowledge Prof. Xiaoliang Zhang, Dr. Xiaoyu Zhang and Dr. Zaiwei Wang for fruitful discussion, and acknowledge Dr. Byeong Jo Kim for great help on stability test setup.
H. Wu, A. Erbing, M. B. Johansson, J. Wang, C. Kamal, M. Odelius, E. M. J. Johansson, ChemSusChem 2021, 14, 4507.
Contributor Information
Dr. Michael Odelius, Email: odelius@fysik.su.se.
Prof. Erik M. J. Johansson, Email: erik.johansson@kemi.uu.se.
References
- 1.
- 1a. Lee M. M., Teuscher J., Miyasaka T., Murakami T. N., Snaith H. J., Science 2012, 338, 643–647; [DOI] [PubMed] [Google Scholar]
- 1b. Kim H.-S., Lee C.-R., Im J.-H., Lee K.-B., Moehl T., Marchioro A., Moon S.-J., Humphry-Baker R., Yum J.-H., Moser J. E., Grätzel M., Park N.-G., Sci. Rep. 2012, 2, 591; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Tan H., Jain A., Voznyy O., Lan X., García de Arquer F. P., Fan J. Z., Quintero-Bermudez R., Yuan M., Zhang B., Zhao Y., Fan F., Li P., Quan L. N., Zhao Y., Lu Z.-H., Yang Z., Hoogland S., Sargent E. H., Science 2017, 355, 722–726; [DOI] [PubMed] [Google Scholar]
- 1d. Xiao Z., Dong Q., Bi C., Shao Y., Yuan Y., Huang J., Adv. Mater. 2014, 26, 6503–6509; [DOI] [PubMed] [Google Scholar]
- 1e. Jiang Q., Zhao Y., Zhang X., Yang X., Chen Y., Chu Z., Ye Q., Li X., Yin Z., You J., Nat. Photonics 2019, 13, 460–466; [Google Scholar]
- 1f. Tan Z.-K., Moghaddam R. S., Lai M. L., Docampo P., Higler R., Deschler F., Price M., Sadhanala A., Pazos L. M., Credgington D., Hanusch F., Bein T., Snaith H. J., Friend R. H., Nat. Nanotechnol. 2014, 9, 687–692; [DOI] [PubMed] [Google Scholar]
- 1g. Wu H., Zhang Y., Lu M., Zhang X., Sun C., Zhang T., Colvin V. L., Yu W. W., Nanoscale 2018, 10, 4173–4178; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1h. Kojima A., Teshima K., Shirai Y., Miyasaka T., J. Am. Chem. Soc. 2009, 131, 6050–6051. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Rong Y., Hu Y., Mei A., Tan H., Saidaminov M. I., Seok S. I., McGehee M. D., Sargent E. H., Han H., Science 2018, 361, eaat8235; [DOI] [PubMed] [Google Scholar]
- 2b. Meng L., You J., Yang Y., Nat. Commun. 2018, 9, 5265; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2c. Saliba M., Matsui T., Seo J.-Y., Domanski K., Correa-Baena J.-P., Nazeeruddin M. K., Zakeeruddin S. M., Tress W., Abate A., Hagfeldt A., Grätzel M., Energy Environ. Sci. 2016, 9, 1989–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Meyer E., Mutukwa D., Zingwe N., Taziwa R., Metals 2018, 8, 667; [Google Scholar]
- 3b. Luo J., Li S., Wu H., Zhou Y., Li Y., Liu J., Li J., Li K., Yi F., Niu G., Tang J., ACS Photonics 2017, 5, 398–405; [Google Scholar]
- 3c. Zhao X.-G., Yang D., Ren J.-C., Sun Y., Xiao Z., Zhang L., Joule 2018, 2, 1662–1673; [Google Scholar]
- 3d. Pan W., Wu H., Luo J., Deng Z., Ge C., Chen C., Jiang X., Yin W.-J., Niu G., Zhu L., Yin L., Zhou Y., Xie Q., Ke X., Sui M., Tang J., Nat. Photonics 2017, 11, 726–732. [Google Scholar]
- 4.
- 4a. Meng W., Wang X., Xiao Z., Wang J., Mitzi D. B., Yan Y., J. Phys. Chem. Lett. 2017, 8, 2999–3007; [DOI] [PubMed] [Google Scholar]
- 4b. Volonakis G., Filip M. R., Haghighirad A. A., Sakai N., Wenger B., Snaith H. J., Giustino F., J. Phys. Chem. Lett. 2016, 7, 1254–1259. [DOI] [PubMed] [Google Scholar]
- 5. Khalfin S., Bekenstein Y., Nanoscale 2019, 11, 8665–8679. [DOI] [PubMed] [Google Scholar]
- 6.
- 6a. Slavney A. H., Hu T., Lindenberg A. M., Karunadasa H. I., J. Am. Chem. Soc. 2016, 138, 2138–2141; [DOI] [PubMed] [Google Scholar]
- 6b. Bartesaghi D., Slavney A. H., Gélvez-Rueda M. C., Connor B. A., Grozema F. C., Karunadasa H. I., Savenije T. J., J. Phys. Chem. C 2018, 122, 4809–4816; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Hoye R. L., Eyre L., Wei F., Brivio F., Sadhanala A., Sun S., Li W., Zhang K. H., MacManus-Driscoll J. L., Bristowe P. D., Adv. Mater. Interfaces 2018, 5, 1800464. [Google Scholar]
- 7.
- 7a. Filip M. R., Hillman S., Haghighirad A. A., Snaith H. J., Giustino F., J. Phys. Chem. Lett. 2016, 7, 2579–2585; [DOI] [PubMed] [Google Scholar]
- 7b. McClure E. T., Ball M. R., Windl W., Woodward P. M., Chem. Mater. 2016, 28, 1348–1354. [Google Scholar]
- 8.
- 8a. Igbari F., Wang R., Wang Z. K., Ma X. J., Wang Q., Wang K. L., Zhang Y., Liao L. S., Yang Y., Nano Lett. 2019, 19, 2066–2073; [DOI] [PubMed] [Google Scholar]
- 8b. Wu C., Zhang Q., Liu Y., Luo W., Guo X., Huang Z., Ting H., Sun W., Zhong X., Wei S., Adv. Sci. 2018, 5, 1700759; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Greul E., Petrus M. L., Binek A., Docampo P., Bein T., J. Mater. Chem. A 2017, 5, 19972–19981; [Google Scholar]
- 8d. Wang M., Zeng P., Bai S., Gu J., Li F., Yang Z., Liu M., Solar RRL 2018, 2, 1800217; [Google Scholar]
- 8e. Pantaler M., Cho K. T., Queloz V. I., García Benito I. S., Fettkenhauer C., Anusca I., Nazeeruddin M. K., Lupascu D. C., Grancini G., ACS Energy Lett. 2018, 3, 1781–1786; [Google Scholar]
- 8f. Ning W., Wang F., Wu B., Lu J., Yan Z., Liu X., Tao Y., Liu J. M., Huang W., Fahlman M., Adv. Mater. 2018, 30, 1706246; [DOI] [PubMed] [Google Scholar]
- 8g. Wang B., Li N., Yang L., Dall'Agnese C., Jena A. K., Sasaki S.-i., Miyasaka T., Tamiaki H., Wang X.-F., J. Am. Chem. Soc. 2021, 143, 2207–2211. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Jesper Jacobsson T., Correa-Baena J.-P., Pazoki M., Saliba M., Schenk K., Grätzel M., Hagfeldt A., Energy Environ. Sci. 2016, 9, 1706–1724; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Zhang X., Sun C., Zhang Y., Wu H., Ji C., Chuai Y., Wang P., Wen S., Zhang C., Yu W. W., J. Phys. Chem. Lett. 2016, 7, 4602–4610. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Du K. Z., Meng W., Wang X., Yan Y., Mitzi D. B., Angew. Chem. Int. Ed. 2017, 56, 8158–8162; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 8270–8274; [Google Scholar]
- 10b. Slavney A. H., Leppert L., Bartesaghi D., Gold-Parker A., Toney M. F., Savenije T. J., Neaton J. B., Karunadasa H. I., J. Am. Chem. Soc. 2017, 139, 5015–5018. [DOI] [PubMed] [Google Scholar]
- 11. Gray M. B., McClure E. T., Woodward P. M., J. Mater. Chem. C 2019, 7, 9686–9689. [Google Scholar]
- 12.
- 12a. Kubicki D. J., Saski M., MacPherson S., Galokowski K., Lewiński J., Prochowicz D., Titman J. J., Stranks S. D., Chem. Mater. 2020, 32, 8129–8138; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12b. Xiao Z., Meng W., Wang J., Mitzi D. B., Yan Y., Mater. Horiz. 2017, 4, 206–216; [Google Scholar]
- 12c. Savory C. N., Walsh A., Scanlon D. O., ACS Energy Lett. 2016, 1, 949–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.
- 13a. Jang D. M., Park K., Kim D. H., Park J., Shojaei F., Kang H. S., Ahn J.-P., Lee J. W., Song J. K., Nano Lett. 2015, 15, 5191–5199; [DOI] [PubMed] [Google Scholar]
- 13b. Guhrenz C., Benad A., Ziegler C., Haubold D., Gaponik N., Eychmüller A., Chem. Mater. 2016, 28, 9033–9040; [Google Scholar]
- 13c. Fu F., Pisoni S., Weiss T. P., Feurer T., Wäckerlin A., Fuchs P., Nishiwaki S., Zortea L., Tiwari A. N., Buecheler S., Adv. Sci. 2018, 5, 1700675; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13d. Chen J., Kim S. G., Park N. G., Adv. Mater. 2018, 30, 1801948; [DOI] [PubMed] [Google Scholar]
- 13e. Solis-Ibarra D., Smith I., Karunadasa H., Chem. Sci. 2015, 6, 4054–4059; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13f. Guhrenz C., Benad A., Ziegler C., Haubold D., Gaponik N., Eychmüller A., Chem. Mater. 2016, 28, 9033–9040. [Google Scholar]
- 14. Yang B., Chen J., Yang S., Hong F., Sun L., Han P., Pullerits T., Deng W., Han K., Angew. Chem. 2018, 130, 5457–5461; [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2018, 57, 5359–5363. [DOI] [PubMed] [Google Scholar]
- 15. Creutz S. E., Crites E. N., De Siena M. C., Gamelin D. R., Nano Lett. 2018, 18, 1118–1123. [DOI] [PubMed] [Google Scholar]
- 16.
- 16a. Li Z., Yang M., Park J.-S., Wei S.-H., Berry J. J., Zhu K., Chem. Mater. 2016, 28, 284–292; [Google Scholar]
- 16b. Goldschmidt V. M., Naturwissenschaften 1926, 14, 477–485. [Google Scholar]
- 17. Shannon R. D., Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar]
- 18. Bekenstein Y., Dahl J. C., Huang J., Osowiecki W. T., Swabeck J. K., Chan E. M., Yang P., Alivisatos A. P., Nano Lett. 2018, 18, 3502–3508. [DOI] [PubMed] [Google Scholar]
- 19. Xiao Z., Meng W., Wang J., Yan Y., ChemSusChem 2016, 9, 2628–2633. [DOI] [PubMed] [Google Scholar]
- 20. Noh J. H., Im S. H., Heo J. H., Mandal T. N., Seok S. I., Nano Lett. 2013, 13, 1764–1769. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a.CP2K, Version 7.1 the CP2K developers group. http://www.cp2k.org 2018;
- 21b. VandeVondele J., Hutter J., J. Chem. Phys. 2007, 127, 114105. [DOI] [PubMed] [Google Scholar]
- 22. Perdew J. P., Burke K., Ernzerhof M., Phys. Rev. Lett. 1996, 77, 3865. [DOI] [PubMed] [Google Scholar]
- 23. Grimme S., Antony J., Ehrlich S., Krieg H., J. Chem. Phys. 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- 24. Goedecker S., Teter M., Hutter J., Phys. Rev. B 1996, 54, 1703. [DOI] [PubMed] [Google Scholar]
- 25. Monkhorst H. J., Pack J. D., Phys. Rev. B 1976, 13, 5188. [Google Scholar]
- 26. Giannozzi P., Andreussi O., Brumme T., Bunau O., Nardelli M. B., Calandra M., Car R., Cavazzoni C., Ceresoli D., Cococcioni M., Colonna N., Carnimeo I., Dal Corso A., Gironcoli D. S., Delugas P., Di Stasio R. A., Ferretti A., Floris A., Fratesi G., Fugallo G., Gebauer R., Gerstmann U., Giustino F., Gorni T., Jia J., Kawamura M., Ko H.-Y., Kokalj A., Kucukbenli E., Lazzeri M., Marsili M., Marzari N., Mauri F., Nguyen N. L., Nguyen H.-V., Otero de-la Roza A., Paulatto L., Ponc'e S., Rocca D., Sabatini R., Santra B., Schlipf M., Seitsonen A. P., Smogunov A., Timrov I., Thonhauser T., Umari P., Vast N., Wu X., Baroni S., J. Phys. Condens. Matter 2017, 29, 465901. [DOI] [PubMed] [Google Scholar]
- 27. Rappe A. M., Rabe K. M., Kaxiras E., Joannopoulos J., Phys. Rev. B 1990, 41, 1227. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Perdew J. P., Levy M., Phys. Rev. Lett. 1983, 51, 1884; [Google Scholar]
- 28b. Perdew J. P., Int. J. Quantum Chem. 1985, 28, 497–523. [Google Scholar]
- 29. Nakajima H., JOM 1997, 49, 15–19. [Google Scholar]
- 30. Li G., Ho J. Y.-L., Wong M., Kwok H. S., J. Phys. Chem. C 2015, 119, 26883–26888. [Google Scholar]
- 31. Yuan Y., Huang J., Acc. Chem. Res. 2016, 49, 286–293. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Min H., Kim G., Paik M. J., Lee S., Yang W. S., Jung M., Seok S. I., Adv. Energy Mater. 2019, 9, 1803476; [Google Scholar]
- 32b. Wang X., Fan Y., Wang L., Chen C., Li Z., Liu R., Meng H., Shao Z., Du X., Zhang H., Chem 2020, 6, 1369–1378; [Google Scholar]
- 32c. Juarez-Perez E. J., Ono L. K., Qi Y., J. Mater. Chem. A 2019, 7, 16912–16919. [Google Scholar]
- 33. Shao Z., Wang Z., Li Z., Fan Y., Meng H., Liu R., Wang Y., Hagfeldt A., Cui G., Pang S., Angew. Chem. Int. Ed. 2019, 58, 5587–5591; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 5643–5647. [Google Scholar]
- 34. Brennan M. C., Draguta S., Kamat P. V., Kuno M., ACS Energy Lett. 2018, 3, 204–213. [Google Scholar]
- 35. Ning W., Zhao X. G., Klarbring J., Bai S., Ji F., Wang F., Simak S. I., Tao Y., Ren X. M., Zhang L., Adv. Funct. Mater. 2019, 29, 1807375. [Google Scholar]
- 36.
- 36a. Zhu H., Pan M., Johansson M. B., Johansson E. M. J., ChemSusChem 2017, 10, 2592–2596; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36b. Zhu H., Erbing A., Wu H., Man G. J., Mukherjee S., Kamal C., Johansson M. B., Rensmo H., Odelius M., Johansson E. M. J., ACS Appl. Mater. Interfaces 2020, 3, 7372–7382; [Google Scholar]
- 36c. Wu H., Zhu H., Erbing A., Johansson M. B., Mukherjee S., Man G. J., Rensmo H., Odelius M., Johansson E. M. J., ACS Appl. Mater. Interfaces 2019, 2, 5356–5362. [Google Scholar]
- 37. Hutter J., Iannuzzi M., Schiffmann F., VandeVondele J., Wiley Interdiscip. Rev.: Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information