

Abstract
Protein glycosylation is crucial for the central nervous system and brain functions, including processes that are defective in Alzheimer disease (AD) such as neurogenesis, synaptic function, and memory formation. Still, the roles of glycans in the development of AD are relatively unexplored. Glycomics studies of cerebrospinal fluid (CSF) have previously shown altered glycosylation pattern in patients with different stages of cognitive impairment, including AD, compared to healthy controls. As a consequence, we hypothesized that the glycan profile is altered in the brain of patients with AD and analyzed the asparagine‐linked (N‐linked) glycan profile in hippocampus and cortex in AD and control brain. Glycans were enzymatically liberated from brain glycoproteins and analyzed by liquid chromatography‐tandem mass spectrometry (LC‐MS/MS). Eleven glycans showed significantly different levels in hippocampus compared to cortex in both control and AD brain. Two glycans in cortex and four in hippocampus showed different levels in AD compared to control brain. All glycans that differed between controls and AD brain had similar structures with one sialic acid, at least one fucose and a confirmed or potential bisecting N‐acetylglucosamine (GlcNAc). The glycans that were altered in AD brain differed from those that were altered in AD CSF. One glycan found to be present in significantly lower levels in both hippocampus and cortex in AD compared to control contained a structurally and functionally interesting epitope that we assign as a terminal galactose decorated with fucose and sialic acid. Altogether, these studies suggest that protein glycosylation is an important component in the development of AD and warrants further studies.
Keywords: Alzheimer disease, frontal cortex, glycomics, hippocampus, mass spectrometry, N‐glycan
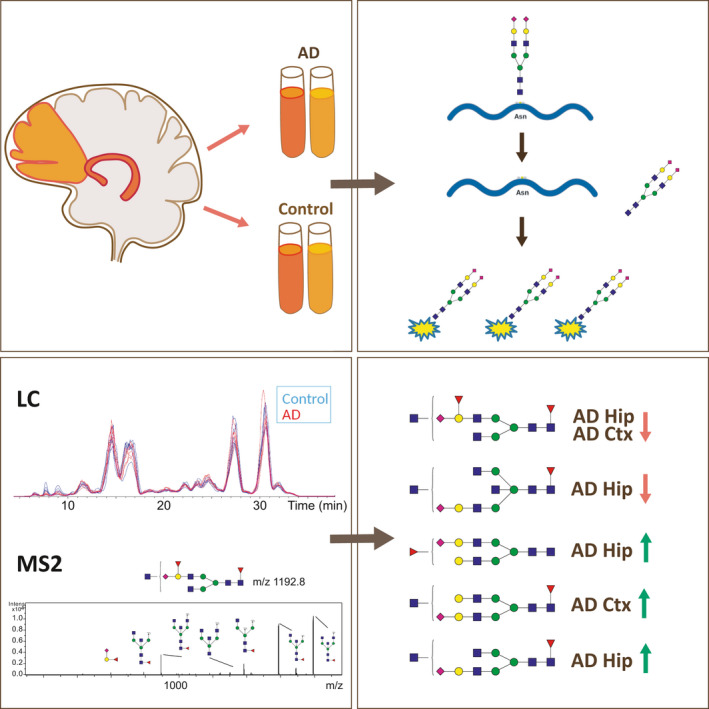
The N‐glycan profile of brain in Alzheimer‐ disease (AD) was studied by glycomics analysis of homogenates from hippocampus (Hip) and frontal cortex (Ctx) and compared with nondemented controls. N‐glycans were enzymatically released and separated from the glycoproteins, labeled with 2‐anthranilic acid, purified and analyzed by liquid chromatography‐tandem mass spectrometry. The levels of four N‐glycans in hippocampus and two in frontal cortex, all with confirmed structures from mass spectrometric fragmentation, were significantly altered in AD compared to control. These findings may lay ground for novel potential treatment strategies.
Abbreviations
- 2‐AA
2‐anthranilic acid
- AD
Alzheimer disease
- APP
amyloid precursor protein
- Asn
asparagine
- Aβ
amyloid β‐peptide
- BACE1
beta‐secretase 1
- BCA
bicinchoninic acid assay
- CSF
cerebrospinal fluid
- EIC
extracted ion chromatogram
- Fuc
fucose
- Gal
galactose
- GlcNAc
N‐acetylglucosamine
- Hex
hexose
- LC‐MS/MS
liquid chromatography‐tandem mass spectrometry
- Man
mannose
- MS
mass spectrometry
- m/z
mass/charge
- N‐linked
asparagine‐linked
- PNGase F
peptide N‐glycosidase F
- Ser
serine
- Sia
sialic acid
- Thr
threonine
1. INTRODUCTION
Protein glycosylation, the covalent linkage of oligosaccharides to proteins, is the most complex form of posttranslational modifications of proteins and provides a higher level of complexity to the proteome in higher organisms. The two most common types are O‐linked (linked to OH‐groups of for instance Ser/Thr) and N‐linked (Asn linked), but also other types of protein glycosylation exist (Sytnyk et al., 2020). In this study we have focused on N‐linked glycans, which share a core sequence that can be extended and classified into three types: oligo‐mannose (also called high mannose), complex and hybrid. The latter two types can be decorated with unique features such as fucose, sialic acid and bisecting GlcNAc (Figure 1). Glycans are crucial for the development and function of the highly specialized brain and nervous system in human. A multitude of different glycan epitopes regulate the development and activities of neurons, including neuritogenesis, synaptic plasticity and signal transduction (Sytnyk et al., 2020) and an interest in the involvement of glycans in neurodegenerative disorders has recently begun to emerge.
FIGURE 1.
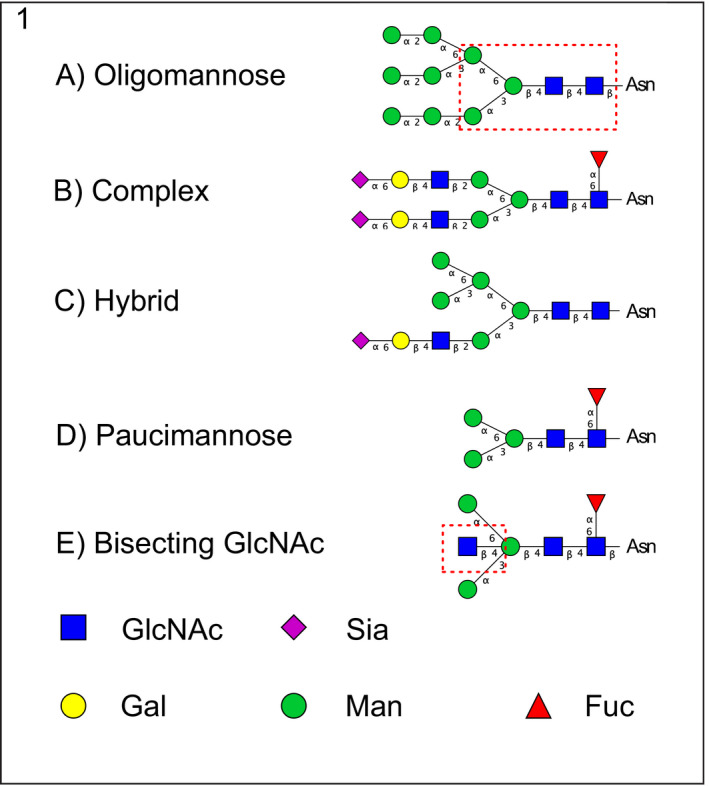
Outline of N‐glycan classes. N‐glycans share a core sequence (Manα1–3(Manα1–6)Manβ1‐4GlcNAcβ1–4GlcNAcβ1–Asn) that can be extended and classified into three different types (a–c). Examples representing (a) Oligo‐mannose (also called high mannose); (b) complex; (c) hybrid; (d) paucimannose and (e) bisecting GlcNAc (the first mannose in the core extended with a single GlcNAc) structures are shown. In the oligomannose structure the N‐glycan core is marked inside a dashed red square (a). The bisecting GlcNAc is also marked with a red dashed square for clarity (e). Symbol nomenclature as suggested in (Varki et al., 2009). GlcNAc, N‐acetylglucosamine; Sia, sialic acid; Gal, galactose; Man, mannose; Fuc, fucose
Alzheimer disease (AD) is a devastating neurodegenerative disorder and the most common form of dementia, with loss of memory being one of the earliest clinical signs. The disease pathology starts decades before clinical diagnosis can be made (Jack & Holtzman, 2013) and the available treatments are only symptomatic. Pathologically, the brain in AD is characterized by extracellular amyloid plaques composed of fibrils of the aggregation‐prone amyloid β‐peptide (Aβ) consisting of 42 amino acids (Aβ42) (Söderberg et al., 2006) and intraneuronal neurofibrillary tangles composed of hyperphosphorylated tau protein (Spillantini & Goedert, 2013). Aβ is formed by a two‐step proteolytical cleavage of the Aβ precursor protein (APP) by the transmembrane secretases BACE1 and γ‐secretase (De Strooper et al., 2010). Several studies during the past decades have shown that intracellular Aβ42 is neurotoxic and correlates with AD progression, including synaptic loss and cognitive decline (Aoki et al., 2008; Hashimoto et al., 2012; Nilsson et al., 2013; Takahashi et al., 2002). The involvement of Aβ in synapse biology and/or pathology in AD is underscored by our recent super‐resolution microscopy study, which revealed that Aβ42 is enriched in small vesicles at the presynaptic side of hippocampal neurons (Yu et al., 2018).
In addition to the classical pathological hallmarks, alterations in protein glycosylation have been observed in AD (for reviews, see Kizuka et al., 2017; Regan et al., 2019; Schedin‐Weiss et al., 2014). For instance, tau, which is a cytosolic protein and not expected to be glycosylated, was found to be N‐glycosylated in AD brain (Wang et al., 1996). Furthermore, BACE‐1, APP, cholinesterases, and other proteins display changes in the glycosylation pattern in AD compared to control brain (Halim et al., 2011; Kizuka et al., 2015; Saez‐Valero & Small, 2001). The mRNA levels for several glycosylation‐related genes are altered in AD brain, suggesting global defects in protein glycosylation (Frenkel‐Pinter, et al., 2017), which is in line with the observed overall alterations in protein glycosylation pathways in brain and serum in AD (Frenkel‐Pinter, et al., 2017).
The determination of the glycosylation of each protein (glycoproteomics) in a complex sample such as a tissue has been hampered by technical and methodological hurdles that only recently have started to be overcome with improved sample enrichment and complementary fragmentation of glycan and peptide moieties of glycopeptides (reviewed in (Thaysen‐Andersen et al., 2016). N‐glycomics, the analysis of the complete set of N‐glycans in a cell or organism, is greatly facilitated by the use of specific enzymes that can liberate N‐glycans from proteins. While MS based proteomics is nowadays routine analysis, detailed sequence information from glycomics is more difficult to obtain because of the presence of various monosaccharides with the same mass and multiple branching and linkage possibilities that are not easily resolved, requiring tandem MS along with extensive data analysis.
We recently performed an N‐glycomics study of cerebrospinal fluid (CSF), which revealed increased levels of several N‐glycans containing bisecting GlcNAc (see structural explanation in Figure 1) in AD compared to control cases (Schedin‐Weiss, et al., 2020). Certain glycans were increased in some but not all of the AD cases, suggesting that there are differences in the glycosylation pattern in subpopulations of AD, in line with another study (Palmigiano et al., 2016). Thus, CSF levels of glycans may be used as biomarkers for diagnosis and, possibly, prognosis of AD as well as treatment outcome. Further studies on why these alterations occur, and how glycosylation is affected in different regions in AD brain, are necessary for understanding how protein glycosylation is related to AD pathology and may potentially be used to develop novel treatment strategies. Here, we have analyzed the N‐glycome in homogenates from hippocampus and frontal cortex of postmortem AD and control brain by LC‐MS/MS of 2‐anthranilic acid (AA)‐labeled N‐glycans and found differences in the glycosylation pattern both in hippocampus and frontal cortex in AD compared to control.
2. MATERIALS AND METHODS
2.1. Preparation of postmortem brain homogenates
Frozen brain pieces of hippocampus and frontal cortex from AD patients (n = 5) and control individuals (n = 5) were obtained from the Netherlands Brain Bank (NBB), Netherlands Institute for Neuroscience, Amsterdam. The sample size, determined by the number of samples that were experimentally realistic to perform in parallel, was sufficient to show significant differences between the groups. All AD subjects met the criteria for definitive AD according to the Consortium to Establish a Registry for AD (Jack et al., 2018). All procedures performed in studies involving the postmortem human samples were in accordance with the ethical standards of the institutional and national regional research committee “Regionala etikprövningsnämnden i Stockholm” (ethic permit nr 2013/1301‐31/2) and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. The control subjects had no known psychiatric or neurological disorders. Samples were selected according to Braak stages and short postmortem times, and all samples were age‐ and gender‐matched (Table 1). No blinding was performed. Brain homogenates were prepared from frozen pieces of postmortem human hippocampus or frontal cortex in 8 M urea (SigmaAldrich U4884), 500 mM ammonium bicarbonate (Fluka 40,867) at a final concentration of 100 mg/ml. The tissue was homogenized for 15 times up and down at 1,500 rpm in a teflon piston homogenizer and snap‐frozen in liquid N2 and stored at −80°C until glycan analysis was performed. The protein concentration was determined with bicinchoninic acid assay (Pierce™ BCA) assay (ThermoFisher 23,227) according to the manufacturer's instructions.
TABLE 1.
AD and control brain patient information
| Variable | Non‐demented controls (n = 5) | AD (n = 5) | |
|---|---|---|---|
| Age of death | 79 ± 6 | 81 ± 5 | |
| Number (%) of females | 3 (60%) | 3 (60%) | |
| PMD (hours) | 6.8 ± 2.0 | 6.3 ± 1.1 | |
| Braak scores | 0 | 1 | 0 |
| I/II | 4 | 0 | |
| III/IV | 0 | 1 | |
| V/VI | 0 | 4 |
2.2. Release of N‐glycans from brain extract glycoproteins
Homogenates containing 100 µg protein denatured in urea (determined with the BCA assay) was taken from each sample and diluted to a urea concentration where PNGase F would be active. Briefly, the sample was diluted in 20 µl 6 M urea, 70 µl 100 mM ammonium bicarbonate buffer, and 5 µl 2% Rapigest (Waters Corporation, cat no 186001861) and heated at 95°C for 10 min. After the sample cooled to room temperature, 5 units of PNGase F (Roche Applied Science, cat no 11365177001) was added and the sample was incubated over‐night at 37°C. Additionally 3 units of PNGase F were added the next day and incubated for 3 hr. The released N‐glycans and proteins were separated with C18 tips as described below.
2.3. N‐glycan Isolation and Cleanup
The released N‐glycans were separated from the brain extract proteins with a C18 OMIX tip (Agilent Technologies, part no A57003100). Proteins and peptides bind to the column while the enzymatically released N‐glycans are collected in the flowthrough. The pipette was set to the corresponding volume of the sample, here 100 µl. The C18 tip was conditioned two times with 60% acetonitrile (SigmaAldrich 34851) and two times with 0.2% formic acid (SigmaAldrich F0507) in H20. After conditioning of the tip, 100 µl brain extract was aspirated/dispensed five times, and the effluent containing the released N‐glycans was collected. The tip was washed two times with 0.2% formic acid in H20 and the effluent was pooled with the first flow‐through from the column. The pooled sample containing salts and glycans was lyophilized to dryness in a vacuum centrifuge. No desalting was performed prior to the 2‐AA derivatization, see below.
2.4. 2‐AA labeling of N‐glycans
Reductive amination of the N‐glycans with Anthranilic acid (2‐AA, SigmaAldrich 10680) was performed to increase the MS signal and enable chromatographic separation on C18 columns. The labeling is highly efficient and the reduction of the sample also solves the problem of peak splitting of α and β anomers observed when analyzing native glycans. The labeling was performed as described by SigmaAldrich with some modifications. Briefly, a labeling solution with 60 mg/ml 2‐AA and 60 mg/ml sodium cyanoborohydride (SigmaAldrich 42077) in DMSO (SigmaAldrich 276855):acetic acid (70:30) was prepared. The solution was briefly heated in a heating block to 65°C and water was added to a final concentration of 10%. Labeling solution (5 µl) was added to the lyophilized N‐glycans and incubated at 65°C in a heating block under aluminum foil for 3 hr. Condensed liquid in the lid was briefly spun down a few times during the incubation. The reaction was stopped with the addition of 95 µl water and immediately applied to disposable size exclusion columns (SEC), see below.
2.5. Removal of salts and excess label with size exclusion chromatography (SEC)
This protocol has been adapted from the manufacturer's protocol. The washing and elution volumes of the protocol were optimized by measuring the fluorescence intensity of eluted fractions in a fluorescence plate reader (data not shown). The PD minitrap G‐10 columns (GE Healthcare 28‐9180‐10) were equilibrated with 10 ml H20 with gravity flow. The sample was added in 100 µl volume (5 µl sample + 95 µl H20), and the flow‐through collected. The column was washed with 850 µl H20, which was pooled with the first flow‐through. Labeled N‐glycans were eluted with 250 µl H20 and collected in a new marked vial. The sample was stored at −20°C in darkness until analyzed with LC‐MS.
2.6. LC‐MS analysis
The AA‐labeled N‐glycans were analyzed with an Agilent 6,300 MS LC‐MS system consisting of a 1,200 LC system connected to an Agilent chipcube with integrated ionsource serving as the interface to an Agilent 6,300 iontrap. The AA label increases the MS signal and allows chromatographic separation on regular C18 columns. Briefly, the samples (6 µl/injection, corresponding to 0.8 µl CSF starting material) were concentrated on a 160 nl trap column and separated on a 15 cm capillary C18 chip column packed with 5 µm 300 Å C18 Zorbax particles (G4240‐62010) with increasing concentration of acetonitrile (buffer A: 0.2% formic acid in H2O; buffer B: 0.2% formic acid in acetonitrile). With a flow‐rate of 0.3 µl/min the gradient was performed in a stepwise manner: 0–1 min 1% buffer B, 1–2 min 1%–4.5% buffer B, 2–34 min 4.5%–8.5% buffer B, 35–38 min 95% buffer B, 40–50 min 1% buffer B. The MS detection was performed in positive Ultrascan mode in the scan range of 700–1510 m/z with averages of three spectra. Smart ICC target was set to 200 000 with a maximum accumulation time of 100 ms. Tune settings were set with capillary voltage of 1950 V, drying temperature of 320°C. Tune SPS was active and set to target mass m/z 1,200, compound stability 50% and trap drive level 100%. The decrease of compound stability setting from 100% to 50% greatly increase the stability of the glycans. Tandem MS was performed in an automatic manner on the top four ions with active exclusion after two positive spectra for half a minute.
2.7. Data processing of LC‐MS data
The MS data were viewed and processed in Agilent Data Analysis for 6,300 Series Ion Trap LC/MS version 3.4 (build 175). For each sample, extracted ion chromatograms (EIC) from 21 different m/z ions were plotted representing the major peaks in the total basepeak chromatograms. The area of 31 peaks was manually integrated in the DataAnalysis software. The relative area % of each peak was calculated and normalized by dividing the peak area with the total area of all 31 peaks. Bar graph plots of relative % area for each peak (average from five samples) was performed in Microsoft Excel 2013 with the error bars indicating the standard deviation calculated from five different samples. The structural elucidation, including both composition and sequence, was facilitated by the free cross platform software GlycoWorkBench (RRID SCR_000782), which can be used to rapidly draw and calculate MS fragments in‐silico (Ceroni et al., 2008; Damerell et al., 2012). For each m/z value several possible glycan structures were drawn in GlycoWorkBench, their fragments were generated in‐silico and compared to our recorded MS/MS spectra. The tentative structures and compositions suggested in this report are based on knowledge of biological constraints of human cells, which limited the number of possible structures that we tested in‐silico against the MS/MS data.
2.8. Statistical analysis
In order to determine if there were some statistically significant differences in the N‐glycan profile, a pairwise comparison with T‐test was performed between control cortex and control hippocampus, control cortex and AD cortex and, finally, control hippocampus and AD hippocampus. Student's T‐test was done in Microsoft Excel 2013 with the function T‐test (T.Test) with two tails distribution (tails 2) and assuming two‐sample equal variance (type 2). Data were not assessed for normality. No data points were excluded and no test for outliers was conducted. A P‐value filter of at or below 0.05 was considered significant. Boxplots were performed in OriginPro 2018 SR1 on peaks that were deemed significant in the Student's T‐test performed above. The Boxplots were displaying Max, Min, 99%, 1%, and mean in the plot in addition to the individual data points from the control cortex and control hippocampus, control cortex and AD cortex and, finally, control hippocampus and AD hippocampus. Error bars in the bar graphs were calculated from five experiments with the excel function STDEV.
3. RESULTS
In this study, the N‐glycan profile in cortex and hippocampus from five AD brains and five control brains was explored. The study was not preregistered. The brain specimens were homogenized and the N‐glycans were enzymatically released with PNGase F prior to 2‐AA derivatization of the reducing end and LC‐MS/MS analysis. The AA label increased the MS signal and allowed chromatographic separation of the glycans on C18 columns. The brain chromatograms of different brain regions were compared and the identified N‐glycans were quantified by integrating the extracted ion chromatograms.
3.1. Complex glycans are the most prevalent type in cortex and hippocampus
The MS base peak chromatograms of cortex and hippocampus in control brains were compared (Figure 2a). The main differences were observed at around 8, 9, 23–24, and 30–32 min, where cortex rendered higher peaks. Next, the chromatograms of controls and AD were compared. Overall, the chromatograms were similar (Figure 2b and c). However, differences were observed in early eluting peaks at around 7–9 min in cortex samples (Figure 1b). In the samples from hippocampus, the peaks eluting at around 7–10 and 30–32 min were higher in the control samples.
FIGURE 2.
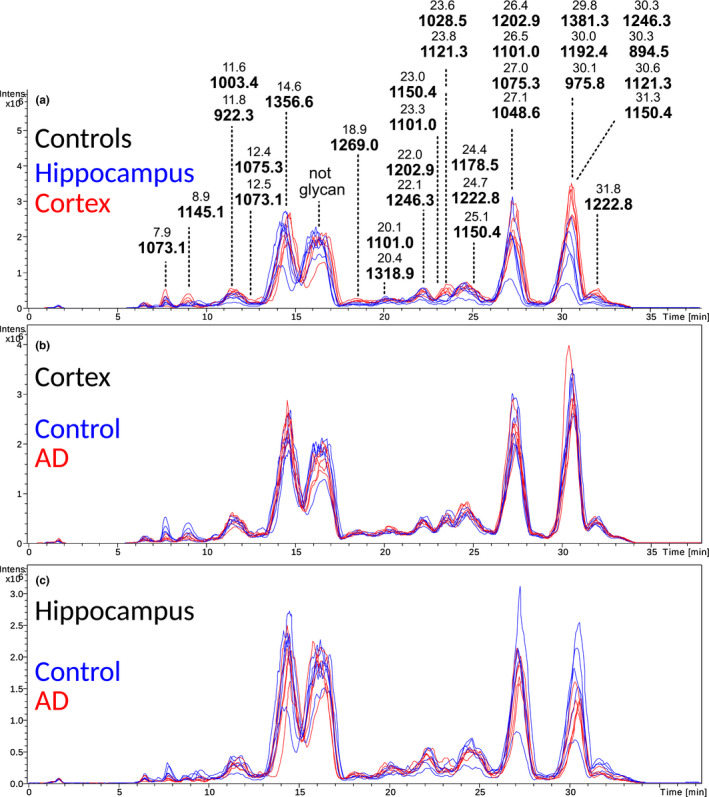
Basepeak chromatograms of the N‐glycome of frontal cortex and hippocampus from control and AD brain. (a) Overlayed control cortex (red) and control hippocampus (blue) basepeak chromatograms of released and 2‐AA‐labeled N‐glycans separated on C18 prior to MS detection. The chromatogram has been annotated with m/z and retention time for the 31 structures reported in this study. (b and c) Overlay of ten base peak chromatograms of N‐glycans released from the cortex (b) and hippocampus (c) of AD (n = 5, red) and control (n = 5, blue) brains (with n referring to the number of individual cases included in the study). The profiles are markedly similar between AD and control brains while in hippocampus the AD and control profiles are slightly less similar
Next, the N‐glycans accounting for the observed differences in the chromatograms were explored by calculating the extracted ion chromatogram (EICs) peak areas of the 21 major compounds. Some EIC traces contained several chromatographic peaks (i.e. different compounds with similar m/z or isomeric separation of glycans). In total 31 peaks were selected for relative abundance comparison and structural analysis.
Several different glycan classes including mannose, complex and tentative hybrid type were identified (the most abundant classes are plotted in Figure S1). These classes can further be divided regarding their specific decoration such as addition of variable number of fucose, sialic acid, and bisecting GlcNAc monosaccharides (Figure S2). Complex type glycans accounted for approximately 70% of the relative abundance in both cortex and hippocampus, while high mannose glycans accounted for approximately 20%. The total amount of mannosylated glycans made up 24%–27% (cortex‐hippocampus) of the relative profile. Non‐determined structures accounted for only 3% of the total profile.
3.2. Glycan structures in cortex and hippocampus
The glycan composition was determined for 27 of 31 peaks. In Table 2 the elution peak number, retention time, m/z value, composition (composition key: H, hexose; N, N‐acetylhexosamine; F, fucose; S, sialic acid), glycan classes and structural elements (bisecting GlcNAc, fucose, and sialic acid) are listed for the N‐glycans. In summary, we found three glycans of high mannose type (#3, #7, #4), one paucimannose and one truncated type glycan (#17 and #24), 24 complex type glycans (5, #8–16, 18–23, 25–31), and potentially a single hybrid type structure (#21). Four peaks could not be identified (#1 and 6 do not seem to represent any glycan and m/z 1,145.1 (#2) and m/z 1,075.3 (#5) lacked informative MS/MS fragments) (Table 2).
TABLE 2.
The major EIC peaks analyzed in this study is listed in chromatographic elution order
| Unique ID | Retention time (min) | m/z | Composition | Glycan class | Fucose | Sialic acid |
|---|---|---|---|---|---|---|
| 1 | 7.9 | 1,073.1 | not glycan | Not applicable | ||
| 2 | 8.9 | 1,145.1 | not determined | Not determined | ||
| 3 | 11.6 | 1,003.4 | H9N2 | Oligo mannose | – | – |
| 4 | 11.8 | 922.3 | H8N2 | Oligo mannose | – | – |
| 5 | 12.4 | 1,075.3 | Not determined | Not determined | ||
| 6 | 12.5 | 1,073.1 | not glycan | Not applicable | – | – |
| 7 | 14.6 | 1,356.6 | H5N2 | Oligo mannose | – | – |
| 8 | 18.9 | 1,269.0 | H7N6F1S4 | complex | yes | Yes |
| 9 | 20.1 | 1,101.0 | H5N4F1S1 | Complex | Yes | Yes |
| 10 | 20.4 | 1,318.9 | H5N4F2S2 | Complex | Yes | Yes |
| 11 | 22 | 1,202.9 | H5N5F1S1 | Complex | Yes | Yes |
| 12 | 22.1 | 1,246.3 | H5N4F1S2 | Complex | Yes | Yes |
| 13 | 23 | 1,150.4 | H4N6F2 | Complex | Yes | |
| 14 | 23.3 | 1,101.0 | H5N4F1S1 | Complex | Yes | Yes |
| 15 | 23.6 | 1,028.5 | H5N4F2 | Complex | Yes | |
| 16 | 23.8 | 1,121.3 | H4N5F1S1 | Complex | Yes | Yes |
| 17 | 24.4 | 1,178.5 | H3N2F1 | Paucimannose | Yes | |
| 18 | 24.7 | 1,222.8 | H4N6F1S1 | Complex | Yes | Yes |
| 19 | 25.1 | 1,150.4 | H4N6F2 | Complex | Yes | |
| 20 | 26.4 | 1,202.9 | H5N5F1S1* | Complex | Yes | Yes |
| 21 | 26.5 | 1,101.0 | H5N4F1S1 | Hybrid | Yes | Yes |
| 22 | 27 | 1,075.3 | Not determined | Not determined | ||
| 23 | 27.1 | 1,048.6 | H4N5F2 | Complex | Yes | |
| 24 | 29.8 | 1,381.3 | H3N2F1 | Truncated | Yes | |
| 25 | 30 | 1,192.4 | H4N5F2S1 | Complex | Yes | Yes |
| 26 | 30.1 | 975.8 | H3N5F1* | Complex | Yes | |
| 27 | 30.3 | 1,246.3 | H5N4F1S2 | Complex | Yes | Yes |
| 28 | 30.3 | 894.5 | H3N5F1* | Complex | Yes | |
| 29 | 30.6 | 1,121.3 | H4N5F1S1 | Complex | Yes | Yes |
| 30 | 31.3 | 1,150.4 | H4N6F2 | Complex | yes | |
| 31 | 31.8 | 1,222.8 | H4N6F1S1* | Complex | yes | Yes |
Some of the compositions have been separated on the C18 column into several peaks eluting at different time points. Compositions labeled with “*" also carry bisecting GlcNAc structure confirmed with MS/MS. Composition key: H (hexose), N (N‐acetylhexosamine), F (fucose), S (sialic acid)
The relative amount of the glycans is plotted in elution order in Figure 3 and descending order in Figure S3. The major peaks in both cortex and hippocampus were m/z 1,048.6 (#23, H4N5F2), which carries a LewisX epitope, 894.5 (#28, H3N5F1), which carries a bisecting GlcNAc, m/z 1,356.6 (#7, H5N2), corresponding to a high mannose structure and 1,192.4 (#25, H4N5F2S1), for which the most probable structure contains a terminal fucosylated, sialylated galactose.
FIGURE 3.
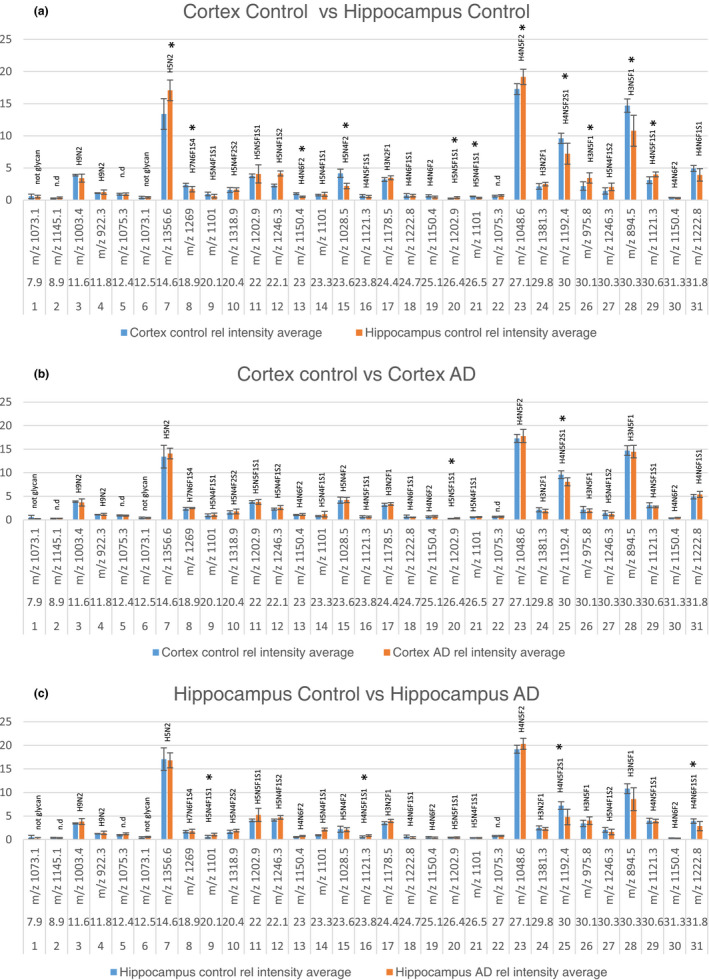
The relative abundance of 31 extracted chromatogram (EIC) peaks was compared between control cortex (n = 5) and control hippocampus (n = 5) (a), control cortex (n = 5) and AD cortex (n = 5) (b) and control hippocampus (n = 5) and AD hippocampus (n = 5) (c) (with n referring to the number of individual cases included in the study). The graphs show EIC # (number), elution time (minutes), and m/z value for each peak. Error bars denote the standard deviation from five biological replicates. Starred peaks denote significant differences between the groups tested with student's T‐test
All complex type glycans were decorated with fucose, and 13 glycans (#8‐12, 14, 16, 18–21, 25, 27, 29 and 31) carried sialic acid on one or two of the antennas. LC‐MS/MS allowed sequencing of the glycans as illustrated in Figures 4, 5, 6. Diagnostic fragment ions revealing the position of bisecting GlcNAc, sialic acid, and antennary fucose are indicated in MS‐MS Figure 4a‐c, respectively within red boxes. In this study, bisecting GlcNAc was only reported if a diagnostic bisecting GlcNAc fragment was found ( H3N5F1 (m/z 975.8 (#26)), H3N5F1 (m/z 894.5 (#28)), and H4N6F1S1 (m/z 1,222.8 peak 2 (#31)) and for the remaining structures the position of terminal HexNAc was undefined. In Figure 4a–d, bisecting GlcNAc has been drawn in all four structures but only Figure 4a include a diagnostic fragment supporting the epitope. The assignment of fucose can also be challenging and was therefore undefined in several structures where conclusive MS/MS fragments were lacking. We suggest that the H4N5F2S1 structure (#25, m/z 1,192.4) most likely carries a terminal epitope with sialic acid, hexose, and fucose as determined with MS/MS in Figure 4d. The MS/MS spectrum lacks any diagnostic fragments previously seen in red squares in Figure 4b and c corresponding to Sia‐Hex‐HexNAc (m/z 657) or Hex‐(Fuc)‐HexNAc (m/z 512) respectively. Instead, major diagnostic fragments include m/z 1787.3 (M—Sia‐Hex‐Fuc) and m/z 598.8 (Sia‐Hex‐Fuc), 4d. The simplest annotation of the MS/MS spectrum is the Sia‐Hex‐Fuc epitope. The relatively low mass fidelity of the m/z 598.8 fragment (expected mass m/z 600.2) makes this annotation only tentative.
FIGURE 4.
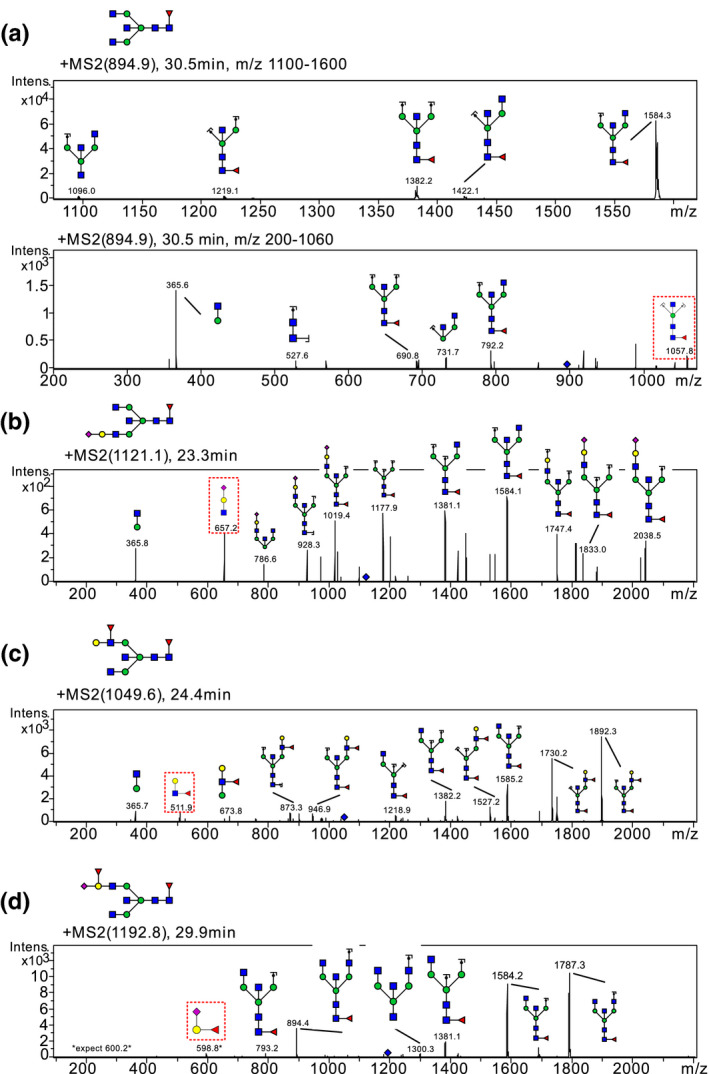
MS/MS spectra including diagnostic protonated (M+H and M+2H) fragment ions indicating bisecting GlcNAc (a), antennary sialic acid (b), antennary fucose (c), and fucosylated and sialylated terminal hexose (d). Bisecting GlcNAc has been drawn in all four structures but only (a) includes a diagnostic fragment supporting the epitope. The bisecting GlcNAc diagnostic ion is a minor m/z 1,057.8 fragment ion of H3N5F1 (m/z 894.5, #28). The antennary sialic acid fragment m/z 657 is easily seen in most glycans with a sialylated antenna, representing Sia‐Hex‐HexNAc. In Figure 4b we observe this ion in the MS/MS spectrum of H4N5F1S1 (m/z 1,121.1 Peak 1, #16). Antennary fucose is easily confirmed with the diagnostic MS/MS ion m/z 512 representing Hex‐(Fuc)‐HexNAc. In (c) we observe this ion in the MS/MS spectrum of H4N5F2 (m/z 1,049.6, #16). The MS/MS spectrum of H4N5F2S1 m/z 1,192.4 in (d) has fragments that are compatible with the Sia‐(Fuc)‐Hex epitope. The diagnostic fragment ions described in b and c, m/z 512 and m/z 657, are not observed, suggesting other terminal epitopes. The major fragment m/z 1787.3 is most likely explained with a single fragment loss (M‐Sia‐(Fuc)‐Hex) suggesting that the fucose is not placed on the terminal HexNAc. The fragment m/z 598.8 may represent Sia‐(Fuc)‐Hex but the relatively low mass fidelity (expected mass m/z 600.2) makes this annotation only tentative
FIGURE 5.
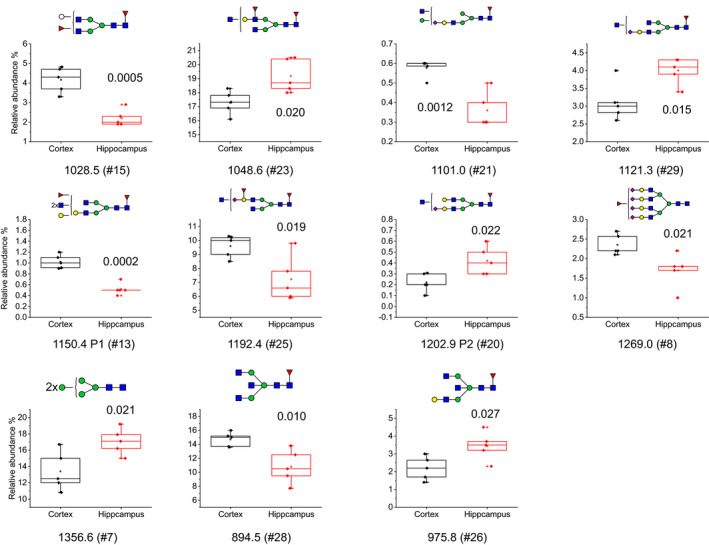
Relative abundance boxplots of structures that were significantly different in control hippocampus (n = 5) and control cortex (n = 5) using student's T‐test (with n referring to the number of individual cases included in the study). The P‐value for each structure is shown in each plot. The boxplots are displaying Max, Min, 99%, 1% and mean in the plot, in addition to the individual data points
FIGURE 6.
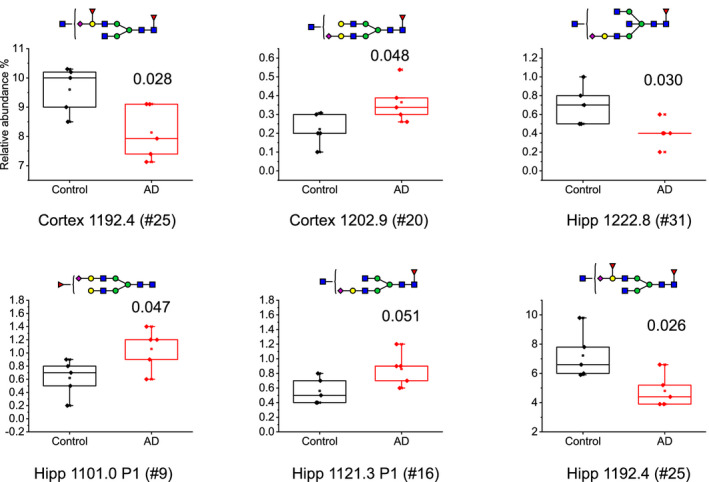
Relative abundance boxplots of structures that were significantly different in AD cortex and hippocampus compared to controls (n = 5 in each group, with n referring to the number of individual cases included in the study) using student's T‐test. The P‐value for each structure is shown in each plot. The boxplots are displaying Max, Min, 99%, 1% and mean in the plot in addition to the individual data points
3.3. N‐glycan expression differs between cortex and hippocampus
A student's t‐test was performed to determine if the differences observed between control cortex and control hippocampus were significant. Figure 5 shows boxplots of the relative abundance of the N‐glycans that displayed significant differences in the brain regions with individual data points, annotated with tentative glycan structures and P‐values. Significantly higher amounts of six N‐glycans were observed in cortex compared to hippocampus (#15, H5N4F2; #21, H5N4F1S1; #13, H4N6F2; #25, H4N5F2S1; #8, H7N6F1S4 and #28, H3N5F1), while five N‐glycans were significantly lower in cortex (#23, H4N5F2; #29, H4N5F1S1; #20, H5N5F1S1; #7, H5N2 and #26, H3N5F1). Nine of these structures were of complex type, one was a tentative hybrid glycan, and one of high mannose type.
3.4. The expression of N‐glycans are significantly different in AD compared to control
Comparing AD and control brains, significant differences were observed in both cortex and hippocampus (Figure 6). One unique glycan structure (#20, H5N5F1S1) was significantly higher in AD cortex compared to control. Another structure (#25, H4N5F2S1) was lower in AD brain in both cortex and hippocampus compared to control. Finally, two structures (#9, H5N4F1S1 and #16, H4N5F1S1) were higher in AD hippocampus, and one was lower in AD hippocampus (#31, H4N6F1S1) compared to control. Interestingly, all of the significantly altered glycans observed were structurally very similar: complex glycans decorated with one sialic acid and at least one fucose and carrying a confirmed (#31) or potential bisecting GlcNAc.
4. DISCUSSION
N‐glycans are crucial for brain functions and play important roles in neuronal activities, including neurogenesis, neurite outgrowth, synapse function, and memory formation, all of which are defective in AD. Therefore, it is highly warranted to analyze the N‐glycan profile in AD brain. Here, we analyzed the N‐glycome of hippocampus and cortex, two brain regions known to be affected in AD. Using 2‐AA‐derivatization of N‐glycans and LC‐MS/MS, we identified 26 N‐glycans based on informative MS/MS data. It is possible that we have not been able to detect all N‐glycans present in the two brain regions because of potentially low abundance of some glycans and the possibility that some glycans may not be extracted by our method. While the linkage site to proteins has been lost in glycomics, one advantage is that the structural elucidation of the glycans can be more complete, such as the sequence of the molecules, compared to glycoproteomics that usually only can resolve the composition. A common problem in proteomics and glycoproteomics is that peptides from major proteins can shield the signal from low abundant proteins during the LC separation, which has been exemplified in serum, where the 20 most abundant proteins account for 99% of all protein mass (Schwenk et al., 2017). In glycomics, this is not a major problem since unique N‐glycans are usually detected even from low abundant proteins, thus conferring a greater dynamic range.
Overall, our data are in line with previous studies reporting high extent of fucosylation, low degree of sialylation, and high degree of high mannose in brain (Barone et al., 2012; Gizaw et al., 2016). In our study, the four most abundant glycans, both in frontal cortex and hippocampus, had the compositions H4N5F2, H5N2, H3N5F1, and H4N5F2S1, three of which were reported to be most abundant also in a previous study of human brain cortex (Gizaw et al., 2016). Here, we used MS/MS to determine the structures and found that they correspond to a glycan carrying LewisX, a high mannose glycan, a glycan carrying bisecting GlcNAc and a glycan carrying a terminal galactose decorated with fucose and sialic acid respectively. All these glycan epitopes play significant roles in the nervous system (reviewed in (Sytnyk et al., 2020)). Glycans carrying the LewisX epitope are abundant at synapses and have been suggested to be involved in nervous system development, cell proliferation, neurite outgrowth, and synaptic plasticity (Sytnyk et al., 2020). High mannose glycans are also involved in brain development and are present in neuronal synapses. It was reported that the predominant high‐mannose glycan species in synapses of adult neurons carry five mannoses, which is consistent with the high‐mannose glycan identified in this study. The bisecting GlcNAc epitope plays a role in regulating neuronal development. Deficiency of N‐acetylglucosaminyltransferase III (GnT‐III) ‐ the enzyme responsible for adding bisecting GlcnAc to N‐glycans ‐ leads to neurological problems, while over‐expression promotes neurite outgrowth. The fucosylated terminal galactose epitope is well‐known for its presence in the ABO blood group antigens. However, several more recent studies have implicated the importance of Fucα(1–2)Gal for neuronal development, learning, and memory. For instance, reports from rodent models have shown that it can suppress hippocampal LTP maintenance (Krug et al., 1991), is enriched in certain synaptic proteins (Smalla et al., 1998) (Murrey et al., 2006) and promotes outgrowth of hippocampal neurons (Kalovidouris et al., 2005). Importantly, Fucα(1–2)Gal has also been confirmed to be present in human brain by in vivo two‐dimensional magnetic resonance correlated spectroscopy (2D L‐COSY), which has been used to assign seven different glycans containing Fucα(1–2)Gal in human brain (Mountford et al., 2015; Tosh et al., 2019).
Of the four most abundant N‐glycans in frontal cortex, the one with composition H4N5F2S1, carrying a fucosylated and sialylated terminal galactose (#25 in Figure 6), was significantly less abundant in AD compared to control. Another glycan with composition H5N4F1S1, decorated with a core fucose, a terminal sialic acid and an unassigned GlcNAc (#20 in Figure 6), was significantly more abundant in AD compared to control cortex. The finding that one glycan was more abundant, while another glycan was less abundant in frontal cortex in AD compared to control, suggests that the altered N‐glycan profile is not because of defects in single glycoproteins or glycosyltransferases.
In nondemented controls, the overall N‐glycan profile of hippocampus was similar to that of frontal cortex, with the same set of 26 N‐glycans identified in the two subregions, although there were some differences in the relative abundance of 11 of the glycans. Comparing the levels of glycans in hippocampus in AD with nondemented controls revealed four glycans with significantly altered levels. As in frontal cortex, the glycan with composition H4N5F2S1, carrying a fucosylated and sialylated terminal galactose (#25 in Figure 6), was significantly less abundant in AD. Another glycan that was less abundant in AD hippocampus had the composition H5N5F1S1 and was confirmed by MS/MS to be decorated with a bisecting GlcNAc (#31 in Figure 6). While several other reports have suggested structural compositions of N‐glycans without using MS/MS fragmentation to confirm the structures, we have unambiguously confirmed this and other specific glycan epitopes. It is, however, possible that some of the GlcNAcs that were not possible to annotate in other glycans in cortex and hippocampus are of the bisecting type. Bisecting GlcNAc has been extensively reported in human brain even though MS/MS evidence have not always been included. Here, we can confirm the bisecting structure for three glycans with diagnostic MS/MS. The two glycans with higher levels in AD compared to control hippocampus are complex glycans and carry one fucose and one sialic acid each and one of them has an unannotated GlcNAc (#9 and #16, respectively, in Figure 6).
The decreased level of a glycan carrying a bisecting GlcNAc in AD hippocampus is in contrast with the pattern reported in our recent study comparing the LC‐MS/MS profile of CSF from AD patients and controls, which showed higher levels of several N‐glycans carrying bisecting GlcNAc in CSF from AD patients compared to controls (Schedin‐Weiss, et al., 2020). Such difference between brain and CSF may reflect that the majority of this glycan epitope is expressed on different proteins in the brain and CSF. Alternatively, the glycosylation pattern of proteins secreted into CSF could be altered in AD. This could be accomplished for instance if the secretion of the proteins is affected by the disease or if the proteins are affected by the neurodegeneration, resulting in degradation products in CSF.
Bisecting GlcNAc has previously been reported to be involved in AD. For instance, expression of GnT‐III has been reported to be up‐regulated in AD brain (Akasaka‐Manya et al., 2010). Moreover, bisecting GlcNAc is present on BACE1 and the presence of this epitope on BACE1 has been suggested to be increased in AD brain, leading to a shift in its subcellular location from lysosomes to endosomes, which in turn leads to increased generation of Aβ (Kizuka et al., 2015). Bisecting GlcNAc is present also on other AD‐related proteins. For instance, tau, which is a cytosolic protein and under normal circumstances considered not to be N‐glycosylated, was reported to be N‐glycosylated in AD brain (Wang et al., 1996) and glycan analysis suggested that tau isolated from AD brain carry bisecting GlcNAc (Liu et al., 2002). Furthermore, it has been shown that the hereditary AD mutations called the Swedish and London mutations cause an increased expression of bisecting GlcNAc on APP in cell culture (Akasaka‐Manya et al., 2008). Clearly, further studies are needed to pinpoint the causes and effects of altered levels of bisecting GlcNAc in CSF and brain in AD.
It is interesting to note that the levels of the glycan with composition H4N5F2S1, carrying a fucosylated and sialylated terminal galactose, were lower in both frontal cortex and hippocampus in AD compared to control brain. Glycans containing fucose α‐(1,2)‐galactose have previously been found to be important for neuronal growth and have been implicated in communication that underlies long‐term memory storage (Kalovidouris et al., 2005). It will be interesting to determine in future studies how the sialic acid influences this epitope and how the glycan is related to AD pathology.
Since AD starts to develop decades before clinical diagnosis can be made, it is important to study the process early, pre‐symptomatically, to enable early diagnosis and treatment, before irreversible damage occurs to the brain. Such studies are difficult to perform in humans, although biomarker modeling can be used to estimate at which disease stages alterations start to occur (Jack & Holtzman, 2013; Jack et al., 2013). One way to study pre‐symptomatic processes is to use animal models. Interestingly, our recent time‐course study of an App knock‐in mouse model for AD, AppNL/F , showed that a selective increase in Aβ42 leads to alterations in several proteins and pathways ‐ affecting for instance synaptic functions and neuritogenesis – at three months of age, i.e. several months before amyloidosis and memory impairment appears (Schedin‐Weiss, et al., 2020). Intriguingly, we observed trends in decreased levels of some enzymes involved in the attachment of N‐glycans to the growing polypeptide chains at the same early age of these mice. The notion that changes in N‐glycosylation occur early during AD development is also supported by glycomics studies of CSF. Indeed, it was found that levels of bisecting GlcNAc correlate with CSF levels of total tau (t‐tau) and phospho‐tau (p‐tau) in cases with subjective cognitive impairment, which may reflect presymptomatic AD cases.
In conclusion, the altered N‐glycan profiles in frontal cortex and hippocampus support substantial alterations in the N‐glycosylation pathway in these subregions of AD brain. One of the most abundant N‐glycans in both hippocampus and cortex contains a terminal galactose decorated with fucose and sialic acid. The levels of this glycan were decreased in both subregions in AD brain. Another glycan carrying a bisecting GlcNAc was decreased in hippocampus in AD compared to control. These glycan epitopes have been suggested to be important for several brain functions including cell proliferation, neurite outgrowth and memory formation. Altogether, these findings strengthen previous propositions that N‐glycans are relevant potential biomarkers for AD, that glycosylation is affected in AD pathology and that glycans or enzymes generating or degrading them may be potential novel targets for AD treatments.
CONFLICTS OF INTEREST
The authors have no conflict of interest to declare.
Supporting information
Fig S1‐S3
ACKNOWLEDGMENTS
This work was supported by grant AF‐745091 from The Swedish Alzheimer Foundation, grant 20180373 from the Swedish County Council (ALF), grants 2018‐00733, 2017‐0544 and 2016‐00426 from Stiftelsen för gamla tjänarinnor, grants from Stohnes foundation, grants from Margaretha af Ugglas foundation, grant SLS501451from Svenska Läkaresällskapet, grant no 94 from Demensfonden, grant from Eva och Oscar Ahréns stiftelse and grants 2014alde42401, 2014fobi41199 and 2015alde45589 from KI fonder, KI forskningsstiftelser.
All experiments were conducted in compliance with the ARRIVE guidelines.
Gaunitz S, Tjernberg LO, Schedin‐Weiss S. The N‐glycan profile in cortex and hippocampus is altered in Alzheimer disease. J Neurochem.2021;159:292–304. 10.1111/jnc.15202
This article is part of the special issue "Mass spectrometry in Alzheimer disease"
Contributor Information
Stefan Gaunitz, Email: rolf.gaunitz@sll.se.
Sophia Schedin‐Weiss, Email: sophia.schedin.weiss@ki.se.
REFERENCES
- Akasaka‐Manya K., Manya H., Sakurai Y., Wojczyk B. S, Kozutsumi Y., Saito Y., Taniguchi N., Murayama S., Spitalnik S. L, Endo T. (2010). Protective effect of N‐glycan bisecting GlcNAc residues on ‐amyloid production in Alzheimer's disease. Glycobiology, 20, (1), 99–106. 10.1093/glycob/cwp152. [DOI] [PubMed] [Google Scholar]
- Akasaka‐Manya, K. , Manya, H. , Sakurai, Y. , Wojczyk, B. S. , Spitalnik, S. L. , & Endo, T. (2008). Increased bisecting and core‐fucosylated N‐glycans on mutant human amyloid precursor proteins. Glycoconjugate Journal, 25, 775–786. 10.1007/s10719-008-9140-x [DOI] [PubMed] [Google Scholar]
- Aoki Mikio, Volkmann Inga, Tjernberg Lars O., Winblad Bengt, Bogdanovic Nenad (2008). Amyloid β‐peptide levels in laser capture microdissected cornu ammonis 1 pyramidal neurons of Alzheimerʼs brain. NeuroReport, 19, (11), 1085–1089. 10.1097/wnr.0b013e328302c858. [DOI] [PubMed] [Google Scholar]
- Barone, R. , Sturiale, L. , Palmigiano, A. , Zappia, M. , & Garozzo, D. (2012). Glycomics of pediatric and adulthood diseases of the central nervous system. Journal of Proteomics, 75, 5123–5139. 10.1016/j.jprot.2012.07.007 [DOI] [PubMed] [Google Scholar]
- Ceroni Alessio, Maass Kai, Geyer Hildegard, Geyer Rudolf, Dell Anne, Haslam Stuart M. (2008). GlycoWorkbench: A Tool for the Computer‐Assisted Annotation of Mass Spectra of Glycans†. Journal of Proteome Research, 7, (4), 1650–1659. 10.1021/pr7008252. [DOI] [PubMed] [Google Scholar]
- Damerell, D. , Ceroni, A. , Maass, K. , Ranzinger, R. , Dell, A. , & Haslam, S. M. (2012). The GlycanBuilder and GlycoWorkbench glycoinformatics tools: Updates and new developments. Biological Chemistry, 393, 1357–1362. 10.1515/hsz-2012-0135 [DOI] [PubMed] [Google Scholar]
- De Strooper Bart, Vassar Robert, Golde Todd (2010). The secretases: enzymes with therapeutic potential in Alzheimer disease. Nature Reviews Neurology, 6, (2), 99–107. 10.1038/nrneurol.2009.218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel‐Pinter Moran, Shmueli Merav Daniel, Raz Chen, Yanku Michaela, Zilberzwige Shai, Gazit Ehud, Segal Daniel (2017). Interplay between protein glycosylation pathways in Alzheimer’s disease. Science Advances, 3, (9), e1601576 10.1126/sciadv.1601576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frenkel‐Pinter, M. , Stempler, S. , Tal‐Mazaki, S. , Losev, Y. , Singh‐Anand, A. , Escobar‐Álvarez, D. , Lezmy, J. , Gazit, E. , Ruppin, E. , & Segal, D. (2017). Altered protein glycosylation predicts Alzheimer's disease and modulates its pathology in disease model Drosophila. Neurobiology of Aging, 56, 159–171. 10.1016/j.neurobiolaging.2017.04.020 [DOI] [PubMed] [Google Scholar]
- Gizaw, S. T. , Ohashi, T. , Tanaka, M. , Hinou, H. , & Nishimura, S. (2016). Glycoblotting method allows for rapid and efficient glycome profiling of human Alzheimer's disease brain, serum and cerebrospinal fluid towards potential biomarker discovery. Biochimica Et Biophysica Acta, 1860, 1716–1727. 10.1016/j.bbagen.2016.03.009 [DOI] [PubMed] [Google Scholar]
- Halim A., Brinkmalm G., Ruetschi U., Westman‐Brinkmalm A., Portelius E., Zetterberg H., Blennow K., Larson G., Nilsson J. (2011). Site‐specific characterization of threonine, serine, and tyrosine glycosylations of amyloid precursor protein/amyloid ‐peptides in human cerebrospinal fluid. Proceedings of the National Academy of Sciences, 108, (29), 11848–11853. 10.1073/pnas.1102664108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto, M. , Bogdanovic, N. , Nakagawa, H. , Volkmann, I. , Aoki, M. , Winblad, B. , Sakai, J. , & Tjernberg, L. O. (2012). Analysis of microdissected neurons by 18O mass spectrometry reveals altered protein expression in Alzheimer's disease. Journal of Cellular and Molecular Medicine, 16, 1686–1700. 10.1111/j.1582-4934.2011.01441.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack, C. R. , Bennett, D. A. , Blennow, K. , Carrillo, M. C. , Dunn, B. , Haeberlein, S. B. , Holtzman, D. M. , Jagust, W. , Jessen, F. , Karlawish, J. , Liu, E. , Molinuevo, J. L. , Montine, T. , Phelps, C. , Rankin, K. P. , Rowe, C. C. , Scheltens, P. , Siemers, E. , Snyder, H. M. , … Silverberg, N. (2018). NIA‐AA research framework: Toward a biological definition of Alzheimer's disease. Alzheimer's & Dementia : The Journal of the Alzheimer's Association, 14, 535–562. 10.1016/j.jalz.2018.02.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack, C. R. Jr , & Holtzman, D. M. (2013). Biomarker modeling of Alzheimer's disease. Neuron, 80, 1347–1358. 10.1016/j.neuron.2013.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack C.R, Knopman D. S, Jagust W. J, Petersen R. C, Weiner M. W, Aisen P. S, … Trojanowski J. Q (2013). Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. The Lancet Neurology, 12, (2), 207–216. 10.1016/s1474-4422(12)70291-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalovidouris Stacey A., Gama Cristal I., Lee Lori W., Hsieh‐Wilson Linda C. (2005). A Role for Fucose α(1−2) Galactose Carbohydrates in Neuronal Growth. Journal of the American Chemical Society, 127, (5), 1340–1341. 10.1021/ja044631v. [DOI] [PubMed] [Google Scholar]
- Kizuka Y., Kitazume S, Fujinawa R, Saito T, Iwata N, Saido T. C, Nakano M., Yamaguchi Y., Hashimoto Y., Staufenbiel M., Hatsuta H., Murayama S., Manya H., Endo T., Taniguchi N. (2015). An aberrant sugar modification of BACE 1 blocks its lysosomal targeting in A lzheimer's disease. EMBO Molecular Medicine, 7, (2), 175–189. 10.15252/emmm.201404438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kizuka, Y. , Kitazume, S. , & Taniguchi, N. (2017). N‐glycan and Alzheimer's disease. Biochimica Et Biophysica Acta ‐ General Subjects, 1861, 2447–2454. 10.1016/j.bbagen.2017.04.012 [DOI] [PubMed] [Google Scholar]
- Krug, M. , Jork, R. , Reymann, K. , Wagner, M. , & Matthies, H. (1991). The amnesic substance 2‐deoxy‐D‐galactose suppresses the maintenance of hippocampal LTP. Brain Research, 540, 237–242. 10.1016/0006-8993(91)90513-U [DOI] [PubMed] [Google Scholar]
- Liu, F. , Zaidi, T. , Iqbal, K. , Grundke‐Iqbal, I. , & Gong, C. X. (2002). Aberrant glycosylation modulates phosphorylation of tau by protein kinase A and dephosphorylation of tau by protein phosphatase 2A and 5. Neuroscience, 115, 829–837. 10.1016/S0306-4522(02)00510-9 [DOI] [PubMed] [Google Scholar]
- Mountford C, Quadrelli S, Lin A, Ramadan S (2015). Six fucose‐α(1‐2) sugars and α‐fucose assigned in the human brain usingin vivotwo‐dimensional MRS. NMR in Biomedicine, 28, (3), 291–296. 10.1002/nbm.3239. [DOI] [PubMed] [Google Scholar]
- Murrey, H. E. , Gama, C. I. , Kalovidouris, S. A. , Luo, W. I. , Driggers, E. M. , Porton, B. , & Hsieh‐Wilson, L. C. (2006). Protein fucosylation regulates synapsin Ia/Ib expression and neuronal morphology in primary hippocampal neurons. Proceedings of the National Academy of Sciences of the United States of America, 103, 21–26. 10.1073/pnas.0503381102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, Tsubuki S, Tanaka M, Iwata N, Saito T, Saido T. C. (2013). Aβ Secretion and Plaque Formation Depend on Autophagy. Cell Reports, 5, (1), 61–69. 10.1016/j.celrep.2013.08.042. [DOI] [PubMed] [Google Scholar]
- Palmigiano, A. , Barone, R. , Sturiale, L. , Sanfilippo, C. , Bua, R. O. , Romeo, D. A. , Messina, A. , Capuana, M. L. , Maci, T. , Le Pira, F. , Zappia, M. , & Garozzo, D. (2016). CSF N‐glycoproteomics for early diagnosis in Alzheimer's disease. Journal of Proteomics, 131, 29–37. 10.1016/j.jprot.2015.10.006 [DOI] [PubMed] [Google Scholar]
- Regan, P. , McClean, P. L. , Smyth, T. , & Doherty, M. (2019). Early stage glycosylation biomarkers in Alzheimer's disease. Medicines (Basel), 6, (3). 10.3390/medicines6030092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saez‐Valero, J. , & Small, D. H. (2001). Acetylcholinesterase and butyrylcholinesterase glycoforms are biomarkers of Alzheimer's disease. Journal of Alzheimer's Disease: JAD, 3, 323–328. 10.3233/JAD-2001-3307 [DOI] [PubMed] [Google Scholar]
- Schedin‐Weiss, S. , Gaunitz, S. , Sui, P. , Chen, Q. , Haslam, S. M. , Blennow, K. , Winblad, B. , Dell, A. , & Tjernberg, L. O. (2020). Glycan biomarkers for Alzheimer disease correlate with T‐tau and P‐tau in cerebrospinal fluid in subjective cognitive impairment. The FEBS Journal, 287, 3221–3234. 10.1111/febs.15197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schedin‐Weiss S, Nilsson P, Sandebring‐Matton A, Axenhus M, Sekiguchi M, Saito T, Winblad B, Saido T, Tjernberg L O. (2020). Proteomics Time‐Course Study of App Knock‐In Mice Reveals Novel Presymptomatic Aβ42‐Induced Pathways to Alzheimer’s Disease Pathology. Journal of Alzheimer's Disease, 75, (1), 321–335. 10.3233/jad-200028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schedin‐Weiss, S. , Winblad, B. , & Tjernberg, L. O. (2014). The role of protein glycosylation in Alzheimer disease. The FEBS Journal, 281, 46–62. 10.1111/febs.12590 [DOI] [PubMed] [Google Scholar]
- Schwenk, J. M. , Omenn, G. S. , Sun, Z. , Campbell, D. S. , Baker, M. S. , Overall, C. M. , Aebersold, R. , Moritz, R. L. , & Deutsch, E. W. (2017). The human plasma proteome draft of 2017: Building on the human plasma PeptideAtlas from mass spectrometry and complementary assays. Journal of Proteome Research, 16, 4299–4310. 10.1021/acs.jproteome.7b00467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smalla K., Angenstein F., Richter K., Gundelfinger E. D., & Staak S. (1998). Identification of fucose α(1‐2) galactose epitope‐containing glycoproteins from rat hippocampus. NeuroReport, 9, (5), 813–817. 10.1097/00001756-199803300-00009. [DOI] [PubMed] [Google Scholar]
- Söderberg, L. , Bogdanovic, N. , Axelsson, B. , Winblad, B. , Naslund, J. , & Tjernberg, L. O. (2006). Analysis of single Alzheimer solid plaque cores by laser capture microscopy and nanoelectrospray/tandem mass spectrometry. Biochemistry, 45, 9849–9856. 10.1021/bi060331+ [DOI] [PubMed] [Google Scholar]
- Spillantini M. G., & Goedert M. (2013). Tau pathology and neurodegeneration. The Lancet Neurology, 12, (6), 609–622. 10.1016/s1474-4422(13)70090-5. [DOI] [PubMed] [Google Scholar]
- Sytnyk V., Leshchyns’ka I., Schachner M. (2020). Neural glycomics: the sweet side of nervous system functions. Cellular and Molecular Life Sciences, 10.1007/s00018-020-03578-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Reisuke H., Milner Teresa A., Li Feng, Nam Ellen E., Edgar Mark A., Yamaguchi Haruyasu, Beal M. Flint, Xu Huaxi, Greengard Paul, Gouras Gunnar K. (2002). Intraneuronal Alzheimer Aβ42 Accumulates in Multivesicular Bodies and Is Associated with Synaptic Pathology. The American Journal of Pathology, 161, (5), 1869–1879. 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaysen‐Andersen M., Packer N. H., & Schulz B. L. (2016). Maturing Glycoproteomics Technologies Provide Unique Structural Insights into theN‐glycoproteome and Its Regulation in Health and Disease. Molecular & Cellular Proteomics, 15, (6), 1773–1790. 10.1074/mcp.o115.057638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosh N., Quadrelli S., Galloway G., & Mountford C. (2019). Two New Fucose‐α (1–2)‐Glycans Assigned In The Healthy Human Brain Taking The Number To Seven. Scientific Reports, 9, (1), 10.1038/s41598-019-54933-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki, A. , Cummings, R. D. , Esko, J. D. , Freeze, H. H. , Stanley, P. , Marth, J. D. , Bertozzi, C. R. , Hart, G. W. , & Etzler, M. E. (2009). Symbol nomenclature for glycan representation. Proteomics, 9, 5398–5399. 10.1002/pmic.200900708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, J. Z. , Grundke‐Iqbal, I. , & Iqbal, K. (1996). Glycosylation of microtubule‐associated protein tau: An abnormal posttranslational modification in Alzheimer's disease. Nature Medicine, 2, 871–875. 10.1038/nm0896-871 [DOI] [PubMed] [Google Scholar]
- Yu Y., Jans D. C., Winblad B., Tjernberg L. O., & Schedin‐Weiss S. (2018). Neuronal Aβ42 is enriched in small vesicles at the presynaptic side of synapses. Life Science Alliance, 1, (3), e201800028. 10.26508/lsa.201800028. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S3