

Abstract
Radical‐mediated gas‐phase reactions play an important role in the conversion of methane under non‐oxidative conditions into olefins and aromatics over iron‐modified silica catalysts. Herein, we use operando photoelectron photoion coincidence spectroscopy to disentangle the elusive C2+ radical intermediates participating in the complex gas‐phase reaction network. Our experiments pinpoint different C2‐C5 radical species that allow for a stepwise growth of the hydrocarbon chains. Propargyl radicals (H2C−C≡C−H) are identified as essential precursors for the formation of aromatics, which then contribute to the formation of heavier hydrocarbon products via hydrogen abstraction–acetylene addition routes (HACA mechanism). These results provide comprehensive mechanistic insights that are relevant for the development of methane valorization processes.
Keywords: heterogeneous catalysis, iron, methane conversion, reaction mechanisms, silica
The complex gas‐phase reaction network present during methane conversion under non‐oxidative conditions over iron‐modified silica is unraveled, shedding light on the formation mechanism of C2+ hydrocarbons.

Methane (CH4) conversion under non‐oxidative conditions at elevated temperatures and ambient pressure represents an attractive one‐step valorization route of the abundant natural gas into C2+ hydrocarbons and hydrogen (H2), which are essential platforms for the production of chemicals, polymers, and liquid fuels. [1] Different types of catalysts can be used for this process, including metal‐exchanged zeolites and metal‐modified oxides.[ 1d , 2 ] Several recent reports pinpointed exceptional performance of iron‐modified silica catalysts in this process, which displayed markedly different activity and product distribution patterns as compared to non‐catalyzed methane pyrolysis under the same conditions (Table S1).[ 2c , 2d , 2e , 2f , 2g , 2h , 2i , 2j , 2k , 2l , 2m ] Nonetheless, the mechanistic understanding that is essential to derive the rationale for tuning the product distribution and enhancing the catalyst lifetime is still elusive. This is mainly caused by the complexity of the process, which likely involves contributions from both surface‐catalyzed and radical‐mediated gas‐phase pathways (Table S2).[ 2c , 2d , 2e , 2f , 2g , 2h , 3 ] In particular, for a silica‐confined single iron sites catalyst, Guo et al. [2c] proposed a reaction mechanism in which C−C bonds are formed via sequential homolytical bond dissociation reactions and stepwise chain growth. Herein, methane is activated into methyl radical (.CH3), the coupling of which enables the formation of C2 species (vinyl (.C2H3), ethyl (.C2H5), ethylene (C2H4), and ethane (C2H6)). [2c] Ethylene plays a key role in the next steps, as it participates in chain growth that together with dehydrogenation and cyclization reactions lead to the formation of benzene (C6H6) and naphthalene (C10H8). [2c] However, some of the elementary reaction steps, such as the formation of ethyl and vinyl radicals, lack direct experimental evidence. Furthermore, the reaction pathways and corresponding intermediates enabling the formation of C2+ products are still poorly understood.
Herein, we report a detailed study of the gas‐phase intermediates involved in methane conversion under non‐oxidative conditions using imaging photoelectron photoion coincidence (iPEPICO) spectroscopy (Figure S1). This technique applies tunable vacuum ultraviolet (VUV) synchrotron radiation in a collision‐reduced environment to soft‐ionize the gas beam entailing unconverted reactants, intermediates, and products that leave the catalytic reactor and expand into high vacuum. The ionization yields pairs of photoelectrons and photoions that are detected in delayed coincidence. [4] As a result, iPEPICO provides unprecedented potential to unravel complex mixtures of highly reactive gas‐phase intermediates in various catalytic processes that are inaccessible by conventional analytical tools, such as gas chromatography coupled to mass spectrometry (GC‐MS), due to their short lifetimes. [5]
We commence our experiments by analyzing the time‐of‐flight (TOF) mass spectra of the reactor effluent formed during the conversion of methane under non‐oxidative conditions at ca. 945–1400 °C over iron‐modified silica (Fe@SiO2) catalyst (Figures 1 and 2), which has been extensively characterized in our previous study. [2i] In addition to the products that can be detected in conventional catalytic tests using reactor designs typically used to study the conversion of methane under non‐oxidative conditions such as acetylene (C2H2, m/z 26), ethylene (C2H4, m/z 28), ethane (C2H6, m/z 30), propyne or allene (C3H4, m/z 40), propene (C3H6, m/z 42), propane (C3H8, m/z 44), 1,3‐butadiyne (C4H2, m/z 50), 1‐buten‐3‐yne (C4H4, m/z 52), 1,3‐butadiene (C4H6, m/z 54), butene (C4H8, m/z 56), cyclopentadiene (C5H6, m/z 66), and fulvene or benzene (C6H6, m/z 78), the iPEPICO TOF mass spectra reveal a rich radical chemistry over iron‐modified silica catalyst. Besides methyl radicals (.CH3, m/z 15), we evidence for the first time the formation of C2+ radical intermediates, including vinyl (.C2H3, m/z 27), ethyl (.C2H5, m/z 29), propargyl (.C3H3, m/z 39), allyl (.C3H5, m/z 41), .C4H3 (m/z 51), butadienyl (.C4H5, m/z 53), and cyclopentadienyl (.C5H5, m/z 65) radicals. These short‐lived intermediates are unequivocally assigned based on their m/z values, ionization energies (IE), mass‐selected threshold photoelectron spectra (ms‐TPES) supported by Franck‐Condon (FC) simulations and photoionization (PI) spectra (Figure 1, Figures S6 and S7, and Table S4). Comparatively, the experiment performed over pure silica (SiO2) catalyst shows only the presence of methyl radicals (Figure 1 b), the concentration of which is more than one order of magnitude lower than that recorded using iron‐modified silica. This result, which is consistent with a long induction period for pure silica (Figure S5), [2i] indicates that the formation of C2+ radicals in the early reaction stages is enabled by the iron sites. Notably, the signals of methyl, ethyl, propargyl, allyl, and cyclopentadienyl radicals together with stable products (ethylene, propyne or allene, propene, 1,3‐butadiyne, 1‐buten‐3‐yne, 1,3‐butadiene, cyclopentadiene, and fulvene or benzene) are clearly visible in the TOF mass spectra acquired during methane conversion under non‐oxidative conditions over pre‐coked iron‐modified silica (Figure 1 b), implying the important role of radical intermediates during the whole course of reaction. Moreover, methyl, ethyl, and allyl are also observed along with stable hydrocarbons (ethylene, propyne or allene, propene, 1,3‐butadiyne, 1‐buten‐3‐yne, 1,3‐butadiene, and fulvene or benzene) in the molecular beam stemming from the active form of pure silica catalyst, i.e., pre‐coked pure silica, which also proves the participation of these radical species in the product formation.
Figure 1.
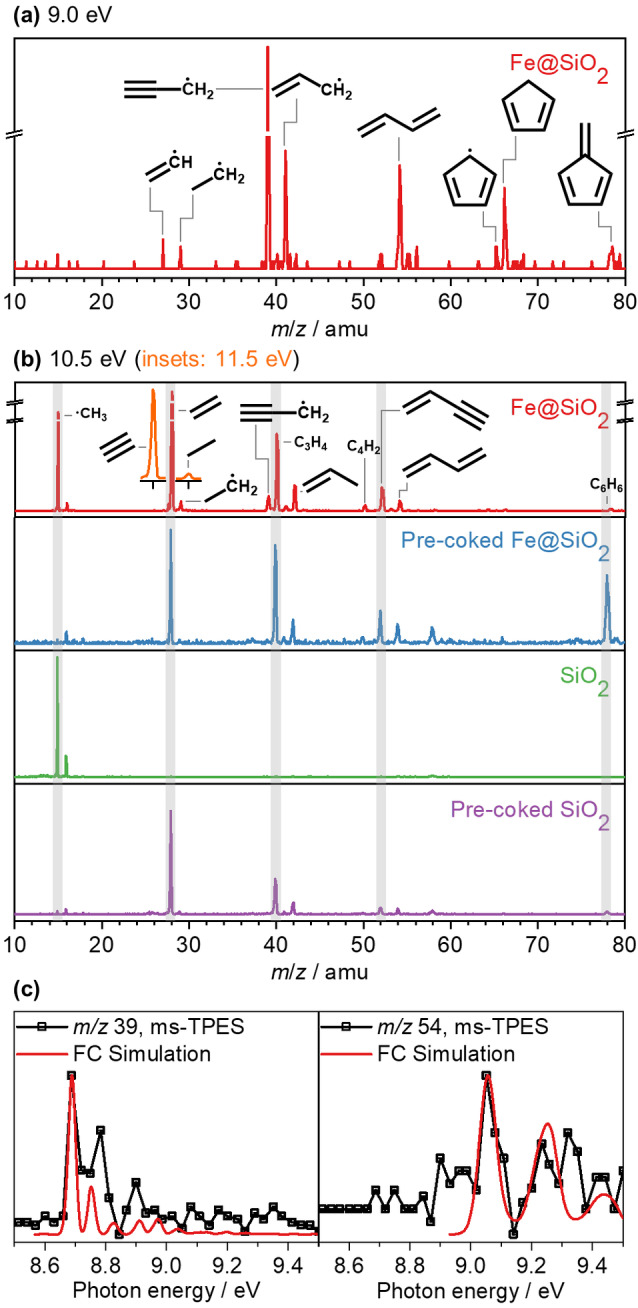
iPEPICO analysis upon conversion of methane under non‐oxidative conditions over Fe@SiO2 and SiO2 catalysts: a),b) time‐of‐flight mass spectra for particular catalysts and photon energies (a: 9.0 eV; b: 10.5 eV and insets at 11.5 eV), and c) mass‐selected threshold photoelectron spectra for selected m/z channels assigned isomer‐specifically with the help of Franck‐Condon simulations as propargyl radical (m/z 39) and 1,3‐butadiene (m/z 54). Reaction conditions: F =2 sccm, c =ca. 100 mol %, W cat=9.5±3.8 mg, T=ca. 1100 °C, and P=100±62 mbar.
Figure 2.

Evolution of radical and molecular species obtained upon conversion of methane over Fe@SiO2 as a function of temperature analyzed by iPEPICO (photon energies in parentheses). Reaction conditions: F =2 sccm, c =ca. 100 mol %, W cat=ca. 8.0 mg, T=ca. 945–1400 °C, and P=199.1±6.8 mbar. *Species ionized with residual non‐filtered high‐order radiation.
Further insights into the role of the C2+ radicals are acquired by inspecting the temperature profiles (ca. 945–1400 °C) of these species in relation to the evolution of various reaction products (Figure 2 and Figure S8). At ca. 945 °C, apart from methyl, no radicals nor stable products are observed. Starting from ca. 1100 °C, the signal of methyl decreases with temperature, while ethyl presents a slight decrease in the range of ca. 1100–1230 °C and then it decreases more pronouncedly. Notably, a decrease in methyl and ethyl signals at ca. 1100–1230 °C correlates with the increase in the signals originating from vinyl, propargyl, and allyl radicals, as well as with the increase in the signals stemming from the acetylene, ethylene, ethane, propyne or allene, propene, and propane products. At higher temperature (ca. 1230–1400 °C), the intensity of methyl, vinyl, ethyl, and allyl radicals and the light products ethylene, ethane, and propene decrease, while the intensities of propargyl radical and propyne or allene increase. This is accompanied with increased production of 1‐buten‐3‐yne, fulvene or benzene, as well as high‐molecular weight and dehydrogenation products. In addition, a decrease in butadienyl and 1,3‐butadiene at high temperature is coupled to the higher intensities of the 1‐buten‐3‐yne and C6H6 species. Higher‐molecular weight products such as m/z 102, 128, and 152, which could be phenylacetylene (C8H6), naphthalene (C10H8), and acenaphthylene (C12H8), constantly increase with temperature, suggesting their lower reactivity in the overall reaction scheme.
Based on this experimental evidence, the following mechanism is proposed (Figure 3 and Figure S9). Consistent with the proposal of Guo et al., [2c] the reaction sequence starts with methane (1) activation by methyl radical (2) formation, which then couple into ethane (3). This initial step is facilitated by iron sites, as also corroborated by our observations on the differences in methyl radical and product concentrations between iron‐modified silica and pure silica catalysts (Figure 1). Thereafter, ethane is transformed into ethyl (4) and ethylene (5) in two consecutive dehydrogenation reactions. In a similar way, ethylene is the precursor of vinyl (6) and acetylene (7). Reactions of vinyl, ethyl, and acetylene with C1 species produce propene (13), propane (12), and allyl radical (8), respectively. The C3 species propene and allyl radical can also be produced by successive dehydrogenation reactions starting from propane. Further dehydrogenations produce propyne or allene (9) and propargyl radical (10). In a side pathway, propene can also react with methyl radical and dehydrogenate to produce butene (C4H8, m/z 56) and 1,3‐butadiene (16), which can continue with dehydrogenation processes down to 1,3‐butadiyne.
Figure 3.

The mechanism of the main products formation via radical‐mediated gas‐phase reaction pathways in the conversion of methane under non‐oxidative conditions over Fe@SiO2 catalyst. Extended mechanism in Figure S9.
The propargyl radical is a key species that can react with (i) C1 species to produce 1,3‐butadiene, (ii) acetylene to produce cyclopentadienyl, or (iii) with another propargyl to produce the benzene (11) and fulvene (15). [6]
The tunability of the VUV photon energy in iPEPICO allows us to confirm the formation of both C6H6 isomers based on their IE (9.24 eV for benzene and 8.36 eV for fulvene). [7] The formation of C3 species (.C3H3, .C3H5, .C3H7, C3H4, C3H6, and C3H8) as well as C4H6 are plausible mass growth pathways in methane conversion under non‐oxidative conditions. Moreover, from 1,3‐butadiene, subsequent events involving homolytic C−H bonds cleavage may produce lower‐mass C4 species down to .C4H3 and 1,3‐butadiyne, as observed in minor concentrations.
The presence of cyclopentadienyl and cyclopentadiene is expected for high‐temperature hydrocarbon reactions. [8] Cyclopentadiene (14) can be produced by reaction of allyl with acetylene, followed by dehydrogenation.[ 2j , 9 ] Decomposition reactions of cyclopentadiene or reaction of propargyl with acetylene followed by successive dehydrogenations could lead to acyclic C5H4 species (m/z 64) or their respective radicals (.C5H3, m/z 63). [2l] These reaction pathways are corroborated by the observation of peaks at m/z values of 66, 65, 64, and 63 (Figure 4 and Figure S8).
Figure 4.

Time‐of‐flight mass spectrum obtained by iPEPICO using 10.5 eV of ionization energy upon conversion of methane under non‐oxidative conditions over Fe@SiO2 catalyst. Reaction conditions: F =2 sccm, c =ca. 100 mol %, W cat=ca. 8.0 mg, T=ca. 1400 °C, and P=ca. 204 mbar.
Considering C6H6 species, benzene is preferred at high temperature over fulvene. [10] Benzene is not only a reaction product; it also plays an important role as a precursor for heavier species involved in the deactivation of catalysts. [11] Even though there are many proposals for benzene formation in methane conversion under non‐oxidative conditions, none of them have been verified experimentally.[ 2j , 2l , 6a , 12 ] Thus, detecting benzene precursors, such as propargyl and fulvene, and understanding their production as well as consumption mechanisms is fundamental.
Miller et al. [6a] reported that propargyl can directly recombine to benzene, but also form fulvene, which then isomerizes to benzene. Our observation of both fulvene (IE=8.36 eV) and benzene (IE=9.24 eV) corroborates these mechanisms. [7] Scherer et al. [13] and Shafir et al. [14] pointed out that propargyl dimerizes to produce benzene or phenyl (.C6H5, m/z 77). Alternatively, fulvene can be formed by cyclopentadiene methylation and dehydrogenation, and may also subsequently isomerize to benzene. [12] Cyclopentadiene also decomposes to cyclopentadienyl and propargyl at high temperature, but methylation should dominate once cyclopentadienyl is formed, considering the rate constants of both processes (Table S5). Another plausible route reported by Dean suggests the reaction of butadienyl radical with acetylene leading to linear C6 species, which cyclize and dehydrogenate to benzene. [2j] These routes are confirmed by our detection of the radical intermediates methyl, propargyl, butadienyl, cyclopentadienyl, and phenyl, as well as the products acetylene, cyclopentadiene, benzene and fulvene (Figures 1 and 2 and 4, and Figure S8). Even with multiple plausible pathways to produce benzene, the routes involving propargyl are the most relevant, considering the important contribution of such species evidenced experimentally (see intensity of signal at m/z 39 in Figures 1 and 2). Benzene and phenyl can provide access to other species with lower molecular weight such as C6H4 (m/z 76) and C6H2 (m/z 74) (Figure 4 and Figure S8).
High‐molecular‐weight species with m/z>78 gain importance at higher temperature (Figures 2 and 4, and Figure S8). Their production can proceed via methylations, dehydrogenations, and the hydrogen abstraction–acetylene addition (HACA) mechanism, which are plausible at high reaction temperatures (extended description in Supporting Information).
In summary, by exploiting the unprecedented detection capabilities of the iPEPICO technique, it is possible to unveil elusive C2+ radical intermediates in methane conversion under non‐oxidative conditions over iron‐modified silica catalysts and to correlate their formation with the generation of different reaction products. Based on such experimental evidence, we postulate the reaction mechanisms for the formation of C2‐C6 as well as higher molecular weight products. Resonantly stabilized propargyl radicals possess a long lifetime as shown by their large abundance and thus play a key role in the formation of the first aromatic ring, benzene. In addition, the observation of phenyl, acetylene, phenylacetylene, naphthalene, and acenaphthylene evidences the HACA reaction mechanism. Our work shows that, besides the stepwise chain growth mechanisms proposed in the literature, alternative reaction channels exist, thus provides important insights into the role of radical‐mediated gas‐phase reactions in conversion of methane under non‐oxidative conditions over heterogeneous catalysts. The findings could help to improve the performance of this process both from a chemical and engineering point of view, as well as serve as a basis to understand other relevant catalysts such as metal‐exchanged zeolites.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
The authors thank the support of the VUV beamline of the Swiss Light Source located at the Paul Scherrer Institute, Villigen, Switzerland, where iPEPICO measurements were performed under the proposals 20191009 and 20200976. Prof. Dr. Christophe Copéret and Shell Global Solutions International B.V. are acknowledged for their collaboration and funding in previous stages of the project in which the catalysts used in this research were prepared. A.P.‐U. thanks the Ministry of Science, Technology, and Telecommunications of Costa Rica, and the Costa Rica Institute of Technology for their support. P.H. and Z.P. are grateful for the funding by Swiss National Science Foundation (SNSF, 200021_178952). P.H., Z.P., and A.P.‐U. thank Patrick Ascher for technical assistance. Open access funding provided by Eidgenossische Technische Hochschule Zurich.
A. Puente-Urbina, Z. Pan, V. Paunović, P. Šot, P. Hemberger, J. A. van Bokhoven, Angew. Chem. Int. Ed. 2021, 60, 24002.
References
- 1.
- 1a. Wang L., Tao L., Xie M., Xu G., Huang J., Xu Y., Catal. Lett. 1993, 21, 35–41; [Google Scholar]
- 1b. Ismagilov Z. R., Matus E. V., Tsikoza L. T., Energy Environ. Sci. 2008, 1, 526–541; [Google Scholar]
- 1c. Alvarez-Galvan M. C., Mota N., Ojeda M., Rojas S., Navarro R. M., Fierro J. L. G., Catal. Today 2011, 171, 15–23; [Google Scholar]
- 1d. Spivey J. J., Hutchings G., Chem. Soc. Rev. 2014, 43, 792–803; [DOI] [PubMed] [Google Scholar]
- 1e. Karakaya C., Kee R. J., Prog. Energy Combust. Sci. 2016, 55, 60–97; [Google Scholar]
- 1f. Huang K., Miller J. B., Huber G. W., Dumesic J. A., Maravelias C. T., Joule 2018, 2, 349–365. [Google Scholar]
- 2.
- 2a. Vollmer I., Kosinov N., Szécsényi Á., Li G., Yarulina I., Abou-Hamad E., Gurinov A., Ould-Chikh S., Aguilar-Tapia A., Hazemann J.-L., Pidko E., Hensen E., Kapteijn F., Gascon J., J. Catal. 2019, 370, 321–331; [Google Scholar]
- 2b. Weckhuysen B. M., Wang D., Rosynek M. P., Lunsford J. H., Angew. Chem. Int. Ed. Engl. 1997, 36, 2374–2376; [Google Scholar]; Angew. Chem. 1997, 109, 2471–2473; [Google Scholar]
- 2c. Guo X., Fang G., Li G., Ma H., Fan H., Yu L., Ma C., Wu X., Deng D., Wei M., Tan D., Si R., Zhang S., Li J., Sun L., Tang Z., Pan X., Bao X., Science 2014, 344, 616; [DOI] [PubMed] [Google Scholar]
- 2d. Sakbodin M., Wu Y., Oh S. C., Wachsman E. D., Liu D., Angew. Chem. Int. Ed. 2016, 55, 16149–16152; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16383–16386; [Google Scholar]
- 2e. Oh S. C., Schulman E., Zhang J., Fan J., Pan Y., Meng J., Liu D., Angew. Chem. Int. Ed. 2019, 58, 7083–7086; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2019, 131, 7157–7160; [Google Scholar]
- 2f. Han S. J., Lee S. W., Kim H. W., Kim S. K., Kim Y. T., ACS Catal. 2019, 9, 7984–7997; [Google Scholar]
- 2g. Hao J., Schwach P., Fang G., Guo X., Zhang H., Shen H., Huang X., Eggart D., Pan X., Bao X., ACS Catal. 2019, 9, 9045–9050; [Google Scholar]
- 2h. Hao J., Schwach P., Li L., Guo X., Weng J., Zhang H., Shen H., Fang G., Huang X., Pan X., Xiao C., Yang X., Bao X., J. Energy Chem. 2021, 52, 372–376; [Google Scholar]
- 2i. Šot P., Newton M. A., Baabe D., Walter M. D., van Bavel A. P., Horton A. D., Copéret C., van Bokhoven J. A., Chem. Eur. J. 2020, 26, 8012–8016; [DOI] [PubMed] [Google Scholar]
- 2j. Dean A. M., J. Phys. Chem. 1990, 94, 1432–1439; [Google Scholar]
- 2k. Olsvik O., Billaud F., Thermochim. Acta 1994, 232, 155–169; [Google Scholar]
- 2l. Matheu D. M., Dean A. M., Grenda J. M., Green W. H., J. Phys. Chem. A 2003, 107, 8552–8565; [Google Scholar]
- 2m. Keramiotis C., Vourliotakis G., Skevis G., Founti M. A., Esarte C., Sánchez N. E., Millera A., Bilbao R., Alzueta M. U., Energy 2012, 43, 103–110. [Google Scholar]
- 3.
- 3a. Schwach P., Pan X., Bao X., Chem. Rev. 2017, 117, 8497–8520; [DOI] [PubMed] [Google Scholar]
- 3b. Li T.-H., Yan M., Liu Y., Huang Z.-Q., Chang C.-R., Li J., J. Phys. Chem. C 2020, 124, 13656–13663; [Google Scholar]
- 3c. Liu Y., Liu J.-C., Li T.-H., Duan Z.-H., Zhang T.-Y., Yan M., Li W.-L., Xiao H., Wang Y.-G., Chang C.-R., Li J., Angew. Chem. Int. Ed. 2020, 59, 18586–18590; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 18745–18749; [Google Scholar]
- 3d. Kim S. K., Kim H. W., Han S. J., Lee S. W., Shin J., Kim Y. T., Commun. Chem. 2020, 3, 58; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e. Olivos-Suarez A. I., Szécsényi À., Hensen E. J. M., Ruiz-Martinez J., Pidko E. A., Gascon J., ACS Catal. 2016, 6, 2965–2981. [Google Scholar]
- 4.
- 4a. Bodi A., Sztáray B., Baer T., Johnson M., Gerber T., Rev. Sci. Instrum. 2007, 78, 084102; [DOI] [PubMed] [Google Scholar]
- 4b. Bodi A., Johnson M., Gerber T., Gengeliczki Z., Sztáray B., Baer T., Rev. Sci. Instrum. 2009, 80, 034101; [DOI] [PubMed] [Google Scholar]
- 4c. Johnson M., Bodi A., Schulz L., Gerber T., Nucl. Instrum. Methods Phys. Res. Sect. A 2009, 610, 597–603. [Google Scholar]
- 5. Hemberger P., van Bokhoven J. A., Pérez-Ramírez J., Bodi A., Catal. Sci. Technol. 2020, 10, 1975–1990. [Google Scholar]
- 6.
- 6a. Miller J. A., Klippenstein S. J., J. Phys. Chem. A 2003, 107, 7783–7799; [Google Scholar]
- 6b. Zhao L., Lu W., Ahmed M., Zagidullin M. V., Azyazov V. N., Morozov A. N., Mebel A. M., Kaiser R. I., Sci. Adv. 2021, 7, eabf0360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.
- 7a. Holzmeier F., Fischer I., Kiendl B., Krueger A., Bodi A., Hemberger P., Phys. Chem. Chem. Phys. 2016, 18, 9240–9247; [DOI] [PubMed] [Google Scholar]
- 7b. Bieri G., Burger F., Heilbronner E., Maier J. P., Helv. Chim. Acta 1977, 60, 2213–2233. [Google Scholar]
- 8.
- 8a. Scheer A. M., Mukarakate C., Robichaud D. J., Nimlos M. R., Carstensen H.-H., Barney Ellison G., J. Chem. Phys. 2012, 136, 044309; [DOI] [PubMed] [Google Scholar]
- 8b. Hemberger P., da Silva G., Trevitt A. J., Gerber T., Bodi A., Phys. Chem. Chem. Phys. 2015, 17, 30076–30083. [DOI] [PubMed] [Google Scholar]
- 9. Bouwman J., Bodi A., Oomens J., Hemberger P., Phys. Chem. Chem. Phys. 2015, 17, 20508–20514. [DOI] [PubMed] [Google Scholar]
- 10. Zichittella G., Hemberger P., Holzmeier F., Bodi A., Pérez-Ramírez J., J. Phys. Chem. Lett. 2020, 11, 856–863. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Kislov V. V., Islamova N. I., Kolker A. M., Lin S. H., Mebel A. M., J. Chem. Theory Comput. 2005, 1, 908–924; [DOI] [PubMed] [Google Scholar]
- 11b. Mebel A. M., Landera A., Kaiser R. I., J. Phys. Chem. A 2017, 121, 901–926; [DOI] [PubMed] [Google Scholar]
- 11c. Parker D. S. N., Kaiser R. I., Troy T. P., Ahmed M., Angew. Chem. Int. Ed. 2014, 53, 7740–7744; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 7874–7878. [Google Scholar]
- 12.
- 12a. Marinov N. M., Pitz W. J., Westbrook C. K., Vincitore A. M., Castaldi M. J., Senkan S. M., Melius C. F., Combust. Flame 1998, 114, 192–213; [Google Scholar]
- 12b. Gueniche H. A., Biet J., Glaude P. A., Fournet R., Battin-Leclerc F., Fuel 2009, 88, 1388–1393. [Google Scholar]
- 13. Scherer S., Just T., Frank P., Proc. Combust. Inst. 2000, 28, 1511–1518. [Google Scholar]
- 14. Shafir E. V., Slagle I. R., Knyazev V. D., J. Phys. Chem. A 2003, 107, 8893–8903. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information