

Abstract
This pharmacokinetic (PK) drug‐interaction trial investigated the effects of repeated dosing of a plant‐derived pharmaceutical formulation of highly purified cannabidiol (CBD; Epidiolex in the United States and Epidyolex in Europe; 100 mg/mL oral solution) on caffeine clearance via modulation of cytochrome P450 (CYP) 1A2 activity in healthy adults. In this phase 1 open‐label, fixed‐sequence trial, all subjects received a single 200 mg caffeine dose and placebo on day 1. Subjects then titrated CBD from 250 mg once daily to 750 mg twice daily between days 3 and 11 and took 750 mg CBD twice daily between days 12 and 27. On day 26, subjects received a single 200‐mg caffeine dose with their morning CBD dose. Plasma concentrations of caffeine and its CYP1A2‐mediated metabolite, paraxanthine, were determined on days 1 and 26 and PK parameters derived using noncompartmental analysis. Safety was monitored throughout. Sixteen subjects enrolled, and 9 completed treatment. When caffeine was administered with steady‐state CBD, caffeine exposure increased by 15% for Cmax and 95% for AUC0‐∞, tmax increased from 1.5 to 3.0 hours, and t1/2 increased from 5.4 to 10.9 hours compared with caffeine administered with placebo. Under the same conditions, paraxanthine exposure decreased by 22% for Cmax and increased by 18% for AUC0‐∞, tmax increased from 8.0 to 14.0 hours, and t1/2 increased from 7.2 to 13.7 hours. Overall, there were no unexpected adverse events; diarrhea was most common, and 6 subjects discontinued because of elevated liver transaminases. These data suggest that CBD is an inhibitor of CYP1A2.
Keywords: cannabinoid, cannabidiol, caffeine, pharmacokinetics
Across 5 randomized placebo‐controlled trials, highly purified cannabidiol (CBD), approved in the United States as Epidiolex (Greenwich Biosciences, Inc., Carlsbad, California), was significantly superior to placebo in reducing seizures associated with Lennox‐Gastaut syndrome (LGS), Dravet syndrome (DS), or tuberous sclerosis complex; it is approved in the European Union as Epidyolex (GW Pharma [International] B.V.) for use in conjunction with clobazam for LGS and DS. 1 , 2 , 3 , 4 , 5 Given its approved uses, it is important to consider the propensity for potential drug‐drug interactions (DDIs).
CBD is metabolized in the liver by the 5’‐diphospho‐glucuronosyltransferase (UGT) and cytochrome P450 (CYP) enzyme families. 6 , 7 , 8 CBD can also be directly conjugated by UGTs, particularly UGT2B17, UGT1A9, and UGT2B7. 8 In human liver microsomes, CBD is metabolized by CYP2C19 to its active metabolite 7‐hydroxy‐cannabidiol and is further metabolized by CYP3A4 to its inactive metabolite, 7‐carboxy‐cannabidiol. 6 , 7 In vitro studies suggest CBD is a potent inhibitor of both CYP2C19 and CYP3A4 and a weaker CYP2D6 inhibitor 6 , 9 , 10 ; however, results from a recent phase 1 healthy volunteer trial found CBD had no clinically relevant effects on CYP3A4 activity, as steady‐state CBD administration did not affect the systemic exposure, terminal half‐life, or plasma clearance of the CYP3A4 probe midazolam. 11 The ratio of geometric least‐squares (LS) means (90% confidence interval [CI]) comparing midazolam + placebo versus midazolam + CBD was 0.922 (0.778‐1.09) for area under the plasma concentration‐time curve up to time t, where t is the last point with a concentration above the lower limit of quantification (AUC0‐t); 0.921 (0.776‐1.09) for area under the concentration‐time curve from time zero to infinity (AUC0‐∞); and 0.779 (0.667‐0.956) for maximum (peak) concentration of drug in blood plasma (Cmax). 11 A review of a further 5 trials of CBD Epidiolex/Epidyolex; 100 mg/mL oral solution) in healthy volunteers (looking at interactions with a CYP3A4/CYP2C19 inducer and a CYP3A4 inhibitor) or patients with epilepsy (looking at interactions with clobazam, stiripentol, and valproic acid) suggested an overall low potential for DDIs between CBD and other antiepileptic drugs, except for clobazam. 12
In vitro metabolism studies conducted by GW Research Ltd predicted that CBD may act as an inhibitor or inducer of CYP1A2. 5 Therefore, CBD may alter the pharmacokinetics (PK) of commonly concomitantly administered drugs that are substrates for this isoform. To determine the extent of any interaction between CBD and CYP1A2 substrates, a clinical DDI trial in healthy subjects was performed. The main objective was to investigate the effects of repeated doses of CBD on the PK of a single dose of caffeine. Additional objectives were to evaluate the safety and tolerability of CBD when administered with a single dose of caffeine. Caffeine is considered a prototypical CYP1A2 substrate and is commonly used for DDI trials investigating PK interactions affecting CYP1A2. 13
Methods
Trial Design
All relevant trial‐related documents, including the protocol, were reviewed by a local independent ethics committee (Medical Ethics Review Committee [METC Assen], The Netherlands). All subjects provided written informed consent for participation in the trial, which was performed in full conformity with the current Declaration of Helsinki, the International Council for Harmonisation guidelines for Good Clinical Practice, and all other applicable regulations. This trial was performed at a single specialist phase 1 unit (PRA‐EDS in The Netherlands) and took place between April and July 2019.
This was a phase 1 open‐label, fixed single‐sequence DDI trial that enrolled healthy subjects to investigate the effect of repeat‐dose administration of CBD on CYP1A2 activity using caffeine as a probe CYP1A2 substrate.
The trial design is presented in Figure 1. Potential subjects were screened to assess their eligibility to enter the trial within 28 days prior to the first dose of trial drug. The subjects were resident at the trial site for 2 periods. They were admitted in the afternoon of day −1, which was the day prior to day 1, the first day of administration of the trial drug (200 mg caffeine as an oral tablet + placebo as an oral solution). The placebo solution was identical to the CBD solution except it did not contain CBD. On day 3, the first dose of 250 mg CBD was taken in the morning at the site. Subjects were discharged after completion of the day 3 assessments and instructed to escalate their CBD doses at home from the next day, as follows:
Figure 1.
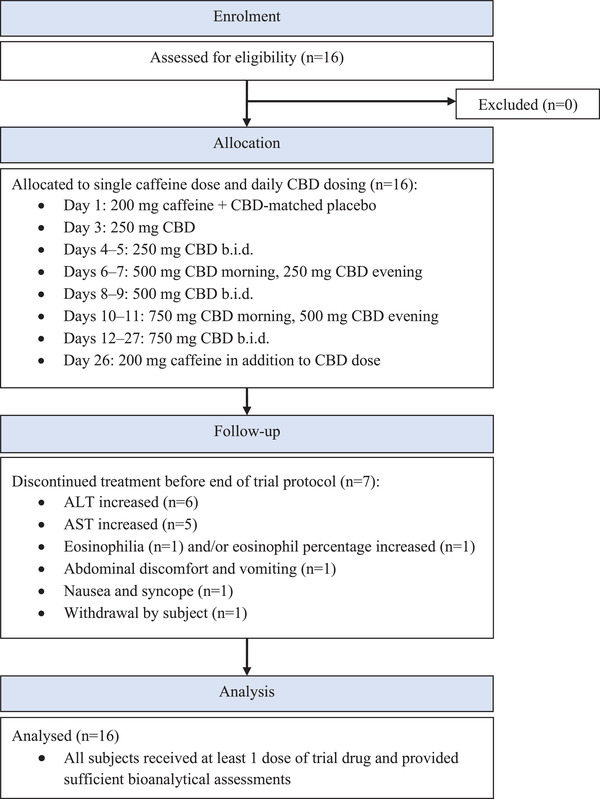
Trial design.ALT, alanine aminotransferase; AST, aspartate aminotransferase; b.i.d., twicedaily; CBD, cannabidiol.
Days 4‐5: 250 mg twice daily;
Days 6‐7: 500 mg morning and 250 mg evening;
Days 8‐9: 500 mg twice daily;
Days 10‐11: 750 mg morning and 500 mg evening;
Days 12‐27: maintenance dosing with 750 mg twice daily.
Subjects were instructed to take their CBD doses 30 minutes after the start of a meal. Ambulatory visits took place on days 12, 18, and 23. Subjects were admitted again to the trial site on day 25. On day 26, subjects received another single oral dose of 200 mg caffeine concurrently with the morning dose of CBD. Subjects were discharged on day 28 after completion of the assessments. A follow‐up visit took place 14‐16 days after the last dose of trial drug.
Trial Population
Healthy male and female subjects aged 18‐60 years inclusive with a body mass index between 18 and 32 kg/m2 and weighing ≥50.0 kg were eligible for this trial. Subjects had no clinically significant medical history, no history of substance abuse, and normal physical examination, 12‐lead electrocardiogram, vital signs, and clinical laboratory findings, as judged by the principal investigator. Female subjects were nonpregnant and nonlactating, and participants of childbearing potential or with a partner of childbearing potential agreed to use effective contraception throughout the trial and for 90 days after the follow‐up visit. The use of all prescribed medication and all over‐the‐counter medication, vitamin preparations and other food supplements, or herbal medications was prohibited from first admission to the trial site until the follow‐up visit. An exception was made for hormonal contraceptives and for acetaminophen (without caffeine; up to 2 g/day for up to 3 consecutive days). The use of >1 dose of CBD or any other cannabinoid within 6 months of the trial was prohibited. The use of methylxanthine‐containing beverages or food (coffee, [iced] tea, cola, chocolate [milk], mocha drinks/sweets, energy drinks) was not allowed from first admission to the trial site until discharge on day 28. The use of decaffeinated coffee and tea was not allowed from 96 hours prior to day −1 until discharge on day 3 and from 96 hours prior to day 25 until discharge on day 28.
Trial Assessments
Materials
Reference and internal standards for CBD (cannabidiol‐d3), caffeine (caffeine˗d3), and paraxanthine (paraxanthine˗d3) were supplied by GW Research Ltd (Sittingbourne, UK), Sigma Aldrich (Zwijndrecht, The Netherlands), CDN Isotopes (Pointe‐Claire, Quebec, Canada), and Toronto Research Chemicals (Toronto, Ontario, Canada).
Plasma Sample Preparation
Blood samples were taken via indwelling intravenous catheter or by direct venipuncture.
Trough concentrations of CBD were measured in plasma from subjects prior to dosing on trial days 23, 25, and 26.
Blood samples for caffeine and paraxanthine PK analysis were taken at predose and 0.5, 1, 1.5, 2, 3, 4, 5, 6, 8, 10, 14, 18, 24, 36, and 48 hours postdose on trial days 1 and 26.
PK parameters evaluated included AUC0‐∞, AUC0‐t, Cmax, and (observed) time after drug administration at which peak plasma concentration occurs (tmax) for caffeine. Additional PK parameters included other PK parameters for caffeine (such as terminal‐phase half‐life [t1/2], time to the observation prior to the first observation with a quantifiable plasma concentration [tlag], time of Ĉlast [the estimated last plasma concentration; tlast], and oral clearance of drug from plasma [CL/F]), all PK parameters for the metabolite paraxanthine (AUC0‐∞, AUC0‐t, Cmax, tmax, tlag, tlast, and t1/2), and metabolite‐to‐parent ratio of AUC (MRAUC) and metabolite‐to‐parent ratio of Cmax (MRCmax) for caffeine and paraxanthine.
Bioanalysis and Pharmacokinetic Assessment
Validated liquid chromatographic‐tandem mass spectrometric bioanalytical methods were used to quantify trough concentrations of CBD in a sample volume of 50 μL and caffeine and paraxanthine in a sample volume of 25 μL.
Sample processing for CBD was performed by liquid‐liquid extraction. Plasma extracts were injected into an ultra‐high‐performance liquid chromatography (UHPLC) machine with a Waters Acquity UPLC BEH C18 column (2.1 × 50 mm, 1.7 μm [Milford, Massachusetts]) and eluted using a step gradient with mobile phases comprising 10 mM ammonium acetate in 100:0.5 (v/v) water:ammonia (mobile phase A) and acetonitrile (mobile phase B) at a flow rate of 1.0 mL/min. Detection was achieved using a Sciex 6500 UHPLC mass spectrometer (Framingham, Massachusetts) equipped with an atmospheric pressure chemical ionization source in negative ionization mode. Quantification was based on multiple reaction monitoring (MRM) of the transitions of m/z 313.1‐245.0 for CBD and m/z 316.1‐248.0 for CBD‐d3. A linear calibration curve with a 1/concentration2 weighting factor was used with an assay range of 1‐1000 ng/mL.
Sample processing for caffeine and paraxanthine was performed by protein precipitation. Caffeine and paraxanthine were separated using a Shimadzu Shim‐pack XR‐ODS III column (Kyoto, Japan) and gradient elution using 0.1% formic acid (mobile phase A) and methanol (mobile phase B) at a flow rate of 0.700 mL/min. A Sciex 5500 UHPLC triple quadrupole mass spectrometer equipped with a turbo ion spray source was used for detection in positive ionization mode. Quantification was based on MRM of the transitions of m/z 195.2‐138.1 for caffeine, 181.2‐124.1 for paraxanthine, 198.2‐138.1 for caffeine˗d3, and 184.2‐127.1 for paraxanthine˗d3. A linear calibration curve, with a 1/concentration2 weighting factor was used with an assay range of 50‐5000 ng/mL for caffeine and paraxanthine.
The precision (coefficient of variation [%CV]) and accuracy (relative error [RE%]/mean % different [Bias%]) of the UHPLC method was acceptable for all analytes (≤15% [≤20% at the lower limit of quantification]). Recovery was 45.4%‐48.8% for CBD, 88.3%‐94.8% for caffeine, and 88.4%‐95.6% for paraxanthine.
Safety Assessments
Safety and tolerability were evaluated by treatment‐emergent adverse event (AE) review, vital sign measurements, 12‐lead electrocardiograms (ECGs), clinical laboratory evaluations, physical examinations, and Columbia‐Suicide Severity Rating Scale assessment.
Statistical Analysis
All subjects who received at least 1 dose of CBD, caffeine, or placebo were included in the safety set. All subjects who received at least 1 dose of caffeine and provided sufficient bioanalytical assessments to calculate reliable estimates of the PK parameters were included in the PK analysis set. The PK parameters for caffeine and its metabolite paraxanthine were calculated using noncompartmental methods in Phoenix WinNonlin version 8.1 (Certara USA, Inc., Princeton, New Jersey).
The effect of CBD on Cmax, AUC0‐t, and AUC0‐∞ of caffeine and paraxanthine was assessed with a linear mixed‐effects model. Caffeine administered with CBD on day 26 was considered test treatment, whereas caffeine administered with placebo on day 1 was considered reference treatment. The PK parameters were natural log‐transformed prior to the analysis. Treatment was used as a fixed effect and subject as a random effect. Point estimates for the means and point estimates and corresponding 90%CIs for the differences in means between the 2 treatments were obtained from the linear mixed‐effects model and exponentiated to obtain geometric LS means, geometric mean ratios, and respective 90%CIs on the original scale.
This analysis was performed separately on 2 data selections: primary analysis and sensitivity analysis.
The primary analysis excluded data for subjects who missed any of the planned 4 CBD doses on days 26‐27 (during caffeine + CBD treatment) and excluded data for specific analytes from analysis periods in which a subject had a predose concentration higher than 5% of Cmax for either caffeine of paraxanthine. Data from analysis periods in which a subject had a predose concentration between 5% and 10% of Cmax were included in the analysis at the discretion of the pharmacokineticist. The sensitivity analysis excluded data for subjects who missed any of the planned 4 CBD doses on days 26‐27 (during caffeine + CBD treatment) and excluded data for specific analytes from analysis periods in which a subject had a predose concentration higher than 5% of Cmax for either caffeine of paraxanthine.
Geometric LS mean ratios (test/reference) and 90%CIs for caffeine were used to estimate the magnitude of any interaction.
For tmax, nonparametric methods of the same comparisons were performed using a Wilcoxon signed rank test. The median tmax for each treatment and the median of the pairwise differences between the treatments (test‐reference) were presented along with the approximate 90%CI.
Results
Subject Demographics
A total of 16 subjects (100%) enrolled and 9 subjects (56.3%) completed the trial (Figure 1). Seven subjects (43.8%) discontinued treatment before the end of the trial; 6 (37.5%) discontinued because of AEs and 1 subject (6.3%) had their reason documented as withdrawal by subject. All subjects were included in the safety and PK analysis sets. Demographics information is presented in Table 1.
Table 1.
Subject Baseline Characteristics; Safety Analysis Set
| Parameter | CBD (n = 16) |
|---|---|
| Age, y | |
| Mean (SD) | 32.6 (12.9) |
| Median (Q1, Q3) | 29 (23.5, 37.8) |
| Sex, n (%) | |
| Male | 6 (37.5) |
| Female | 10 (62.5) |
| Race, n (%) | |
| White | 13 (81.3) |
| Black or African American | 1 (6.3) |
| Asian | 1 (6.3) |
| American Indian or Alaska Native | 1 (6.3) |
| BMI, kg/m2 | |
| Mean (SD) | 22.9 (2.2) |
| Median (Q1, Q3) | 22.7 (21.1, 24.6) |
BMI, body mass index; CBD, cannabidiol; Q1, quartile 1; Q3, quartile 3; SD, standard deviation.
Seven female subjects took ongoing oral contraceptives during the trial. Eight subjects (50.0%) received concomitant medication to treat ≥1 AE. Seven subjects (43.8%) received paracetamol, 2 (12.5%) for AEs of abdominal discomfort, 1 (6.3%) for influenza‐like illness, 1 (6.3%) for pyrexia, 1 (6.3%) for abdominal pain and back pain, 1 (6.3%) for oropharyngeal pain, and 1 (6.3%) for headache. Two subjects (12.5%) received ibuprofen, 1 (6.3%) for an AE of influenza‐like illness and 1 (6.3%) for abdominal pain and back pain. One subject (6.3%) received loperamide hydrochloride for diarrhea.
Pharmacokinetics
CBD
Trough levels of CBD (on days 23, 25, and 26) confirmed that CBD had reached steady state before caffeine and active CBD were coadministered on day 26 (data not shown).
Caffeine
Plasma‐concentration curves for caffeine following administration of caffeine and placebo compared with caffeine and steady‐state CBD are presented in Figure 2A. PK parameters are presented in Table 2 and analysis of PK parameters in Table 3. Geometric LS mean ratios and 90%CIs showing the effect of steady‐state CBD on exposure to caffeine and paraxanthine are presented in Figure 3.
Figure 2.
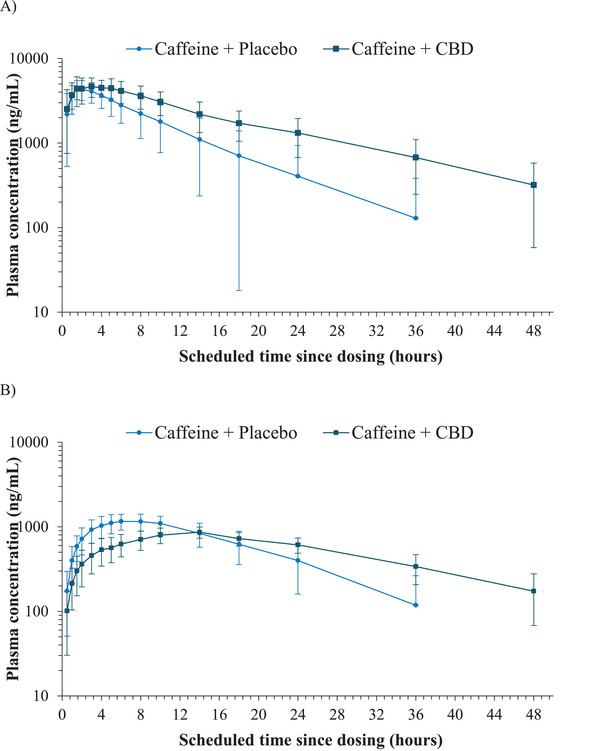
Mean (SD) plasma concentrations of (A) caffeine and (B) paraxanthine following administration of caffeine + placebo (day 1) and of caffeine + CBD (day 26) — semilogarithmic; PK analysis set. CBD, cannabidiol; PK, pharmacokinetic; SD, standard deviation.
Table 2.
Summary of the PK Parameters of Caffeine and Paraxanthine Following Administration of Caffeine + Placebo (Day 1) and of Caffeine + CBD (Day 26) — PK Analysis Set
| Primary Analysis | Sensitivity Analysis | ||
|---|---|---|---|
| Parameter | Caffeine + Placebo (Day 1) n = 16 | Caffeine + CBD (Day 26) n = 9 | Caffeine + CBD (Day 26) n = 6 |
| Caffeine | |||
| AUC0‐t (ng∙h/mL) | 45 600 (55.5) | 82 800 (35.4) | 89 600 (33.5) |
| AUC0‐∞ (ng∙h/mL) | 46 900 (57.4) | 89 200 (38.2) | 97 900 (36.1) |
| Cmax (ng/mL) | 4710 (22.1) | 5270 (20.8) | 5520 (18.7) |
| tmax a (h) | 1.51 (0.50‐3.00) | 3.00 (0.50‐5.00) | 3.00 (0.50‐4.13) |
| tlast a (h) | 35.99 (18.00‐47.92) | 48.00 (35.92‐48.00) | 48.00 (35.92‐48.00) |
| t1/2 (h) | 5.40 (42.5) | 10.9 (32.5) | 11.7 (35.1) |
| tlag a (h) | 0.00 (0.00‐0.00) | 0.00 (0.00‐0.00) | 0.00 (0.00‐0.00) |
| CL/F (L/h) | 5.55 (50.2) | 2.57 (39.4) | 2.30 (37.9) |
| Paraxanthine | |||
| Parameter | Caffeine + Placebo (Day 1) n = 16 |
Caffeine + CBD (Day 26) n = 7 |
Caffeine + CBD (Day 26) n = 6 |
| Caffeine | 22 200 (26.1) | 24 100 (13.0) | 24 100 (14.2) |
| AUC0‐∞ (ng∙h/mL) | 23 900 (29.9) | 28 300 (18.4) | 28 600 (19.8) |
| Cmax (ng/mL) | 1250 (19.9) | 895 (16.4) | 905 (17.5) |
| tmax a (h) | 7.99 (4.00‐18.02) | 14.00 (5.97‐18.00) | 14.00 (10.00‐18.00) |
| tlast a (h) | 36.00 (24.00‐47.92) | 48.00 (35.92‐48.00) | 48.00 (35.92‐48.00) |
| t1/2 (h) | 7.15 (55.3) | 13.7 (30.4) | 13.8 (33.0) |
| tlag a (h) | 0.00 (0.00‐0.52) | 0.00 (0.00‐0.50) | 0.00 (0.00‐0.50) |
| MRAUC0‐t | 0.603 (27.0) | 0.323 (26.1) | 0.311 (27.2) |
| MRAUC0‐∞ | 0.627 (25.5) | 0.349 (23.6) | 0.336 (24.6) |
| MRCmax | 0.299 (28.5) | 0.186 (18.4) | 0.180 (18.2) |
AUC0‐t, area under the plasma concentration‐time curve up to time t, where t is the last point with a concentration above the lower limit of quantification; AUC0‐∞, area under the plasma concentration‐time curve from time 0 to infinity; CBD, cannabidiol; Cmax, maximum (peak) concentration of drug in blood plasma; CV%, coefficient of variation; FU, follow‐up; max, maximum; min, minimum; MRAUC0‐t, ratio of metabolite AUC0‐t to parent AUC0‐t; MRAUC0‐∞, ratio of metabolite AUC0‐∞ to parent AUC0‐∞; MRCmax, ratio of metabolite Cmax to parent Cmax; PK, pharmacokinetic; t1/2, terminal‐phase half‐life; tlag, time to the observation prior to the first observation with a quantifiable plasma concentration; tlast, time of Ĉlast (the estimated last plasma concentration); tmax, (observed) time after drug administration at which peak plasma concentration occurs.
Note: For the treatment of caffeine + placebo, the primary analysis and sensitivity analysis are equal.
Note: Arithmetic mean (%CV) is presented unless otherwise noted.
Median (min‐max).
Table 3.
Statistical Analysis of the Pharmacokinetic Parameters of Caffeine and Paraxanthine Following Administration of Caffeine + Placebo (Day 1) and of Caffeine + CBD (Day 26) — PK Analysis Set
| Geometric LS Means | Ratio Test/Reference | ||||||
|---|---|---|---|---|---|---|---|
| Analyte | PK Parameter | n | Reference | n | Test | Estimate | 90%CI |
| Primary analysis | |||||||
| Caffeine | Cmax a (ng/mL) | 16 | 4600 | 9 | 5269 | 1.15 | 1.04‐1.26 |
| AUC0‐t a (ng·h/mL) | 16 | 39 969 | 9 | 75 237 | 1.88 | 1.56‐2.27 | |
| AUC0‐∞ a (ng·h/mL) | 16 | 40 856 | 9 | 79 718 | 1.95 | 1.62‐2.35 | |
| tmax b (h) | 9 | 1.5 | 9 | 3.0 | 0.58 | 0.01‐1.50 | |
| Paraxanthine | Cmax a (ng/mL) | 16 | 1227 | 7 | 961 | 0.78 | 0.72‐0.86 |
| AUC0‐t a (ng·h/mL) | 16 | 21 454 | 7 | 23 579 | 1.10 | 0.96‐1.26 | |
| AUC0‐∞ a (ng·h/mL) | 16 | 22 972 | 7 | 27 038 | 1.18 | 1.03‐1.35 | |
| tmax b (h) | 7 | 8.00 | 7 | 14.00 | 3.49 | 0.48‐6.00 | |
| Sensitivity analysis | |||||||
| Caffeine | Cmax a (ng/mL) | 16 | 4600 | 6 | 5460 | 1.19 | 1.03‐1.36 |
| AUC0‐t a (ng·h/mL) | 16 | 39 969 | 6 | 71 265 | 1.78 | 1.39‐2.29 | |
| AUC0‐∞ a (ng·h/mL) | 16 | 40 856 | 6 | 76 272 | 1.87 | 1.45‐2.41 | |
| tmax b (h) | 6 | 2.3 | 6 | 3.0 | 0.57 | 0.00‐1.25 | |
| Paraxanthine | Cmax a (ng/mL) | 16 | 1227 | 6 | 984 | 0.80 | 0.73‐0.88 |
| AUC0‐t a (ng·h/mL) | 16 | 21 454 | 6 | 22 526 | 1.05 | 0.93‐1.18 | |
| AUC0‐∞ a (ng·h/mL) | 16 | 22 972 | 6 | 26 023 | 1.13 | 0.99‐1.30 | |
| tmax b (h) | 6 | 8.0 | 6 | 14.0 | 3.99 | −0.02 to 7.00 | |
AUC0‐t, area under the plasma concentration‐time curve up to time t, where t is the last point with a concentration above the lower limit of quantification; AUC0‐∞, area under the plasma concentration‐time curve from time 0 to infinity; CBD, cannabidiol; CI, confidence interval; Cmax, maximum (peak) concentration of drug in blood plasma; LS, least squares; PK, pharmacokinetic; tmax, (observed) time after drug administration at which peak plasma concentration occurs.
Note: Reference, caffeine + placebo treatment; test, caffeine + CBD treatment.
AUC and Cmax, the interaction effect was explored using a mixed‐effects (analysis of variance) model with treatment as fixed factor and subject as a random effect.
tmax, nonparametric Wilcoxon signed rank test presenting the Hodges‐Lehman estimate and 90%CI based on the Tukey method. Median, median of the difference, and approximate 90%CI for the difference are presented.
Figure 3.
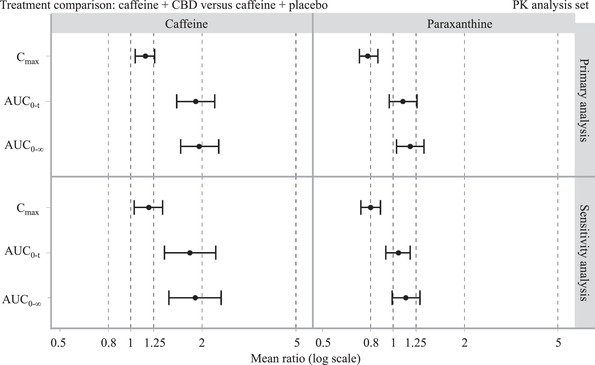
Geometric LS mean ratio and 90%CI showing the effect of steady‐state CBD on exposure to caffeine and paraxanthine; PK analysis set. AUC0‐t, area under the plasma concentration‐time curve up to time t, where t is the last point with a concentration above the lower limit of quantification; AUC0‐∞, area under the plasma concentration‐time curve from time 0 to infinity; CBD, cannabidiol; CI, confidence interval; Cmax, maximum (peak) concentration of drug in blood plasma.
On day 1, following administration of 200 mg caffeine and placebo, maximum postdose geometric mean plasma concentrations were reached at 1.5 hours for caffeine. On day 26, following coadministration of 200 mg caffeine and 750 mg CBD, maximum postdose geometric mean plasma concentrations were reached at 3.0 hours for caffeine. The elimination phase of caffeine was multiphasic following 200 mg caffeine and 750 mg CBD coadministration on day 26, and caffeine concentrations tended to be higher compared with administration of 200 mg caffeine and placebo on day 1 (Figure 2A).
When compared with caffeine and placebo on day 1, coadministration of caffeine and CBD on day 26 resulted in an increase in caffeine Cmax (1.15; 90%CI, 1.04‐1.26) and a larger increase in its AUC0‐t (1.88; 90%CI, 1.56‐2.27) and AUC0‐∞ (1.95; 90%CI, 1.62‐2.35). The tmax for caffeine was later after administration of caffeine and CBD (day 26) compared with caffeine and placebo administration (day 1) — difference Hodges‐Lehman estimate, 0.58; 90%CI, 0.01‐1.50. Also, the t1/2 for caffeine was longer after administration of caffeine and CBD (day 26; 10.9 hours) compared with caffeine and placebo administration (day 1; 5.4 hours), although the t1/2 was not tested statistically. Similar results were seen for the primary and sensitivity analyses (Tables 2 and 3).
Reflecting the slight increase in bioavailability, CL/F of caffeine was reduced when caffeine was administered with CBD (day 26) compared to with placebo (day 1). Between‐subject variability for caffeine, based on arithmetic %CV, was low to moderate, whether caffeine was administered with placebo or in combination with CBD, ranging from 33.5% to 55.5% for AUC0‐t, from 36.1% to 57.4% for AUC0‐∞, and from 18.7% to 22.1% for Cmax (Table 2).
Paraxanthine
Plasma‐concentration curves for paraxanthine following administration of caffeine and placebo compared with caffeine and steady‐state CBD are presented in Figure 2B. PK parameters are presented in Table 2, and analysis of PK parameters in Table 3. Geometric LS mean ratios and 90%CIs showing the effect of steady‐state CBD on exposure to caffeine and paraxanthine are presented in Figure 3.
On day 1, following administration of 200 mg caffeine and placebo, maximum postdose geometric mean plasma concentrations were reached at 6.0 hours for paraxanthine. On day 26, following coadministration of 200 mg caffeine and 750 mg CBD, maximum postdose geometric mean plasma concentrations were reached at 14.0 hours for paraxanthine. The elimination phase of paraxanthine was multiphasic following 200 mg caffeine and 750 mg CBD coadministration on day 26, and paraxanthine concentrations were lower compared with administration of 200 mg caffeine and placebo on day 1 (Figure 2B).
When compared with caffeine and placebo (day 1), coadministration of caffeine and CBD (day 26) resulted in a decrease in Cmax (0.78; 90%CI, 0.72‐0.86) and a slight increase in AUC0‐t (1.10; 90%CI, 0.96‐1.26) and AUC0‐∞ (1.18; 90%CI, 1.03‐1.35). The tmax for paraxanthine tended to be later after administration of caffeine and CBD (day 26) compared with caffeine and placebo administration (day 1) — difference Hodges‐Lehman estimate, 3.49; 90%CI, 0.48‐6.00; Table 3. Also, the t1/2 for paraxanthine was longer after administration of caffeine and CBD (day 26; 13.7 hours) compared with caffeine and placebo administration (day 1; 7.15 hours), although the t1/2 was not tested statistically. These changes in caffeine and paraxanthine exposures in the presence of CBD were reflected in the metabolite‐to‐parent ratios. Similar results were seen for the primary and sensitivity analyses (Tables 2 and 3). Between‐subject variability for paraxanthine was low, ranging from 12.8% to 27.8% for AUC0‐t, from 18.4% to 30.2% for AUC0‐∞, and from 15.8% to 20.6% for Cmax (Table 2).
Safety
CBD was tolerated by most subjects when administered as multiple doses, alone or concomitantly with caffeine. All‐causality AEs were reported by 14 subjects (87.5%), and the most frequently reported AE by preferred term was diarrhea, reported by 8 subjects (50.0%) overall and most commonly when taking titration doses of CBD alone (Table 4). One subject experienced a severe AE (alanine aminotransferase [ALT] increased 8.1× the upper limit of normal [ULN]), 8 subjects (50.0%) reported moderate AEs, and 5 subjects (31.3%) reported mild AEs. There were no deaths or treatment‐emergent serious adverse events (SAEs).
Table 4.
Treatment‐Emergent Adverse Events Reported In >1 Subject Overall by System Organ Class and MedDRA Preferred Term and Treatment; Safety Analysis Set
| System Organ Class MedDRA Preferred Term | Caffeine + Placebo n= 16 | Titration CBD Alone (Days 3‐11) n = 16 | CBD Alone (Days 12‐25) n = 16 | Caffeine + CBD (Day 26) n = 11 | Total n = 16 |
|---|---|---|---|---|---|
| Number of subjects (%) | |||||
| Subjects experiencing any AEs | 7 (43.8) | 8 (50.0) | 8 (50.0) | 6 (54.5) | 14 (87.5) |
| General disorders and administration‐site conditions | 3 (18.8) | 0 | 6 (37.5) | 2 (18.2) | 11 (68.8) |
| Catheter site‐related reaction | 2 (12.5) | 0 | 0 | 1 (9.1) | 3 (18.8) |
| Fatigue | 1 (6.3) | 0 | 2 (12.5) | 0 | 3 (18.8) |
| Pyrexia | 0 | 0 | 2 (12.5) | 0 | 2 (12.5) |
| Gastrointestinal disorders | 1 (6.3) | 6 (37.5) | 5 (31.3) | 2 (18.2) | 9 (56.3) |
| Diarrhea | 1 (6.3) | 6 (37.5) | 4 (25.0) | 0 | 8 (50.0) |
| Abdominal discomfort | 0 | 3 (18.8) | 1 (6.3) | 2 (18.2) | 5 (31.3) |
| Abdominal pain | 0 | 1 (6.3) | 1 (6.3) | 0 | 2 (12.5) |
| Investigations | 0 | 0 | 6 (37.5) | 1 (9.1) | 7 (43.8) |
| Gamma‐glutamyltransferase increased | 0 | 0 | 6 (37.5) | 1 (9.1) | 7 (43.8) |
| Alanine aminotransferase increased | 0 | 0 | 5 (31.3) | 1 (9.1) | 6 (37.5) |
| Aspartate aminotransferase increased | 0 | 0 | 5 (31.3) | 1 (9.1) | 6 (37.5) |
| Nervous system disorders | 3 (18.8) | 0 | 3 (18.8) | 0 | 6 (37.5) |
| Headache | 3 (18.8) | 0 | 2 (12.5) | 0 | 5 (31.3) |
| Musculoskeletal and connective tissue disorders | 1 (6.3) | 1 (6.3) | 1 (6.3) | 1 (9.1) | 4 (25.0) |
| Back pain | 0 | 1 (6.3) | 1 (6.3) | 0 | 2 (12.5) |
FU, follow‐up; MedDRA, Medical Dictionary for Regulatory Activities; AE, treatment‐emergent adverse event.
Six subjects (37.5%) were discontinued from trial drug because of AEs: 3 subjects because ALT and aspartate aminotransferase (AST) increased; 1 subject because of increased ALT and AST and eosinophilia; 1 subject because of increased ALT, abdominal discomfort, vomiting, and increased percentage of eosinophils; and 1 subject because of increased ALT and AST, nausea, and syncope.
During both maintenance dosing of CBD only and caffeine plus CBD dosing, ALT, AST, and/or gamma‐glutamyltransferase (GGT) elevations of ≥3× ULN were noted in 6 subjects (37.5%) who were considered clinically significant by the investigator. Four of these subjects had clinically significant ALT, AST, and GGT elevations; 1 subject had clinically significant ALT and AST elevations; and 1 subject had clinically significant GGT elevation. These changes in liver enzymes were not associated with a rise in total bilirubin > 2× ULN (Hy's law) in any of the subjects. Two subjects (12.5%) had an increase in the percentage of eosinophils that was considered clinically significant by the investigator during maintenance dosing of CBD (1 subject) and during caffeine plus CBD dosing (1 subject). The elevations of ALT, AST, GGT, and percentage of eosinophils were transient, and values generally returned to within the normal reference range during the trial (between 9 and 49 days after onset), except for 1 case of GGT increase that was still ongoing by the end of the trial (last known GGT was 2.4× ULN).
There were no clinically significant physical examination, vital signs, or ECG findings during the trial and no evidence of suicidal behavior or suicidal ideation.
Discussion
Overall, there was an effect of CBD on the exposure of caffeine, a CYP1A2 substrate, leading to an elevation of 15% for Cmax, 88% for AUC0‐t, and 95% for AUC0‐∞. In addition, there was a slight effect of CBD on exposure of paraxanthine, also a substrate for CYP1A2, 14 leading to an elevation of 10% for AUC0‐t and 18% for AUC0‐∞ and a reduction of 22% for Cmax. Total variability in the PK of caffeine and paraxanthine was low to moderate.
Previous phase 1 trials investigating the PK of CBD have demonstrated an effect of food on CBD and metabolite exposures. 15 , 16 In one trial, a single dose of 1500 mg CBD administered with a standardized US Food and Drug Administration (FDA) high‐fat meal increased CBD exposure by 4.2‐fold for AUC0‐t and by 4.9‐fold for Cmax. In a subsequent trial, a single dose of 750 mg CBD administered with a standardized FDA high‐fat meal increased CBD exposure by 3.8‐fold for AUC0‐∞ and by 5.2‐fold for Cmax. 16 Increases in CBD exposure were also observed with both a low‐fat meal and milk, with respective increases in exposure of 2.7‐ and 2.4‐fold for AUC0‐∞ and 3.8‐ and 3.1‐fold for Cmax. 15 , 16 Although food‐effect trials with high‐fat meals are designed to explore extreme scenarios in terms of food intake 16 to account for potential confounding by food in the current trial, subjects were dosed under fed conditions 30 minutes after starting a regular meal. Meals were standardized while subjects were resident at the trial site, and subjects had to complete their meals prior to dosing.
The results of this trial suggest that CBD is an inhibitor of CYP1A2. As such, there is a potential risk that a person who consumes high doses of caffeine over a short period alongside CBD may experience clinically significant caffeine‐related side effects. A previous study investigating the effects of fluvoxamine on the PK of caffeine found that despite a significant reduction in caffeine clearance with fluvoxamine versus placebo (105 versus 9.1 mL/min, P < .01; mean difference, 95.7 mL/min; 95%CI, 54.9‐135.6 mL/min), psychomotor performance, alertness, and electroencephalogram effects attributable to caffeine were not augmented by coadministration of fluvoxamine. 17 Regardless, clinicians should be alert to this potential effect of CBD. Interactions between CBD and other drugs metabolized by CYP1A2 cannot be ruled out. Previous research has shown CYP1A2 induction to be associated with chemotherapy resistance 18 and drug‐induced adverse reactions, including liver toxicity. 19 Reassuringly, the results of this trial suggest that CBD is highly unlikely to be an inducer of CYP1A2.
Safety
CBD was tolerated by most subjects when administered as multiple doses, alone or concomitantly with caffeine. The most frequently reported AE was diarrhea, which affected half the subjects and was most often experienced during CBD titration. One subject experienced a severe AE of increased ALT. There were no deaths or SAEs. Six subjects discontinued treatment in line with protocol‐defined criteria for withdrawal, all because of liver enzyme elevations. None of the changes in liver enzymes experienced during the trial were associated with a rise in total bilirubin levels > 2× ULN (Hy's law) in any of the subjects. In addition, elevations in ALT, AST, GGT, and percentage of eosinophils were transient, and values generally returned to within the normal reference range during the trial (between 9 and 49 days after onset). The abnormal liver chemistry findings in this and other healthy volunteer and patient trials with CBD are described in more detail in a separate publication. 20 The authors of that publication concluded that healthy adults consuming CBD may experience elevations in serum ALT consistent with drug‐induced liver injury.
There were no clinically significant physical examination, vital signs, or ECG findings during the trial and no evidence of suicidal behavior or suicidal ideation.
Limitations
A limitation of this trial is that only 9 of the 16 enrolled subjects completed treatment; however, this did not affect the results, as the sensitivity analysis was well aligned with the primary analysis.
Conclusion
Caffeine and paraxanthine exposure increased when a single dose of caffeine was coadministered with steady‐state CBD. These findings suggest CBD is an inhibitor of CYP1A2. There is the potential for an interaction between CBD and drugs metabolized by CYP1A2. There were no SAEs in this trial, and no new safety concerns were identified.
Conflicts of Interest
D.C., B.T., and C.T. are employees of GW Research Ltd and own share options in GW Pharmaceuticals plc. This trial was sponsored by GW Research Ltd. The authors also acknowledge and thank Dr Lesley Taylor of Alchemy Medical Writing Ltd for medical writing and editorial support, which was funded by Greenwich Biosciences, Inc.
Funding
This trial was sponsored and funded by GW Research Ltd. Medical writing support was provided by Dr Lesley Taylor of Alchemy Medical Writing Ltd., which was funded by Greenwich Biosciences, Inc.
Data Accessibility Statement
The sponsor is adhering to current US and EU requirements and so will not make individual deidentified participant data available; however, the protocol and statistical analysis plan will be made available on request to the corresponding author.
Acknowledgments
The authors thank the volunteers who took part in the trial, as well as the staff that assisted with the trial at each site. We also thank the clinical project manager, Sue Ditton, and PRA Health Sciences for the development of the caffeine and paraxanthine bioanalytical method.
References
- 1. Devinsky O, Cross JH, Laux L, et al. Cannabidiol in Dravet Syndrome Study Group. Trial of cannabidiol for drug‐resistant seizures in the Dravet syndrome. N Engl J Med. 2017;376(21):2011‐2020. [DOI] [PubMed] [Google Scholar]
- 2. Devinsky O, Patel AD, Thiele EA, et al. Randomized, dose‐ranging safety trial of cannabidiol in Dravet syndrome. Neurol. 2018;90(14):e1204‐e1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Devinsky O, Patel AD, Cross JH, et al. Effect of cannabidiol on drop seizures in the Lennox‐Gastaut syndrome. N Engl J Med. 2018;378(201):1888‐1897. [DOI] [PubMed] [Google Scholar]
- 4. Thiele EA, Marsh ED, French JA, et al. Cannabidiol in patients with seizures associated with Lennox‐Gastaut syndrome (GWPCARE4): a randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet. 2018;391(10125):1085‐1096. [DOI] [PubMed] [Google Scholar]
- 5. Thiele E, Bebin ME, Bhathal H, et al. Add‐on cannabidiol treatment for drugresistant seizures in tuberous sclerosis complex. JAMA Neurol. 2020;78(36):285‐292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jiang R, Yamaori S, Takeda S, Yamamoto I, Watanabe K. Identification of cytochrome P450 enzymes responsible for metabolism of cannabidiol by human liver microsomes. Life Sci. 2011;89(5‐6):165‐170. [DOI] [PubMed] [Google Scholar]
- 7. Whalley BJ, Stott C, Gray RA, Jones NA. The human metabolite of cannabidiol, 7‐hydroxyl cannabidiol, but not 7‐carboxy cannabidiol, is anticonvulsant in the maximal electroshock seizure threshold test (MEST) in mouse. American Epilepsy Society 2017 Annual Meeting Abstract Database.
- 8. Mazur A, Lichti CF, Prather PL, et al. Characterization of human hepatic and extrahepatic UDP‐glucuronosyltransferase enzymes involved in the metabolism of classic cannabinoids. Drug Metab Dispos. 2009;37(7):1496‐1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Yamaori S, Ebisawa J, Okushima Y, Yamamoto I, Watanabe K. Potent inhibition of human cytochrome P450 3A isoforms by cannabidiol: role of phenolic hydroxyl groups in the resorcinol moiety. Life Sci. 2011;88(15‐16):730‐736. [DOI] [PubMed] [Google Scholar]
- 10. Yamaori S, Okamoto Y, Yamamoto I, Watanabe K. Cannabidiol, a major phytocannabinoid, as a potent atypical inhibitor for CYP2D6. Drug Metab Dispos. 2011;39(11):2049‐2056. [DOI] [PubMed] [Google Scholar]
- 11. Morrison G, Taylor L, Crockett J, Critchley D, Tayo B. A phase 1 investigation into the potential effects of cannabidiol on CYP3A4‐mediated drug‐drug interactions in healthy volunteers. American Epilepsy Society 2018 Annual Meeting Abstract Database.
- 12. Patsalos PN, Szaflarski JP, Gidal B, VanLandingham K, Critchley D, Morrison G. Clinical implications of trials investigating drug‐drug interactions between cannabidiol and enzyme inducers or inhibitors or common antiseizure drugs. Epilepsia. 2020;61(9):1854‐1868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kot M, Daniel WA. Caffeine as a marker substrate for testing cytochrome P450 activity in human and rat. Pharmacol Rep. 2008;60(6):789‐797. [PubMed] [Google Scholar]
- 14. Thorn CF, Aklillu E, McDonagh EM, Klein TE, Altman RB. PharmGKB summary: caffeine pathway. Pharmacogenet Genomics. 2012;22(5):389‐395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Taylor L, Gidal B, Blakey G, Tayo B, Morrison G. A phase I, randomized, double‐blind, placebo‐controlled, single ascending dose, multiple dose, and food effect trial of the safety, tolerability and pharmacokinetics of highly purified cannabidiol in healthy subjects. CNS Drugs. 2018;32(11):1053‐1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Crockett J, Critchley D, Tayo B, Berwaerts J, Morrison G. A phase 1, randomized, pharmacokinetic trial of the effect of different meal compositions, whole milk, and alcohol on cannabidiol exposure and safety in healthy subjects. Epilepsia. 2020;61(2):267‐277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Culm‐Merdek KE, von Moltke LL, Harmatz JS, Greenblatt DJ. Fluvoxamine impairs single‐dose caffeine clearance without altering caffeine pharmacodynamics. BJCP. 2005;60(5):486‐493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. AbuHammad S, Zihlif M. Gene expression alterations in doxorubicin resistant MCF7 breast cancer cell line. Genomics. 2013;101(6 Pt 8):213‐220. [DOI] [PubMed] [Google Scholar]
- 19. Gomez JL, Dupont A, Cusan L, Tremblay M, Suburu R, Lemay M, et al. Incidence of liver toxicity associated with the use of flutamide in prostate cancer patients. Am J Med. 1992;92(5):465‐470. [DOI] [PubMed] [Google Scholar]
- 20. Watkins PB, Church RJ, Li J, Knappertx V. Cannabidiol and abnormal liver chemistries in healthy adults: Results of a phase I clinical trial. Clin Pharmacol Ther. 2020; 10.1002/cpt.2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The sponsor is adhering to current US and EU requirements and so will not make individual deidentified participant data available; however, the protocol and statistical analysis plan will be made available on request to the corresponding author.