

Abstract
We report a simple and rapid saliva‐based SARS‐CoV‐2 antigen test that utilizes a newly developed dimeric DNA aptamer, denoted as DSA1N5, that specifically recognizes the spike proteins of the wildtype virus and its Alpha and Delta variants with dissociation constants of 120, 290 and 480 pM, respectively, and binds pseudotyped lentiviruses expressing the wildtype and alpha trimeric spike proteins with affinity constants of 2.1 pM and 2.3 pM, respectively. To develop a highly sensitive test, DSA1N5 was immobilized onto gold electrodes to produce an electrochemical impedance sensor, which was capable of detecting 1000 viral particles per mL in 1:1 diluted saliva in under 10 min without any further sample processing. Evaluation of 36 positive and 37 negative patient saliva samples produced a clinical sensitivity of 80.5 % and specificity of 100 % and the sensor could detect the wildtype virus as well as the Alpha and Delta variants in the patient samples, which is the first reported rapid test that can detect any emerging variant of SARS‐CoV‐2.
Keywords: aptamers, COVID-19, electrochemical biosensors, rapid tests, saliva
Rapid and simple: an electrochemical sensor, based on a high‐affinity dimeric DNA aptamer for the SARS‐CoV‐2 spike protein was developed to detect the wildtype and Alpha and Delta variant in unprocessed saliva samples in 10 minutes, providing a detection limit of 1000 copies mL−1, clinical sensitivity of 80.5 % and specificity of 100 %.
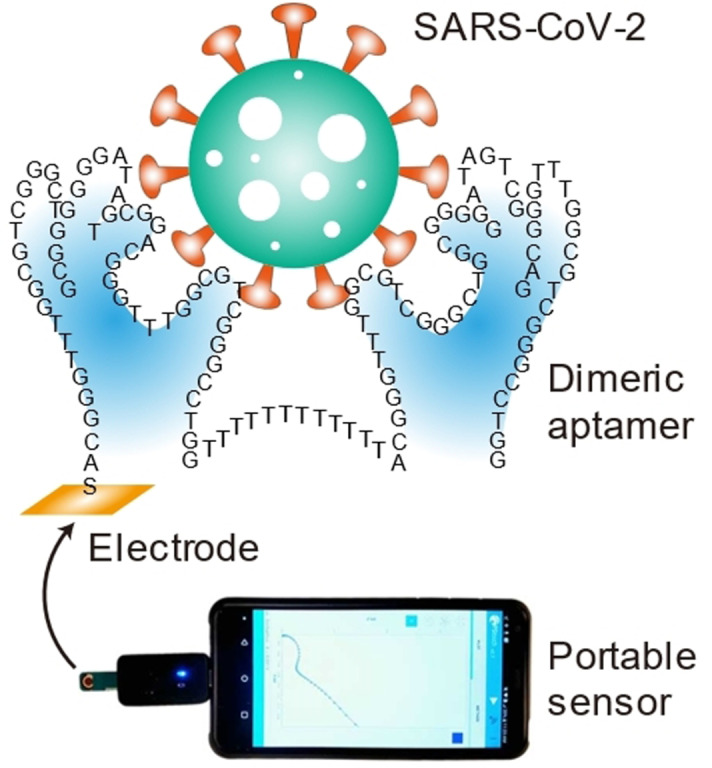
Introduction
As countries around the world strive to re‐open their economies, there is a need to significantly enhance COVID‐19 testing to both monitor and ideally prevent outbreaks and to allow contact tracing. The current testing regime in most countries is based on reverse transcription‐polymerase chain reaction (RT‐PCR) methods performed at centralized laboratories. Such tests are sensitive but are costly, too complicated for operation by non‐skilled laboratory technicians, and have long “sample‐to‐answer” times. [1] These tests are now being supplemented with rapid nucleic acid (e.g., Abbott ID NOWTM) and antigen tests (e.g., Abbott PanBioTM and Abbott BINAX NOWTM among others). However, these still require collection of nasal pharyngeal swabs (NPS) and have moderate performance metrics (sensitivity and specificity) relative to RT‐PCR. [2] The rapid nucleic acid tests also require nucleic acid amplification, resulting in testing times of at least 30 minutes, [3] which makes them unsuitable for testing at points‐of‐entry, workplaces or congregate settings.
Existing rapid antigen tests provide fast sample‐to‐result times (5 to 13 min), [4] and because they detect the nucleocapsid protein, they can identify both the original SARS‐CoV‐2 virus as well as current variants of concern (VoCs) such as the B.1.1.7 (Alpha), B.1.351 (Beta) and P.1 (Gamma) variants. [5] However, while such tests show relatively good sensitivity and specificity for high viral loads, [6] they show poor performance when the viral load is low[ 7 , 8 ] and have been shown to have very poor performance with saliva samples (sensitivity of 2–23 %),[ 9 , 10 ] restricting them to NPS samples, and thus making it necessary to have samples collected by a health‐care professional. [11] Even using NPS samples, such tests are reported to be unsuitable for screening of asymptomatic patients and are not recommended for screening or surveillance in either home or congregate settings, such as schools or long‐term care homes due to high false‐negative rates.[ 9 , 10 ]
To enable rapid tests that can be performed either by the user or those without technical lab skills, several saliva‐based test prototypes have been reported. There are several examples of both nucleic acid and antigen tests (typically nucleocapsid (N)‐protein or spike (S)‐protein), including lateral flow assays for genomic RNA[ 12 , 13 , 14 , 15 , 16 ] and S‐protein, [17] as well as electrochemical sensors for detection of genomic RNA, [18] N‐protein [19] or S‐protein.[ 20 , 21 , 22 ] However, at present these rapid saliva tests either require complicated and time‐consuming separation, [22] RNA extraction[ 15 , 16 ] or amplification steps,[ 13 , 18 , 23 ] have not been validated with clinical samples, [21] require long assay times (>1 h),[ 12 , 13 , 14 , 15 , 16 ] or do not provide adequate detection limits (<1000 viral copies per mL,[ 11 , 17 , 19 , 20 , 21 , 22 , 24 , 25 , 26 , 27 , 28 ] corresponding to a RT‐PCR cycle threshold (Ct ) of 36). [29] Given these issues, there remains a need for simple, rapid and sensitive SARS‐CoV‐2 tests, which should be possible to achieve via detection of viral surface proteins (i.e., spike proteins) directly in unprocessed saliva.
Current saliva‐based antigen tests targeting the S‐protein typically show poor sensitivity, which in part is due to the affinity of monoclonal antibodies used as the capture and detection agents, with dissociation constants (K d) ranging from 1–10 nM.[ 30 , 31 , 32 ] To address this issue, our group and others have reported on the selection of a number of DNA aptamers for the S1 subunit or receptor binding domain (RBD) of the SARS‐CoV‐2 spike protein.[ 33 , 34 , 35 , 36 , 37 , 38 ] However, all aptamers reported to date show affinity similar to antibodies (K d ranges from 2–85 nM), and as such only a few aptamer‐based rapid saliva tests for SARS‐CoV‐2 have been reported to date.[ 28 , 39 ] We hypothesized that these issues could be solved from two approaches: (1) deriving a spike‐binding DNA aptamer with extremely high affinity as the molecular recognition element, and (2) utilizing an extremely sensitive signal transduction strategy to report the viral binding by the aptamer. This rational approach was expected to overcome the loss of sensitivity reported for patient saliva samples, resulting in a rapid, simple and sensitive test for detecting SARS‐CoV‐2 from human saliva without amplification.
To derive a high‐affinity DNA aptamer, we took advantage of a well‐established strategy of engineering bivalent or multivalent aptamers through ligation of two or more monomers,[ 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 ] given the fact that the full spike protein is a homotrimeric protein and each SARS‐CoV‐2 virus expresses ≈30 spikes on its surface.[ 48 , 49 ] We hypothesized that generation of dimeric aptamers from the best members in the collection of the diverse monomeric S1 protein‐binding aptamers developed recently by our groups [38] may provide an aptamer with sufficient affinity to achieve this goal.
To develop a highly sensitive rapid test that works with real saliva samples, we chose to use Electrochemical Impedance Spectroscopy (EIS) as it is one of the most sensitive signal transduction strategies available.[ 50 , 51 ] While several methods exist to produce electrochemical signals using aptamers,[ 52 , 53 , 54 ] the EIS readout provides a rapid, single‐step detection method with high detection sensitivity using simple handheld instrumentation[ 55 , 56 ] that can be scaled up for widespread use, making it ideally suited for developing rapid COVID‐19 tests for use at home or in congregate settings.
Herein, we report on a rapid electrochemical test for SARS‐CoV‐2 using a dimeric aptamer for direct detection of the spike protein in unprocessed saliva. The rapid test, denoted as the Cov‐eChip, has been evaluated with the spike proteins of the original SARS‐CoV‐2 virus as well as its Alpha and Delta variants, and has been validated using a large number (>70) of clinical saliva samples, demonstrating performance exceeding any currently reported rapid test.
Results and Discussion
Construction of Dimeric Aptamers and Binding to the Trimeric Spike Protein of Wild‐type SARS‐CoV‐2
A number of DNA aptamers were selected against the S1 subunit of the spike protein of the wildtype SARS‐CoV‐2 virus, as reported in our previous work, [38] several of which had dissociation constants (K d) in the range of 1–10 nM. Given the fact that (1) the spike protein of SARS‐CoV‐2 is a trimeric protein made of 3 S1‐S2 monomers, (2) each viral particle of SARS‐CoV‐2 contains ≈30 spike proteins,[ 48 , 49 ] and (3) our aptamers were selected using the monomeric S1 subunit, we hypothesized that dimeric aptamers could display much higher affinity for both the full‐length trimeric spike protein and the intact virus.
We chose two M onomeric S pike‐protein binding A ptamers, denoted as MSA1 and MSA5, to build dimeric aptamers as they exhibited the highest affinity to the S1 protein. Specifically we constructed two homodimeric aptamers and one heterodimeric aptamer using MSA1T and MSA5T, the truncated minimal sequences of the two aptamers that retained full activity (sequences and proposed secondary structures are shown in Figure 1 A; the sequences of all DNA oligonucleotides are shown in the Table S1). [38] These dimeric aptamers were generated by linking the MSA1T and MSA5T with a polythymidine (polyT) linker and are named DSA1N1, DSA5N5 and DSA1N5 (DSA: D imeric S pike binding DNA A ptamer; N: and; 1: MSA1T; 5: MSA5T).
Figure 1.

A) The secondary structures of truncated aptamers MSA1T and MSA5T, as well as dimeric aptamers DSA1N1, DSA1N5 and DSA5N5 built with MSA1T, MSA5T and 30‐mer polythymidine linker (T30). B) Affinity tests and K d (nM) values of the aptamers binding to the wild‐type trimeric spike protein (Wuhan variant, WHTS), the UK variant trimeric spike protein (UKTS) and the Indian variant trimeric protein (INTS). The scrambled sequences of these aptamers (M1C, M5C and DMC) were also tested as negative controls.
We first examined the effect of the linkers containing 10, 15, 20, 30 and 40 thymidines on the binding affinity of DSA1N5 to the wild‐type trimeric spike protein (Wuhan variant, denoted as WHTS) using dot‐blot assays. The dimeric aptamers containing a linker of 30, 40, and 20 thymidines showed similar binding affinity (K d of 0.12, 0.14 and 0.17 nM, respectively, Figure S1), but those with 15‐ and 10‐thymidine linkers had poorer affinity (K d of 0.58 and 12.6 nM, respectively). DSA1N5 with the 30‐T linker had a binding affinity for the trimeric spike protein that is ≈99‐ and 28‐fold higher than the monomeric aptamers MSA1T and MSA5T, respectively (Table S2). The results indicate that the dimeric aptamer approach can produce aptamers with significantly enhanced affinity for the trimeric spike protein, presumably via bivalent recognition.[ 40 , 41 , 42 , 43 , 44 , 45 , 46 , 47 ] Figure 1 B shows binding curves derived from dot‐blot assays using the full trimeric spike proteins of SARS‐CoV‐2 (representative dot blot assay results are provided in Figure S2). In comparison to the substantial affinity enhancement observed for the heterodimeric aptamer DSA1N5 (≈99‐fold; Table S2), the two homodimeric aptamers showed much reduced affinity enhancements relative to their monomeric counterparts (Table S2). As controls we also tested the scrambled sequences of the dimeric and monomeric aptamers (named as DMC, M1C and M5C) and no binding was observed for any of these controls (Figure 1 B and S2).
It is interesting to note that the affinity of MSA1T for the trimeric spike protein decreased 4.2‐fold relative to the S1 subunit (K d of 11.9 nM vs. 2.8 nM; Figure S3A), but MSA5T had 2.9‐fold higher affinity for the trimeric protein (K d of 3.4 nM vs. 10.1 nM; Figure S3B), suggesting that the trimer formation affects the MSA1 binding epitope, and that MSA1 and MSA5 bind slightly different epitopes. To determine if MSA1T and MSA5T recognize the same epitope or different epitopes of the spike protein, we conducted a competition assay that used non‐radioactive MSA5T to compete with radioactive MSA1T (Figure S4). We first incubated radioactive MSA1T with S1 under the condition where MSA1T was fully bound to S1, followed by the addition of increasing concentrations of MSA5T. The results provided in Figure S4 clearly indicate that MSA5T can successfully compete with MSA1T, suggesting that they recognize the same epitope of the S1 subunit, though, as noted above, it is likely that the exact epitopes may differ slightly.
Dimeric Aptamers Binding with Spike Proteins of SARS‐Cov‐2 Variants
In addition to the spike protein from the wild‐type virus, we also tested the binding of the dimeric aptamer DSA1N5 with the spike protein of the B.1.1.7 Alpha variant that first emerged in the UK. DSA1N5 retained high affinity to the Alpha variant spike protein (denoted as UKTS, K d=0.29 nM, Figure 1 B), suggesting that the 501Y mutation did not alter the binding epitope(s) of the dimeric aptamer. [57]
Another current variant of concern is B.1.617.2 Delta variant, (also known as the Indian variant), which was first detected in India but now is in circulation around the world. [58] We investigated the binding of DSA1N5 with the spike protein of this variant and found that the dimeric aptamer still exhibited high affinity for the spike protein of this variant (denoted as INTS, K d=0.48 nM, Figure 1 B). Because the S1 subunit of this variant was also available, we carried out comparison studies to examine the binding profiles of MSA1, MSA5 and DSA1N5 for binding to the S1 subunit of the wildtype SARS‐CoV‐2 (named WH‐S1) and the Indian variant (named IN‐S1). The data provided in Figure S5 indicates that the two monomeric aptamers and the dimeric aptamer exhibited very similar binding affinities for WH‐S1 and IN‐S1. These results suggest that the dimeric aptamer can detect this hyper‐transmissible and more immune‐evasive variant. [59]
Dimeric Aptamer Binding with Pseudotyped Lentiviruses of SARS‐CoV‐2
Each viral particle of SARS‐CoV‐2 carries multiple trimeric spike proteins and the average spacing distance of adjacent spike proteins on coronaviruses has been reported to be 13–15 nm.[ 60 , 61 ] The predicted length of the dimeric aptamer DSA1N5 is greater than 15 nm. Therefore, it was expected that this aptamer would show enhanced affinity for viral particles.
To test for viral binding, we used a pseudotyped lentivirus that was engineered to express the full trimeric S‐protein of SARS‐CoV‐2.[ 62 , 63 , 64 ] Because the surface of this virus resembles that of SARS‐CoV‐2 and it can enter human cells but cannot replicate itself, it has been used as a substitute for SARS‐CoV‐2. [62] The same lentivirus that lacks the S‐protein was used as a control virus for this experiment.
Using dot blot assays, we evaluated DSA1N5 for viral recognition (Figure 2). The analysis of binding affinity showed that DSA1N5 had high affinity binding to the pseudotyped viruses carrying the wild‐type Wuhan variant spike protein (denoted as WHPV), with a K d of 2.1 pM, a 57‐fold improvement relative to the K d for the trimeric spike protein (Figure 2). The additional affinity improvement was attributed to the bivalent binding of the same aptamer to two different spike proteins on the viral surface. As controls, no binding was observed between WHPV and DMC (an inactive mutant dimeric aptamer) or between DSA1N5 with the control lentivirus that does not express the spike protein of SARS‐CoV‐2 (CV).
Figure 2.

Binding between DSA1N5 and pseudotyped lentiviruses expressing the spike of SARS‐CoV‐2. CV: control lentivirus, WHPV and UKPV: lentiviruses pseudotyped with the spike protein of the original Wuhan virus and the UK variant. WHTS and UKTS: trimeric spike protein of the wild‐type Wuhan virus and the UK variant. DMC: inactive mutant dimeric aptamer control.
DSA1N5 showed similar affinity (2.3 pM) for the pseudotyped lentiviruses expressing the B.1.1.7 Alpha variant of the spike protein (UKPV), demonstrating that the dimeric DSA1N5 aptamer should be able to bind both the Wuhan and Alpha variants of SARS‐CoV‐2, producing a 126‐fold improvement relative to the K d for the trimeric Alpha variant spike protein (Figure 2).
Generation and Characterization of Aptamer‐Modified Electrodes
To produce Cov‐eChips, terminal thiol modified aptamer DSA1N5‐SH was bound to a gold electrode, which was backfilled with thiol terminated PEG 6000 to minimize non‐specific adsorption on the electrode surface. The data on the characterization of the electrode are shown in Figure S6. Gold electrodes were first cleaned and evaluated for reproducibility using cyclic voltammetry (Figure S6A). The overlapping reduction and oxidation peaks demonstrate the reproducibility of the electrodes. Following each step of derivatization, the electrodes were evaluated using EIS and analyzed using the equivalent circuit model shown in Figure S7A, with the addition of the aptamer and PEG both showing an increase in the charge transfer resistance (R ct) due to the presence of passivating materials blocking the surface diffusion of redox reagents in the measurement solution (Figure S6B and Figure S7B). Note that the increase in R ct validates that the functionalization occurred, and all Cov‐eChips were validated by this method prior to use. EIS was further utilized to optimize the concentration of the aptamer (Figure S6C), demonstrating an optimal concentration of 2 μM for the aptamer solution. The density of the aptamer on the electrode surface was determined using chrono‐coulometry (Figure S6D), providing a surface density value of (1.3±0.2)×1014 aptamers per cm2. Note that the overlap in the graphs used for measuring the aptamer surface density also indicates that the aptamer functionalization is very uniform.
Electrochemical Detection of Spike Proteins
The Cov‐eChip sensor functionalized with DSA1N5 on the working electrode was used to measure the change in charge transfer resistance (denoted as the change in resistance over the initial resistance (ΔR ct/R ct)) of the Cov‐eChip at different concentrations of the spike protein WHTS. In this case, the functionalized Cov‐eChip acts as an electrochemical transducer that translates the binding of the spike protein to the aptamer‐modified electrode to an increase in electrochemical impedance (Figure 3 A). Initial studies evaluated the incubation time required for optimal detection of WHTS (Figure 3 B), indicating an incubation time of as little as 5 min could produce a measurable signal with minimal background. Longer incubation times produced higher signals but also increased the background, and thus a 5 min incubation time was selected.
Figure 3.

A) Schematic of the electrochemical assay for the detection of SARS‐CoV‐2 using spike protein aptamer. After incubation with the viral target, the charge transfer resistance increases due to surface blocking of the redox reaction of the Fe2+/Fe3+ ions. B) The changes in the charge transfer resistance measured in the redox solution containing Fe+2/Fe+3 ions at different time interval (5, 10, 20 minutes) tested with and without 40 fM trimeric spike protein. C) Nyquist plot of the different concentrations of trimeric spike protein spiked in buffer incubated on the chip for 5 min. D) Calibration plot of the different concentrations of three trimeric spike proteins, WHTS, UKTS and INTS. Dotted line indicates the mean signal change for the buffer without protein load (n=3). The points represent the mean of the signal change calculated using charge transfer resistance (R ct) extracted from Nyquist plot (R ctf−R cti)/R cti) for a given sample. The error bars represent the standard deviation from the mean obtained using three (n=3) separate devices per sample.
We then tested the detection of WHTS in binding buffer, which resulted in an enhanced R ct as WHTS concentrations increased, indicating reduced access of the redox reagents to the working electrode surface (Figure 3 C). Figure 3 D shows the changes in ΔR ct/R ct versus the concentration of WHTS, UKTS and INTS. For these three proteins, the log‐linear range was 4 fM to 4.4 pM with a limit of detection (LOD) of 1 fM for WHTS, 2.8 fM for UKTS and 3.6 fM for INTS (Figure 3 D). The sensitivity of the device for the WHTS (0.93), measured as the signal change (ΔR ct/R ct) per log of concentration, however, is considerably higher than for UKTS (0.50) and INTS (0.65). Even so, the results clearly show that the electrochemical assay can detect WHTS, UKTS and INTS. More importantly, the excellent LOD values produced by DSA1N5‐Cov‐eChip suggest that the dimeric sensor system should allow for detection of the SARS‐CoV‐2 virus in the clinically relevant concentration range.
We also examined the ability of the Cov‐eChip functionalized with monomeric aptamer MSA1T or MSA5T to detect WHTS (Figure S8). The LOD values were found to be 462 fM and 52 fM respectively, which were much worse than the LOD of 1 fM for the dimeric aptamer Cov‐eChip. Additionally, the sensitivity for MSA1T‐Cov‐eChip and MSA5T‐Cov‐eChip is 0.11 and 0.17, respectively, compared to 0.94 observed for DSA1N5‐Cov‐eChip. These results clearly indicate that the dimeric aptamer provides far superior detection sensitivity relative to its composite monomeric aptamers.
Electrochemical Detection of Pseudotyped Lentiviruses in Saliva Samples
The performance of DSA1N5‐Cov‐eChip was further validated using EIS to test human saliva samples (with a 1:1 dilution) spiked with various concentrations of WHPV (Nyquist plots and electrochemical parameters are shown in Figures S9, panel A and B). A log‐linear dynamic range was exhibited in the 103 cp mL−1 to 105 cp mL−1 range and an LOD of 1000 cp mL−1 was calculated based on 3σ of the background error (Figure 4 A), again demonstrating superior detection sensitivity relative to the dot‐blot assay. We also assessed the ability of DSA1N5‐Cov‐eChip to detect UKPV (Figure 4 A). The result indicates that DSA1N5‐Cov‐eChip was capable of recognizing this variant PV, but with a reduced detection limit (5000 cp mL−1; a loss of over 5‐fold), but still on par with the LOD of other rapid tests for the WH variant.[ 17 , 19 , 20 , 21 , 24 , 25 , 28 , 65 ] The LOD of DSA1N5‐Cov‐eChip for WHPV was 2–3 orders of magnitude better than the monomeric aptamer sensors MSA1T‐Cov‐eChip (106 cp mL−1) and MSA5T‐Cov‐eChip (105 cp mL−1; Figure S10).
Figure 4.

A) Calibration plot of the different concentrations of the WH and UK pseudotyped virus spiked in diluted pooled saliva. Dotted line indicates the signal change for the diluted pooled saliva without viral load. B) Control experiments done with diluted pooled saliva, mutant aptamer functionalized chip incubated with 103 cp mL−1 pseudotyped virus spiked diluted pooled saliva, Interleukin‐6–1000 pg mL−1 spiked diluted pooled saliva, Streptavidin‐1000 pg mL−1 spiked diluted pooled saliva, S. aureus‐105 CFU mL−1 spiked diluted pooled saliva, control lentivirus‐105 cp mL−1 spiked diluted pooled saliva, pseudotyped virus (103–104 cp mL−1) spiked diluted pooled saliva. C) Method for collection of patient saliva, patient sample treatment was done by heating the collected sample at 65 °C for 30 min followed by diluting in binding buffer (1:1 v/v %). D) Charge transfer resistance for 25 COVID‐19 negative saliva samples before and after spiking with 104 cp mL−1 Wuhan pseudotyped virus. The error bars represent the standard deviation from the mean obtained using three (n=3) separate devices per sample.
DSA1N5‐Cov‐eChip was also challenged with other proteins (IL‐6, IgG, Streptavidin), a control lentivirus not expressing spike proteins, and a bacterium (S. aureus) spiked in human saliva to study the non‐specific interaction of the Cov‐eChip with non‐target samples (Figure 4 B). The signal measured in response to the WHPV (≥103 cp mL−1) was significantly higher than measured with potentially interfering species, demonstrating the capability of the aptamer to bind with the SARS‐CoV‐2 spike protein with high selectivity.
Another cross‐reactivity study was done using a series of negative patient saliva samples that were collected as shown in Figure 4 C. In this case, the saliva samples were first spiked with 104 cp mL−1 of pseudotyped virus and then heat inactivated by treating them at 65 °C for 30 min to ensure operator safety and diluted 1:1 with binding buffer. As shown in Figure S11, the heating step causes a small decrease in the measured signal (≈20 %) and an increase in measurement error but enables safer sample handling of clinical samples in the laboratory. While the negative saliva samples showed a relatively variable initial charge transfer resistance (0.8±0.3), all samples showed substantial increases in the signal after spiking, with an average increase in charge transfer resistance of 0.9±0.4 observed following the spiking of the saliva with the pseudotyped virus (Figure 4 D). These results clearly show that, although the variability of patient samples impacts the measured background signals, it is feasible to use DSA1N5‐Cov‐eChip assay for detecting low levels of virus even in heterogeneous clinical saliva specimens.
Validation of the Cov‐eChip Saliva Assay with Clinical Samples
To assess the clinical sensitivity and specificity of the Cov‐eChip saliva assay, we examined a panel of 73 patient saliva samples (Table S3; 36 positives including 3 Alpha variants and 2 Delta variants; 37 negatives according to the nasopharyngeal swab RT‐PCR results, and in line with the sample size requirements for obtaining emergency use authorization [66] ) in a blinded study. The test procedure included: (1) sample dilution in binding buffer to achieve 50 % saliva content; (2) sample incubation on DSA1N5‐Cov‐eChip (5 min); (3) washing the chip with binding buffer (1 min); (4) acquiring the EIS signal readout in redox buffer (2 min) using a mobile‐operated potentiostat the size of a USB stick (Figure 5 A). Hence, the sample‐to‐readout time, including sample processing, is under 10 min.
Figure 5.

A) Schematic of the assay. Following sample dilution, it is added to the electrode and incubated for 5 min at room temperature, washed for 1 min and scanned for 2 min using a handheld mobile‐operated potentiostat. B) Signal change measured on the Cov‐eChip using 36 clinically‐obtained COVID‐19 positive (red‐Wuhan variant, purple‐ UK variant) and 37 negative (green) patient saliva samples. Dotted line indicates the cut‐off point for the assay. The bars represent the mean of the signal change calculated using charge transfer resistance (R ct) extracted from Nyquist plot (R ctf−R cti)/R cti) for a given sample. The error bars represent the standard deviation from the mean obtained using three (n=3) separate devices per sample. C) Box and whisker plot showing distribution of the COVID‐19 positive (red, purple, black, representing the original virus, Alpha and Delta variants, respectively) and negative (green) patient saliva samples presented in (B).
Testing of the patient saliva samples collected from patients diagnosed with SARS‐CoV‐2 (positive) and those without the infection (negative) on the Cov‐eChip showed that the ΔR CT/R CT values were clearly different between these groups: 0.5–1.2 for the negative group and 0.5–3.8 for the positive group (Figure 5 B; the ΔR CT/R CT values measured for the saliva sample from each of the 73 patients are presented in Figure 5 C). We determined the clinical performance characteristics of this assay using the Receiver Operator Characteristics (ROC) curve (See Figure S12). [67] The overall accuracy, or area under the curve (AUC) was 0.923 (CI, 0.860–0.985) with an optimum sensitivity of 80.5 % (true positive cases detected) at a threshold of 1.27 ΔR ct/R ct and a corresponding specificity of 100 % (no false positive cases detected). These diagnostic performance characteristics meet the FDA requirements for sensitivity (≥80 %) and specificity (99 %) for home‐based antigen tests[ 68 , 69 ] and surpasses the performance of many other antigen based rapid tests for detecting SARS‐CoV‐2 in saliva (Table S4), none of which tested a minimum of 30 positive and negative samples as required by FDA regulations. Furthermore, this is the first report of any rapid tests being able to detect patients infected with the Alpha and Delta variants as positive for SARS‐CoV‐2.
The determination of all our COVID‐19 positive samples was made based on nasopharyngeal swab (NPS) RT‐PCR data, which tests for viral RNA, while our test is designed to detect the spike protein on the viral particles in saliva. Therefore, it might be possible that even though the NPS test was positive, there were not sufficient viruses in the saliva to be detected by the Cov‐eChip assay. Although saliva samples were collected a median of five days post NPS collection, this was not associated with the false‐negative results. Another possibility is that there were unknown inhibitory factors in the saliva that prevented the recognition of the spike protein by the aptamer in those samples that were determined to be positive by NPS RT‐PCR. To test the latter hypothesis, we further examined six false‐negative samples (16, 26, 54, 66, 62 and 159) by spiking the WHPV into these samples at a concentration of 106 cp mL−1. At this concentration, the Cov‐eChip assay should provide a convincing positive signal. However, as the data provided in Figure S13 show, none of these spiked samples produced a significantly enhanced signal over the unspiked samples. These results strongly suggest that there were indeed inhibitory factors in these samples that prevented the aptamer recognition. Since these saliva samples were donated by hospitalized COVID patients, there is a good possibility that these saliva samples may contain spike‐binding neutralizing antibodies, which can prevent the interaction of the aptamer with the spike protein.[ 70 , 71 ] Saliva is also a highly diverse and variable fluid containing many proteins with a principal one being mucin, a heavily glycosylated protein. [72] Functionally, mucins provide lubrication and a barrier in the oral cavity against bacteria, viruses and environmental contaminants. These virus traps could therefore be another mechanism of interference to hinder aptamer binding. [73]
Conclusion
Given the ongoing surges in COVID‐19 cases coupled with increasing reports of more highly transmissible and infective variants of concern, the need for a simple and rapid saliva‐based SARS‐CoV‐2 test is more critical than ever. Herein, we have addressed this need by developing a new electrochemical sensor based on a dimeric DNA aptamer that can selectively bind the spike protein of both the Wuhan, Indian Delta and UK Alpha variants of the virus with high affinity, allowing for detection of the SARS‐CoV‐2 virus directly in patient saliva samples in under 10 min with a clinical sensitivity of 80.5 % and specificity of 100 %. To the best of our knowledge, this is the first study that integrates a high affinity dimeric DNA aptamer with an electrochemical test to produce ultrasensitive rapid antigen tests, the first example of an aptamer‐based antigen test for SARS‐CoV‐2 that is validated with a large number of unprocessed, real clinical saliva samples, and the first antigen test for detection of key variants of concern of SARS‐CoV‐2 including the Alpha and Delta variants. The aptamer‐based electrochemical sensor outperforms all current commercial and published rapid tests for SARS‐CoV‐2, including those that utilize NPS samples. The ability to detect both the original as well as Alpha and Delta variants of the virus in an easily obtained saliva sample and to run the test without the need for complicated sample processing steps should allow implementation of the test in a variety of settings, including congregate settings (schools, long‐term care homes), airports, arenas, or even as a home‐based test run by the test subject. Another advantage of the rapid test is the ability to perform diagnostic tests over time for the same patient to follow disease progression and determine when the virus is cleared. [74] It is well known that the viral load peaks several days post‐infection and can remain above the infectious threshold for up to a month following infection, [75] hence such a longitudinal assessment can be useful both for catching infections early when viral load is still low, and also ensuring that patients isolate for a sufficient period so as to not prematurely re‐enter the community. The use of DNA aptamers as recognition elements is expected to make it relatively easy to adapt the test for future variants of concern by selecting new aptamers against variant S1 proteins followed by evaluation of the highest affinity dimeric constructs, allowing the test to be rapidly deployed to allow screening of new variants.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
Funding for this work was provided by the Canadian Institutes for Health Research (CIHR; Grant No. 446898), the Canadian Foundation for Innovation (CFI, grant No. 20339), the Ontario Ministry for Research and Innovation (ORF‐RE; Grant No. RE07‐045), the Weston Foundation Microbiome Initiative and the McMaster University COVID‐19 Research Fund. MSM was supported, in part, by a CIHR New Investigator Award and an Ontario Research Fund Early Researcher Award. LS holds the Canada Research Chair (CRC) in Miniaturized Biomedical Devices and an Ontario Early Researcher Award. JDB holds the CRC in Point‐of‐Care Diagnostics.
Z. Zhang, R. Pandey, J. Li, J. Gu, D. White, H. D. Stacey, J. C. Ang, C.-J. Steinberg, A. Capretta, C. D. M. Filipe, K. Mossman, C. Balion, M. S. Miller, B. J. Salena, D. Yamamura, L. Soleymani, J. D. Brennan, Y. Li, Angew. Chem. Int. Ed. 2021, 60, 24266.
Contributor Information
Prof. Dr. Leyla Soleymani, Email: soleyml@mcmaster.ca.
Prof. Dr. John D. Brennan, Email: brennanj@mcmaster.ca.
Prof. Dr. Yingfu Li, Email: liying@mcmaster.ca.
References
- 1. Mina M. J., Andersen K. G., Science 2021, 371, 126–127. [DOI] [PubMed] [Google Scholar]
- 2. Dinnes J., Deeks J. J., Berhane S., Taylor M., Adriano A., et al., Cochrane Database Syst. Rev. 2021, 3, CD013705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hosseini A., Pandey R., Osman E., Victorious A., Li F., Didar T., Soleymani L., ACS Sens. 2020, 5, 3328–3345. [DOI] [PubMed] [Google Scholar]
- 4. Basu A., Zinger T., Inglima K., Woo K., Atie O., Yurasits L., See B., Aguero-Rosenfeld M. E., J. Clin. Microbiol. 2020, 58, e01136-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.O. L. Akingba, K. Sprong, D. R. Hardie, medRxiv 2021, 2021.02.03.21251057.
- 6. Gremmels H., Winkel B. M. F., Schuurman R., Rosingh A., Rigter N. A. M., Rodriguez O., Ubijaan J., Wensing A. M. J., Bonten M. J. M., Hofstra L. M., EClinicalMedicine 2021, 31, 100677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bulilete O., Lorente P., Leiva A., Carandell E., Oliver A., Rojo E., Pericas P., Llobera J., J. Infect. 2021, 82, 391–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Linares M., Pérez-Tanoira R., Carrero A., Romanyk J., Pérez-García F., Gómez-Herruz P., Arroyo T., Cuadros J., J. Clin. Virol. 2020, 133, 104659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Stokes W., Berenger B. M., Portnoy D., Scott B., Szelewicki J., et al., Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 1721–1726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Agulló V., Fernández-González M., de la Tabla V. O., Gonzalo-Jiménez N., García J. A., Masiá M., Gutiérrez F., J. Infect. 2021, 82, 186–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schwartz K. L., McGeer A. J., Bogoch I. I., CMAJ 2021, 193, E449-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu S., Nimse S. B., Kim J., Song K.-S., Kim T., Anal. Chem. 2020, 92, 14139–14144. [DOI] [PubMed] [Google Scholar]
- 13. Azmi I., Faizan M. I., Kumar R., Raj Yadav S., Chaudhary N., et al., Front. Cell. Infect. Microbiol. 2021, 11, 632646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang D., He S., Wang X., Yan Y., Liu J., et al., Nat. Biomed. Eng. 2020, 4, 1150–1158. [DOI] [PubMed] [Google Scholar]
- 15. Zhang C., Zheng T., Wang H., Chen W., Huang X., Liang J., Qiu L., Han D., Tan W., Anal. Chem. 2021, 93, 3325–3330. [DOI] [PubMed] [Google Scholar]
- 16. Zhu X., Wang X., Li S., Luo W., Zhang X., Wang C., Chen Q., Yu S., Tai J., Wang Y., ACS Sens. 2021, 6, 881–888. [DOI] [PubMed] [Google Scholar]
- 17. Lee J.-H., Choi M., Jung Y., Lee S. K., Lee C.-S., Kim J., Kim J., Kim N. H., Kim B.-T., Kim H. G., Biosens. Bioelectron. 2021, 171, 112715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chaibun T., Puenpa J., Ngamdee T., Boonapatcharoen N., Athamanolap P., O'Mullane A. P., Vongpunsawad S., Poovorawan Y., Lee S. Y., Lertanantawong B., Nat. Commun. 2021, 12, 802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Torrente-Rodríguez R. M., Lukas H., Tu J., Min J., Yang Y., Xu C., Rossiter H. B., Gao W., Matter 2020, 3, 1981–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yousefi H., Mahmud A., Chang D., Das J., Gomis S., et al., J. Am. Chem. Soc. 2021, 143, 1722–1727. [DOI] [PubMed] [Google Scholar]
- 21. Zakashansky J. A., Imamura A. H., Salgado D. F., Mercieca H. C. R., Aguas R. F. L., Lao A. M., Pariser J., Arroyo-Currás N., Khine M., Anal. Methods 2021, 13, 874–883. [DOI] [PubMed] [Google Scholar]
- 22. Fabiani L., Saroglia M., Galatà G., De Santis R., Fillo S., et al., Biosens. Bioelectron. 2021, 171, 112686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhu X., Wang X., Han L., Chen T., Wang L., Li H., Li S., He L., Fu X., Chen S., Xing M., Chen H., Wang Y., Biosens. Bioelectron. 2020, 166, 112437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Seo G., Lee G., Kim M. J., Baek S.-H., Choi M., et al., ACS Nano 2020, 14, 5135–5142. [DOI] [PubMed] [Google Scholar]
- 25.S. Mahari, A. Roberts, D. Shahdeo, S. Gandhi, bioRxiv 2020, 2020.04.24.059204.
- 26. Jääskeläinen A. E., Ahava M. J., Jokela P., Szirovicza L., Pohjala S., Vapalahti O., Lappalainen M., Hepojoki J., Kurkela S., J. Clin. Virol. 2021, 137, 104785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mojsoska B., Larsen S., Olsen D. A., Madsen J. S., Brandslund I., Alatraktchi F. A., Sensors 2021, 21, 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Singh N. K., Ray P., Carlin A. F., Magallanes C., Morgan S. C., Laurent L. C., Aronoff-Spencer E. S., Hall D. A., Biosens. Bioelectron. 2021, 180, 113111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Broughton J. P., Deng X., Yu G., Fasching C. L., Servellita V., et al., Nat. Biotechnol. 2020, 38, 870–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tian X., Li C., Huang A., Xia S., Lu S., Shi Z., Lu L., Jiang S., Yang Z., Wu Y., Ying T., Emerging Microbes Infect. 2020, 9, 382–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hurlburt N. K., Seydoux E., Wan Y.-H., Edara V. V., Stuart A. B., Feng J., Suthar M. S., McGuire A. T., Stamatatos L., Pancera M., Nat. Commun. 2020, 11, 5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Guo Y., Kawaguchi A., Takeshita M., Sekiya T., Hirohama M., Yamashita A., Siomi H., Murano K., J. Biol. Chem. 2021, 296, 100346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Song Y., Song J., Wei X., Huang M., Sun M., Zhu L., Lin B., Shen H., Zhu Z., Yang C., Anal. Chem. 2020, 92, 9895–9900. [DOI] [PubMed] [Google Scholar]
- 34. Sun M., Liu S., Wei X., Wan S., Huang M., et al., Angew. Chem. Int. Ed. 2021, 60, 10266–10272; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 10354–10360. [Google Scholar]
- 35. Schmitz A., Weber A., Bayin M., Breuers S., Fieberg V., Famulok M., Mayer G., Angew. Chem. Int. Ed. 2021, 60, 10279–10285; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 10367–10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu X., Wang Y., Wu J., Qi J., Zeng Z., et al., Angew. Chem. Int. Ed. 2021, 60, 10273–10278; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 10361–10366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liu R., He L., Hu Y., Luo Z., Zhang J., Chem. Sci. 2020, 11, 12157–12164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li J., Zhang Z., Gu J., Stacey H. D., Ang J. C., et al., Nucleic Acids Res. 2021, 49, 7267–7279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Deng J., Tian F., Liu C., Liu Y., Zhao S., Fu T., Sun J., Tan W., J. Am. Chem. Soc. 2021, 143, 7261–7266. [DOI] [PubMed] [Google Scholar]
- 40. Zhou J., Rossi J. J., Chem. Biol. 2008, 15, 644–645. [DOI] [PubMed] [Google Scholar]
- 41. McNamara J. O., Kolonias D., Pastor F., Mittler R. S., Chen L., Giangrande P. H., Sullenger B., Gilboa E., J. Clin. Invest. 2008, 118, 376–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Hughes Q., Le B., Gilmore G., Baker R., Veedu R., Molecules 2017, 22, 1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kuai H., Zhao Z., Mo L., Liu H., Hu X., Fu T., Zhang X., Tan W., J. Am. Chem. Soc. 2017, 139, 9128–9131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Zhou Y., Qi X., Liu Y., Zhang F., Yan H., ChemBioChem 2019, 20, 2494–2503. [DOI] [PubMed] [Google Scholar]
- 45. Riccardi C., Napolitano E., Musumeci D., Montesarchio D., Molecules 2020, 25, 5227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Torabi R., Ranjbar R., Halaji M., Heiat M., Mol. Cell. Probes 2020, 53, 101636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lin M., Zhang J., Wan H., Yan C., Xia F., ACS Appl. Mater. Interfaces 2021, 13, 9369–9389. [DOI] [PubMed] [Google Scholar]
- 48. Ke Z., Oton J., Qu K., Cortese M., Zila V., et al., Nature 2020, 588, 498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yao H., Song Y., Chen Y., Wu N., Xu J., et al., Cell 2020, 183, 730–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chiriacò M. S., Primiceri E., Monteduro A. G., Bove A., Leporatti S., Capello M., Ferri-Borgogno S., Rinaldi R., Novelli F., Maruccio G., Lab Chip 2013, 13, 730–734. [DOI] [PubMed] [Google Scholar]
- 51. Bertok T., Lorencova L., Chocholova E., Jane E., Vikartovska A., Kasak P., Tkac J., ChemElectroChem 2019, 6, 989–1003. [Google Scholar]
- 52. Xiao Y., Piorek B. D., Plaxco K. W., Heeger A. J., J. Am. Chem. Soc. 2005, 127, 17990–17991. [DOI] [PubMed] [Google Scholar]
- 53. Zuo X., Song S., Zhang J., Pan D., Wang L., Fan C., J. Am. Chem. Soc. 2007, 129, 1042–1043. [DOI] [PubMed] [Google Scholar]
- 54. Xiang Y., Lu Y., Nat. Chem. 2011, 3, 697–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cai H., Lee T. M.-H., Hsing I.-M., Sens. Actuators B 2006, 114, 433–437. [Google Scholar]
- 56. Traynor S. M., Pandey R., Maclachlan R., Hosseini A., Didar T. F., Li F., Soleymani L., J. Electrochem. Soc. 2020, 167, 037551. [Google Scholar]
- 57.F. Tian, B. Tong, L. Sun, S. Shi, B. Zheng, Z. Wang, X. Dong, P. Zheng, bioRxiv 2021, 2021.02.14.431117.
- 58. Campbell F., Archer B., Laurenson-Schafer H., Jinnai Y., Konings F., et al., Euro Surveill 2021, 26, pii=2100509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Wall E. C., Wu M., Harvey R., Kelly G., Warchal S., et al., Lancet 2021, 397, 2331–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Bar-On Y. M., Flamholz A., Phillips R., Milo R., eLife 2020, 9, e57309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Neuman B. W., Adair B. D., Yoshioka C., Quispe J. D., Orca G., Kuhn P., Milligan R. A., Yeager M., Buchmeier M. J., J. Virol. 2006, 80, 7918–7928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Crawford K. H. D., Eguia R., Dingens A. S., Loes A. N., Malone K. D., et al., Viruses 2020, 12, 513. [Google Scholar]
- 63. Chen X., Li R., Pan Z., Qian C., Yang Y., et al., Cell. Mol. Immunol. 2020, 17, 647–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ou X., Liu Y., Lei X., Li P., Mi D., et al., Nat. Commun. 2020, 11, 1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Mahshid S. S., Flynn S. E., Mahshid S., Biosens. Bioelectron. 2021, 176, 112905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.“COVID-19 antigen testing devices: Notice on minimum value for sensitivity,” can be found under https://www.canada.ca/en/health-canada/services/drugs-health-products/covid19-industry/medical-devices/testing/antigen.html.
- 67. Zweig M. H., Campbell G., Clin. Chem. 1993, 39, 561–577. [PubMed] [Google Scholar]
- 68.“Policy for Coronavirus Disease-2019 Tests During the Public Health Emergency (Revised),” can be found under https://www.fda.gov/regulatory-information/search-fda-guidance-documents/policy-coronavirus-disease-2019-tests-during-public-health-emergency-revised.
- 69.“Emergency Use Authorization of Medical Products and Related Authorities,” can be found under https://www.fda.gov/regulatory-information/search-fda-guidance-documents/emergency-use-authorization-medical-products-and-related-authorities.
- 70. Wajnberg A., Amanat F., Firpo A., Altman D. R., Bailey M. J., et al., Science 2020, 370, 1227–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Isho B., Abe K. T., Zuo M., Jamal A. J., Rathod B., et al., Sci. Immunol. 2020, 5, 10.1126/sciimmunol.abe5511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.“Human Salivary Proteome Wiki,” can be found under https://salivaryproteome.nidcr.nih.gov/public/index.php/Main Page.
- 73. Ojha Y. R., Giovannucci D. R., Cameron B. D., J. Biotechnol. 2021, 327, 9–17. [DOI] [PubMed] [Google Scholar]
- 74. Whitman J. D., Hiatt J., Mowery C. T., Shy B. R., Yu R., et al., Nat. Biotechnol. 2020, 38, 1174–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Wyllie A. L., Fournier J., Casanovas-Massana A., Campbell M., Tokuyama M., et al., N. Engl. J. Med. 2020, 383, 1283–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information