

Abstract
Aims
HTL0009936 is a selective M1 muscarinic receptor agonist in development for cognitive dysfunction in Alzheimer's disease. Safety, tolerability and pharmacokinetics and exploratory pharmacodynamic effects of HTL0009936 administered by continuous IV infusion at steady state were investigated in elderly subjects with below average cognitive functioning (BACF).
Methods
Part A was a four‐treatment open label sequential study in healthy elderly investigating 10–83 mg HTL0009936 (IV) and a 24 mg HTL0009936 single oral dose. Part B was a five‐treatment randomized, double‐blind, placebo and physostigmine controlled cross‐over study with IV HTL0009936 in elderly subjects with BACF. Pharmacodynamic assessments were performed using neurocognitive and electrophysiological tests.
Results
Pharmacokinetics of HTL0009936 showed dose‐proportional increases in exposure with a mean half‐life of 2.4 hours. HTL0009936 was well‐tolerated with transient dose‐related adverse events (AEs). Small increases in mean systolic blood pressure of 7.12 mmHg (95% CI [3.99–10.24]) and in diastolic of 5.32 mmHg (95% CI [3.18–7.47]) were noted at the highest dose in part B. Overall, there was suggestive, but no definitive, positive or negative pharmacodynamic effects. Statistically significant effects were observed on P300 with HTL0009936 and adaptive tracking with physostigmine.
Conclusions
HTL0009936 showed well‐characterized pharmacokinetics and single doses were safe and generally well‐tolerated in healthy elderly subjects. Due to physostigmine tolerability issues and subject burden, the study design was changed and some pharmacodynamic assessments (neurocognitive) were performed at suboptimal drug exposures. Therefore no clear conclusions can be made on pharmacodynamic effects of HTL0009936, although an effect on P300 is suggestive of central target engagement.
Keywords: Alzheimer's disease, cholinergic system, elderly, M1 receptor, muscarinic receptors, pharmacokinetics, safety
What is already known about this subject
Degeneration of cholinergic neurons contributes to cognitive dysfunction in Alzheimer's disease (AD).
The M1 muscarinic receptor plays a key role in cognitive function.
The M1/M4 receptor agonist xanomeline showed efficacy in AD but was withdrawn due to adverse effects. Selectively targeting M1 receptors may be a more promising approach to improve cognition without adverse events.
What this study adds
The intravenously administered selective M1 mAChR agonist HTL0009936 was well‐tolerated by elderly subjects up to doses of 83 mg.
HTL0009936 showed dose‐proportional exposures with a half‐life between 2.2 and 2.6 hours and modest variability.
No clear positive or negative effects could be detected for both drugs due to study limitations.
1. INTRODUCTION
Alzheimer's Disease (AD) and Dementia with Lewy Body (DLB) are the most common cause of dementia. 1 Clinically, AD and DLB are characterized by the progressive decline of cognitive functions. Research has shown that AD is characterized by a significant and progressive loss of cholinergic neurons, especially in the nucleus basalis of Meynert, along with their cortically projecting axons, 2 and this cholinergic degeneration is correlated with cognitive decline. 3 , 4 To date, no curative treatment is available and patients can only benefit from symptomatic treatments, such as the acetylcholinesterase inhibitors (AChEIs) galantamine, donepezil and rivastigmine. 5 However, the efficacy of treatment with AChEIs is moderate 6 , 7 , 8 due to only partial central inhibition of AChEIs 9 , 10 and it often leads to gastrointestinal side effects (e.g. nausea, vomiting and diarrhoea) associated with increased activation of peripherally located muscarinic receptors, causing dose limitations and a significant burden for patients. 6 , 7 , 8
The cholinergic receptors comprise two broad classes; the ionotropic nicotinic receptors and metabotropic muscarinic receptors. The muscarinic receptors are a group of Class I G‐protein‐coupled receptors (GPCRs) comprising five distinct sub‐types, termed M1, M2, M3, M4 and M5. 11 Drugs that selectively target specific muscarinic receptor type(s) may enhance cognitive and behavioural function in AD and DLB patients while minimizing the negative side‐effects associated with non‐selective activation of all muscarinic receptor types, in particular M2 and M3 receptors that have been predominantly linked to the gastrointestinal and cardiovascular side effects. 12 The muscarinic M1 receptor (M1 AChR) is predominant in the central nervous system (CNS) and found to be expressed in the prefrontal cortex, striatum and hippocampus. These brain areas are known to be associated with cognitive processes. 13 , 14 The M1 AChR is relatively well preserved in AD and DLB patients. 15 , 16 Drugs that selectively target M1 AChR could be potential treatment for cognitive and behavioural dysfunction in AD and DLB. 12 , 17 Additionally, the effects of selective M1 AChR agonists are independent of the existence of cholinergic tone in the CNS, and their benefit may be sustained further into disease progression than the benefit of cholinesterase inhibitors or M1 receptor‐positive allosteric modulators which rely on pre‐synaptic cholinergic tone.
HTL0009936 ((S)‐Ethyl 4‐(4‐[1‐methylcyclobutylcarbamoyl]piperidin‐1‐yl)azepane‐1‐carboxylate) 18 is a potent and selective M1 AChR agonist that is currently under development for the symptomatic treatment of the cognitive symptoms of dementias including AD and DLB. HTL0009936 has no detectable activity at M2 and M3 AChRs, and a seven‐fold margin of functional selectivity over M4 AChR in vitro. It has been investigated in an oral solution formulation, dosed at 1–175 mg in a phase I trial in young adults and elderly subjects (in preparation). Pharmacokinetics (PK) of oral HTL0009936 showed a low oral bioavailability and a significant degree of variability between subjects. In order to reduce this variability and to ensure sustained exposure within the central nervous system (CNS) over the period of cognitive testing, HTL0009936 was given as an intravenous infusion in the current study.
This study was conducted in two parts. The aim of part A was to evaluate the safety, tolerability and PK in elderly subjects in order to identify a well‐tolerated dosing regimen to take forward into part B, and to determine the absolute oral bioavailability of HTL0009936. In part B safety, tolerability, PK and exploratory PD of IV HTL0009936 were investigated in elderly subjects with below average cognitive functioning (BACF). These subjects had no evidence of progressive cognitive deterioration.
2. METHODS
This study was approved by the medical ethics review board Stichting Beoordeling Ethiek Biomedisch Onderzoek (BEBO, Assen, The Netherlands) and was conducted according to the Dutch Act on Medical Research Involving Human Subjects (WMO) and in compliance with Good Clinical Practice (ICH‐GCP) and the Declaration of Helsinki. 19
2.1. Trial design and subjects
This study consisted of part A and B. Part A was an initial pilot phase administering 0.1 and 1 mg HTL009936 given as a 30 minute infusion followed by a four‐treatment open label sequential study with IV and oral administration of HTL0009936 in elderly subjects (n = 10). The objectives of part A were to evaluate the safety, tolerability and the PK profile of HTL0009936, to identify a well‐tolerated dosing regimen for part B and to determine the absolute oral bioavailability of HTL0009936. Part B was a five‐treatment randomized, double‐blind, placebo and positive comparator‐controlled crossover study with IV HTL0009936 in elderly subjects with BACF (n = 33). The objectives of part B were to evaluate safety, tolerability and PK of HTL0009936 and to evaluate PD in comparison to placebo and a positive comparator.
In both parts A and B, subjects were healthy male and female elderly (65+ years) with a maximum blood pressure of 140/90 mm Hg and a heart rate between 45–100 bpm at screening. Use of antihypertensive drugs was not allowed. Consumption of alcohol and caffeine‐containing products, use of nicotine‐containing products and drugs influencing CYP3A4 and CYP2D6 activity were not allowed prior to and during the study. Subjects were defined as intermediate (IM) or extensive (EM) CYP2D6 metabolizers based on their genotype and were excluded if they were poor or ultra‐rapid metabolizers in order to minimize variability in the steady state plasma concentrations in part B.
Subjects in part B functioned below average on tests of cognitive functioning based on one of their scores on three tests: the auditory verbal learning test (AVLT) (memory), the word fluency test category (executive function), and the adaptive tracking test (attention). Below average cognitive functioning was defined as a score of ≤−1 SD on at least one of the tests. The reference values for the AVLT and word fluency test were based on available norms. 20 The mean score of the adaptive tracking test was calculated from data from previously performed studies in healthy elderly. Age and education level were taken into account in the calculation of the score. Per cognitive domain, a minimum of eight subjects showed below average functioning. Subjects were excluded if they had a Clinical Dementia Rating scale (CDR) score of >0, a mini‐mental state examination (MMSE) score of <24 or a Becks Depression Index‐II (BDI‐II) score of >13. Thus, subjects did not have MCI (mild cognitive impairment) and did not have evidence of progressive cognitive deterioration and it was therefore unknown whether they were cholinergically deficient.
2.2. Materials
In part A, HTL0009936 was administered as an IV solution and as an oral solution. In the first treatment session, two subjects were dosed 0.1 mg HTL0009936 IV according to a sentinel procedure, followed by two subjects dosed 1 mg HTL0009936 IV, followed by six subjects dosed 10 mg HTL0009936 IV. The latter six subjects were administered 49.2 mg HTL0009936 IV during the second treatment session, 83 mg HTL0009936 IV during the third treatment session, and 24 mg HTL0009936 orally during the fourth treatment session to determine the absolute oral bioavailability. The IV administration lasted up to 5 hours including the loading phase that varied per dose from 30 minutes to 2 hours. Safety, tolerability and PK data of part A was used to find a well‐tolerated dosing regimen for part B.
In part B, subjects received the following IV treatments in random sequence (30 sequences were used): 13.5 mg HTL0009936 in order to target an average concentration of HTL0009936 in plasma during infusion of the maintenance dose (C mean) of 25 ng/mL, 40 mg HTL0009936 in order to target a C mean of 75 ng/mL, 79.5 mg HTL0009936 in order to target a C mean of 150 ng/mL, placebo (saline solution [sodium chloride 0.9%]), and physostigmine salicylate at a rate of 1 mg/h for 50 minutes as positive comparator in combination with an IV bolus administration of 0.2 mg glycopyrrolate bromide (a peripheral muscarinic antagonist) administered immediately prior to physostigmine administration. 21 Physostigmine salicylate has reversed temporary cognitive impairment in cognitively normal subjects that was induced by administration of the anticholinergic drug scopolamine. 22 , 23 The dual infusion of HTL0009936 in part B consisted of a 1 hour loading dose in order to reach the C mean followed by a 4 hour maintenance dose designed to maintain the target C mean. As the infusion regimens for the study drug and the positive comparator were different, this study comprised a double‐dummy condition.
2.3. Safety and tolerability assessments
For parts A and B, all subjects underwent medical screening, including assessment of medical history, physical examination, urine drug screen, vital signs, ECG and safety laboratory measurements. During treatment periods, safety was assessed by monitoring of adverse events (AEs), vital signs, ECG, 5‐hour Holter monitoring, and safety chemistry and haematology blood sampling. Following a protocol amendment, subjects were to be withdrawn when a rise of >40% in systolic or diastolic blood pressure was measured as compared to the mean of three pre‐dose vital signs measurements and blood pressure >150/90 mm Hg or when the blood pressure was >180/115 mm Hg regardless of the change from baseline.
2.4. Pharmacokinetic assessments
In part A, venous blood samples were collected pre‐dose and post‐dose at different times during the different treatment sessions because of varying loading times. During all treatment sessions in part B, PK was sampled according to the same schedule pre dose, 9–15 times within the first 8 hours after starting the administration and at 12 and 24 hours post dose. Urine was collected continuously for PK determination of HTL0009936 (Supplementary Table S1).
All HTL0009936 plasma and urine concentrations were analysed using an achiral liquid chromatography with tandem mass spectrometric detection (LC–MS/MS) assay validated according to current guidelines. The detection range was 0.5–1000 ng/mL. Physostigmine plasma concentrations were determined using a validated LC–MS/MS assay with a quantification range of 0.10–10 ng/mL.
PK non‐compartmental analysis was performed to determine the maximum plasma concentration (C max), time to reach C max (T max), area under the concentration–time curve from time of dosing to the last quantifiable concentration measurement (AUC0‐last), apparent terminal elimination rate constant (lambda‐z), AUC from time of dosing to infinity (AUC0‐∞), apparent terminal half‐life (t ½), total plasma clearance (CLp), volume of distribution (Vd), absolute bioavailability (F), amount unchanged in urine (Ae), fraction excreted in urine (fe) and renal clearance (CLr). The AUC was calculated using the linear‐logarithmic trapezoidal method. Dose‐proportionality was evaluated by making pair‐wise comparisons of the increase in dose and the corresponding increase in exposure between dose levels. However, in part A, the loading dose was not a constant fraction of the total dose. Therefore dose‐exposure proportionality of C max was determined by relating the C max to the loading dose only. The software used for non‐compartmental analysis was R version 2.14.1. 24
2.5. Pharmacodynamic assessments
Only in part B of this study were PD assessments using both the NeuroCart 25 and the Cambridge Neuropsychological Test Automated Battery (CANTAB) 26 performed. The NeuroCart and CANTAB are test batteries that include cognitive tests that can be used to examine effects of CNS‐active drugs on a wide range of cognitive domains. NeuroCart and CANTAB tests have previously been shown to be sensitive to cholinergic modulation. 27 , 28 , 29 The NeuroCart also includes neurophysiological measurements. Blood pressure and pulse rate were considered both as safety and PD measurements.
The following NeuroCart tests were performed: the adaptive tracking test measured attention and visuomotor coordination, 25 , 30 , 31 the Milner maze test was used to evaluate spatial working memory, learning and executive function, 32 the n‐back task was used to assess (short‐term) working memory, 33 , 34 , 35 pupil size was measured to monitor any drug effects on the sympathetic nervous system, 36 , 37 synaptic activity was assessed using electrophysiology and included resting electroencephalography (EEG, power in delta, theta, alpha, beta and gamma bands) and the event‐related potentials (ERP) P300 and Mismatch negativity (MMN). 38 , 39 P300 is related to an early attention process and is used as marker for attention 40 and memory. 40 , 41 MMN is related to central auditory processing and is used as a marker for auditory memory. 42 Visual verbal learning test (VVLT) measured the whole scope of learning behaviour (i.e., acquisition, consolidation, storage and retrieval), 25 and a visual analogue scale was used to evaluate subjective nausea. The Leeds Sleep Evaluation Questionnaire (LSEQ) was used to assess changes in sleep quality. 43 The following CANTAB tests were performed: the paired associates learning test assessed visual memory, new learning and evaluated episodic memory, 44 the rapid visual information processing test was used to measure sustained attention, 45 and the spatial working memory test required retention and manipulation of visuospatial information. 46 Detailed task descriptions are provided in the Supplementary Information.
PD tests were performed repeatedly and the timing was based on PK characteristics of HTL0009936 measured in a previous study in humans (maximum drug levels were measured in the CSF 1–2 hours after plasma T max). PD assessments were conducted at baseline (pre‐dose) and between 1 hour and 8 hours post treatment. While the electrophysiological assessments ERPs MMN and P300, and EEG and NeuroCart assessments were performed during steady‐state levels of HTL0009936, due to heavy study burden, the three CANTAB assessments were performed at 5 hours post start of treatment when infusion was stopped and plasma levels of HTL0009936 were declining below target exposure levels. All post‐drug assessments for physostigmine were performed after infusion was stopped at 50 minutes post dose when plasma levels were declining and low.
2.6. Statistics
No formal power calculations were performed to assess sample size in part A. The sample size of ten subjects was considered adequate and a compromise between minimizing exposure and the need to provide sufficient data in order to find a well‐tolerated dosing regimen for part B and assess the bioavailability of oral HTL0009936. In part B, a sample size of 30 elderly subjects was defined to have 80% power to detect a 1.53%‐point difference in the adaptive tracking task, assuming a standard deviation of 2.9, using a paired t‐test with a two‐sided significance level of 0.05. Adaptive tracking was chosen to set the sample size in this exploratory study because it was the task shown previously to be most sensitive to cholinergic stimulation in studies of donepezil. 29
The PD analysis population per treatment session comprised all subjects who had at least one post‐baseline assessment of any parameter being analysed. Repeatedly measured PD variables (NeuroCart tests, CANTAB tests, blood pressure and pulse rate) were analysed with a mixed model analysis of covariance with treatment, period, time and treatment by time as fixed factors, and subject, subject by treatment and subject by time as random factors, and the average baseline measurement as covariate. The single measured PD variables were analysed with a mixed model analysis of variance with treatment and period as fixed factors and subject as random factor and the baseline measurement, if available, as covariate. The mean outcomes are presented as least square means (LSMs). Only PD data that was measured within 8 hours after starting the HTL0009936 administration and within 2 hours after start of the physostigmine administration were included in the analyses. PD tests performed within 2 hours after start of physostigmine were adaptive tracking test, VAS nausea, n‐back test, pupillometry, EEG and ERP (P300 and MMN). The following contrasts were calculated: HTL0009936 vs placebo and physostigmine vs placebo. All calculations were performed using SAS (version 9.4, SAS, Cary, NC).
3. RESULTS
3.1. Subjects
Subject demographics and baseline characteristics are summarized in Table 1. A total of ten subjects participated in part A. No subjects dropped out of part A after drug administration.
TABLE 1.
Summary demographics and baseline characteristics, mean (SD)
| Part A (n = 10) | Part B (n = 33) | |
|---|---|---|
| Age, years | 70.2 (3.6) | 70 (5.0) |
| Weight, kg | 74.8 (12.3) | 74.2 (8.7) |
| BMI, kg/m2 | 25.5 (3.7) | 25.5 (2.5) |
| Gender, n (%) | ||
| Female | 5 (50) | 17 (52) |
| Male | 5 (50) | 16 (48) |
| CYP2D6 predicted phenotype, n (%) | ||
| Extensive metabolizer | 10 (100) | 27 (82) |
| Intermediate metabolizer | 0 | 6 (18) |
| Cognitive score at screening < 1 SD, n (%) | ||
| Word fluency | N/A | 12 (36) |
| AVLT | N/A | 13 (39) |
| Adaptive tracking test | N/A | 14 (42) |
In part B, 33 subjects were enrolled. Eight subjects withdrew or were withdrawn before the end of Part B for personal reasons (n = 4) and safety reasons (n = 4) and (as per protocol) three of them were replaced. Of the four subjects that were withdrawn due to safety reasons, one subject presented with a raised serum creatinine after completing the 13.5 mg dose before starting the second dosing day; one subject completed three dosing days (placebo, physostigmine and 79.5 mg HTL0009936 respectively) before withdrawal due to a second degree atrioventricular block on the Holter registration; one subject was withdrawn after completing the placebo and 13.5 mg HTL0009936 dosing day because of ST‐segment depression seen on Holter registration; one subject completed the 40 mg, 79.5 mg, physostigmine and placebo dosing days before withdrawal due to ST segment depression on the Holter registration.
All treatment infusions were started by at least 28 subjects and completed by at least 26 subjects (Figure 1).
FIGURE 1.
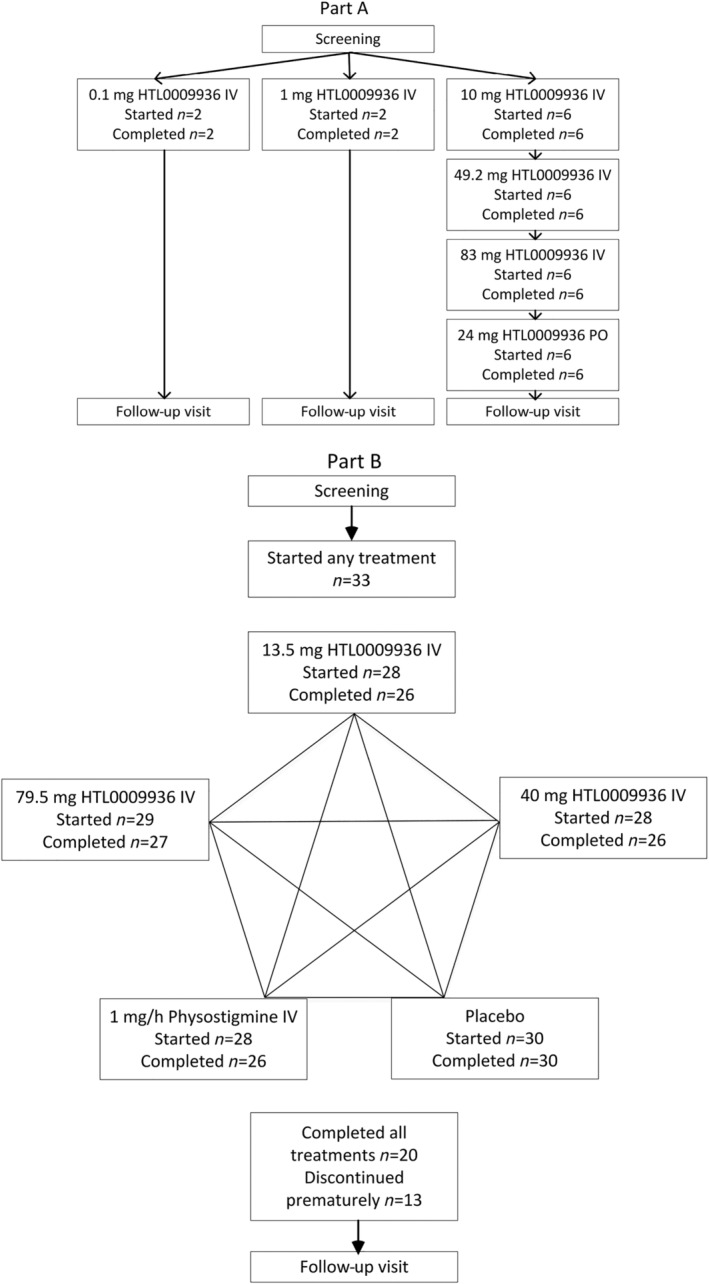
Study design of part A (four‐treatment open label sequential design) and B (five‐treatment randomized, placebo and positive comparator‐controlled crossover design) and the number of subjects that started and completed the treatment
3.2. Safety and tolerability
In seven cases, study drug administration had to be prematurely stopped due to a clinically significant rise in blood pressure. In part A there was one such case. Of the six cases of clinically significant rises in blood pressure in part B, one was related to administration of physostigmine, the remaining five were attributed to administration of HTL0009936 (three of which were experienced in the same subject). No subject was withdrawn from the study as a result of increased blood pressure.
In both parts A and B, only mild or moderate self‐limiting treatment emergent adverse events (TEAEs) were reported and there were no serious adverse events. The most frequently reported TEAEs in part B following HTL0009936 administration were headache (14 AEs), hyperhidrosis (6 AEs) and nausea (6 AEs).
One subject was withdrawn from the study because an ST‐depression was recorded during the Holter monitoring between 2 and 3 hours after starting the 13.5 mg HTL0009936 dose. There were no relevant changes in ECG, physical examination findings or laboratory values.
3.3. Pharmacokinetics
The PK profile of HTL0009936 was well‐characterized after IV infusion and oral dosing in elderly subjects (Figure 2 and Tables 2, 3, 4). In part B, targeted C mean were reached. Systemic exposure after IV dosing was dose‐proportional over a wide dose range and showed an inter‐subject variability of ~30%CV, irrespective of CYP2D6 intermediate or extensive metabolizer predicted phenotype. Plasma clearance was 68–81 L/hr with a volume of distribution of 222–262 L consistent with a short half‐life (2.2–2.6 h). Renal clearance was a significant route of elimination of unchanged HTL0009936 (CLr 8.0 L/h, range 3.4–14.2 L/h) with about 10% of the dose excreted unchanged after IV dosing. Absolute oral bioavailability was established to be about 15% ranging from 8.7 to 27%. Variability after oral administration (~50%CV) was higher compared to IV infusion and CYP2D6 predicted phenotype was found to be related to systemic exposure and clearance of HTL0009936, with higher clearance and lower exposure in EM subjects compared with IM subjects (Supplementary Table S4).
FIGURE 2.
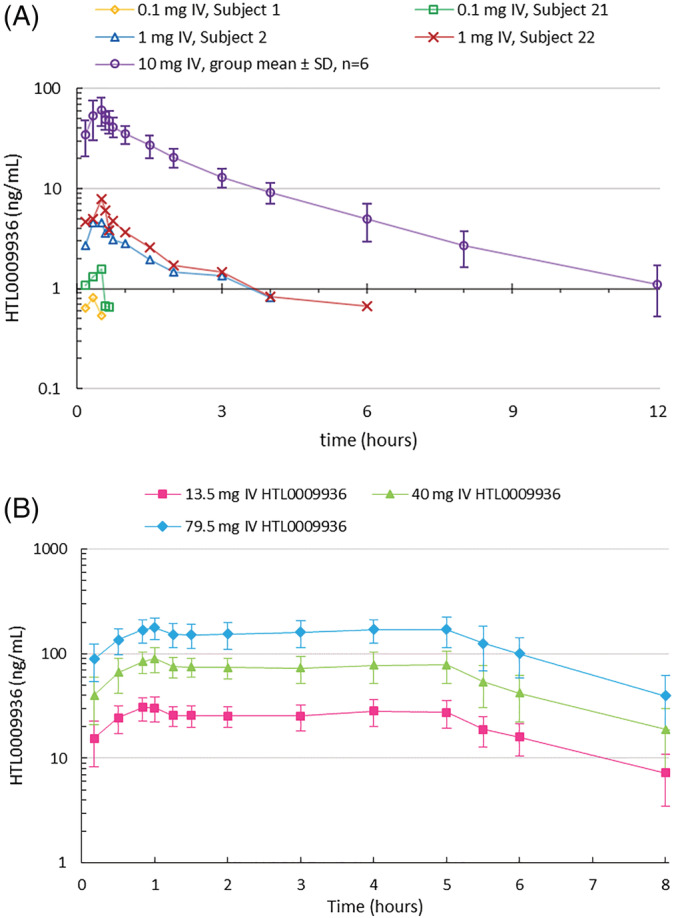
A. Concentration–time profiles of HTL0009936 single IV infusion at 0.1 mg (n = 2), 1 mg (n = 2) and 10 mg in part A (mean ± SD for n = 6). B. Concentration–time profiles at 13.5, 40 and 79.5 mg HTL0009936 by dual IV infusion in part B (arithmetic mean ± SD; n = 28–29). Profile truncated at 8 hours to show plateau during maintenance dose
TABLE 2.
Summary of HTL0009936 exposures after IV infusion in part A, mean (%CV) or [range]
| Dose (mg) | Observed C mean (ng/mL) | T max (hr) | C max (ng/mL) | AUC0–24 (hr.ng/mL) | AUC0‐∞ (hr.ng/mL) | t½ (hr) | CLp (L/hr) | CLr (L/hr) |
|---|---|---|---|---|---|---|---|---|
| 10 a | n/a | 0.50 [0.33–0.58] | 59.5 (35) | 120 (24) | 124 (24) | 2.2 (12) | 81 (24) | 8.7 (27) |
| 49.2 b | 97 (22) | 0.50 [0.17–5.5] | 125 (33) | 684 (24) | 691 (24) | 2.3 (35) | 71 (24) | 7.2 (41) |
| 83 c | 172 (17) | 2.0 [2.0–3.0] | 197 (20) | 1130 (17) | 1140 (16) | 2.4 (25) | 73 (17) | 7.8 (25) |
Geometric mean and (geometric %CV) except T max median [minimum − maximum] for n = 6 per dose except n = 5 at 83 mg. AUC0‐∞, area under the plasma concentration–time curve from zero extrapolated to infinity; AUC0–24, area under the plasma concentration–time curve from zero to 24 hours post dose; C max, maximum plasma concentration; C mean, mean plasma concentration during maintenance infusion; CLp, total plasma clearance; CLr, renal clearance; T max, time to C max; t½, apparent terminal half‐life.
10 mg over 0.5 hr at 33.2 mL/h.
14.1 mg over 0.5 hr at 47 mL/hr + 35.1 mg over 4.5 hr at 13 mL/hr.
43 mg over 2 hr at 64.8 mL/hr + 40 mg over 3 hr at 40.2 mL/hr.
TABLE 3.
Oral PK of HTL0009936 at 24 mg, mean (%CV) or [range] for n = 6
| Dose (mg) | T max (hr) | C max (ng/mL) | AUC0–24 (h.ng/mL) | AUC0‐∞ (hr.ng/ml) | t½ po (hr) | Fpo (%) a |
|---|---|---|---|---|---|---|
| 24 | 1.0 [0.50–1.5] | 14.1 (49) | 44.1 (48) | 47.2 (41) | 2.4 (28) | 14.8 (44) [8.7–27] |
Geometric mean and (geometric %CV) except T max median [minimum − maximum] for n = 6. AUC0‐∞, area under the plasma concentration–time curve from zero extrapolated to infinity; AUC0–24, area under the plasma concentration–time curve from zero to 24 hours post dose; C max, maximum plasma concentration; Fpo, oral bioavailability and [minimum − maximum]; T max, time to C max; t½ po, apparent terminal half‐life after oral administration.
Oral bioavailability estimated in comparison with 10 mg IV single infusion.
TABLE 4.
Summary table of HTL0009936 exposures in part B (CYP2D6 EM and IM subjects combined), mean (%CV) and [range]
| Dose (mg) a | C mean (ng/mL) b | T max (hr) | C max (ng/mL) | AUC0–24hr (hr.ng/mL) | AUC0‐∞ (hr.ng/mL) | t½ IV (hr) | CLp (L/hr) | CLr (L/hr) |
|---|---|---|---|---|---|---|---|---|
| 13.5 (4.5 + 9) | 27.1 (20) | 1.0 [0.52–5.1] | 33.8 (21) | 192 (27) | 197 (26) | 2.2 (28) | 69 (26) | 8.6 (23) |
| 40 (13.3 + 26.7) | 78.2 (18) | 1.0 [0.58–5.3] | 97.6 (21) | 550 (24) | 564 (24) | 2.3 (33) | 71 (24) | 8.2 (27) |
| 79.5 (26.5 + 53) | 166 (20) | 1.1 [0.83–5.6] | 203 (20) | 1200 (31) c | 1170 (25) | 2.6 (27) | 68 (25) | 7.3 (30) |
Geometric mean and (geometric %CV) except T max median [minimum − maximum] for n = 25–28 observations excluding subjects where infusion was stopped early or interrupted. AUC0‐∞, area under the plasma concentration–time curve from zero extrapolated to infinity; AUC0–24, area under the plasma concentration–time curve from zero to 24 hours post dose; C max, maximum plasma concentration; C mean, mean plasma concentration during 4 hour maintenance infusion; CLp, total plasma clearance; CLr, renal clearance; T max, time to C max; t½ IV, post‐infusion intravenous apparent half‐life;
Loading dose (1 hr at 83.3 mL/hr) + maintenance dose (4 hr at 41.7 mL/hr).
Steady‐state concentration maintained between 1 and 5 hours after the start of dosing.
Includes a subject with a large value of AUC0‐t due to limited available PK sampling times but for whom a value of AUC0‐∞ could not be estimated, therefore the group mean value of AUC0‐t was greater than AUC0‐∞.
Physostigmine plasma concentrations increased immediately after dosing, with the mean T max at 50 minutes. It was rapidly eliminated from plasma with a mean t ½ of 0.37 hours (CV 31%) with observed concentrations ≤ 1 ng/mL and typically < 0.5 ng/mL by 1.5 hours after the start of infusion (see Supplementary Figure S5).
3.4. Pharmacodynamics
Dose‐related increases in both systolic and diastolic blood pressure were observed following administration of 40 mg and 79.5 mg HTL0009936 compared to placebo (Figure 3). There were no increases in systolic or diastolic blood pressure at the 13.5 mg dose. The mean systolic blood pressure increased 3.87 mm Hg following 40 mg HTL0009936 (95% CI [0.70–7.05]) and 7.12 mm Hg after 79.5 mg HTL0009936 (95% CI [3.99–10.24]) compared with placebo. Mean diastolic blood pressure increased 3.83 mm Hg following 40 mg HTL0009936 (95% CI [1.64–6.01]) and 5.32 mm Hg after 79.5 mg HTL0009936 (95% CI [3.18–7.47]) compared with placebo. Similarly, there was a dose‐related increase in heart rate. There were no significant increases in pulse rate at the 13.5 mg and 40 mg doses. Administration of 79.5 mg HTL0009936 resulted in increased pulse rate of 4.75 bpm when compared with placebo (95% CI [3.14–6.36]).
FIGURE 3.
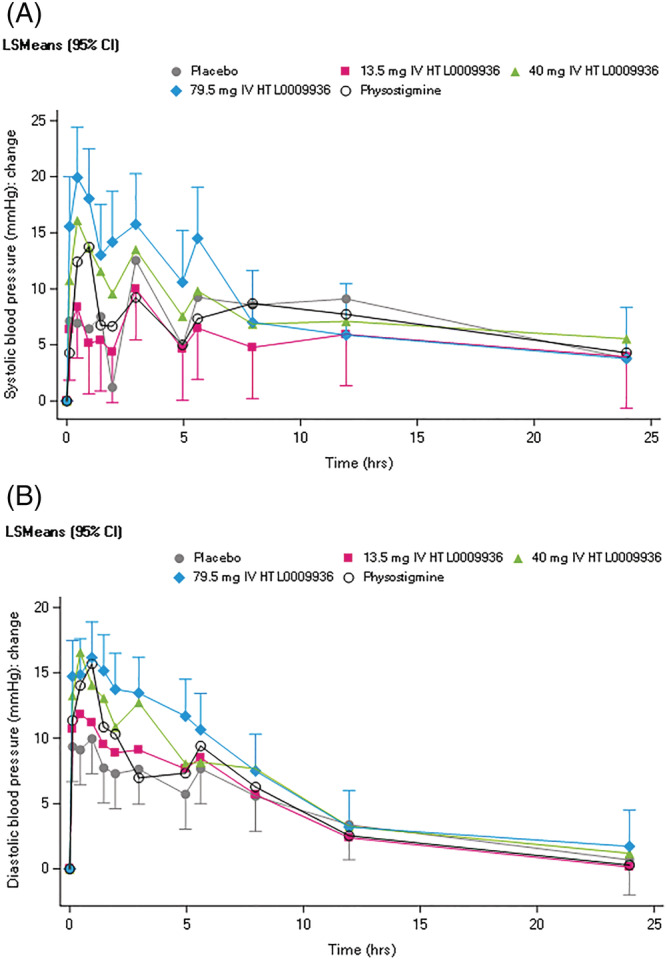
A. Systolic blood pressure (mm Hg) shown as change from baseline and B. Diastolic blood pressure (mm Hg) shown as change from baseline
Overall, single doses of HTL0009936 showed no consistent acute effects on measures of cognitive or neurophsyiological function as measured by NeuroCart, CANTAB, EEG and ERPs compared with placebo (Supplementary Table S6). However, 13.5 mg HTL0009936 resulted in a mean increase in P300 maximum amplitude of 0.56 uV over the Cz lead compared to placebo administration (95% CI [0.139–0.971]), although similar increases were not observed at the Fz and Pz leads (Figure 4). No clinically relevant effects were observed on the VAS nausea scale and the LSEQ compared with placebo.
FIGURE 4.
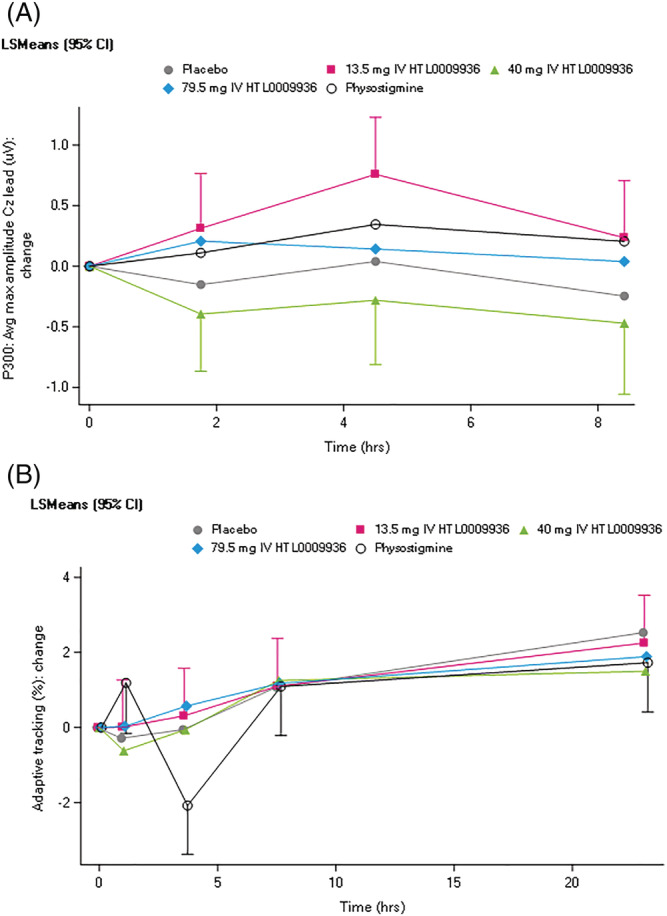
A. P300 results shown as change from baseline and B. Adaptive tracking test results shown as change from baseline
Physostigmine administration led to an improvement of 1.5%‐poinst (95% CI 0.216–2.734) on the adaptive tracking test performance within 2 hours post dose (Figure 4). No improvements in adaptive tracking were observed with HTL0009936.
4. DISCUSSION
The objective of the study was to assess safety, tolerability and PK in elderly subjects and the effect of HTL0009936 on cognitive performance in elderly subjects with below average cognitive function. In part A, focusing on safety, tolerability and PK in normal healthy elderly, HTL0009936 was administered IV over a dose range of 0.1 mg (over 30 min) up to 83 mg (over 5 h) and 24 mg orally. In part B, focusing on safety, tolerability, PK and PD in elderly with below average cognitive function, HTL0009936 was administered IV over a dose range of 13.5 to 79.5 mg and compared to placebo and physostigmine infusions in a double dummy manner. The infusion in part B consisted of a 1 hour loading dose in order to reach the target steady‐state plasma concentration followed by a 4 hour maintenance dose designed to maintain the target steady‐state concentration to ensure sustained exposure within the CNS over the period of cognitive testing.
All doses of HTL0009936 were associated with mild to moderate self‐limiting TEAEs. Fewer subjects reported TEAEs after HTL0009936 (50–56.7% of the subjects) than after physostigmine (85.7% of the subjects) (Supplementary Information S3). The observed small increases in systolic (3.87 mm Hg) and diastolic (5.32 mm Hg) blood pressure and pulse rate (4.75 bpm) were dose‐dependent and consistent with expected effects of M1 mAChR stimulation on the peripheral cardiovascular system. 47 Importantly, the effects of blood pressure and heart rate were acute, returning to normal soon after HTL0009936 infusion was stopped suggesting there were no persistent effects. Overall, HTL0009936 was considered safe and well‐tolerated in elderly subjects at exposures predicted to have central physiological effects.
The PK of HTL0009936 were well‐characterized up to single doses of 83 mg. IV infusion in part B resulted in stable and sustained exposure of HTL0009936. The PK variability after IV administration was lower than after oral administration (i.e., 30% vs 50% respectively).
Overall, no definitive positive or negative PD effects were observed on behavioural and electrophysiological biomarkers of cognitive function. Potential reasons for a lack of a clear PD effect are discussed below, which impacts the conclusions that can be drawn on the PD effects of HTL0009936. However, HTL009936 showed a selective pro‐cognitive effect as shown by an increase in P300 amplitude at the 13.5 mg doses, suggesting an improvement in early attentional processing. However, these data need to be interpreted with caution as the effects were only noted at the Cz lead, and not at the Fz lead (leads with the greatest signal change with P300 generated using a passive odd ball task).
In order to reduce the ceiling effects that cognitive tests have in healthy optimal cognitive functioning subjects, we aimed to investigate HTL0009936 in a study population in which the ceiling effects could be expected to be more limited, based on lower cognitive test scores. The percentage of subjects with impairments were 39% for memory, 36% for executive function and 42% for attention. One limitation of using this approach is that not all subjects were impaired on all tests and the percentage of subjects impaired in any one test or on all tests was low. This may have led to a variable cognitive baseline for the study population. Hence detecting drug effects may have been difficult for some domains of cognition. Alternatively, as subjects had no evidence of cholinergic deficiency, it is possible that they were not an appropriate population for study of this mechanism of action.
In addition to the potential limitation discussed above, the study was powered to detect a significant change in the adaptive tracking and therefore not to detect statistically significant changes in EEG/ERP or other cognitive tests in which either smaller treatment effects or larger variability could have been present. In addition, multiple PD assessments were not performed at the optimal time of target concentration of HTL0009936 (for the CANTAB tests performed at 5 h post dose) and physostigmine (for EEG and all cognitive tests performed after 1 h post dose). This was due to stopping the infusion of HTL0009936 at 5 hours and physostigmine at 50 minutes and the rapid drop in exposures of both drugs post cessation of infusion during the time of these assessments. The main reason for the latter was concerns with side effects associated with prolonged exposure to physostigmine. Additionally, subject discontinuation in the study, due to the significant burden of the number of assessments, required a change to the protocol in order to reduce the frequency of CANTAB tests. These limitations in the execution of the study are likely to have contributed to the lack of clear PD effects on the neurophysiological and neurocognitive tests after administration of HTL0009936 or physostigmine. However, physostigmine was associated with a significant but small improvement in adaptive tracking (reflecting psychomotor function and sustained attention). The improvement in adaptive tracking and the lack of effect on other tests may be due to the adapting tracking being performed close to the time when the physostigmine infusion was stopped (i.e., 10 min after infusion was stopped). As this study was powered on the adaptive tracking test, it is likely that this is a cholinergic relevant pharmacological effect of physostigmine and supports previous studies that have similarly shown positive effects of a cholinesterase inhibitor galantamine. 35 The absence of an effect on adaptive tracking performance during HTL0009936 exposure based on visual inspection of the graphs might be due to specificity of the cognitive processes modulated by M1 receptor modulation. It is possible psychomotor/attentional processes are less affected whereas memory is more affected by M1 receptor modulation. In support, a study with the M1 agonist GSK1034702 showed improvement in episodic memory but not psychomotor speed or attention. 48 Furthermore, preclinical studies with HTL0009936 showed reversal of scopolamine‐induced impairment in the novel object recognition and passive avoidance tests of memory and improvement in working memory in aged Beagle dogs. 49 On the other hand, the M1/M4 muscarinic antagonist biperiden led to a decrease in performance in the adaptive tracking task at dose levels that did not lead to clinically overt (subjective or objective) sedation (results in preparation to be published). Given the limitations discussed, which may have impacted the ability of HTL0009936 to exert effects of cognitive and neurophysiological function, no clear conclusions can be drawn with regard to the PD effects of HTL0009936 in this study. This would require further investigation in an appropriately designed and adequately powered study.
In summary, this safety, tolerability, PK and exploratory PD study of HTL0009936 showed that the drug had well‐characterized PK and was generally well‐tolerated in the dose range studied in elderly subjects. The incidence of adverse events was mild and dose‐related. No clear PD effects of HTL0009936 could be observed, except a potential increase (i.e., improvement) in P300 amplitude, a measure of cognitive function, and a lack of effect of attention and psychomotor speed as measured by the adaptive tracking test. However, overall, no conclusions can be drawn with regard to positive or negative effects of HTL0009936 on neurophysiological and neurocognitive function, given the limitations in the execution of this study, including multiple cognitive tests performed at suboptimal exposures which may have impacted the ability to detect a drug effect. While the PD effects of HTL0009936 require further investigation, the good safety profile of HTL0009936 supports further safety and PD investigation in patients with AD and other dementias.
COMPETING INTERESTS
This study was sponsored by Sosei Heptares. J.L., G.A.B., A.B., M.C., M.W., F.H.M., T.T. and P.J.N. are employees of Sosei Heptares and hold shares in the company.
CONTRIBUTORS
G.A.B., A.B., M.C., M.W. and F.H.M. developed the compound. T.T., J.L., E.P.H., S.P. and G.J.G. contributed to design of the study. S.P., E.P.H. and C.B. performed the study. E.S.K., J.S. and D.M.C. contributed to data analysis. S.P., T.T., J.L., G.J.G., C.B., D.M.C. and P.J.N. contributed to writing and critical revision of the manuscript. All authors have read and approved the final version.
5.
Supporting information
Table S1. Schedule of assessments study part B
Table S4. Part B: Mean (%CV) HTL0009936 IV PK by CYP2D6 predicted phenotype
Figure S5. Physostigmine plasma concentration–time profile
Table S6. Pharmacodynamic results
ACKNOWLEDGEMENTS
The bioanalysis of HTL0009936 and physostigmine was performed by Irene Morelli (York Bioanalytical Solutions Limited, York, UK). The script for the N‐back task was downloaded from the E‐prime website (http://step.psy.cmu.edu/scripts-plus) and was adapted for the use in this study.
Bakker C, Prins S, Liptrot J, et al. Safety, pharmacokinetics and pharmacodynamics of HTL0009936, a selective muscarinic M1‐acetylcholine receptor agonist: A randomized cross‐over trial. Br J Clin Pharmacol. 2021;87(11):4439-4449. 10.1111/bcp.14872
Charlotte Bakker MD and Samantha Prins MSc shared first authorship.
ISRCTN.org Identifier: ISRCTN12371179 (retrospective registration).
PI statement: The authors confirm that the PI for this paper is Geert Jan Groeneveld and that he had direct clinical responsibility for patients.
DATA AVAILABILITY STATEMENT
Research data are not shared.
REFERENCES
- 1. Riedel WJ. Preventing cognitive decline in preclinical Alzheimer's disease. Curr Opin Pharmacol. 2014;14:18‐22. [DOI] [PubMed] [Google Scholar]
- 2. Potter PE, Rauschkolb PK, Pandya Y, et al. Pre‐ and post‐synaptic cortical cholinergic deficits are proportional to amyloid plaque presence and density at preclinical stages of Alzheimer's disease. Acta Neuropathol. 2011;122(1):49‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Terry RD, Masliah E, Salmon DP, et al. Physical basis of cognitive alterations in Alzheimer's disease: synapse loss is the major correlate of cognitive impairment. Ann Neurol. 1991;30(4):572‐580. [DOI] [PubMed] [Google Scholar]
- 4. Whitehouse PJ, Price DL, Clark AW, Coyle JT, DeLong MR. Alzheimer disease: evidence for selective loss of cholinergic neurons in the nucleus basalis. Ann Neurol. 1981;10(2):122‐126. [DOI] [PubMed] [Google Scholar]
- 5. Galimberti D, Scarpini E. Treatment of Alzheimer's disease: symptomatic and disease‐modifying approaches. Curr Aging Sci. 2010;3(1):46‐56. [DOI] [PubMed] [Google Scholar]
- 6. Birks J. Cholinesterase inhibitors for Alzheimer's disease. Cochrane Database Syst Rev. 2006;1:Cd005593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Blanco‐Silvente L, Castells X, Saez M, et al. Discontinuation, efficacy, and safety of cholinesterase inhibitors for Alzheimer's disease: a meta‐analysis and meta‐regression of 43 randomized clinical trials enrolling 16 106 patients. Int J Neuropsychopharmacol. 2017;20(7):519‐528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Loy C, Schneider L. Galantamine for Alzheimer's disease and mild cognitive impairment. Cochrane Database Syst Rev. 2006;1:Cd001747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kuhl DE, Minoshima S, Frey KA, Foster NL, Kilbourn MR, Koeppe RA. Limited donepezil inhibition of acetylcholinesterase measured with positron emission tomography in living Alzheimer cerebral cortex. Ann Neurol. 2000;48(3):391‐395. [PubMed] [Google Scholar]
- 10. Ota T, Shinotoh H, Fukushi K, et al. Estimation of plasma IC50 of donepezil for cerebral acetylcholinesterase inhibition in patients with Alzheimer disease using positron emission tomography. Clin Neuropharmacol. 2010;33(2):74‐78. [DOI] [PubMed] [Google Scholar]
- 11. Caulfield MP, Birdsall NJ. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol Rev. 1998;50(2):279‐290. [PubMed] [Google Scholar]
- 12. Erskine D, Taylor JP, Bakker G, Brown AJH, Tasker T, Nathan PJ. Cholinergic muscarinic M1 and M4 receptors as therapeutic targets for cognitive, behavioural, and psychological symptoms in psychiatric and neurological disorders. Drug Discov Today. 2019;24(12):2307‐2314. [DOI] [PubMed] [Google Scholar]
- 13. Foster DJ, Choi DL, Conn PJ, Rook JM. Activation of M1 and M4 muscarinic receptors as potential treatments for Alzheimer's disease and schizophrenia. Neuropsychiatr Dis Treat. 2014;10:183‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Langmead CJ, Watson J, Reavill C. Muscarinic acetylcholine receptors as CNS drug targets. Pharmacol Ther. 2008;117(2):232‐243. [DOI] [PubMed] [Google Scholar]
- 15. Mash DC, Flynn DD, Potter LT. Loss of M2 muscarine receptors in the cerebral cortex in Alzheimer's disease and experimental cholinergic denervation. Science. 1985;228(4703):1115‐1117. [DOI] [PubMed] [Google Scholar]
- 16. Shiozaki K, Iseki E, Uchiyama H, et al. Alterations of muscarinic acetylcholine receptor subtypes in diffuse Lewy body disease: relation to Alzheimer's disease. J Neurol Neurosurg Psychiatry. 1999;67(2):209‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Melancon BJ, Tarr JC, Panarese JD, Wood MR, Lindsley CW. Allosteric modulation of the M1 muscarinic acetylcholine receptor: improving cognition and a potential treatment for schizophrenia and Alzheimer's disease. Drug Discov Today. 2013;18(23‐24):1185‐1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brown GA, Congreve M, Cansfiels J, Tehan BG, inventors; Heptares Therapeutics Limited, assignee . Muscarinic M1 receptor agonists. US Patent 9,907,805 B2. March 6, 2018.
- 19. World Medical Association Declaration of Helsinki: ethical principles for medical research involving human subjects. JAMA. 2013;310(20):2191‐2194. [DOI] [PubMed] [Google Scholar]
- 20. Schmand B, Houx P, de Koning I. Normen van psychologische tests voor gebruik in de klinische neuropsychologie. 2012. Available at: https://www.psynip.nl/wp‐content/uploads/2016/07/Normen‐Np‐tests‐2012.xls
- 21. Bentley P, Driver J, Dolan RJ. Cholinesterase inhibition modulates visual and attentional brain responses in Alzheimer's disease and health. Brain. 2008;131(Pt 2):409‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bartus RT, Dean RL 3rd, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217(4558):408‐414. [DOI] [PubMed] [Google Scholar]
- 23. Prohovnik I, Arnold SE, Smith G, Lucas LR. Physostigmine reversal of scopolamine‐induced hypofrontality. J Cereb Blood Flow Metab. 1997;17(2):220‐228. [DOI] [PubMed] [Google Scholar]
- 24. R. Core Team . A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2010. [Google Scholar]
- 25. Hart EP, Alvarez‐Jimenez R, Davidse E, et al. A computerized test battery to study pharmacodynamic effects on the central nervous system of cholinergic drugs in early phase drug development. J Vis Exp. 2019;144:e56569. [DOI] [PubMed] [Google Scholar]
- 26. Egerhazi A, Berecz R, Bartok E, Degrell I. Automated Neuropsychological Test Battery (CANTAB) in mild cognitive impairment and in Alzheimer's disease. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(3):746‐751. [DOI] [PubMed] [Google Scholar]
- 27. Kuzmickiene J, Kaubrys G. Selective ability of some CANTAB battery test measures to detect cognitive response to a single dose of donepezil in Alzheimer disease. Med Sci Monit. 2015;21:2572‐2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Silver JM, Koumaras B, Chen M, et al. Effects of rivastigmine on cognitive function in patients with traumatic brain injury. Neurology. 2006;67(5):748‐755. [DOI] [PubMed] [Google Scholar]
- 29. Baakman AC, Hart ET, Kay DG, et al. First in human study with a prodrug of galantamine: improved benefit‐risk ratio? Alzheimers Dement (N Y). 2016;2(1):13‐22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Borland RG, Nicholson AN. Visual motor co‐ordination and dynamic visual acuity. Br J Clin Pharmacol. 1984;18(Suppl 1):69S‐72S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. van Steveninck AL, Schoemaker HC, Pieters MS, Kroon R, Breimer DD, Cohen AF. A comparison of the sensitivities of adaptive tracking, eye movement analysis and visual analog lines to the effects of incremental doses of temazepam in healthy volunteers. Clin Pharmacol Ther. 1991;50(2):172‐180. [DOI] [PubMed] [Google Scholar]
- 32. Milner B. Visually‐guided maze learning in man: effects of bilateral hippocampal, bilateral frontal, and unilateral cerebral lesions. Neuropsychologia. 1965;3(4):317‐338. [Google Scholar]
- 33. Lim HK, Juh R, Pae CU, et al. Altered verbal working memory process in patients with Alzheimer's disease: an fMRI investigation. Neuropsychobiology. 2008;57(4):181‐187. [DOI] [PubMed] [Google Scholar]
- 34. Rombouts SA, Barkhof F, Van Meel CS, Scheltens P. Alterations in brain activation during cholinergic enhancement with rivastigmine in Alzheimer's disease. J Neurol Neurosurg Psychiatry. 2002;73(6):665‐671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sweet LH, Rao SM, Primeau M, Durgerian S, Cohen RA. Functional magnetic resonance imaging response to increased verbal working memory demands among patients with multiple sclerosis. Hum Brain Mapp. 2006;27(1):28‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Twa MD, Bailey MD, Hayes J, Bullimore M. Estimation of pupil size by digital photography. J Cataract Refract Surg. 2004;30(2):381‐389. [DOI] [PubMed] [Google Scholar]
- 37. Baakman AC, Alvarez‐Jimenez R, Rissmann R, et al. An anti‐nicotinic cognitive challenge model using mecamylamine in comparison with the anti‐muscarinic cognitive challenge using scopolamine. Br J Clin Pharmacol. 2017;83(8):1676‐1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Groeneveld GJ, Hay JL, Van Gerven JM. Measuring blood‐brain barrier penetration using the NeuroCart, a CNS test battery. Drug Discov Today Technol. 2016;20:27‐34. [DOI] [PubMed] [Google Scholar]
- 39. van Steveninck AL, van Berckel BN, Schoemaker RC, Breimer DD, van Gerven JM, Cohen AF. The sensitivity of pharmacodynamic tests for the central nervous system effects of drugs on the effects of sleep deprivation. J Psychopharmacol. 1999;13(1):10‐17. [DOI] [PubMed] [Google Scholar]
- 40. Polich J. Updating P300: an integrative theory of P3a and P3b. Clin Neurophysiol. 2007;118(10):2128‐2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Fabiani M, Karis D, Donchin E. P300 and recall in an incidental memory paradigm. Psychophysiology. 1986;23(3):298‐308. [DOI] [PubMed] [Google Scholar]
- 42. Näätänen R, Sussman ES, Salisbury D, Shafer VL. Mismatch negativity (MMN) as an index of cognitive dysfunction. Brain Topogr. 2014;27(4):451‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Parrott AC, Hindmarch I. The Leeds Sleep Evaluation Questionnaire in psychopharmacological investigations—a review. Psychopharmacology (Berl). 1980;71(2):173‐179. [DOI] [PubMed] [Google Scholar]
- 44. Barnett JH, Blackwell AD, Sahakian BJ, Robbins TW. The Paired Associates Learning (PAL) Test: 30 years of CANTAB translational neuroscience from laboratory to bedside in dementia research. Curr Top Behav Neurosci. 2016;28:449‐474. [DOI] [PubMed] [Google Scholar]
- 45. Gau SS, Huang WL. Rapid visual information processing as a cognitive endophenotype of attention deficit hyperactivity disorder. Psychol Med. 2014;44(2):435‐446. [DOI] [PubMed] [Google Scholar]
- 46. Cacciamani F, Salvadori N, Eusebi P, et al. Evidence of practice effect in CANTAB spatial working memory test in a cohort of patients with mild cognitive impairment. Appl Neuropsychol Adult. 2018;25(3):237‐248. [DOI] [PubMed] [Google Scholar]
- 47. Hardouin SN, Richmond KN, Zimmerman A, Hamilton SE, Feigl EO, Nathanson NM. Altered cardiovascular responses in mice lacking the M(1) muscarinic acetylcholine receptor. J Pharmacol Exp Ther. 2002;301(1):129‐137. [DOI] [PubMed] [Google Scholar]
- 48. Nathan PJ, Watson J, Lund J, et al. The potent M1 receptor allosteric agonist GSK1034702 improves episodic memory in humans in the nicotine abstinence model of cognitive dysfunction. Int J Neuropsychopharmacol. 2013;16(4):721‐731. [DOI] [PubMed] [Google Scholar]
- 49. Brown A, Bradley S. From structure to clinic: The discovery of a selective M1 muscarinic acetylcholine receptor agonist for the treatment of memory loss in Alzheimer's disease. In preparation.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Schedule of assessments study part B
Table S4. Part B: Mean (%CV) HTL0009936 IV PK by CYP2D6 predicted phenotype
Figure S5. Physostigmine plasma concentration–time profile
Table S6. Pharmacodynamic results
Data Availability Statement
Research data are not shared.