

Abstract
Haemophilia A and B are rare bleeding disorders. Over the past decades, they have been transformed from debilitating diseases to manageable conditions in the Western world. However, optimizing haemophilia care remains challenging in developing countries. Several challenges and unmet needs remain in the treatment of the haemophilia limiting the QoL of patients. These challenges are now being addressed by extended half‐life recombinant factors, rebalancing and substitution therapies. Gene therapy and genome editing show promise for a definite clinical cure. Here, we provide an overview of new therapeutic opportunities for haemophilia and their advances and limitations from a regulatory perspective. The database on human medicines from the European Medicines Agency (EMA) was used and data from rare disease (orphan) designations and EPARs were retrieved for the analysis. Clinical trial databases were used to query all active studies on haemophilia. Gene therapy medicinal products based on AAV and lentiviral vectors are in development and clinical trials have reported substantial success in ameliorating bleeding tendency in haemophilia patients. The prospect of gene editing for correction of the underlying mutation is on the horizon and has considerable potential. With regard to the benefit of the gene therapy medicinal products, more long‐term efficacy and safety data are awaited. We are entering an era of innovation and abundance in treatment options for those affected by bleeding disorders, but issues remain about the affordability and accessibility to patients.
Keywords: gene therapy, haemophilia, marketing authorization, orphan designation, orphan drugs, rebalancing therapy, replacement therapy
1. INTRODUCTION
Haemophilia A and B are rare X chromosome‐linked monogenic disorders resulting from a deficiency in a coagulation factor in the intrinsic pathway of blood coagulation. These two diseases affect approximately 210 000 persons worldwide. 1 Developing medicines intended for small numbers of patients has little commercial incentive under normal market conditions. Therefore, the EU offers a range of incentives to encourage the development of designated orphan medicines. 2 The scope of this review is to provide an overview of the clinical development of medicinal products currently holding an orphan designation in the EU for the treatment of haemophilia A and B based on current experience and knowledge gained in the regulatory field.
Haemophilia is caused by mutations in the genes encoding for Factor VIII (haemophilia A, the more prevalent form) and Factor IX (haemophilia B), resulting in decreased production and/or function of Factor VIII and Factor IX proteins. 3 Both FVIII and FIX are naturally synthesized in the liver: FVIII in the liver sinusoidal endothelial cells (LSEC) and FIX in hepatocytes. Patients with severe haemophilia have an absence of circulating plasma FVIII or FIX activity (<1%), resulting in spontaneous or excessive bleeding into joints and muscles. If left untreated, recurrent bleedings result in the development of chronic arthropathy (knee, ankle and elbow) and early mortality from spontaneous or trauma‐induced bleeds.
Haemophilia care has undergone remarkable improvements over the past decades and haemophilia has been transformed from a debilitating disease into a manageable condition. Despite the diffused availability of safe and effective replacement therapies, haemophilia patients continue to experience enormous burden of treatment, spontaneous bleeding and progressive joint disease, as well as high rates of development of inhibitors to conventional factor products. In the past 20 years, recombinant bioengineering has led to extended half‐life (EHL) therapies with easier modes of administration. 4 There is now a change in prophylaxis for haemophilia through the development of the first substitution therapy, as well as innovative rebalancing therapies. Investigational gene therapy holds the promise for a definitive clinical cure. 5 , 6
1.1. Prevalence of haemophilia in the EU
To obtain prevalence of individuals living with haemophilia A and B in Europe, we used the World Federation of Haemophilia (WFH) 2018 survey, complemented by literature data for missing countries (Table 1). A recent meta‐analysis 7 proposed an estimate of 2.46 cases per 10 000 males for all severities of haemophilia A and 0.5 cases per 10 000 males for all severities of haemophilia B. These estimates are higher than those historically cited but still characterize haemophilia as rare diseases on the basis of definitions used in the EU (<5 cases per 10 000 persons; Regulation [EC] No. 141/2000 of the European Parliament and of the Council). 2 The management of haemophilia continues to advance with technological developments and new treatment‐related research. There is currently a general agreement on the definitions of prophylactic therapy, on‐demand therapy and immune tolerance induction therapy for haemophilia. The introduction of prophylactic treatment of haemophilia patients at an early age has gained acceptance as the optimal therapeutic option; however, prophylactic treatment as a standard has not been widely implemented in many countries because of a combination of supply limitations, access to care and financial constraints. The reported prevalence of haemophilia varies considerably between countries, especially in the developing world where some countries do not have reliable diagnosis and reporting procedures that allow for accurate identification of patients with severe, moderate or mild haemophilia.
TABLE 1.
Haemophilia A and B prevalence in European countries
| Country | Population | People with haemophilia A | Prevalence (per 10 000) | People with haemophilia B | Prevalence (per 10 000) |
|---|---|---|---|---|---|
| Albania | 2 876 101 | 161 | 0.56 | 33 | 0.11 |
| Austria | 8 747 358 | 658 | 0.75 | 117 | 0.13 |
| Belgium | 11 348 159 | 970 | 0.85 | 242 | 0.21 |
| Bulgaria | 6 981 642 | 560 | 0.80 | 68 | 0.10 |
| Cyprus | 1 172 458 | 43 | 0.37 | ‐ | ‐ |
| Czech Republic | 10 561 633 | 937 | 0.89 | 136 | 0.13 |
| Denmark | 5 731 118 | 410 | 0.72 | 102 | 0.18 |
| Estonia | 1 316 481 | 97 | 0.74 | 10 | 0.08 |
| Finland | 5 495 096 | 150 | 0.27 | 33 | 0.06 |
| France (metropolitan) | 66 896 109 | 5864 | 0.88 | 1498 | 0.22 |
| Germany | 82 667 685 | 3686 | 0.45 | 628 | 0.08 |
| Greece | 10 746 740 | 873 | 0.81 | 184 | 0.17 |
| Hungary | 9 817 958 | 893 | 0.91 | 230 | 0.23 |
| Ireland | 4 773 095 | 617 | 1.38 | 243 | 0.51 |
| Italy | 61 680 122 | 3992 | 0.65 | 886 | 0.14 |
| Latvia | 1 960 424 | 129 | 0.66 | 21 | 0.11 |
| Lithuania | 2 872 298 | 147 | 0.51 | 24 | 0.08 |
| Netherlands | 16 877 351 | 1026 | 0.61 | 125 | 0.07 |
| Norway | 5 232 929 | 325 | 0.62 | 90 | 0.17 |
| Poland | 37 948 016 | 2413 | 0.64 | 428 | 0.11 |
| Portugal | 10 324 611 | 539 | 0.52 | 112 | 0.11 |
| Romania | 19 705 301 | 1615 | 0.82 | 210 | 0.11 |
| Slovakia | 5 428 704 | 521 | 1.00 | 79 | 0.15 |
| Slovenia | 2 064 845 | 207 | 1.00 | 30 | 0.15 |
| Spain | 47 042 984 | 1679 | 0.36 | ‐ | ‐ |
| Sweden | 9 798 871 | 860 | 0.88 | 195 | 0.20 |
| United Kingdom | 65 637 239 | 6559 | 1.00 | 1518 | 0.23 |
1.2. Treatments for haemophilia
The current ‘gold‐standard’ treatment for haemophilia is prophylactic factor replacement therapy, intravenous injection of Factor VIII/IX concentrates. For prophylaxis, several medicines are available in Europe (Table 2), with the aim of raising FVIII and FIX activity above a detectable level (>1%) in order to prevent bleedings. The short biological half‐lives of FVIII and FIX proteins require frequent infusions, three times a week (haemophilia A) or twice a week (haemophilia B). 8 A major complication of factor replacement therapy is the formation of inhibitory antibodies against the coagulation factors, rendering these ineffective. 9
TABLE 2.
Licensed products for the treatment of haemophilia A and B
| Product type | Licensed products a | MA holder/company name | Indication (abbreviated) |
|---|---|---|---|
|
Recombinant FVIII concentrate (rFVIII) |
Advate | Takeda Manufacturing Austria AG | Treatment and prophylaxis of bleeding in patients with haemophilia A (congenital factor VIII deficiency) |
| Adynovi | Baxalta Innovations GmbH | ||
| Afstyla | CSL Behring GmbH | ||
| Elocta | Swedish Orphan Biovitrum AB (publ) | ||
| Esperoct | Novo Nordisk A/S | ||
| Jivi | Bayer AG | ||
| Kogenate Bayer | Bayer AG | ||
| Kovaltry | Bayer Pharma AG | ||
| Helixate NexGen | Bayer AG | ||
| NovoEight | Novo Nordisk A/S | ||
| Nuwiq | Octapharma AB | ||
| Obizur | Baxalta Innovations GmbH | ||
| Recombinate | Baxter Healthcare | ||
| ReFacto AF | Pfizer Europe MA EEIG | ||
| Vihuma | Octapharma AB | ||
|
Plasma‐derived FVIII concentrate (pdFVIII) |
Beriate | CSL Behring GmbH | |
| Haemoctin | Biotest (UK) Ltd | ||
| Hemofil | Baxter U.S. | ||
| Immunate | Baxter | ||
| Monoclate | CSL Behring GmbH | ||
| Octanate | Octapharma AB | ||
| Wilate | Octapharma AB | ||
|
Recombinant FIX concentrate (rFIX) |
Alprolix | Swedish Orphan Biovitrum AB (publ) | Treatment and prophylaxis of bleeding in patients with haemophilia B (congenital factor IX deficiency) |
| BeneFIX | Pfizer Europe MA EEIG | ||
| Idelvion | CSL Behring GmbH | ||
| Reflixia | Novo Nordisk A/S | ||
| Rixubis | Baxalta Innovations GmbH | ||
|
Plasma‐derived FIX concentrate (pdFIX) |
AlphaNine SD | Grifols | |
| Mononine | CSL Behring GmbH |
For plasma‐derived products, the list is only partial, especially regarding nationally licensed products in the EU. Adapted from “Jivi Report on the Maintenance of the Orphan Designation”. Available at: https://www.ema.europa.eu/en/documents/orphan-maintenance-report/jivi-orphan-designation-withdrawal-assessment-report-initial-authorisation_en.pdf.
Inhibitors develop in approximately 25% to 30% of haemophilia A patients and, less frequently, in 3% to 5% of haemophilia B patients. Treatment with bypassing agents (BPAs) and immune tolerance induction (ITI) is required in such patients to eradicate inhibitors; nonetheless, patients continue to exhibit increased morbidity and mortality. 9 , 10 In non‐inhibitor patients, prophylaxis can decrease the frequency of bleeding and slow the progression of joint disease, thus improving the quality of life. However, studies have shown that bleeding events are not eliminated for all patients, and joint disease still appears in young adults. 10 , 11 , 12
The mainstay for treatment of bleeding episodes in patients with high‐responding inhibitors is the so‐called ‘by‐passing agents’, recombinant FVIIa and anti‐inhibitor coagulant complex (FEIBA). In patients with inhibitors to FIX, prophylaxis with FIX products is not possible.
A bispecific monoclonal antibody (emicizumab) has recently been approved for prophylaxis of haemophilia A patients with or without FVIII inhibitors. The drug is designed to bridge activated Factor IX and coagulation Factor X, in order to restore the function of the missing activated FVIII necessary for effective haemostasis. Emicizumab has no structural correlation or sequential homology with Factor VIII and therefore does not induce or potentiate the development of FVIII inhibitors. Recent studies evaluating emicizumab prophylaxis showed clinically meaningful bleed control. 13 , 14 , 15
However, several challenges and unmet needs remain in the treatment of haemophilia, since patients continue to experience breakthrough bleedings, progressive joint disease and high rate of inhibitor development. These numerous challenges could be possibly addressed by emerging technologies such as gene replacement therapies (advanced therapy medicinal products or ATMPs).
2. METHODS
The database on human medicines from the European Medicines Agency was used and data from Rare disease (orphan) designations (https://www.ema.europa.eu/en/medicines/download-medicine-data#rare-disease-(orphan)‐designations‐section) and European public assessment reports (EPARs) (https://www.ema.europa.eu/en/medicines/download-medicine-data#european-public-assessment-reports-(epar)‐section) were retrieved on May 20, 2020 for the analysis.
We retrieved the list of all rare disease (orphan) designations from 2000 (start of the Orphan regulation) to the beginning of 2020, which are found on the EMA's website, resulting in a total of positive 2289 orphan designations. Data extraction forms were designed as: medicine name, active substance, Agency product number, date of first decision, disease/condition, EU designation number, status of orphan designation, first published, revision date, URL.
In order to understand which products had received a central market authorization for haemophilia A and B, we carried out an investigation in EMA's EPARs (European public assessment reports) database, which contains all scientific assessment reports of medicines authorized at a European Union level. We downloaded the table of all EPARs for human and veterinary medicines from EMA's website. A total of 1694 medicinal products resulted from the research.
A control with the data obtained from the EMA database was performed for consistency with the Community Register of orphan medicinal products from the European Commission (https://ec.europa.eu/health/documents/community-register/html/reg_od_act.htm?sort=a) in which all ODs are registered (accessed May 20, 2020).
The ClinicalTrials.gov database and The European Union Clinical Trials Register were used to query all active studies on haemophilia (accessed May 20, 2020). ClinicalTrials.gov provides information on medical studies in human volunteers. Most of the records on ClinicalTrials.gov describe clinical trials. The EU Clinical Trials Register contains information on interventional clinical trials on medicines conducted in the EU or the European Economic Area (EEA).
3. RESULTS
3.1. Orphan designation of haemophilia A and B
New legislation was introduced by the EU in 2000 in order to provide incentives for the development of medicines for rare diseases called orphan medicinal products. Orphan designation (OD) through the European Orphan Regulation fosters research in new therapeutic approaches in the treatment of rare diseases and sets the criteria for receiving an OD. Among all the orphan designations resulting from the search strategy (n = 2289), we selected only ODs for the treatment of haemophilia A and B: thus, 41 ODs resulted from the filtered research.
Since 14 medicinal products were withdrawn from the Community Register of designated Orphan Medicinal Products and one OD expired (Figure 1) only 26 ‘active’ ODs resulted from the search.
FIGURE 1.
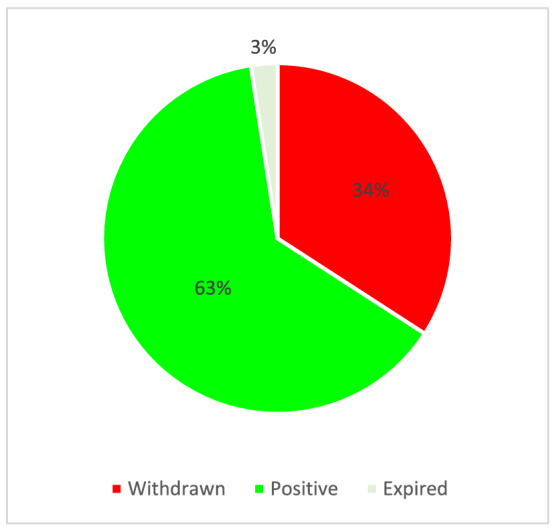
ODs for the treatment of haemophilia A and B (n = 41)
3.1.1. Classification of haemophilia ODs
Of all active ODs, treatment of haemophilia A and haemophilia B are almost equally represented (46% and 54% respectively). It was noted that factor replacement therapy medicinal products represent the largest group (42%) followed by gene therapy medicinal products (GTMPs) and rebalancing therapy medicinal products (39% and 19% respectively) (Figure 2). Replacement therapy involves the supply of the missing clotting factor to the patient from an external source, which is slowly injected intravenously. Rebalancing therapy aims to restore or rebalance the coagulation system targeting natural anticoagulant pathways.
FIGURE 2.
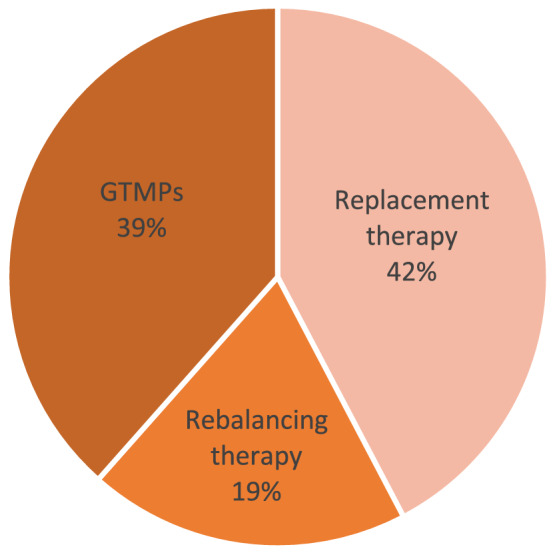
Haemophilia ‘active’ ODs classification according to MOA (n = 26)
3.2. Authorized medicinal products for haemophilia in the EU
Among all authorized medicinal products for human use, we selected only those for the treatment of haemophilia A and B. Twenty‐four medicinal products resulted from the filtered research. Among them, two (Iblias and Nonafact) were withdrawn from use in the European Union (both at the request of the marketing authorization holder). Currently, there are 22 authorized medicinal products for the treatment of haemophilia A and B. Among them, only Alprolix and Idelvion were designated orphan medicinal products and received a 10‐year market exclusivity. The remaining authorized medicines are no longer orphan medicines and were withdrawn from the Community register of orphan medicinal products at the time of the granting of a marketing authorization. These products did not provide any advantage compared to the current state of art, and did not bring the innovation that was hoped to transform the treatment of patients with haemophilia A and B.
3.3. Novel therapeutic opportunities in haemophilia care
Novel therapeutic opportunities to improve treatment of haemophilia patients are rapidly evolving. These include novel FVIII/FIX products, substitution therapies, haemostatic rebalancing therapy and gene therapy/editing. Figure 3 provides an overview of the coagulation cascade and the site of action of novel therapeutics in haemophilia care.
FIGURE 3.
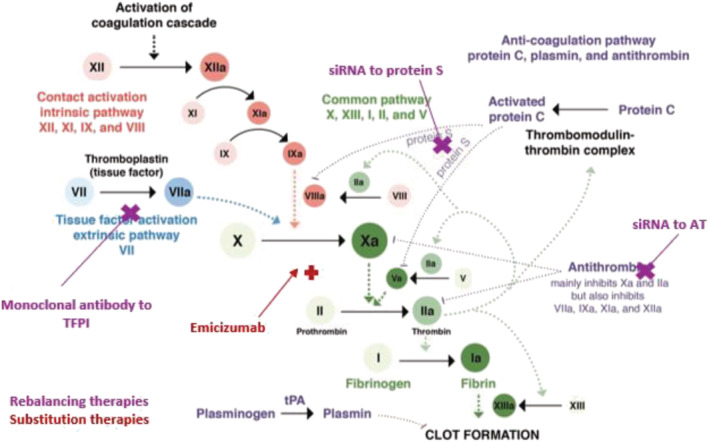
Mechanisms of action of novel nonfactor therapeutics for haemophilia. Haemostatic nonfactor agents in varying phases of development include substitution therapies (Emicizumab) for FVIII, which can restore Factor Xa generation, and rebalancing therapies, which knock down or disrupt the natural anticoagulants (small interfering RNA to AT and PS, monoclonal antibodies to TFPI), to augment haemostasis
3.3.1. Recombinant and modified release FVIII and FIX products
The short half‐life of the standard therapies for haemophilia requires frequent administration within prophylaxis regimens. Extended half‐life FVIII/FIX recombinant concentrates have been obtained by fusion to polyethylene glycol (FVIII and FIX), IgG1‐Fc (FVIII and FIX) or albumin. These modifications have provided prominent decrease in injection frequency—every 1 to 2 weeks for prophylaxis, 4 thus improving patients' quality of life. EHL clotting factors may not provide advantage over non‐EHL products in terms of efficacy in preventing and treating bleeds, but they increase treatment adherence, improve clinical outcomes and provide an opportunity for improved individualized treatment for haemophilia. Idelvion and Alprolix, for haemophilia B, are examples of EHL products (both orphan medicines), which have recently been authorized for use in the EU.
3.3.2. Substitution therapy
A subcutaneously administered bispecific monoclonal antibody emicizumab (Hemlibra) has recently been approved for the treatment of haemophilia A patients with or without FVIII inhibitors. It bridges FX and FIXa and acts as a partial functional mimic to FVIIIa to restore the missing function of FVIIIa. Because of its unique structure, emicizumab is not expected to induce or be affected by Factor VIII inhibitors. HAVEN 1, a phase 3 non‐interventional study in severe haemophilia A patients with inhibitors, showed an 87% reduction in bleed rates compared with no prophylaxis, and a 79% reduction compared with prior BPA prophylaxis. 13 The HAVEN 3 study evaluated emicizumab prophylaxis in haemophilia A patients without inhibitors, using weekly dosing, and demonstrated a 68% reduction in treated bleeds compared with prior Factor VIII prophylaxis. 14 These results support the hypothesis that the level of steady‐state maintenance of haemostasis achieved with emicizumab prophylaxis can result in superior efficacy with respect to traditional prophylaxis. A recently completed open‐label, phase 3 trial (HAVEN 4), evaluated emicizumab prophylaxis (6 mg/kg every 4 weeks for 24 weeks) in patients aged 12 years and older with severe congenital haemophilia A (<1% of normal FVIII activity in blood) or haemophilia A with FVIII inhibitors, undergoing treatment with either FVIII concentrates or bypassing agents. 15 The results showed clinically meaningful bleed control while being well tolerated. Hemlibra could improve patient care by decreasing treatment burden and increasing adherence to effective prophylaxis, potentially decreasing the development of secondary complications for people with haemophilia A.
3.3.3. Rebalancing therapy
Haemostasis is a complex physiological process that maintains a balance between the normal blood flow within the vasculature and the induction of blood clot formation following injury. 16 In the presence of a coagulation factor deficiency, such as haemophilia, this delicate balance is shifted towards bleeding. In contrast, derangements in the natural anticoagulant pathways can lead to thrombosis. Evidence from current knowledge demonstrate that targeting these natural anticoagulant pathways (anti‐thrombin and tissue factor pathway inhibitors, protein S and protein C) can restore the haemostatic equilibrium in the presence of a bleeding disorder. 17 Fitusiran is an RNA interference (RNAi) therapy which targets antithrombin (AT) in the liver and interferes with AT translation by binding and degrading messenger RNA‐AT to prevent AT synthesis and promote haemostasis. In both preclinical and clinical studies, AT knockdown led to dose‐dependent lowering of AT levels and reduced the bleeding phenotypes in haemophilia patients. 18 , 19 Fitusiran has been evaluated in haemophilia patients with or without inhibitors in phase 1 and phase 3 trials and is currently under evaluation in phase 3 trials. A phase 1 study sponsored by Alnylam Pharmaceuticals (NCT02035605) demonstrated dose‐dependent decrease in AT of 61% to 89%, which correlated with increased thrombin generation in haemophilia A and haemophilia B patients without inhibitors. Results from a Phase 1/2 long‐term safety and efficacy extension study (NCT02554773) showed that Fitusiran lowered the levels of antithrombin by about the 80%. A post‐hoc exploratory analysis of bleed events demonstrated a median annual bleeding rate (ABR) of 1 in all patients, thus Fitusiran reduced the frequency of annual bleeding episodes. Treatment was generally well tolerated. Elevation of liver enzymes was reported. One trial subject suffered a fatal cerebral venous sinus thrombosis. 20 Tissue factor pathway inhibitor (TFPI) is the primary inhibitor of the initiation of blood coagulation and modulates the severity of a wide variety of bleeding and clotting disorders. Inhibition of TFPI has been shown to reduce bleeding in multiple haemophilia animal models. 21 Different approaches have been attempted to inhibit TFPI, including aptamers, fucoidan, monoclonal antibodies and peptide agents. 4 Concizumab, a humanized monoclonal antibody against TFPI, is the most advanced in development. Two phase 2/3 trials are ongoing in haemophilia A and haemophilia B patients with and without inhibitors. Two other products PF‐06741086 in phase 3 and BAY1093884 in phase 2 are also in development; the latter was recently discontinued due to multiple thrombotic events. Specifically, TFPI inhibits FXa whereas APC degrades FVa and FVIIIa. Activated protein C (APC) is a serine protease that can bind the endothelial protein C receptor, which approximates it to thrombin for its activation, whereby it functions to downregulate the amplification of FXa generation and, in turn, thrombin generation via the intrinsic pathway, through proteolysis of FVa and FVIIIa. APC is regulated by serpins, protein C inhibitor (PCI), Protein S and α1‐antitrypsin. Currently non‐clinical data support the use of serin protease inhibitor, monoclonal antibody against APC and siRNA targeting Protein S. In particular, serine protease inhibitor (SerpinPC) is currently in the early phase of the clinical program.
3.4. Emerging advanced therapeutic medicinal products: gene therapy for haemophilia
Gene replacement therapies represent a frequently submitted category of medicinal products for orphan designation. 22 In this form of ATMPs, a functional copy of a defective gene in the target condition, which is either absent or expressed as a non‐functional protein, is substituted by a fully functional gene, which is inserted using a viral vector thereby offering a highly effective means for overcoming diseases, such as haemophilia. The gene correction in haemophilia is well suited because it is associated with a well‐understood mutated gene sequence which can be substituted by a corrected DNA sequence leading to the endogenous production of functional Factor VIII or IX. 5 This would therefore alleviate the need for exogenous sources of the normal clotting factor to overcome the defective factors. An important characteristic of haemophilia treatment is that there is no need to normalize circulating clotting factor levels to obtain a therapeutic effect since a slight increase in plasma clotting factor levels (above 1%) is sufficient to decrease the risk of morbidity and mortality. As a consequence, a small increase in clotting factor levels by gene therapy can greatly improve the clinical symptoms. In addition, therapeutic efficacy of GTMPs can be determined by clear‐cut clinical endpoints such as circulating clotting factor levels and bleeding frequency. 23
Initial clinical gene therapy studies using retroviral, adenoviral and non‐viral (ex vivo) approaches were associated with transient low‐level factor expression. 24 , 25 These studies have been unsuccessful and have been terminated for both efficacy and safety concerns. As an example, a trial for haemophilia A, in which adenovirus was used to deliver the Factor VIII gene, was terminated owing to toxic events in a single patient.
This led to a shift toward usage of “non‐integrating” recombinant adeno‐associated viral (AAV) vectors. AAV vectors are derived from wild‐type AAV, 26 a member of the parvovirus family. Wild‐type AAV is non‐pathogenic, weakly immunogenic and replication‐deficient, requiring a helper virus for replication. AAV vectors can deliver a therapeutic transgene cassette up to 5 kb into both dividing and nondividing cells. The DNA sequences carried by recombinant AAV vectors are stabilized predominantly in an episomal form so that long‐term expression can occur only with delivery into long‐lived, post mitotic cell types; the vector DNA integrates at a very low frequency and is typically lost from replicating cells. The recombinant vector has tropism for a range of target tissues including the liver, cell types in the retina and the central nervous system, skeletal muscle, and cardiac muscle, among others. AAV vectors are the most frequently used viral vectors for gene therapy for haemophilia (90%), followed by in vivo and ex vivo lentiviral vectors (10%).
Table 3 provides a detailed list of GTMPs for haemophilia, which were granted an orphan designation by the European Commission.
TABLE 3.
List of gene therapy medicinal products for haemophilia
| Active substance | Disease/condition | EU designation number |
|---|---|---|
| Adeno‐associated viral vector serotype 5 containing a B‐domain‐deleted variant of human coagulation factor VIII gene (also called BMN 270) | Haemophilia A | EU/3/16/1622 |
| Adeno‐associated viral vector serotype 8 containing a functional copy of the codon‐optimized F8 cDNA encoding the B‐domain‐deleted human coagulation factor VIII | Haemophilia A | EU/3/18/2015 |
| Recombinant adeno‐associated viral vector serotype 6 encoding the B‐domain‐deleted human factor VIII (also known as SB‐525) | Haemophilia A | EU/3/17/1874 |
| Recombinant adeno‐associated viral vector containing a bioengineered capsid and a codon‐optimized expression cassette to drive the expression of the SQ form of a B‐domain‐deleted human coagulation factor VIII (also known as SPK‐8011) | Haemophilia A | EU/3/18/2079 |
| Adeno‐associated viral vector serotype rh10 containing the human factor IX gene for the treatment of haemophilia B | Haemophilia B | EU/3/15/1599 |
| A non‐replicating adeno‐associated virus serotype 2 (AAV2) expressing the Padua variant (R338L) of human coagulation factor IX (F9, Factor IX, FIX), under the control of the liver‐specific apolipoprotein E (Apo E) enhancer/alpha1‐antitrypsin (hAAT) promoter (ApoE/hAAT) | Haemophilia B | EU/3/18/2090 |
| Adeno‐associated viral vector containing the human factor‐IX gene | Haemophilia B | EU/3/11/938 |
| Recombinant adeno‐associated viral vector containing a codon‐optimized Padua derivative of human coagulation factor IX cDNA (also known as AMT‐061) | Haemophilia B | EU/3/18/1999 |
| Recombinant adeno‐associated viral vector serotype S3 containing codon‐optimized expression cassette encoding human coagulation factor IX variant (also known as FLT180a) | Haemophilia B | EU/3/18/2080 |
| Lentiviral vector encoding human coagulation factor IX | Haemophilia B | EU/3/19/2141 |
FVIII and FIX synthesis takes place primarily in the liver, from where the proteins can easily enter the bloodstream. This makes hepatocytes a suitable target for gene therapy in haemophilia. Over the years, hepatic in vivo gene transfer using AAV vectors has shown the best success in preclinical and clinical studies, with several clinical studies for both haemophilia A and B enrolling patients for phase 3 trials. We analysed the studies listed in the ClinicalTrials.gov and the EU Clinical Trials Register repositories. 27 , 28
Many AAV vectors have progressed into clinical studies for both haemophilia A and B. The ClinicalTrials.gov database presently lists 28 active gene therapy clinical trials for haemophilia, 11 clinical trials to evaluate different AAV‐based GTMPs for haemophilia A and 12 trials to evaluate AAV‐FVIII gene therapy for haemophilia B (Table 4), with some overlap, as the same vector is being evaluated in both phase 1/2 and phase 3 studies.
TABLE 4.
Summary of active gene therapy clinical trials for haemophilia registered on ClinicalTrials.gov
| NCT Number | Title | Status | Interventions | Sponsor/ | Phases | Funder type |
|---|---|---|---|---|---|---|
| NCT03001830 | Gene Therapy for Haemophilia A | Recruiting | Novel serotype 8 capsid pseudotyped adeno‐associated viral vector encoding Factor VIII‐V3 (AAV2/8‐HLP‐FVIII‐V3) | University College, London/Medical Research Council | Phase 1 | Other |
| NCT03520712 | Gene Therapy Study in Severe Hemophilia A Patients with Antibodies against AAV5 | Enrolling by invitation | Adeno‐associated viral vector serotype 5 containing a B‐domain deleted variant of human coagulation factor VIII gene (Valoctocogene Roxaparvovec) | BioMarin Pharmaceutical | Phase 1/Phase 2 | Industry |
| NCT04323098 | Study to Evaluate the Efficacy and Safety of Volactocogene Roxaparvovec, with Prophylactic Steroids in Hemophilia A | Not yet recruiting | Valoctocogene Roxaparvovec | BioMarin Pharmaceutical | Phase 1/Phase 2 | Industry |
| NCT02576795 | Gene Therapy Study in Severe Haemophilia A patients | Active, not recruiting | Valoctocogene Roxaparvovec – BMN 270 | BioMarin Pharmaceutical | Phase 1/Phase 2 | Industry |
| NCT03370913 | Study to Evaluate the Efficacy and Safety of Valoctocogene Roxaparvovec in Hemophilia A Patients | Active, not recruiting | Valoctocogene Roxaparvovec | BioMarin Pharmaceutical | Phase 3 | Industry |
| NCT03391974 | Single‐Arm Study to Evaluate the Efficacy and Safety of Valoctocogene Roxaparvovec in Hemophilia A patients at a Dose of 4E13 vg/kg | Valoctocogene Roxaparvovec | BioMarin Pharmaceutical | Phase 3 | Industry | |
| NCT03370172 | Safety and Dose Escalation Study of an Adeno‐Associated Viral Vector for Gene Transfer in Hemophilia A | Active, not recruiting | Adeno‐associated virus serotype 8 (AAV8) vector expressing B‐domain‐deleted Factor VIII (BDD‐FVIII) (BAX 888) | Baxalta now part of Shire | Phase 1/Phase 2 | Industry |
| NCT03588299 | Study to Test the Safety in Patients with Severe Hemophilia a Drug Therapy that Delivers a Healthy Version of the Defective Factor VIII Gene into the Nucleus of Liver Cells Using an Altered, Non‐infectious Virus (AAV) as a “Shuttle”. | Recruiting | Adeno‐associated Virus (AAV) hu37‐mediated Gene Transfer of B‐domain‐deleted Human Factor VIII (BAY2599023 [DTX201]) | Baye/Ultragenix pharmaceutical | Phase 1/Phase 2 | Industry| Other |
| NCT03734588 | Study of SPK‐8016 Gene Therapy in Patients with Hemophilia A to Support Evaluation in Individuals with FVIII Inhibitors | Active, not recruiting | Adeno‐associated virus (AAV) that carries a bioengineered gene whose protein product can suppress factor VIII inhibitors (SPK‐8016) | Spark Therapeutics | Phase 1/Phase 2 | Industry |
| NCT04370054 | Study to Evaluate the Efficacy and Safety of PF‐07055480 in Moderately Severe to Severe Hemophilia A adults | Not yet recruiting | Recombinant AAV2/6 encoding the complementary deoxyribonucleic acid for B‐domain‐deleted human FVIII | UniQure Biopharma B.V. | Phase 1/Phase 2 | Industry |
| NCT03061201 | Study of Recombinant AAV2/6 Human Factor 8 Gene Therapy SB‐525 in Subjects with Severe Hemophilia A | Recruiting | Recombinant adeno‐associated virus serotype 6 vector (AAV6) encoding the complementary deoxyribonucleic acid for B‐domain‐deleted human FVIII | Pfizer | Phase 2 | Industry |
| NCT03003533 | A Gene Transfer Study for Hemophilia A | Recruiting | Recombinant adeno‐associated viral vector carrying human factor VIII gene (SPK‐8011) | Spark Therapeutics | Phase 1/Phase 2 | Industry |
| NCT04418414 | Hematopoietic Stem Cell Transplantation Gene Therapy for Treatment of Severe Hemophilia A | Not yet recruiting | CD34+ hematopoietic stem cells transduced with CD68‐ET3 lentiviral vector encoding human factor VIII gene | Expression Therapeutics, LLC | Phase 1 | Industry |
| NCT03818763 | Gene Therapy Trial for Platelet Derived Factor VIII Production in Hemophilia A | Recruiting | Auto CD34+PBSC, transduced with a lentiviral vector encoding the B‐domain‐deleted from of human coagulation factor VIII | Parameswaran Hari/Medical College of Wisconsin | Phase 1 | Other |
| NCT03217032 | Lentiviral FVIII Gene Therapy | Not yet recruiting | Lentiviral factor VIII gene modified autologous hematopoietic stem cells and mesenchymal stem cells (YUVA‐GT‐F801) | Shenzhen Geno‐Immune Medical Institute | Phase 1 | Other |
| NCT01687608 | Open‐Label Single Ascending Dose of Adeno‐associated Virus Serotype 8 Factor IX Gene Therapy in Adults with Hemophilia B | Active, not recruiting | Adeno‐associated virus serotype 8 Factor IX Gene Therapy (AskBio009) | Baxalta now part of Shire | Phase 1/Phase 2 | Industry |
| NCT04135300 | Gene Therapy for Chinese Hemophilia B | Recruiting | Adeno‐associated viral (AAV) vector designed to drive expression of the human factor IX (hFIX) transgene in liver (BBM‐H901) | Institute of Hematology & Blood Diseases Hospital/East China University of Science and Technology | Not Applicable | Other |
| NCT02396342 | Trial of AAV5‐hFIX in Severe or Moderately Severe Hemophilia B | Active, not recruiting | AAV5 containing a codon‐optimized human factor IX gene (AAV5‐hFIX) | UniQure Biopharma BV | Phase 1/Phase 2 | Industry |
| NCT03489291 | Dose Confirmation Trial of AAV5‐hFIXco‐Padua | Active, not recruiting | Recombinant adeno‐associated viral vector of serotype 5 (AAV5) containing the Padua variant of a codon‐optimized human FIX complementary deoxyribonucleic acid (cDNA) under the control of a liver‐specific promoter (AAV5‐hFIXco‐Padua, AMT‐061) | UniQure Biopharma BV | Phase 2 | Industry |
| NCT03569891 | HOPE‐B: Trial of AMT‐061 in Severe or Moderately Severe Hemophilia B Patients | Active, not recruiting | AAV5‐hFIXco‐Padua, AMT‐061 | UniQure Biopharma BV | Phase 3 | Industry |
| NCT04394286 | A Phase 1/2 Study of SHP648, an Adeno‐Associated Viral Vector for Gene Transfer in Hemophilia B Subjects | Recruiting | Adeno‐Associated Virus Serotype 8 (AAV8) Vector Expressing FIX Padua (SHP648) | Baxalta now part of Shire | Phase 1/Phase 2 | Industry |
| NCT00979238 | Dose‐Escalation Study of a Self Complementary Adeno‐Associated Viral Vector for Gene Transfer in Hemophilia B | Active, not recruiting | Self‐complementary AAV (AAV8) vector expressing a codon‐optimized factor IX transgene (scAAV2/8‐LP1‐hFIXco) | St. Jude Children's Research Hospital/National Heart, Lung, and Blood Institute (NHLBI)/Hemophilia of Georgia, Inc./Children's Hospital of Philadelphia/University College, London | Phase 1 | Other/NIH |
| NCT03307980 | Long‐term Safety and Efficacy Study of SPK‐9001 in Individuals with Hemophilia B | Recruiting | A non‐replicating adeno‐associated virus serotype 2 (AAV2) expressing the Padua variant (R338L) of human coagulation factor IX (F9, Factor IX, FIX), under the control of the liver‐specific apolipoprotein E (Apo E) enhancer/alpha1‐antitrypsin (hAAT) promoter (ApoE/hAAT) (PF‐06838435/fidanacogene elaparvovec) | Pfizer | Phase 2 | Industry |
| NCT03861273 | A Study to Evaluate the Efficacy and Safety of Factor IX Gene Therapy with PF‐06838435 in Adult Males with Moderately Severe to Severe Hemophilia B | Recruiting | Fidanacogene elaparvovec | Pfizer | Phase 3 | Industry |
| NCT03369444 | A Factor IX Gene Therapy Study (FIX‐GT) | Recruiting | A recombinant non‐replicating adeno‐associated virus of a modified liver‐tropic serotype (S3) (rAAV Rep2‐CapS3) vector, encoding a gain‐of‐function variant (R338L; Padua) of codon‐optimized human coagulation factor IX (hFIX) under the control of a liver‐specific promoter (FRE1) comprising a truncated version of the human apolipoprotein E locus control region with a truncated version of the human alpha‐1‐antitrypsin promoter (FLT180a) | University College, London | Phase 1 | Other |
| NCT03641703 | A Long‐Term Follow‐Up Study of Haemophilia B Patients Who Have Undergone Gene Therapy | Recruiting | FLT180a | Freeline Therapeutics | Phase 2/Phase 3 | Industry |
| NCT03961243 | Lentiviral FIX Gene Therapy | Not yet recruiting | Lentiviral factor IX gene‐modified autologous hematopoietic and mesenchymal stem cells (YUVA‐GT‐F901) | Shenzhen Geno‐Immune Medical Institute | Phase 1 | Other |
| NCT02695160 | Ascending Dose Study of Genome Editing by Zinc Finger Nuclease Therapeutic SB‐FIX in Subjects with Severe Hemophilia B | Active, not recruiting | Zinc Finger Nuclease (ZFN) Therapeutic for genome editing (SB‐FIX) | Sangamo Therapeutics | Phase 1 | Industry |
Of the ongoing haemophilia A and B AAV clinical trials, the results are promising. The trial sponsored by St. Jude Children's Research Hospital in haemophilia B patients, using an scAAV2/8‐LP1‐hFIXco vector, was the first confirming long‐term benefit of haemophilia gene therapy. 29 This study evaluated the stability of transgene expression and long‐term safety in 10 patients with severe haemophilia B. The infusion of a single dose of AAV8 vector in all 10 patients resulted in a vector dose‐dependent manner increase in circulating Factor IX to a level that was 1–6% of the normal value over a median period of 3 years, allowing either the discontinuation of prophylactic FIX protein infusions or a significant reduction in frequency. However, capsid‐specific antibodies arose in all participants, which would likely block future re‐administration of the vector. The main adverse effect observed was the rise of liver enzyme levels (alanine aminotransferase, ALT), that was treated with a tapering dose of prednisolone. The University College of London has initiated a new phase 1/2 trial for haemophilia B, using AAV‐F9 vector, FLT180a (NCT03369444). As of this review, no data have been released.
UniQure has recently published data on their clinical trial for haemophilia B with an AAV5 vector using the same F9 expression cassette as the UCL and St. Jude Children's Research Hospital trial. AMT‐060, indeed, combines the previously tested human wild‐type FIX gene cassette with an AAV5 capsid. 30 Similar levels of steady‐state FIX activity were achieved in this study compared with the previous study, in which Nathwani et al. used an AAV8 vector, although the highest dose of AAV5 vector used was greater than the highest dose of the AAV8 vector used. Several patients displayed transient transaminitis without indication of a capsid T‐cell response. The investigators' conclusion was that scAAV5‐pseudotyped vectors should be effective for treating individuals with pre‐existing immunity to AAV5. 31 A study of prevalence of naturally occurring antibodies against AAV serotypes conducted in a healthy European population documented neutralizing antibody seroprevalence of 59% for AAV2, 19% for AAV8 and 3% for AAV5. 32 In addition, it should be noted that there is a wide range in the reported prevalence of NAbs to AAV, including AAV5, in the general as well as haemophilia populations due to variability in populations but also likely from differences in the assay techniques. 33 Thus AAV5 may offer a preferential immune profile.
The development of AAV‐based therapies for the treatment of haemophilia A is still at an early stage. One of the main challenges has been fitting the FVIII expression cassette within the restricted packaging limits of AAV, as the ~7 kb cDNA for FVIII exceeds the packaging capacity of AAV (~5 kb). BioMarin Pharmaceutical initiated the first clinical trial for haemophilia A, using liver gene transfer of and AAV5 vector expressing a codon‐optimized BDD‐FVIII‐DQ protein, BMN 270 (NCT02576795). Similar to in other clinical trials, some patients (88.8%) experienced mild elevation in ALT level, accompanied by a decline in the Factor VIII activity level only in one patient. Two additional phase 3 studies have been initiated by BioMarin Pharmaceutical, with target vector dose of 4 × 1013 vg/kg and 6 × 1013 vg/kg (NCT03392974/NCT03370913). Pasi and colleagues reported a durable efficacy, long‐term safety, and clinical and biologic results in 15 adults with severe haemophilia A (Factor VIII level, <1% of normal coagulation activity) who had received a single infusion of AAV5‐hFVIII‐SQ at various dose levels. Three years after infusion, patients in the highest dose cohort (6 × 1013 vg/kg), had Factor VIII expression of 20% of normal; the median number of annualized treated bleeding events was 0, and the median use of exogenous Factor VIII was reduced from 138.5 infusions to 0 infusions per year. 6
BioMarin is enrolling by invitation a phase 1/2 study to evaluate high‐dose Valoctocogene Roxaparvovec gene delivery in patients with haemophilia A with pre‐existing AAV5 capsid antibodies (ClinicalTrials.gov number NCT03520712). As of this review, no results are available. The findings with respect to the safety of AAV‐based gene therapy are encouraging, with the main vector‐related adverse event being an elevated serum ALT level, an effect that appears to be readily attenuated by a short, tapering course of prednisolone. Nevertheless, the complication limits the ability to offer the therapy to adults with haemophilia who are active carriers of hepatitis or to increase the vector dose to achieve truly normal haemostasis.
However, an important limitation to successful AAV‐based gene therapy is the pre‐existing humoral capsid immunity. Immune recognition by cytotoxic CD8+ T cells or antibody responses to the vector capsid, the transgene product, or both, can compromise the therapeutic expression of the transgene. 34 , 35 Wild‐type AAV infection occurs during childhood, and thus patients may develop neutralizing antibodies (nAb) that prevent gene transfer with AAV vectors. Due to the high level of conservation in the amino acid sequence among AAV capsids, anti‐AAV antibodies show cross‐reactivity with multiple serotypes. This has resulted in exclusion of many patients from recent studies. The main toxicity observed in AAV‐based clinical studies has been dose‐related elevation of liver transaminase following vector infusion. Within some studies, this coincides with the demonstration of cell‐mediated AAV capsid immunity. Most studies have used either early intervention or prophylaxis with corticosteroids in order to protect transduced hepatocytes. Although the majority of episodes have been managed effectively with this approach, some episodes have been associated with partial or complete loss of transgene expression, despite intervention. 36 The pre‐existing AAV humoral immunity and the potential loss of vector transduction with time can render the therapy ineffective. Due to the pre‐existing AAV humoral immunity and the potential loss of vector transduction with time, there is need for other viral and non‐viral‐based vectors for transgene delivery. Lentiviral vectors may complement the therapeutic reach of AAV vectors due to lower incidence of pre‐existing humoral immunity and greater packaging capacity. Pre‐clinical studies using in‐vivo or ex‐vivo stem cell (haematopoietic or induced pluripotent) transduction or blood outgrowth endothelial cells have recently been reviewed. 36 , 37 Two phase 1 studies are registered with plans to evaluate ex‐vivo lentiviral stem cell transduction for FVIII (NCT03818763) or FVIII/FIX (NCT03217032/NCT03961243).
3.5. Genome editing
The most advanced approach to genome editing in haemophilia is usage of zinc finger nuclease (ZFN). There have been several recent orphan designations of products with this alternative approach to overcome the defective gene. The gene editing technique can correct the endogenous genetic defect or direct the integration site of a therapeutic gene through nuclease‐targeted double‐strand DNA breaks and homology‐directed repair. Thus, gene‐editing technologies are based on harnessing the natural repair machinery of the cell to modify DNA. A phase 1 study (NCT02695160) is currently recruiting using AAV‐directed correction using ZFN‐targeted insertion of FIX gene into the first intron of the albumin locus. Although gene editing holds significant promise for haemophilia, in its current state, the approach still requires AAV for delivery of the nuclease or transgene. 38 Therefore, the same limitations for AAV gene therapy are present for gene‐editing approaches.
4. DISCUSSION
Current haemophilia treatment consists of parenterally administered plasma‐derived or recombinant clotting factor concentrates. Patients have benefited significantly from replacement therapy, as it greatly improves their quality of life and prolongs life‐expectancy. It has been observed that the recently introduced modified release formulations, which can be applied subcutaneously, have improved the dosing regimen for haemophilia patients.
Although current coagulation factor replacement prophylaxis regimens are effective, they are expensive, challenging to adhere to, result in regular subtherapeutic trough factor levels and are potentially immunogenic. As such, nearly all haemophilia patients would benefit from an intervention that induces more stable factor expression. Such therapies are needed both for adult patients with increasing life expectancy and comorbidities, and for children in need of stable prophylaxis to prevent sequelae of the disease.
In recent studies, gene therapies have induced sustained levels of FVIII and FIX, prevented bleeding and reduced or eliminated the use of factor replacement products. This approach could also alleviate the heavy burden of the need for frequent factor infusions, concerns about inhibitor development and limited global access to factor products. However, while short‐term risks appear to be low, it has been noted that long‐term safety and efficacy are not yet established.
Some additional considerations are related to the type of vector used (namely AAV‐based or lentiviral) and their applicability in different age groups of patients with haemophilia. Humoral immunity to wild‐type AAV represents one of the most important limitations to successful systemic transduction with AAV vectors. The main toxicity seen in clinical trials has been dose‐related elevation of liver transaminase following vector infusion and, in some studies, this coincides with the demonstration of cell‐mediated AAV capsid immunity. Early intervention or prophylaxis with corticosteroids with the aim of protecting transduced hepatocytes have been used in most studies, but not all episodes have been managed effectively and partial/complete loss of transgene expression occurred. Pre‐existing AAV humoral immunity and the potential loss of vector transduction with time represent important and limiting issues in the AAV‐based gene therapy and can render the therapy ineffective. Thus, there is a requirement for other viral and non‐viral‐based vectors for transgene delivery. The predominant methods studied in haemophilia have used lentiviral vectors, for which there is a lower incidence of pre‐existing humoral immunity and which have a greater packaging capacity than AAV vectors.
A specific concern is the external validity of the results obtained from the clinical trials with a selected patient population. Although the clinical data supporting gene therapy for haemophilia have been rather positive, the enrolled subjects are a selected group, and thus some caution is advised in extending the benefits of gene therapy to the general population of patients with haemophilia due to potential selection bias of the results from the clinical studies.
The use of Hemlibra in haemophilia A patients with inhibitors constitutes a substantial achievement as this medicine, approved in the EU in January 2018, allows the patients to have one subcutaneous administration per week, rather than one every 2 or 3 days. This is a clear improvement in the treatment of haemophilia A, especially in terms of QoL, but there is still the need to know how and when it will be possible to switch the patients without inhibitors to this treatment option. Gene therapies using AAVs look very promising in reducing the need for replacement therapy almost completely and reducing the ABR very close to 0. However, patients still have some concerns that need to be elucidated, particularly many unanswered questions about the AAVs, the increase of the liver enzymes level and hepatotoxicity, the reduction in the FVIII expression over time and the possibility of re‐treatment, if needed. Further, some ethical considerations have to be taken into account concerning the use of gene therapies in children. The hot topic for the patient community is the current lack of long‐term data on the gene replacement therapy. The latter is an urgent challenge to be addressed in the upcoming months in order to allow haemophilia patients to be much more aware about the therapeutic options available for the whole community.
5. CONCLUSIONS
The management of haemophilia has been evolving. It started with filtered exogenous factor replacement, followed by improvements in the delivery and pharmacokinetic properties of these exogenous replacement factors. Currently gene replacement medicinal products have emerged, and these may represent a potential cure for people with haemophilia A and B.
Careful consideration should be given to the advantages and disadvantages of the different types of vectors and their suitability for the different haemophilia patients depending on their age, availability or risk for inhibitors, and other factors.
All the above‐mentioned factors play a role in the decision making for each individual patient with haemophilia. In addition to gene therapy, novel replacement therapies and other non‐factor‐based therapy options are being developed.
Still, many questions remain: what will be the eligibility criteria to qualify for this therapy, will it be affordable and accessible to all patients, and what will be the long‐term durability, efficacy and safety? Although the choice of therapy available in the future may in part be directed by financial implications, this may eventually allow for more personalized care with improvement in quality of life for the next generation of patients with haemophilia.
5.1. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20). 39
ACKNOWLEDGEMENT
This research received no external funding.
COMPETING INTERESTS
The authors declare no conflict of interest.
CONTRIBUTORS
V.S.B. and A.M. conceptualized the study. Formal analysis was conducted by F.T. and S.M. F.T. prepared the original draft of the paper, and all authors reviewed and edited the paper. A.M. supervised the study. All authors have read and agreed to the published version of the manuscript.
Tomeo F, Mariz S, Brunetta AL, Stoyanova‐Beninska V, Penttila K, Magrelli A. Haemophilia, state of the art and new therapeutic opportunities, a regulatory perspective. Br J Clin Pharmacol. 2021;87(11):4183–4196. 10.1111/bcp.14838
REFERENCES
- 1. World Federation of Haemophilia . Report on the Annual Global Survey 2018. Montreal, Canada: WFH; 2019. http://www1.wfh.org/publications/files/pdf-1731.pdf. Accessed May 20, 2020. [Google Scholar]
- 2. Regulation (EC) No. 141/2000 of the European Parliament and of the Council. https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:32000R0141%26from=EN. Accessed May 20, 2020.
- 3. Giannelli F, Green PM. The molecular basis of haemophilia A and B. Baillieres Clin Haematol. 1996;9(2):211–228. 10.1016/s0950-3536(96)80059-x [DOI] [PubMed] [Google Scholar]
- 4. Weyand AC, Pipe SW. New therapies for hemophilia. Blood. 2019;133(5):389–398. 10.1182/blood-2018-08-872291 [DOI] [PubMed] [Google Scholar]
- 5. Perrin GQ, Herzog RW, Markusic DM. Update on clinical gene therapy for hemophilia. Blood. 2019;133(5):407–414. 10.1182/blood-2018-07-820720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pasi KJ, Rangarajan S, Mitchell N, et al. Multiyear follow‐up of AAV5‐hFVIII‐SQ gene therapy for hemophilia A. N Engl J Med. 2020;382(1):29–40. 10.1056/NEJMoa1908490 [DOI] [PubMed] [Google Scholar]
- 7. Iorio A, Stonebraker JS, Chambost H, et al. Establishing the prevalence and prevalence at birth of hemophilia in males: a meta‐analytic approach using national registries. Ann Intern Med. 2019;171(8):540–546. 10.7326/M19-1208 [DOI] [PubMed] [Google Scholar]
- 8. Srivastava A, Brewer AK, Mauser‐Bunschoten EP, et al. Guidelines for the management of hemophilia. Haemophilia. 2013;19(1):e1–e47. 10.1111/j.1365-2516.2012.02909.x [DOI] [PubMed] [Google Scholar]
- 9. White GC 2nd, Kempton CL, Grimsley A, Nielsen B, Roberts HR. Cellular immune responses in hemophilia: why do inhibitors develop in some, but not all hemophiliacs? J Thromb Haemost. 2005;3(8):1676–1681. 10.1111/j.1538-7836.2005.01375.x [DOI] [PubMed] [Google Scholar]
- 10. Oldenburg J. Optimal treatment strategies for hemophilia: achievements and limitations of current prophylactic regimens. Blood. 2015;125(13):2038–2044. 10.1182/blood-2015-01-528414 [DOI] [PubMed] [Google Scholar]
- 11. Mazepa MA, Monahan PE, Baker JR, Riske BK, Soucie JM, US Hemophilia Treatment Center Network . Men with severe hemophilia in the United States: birth cohort analysis of a large national database. Blood. 2016;127(24):3073–3081. 10.1182/blood-2015-10-675140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Di Minno MND, Pasta G, Airaldi S, et al. Ultrasound for early detection of joint disease in patients with hemophilic arthropathy. J Clin Med. 2017;6(8):77. 10.3390/jcm6080077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Shima M, Hanabusa H, Taki M, et al. Factor VIII‐mimetic function of humanized bispecific antibody in hemophilia A. N Engl J Med. 2016;374(21):2044–2053. 10.1056/NEJMoa1511769 [DOI] [PubMed] [Google Scholar]
- 14. Mahlangu J, Oldenburg J, Paz‐Priel I, et al. Emicizumab prophylaxis in patients who have hemophilia A without inhibitors. N Engl J Med. 2018;379(9):811–822. 10.1056/NEJMoa1803550 [DOI] [PubMed] [Google Scholar]
- 15. Pipe SW, Shima M, Lehle M, et al. Efficacy, safety, and pharmacokinetics of emicizumab prophylaxis given every 4 weeks in people with haemophilia A (HAVEN 4): a multicentre, open‐label, non‐randomised phase 3 study. Lancet Haematol. 2019;6(6):e295–e305. 10.1016/S2352-3026(19)30054-7 [DOI] [PubMed] [Google Scholar]
- 16. Miyazaki K. Coagulation of blood. In: Gellman MD, Turner JR, eds. Encyclopedia of Behavioral Medicine. New York, NY: Springer; 2013. [Google Scholar]
- 17. Shetty S, Vora S, Kulkarni B, et al. Contribution of natural anticoagulant and fibrinolytic factors in modulating the clinical severity of haemophilia patients. Br J Haematol. 2007;138(4):541–544. 10.1111/j.1365-2141.2007.06693.x [DOI] [PubMed] [Google Scholar]
- 18. Pasi KJ, Rangarajan S, Georgiev P, et al. Targeting of antithrombin in hemophilia A or B with RNAi therapy. N Engl J Med. 2017;377(9):819–828. 10.1056/NEJMoa1616569 [DOI] [PubMed] [Google Scholar]
- 19. Machin N, Ragni MV. An investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia A and B. J Blood Med. 2018;9:135–140. 10.2147/JBM.S159297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Pasi KJ, Georgiev P, Mant T, et al. Fitusiran, an investigational RNAi therapeutic targeting antithrombin for the treatment of hemophilia: updated results from a phase 1 and phase 1,2 extension study in patients with inhibitors. Blood. 2016;128(22):1397. [Google Scholar]
- 21. Dockal M, Hartmann R, Fries M, et al. Small peptides blocking inhibition of factor Xa and tissue factor‐factor VIIa by tissue factor pathway inhibitor (TFPI). J Biol Chem. 2014;289(3):1732–1741. 10.1074/jbc.M113.533836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Farkas AM, Mariz S, Stoyanova‐Beninska V, et al. Advanced therapy medicinal products for rare diseases: state of play of incentives supporting development in Europe. Front Med (Lausanne). 2017;4:53. 10.3389/fmed.2017.00053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chuah MK, Evens H, VandenDriessche T. Gene therapy for hemophilia. J Thromb Haemost. 2013;11(Suppl 1):99–110. 10.1111/jth.12215 [DOI] [PubMed] [Google Scholar]
- 24. Swystun LL, Lillicrap D. Gene therapy for coagulation disorders. Circ Res. 2016;118(9):1443–1452. 10.1161/CIRCRESAHA.115.307015 [DOI] [PubMed] [Google Scholar]
- 25. Chapin JC, Monahan PE. Gene therapy for hemophilia: progress to date. BioDrugs. 2018;32(1):9–25. 10.1007/s40259-017-0255-0 [DOI] [PubMed] [Google Scholar]
- 26. Atchison RW, Casto BC, Hammon WM. Adenovirus‐associated defective virus particles. Science. 1965;149(3685):754–756. 10.1126/science.149.3685.754 [DOI] [PubMed] [Google Scholar]
- 27. ClinicalTrials.gov. https://clinicaltrials.gov/ct2/home. Accessed on May 20, 2020.
- 28. EU Clinical Trials Register. https://www.clinicaltrialsregister.eu/. Accessed on May 20, 2020.
- 29. Nathwani AC, Reiss UM, Tuddenham EG, et al. Long‐term safety and efficacy of factor IX gene therapy in hemophilia B. N Engl J Med. 2014;371(21):1994–2004. 10.1056/NEJMoa1407309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Miesbach W, Meijer K, Coppens M, et al. Gene therapy with adeno‐associated virus vector 5‐human factor IX in adults with hemophilia B. Blood. 2018;131(9):1022–1031. 10.1182/blood-2017-09-804419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Von Drygalski A, Giermasz A, Castaman G, et al. Etranacogene dezaparvovec (AMT‐061 phase 2b): normal/near normal FIX activity and bleed cessation in hemophilia B. Blood Adv. 2019;3(21):3241–3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Boutin S, Monteilhet V, Veron P, et al. Prevalence of serum IgG and neutralizing factors against adeno‐associated virus (AAV) types 1, 2, 5, 6, 8, and 9 in the healthy population: implications for gene therapy using AAV vectors. Hum Gene Ther. 2010;21(6):704–712. 10.1089/hum.2009.182 [DOI] [PubMed] [Google Scholar]
- 33. Arruda VR, Doshi BS. Gene therapy for hemophilia: facts and quandaries in the 21st century. Mediterr J Hematol Infect Dis. 2020;12(1):e2020069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Herzog RW. Hemophilia gene therapy: caught between a cure and an immune response. Mol Ther. 2015;23(9):1411–1412. 10.1038/mt.2015.135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mingozzi F, High KA. Immune responses to AAV vectors: overcoming barriers to successful gene therapy. Blood. 2013;122(1):23–36. 10.1182/blood-2013-01-306647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Doshi BS, Arruda VR. Gene therapy for hemophilia: what does the future hold? Ther Adv Hematol. 2018;9(9):273–293. 10.1177/2040620718791933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ide LM, Gangadharan B, Chiang KY, Doering CB, Spencer HT. Hematopoietic stem‐cell gene therapy of hemophilia A incorporating a porcine factor VIII transgene and nonmyeloablative conditioning regimens. Blood. 2007;110(8):2855–2863. 10.1182/blood-2007-04-082602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sangamo Therapeutics . Sangamo announces treatment of first patient in phase 1/2 clinical trial of in vivo genome editing therapy for hemophilia B. 2018. https://investor.sangamo.com/news-releases/news-release-details/sangamo-announces-treatment-first-patient-phase-12-clinical-0. Accessed on May 20, 2020.
- 39. Alexander SPH, Kelly E, Mathie A, et al. THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Introduction and Other Protein Targets. Br J Pharmacol. 2019;176(Suppl 1):S1–S20. 10.1111/bph.14747 [DOI] [PMC free article] [PubMed] [Google Scholar]