

Abstract
Victor McKusick's many contributions to medicine are legendary, but his magnum opus is Mendelian Inheritance in Man (MIM), his catalog of Mendelian phenotypes and their associated genes. The catalog, originally published in 1966 in book form, became available on the internet as Online Mendelian Inheritance in Man (OMIM®) in 1987. The first of 12 editions of MIM included 1486 entries; this number has increased to over 25,000 entries in OMIM as of April 2021, which demonstrates the growth of knowledge about Mendelian phenotypes and their genes through the years. OMIM now has over 20,000 unique users a day, including users from every country in the world. Many of the early decisions made by McKusick, such as to maintain MIM data in a computer‐readable format, to separate phenotype entries from those for genes, and to give phenotypes and genes MIM numbers, have proved essential to the long‐term utility and flexibility of his catalog. Based on his extensive knowledge of genetics and vision of its future in the field of medicine, he developed a framework for the capture and summary of information from the published literature on phenotypes and their associated genes; this catalog continues to serve as an indispensable resource to the genetics community.
Keywords: gene‐phenotype relationship, history of medicine, Mendelian Inheritance in Man, OMIM, single‐gene conditions
1. INTRODUCTION
Among the many contributions of Victor A. McKusick to medicine is his magnum opus, Mendelian Inheritance in Man (MIM). First published in book form in 1966 (McKusick, 1966), this curated, authoritative catalog of Mendelian phenotypes, based on comprehensive review of the peer‐reviewed literature, quickly became a necessary reference guide for clinicians and researchers who eagerly awaited the next edition of the book. The first edition of MIM was a modest 344 pages with 1486 entries and served a relatively narrow audience, as the field of medical genetics was just beginning. By the time the last (12th) edition of the book was published (McKusick, 1998), MIM had been translated into Spanish, Russian, and Chinese; had become available online as Online Mendelian Inheritance in Man (OMIM®); and had become an essential resource for the genetics community. The number of entries in OMIM has increased dramatically, with 25,804 entries (16,478 genes and 9326 phenotypes) as of April 6, 2021 (Table1 ). OMIM has been updated daily since it debuted on the internet in 1987, and focuses primarily on the relationship between genes and disease.It now catalogs over 6800 phenotypes with a known molecular basis and over 4400 genes with a phenotype‐causing mutation (Figure 1 ).Today, OMIM has over 20,000 unique users per day from every country of the world and is accessed regularly by a wide range of users including clinicians, diagnosticians, bench scientists, counselors, and informaticians, as well as students of these disciplines. These data illustrate the central role that OMIM plays in the field of genetics.
TABLE 1.
OMIM statistics
| Entry class | Symbol | July 18, 2008 | April 6, 2021 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of entries by category | Number of entries by category | ||||||||||
| Autosomal | X‐linked | Y‐linked | Mitochondrial | TOTAL | Autosomal | X‐linked | Y‐linked | Mitochondrial | TOTAL | ||
| Gene | * | 11,766 | 542 | 48 | 37 | 12,394 | 15,616 | 747 | 51 | 37 | 16,451 |
| Gene and phenotype | + | 354 | 30 | 0 | 0 | 384 | 27 | 0 | 0 | 0 | 27 |
| Phenotype description, molecular basis known | # | 2119 | 200 | 2 | 26 | 2346 | 5642 | 356 | 5 | 34 | 6037 |
| Mendelian phenotype or locus, molecular basis unknown | % | 1483 | 137 | 5 | 0 | 1625 | 1412 | 112 | 4 | 0 | 1528 |
| Mainly phenotypes with suspected Mendelian basis | Null (no symbol) | 1942 | 140 | 2 | 0 | 2084 | 1656 | 102 | 3 | 0 | 1761 |
| Total | 17,667 | 1048 | 57 | 63 | 18,835 | 24,353 | 1317 | 63 | 71 | 25,804 | |
FIGURE 1.
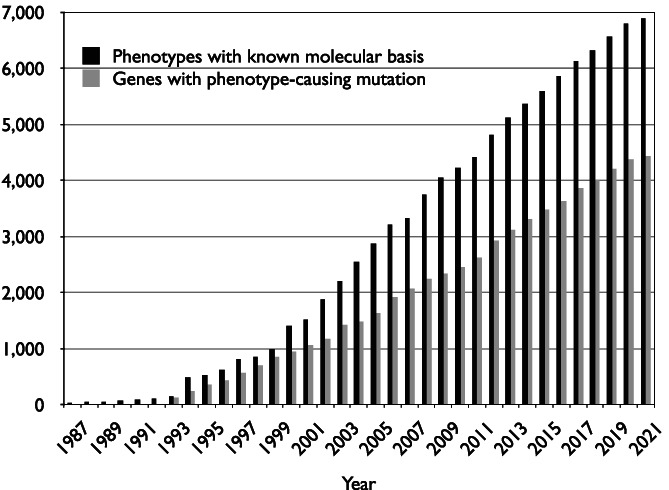
The pace of disease gene discovery as cataloged by the OMIM Morbid Map Scorecard. The current scorecard is available from https://omim.org/statistics/geneMap. As of May 22, 2021, there were over 6800 phenotypes with a known molecular basis and over 4400 genes with a phenotype‐causing mutation
2. DEVELOPMENT OF MENDELIAN INHERITANCE IN MAN/OMIM
Many geneticists might be too young to remember a time before the availability of OMIM and thus might not recognize the huge accomplishment of developing a genetics resource that has maintained its usefulness from the early days of medical genetics to the present day. Many of the early decisions made by McKusick in developing MIM and later OMIM are responsible for its long‐term value to physicians, researchers, and students for more than 50 years.
At a time when medical genetics was a new field and many in medicine saw identification of genetic syndromes as equivalent to “postage stamp collecting” (McKusick, 2006), McKusick recognized the need for an authoritative resource on Mendelian phenotypes and their associated genes. In addition to recognizing the need for such a catalog, McKusick dedicated a substantial amount of his time to author catalog entries. He served as the sole author of the catalog until 1994 when other authors were added (Pearson et al., 1994). At the time of his death in 2008, McKusick had created or contributed to 11,502 (61%) of the 18,839 entries in OMIM. As of April 5, 2021, McKusick had contributed to 45% of the more than 25,000 entries. Today a dedicated staff of writers and curators produces most of the content, although distributed authorship to subject area experts and freelance writers is also used. To date, over 80 people have contributed to OMIM.
The published biomedical literature serves as the source of OMIM information. In the 1950s and 1960s, Dr. McKusick spent many hours in the Welch Medical Library at Johns Hopkins reading articles from a broad spectrum of medical journals. He found articles by surveying IndexMedicus, a monthly serial of indexed medical articles. For many years, he relied heavily on the printed periodical Current Contents: Life Sciences edition. The portability of this periodical meant he could perform literature review anywhere. Today, OMIM uses a custom‐made online literature retrieval tool, LiTrack, which combines journal‐based article reviews reminiscent of Current Contents with automated content markup to prioritize relevant articles.
A critical early decision made by McKusick was to maintain MIM data in a computer‐readable format. Although it now seems impossible to think of OMIM outside of a computer environment, the idea to use computers in the storage of medical data was relatively novel in 1966. McKusick understood that maintaining the catalog in a computer‐readable format would facilitate the assembly of title and author indices and ensure the integrity of the information as it was updated. The first edition of MIM wasprinted from files stored on computer tape.
McKusick also recognized the importance of making the information in MIM easily available as technological advances made online access possible. In 1979, the National Library of Medicine selected MIM as an authoritative, computer‐based knowledgebase to be used as a test‐bed for the development of IRx (Information Retrieval Experiment), a natural language retrieval algorithm. Following the success of this collaboration, the Welch Medical Library at Johns Hopkins provided online access to MIM (Online Mendelian Inheritance in Man or OMIM®) under the IRx system in 1987; thus, OMIM data was available to the scientific community 10 years before Google was founded in 1998 (McKusick, 1998). At the time of McKusick's death in 2008, OMIM was included as a resource in Entrez, the indexing and data retrieval system developed by the National Center for Biotechnology Information (NCBI). Because of funding changes, OMIM was moved to an independent website (OMIM.org) in 2011, but NCBI continues to incorporate daily updates of OMIM into their services. McKusick's organizing principles are evident in the structure of the information in OMIM.org (Figure 2). This website has optimized views and search capabilities that highlight over 50 years of rich and nuanced discussion of genes and disease (Amberger et al., 2015).
FIGURE 2.
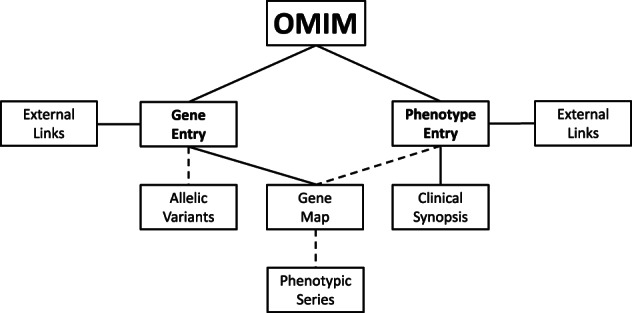
Structure of OMIM.org.Genes and phenotypes have separate entries with associated targeted external links and attributes. Dashed lines indicate that not all genes have allelic variants, not all phenotypes are mapped, and mapped phenotypes are not necessarily part of a phenotypic series
Another of McKusick's visionary decisions was to give a unique identifier (MIM number) to each catalog entry. McKusick recognized the fluid nature of language and the challenges inherent in disease nosology; some names would unavoidably change based on advances in knowledge or for social considerations. MIM numbers were the unifying and stable attribute under which preferred and alternate names could be listed, and they soon became used throughout the diverse medical literature. Today, many journals require inclusion of MIM numbers in publications. McKusick also realized OMIM's role in naming of phenotypes; in his article on the history of OMIM (McKusick, 2007), he discussed the essential nature of disease nosology and the importance of naming of phenotypes as part of OMIM's mission, a role that continues to the present day.
McKusick's adept incorporation of the principles of genetics into the structure of the catalog set a solid and flexible foundation on which MIM could develop. McKusick understood that phenotypes and genes are distinct entities that should reside in separate entries. Though initially MIM focused on Mendelian phenotypes, McKusick noted that,“From the beginning, the gene behind the phenotype was always kept in mind,” despite the fact that, for most of the first 15 years of MIM, specific genes had not been identified as responsible for most phenotypes (McKusick, 2007). While the subtitle of the first edition of MIM was “Catalogs of Autosomal Dominant, Autosomal Recessive and X‐linked Phenotypes” (McKusick, 1966), in 1994 the subtitle became “A Catalog of Human Genes and Genetic Disorders” (McKusick, 1994) to emphasize the inclusion of entries on genes. In 1990, separate entries were created for phenotypes and the genes underlying them, thus resolving issues created by genetic heterogeneity (one phenotype cause by several different genes) and phenotypic diversity (one gene causing several distinct phenotypes). More than one‐third of genes currently known to cause disease result in more than one phenotype. An interesting example of genetic heterogeneity is Weill–Marchesani syndrome (Phenotypic Series [PS]: 277600), which can be caused by pathogenic variants in four different genes and inherited as a dominant or recessive phenotype, depending on the gene. The breadth of phenotypes associated with pathogenic variants in the FBN1 gene (MIM: 134797) is a good example of phenotypic diversity. FBN1‐related disorders range from isolated ectopia lentis (MIM: 129600)to stiff skin syndrome (MIM: 184900), acromicric dysplasia (MIM: 102370), Weill–Marchesani syndrome (MIM: 608328), geleophysic dysplasia (MIM: 614185), Marfan lipodystrophy syndrome (MIM: 616914), and Marfan syndrome (MIM: 154700).
As disease gene discovery dramatically expanded, McKusick's decision to put disease‐causing variants in an allelic variant section in the gene entry was vital to OMIM's organization and structure. Each allelic variant is given a unique number linked to the MIM number for the gene, which can be cross‐referenced between entries. Variants in a single gene may cause many phenotypes depending on the specific variant and its effect on function. OMIM does not incorporate all variants in a gene: only select variants that highlight a unique variant‐phenotype duo, a different mode of inheritance, a different type of variant, or a different functional effect are included. Users of OMIM.org can find additional variant information from links to other resources including ClinVar, locus specific mutation databases, HGMD, and gnomAD.
Linkage mapping revealed genetically heterogeneous phenotypes, and McKusick created separate MIM entries for each phenotype locus, which were retained when the molecular basis was found. As more information on the clinical descriptions were added to these split phenotypes, variations in factors such as age of onset, prognosis, and distinctive features were revealed. The clinical features of each phenotype were summarized anatomically in a Clinical Synopsis table. After OMIM.org was created, the capability to view clinical synopses side‐by‐side was developed to allow a comparison of unique and overlapping clinical features. Further, genetically heterogeneous phenotypes were combined into Phenotypic Series, which allowed easy review of the genes underlying phenotypes to reveal common pathways. To facilitate molecular genetic inquiry, visualizations of phenotype‐gene relationships called PheneGene graphics were created in 2018. These interactive graphical representations are built in real time from current data and can be displayed in linear (Figure 3(a)) or radial (Figure 3(b)) formats. The dynamic views of the connections between phenotypes and genes may suggest novel biological or clinical relationships or inform treatment and prognosis.
FIGURE 3.
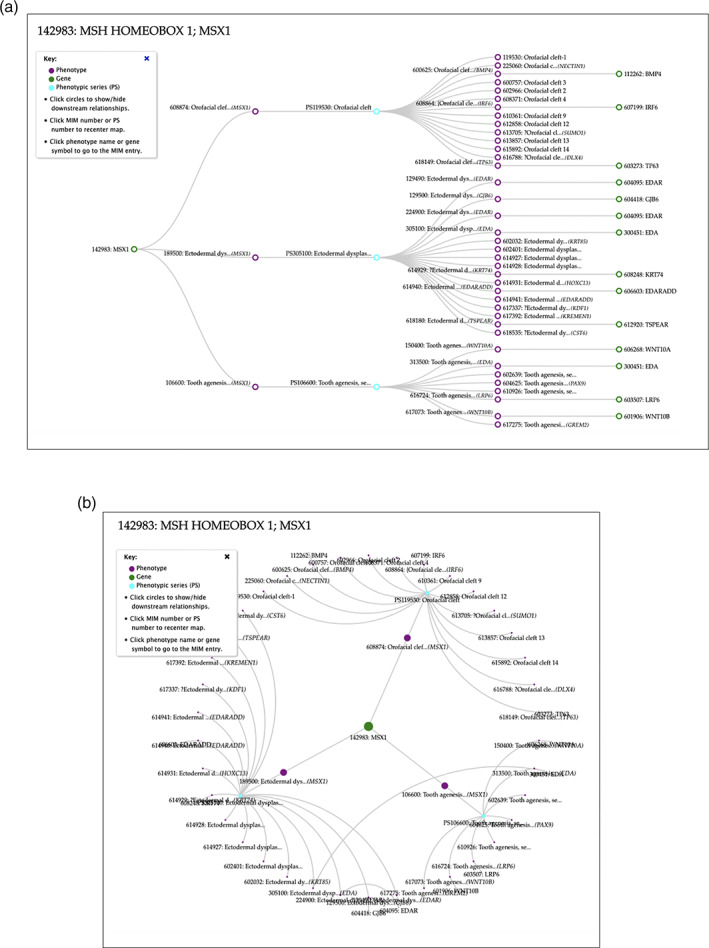
(a/b): OMIM PheneGene graphics.Linear (a) and radial (b) graphics depicting the relationship between phenotypes associated with pathogenic variants in the MSX1 gene and groups of related phenotypes (phenotypic series), and their associated genes.These relationships are not hierarchical
The human genome is made up of more than classically defined protein‐coding genes; the flexible structure of OMIM allows the description of new elements of the genome as their role in biology and disease becomes known. For example, OMIM includes separate entries for noncoding RNAs, five of which have been found to cause a phenotype. OMIM also includes information on DNA regulatory elements. For example, the ZPA regulatory sequence (ZRS), which resides in intron 5 of the LMBR1 gene (MIM: 605522), affects expression of the SHH gene (MIM: 600725). Disruption of ZRS can cause polydactyly (MIM: 174500) or syndactyly (MIM: 186200), whereas pathogenic variants in the LMBR1 gene cause a rare malformation characterized by bilateral congenital amputations of the hands and feet called acheiropody (MIM: 200500). Information on the ZRS is included in the LMBR1 gene entry to indicate where the element resides in the genome. Although deletion/duplication syndromes are not generally inherited, some of them are and are thus included in OMIM (e.g., 16p11.2 duplication syndrome; MIM: 614671).
3. USE STATISTICS
In 2000 when OMIM was exclusively served from NCBI's web services, OMIM had on average 5300 daily users. In 2020, OMIM.org serves over 20,000 daily users and over 2.7million unique users from every country in the world over the course of a year. These users range from high school, college, graduate, genetic counseling, and medical students to residents, geneticists, clinicians in other disciplines, as well as basic, translational, and computational scientists. OMIM.org is also accessed programmatically over 120,000 times a day via its application programming interface (API), which allows automated incorporation of OMIM into exome and genome analysis pipelines.
4. MOST COMMON USES OF OMIM IN 2021
What started as a book used by practitioners of an as‐yet‐to‐be defined field of medical genetics, is now a dynamic knowledgebase of the field. Today, the structured OMIM text and searching capabilities accommodate many diverse uses. Users can explore OMIM's features from the online help pages for both searching and API use or from the YouTube video tutorials for OMIM searching, MIMmatch, and GeneScout. The following are examples of the most common uses of OMIM in 2021.
Creating a differential diagnosis. This is a common use for a clinician who is seeing a patient and suspects a genetic diagnosis. As an example, one of us (Ada Hamosh) was called to see a premature baby with an intracranial hemorrhage whose parents had lost a previous child due to the same cause. In addition to the bleeding, the baby had a very small nose. Entering "“nasal hypoplasia” AND bleeding” into the search box retrieved “Vitamin K‐dependent clotting factors, combined deficiency of” (MIM: 277450). The child's facial features were similar to those seen in warfarin embryopathy (Hall et al., 1980), and while this could not establish an immediate molecular diagnosis, it led to the recognition that the infant might benefit from treatment with vitamin K. Following administration of vitamin K to the patient, the bleeding and need for more blood products resolved.
Learning about a disease. OMIM describes the clinical features of over 7500 Mendelian conditions, many of which are rare. A clinician can learn about a newly described syndrome of cataracts, spastic paraparesis, and speech delay (MIM: 619338) caused by mutations in the FAR1 gene (MIM: 616107) or discover the vast genetic heterogeneity of developmental and epileptic encephalopathy currently caused by mutation in over 95 genes. The clinical entries in OMIM have directed links to additional references in PubMed, genetic testing, newborn screening (if relevant), GeneReviews, clinical trials, and more.
Learning about a gene. With widespread use of exome sequencing to evaluate patients (Rehder et al., 2021), learning about the function of genes called in variant analysis is extremely helpful. OMIM describes over 16,400 genes, with priority given to genes related to a phenotype and those with known function. The text‐based entries with structured headings provide easy access to authoritative information about a gene, its function, and its role in disease with links to appropriate references. At the beginning of each gene entry is a table summarizing known gene‐phenotype relationships, including the phenotype name, link to the phenotype entry in OMIM, and the mode of inheritance. A reference‐plus icon at the end of each paragraph will initiate a keyword search of PubMed. Gene entries have links to genome browsers, protein, and cellular pathway resources, as well as variation databases, ClinGen dosage sensitivity and gene‐disease validity pages (www.clinicalgenome.org), and model organism databases.
Identifying genes associated with a condition. For genetically heterogeneous phenotypes, OMIM's Phenotypic Series feature provides a quick view of the underlying genes. For example, pathogenic variants in 90 genes have been identified as causing retinitis pigmentosa. OMIM can also be helpful for laboratories developing genetic testing panels, or for clinicians or genetic counselors assessing commercial gene sequencing panels offered by genetic testing laboratories. These panels interrogate multiple genes in a single test and can be a helpful tool for identifying the responsible gene for genetically heterogeneous phenotypes (Bean et al., 2020); thus an evaluation of a patient with retinitis pigmentosa might include a commercial gene sequencing panel. A query of OMIM is helpful to determine if a particular panel will identify a genetic cause by providing a list of genes known to be associated with a particular phenotype.
Identifying genes and phenotypes in a genomic interval. OMIM's gene map can be searched by genomic coordinate range to see the genes and phenotypes within an interval. If a user wants to query multiple genomic intervals in a single search, the new GeneScout tool (https://genescout.omim.org) can be used to identify genes and phenotypes residing in the regions of interest. Results from a GeneScout search can be limited to OMIM genes only, OMIM disease genes only, or OMIM genes with dominant or recessive inheritance. Results can be further filtered by clinical features and the Clinical Synopses of the retrievals can be compared. Furthermore, GeneScout can be used to compare two sets of coordinate ranges, and results can be restricted to genes in the overlapping region by selecting Intersection and to genes in the non‐overlapping regions by selecting Subtraction.
Keeping updated on entries and new phenotype‐gene relationships. Creating a MIMmatch account allows users to receive notifications of updates to OMIM on selected genes, phenotypes, or phenotypic series, as well to be informed of new gene‐phenotype relationships. Complex searches can be saved for reuse. Any relevant updates are received in one daily email. MIMmatch can also be used to identify other individuals with an interest in the same phenotype or gene.
Programmatic searching of OMIM. The OMIM website uses a robust API, which allows communication with other software programs. Exome and genome analysis pipelines programmatically interrogate OMIM to include up‐to‐date OMIM data into their interfaces to speed variant prioritization. OMIM.org has integrated medical classification coding including ICD and SNOMED codes to facilitate interoperability with a wide range of emerging medical resources.
5. CONCLUSIONS
Based on McKusick's knowledge of genetics and his vision of its future in medicine, he laid out an enduring framework for the capture and summary of information from the published literature on genes and their relevance to Mendelian phenotypes. Fifty years ago, the prevailing thought was that one gene encodes one protein or one phenotype. We now know it is not as simple as that. The gene‐phenotype relationship continues to evolve and is more nuanced and complex. Mendelian diseases have sometimes been thought of as medical rarities not worthy of study and research funding, a view that has proven to be short‐sighted. Although individually rare, the cumulative burden of genetic disorders is significant. Insights from the discovery of pathogenic variants in genes that cause rare disease inform pathophysiology and biological networks, leading to better diagnosis, treatment, and prognosis of diseases beyond the original ones studied.
Going forward, OMIM will continue to coordinate with existing and emerging efforts at gene‐disease curation, variant classification, and disease ontology, and to adapt to incorporate new discoveries and record the advances in understanding of genetic variation and its role in Mendelian phenotypes. As in the past, OMIM will adapt to ensure that it meets the needs of the ever‐expanding genetics and genomics community, the patients we serve, and the vision of Victor McKusick.
CONFLICT OF INTEREST
The authors have no relevant conflict of interest.
Hamosh, A. , Amberger, J. S. , Bocchini, C. , Scott, A. F. , & Rasmussen, S. A. (2021). Online Mendelian Inheritance in Man (OMIM®): Victor McKusick's magnum opus. American Journal of Medical Genetics Part A, 185A:3259–3265. 10.1002/ajmg.a.62407
Funding information National Human Genome Research Institute, Grant/Award Number: NIH/NHGRI U41HG006627
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.
REFERENCES
- Amberger, J. S. , Bocchini, C. A. , Schiettecatte, F. , Scott, A. F. , & Hamosh, A. (2015). OMIM.org: Online Mendelian Inheritance in Man (OMIM(R)), an online catalog of human genes and genetic disorders. Nucleic Acids Research, 43, D789–D798. 10.1093/nar/gku1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean, L. J. H. , Funke, B. , Carlston, C. M. , Gannon, J. L. , Kantarci, S. , Krock, B. L. , Zhang, S. , Bayrak‐Toydemir, P. , & ACMG Laboratory Quality Assurance Committee . (2020). Diagnostic gene sequencing panels: From design to report‐a technical standard of the American College of Medical Genetics and Genomics (ACMG). Genetics in Medicine, 22, 453–461. 10.1038/s41436-019-0666-z [DOI] [PubMed] [Google Scholar]
- Hall, J. G. , Pauli, R. M. , & Wilson, K. M. (1980). Maternal and fetal sequelae of anticoagulation during pregnancy. American Journal of Medicine, 68, 122–140. 10.1016/0002-9343(80)90181-3 [DOI] [PubMed] [Google Scholar]
- McKusick, V. A. (1966). Mendelian Inheritance in Man:Acatalog of autosomal dominant, autosomal recessive, and X‐linked phenotypes. Johns Hopkins University Press. [Google Scholar]
- McKusick, V. A. (1994). Mendelian Inheritance in Man: A catalog of human genes and genetic disorders. Johns Hopkins University Press. [Google Scholar]
- McKusick, V. A. (1998). Mendelian Inheritance in Man. A catalog of human genes and genetic disorders. The Johns Hopkins University Press. [Google Scholar]
- McKusick, V. A. (2006). A 60‐year tale of spots, maps, and genes. Annual Review of Genomics and Human Genetics, 7, 1–27. 10.1146/annurev.genom.7.080505.115749 [DOI] [PubMed] [Google Scholar]
- McKusick, V. A. (2007). Mendelian Inheritance in Man and its online version, OMIM. American Journal of Human Genetics, 80, 588–604. 10.1086/514346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson, P. , Francomano, C. , Foster, P. , Bocchini, C. , Li, P. , & McKusick, V. (1994). The status of online Mendelian inheritance in man (OMIM) medio 1994. Nucleic Acids Research, 22, 3470–3473. 10.1093/nar/22.17.3470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehder, C. , Bean, L. J. H. , Bick, D. , Chao, E. , Chung, W. , Das, S. , O'Daniel, J. , Rehm, H. , Shashi, V. , Vincent, L. M. , & ACMG Laboratory Quality Assurance Committee . (2021). Next‐generation sequencing for constitutional variants in the clinical laboratory, 2021 revision: A technical standard of the American College of Medical Genetics and Genomics (ACMG). Genetics in Medicine. 10.1038/s41436-021-01139-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analyzed during the current study.