

Abstract
Alzheimer's disease and other Tauopathies are associated with neurofibrillary tangles composed of Tau protein, as well as toxic Tau oligomers. Therefore, inhibitors of pathological Tau aggregation are potentially useful candidates for future therapies targeting Tauopathies. Two hexapeptides within Tau, designated PHF6* (275‐VQIINK‐280) and PHF6 (306‐VQIVYK‐311), are known to promote Tau aggregation. Recently, the PHF6* segment has been described as the more potent driver of Tau aggregation. We therefore employed mirror‐image phage display with a large peptide library to identify PHF6* fibril binding peptides consisting of D‐enantiomeric amino acids. The suitability of D‐enantiomeric peptides for in vivo applications, which are protease stable and less immunogenic than L‐peptides, has already been demonstrated. The identified D‐enantiomeric peptide MMD3 and its retro‐inverso form, designated MMD3rev, inhibited in vitro fibrillization of the PHF6* peptide, the repeat domain of Tau as well as full‐length Tau. Dynamic light scattering, pelleting assays and atomic force microscopy demonstrated that MMD3 prevents the formation of tau β‐sheet‐rich fibrils by diverting Tau into large amorphous aggregates. NMR data suggest that the D‐enantiomeric peptides bound to Tau monomers with rather low affinity, but ELISA (enzyme‐linked immunosorbent assay) data demonstrated binding to PHF6* and full length Tau fibrils. In addition, molecular insight into the binding mode of MMD3 to PHF6* fibrils were gained by in silico modelling. The identified PHF6*‐targeting peptides were able to penetrate cells. The study establishes PHF6* fibril binding peptides consisting of D‐enantiomeric amino acids as potential molecules for therapeutic and diagnostic applications in AD research.
Keywords: Alzheimer's disease, D-peptides, phage display, tau aggregation inhibitors, therapy
Alzheimer's disease and other Tauopathies are associated with neurofibrillary tangles composed of Tau protein, as well as toxic Tau oligomers. Inhibitors of pathological Tau aggregation are potentially useful candidates for future therapies targeting Tauopathies. We employed mirror‐image phage display with a large peptide library to identify Tau binding peptides consisting of D‐enantiomeric amino acids. The suitability of D‐enantiomeric peptides for in vivo applications, which are protease stable and less immunogenic than L‐peptides, has already been demonstrated. The identified D‐enantiomeric peptides prevent the formation of β‐sheet‐rich Tau fibrils by diverting Tau into large amorphous aggregates. They might be interesting for AD therapy.
Introduction
Alzheimer's disease (AD) is an irreversible neurodegenerative disorder associated with a progressive decline of cognitive functions. [1] The neuropathological hallmarks of AD are extracellular Aβ amyloid plaques and intracellular neurofibrillary tangles (NFT), consisting of abnormally folded Tau proteins. [2] The microtubule associated protein Tau plays an essential role in microtubule‐stabilization. In healthy neurons, Tau binds to microtubules in axons and stabilizes them, promoting the neuronal function. [3] In several neurodegenerative diseases, termed Tauopathies, the most common of which is AD, Tau detaches from the axonal microtubules and forms fibrils. [4] Recent data indicated that Tau oligomers are the highly toxic species contributing to tau pathology. [5]
AD clinical progression correlates with the pathological aggregation of Tau. [6] Compounds that target Tau aggregation may thus represent a promising therapeutic strategy. Several Tau aggregation inhibitors have been reported in the literature. This includes the diphenylpyrazole anle138b, which restores hippocampal synaptic and transcriptional plasticity as well as spatial memory in a mouse model for Alzheimer's disease. [7] Another promising small molecule is the methylene blue derivative LMTX, which reached phase III clinical trials after showing its ability to delay disease progression in a phase II clinical trial carried out of the course of one year. [8] However, up to the current day there is no curative treatment for AD or other Tauopathies with proven efficacy.
The use of small D‐enantiomeric peptides to prevent the pathological fibrillization of Tau may provide a reasonable alternative to chemical compounds. D‐peptides have several advantages over L‐peptides. They are stable against proteases and less immunogenic than L‐peptides. In addition, some D‐enantiomeric peptides can reach the brain after oral administration.[ 9 , 10 , 11 , 12 , 13 ] The D‐enantiomeric peptide D3 was selected using mirror image phage display against D‐Aβ peptide. D3 disassembles Aβ plaques and causes improvement in behavioral tests of APP/PS1 double transgenic mice.[ 10 , 11 , 12 , 13 , 14 , 15 ] A derivative of D3, the peptide RD2, completed a phase I clinical study.
It has been shown that Tau aggregation is strongly driven by two hexapeptide fragments within Tau, namely PHF6 (VQIVYK) and PHF6* (VQIINK).[ 16 , 17 ] The PHF6 segment is located at the beginning of the third repeat (R3) and is present in all Tau isoforms. In contrast, the PHF6* segment is located at the beginning of the second repeat (R2) and is present only in four‐repeat (4R) Tau isoforms (Figure 1A). [18] Until recently, it was thought that PHF6, 306‐VQIVYK‐311, is the most potent driver for Tau assembly into paired helical filaments (PHF), and that mutations in this six‐residue segment could decrease or increase the aggregation of Tau. [16] However, in 2018, Seidler et al. suggested that the PHF6* segment 275‐VQIINK‐280 is the more powerful driver of Tau aggregation. [19]
Figure 1.

Scheme for selection of PHF6*‐binding peptides using mirror image phage display. (A) The largest human Tau isoform (Tau 2 N4R) in the central nervous systems contains four microtubule binding repeats. PHF6*, consisting of amino acids 275 to 280, is located at the beginning of repeat two. [18] (B) The D‐enantiomeric form of PHF6* was synthesized. After fibrillization, the D‐enantiomeric fibrils were used for phage display. An L‐peptide, binding to the D‐enantiomeric PHF6* fibrils, was selected and the D‐enantiomeric version of the selected L‐peptide was synthesized, which will bind to the L‐enantiomeric form of the target, regular PHF6* fibrils.
The Eisenberg group designed a D‐enantiomeric peptide, TLKIVW, on PHF6 fibrils as template. The TLKIVW peptide inhibited the aggregation of the Tau peptide PHF6 and of truncated Tau constructs K12 and K19. [20] In addition, our group selected D‐enantiomeric peptides using mirror image phage display against PHF6 fibrils. The identified peptides were able to inhibit PHF6 and full‐length Tau fibrillization in vitro. Moreover, the selected peptides were able to penetrate Tau expressing N2a cells. [21] Recently, the peptide W‐MINK was developed using PHF6* fibrils as template for a structure‐based inhibitor design. W‐MINK reduces the number of inclusions appearing in HEK293 biosensor cells expressing YFP‐labeled tau variants when the cells are exposed to aggregates of full‐length Tau, indicative of induced local accumulations. [19]
Here, we describe the development of specific D‐enantiomeric peptides that inhibit the pathological aggregation of Tau by employing mirror image phage display using D‐PHF6* fibrils as a target. With our selection against PHF6* fibrils, we aimed to target the PHF6* site of Tau rather than any specific conformer like Tau monomers, oligomers or fibrils. We have used fibrils for the selection as PHF6* monomers might not be suitable to be target in a successful phage display ‐ we doubted whether the peptides consisting of six amino acids could be immobilized properly in a way that would allow binding options for the phages. PHF6* oligomers can hardly be isolated and might not be stable enough during a selection process. At the end, the D‐peptides selected against PHF6* fibrils will not be conformer specific and will target PHF6* also in other conformers, like fibrils.
After biophysical and biochemical characterization of the D‐peptide‐Tau interaction, we started to investigate, which of the two hexapeptide sequences within Tau, PHF6 or PHF6*, is a more effective target for the development of inhibitor peptides against Tau aggregation into oligomer species and subsequently fibrils. We compared the inhibitory effects of the D‐peptides identified here with other previously described PHF6/PHF6*‐targeting peptides in vitro. The combined analysis suggests that the inhibitor peptides identified in the current work might present promising candidates for therapeutic and diagnostic applications in AD research.
Results
Mirror image phage display selection against fibrils of D‐PHF6*
As it has recently been reported that the hexapeptide segment PHF6* plays a more powerful role in Tau aggregation than PHF6, and that PHF6* was an excellent target to develop full‐length Tau aggregation inhibitors, [19] we decided to perform a mirror image phage display against D‐PHF6* fibrils to obtain PHF6*‐specific D‐peptides as compounds with potential for AD therapy development. In a previous project from our group, [21] we already selected Tau aggregation inhibiting D‐peptides by employing mirror image phage display using PHF6 fibrils as a target. It was of our interest to investigate which of the hexapeptide segments within Tau, PHF6 or PHF6*, is the more powerful target for developing Tau aggregation inhibitors.
Fibrils of D‐PHF6* were prepared to be used as a target for mirror image phage display. D‐PHF6* was first incubated in NaPi buffer in presence of 10 μM ThT to test fibril formation, which was monitored by measuring the fluorescence of ThT. For the selection process, D‐PHF6* fibrils without thioflavin were immobilized on the plate and a mirror image phage display selection was performed as shown schematically in Figure 1B.
After four panning rounds, single phage ELISA was performed to investigate the binding properties of individual phage clones to the target. The DNA of promising phages, which showed relatively high signal in comparison to the negative controls, was sequenced to identify the amino‐acid sequence of the respective peptide. In total, 29 displayed peptides were identified (see Supporting Information Table S1). Across these 29 sequences, we did not find any dominating peptide sequence. All obtained sequences were screened using the SAROTUP database (an abbreviation of “Scanner And Reporter Of Target‐Unrelated Peptides“) [22] to exclude possible target unrelated‐peptides. After selection, three peptides (MMDP2, MMDP6 and MMD3; Table 1) were chosen to be synthesized as D‐peptides to test their inhibitory effect on PHF6* and full‐length Tau fibril formation using a THT fibrillization assay. MMDP2 and MMDP6 inhibited the aggregation of the PHF6* peptide (data not shown), but not the fibrillization of full‐length Tau. In contrast, the peptide MMD3 inhibited the aggregation of both PHF6* and full‐length Tau. The retro‐inverso form of MMD3, MMD3rev, was also synthesized and inhibited the aggregation of both PHF6* and full‐length Tau (Figure S2, Table 1).
Table 1.
Amino acid sequences of the synthesized D‐peptides selected against D‐PHF6* fibrils by employing mirror image phage display.
|
Name |
Sequences |
Inhibition of PHF6* fibrillization |
Inhibition of full‐length Tau fibrillization[b] |
|---|---|---|---|
|
MMDP2 |
wphdtkrylfpa |
+/−[a] |
− |
|
MMDP6 |
hsdlwrrsfelm |
+ |
− |
|
MMD3 |
dplkarhtsvwy |
+ |
+ |
|
MMD3rev |
ywvsthraklpd |
+ |
+ |
[a] Comparably low inhibition of Tau fibril formation. [b] As tested by ThT assay.
MMD3 and MMD3rev inhibit the aggregation of PHF6* but not of PHF6
As MMD3 and MMD3rev were selected against PHF6*, we asked if MMD3 and MMD3rev were specific inhibitors of PHF6* fibrillization. Therefore, the inhibitory effects of MMD3 and MMD3rev were tested against PHF6* and PHF6 using ThT assays. As PHF6 forms fibrils spontaneously by incubation at RT, a sample containing PHF6 alone was used as positive control. An increase in ThT signal was observed in all samples, which indicates no inhibitory effect of MMD3 or MMD3rev on PHF6 fibril formation. A small shift of the lag phase was observed, which could be due to an interaction of any kind of the D‐peptides with PHF6, but later, fibrillation reaches the same level as the Tau control without D‐peptides (Figure 2A). Next, the influence of MMD3 and MMD3rev on the fibrillization of PHF6* was investigated. To this end, PHF6* samples were incubated with either MMD3 or MMD3rev in the presence of ThT at RT. As shown in Figure 2B, PHF6* fibrillized spontaneously by incubation at RT and the saturation level was reached after 30 h. The fibrillization level of PHF6* was detected by increasing the relative fluorescence signal of ThT. Under the same conditions, PHF6*‐samples were treated with either the MMD3 or the MMD3rev inhibitor peptide in a molar ratio of 1 : 10 (PHF6*: inhibitor peptide). The treated samples showed low ThT signals, which indicates no detectable aggregation.
Figure 2.

PHF6 fibrillizes spontaneously by incubation at room temperature. The assay was performed using 50 μM PHF6 in NaPi buffer with 10 μM ThT (gray column). NaPi and 10 μM ThT without addition of PHF6 was used as control (red column). Peptides MMD3 or MMD3rev were added in concentrations of 500 μM to 50 μM PHF6 samples (green column and blue column, respectively). Fluorescence was measured at 490 nm in relative units (mean +/− standard deviations of results, three replicates per run). Samples treated with D‐peptides do not show a decreasing ThT signal, indicating no inhibition of PHF6 aggregation. (B) PHF6* fibrillization was performed by incubating 100 μM PHF6* in NaPi buffer with 10 μM ThT at room temperature (gray column). NaPi and 10 μM ThT without addition of PHF6 was used as control (red column). Peptides MMD3 or MMD3rev were added in concentrations of 1000 μM to 100 μM PHF6* samples (green column and blue column respectively). Fluorescence was measured at 490 nm in relative units (mean +/− standard deviations of results, three replicates per run). Samples treated with D‐peptides show a decreasing ThT signal, indicating an inhibition of PHF6* aggregation.
ELISAs demonstrate the binding of MMD3 and MMD3rev to PHF6* and Tau fibrils
To investigate the binding properties of MMD3 and MMD3rev to Tau fibrils and PHF6* fibrils, ELISAs were carried out. To this end, FAM‐labeled versions of MMD3 and MMD3rev were synthesized. To test the binding of MMD3 and MMD3rev to Tau fibrils, the wells were coated with Tau fibrils and increasing concentrations of FAM‐MMD3 and FAM‐MMD3rev (1, 5, 10 and 20 μg/mL) were used. The coated wells exhibited higher signals when compared to control wells, indicating a binding of MMD3 and MMD3rev to Tau fibrils (Figure 3A and 3B).
Figure 3.

ELISA was performed with FAM‐labeled versions of MMD3 and MMD3rev. The plate was coated with Tau fibrils in a 10 μg/mL concentration or with PHF6* fibrils at a concentration of 50 μg/mL. As negative control, only coating buffer was added to the wells. The peptides MMD3‐FAM and MMD3rev‐FAM were added in an increasing concentration; the bound peptides were detected with anti‐FAM antibodies. After adding the TMB substrate, the absorption at 450 nm, which represents the binding affinities, was measured. We observed that the absorption signal increased with increasing the concentration of MMD3 or MMD3rev, which indicates that MMD3 and MMD3rev bound to Tau fibrils as well as PHF6* fibrils.
In the case of PHF6*, the plate was coated with PHF6* fibrils. After blocking, FAM‐MMD3 and FAM‐MMD3rev were added to the coated plate at increasing concentration (1, 5, 10, 20, 40, 60, 80 and 100 μg/mL). The detection of bound D‐peptides was performed using anti‐FAM antibodies. As negative control, wells were filled with buffer. Wells coated with PHF6* fibrils showed high signals when compared to buffer wells, which demonstrates binding of MMD3 and MMD3rev to PHF6* fibrils (Figure 3C).
NMR studies reveal that MMD3 binds to Tau monomers with low affinity
To investigate if MMD3 binds to Tau monomers and also in order to detect possible binding sites, NMR titrations were carried out. The 1H‐15N SOFAST HMQC of hTau40 in the presence and absence of MMD3 showed low chemical shift dispersion along the 1H dimension, indicating that hTau40 remains disordered throughout the course of the NMR titrations (Figure 4A). Upon addition of MMD3, no new peaks were observed in the spectra of hTau40. Next, the chemical shift perturbations (CSPs) of 1H/15N pairs were investigated, because they are sensitive to changes in the chemical environment of the amide backbone of hTau40. However, at all tested hTau40 : peptide molar ratios the CSPs of the hTau40 backbone amide protons were very small and did not show a correlation with the doses of MMD3 (Figure 4B). We also note that overnight incubation of the hTau40 reference sample (i. e. without peptide) in a buffer system devoid of reducing agents (e. g. tris(2‐carboxyethyl)phosphine) promotes the oligomerization/aggregate formation of hTau40. Therefore, the samples used for the 1H‐15N SOFAST HMQC reference spectra will contain small amounts of higher‐order structures. Such oligomers/aggregates are NMR‐invisible due to slow tumbling and/or inhomogeneity. Notably, for most of the hTau40 residues a slight (∼2–5 %) increase in NMR signal intensity was observed in the presence of MMD3 (Figure 4C). The increase in NMR signal intensity of hTau40 might be due to decreased oligomerization of hTau40 in the presence on MMD3.
Figure 4.

MMD3 has low affinity for binding to monomeric hTau40. (A) Superposition of 2D 1H‐15N SOFAST‐HMQC of hTau40 in the absence and presence of MMD3 (hTau40 : MMD3 molar ratio of 1 : 30). (B) MMD3‐induced CSPs in hTau40 at hTau40 : MMD3 mole ratios of 1 : 3 (green), 1 : 10 (blue), and 1 : 30 (red). The dotted black line corresponds to a threshold of 0.0019 ppm, which is two standard deviations above the average CSP value for the mole ratio 1 : 10. (C) NMR signal intensity ratios I/I0 from 2D 1H‐15N SOFAST‐HMQC of hTau40 for hTau40 : MMD3 mole ratios of 1 : 3 (green), 1 : 10 (blue), and 1 : 30 (red). The dotted black line corresponds to a threshold of 0.94, which is two standard deviations below the average I/I0 value for the mole ratio 1 : 10.
MMD3 and MMD3rev inhibit the formation of amyloidogenic Tau fibrils by forming amorphous aggregates
To investigate the effect of MMD3 and MMD3rev on Tau aggregation, we studied the change of the size of Tau aggregates in the presence of MMD3 and MMD3rev using dynamic light scattering (DLS) and pelleting assays. The 4‐repeat domain of Tau with the pro‐aggregation mutation ΔK280 (10 μM) (TauRDΔK) was incubated with heparin 16000 (H16 K) to induce filaments in the presence and absence of MMD3 or MMD3rev for 24 h. According to DLS, the hydrodynamic diameter of TauRDΔK monomer was <10 nm. In the absence of the tested D‐peptides, heparin promoted the assembly of TauRDΔK predominantly to PHF‐like filaments with the size of 15–100 nm in diameter. In contrast, TauRDΔK with heparin in the presence of either MMD3 or MMD3rev formed larger oligomers/aggregates with the size ranging from 3000–4000 nm in diameter (Figure 5A). D‐Peptides alone in the presence of heparin did not form larger aggregates confirming that the peptides promote TauRDΔK to form larger aggregates (Figure S3).
Figure 5.

Changes in the size of Tau aggregates in the presence of MMD3 and MMD3rev using dynamic light scattering (DLS) and pelleting assay. TauRDΔK (10 μM) was incubated with 2.5 μM of heparin 16000 (H16 K) and 100 μM of peptides. (A) Hydrodynamic size of TauRDΔK monomer is <10 nm in diameter (red curve). TauRDΔK aggregated in the presence of heparin 16000 (H16 K) formed aggregates in the size of 15–100 nm in diameter (blue curve). TauRDΔK in the presence of both peptides (MMD3 and MMD3rev) forms aggregates with the size ranging from 3000–4000 nm in diameter (green and black curves). Peptides alone in the presence of heparin did not form larger aggregates, see Figure S3. (B) Pelleting assay: Aggregated TauRDΔK centrifuged samples resolved on SDS gels showing supernatant and pellet fractions (lanes 1, 2; S, P). The majority of the high molecular weight aggregated Tau is present in the pellet fractions of TauRDΔK treated with peptides MMD3 and MMD3rev (lanes 4, 6). (C) Quantification of the Tau pellet and supernatant fractions from western blot gels shows differences between the Tau pellets fractions treated with/without peptides (bars 2, 4, 6).
To further investigate the effect of MMD3 and MMD3rev on aggregate size, pelleting assays were carried out. Aggregated TauRDΔK with or without D‐peptides were centrifuged, subsequently the supernatant (mostly monomers of TauRDΔK) and pellet fractions (mostly fibers) were applied on SDS gels and analyzed by western blot and densitometry. In the absence of the inhibitor peptides, the Tau protein was distributed over the supernatant and the pellet fractions, and the supernatant fraction had a high content of TauRDΔK monomers (Figure 5B, C). When treated with the D‐peptides MMD3 and MMD3rev, the majority of the high molecular weight‐aggregated Tau was present in the pellet fractions and the supernatant fractions had drastically reduced content of Tau protein. To confirm the results obtained from DLS and pelleting assays, the samples were analyzed by atomic force microscopy (AFM). Samples of aggregated TauRDΔK with heparin were analyzed in the presence and absence of MMD3 and MMD3rev. AFM images showed that TauRDΔK with heparin aggregated into fine structured PHF‐like filaments with a height of ∼15 nm. In the presence of MMD3 and MMD3rev, the Tau protein formed amorphous aggregates (Figure 6).
Figure 6.
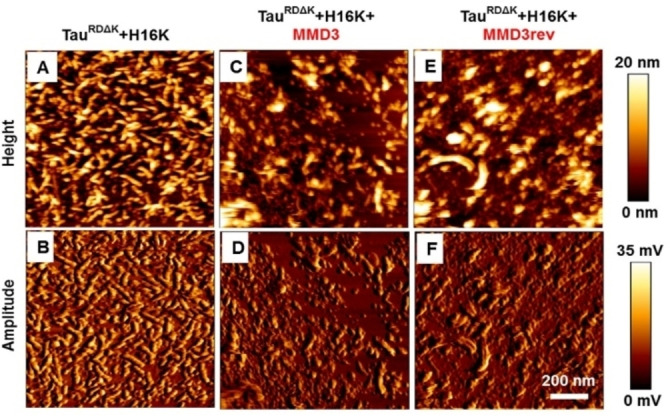
Tau forms amorphous aggregates in the presence of Tau peptides. AFM height and amplitude images reveal that TauRDΔK (with heparin 16000 (H16 K)) aggregates into fine structured filaments with the height of ∼15 nm (image A, B). In presence of peptides MMD3 (images C, D) and MMD3rev (images E, F), Tau aggregates into amorphous aggregates without attaining definite structure.
In silico analysis of binding mode of MMD3 and MMD3rev to the PHF6* fibril
The experimental data of the present study assume that MMD3 and MMD3rev target the PHF6* site of Tau. To gain a structural understanding of their mode of action, molecular modelling was performed. The three‐dimensional structure of PHF6* [19] revealed two polymorphic types of fibrils formed either by residues Lys274‐Asp283 (fibril polymorph 1) or residues Val275‐Lys280 (fibril polymorph 2) (see Supporting Information Figure S4 for details). These structures were exploited previously for the design of L‐peptidic inhibitors, e. g. W‐MINK. [19]
W‐MINK (1‐DVWMINKKRK‐10) resembles MMD3rev (1‐YWVSTHRAKLPD‐12) and MMD3 (1‐DPLKARHTSVWY‐12) with respect to the presence and spacing of aromatic and basic residues (highlighted in bold in the sequences). This similarity prompted us to investigate, whether MMD3 and MMD3rev can interact with PHF6* in a similar fashion as previously described for W‐MINK. [19]
The interaction of W‐MINK with fibril polymorph 1 has been already modelled by Seidler et al. [19] (shown in Figure 7A for comparison). W‐MINK blocks interface A and interface B via steric clashes of Met4 and Arg9 respectively (Figure 7A). The D‐peptide MMD3 causes steric clashes at similar sites, mainly by residues Tyr12 and Leu3 (Figure 7B). In addition, the proline at position 2 hampers fibril growth since this amino acid acts as beta‐sheet breaker. Similar to Lys10 of W‐MINK, Lys4 of MMD3 interacts favorably with Asp283 of the Tau fibril (Figures 7A and B) thereby mediating long‐range attractive electrostatic interaction, which guide the peptide to its binding position and register. Modelling of MMD3rev in the reverse peptide orientation compared to MMD3 reveals that the same overall binding mode and the same type of interference with the fibril interfaces remark possible (Figure 7C). The tryptophan (Trp3) in W‐MINK was engineered to cause steric clashes at interface C of polymorph2 resulting in an enhanced inhibitory activity. [19] Molecular modelling reveals that the tryptophan residue present in MMD3 (Trp11) and MMD3rev (Trp2) can target interface C of the Tau‐fibril (polymorph 2) in a similar fashion as Trp3 of the W‐MINK peptide (Figure 7D–F).
Figure 7.

Interaction of inhibitory peptides with the Tau polymorph 1 and 2. Binding mode of (A) W‐MINK, (B) MMD3, and (C) MMD3rev to the Tau fibril polymorph 1. The peptides are shown in color presentation and key interacting residues described in the text are labelled. Major sites of interference with interface A and B are marked by red and yellow circles, respectively. Binding mode of (D) W‐MINK, (E) MMD3, and (F) MMD3rev to the Tau fibril polymorph 2. The peptides are shown in color presentation and key interacting residues described in the text are labelled. Major sites of interference with interface C are marked by blue circles.
Ability of the peptides to penetrate cells
Next, we tested the ability of the MMD3 and MMD3rev to cross the cell membrane of neurons. N2a cells expressing TauRDΔK were incubated with ALEXA 647‐labeled forms of the D‐peptides MMD3 and MMD3rev at a concentration of 100 μM for 48 h. After incubation, they were fixed on thick glass bottom plates and monitored by confocal microscopy. The localizations of the D‐peptides were detected by their ALEXA 647‐label (excitation: 651 nm, emission: 667 nm). In addition, cell nuclei were stained with DAPI dye (excitation: 358 nm, emission: 461 nm). Representative pictures are shown in Figures 8A and 8B.
Figure 8.

Uptake of fluorescently labelled D‐peptides analyzed by confocal microscopy and flow cytometry. At different concentrations (25, 50 and 100 μM) of fluorescently labelled peptides (MMD3 and MMD3 reverse) were incubated with N2a cells expressing TauRDΔK for 48 h. The uptake of A647 peptides were analyzed by confocal microscopy (A, B) and by flow cytometry (C). As shown in the representative images the peptides are predominantly localized in the cytoplasm. A1, B1: DAPI shown as green; A2, B2: A647‐MMD3, A647‐MMD3 reverse; A3, B3: merged images. (C) The uptake of A647 labelled peptides were monitored by flow cytometry. As shown in the representative flow cytometry data, almost 100 % of the cells take up both the peptides. MMD3 reverse peptides show concentration dependent increase in intensity the fluorescence.
We observed accumulation of MMD3 and MMD3rev in the cytoplasm, visible at the latest after 4 days of incubation. We concluded that all D‐peptides are able to penetrate the cell membrane and are stable over the whole incubation time. In addition, flow cytometry analysis of Tau expressing N2a cells treated with ALEXA 647‐labeled D‐peptides showed that the cells take up the fluorescently labelled peptides (Figure 8C).
Comparison of the potential of PHF6*‐and PHF6 inhibitors to inhibit TauRDΔK aggregation using ThS assay
To test whether PHF6*‐based inhibitors are more powerful in inhibiting the aggregation of the 4‐repeat domain of Tau with ΔK280 mutation (TauRDΔK) than PHF6‐based inhibitors, we compared the inhibitory effect of our PHF6*‐targeted peptides MMD3 and MMD3rev with PHF6‐targeted peptide APT, which was earlier selected by our group against PHF6 fibrils. [21] In addition, we tested the performance of two previously published peptides TLKIVW [20] and W‐MINK, [19] which target PHF6 and PHF6* respectively. In vitro ThS fluorescence assay and the peptides were added in a concentration range of 1 nM to 200 μM to 10 μM TauRDΔK protein in the presence of heparin and the fluorescence over the period of 24 hours was monitored. All tested peptides were able to inhibit the aggregation of TauRDΔK at different rates. The representative plots are shown in the Supporting Information Figure S5 for the respective peptides. Except for the “Sievers” peptide TLKIVW, all other peptides were able to reduce the aggregation starting at ∼1 μM peptide concentration. The ThS fluorescence (as indicator for the extent of Tau aggregation) after 24 h incubation in the presence of peptides was plotted as percentage of the untreated control (Figure S5) to derive IC50 values (Table 2).
Table 2.
Half maximum inhibition concentration.
Discussion
Currently, Alzheimer's disease can be treated only symptomatically. No disease modifying therapies are available yet. In recent years, several trial failures were observed with compounds developed for AD modification. In early 2019 Cummings and colleagues identified 132 compounds in clinical trials, 70 % of them being disease modifying therapies and the majority, especially the ones in later stages of development, targeting amyloid‐β. [23] Among over twenty clinical studies focusing on Aβ, only one drug, Aducanumab, could recently obtain a tentative FDA (Food and Drug Administration in U.S.) approval. However, the decision for approval is highly controversial and the efficacy of the compound will still have to be proven. [24] Treatment options targeting Tau pathology are therefore still valid alternatives. Tau pathology plays a fundamental role in AD, but also in other neurodegenerative diseases. [25] However, the connection between amyloid‐β pathology, Tau pathology and the causes of AD remain an open question. Evidences strongly support that the distribution and spreading pattern of Tau pathology in the brain correlates well with clinical features in AD and can be used for disease staging. [26] In addition, the onset of Tau pathology can be linked to the onset of symptoms, as verified by cerebrospinal fluid testing and positron emission tomography imaging. [27] In the present study, we have demonstrated that D‐enantiomeric peptides targeting aggregated PHF6*, selected by mirror image phage display, inhibited PHF6*, TauRDΔK280 and full‐length Tau fibrilization in vitro. Our DLS, pelleting assay and AFM data suggested that the peptides prevent the formation of Tau β‐sheet‐rich fibrils by building large amorphous aggregates. Furthermore, the peptides were able to penetrate cells and might be interesting for therapeutic and diagnostic applications in AD research.
Phage display selections for this study were performed using fibrils of the D‐enantiomeric hexapeptide VQIINK as a target, representing residues 275 to 280 of the Tau protein. The two hexapeptides within Tau, PHF6* (275‐VQIINK‐280 in R2) and PHF6 (306‐VQIVYK‐311 in R3), are known to strongly promote Tau aggregation into β‐structured fibers.[ 16 , 17 ] In the literature, many PHF6‐targeting peptides were reported and showed their ability to inhibit the aggregation of PHF6 into β‐sheet fibrils. [28] Recently, the PHF6* segment has been described as the more potent driver of Tau aggregation. [19] By addressing PHF6*, we intended to generate a set of starting compounds for Tau directed AD therapy development. Earlier, we had selected peptides binding the PHF6 part of Tau, [21] and with peptides binding PHF6 and PHF6* respectively, we could also start the development of dual specificity (heterodimeric) D‐peptides, which address either PHF6 and PHF6* within one molecule. In addition, we have now the opportunity to investigate which of the two hexapeptide motifs, PHF6 or PHF6*, is the more effective target for the development of Tau aggregation inhibitors. While discussing the most effective target for Tau directed AD therapy development, however, one has to keep in mind that in vivo PHF6* within R2 (Exon 10) is only present in AD patients in 50 % of neuronal Tau due to alternative splicing (3R vs. 4R Tauopathies, see ref. [29]). Studies suggested that inhibitors against R2 might be highly effective in 4R Tauopathies. [19] Our peptide might be developed for therapy of AD, but also for therapy of other neurodegenerative diseases in which Tau aggregation plays a role. We have selected our D‐enantiomeric peptides with the idea to prevent fibrillization rather than to dissolve existing fibrils. Recent Cryo‐EM studies, however, demonstrated that tau fibrils extracted from AD brains have a core that contains R3, R4 and ten residues beyond the end of R4. [30] These findings suggest that PHF6 might be the better target for development of tau aggregation inhibitors. On the other hand, the study of Seidler et al. suggested that PHF6*‐based inhibitors are more potent as tau aggregation inhibitors. They hypothesized that the core of the fibrils is not the primary driver of aggregation, but serves as a solvent‐excluded scaffold that clusters PHF6* together in the fuzzy coat, and poises the solvent‐exposed VQIINK steric zippers for seeding. [19] From this perspective, the localization of PHF6* to the fuzzy coat could make it more accessible to protein monomers in the cells, and explains a possible prominent role of PHF6* in tau fibrillization.
The peptides MMD3 and MMD3rev, selected against PHF6* fibrils, bound to PHF6* fibrils, and full‐length Tau fibrils, as demonstrated by ELISA studies. The interactions between the D‐peptide MMD3 and full‐length Tau monomer were investigated by NMR spectroscopy. The data suggest that MMD3, selected against PHF6* fibrils, binds to Tau monomers only with low affinity. The binding mode of MMD3 and MMD3rev to PHF6* fibrils was illustrated by in silico modeling. The modelling suggests that the D‐peptides MMD3 and MMD3rev identified in the present study exhibit a similar molecular mechanism in disrupting PHF6* fibrils as the previously designed L‐peptide W‐MINK. [19] It is particularly remarkable that this activity can be obtained regardless of the peptide chirality (D‐ vs. L‐peptides) or an inversion of the amino acid sequence (MMD3 vs. MMD3rev). This finding suggests that the spacing of several key residues required to interfere with the fibril interfaces is the key prerequisite for the potency of inhibitory peptides. This observation may allow additional modifications of the scaffold that positions the functional groups in future, e. g. by enhancing rigidity or by using non‐peptogenic connectors.
The selected peptides inhibited the aggregation of PHF6*, but not of PHF6 in agreement with the selection strategy and despite very similar amino acid sequences of the two hexapeptides. In ThS studies, the peptides reduced the formation of regular Tau fibrils in concentrations which are typical for peptide inhibitors.[ 10 , 14 , 20 ] The amorphous aggregates detected by DLS and AFM would not lead to a signal in ThT assays, as ThT only detects regular β‐structured fibrils. [10]
The mechanism of action is not totally clear, yet. Modelling data suggest that the D‐peptides [10] exhibit a similar molecular mechanism in disrupting PHF6* fibrils as the previously designed L‐peptide W‐MINK. W‐MINK blocks interface A and interface B via steric clashes of Met4 and Arg9 respectively, the D‐peptide MMD3 causes steric clashes at these interfaces, mainly by residues Tyr12 and Leu3. The major clash at interface A is Tyr12(MMD3) – Asn179(tau: VQIINK) and at interface B Leu3(MMD3) – Lys280(tau: VQIINK).
In in vitro DLS, pelleting assay and AFM studies, we found that that the peptides prevent the formation of Tau β‐sheet‐rich fibrils by building large amorphous aggregates. In the latter studies, however, relatively high concentrations of the peptide (10× excess) were present.
Similar phenomena were already described for the D‐peptide D3, which was developed to inhibit Aβ aggregation. D3 applied in ratios of more than 1 : 1 inhibits the formation of regular Aβ fibrils, removes Aβ oligomers by formation of large amorphous aggregates and decreases Aβ cytotoxicity in vitro. The amorphous D3‐Aβ‐co‐aggregates were shown to be non‐toxic, non‐amyloidogenic, amorphous and ThT negative. [10] In vivo, D3 was able to reduce plaque load, decrease inflammation and enhance cognition in a transgenic AD mouse model even after oral application. [14] A derivative of D3 with the same amino acid composition in different order, RD2, slows down the secondary structure conversion of Aβ42 and significantly delays the fibril formation at substoichiometric levels. Experimental evidence was provided that RD2 eliminates toxic Aβ assemblies by stabilizing Aβ monomers in their native intrinsically disordered conformation. [31] At higher concentration, a similar mechanism as for D3 was described. [32]
In a recent study, [33] it has been observed that larger Tau aggregates are significantly less toxic in the neuron. Indeed, several pieces of evidence have shown an inverse correlation between the size of the protein aggregate and the neurotoxicity. [34] For our D‐peptides, a more detailed investigation with respect to the mechanism will be needed in future.
To be effective as a therapeutic in the brain, lead compounds need to cross the blood‐brain‐barrier and in addition penetrate neurons. MMD3 and MMD3rev were demonstrated to cross the membranes of N2a cells. The mechanism of penetration is unclear yet. Peptides do in general not cross membranes very well, but several classes of cell penetrating peptides have been discovered, including naturally occurring transcription factor domain as penetratin, HIV‐Tat or synthetic cationic peptides. [35] Whether the peptides also will cross the blood‐brain‐barrier of e. g. AD mouse models, will be subject of a future study. Several strategies could be used improve blood‐brain‐barrier permeability, if needed. For example, current approaches for AD therapy enable the use of designated transporter to facilitate the entry of tau directed drugs into neuronal brain cells. [36]
To investigate the hypothesis that PHF6* is the stronger driver of Tau aggregation, we compared the Tau aggregation inhibiting effects of our newly selected peptides, MMD3 and MMD3rev, with other PHF or PHF* binding peptides, e. g. our peptide designated APT, which was selected against PHF6 fibrils earlier by our group. From our very preliminary data, it seems likely that PHF6 and PHF6* aggregation inhibitors are comparably effective in inhibiting the aggregation of TauRDΔK in vitro. All peptides, except for TLKIVW, were able to inhibit the aggregation above ∼1 μM concentration.
Future research is required to further characterize these peptides using different biophysical and biochemical methods to determine which of the two sequences is more favorable for AD therapy development. Binding affinities and exact binding sequence of all the peptides will have to be determined next to characteristics of in vivo performance, like blood‐brain permeability, therapeutic effects or side effects in mouse models, etc. In future, it might be valuable to develop “heterodimeric” D‐peptides which address PHF6* and PHF6 within one molecule.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
This work was supported by a grant of TechnologieAllianzOberfranken (TAO) to M.M., a scholarship of Landeskonferenz der Frauenbeauftragten, Bavaria, Germany, to M.M., and a grant of Katharina Hardt Foundation to E.M. I.A. was supported by Alzheimer Forschung Initiative e.V. (AFI). M.Z. was supported by the European Research Council (ERC) under the EU Horizon 2020 research and innovation programme (grant agreement No. 787679). Open Access funding enabled and organized by Projekt DEAL.
M. Malhis, S. Kaniyappan, I. Aillaud, R. R. Chandupatla, L. M. Ramirez, M. Zweckstetter, A. H. C. Horn, E. Mandelkow, H. Sticht, S. A. Funke, ChemBioChem 2021, 22, 3049.
References
- 1. Alzheimer A., Stelzmann R. A., Schnitzlein H. N., Murtagh F. R., Clin. Anat. 1995, 8, 429. [DOI] [PubMed] [Google Scholar]
- 2. McKhann G., Drachman D., Folstein M., Katzman R., Price D., Stadlan E. M., Neurology 1984, 34, 939. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Binder L. I., Frankfurter A., Rebhun L. I., J. Cell Biol. 1985, 101, 1371; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3b. Black M. M., Slaughter T., Moshiach S., Obrocka M., Fischer I., J. Neurosci. 1996, 16, 3601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brion J. P., Couck A. M., Passareiro E., Flament-Durand J., J. Submicrosc. Cytol. 1985, 17, 89. [PubMed] [Google Scholar]
- 5.
- 5a. Calafate S., Buist A., Miskiewicz K., Vijayan V., Daneels G., de Strooper B., de Wit J., Verstreken P., Moechars D., Cell Rep. 2015, 11, 1176; [DOI] [PubMed] [Google Scholar]
- 5b. Wu J. W., Hussaini S. A., Bastille I. M., Rodriguez G. A., Mrejeru A., Rilett K., Sanders D. W., Cook C., Fu H., Boonen R. A. C. M., Herman M., Nahmani E., Emrani S., Figueroa Y. H., Diamond M. I., Clelland C. L., Wray S., Duff K. E., Nat. Neurosci. 2016, 19, 1085; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Kaniyappan S., Chandupatla R. R., Mandelkow E.-M., Mandelkow E., Alzheimer's Dementia 2017, 13, 1270; [DOI] [PubMed] [Google Scholar]
- 5d. Kaniyappan S., Tepper K., Biernat J., Chandupatla R. R., Hübschmann S., Irsen S., Bicher S., Klatt C., Mandelkow E.-M., Mandelkow E., Mol. Neurodegener. 2020, 15, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Arriagada P. V., Growdon J. H., Hedley-Whyte E. T., Hyman B. T., Neurology 1992, 42, 631; [DOI] [PubMed] [Google Scholar]
- 6b. Bierer L. M., Haroutunian V., Gabriel S., Knott P. J., Carlin L. S., Purohit D. P., Perl D. P., Schmeidler J., Kanof P., Davis K. L., J. Neurochem. 1995, 64, 749; [DOI] [PubMed] [Google Scholar]
- 6c. Gómez-Isla T., Hollister R., West H., Mui S., Growdon J. H., Petersen R. C., Parisi J. E., Hyman B. T., Ann. Neurol. 1997, 41, 17. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Brendel M., Deussing M., Blume T., Kaiser L., Probst F., Overhoff F., Peters F., von Ungern-Sternberg B., Ryazanov S., Leonov A., et al., Alzheimer's Res. Ther. 2019, 11, 67; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7b. Wagner J., Krauss S., Shi S., Ryazanov S., Steffen J., Miklitz C., Leonov A., Kleinknecht A., Göricke B., Weishaupt J. H., et al., Acta Neuropathol. 2015, 130, 619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Gura T., Nat. Med. 2008, 14, 894; [DOI] [PubMed] [Google Scholar]
- 8b. Huang Y M. L., Cell. 2012, 148, 1204; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8c. Jadhav S., Avila J., Schöll M., Kovacs G. G., Kövari E., Skrabana R., Evans L. D., Kontsekova E., Malawska B., de Silva R., et al., Acta Neuropathol. Commun. 2019, 7, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.
- 9a. Milton D. L., Norqvist A., Wolf-Watz H., J. Bacteriol. 1992, 174, 7235; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. Pappenheimer J. R., Karnovsky M. L., Maggio J. E., J. Pharmacol. Exp. Ther. 1997, 280, 292; [PubMed] [Google Scholar]
- 9c. Chalifour R. J., McLaughlin R. W., Lavoie L., Morissette C., Tremblay N., Boule M., Sarazin P., Stéa D., Lacombe D., Tremblay P., Gervais F., J. Biol. Chem. 2003, 278, 34874–81. [DOI] [PubMed] [Google Scholar]
- 10. Funke S. A., van Groen T., Kadish I., Bartnik D., Nagel-Steger L., Brener O., Sehl T., Batra-Safferling R., Moriscot C., Schoehn G., Horn A. H. C., Müller-Schiffmann A., Korth C., Sticht H., Willbold D., ACS Chem. Neurosci. 2010, 1, 639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Willbold D., Kutzsche J., Molecules 2019, 24, 2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Klein R. M., Christie J., Parkvall M., SSM Popul. Health 2016, 2, 463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Kutzsche J., Schemmert S., Tusche M., Neddens J., Rabl R., Jürgens D., Brener O., Willuweit A., Hutter-Paier B., Willbold D., Molecules 2017, 22, 1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. van Groen T., Wiesehan K., Funke S. A., Kadish I., Nagel-Steger L., Willbold D., ChemMedChem 2008, 3, 1848. [DOI] [PubMed] [Google Scholar]
- 15.
- 15a. van Groen T., Kadish I., Wiesehan K., Funke S. A., Willbold D., ChemMedChem. 2009, 4, 276; [DOI] [PubMed] [Google Scholar]
- 15b. Leithold L., Jiang N., Post J., Ziehm T., Schartmann E., Kutzsche J., Pharmac. Res. 2016, 33, 328; [DOI] [PubMed] [Google Scholar]
- 15c. Groen-van de Ven L., Smits C., de Graaff F., Span M., Eefsting J., Jukema J., Vernooij-Dassen M., BMJ Open 2017, 7, e018337; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15d. Elfgen A., Hupert M., Bochinsky K., Tusche M., de San Román G., Estibaliz Martin, Gering I., Sacchi S., Pollegioni L., Huesgen P. F., Hartmann R., Santiago-Schübel B., Kutzsche J., Willbold D., Sci. Rep. 2019, 9, 5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. von Bergen M., Friedhoff P., Biernat J., Heberle J., Mandelkow E. M., Mandelkow E., Proc. Natl. Acad. Sci. USA 2000, 97, 5129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. von Bergen M., Barghorn S., Li L., Marx A., Biernat J., Mandelkow E. M., Mandelkow E., J. Biol. Chem. 2001, 276, 48165. [DOI] [PubMed] [Google Scholar]
- 18. Himmler A., Drechsel D., Kirschner M. W., Martin D. W., Mol. Cell. Biol. 1989, 9, 1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Seidler P. M., Boyer D. R., Rodriguez J. A., Sawaya M. R., Cascio D., Murray K., Gonen T., Eisenberg D. S., Nat. Chem. 2018, 10, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sievers S. A., Karanicolas J., Chang H. W., Zhao A., Jiang L., Zirafi O., Stevens J. T., Münch J., Baker D., Eisenberg D., Nature 2011, 475, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dammers C., Yolcu D., Kukuk L., Willbold D., Pickhardt M., Mandelkow E., Horn A. H. C., Sticht H., Malhis M. N., Will N., Schuster J., Funke S. A., PLoS One 2016, 11, e0167432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Huang J., Ru B., Li S., Lin H., Guo F.-B., J. Biomed. Biotechnol. 2010, 2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Cummings J., Blennow K., Johnson K., Keeley M., Bateman R. J., Molinuevo J. L., Touchon J., Aisen P., Vellas B., J. Prev. Alzheimer's Dis. 2019, 6, 157; [DOI] [PubMed] [Google Scholar]
- 23b. Lovestone S., Manji H. K., Clin. Pharmacol. Ther. 2020, 107, 79. [DOI] [PubMed] [Google Scholar]
- 24.
- 24a. Walsh S., Merrick R., Milne R., Brayne C., BMJ 2021, 374, n1682; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24b. Knopman D. S., Jones D. T., Greicius M. D., Alzheimer's Dementia 2021, 17, 696; [DOI] [PubMed] [Google Scholar]
- 24c. Mullard A., Nat. Rev. Drug Discovery 2021, 20, 496. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Ballatore C., Lee V. M.-Y., Trojanowski J. Q., Nat. Rev. Neurosci. 2007, 8, 663; [DOI] [PubMed] [Google Scholar]
- 25b. Götz J., Xia D., Leinenga G., Chew Y. L., Nicholas H., Front. Neurol. 2013, 4, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Braak H., Braak E., Acta Neuropathol. 1991, 82, 239. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Johnson K. A., Schultz A., Betensky R. A., Becker J. A., Sepulcre J., Rentz D., Mormino E., Chhatwal J., Amariglio R., Papp K., Marshall G., Albers M., Mauro S., Pepin L., Alverio J., Judge K., Philiossaint M., Shoup T., Yokell D., Dickerson B., Gomez-Isla T., Hyman B., Vasdev N., Sperling R., Ann. Neurol. 2016, 79, 110; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27b. Roe C. M., Fagan A. M., Grant E. A., Hassenstab J., Moulder K. L., Maue Dreyfus D., Sutphen C. L., Benzinger T. L. S., Mintun M. A., Holtzman D. M., Morris J. C., Neurology 2013, 80, 1784; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27c. Schwarz A. J., Yu P., Miller B. B., Shcherbinin S., Dickson J., Navitsky M., Joshi A. D., M. D. Devous, Sr. , Mintun M. S., Brain 2016, 139, 1539. [DOI] [PubMed] [Google Scholar]
- 28.
- 28a. Frenkel-Pinter M., Richman M., Belostozky A., Abu-Mokh A., Gazit E., Rahimipour S., Segal D., Chem. Eur. J. 2016, 22, 5945; [DOI] [PubMed] [Google Scholar]
- 28b. Belostozky A., Richman M., Lisniansky E., Tovchygrechko A., Chill J. H., Rahimipour S., Chem. Commun. 2018, 54, 5980; [DOI] [PubMed] [Google Scholar]
- 28c. Beatty J. W., Douglas J. J., Miller R., McAtee R. C., Cole K. P., Stephenson C. R. J., Chem 2016, 1, 456; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28d. Zheng J., Liu C., Sawaya M. R., Vadla B., Khan S., Woods R. J., Eisenberg D., Goux W. J., Nowick J. S., J. Am. Chem. Soc. 2011, 133, 3144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.
- 29a. Hu W. T., Parisi J. E., Knopman D. S., Boeve B. F., Dickson D. W., Ahlskog J. E., Petersen R. C., Josephs K. A., Alzheimer Dis. Assoc. Disord. 2007, 21, S39–43; [DOI] [PubMed] [Google Scholar]
- 29b. Sealey M. A., Vourkou E., Cowan C. M., Bossing T., Quraishe S., Grammenoudi S., Skoulakis E. M., Mudher A., Neurobiol. Dis. 2017, 105, 74. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Fitzpatrick A. W. P., Falcon B., He S., Murzin A. G., Murshudov G., Garringer H. J., Crowther R. A., Ghetti B., Goedert M., Scheres S. H. W., Nature 2017, 547, 185; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30b. Falcon B., Zhang W., Schweighauser M., Murzin A. G., Vidal R., Garringer H. J., Ghetti B., Scheres S. H. W., Goedert M., Acta Neuropathol. 2018, 136, 699; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30c. Shi Y., Murzin A. G., Falcon B., Epstein A., Machin J., Tempest P., Newell K. L., Vidal R., Garringer H. J., Sahara N., et al., Acta Neuropathol. 2021, 141, 697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang T., Gering I., Kutzsche J., Nagel-Steger L., Willbold D., ACS Chem. Neurosci. 2019, 10, 4800. [DOI] [PubMed] [Google Scholar]
- 32. van Groen T., Schemmert S., Brener O., Gremer L., Ziehm T., Tusche M., Nagel-Steger L., Kadish I., Schartmann E., Elfgen A., Jürgens D., Willuweit A., Kutzsche J., Willbold D., Sci. Rep. 2017, 7, 16275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lo Cascio F., Garcia S., Montalbano M., Puangmalai N., McAllen S., Pace A., Palumbo Piccionello A., Kayed R., J. Biol. Chem. 2020, 295, 14807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.
- 34a. Ahmed R., Akcan M., Khondker A., Rheinstädter M. C., Bozelli J. C., Epand R. M., Huynh V., Wylie R. G., Boulton S., Huang J., Verschoor C. P., Melacini G., Chem. Sci. 2019, 10, 6072; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34b. Mannini B., Mulvihill E., Sgromo C., Cascella R., Khodarahmi R., Ramazzotti M., Dobson C. M., Cecchi C., Chiti F., ACS Chem. Biol. 2014, 9, 2309; [DOI] [PubMed] [Google Scholar]
- 34c. Sengupta U., Nilson A. N., Kayed R., EBioMedicine 2016, 6, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.
- 35a. Futaki S., Adv. Drug Delivery Rev. 2005, 57, 547; [DOI] [PubMed] [Google Scholar]
- 35b. Derossi D., Joliot A. H., Chassaing G., Prochiantz A., J. Biol. Chem. 1994, 269, 10444; [PubMed] [Google Scholar]
- 35c. Gump J. M., Dowdy S. F., Trends Mol. Med. 2007, 13, 443. [DOI] [PubMed] [Google Scholar]
- 36. Lawatscheck C., Pickhardt M., Wieczorek S., Grafmüller A., Mandelkow E., Börner H. G., Angew. Chem. Int. Ed. 2016, 55, 8752. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information