

Abstract
Cyclopropane rings are an important structural motif frequently found in many natural products and pharmaceuticals. Commonly, biocatalytic methodologies for the asymmetric synthesis of cyclopropanes rely on repurposed or artificial heme enzymes. Here, we engineered an unusual cofactor‐independent cyclopropanation enzyme based on a promiscuous tautomerase for the enantioselective synthesis of various cyclopropanes via the nucleophilic addition of diethyl 2‐chloromalonate to α,β‐unsaturated aldehydes. The engineered enzyme promotes formation of the two new carbon‐carbon bonds with excellent stereocontrol over both stereocenters, affording the desired cyclopropanes with high diastereo‐ and enantiopurity (d.r. up to 25:1; e.r. up to 99:1). Our results highlight the usefulness of promiscuous enzymes for expanding the biocatalytic repertoire for non‐natural reactions.
Keywords: biocatalysis, catalytic promiscuity, cyclopropanation, enzyme engineering
Engineering of a promiscuous 4‐oxalocrotonate tautomerase (4‐OT) resulted in an unusual cofactor‐independent cyclopropanation enzyme that promotes the stereoselective addition of diethyl 2‐chloromalonate to substituted cinnamaldehydes to afford various cyclopropanes with high diastereo‐ and enantiopurity.

Enantioenriched cyclopropanes are frequently found in a variety of bioactive natural products as well as pharmaceuticals and face increasing interest owing to their importance in drug development. [1] The unique physical and chemical properties of the cyclopropane ring, featuring unusual short C−C bonds, characteristic ring strain, and inherent high reactivity, pose several challenges for the synthesis of these compounds. [2] The development of facile biocatalytic methodologies for the asymmetric preparation of cyclopropanes, serving as either chiral building blocks or bioactive compounds, is therefore of high academic and industrial interest.
In nature, two distinct mechanisms have evolved for the biosynthesis of cyclopropanes. Typically, these involve the activation of allyl pyrophosphates to form an allylic cation that undergoes cyclization or the addition of the methyl cation of S‐adenosyl methionine to alkenes. [1a] To overcome the limited substrate scope of natural cyclopropanation enzymes, several additional cyclopropanation strategies depending on redesigned heme enzymes, such as cytochrome P450, cytochrome c and myoglobin, as well as artificial heme enzymes, have been developed. [3] Commonly, these cofactor‐dependent biocatalytic strategies rely on an activated carbenoid intermediate, formed between ethyl diazoacetate and the ferrous state (FeII) of the prosthetic heme group.
Inspired by outstanding developments in the organocatalysis field, describing asymmetric cyclopropanation reactions catalyzed by chiral aminocatalysts, [4] we aimed to design an analogous biocatalytic approach for the preparation of enantioenriched cylcopropanes. Herein, we report the engineering of an uncommon cofactor‐independent cyclopropanation enzyme based on a promiscuous 4‐oxalocrotonate tautomerase (4‐OT) that accepts diethyl 2‐chloromalonate to achieve asymmetric cyclopropanations with various α,β‐unsaturated aldehydes to form the corresponding cyclopropanes with excellent stereochemical purities.
Previous work from our group has shown that 4‐OT can promiscuously catalyze the enantioselective Michael addition of nitromethane to cinnamaldehydes, which involves the formation of a reactive enzyme‐bound iminium ion intermediate between the catalytic N‐terminal proline (Pro‐1) of the enzyme and the cinnamaldehyde substrate (Scheme 1 A). [5] We subsequently expanded the catalytic repertoire of 4‐OT to include enantiocomplementary epoxidation reactions, based on hydroperoxide additions to cinnamaldehydes (Scheme 1 B). [6] In the current study, we explored whether the reaction scope of 4‐OT can be enlarged further to include cyclopropanation reactions between diethyl 2‐halomalonates and cinnamaldehydes (Scheme 1 C). We reasoned that diethyl 2‐halomalonates can rather easily deprotonate to form a nucleophilic carbanion, which could attack the enzyme‐bound iminium ion intermediate via a Michael addition reaction. The resulting enamine intermediate could subsequently attack the halogen‐substituted carbon, with the halogen acting as a leaving group, to promote an intramolecular cyclization reaction to generate the desired cyclopropane product.
Scheme 1.

Iminium ion biocatalysis, highlighting the key catalytic role of the N‐terminal proline residue (Pro‐1) of the 4‐OT enzyme. A) Proposed mechanism of the 4‐OT catalyzed Michael addition of nitromethane to cinnamaldehydes. B) Proposed mechanism of the 4‐OT catalyzed epoxidation reaction between hydroperoxides and cinnamaldehydes. C) Proposed mechanism of the 4‐OT catalyzed cyclopropanation reaction between diethyl 2‐halomalonates and cinnamaldehydes.
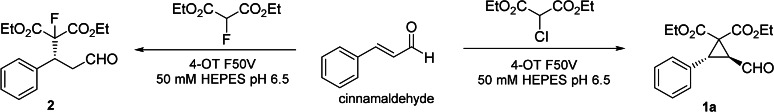
We started our investigation by testing different diethyl 2‐halomalonates (leaving group ability: 2‐Br>2‐Cl≫2‐F) in a reaction with cinnamaldehyde using 4‐OT F50V as the catalyst. This 4‐OT variant was selected as a biocatalyst because it was previously identified to efficiently catalyze the Michael addition of nitromethane to cinnamaldehyde. [5] Analytical‐scale reactions were performed with 1 mM cinnamaldehyde, 5 mM diethyl 2‐halomalonate, and 15 μM 4‐OT F50V in 50 mM HEPES, pH 6.5. After 18 h reaction time, the conversion was determined by GC‐MS analysis. With the nucleophile diethyl 2‐fluoromalonate a conversion of 40 % was reached, but the formed enzymatic product was identified as the enantioenriched fluorinated Michael adduct 2 (Table 1, entry 1). This suggests that although 4‐OT F50V can catalyze the highly enantioselective Michael addition of diethyl 2‐fluoromalonate to cinnamaldehyde, the poor leaving group ability of fluorine does not support the intramolecular cyclization reaction, preventing formation of the cyclopropane ring.
Table 1.
Biocatalytic addition of diethyl 2‐halomalonates to cinnamaldehyde catalyzed by 4‐OT F50V.
|
Entry |
Halomalonate |
Conv. [%][a] |
Product |
|
d.r. |
e.r.[b] |
|---|---|---|---|---|---|---|
|
1 |
diethyl 2‐fluoromalonate |
40 |
|
2 |
n.a. |
99:1[c] |
|
2 |
diethyl 2‐chloromalonate |
95 |
|
1 a |
>25:1[d] |
98:2[e] |
|
3 |
diethyl 2‐bromomalonate |
<5 |
n.d. |
|
n.d. |
n.d. |


[a] Determined by GC‐MS. [b] Determined by chiral‐phase HPLC analysis (Chiralpak IC) after reduction to the corresponding alcohol. The absolute configuration was assigned by comparison with an enantioenriched authentic standard (1 a) or by earlier reported chiral‐phase HPLC‐data (2).[ 4a , 4b , 7 ] [c] R:S. [d] Determined by 1H NMR (syn/anti). [e] 2S,3R:2R,3S. n.a.: not applicable; n.d.: not determined.
However, when diethyl 2‐fluoromalonate was replaced by diethyl 2‐chloromalonate, which harbors chlorine as a good leaving group, the reaction reached 95 % conversion, and the desired cyclopropane 1 a was obtained with very high diastereo‐ and enantiopurity (d.r.: >25:1, e.r.: 98:2; Table 1, entry 2). A control reaction under otherwise similar conditions but without enzyme did not yield 1 a. The reaction with diethyl 2‐bromomalonate only showed trace conversion; given that bromine has a higher leaving group ability than chlorine, this result was somewhat unexpected (an exploration of this phenomenon is provided below).
With a suitable nucleophile for the 4‐OT catalyzed cyclopropanation reaction in hand, we further optimized the enzyme by protein engineering for the addition of diethyl 2‐chloromalonate to cinnamaldehyde. By using a systematic mutagenesis strategy, amino acid positions Met‐45 and Phe‐50 were previously identified as hot spots to enhance the promiscuous Michael addition activity of 4‐OT.[ 5 , 8 ] We reasoned that these positions might similarly be important for the addition of diethyl 2‐chloromalonate to cinnamaldehyde and therefore targeted positions Met‐45 and Phe‐50 by constructing a double‐site saturation library with NNK codon degeneracy to ensure maximal diversity. This library was used to transform Escherichia coli cells and screened by evaluating enzyme activity in cell‐free extracts prepared from ≈3000 transformants. Improved 4‐OT variants were identified by monitoring the depletion of cinnamaldehyde in a spectrophotometric kinetic assay in multiwell plates (see Supporting Information). Selected variants with enhanced activity were purified to homogeneity (Figure S1), and their catalytic activity was assessed by determining specific activities. The single mutant 4‐OT F50V showed a 6‐fold improved specific activity compared to the wild‐type enzyme, while the engineered double mutants 4‐OT M45C/F50A, M45T/F50A, M45L/F50V and M45I/F50A displayed a 12‐ to 23‐fold enhanced specific activity (Figure 1). Gratifyingly, variant 4‐OT M45V/F50A showed a 30‐fold enhanced specific activity for the cyclopropanation of cinnamaldehyde compared to wild‐type 4‐OT, while retaining excellent diastereo‐ and enantioselectivity (d.r. of 1 a>25:1; e.r.=97:3). As the 4‐OT M45V/F50A variant showed the highest specific activity for the cyclopropanation reaction between diethyl 2‐chloromalonate and cinnamaldehyde, it was selected for further study.
Figure 1.

Engineering of 4‐OT for cyclopropanations. A) Reaction of the 4‐OT catalyzed cyclopropanation of diethyl 2‐chloromalonate and cinnamaldehyde to afford 1 a. B) Comparison of the specific activity of wild type 4‐OT and engineered 4‐OT variants for the cyclopropanation reaction between diethyl 2‐chloromalonate and cinnamaldehyde to yield 1 a. The specific activities are as follows: wt (3.0 mU mg−1), F50V (16.7 mU mg−1), M45C/F50A (36.0 mU mg−1), M45T/F50A (40.8 mU mg−1), M45L/F50V (43.7 mU mg−1), M45I/F50A (67.9 mU mg−1), M45V/F50A (89.4 mU mg−1). Reaction conditions: 1 mM cinnamaldehyde, 5 mM diethyl 2‐chloromalonate, 0.1 mg mL−1 4‐OT variant, 50 mM HEPES pH 6.5. Error bars represent the standard deviation of two measurements using the same enzyme batch (n=2).
For the evolved 4‐OT variant M45V/F50A, we also observed the superior performance of diethyl 2‐chloromalonate over diethyl 2‐bromomalonate as a nucleophile in the cyclopropanation of cinnamaldehyde to give 1 a. To obtain insights into this puzzling phenomenon, we performed several additional experiments. Spectrophotometric analysis of the 4‐OT M45V/F50A catalyzed reaction with diethyl 2‐bromomalonate and cinnamaldehyde showed that substrate turnover occurs in the initial phase of the reaction but quickly stops after several minutes (Figure S2). This suggests that the enzyme is rapidly and irreversibly inactivated by diethyl 2‐bromomalonate during the course of the reaction. To confirm the formation of a covalent enzyme‐inhibitor complex, 4‐OT M45V/F50A was isolated from the reaction mixture and analyzed by ESI‐MS. Compared to the unmodified enzyme, 4‐OT M45V/F50A incubated in the presence of diethyl 2‐bromomalonate showed a mass increase of +158.1 Da (Figure S3). This is consistent with the covalent modification of the enzyme with one molecule of diethyl malonate. In contrast, reactions with diethyl 2‐fluoromalonate or diethyl 2‐chloromalonate showed no or only minor modification of the enzyme. To gain further insights into the position of modification, the modified and unmodified 4‐OT M45V/F50A samples were digested with endoproteinase Glu‐C, and the resulting peptide mixtures were analyzed by LC‐MS/MS. A comparison of the detected peptide fragments of 4‐OT M45V/F50A modified with diethyl malonate to those of unmodified 4‐OT M45V/F50A showed that peptides that contain the Pro‐1 residue are the major sites of modification (mass increase: + 158.1 Da; Table S2 and S3). Indeed, analysis of the peptide fragments of 4‐OT M45V/F50A treated with diethyl 2‐fluoromalonate or diethyl 2‐chloromalonate showed no or only minor modifications of Pro‐1 containing peptides with diethyl malonate (Table S4 and S5). Taken together, these results suggest that like other bromo‐substituted compounds, such as 3‐bromopyruvate, [9] diethyl 2‐bromomalonate acts as a potent irreversible inhibitor of the enzyme by covalently modifying its nucleophilic Pro‐1 residue (Scheme S1). Due to the lower leaving group ability of chlorine and fluorine, the 2‐Cl‐ and 2‐F‐substituted diethyl malonates primarily function as substrates in the Michael addition to cinnamaldehyde, and only result in minor enzyme inactivation upon prolonged incubation.
Having engineered a 4‐OT variant (M45V/F50A) with enhanced activity and stereoselectivity, and understanding the reactivity of the diethyl 2‐halomalonates, the substrate scope of this designer enzyme was explored by testing a set of α,β‐unsaturated aldehydes using diethyl 2‐chloromalonate as the nucleophile (Table 2). To avoid enzyme inactivation of 4‐OT by diethyl 2‐chloromalonate, a 2.5‐fold excess of α,β‐unsaturated aldehydes (5 mM) over diethyl 2‐chloromalonate (2 mM) was used in these semi‐preparative scale reactions. Pleasingly, the results showed that 4‐OT M45V/F50A accepts various substituted cinnamaldehydes and catalyzes their cyclopropanation to yield the corresponding products 1 a–i with excellent enantiopurity (e.r. up to 99:1). The attained d.r. values (up to >25:1) indicate that the enzyme has good stereocontrol over both carbons of the double bond of the cinnamaldehyde substrate. 4‐OT M45V/F50A did not promote the synthesis of aliphatic cyclopropane 1 j and tricarboxylate 1 k, indicating that the enzyme has a strong preference for aromatic α,β‐unsaturated aldehydes. All enzymatic cyclopropane products have the syn configuration with the major enantiomer being 2S,3R for products 1 a,d,e,i, 2S,3S for 1 b,c and 2R,3S for 1 f,g,h. Notably, products 1 a–i have the same geometry and the deviant configuration is due to different prioritization of the substituents at the respective chiral center. Formation of the syn product is consistent with the stereochemical outcome of other 4‐OT catalyzed reactions with cinnamaldehydes, in which the nucleophile exclusively attacks from the re‐face of the enzyme‐bound iminium ion.[ 5 , 6 ] A 4‐OT M45V/F50A variant in which the Pro‐1 residue is replaced by an alanine (P1A/M45V/F50A) has almost completely lost its cyclopropanation activity (Figure S36), providing support for a reaction mechanism that proceeds via iminium ion formation between the active site Pro‐1 residue and the cinnamaldehyde substrate.
Table 2.
4‐OT M45V/F50A catalyzed enantioselective synthesis of cyclopropanes.

[a] Reaction conditions: diethyl 2‐chloromalonate (2 mM, 0.154 mmol), α,β‐unsaturated aldehyde (5 mM), and 30 μM 4‐OT M45V/F50A in 10 % EtOH (10 % MeCN for 1 e,i), and 50 mM MES pH 6.2 at 120 rpm, 18 °C, N2 (to avoid potential minor aldehyde oxidation; notably, reactions can also be performed aerobically). Reaction time: 12–71 h; Estimated conversions by 1H NMR: 20 %–>95 %; [b] Isolated yields after silica column purification (based on diethyl 2‐chloromalonate); Yields could be further improved by optimization of the extraction and purification procedures. [c] Determined by chiral‐phase HPLC analysis (Chiralpak IC/ID) after reduction of the aldehyde to the corresponding alcohol. Absolute configurations (2S,3R‐1 a,d,e,i; 2R,3S‐1 f,g,h; 2S,3S‐1 b,c) were assigned by comparison with authentic standards.[ 4a , 4b ] [d] Determined by 1H NMR (syn/anti).
In summary, biocatalytic methodologies for the asymmetric synthesis of difficult to prepare cyclopropanes have gained increasing interest and attention in the literature in the past number of years. Typically, these syntheses depend on repurposed or artificial heme enzymes for the formation of a highly reactive carbenoid intermediate starting from ethyl diazoacetate. [10] Furthermore, redesigned FMN‐dependent ene‐reductases have been exploited for asymmetric reductive cyclopropanations. [11] Inspired by developments in the organocatalysis field, [4] we here present the engineering of an unusual cofactor‐independent biocatalyst for the preparation of various enantioenriched cyclopropanes. To this end, we first demonstrated that the active site of 4‐OT can give rise to synthetically useful promiscuous cyclopropanation activity. We subsequently improved this promiscuous activity of 4‐OT by data‐driven (i.e., mutability‐landscape guided) engineering, [8] yielding variant M45V/F50A that can efficiently promote the addition of diethyl 2‐chloromalonate to α,β‐unsaturated aldehydes, enabling the biocatalytic synthesis of cyclopropanes with excellent stereochemical purities (e.r. up to 99:1; d.r. up to >25:1). Remarkably, this simple biotransformation thus generates two new carbon‐carbon bonds and two new chiral centers with excellent stereocontrol. Given that the reactions proceed via a reactive enzyme‐bound iminium ion intermediate and that the promiscuous cyclopropanation activity of 4‐OT can be significantly enhanced by introducing only two amino acid substitutions, it is feasible that further optimization of the enzyme's activity and selectivity can be achieved by using a random directed evolution approach. We have initiated studies aimed at exploring different electrophiles and nucleophiles (with distinct leaving groups), which could lead to the asymmetric enzymatic synthesis of new cyclopropanes and related Michael addition products.
The enzyme 4‐OT can be easily obtained in large amounts by heterologous expression in Escherichia coli. The 4‐OT catalyzed reactions proceed in environmentally friendly aqueous buffer rather than in organic solvent or two‐phase systems and require lower catalyst loading compared to organocatalytic procedures. [4] Several previously reported heme enzymes used for preparation of cyclopropanes suffer from the need to work in an oxygen free environment to avoid undesired competitive epoxide formation. However, this limitation has been overcome in other heme enzymes by histidine ligation of the heme cofactor, which largely abolishes oxo‐transfer chemistry, enabling aerobic cyclopropanations at preparative scale.[ 3h , 3i ] Although we have used an N2 atmosphere to avoid potential minor aldehyde oxidation, the 4‐OT catalyzed cyclopropanations can also be performed under aerobic conditions, which offers an advantage for preparative scale reactions.
Our results showcase the potential of catalytic promiscuity for the creation of new biocatalysts for non‐natural reactions. [12] The non‐native cyclopropanation activity of 4‐OT, together with its previously reported peroxygenase, [6] Michaelase[ 5 , 8 , 13 ] and aldolase activities, [14] highlights the versatile catalytic repertoire of this promiscuous enzyme for asymmetric carbon‐oxygen and carbon‐carbon bond‐forming reactions.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank M.H. de Vries for support in acquiring mass spectrometry data. We acknowledge financial support from the Netherlands Organisation of Scientific Research (VICI grant 724.016.002).
A. Kunzendorf, G. Xu, M. Saifuddin, T. Saravanan, G. J. Poelarends, Angew. Chem. Int. Ed. 2021, 60, 24059.
References
- 1.
- 1a. Wessjohann L. A., Brandt W., Thiemann T., Chem. Rev. 2003, 103, 1625–1648; [DOI] [PubMed] [Google Scholar]
- 1b. Talele T. T., J. Med. Chem. 2016, 59, 8712–8756. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. de Meijere A., Angew. Chem. Int. Ed. Engl. 1979, 18, 809–826; [Google Scholar]; Angew. Chem. 1979, 91, 867–884; [Google Scholar]
- 2b. Wiberg K. B., Angew. Chem. Int. Ed. Engl. 1986, 25, 312–322; [Google Scholar]; Angew. Chem. 1986, 98, 312–322. [Google Scholar]
- 3.
- 3a. Coelho P. S., Brustad E. M., Kannan A., Arnold F. H., Science 2013, 339, 307–310; [DOI] [PubMed] [Google Scholar]
- 3b. Coelho P. S., Wang Z. J., Ener M. E., Baril S. A., Kannan A., Arnold F. H., Brustad E. M., Nat. Chem. Biol. 2013, 9, 485–487; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Bordeaux M., Tyagi V., Fasan R., Angew. Chem. Int. Ed. 2015, 54, 1744–1748; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 1764–1768; [Google Scholar]
- 3d. Key H. M., Dydio P., Clark D. S., Hartwig J. F., Nature 2016, 534, 534–537; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3e. Oohora K., Meichin H., Zhao L., Wolf M. W., Nakayama A., Hasegawa J., Lehnert N., Hayashi T., J. Am. Chem. Soc. 2017, 139, 17265–17268; [DOI] [PubMed] [Google Scholar]
- 3f. Villarino L., Splan K. E., Reddem E., Alonso-Cotchico L., Gutiérrez de Souza C., Lledós A., Maréchal J.-D., Thunnissen A.-M. W. H., Roelfes G., Angew. Chem. Int. Ed. 2018, 57, 7785–7789; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 7911–7915; [Google Scholar]
- 3g. Stenner R., Steventon J. W., Seddon A., Anderson J. L. R., Proc. Natl. Acad. Sci. USA 2020, 117, 1419–1428; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3h. Renata H., Wang Z. J., Kittoa R. Z., Arnold F. H., Catal. Sci. Technol. 2014, 4, 3640–3643; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3i. Wang Z. J., Renata H., Peck N. E., Farwell C. C., Coelho P. S., Arnold F. H., Angew. Chem. Int. Ed. 2014, 53, 6810–6813; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 6928–6931. [Google Scholar]
- 4.
- 4a. Xie H., Zu L., Li H., Wang J., Wang W., J. Am. Chem. Soc. 2007, 129, 10886–10894; [DOI] [PubMed] [Google Scholar]
- 4b. Rios R., Sundén H., Vesely J., Zhao G.-L., Dziedzic P., Córdova A., Adv. Synth. Catal. 2007, 349, 1028–1032; [Google Scholar]
- 4c. Ibrahem I., Zhao G.-L., Rios R., Vesely J., Sundén H., Dziedzic P., Córdova A., Chem. Eur. J. 2008, 14, 7867–7879; [DOI] [PubMed] [Google Scholar]
- 4d. Companyó X., Alba A.-N., Cárdenas F., Moyano A., Rios R., Eur. J. Org. Chem. 2009, 3075–3080; [Google Scholar]
- 4e. Uria U., Vicario J. L., Badía D., Carrillo L., Reyes E., Pesquera A., Synthesis 2010, 701–713; [Google Scholar]
- 4f. Li W., Li X., Ye T., Wu W., Liang X., Ye J., Tetrahedron Lett. 2011, 52, 2715–2718; [Google Scholar]
- 4g. Rueping M., Sundén H., Hubener L., Sugiono E., Chem. Commun. 2012, 48, 2201–2203; [DOI] [PubMed] [Google Scholar]
- 4h. Martínez J. I., Reyes E., Uria U., Carrillo L., Vicario J. L., ChemCatChem 2013, 5, 2240–2247. [Google Scholar]
- 5. Guo C., Saifuddin M., Saravanan T., Sharifi M., Poelarends G. J., ACS Catal. 2019, 9, 4369–4373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Xu G., Crotti M., Saravanan T., Kataja K. M., Poelarends G. J., Angew. Chem. Int. Ed. 2020, 59, 10374–10378; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 10460–10464. [Google Scholar]
- 7. Companyó X., Hejnová M., Kamlar M., Vesely J., Moyano A., Rios R., Tetrahedron Lett. 2009, 50, 5021–5024. [Google Scholar]
- 8. van der Meer J.-Y., Poddar H., Baas B., Miao Y., Rahimi M., Kunzendorf A., van Merkerk R., Tepper P. G., Geertsema E. M., Thunnissen A. W. H., Quax W. J., Poelarends G. J., Nat. Commun. 2016, 7, 10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Stivers J. T., Abeygunawardana C., Mildvan A. S., Hajipour G., Whitman C. P., Biochemistry 1996, 35, 814–823; [DOI] [PubMed] [Google Scholar]
- 9b. Stivers J. T., Abeygunawardana C., Mildvan A. S., Hajipour G., Whitman C. P., Chen L. H., Biochemistry 1996, 35, 803–813. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Gober J. G., Brustad E. M., Curr. Opin. Chem. Biol. 2016, 35, 124–132; [DOI] [PubMed] [Google Scholar]
- 10b. Kaur P., Tyagi V., Adv. Synth. Catal. 2021, 363, 877–905; [Google Scholar]
- 10c. Roelfes G., J. Inorg. Biochem. 2021, 222, 111523. [DOI] [PubMed] [Google Scholar]
- 11. Heckenbichler K., Schweiger A., Brandner L. A., Binter A., Toplak M., Macheroux P., Gruber K., Breinbauer R., Angew. Chem. Int. Ed. 2018, 57, 7240–7244; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 7360–7364. [Google Scholar]
- 12.
- 12a. Khersonsky O., Tawfik D. S., Annu. Rev. Biochem. 2010, 79, 471–505; [DOI] [PubMed] [Google Scholar]
- 12b. Humble M. S., Berglund P., Eur. J. Org. Chem. 2011, 3391–3401; [Google Scholar]
- 12c. Miao Y., Rahimi M., Geertsema E. M., Poelarends G. J., Curr. Opin. Chem. Biol. 2015, 25, 115–123; [DOI] [PubMed] [Google Scholar]
- 12d. Leveson-Gower R. B., Mayer C., Roelfes G., Nat. Rev. Chem. 2019, 3, 687–705; [Google Scholar]
- 12e. Chen K., Arnold F. H., Nat. Catal. 2020, 3, 203–213. [Google Scholar]
- 13.
- 13a. Zandvoort E., Geertsema E. M., Baas B.-J., Quax W. J., Poelarends G. J., Angew. Chem. Int. Ed. 2012, 51, 1240–1243; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 1266–1269; [Google Scholar]
- 13b. Biewenga L., Saravanan T., Kunzendorf A., van der Meer J.-Y., Pijning T., Tepper P. G., van Merkerk R., Charnock S. J., Thunnissen A.-M. W. H., Poelarends G. J., ACS Catal. 2019, 9, 1503–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Saifuddin M., Guo C., Biewenga L., Saravanan T., Charnock S. J., Poelarends G. J., ACS Catal. 2020, 10, 2522–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information