

Abstract
Compartmentalization of chemical reactions inside cells are a fundamental requirement for life. Encapsulins are self‐assembling protein‐based nanocompartments from the prokaryotic repertoire that present a highly attractive platform for intracellular compartmentalization of chemical reactions by design. Using single‐molecule Förster resonance energy transfer and 3D‐MINFLUX analysis, we analyze fluorescently labeled encapsulins on a single‐molecule basis. Furthermore, by equipping these capsules with a synthetic ruthenium catalyst via covalent attachment to a non‐native host protein, we are able to perform in vitro catalysis and go on to show that engineered encapsulins can be used as hosts for transition metal catalysis inside living cells in confined space.
Keywords: encapsulins, homogeneous catalysis, MINFLUX, self-assembly, single-molecule FRET
A bioorthogonal catalysis strategy has been developed inside a confined space using encapsulins equipped with HaloTag and [CpRu(hydroxyquinoline)(allyl)]PF6. The strategy opens up a toolbox for designing compartmentalized “new‐to‐biology” reactions inside living cells.
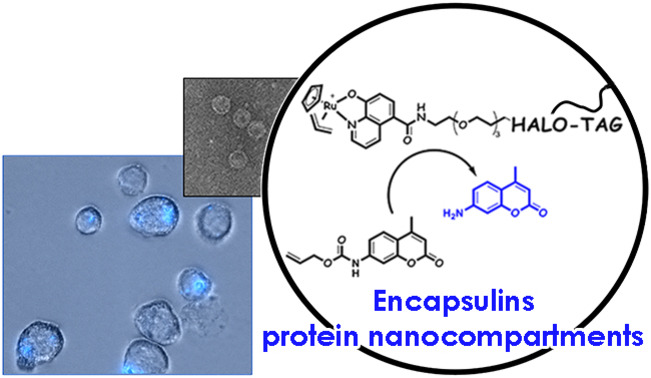
Introduction
A fundamental concept in biology is compartmentalization. This feature of life is not limited to structure but extends to enzyme catalysis, for example to achieve high local concentrations of reaction partners within organelles. Compartmentalized assembly lines limit loss of substrate and/or diffusion of toxic reaction intermediates into the surroundings and enable the generation of dedicated biological “reaction vessels”. Moreover, benefits such as resistance to thermal or proteolytic degradation of substrates or products can be achieved.[ 1 , 2 ] In this context, bacterial micro‐ and nanocompartments and other protein cages, such as vaults, viral capsids or virus‐like particles have gained significant attention as unique self‐assembly platforms for the design and construction of (catalytically) functional nanomaterials[ 3 , 4 , 5 ] or cargo delivery vehicles. [6] Endowing these compartments with the ability to catalyze chemical transformations, provides the opportunity for creating new dynamic confined spaces with tunable reactivity. One highly attractive platform for enzyme compartmentalization are encapsulins (Enc), which are self‐assembling bacterial nanocompartments. Encs are virus‐like particles that have been identified as conserved prokaryotic compartments.[ 2 , 7 ] Encs sort into icosahedral compartments with triangulation numbers of T=1 (60 subunits, Ø≈240 Å), T=3 (180 subunits, Ø≈320 Å), and T=4 (240 subunits, Ø≈420 Å) respectively, where at each point of symmetry uncharged, positively charged, or negatively charged pores are formed (Ø≈5–8 Å). [8] The capsids are temperature and pH‐stable. [4] Typically, the gene encoding for Enc is located in a two‐gene operon with either ferritin (Flps) or hemerythrins‐like proteins, or dye‐decolorizing peroxidases (DyPs).[ 2 , 9 ] Selective packaging of co‐expressed proteins is promoted by a conserved amino‐acid sequence (encapsulin localization sequence, ELS) at their C‐terminus.[ 10 , 11 , 12 ] Importantly, non‐native cargo proteins can be directed inside Enc via simple tagging with ELS, enabling self‐assembly of the new functional compartment.[ 12 , 13 , 14 ] The tremendous possibilities of such functional systems inspired several groups to investigate enzyme catalysis in encapsulin nanocompartments.[ 13 , 15 , 16 , 17 ] However, transition metal catalysis not found in biology and therefore chemically orthogonal has yet to be explored in Encs. In this study, we describe the first targeted incorporation of an organometallic catalyst to the interior of Enc from Mycobacterium smegmatis (M. smegmatis) enabled by a HaloTag‐ELS construct. The approach is versatile, as the HaloTag‐ELS strategy enables the introduction of many different metal/ligand systems new to biology, provided they operate in water. We explore, as a proof of concept reaction, the ruthenium catalyzed deallylation of a profluorophore inside the Enc nanoreactor.
Results and Discussion
Encapsulin from M. smegmatis (in the following StrepEncSM) was expressed and purified from Escherichia coli (E. coli; Supporting Information, Figure S1). Native‐PAGE analysis of the purified construct StrepEncSM revealed a large protein assembly that did not enter the gel (Figure 1 A). However, under denaturing conditions, a single protein band corresponding to 30 kDa, the expected size of one StrepEncSM monomer, was observed. Analysis of purified StrepEncSM by transmission electron microscopy (TEM) and dynamic light scattering (DLS) revealed the assembly of StrepEncSM into nanocompartments of an average size of 20±3 nm, with an inner diameter of 14±1 nm (n=106, Figure 1 B), and with a mean hydrodynamic diameter of 28.5±0.3 nm (Figure S1C). Additionally, the polydispersity index (PDI) with 0.13±0.01 showed a near monodisperse sample. [18]
Figure 1.

Purification and characterization of encapsulin from M. smegmatis. A) Native and denaturing PAGE analysis of purified StrepEncSM. The expected mass of a 60‐mer assembly is 1.8 MDa. B) TEM image (negative staining) of empty StrepEncSM. Number of capsids assessed for diameter calculation (n)=106.
The shell diameter is in good agreement with the previously described 60‐subunit Enc nanocompartment from M. smegmatis (purified from the wild‐type organism) [19] and from Mycobacterium tuberculosis (82 % sequence identity to EncSM), [17] suggesting that StrepEncSM also forms a 60‐subunit assembly.
In order to determine the minimally required ELS for successful incorporation of cargo proteins into StrepEncSM, we engineered several constructs of eGFP (enhanced green fluorescent protein) carrying varying sizes of the putative ELS from M. smegmatis (Figures S1 and S2) for co‐expression and subsequent purification of the assembly via Streptavidin‐tag (Figure 2). Co‐elution of eGFP with the capsid was confirmed by native PAGE analysis (Figure 2A. Presence of eGFP was quantified by in‐gel fluorescence detection, indicating that a minimal length of 12 amino acids of the localization sequence is required for successful incorporation of eGFP into StrepEncSM (Figure 2B. Calculation of loading efficiency of StrepEncSM{eGFP}, based on in‐gel fluorescence analysis and plotting of a standard curve (eGFP only) provided an estimate of 5.7±0.2 cargo proteins per capsid (Figure S2A). Stoichiometry and cargo load of encapsulins has been identified for example, by mass spectrometry or fluorescence‐based means [11] and contributed loading capacities of 1:4–1:12 (Enc:cargo protein), while being potentially constrained by the shape and oligomeric state of the respective cargo enzyme. Subsequent analysis of the StrepEncSM{eGFP} construct by TEM revealed successful assembly into a nanocompartment, displaying a similar outer diameter as the corresponding empty construct of 20±2 nm (n=117, Figure S2B). Negative staining allowed for the visualization of the outer shell, while indicating that the Enc core was now filled with cargo. Analysis of StrepEncSM{eGFP} by DLS identified a mean hydrodynamic diameter of 28.8±0.1 nm (PDI=0.13±0.01, Figure S2C).
Figure 2.

Characterization of StrepEncSM loaded with eGFP. Four cargo protein constructs, tagged with one short peptide of the putative ELS (6 aa, 9 aa, 12 aa and 19aa long; ctrl. 0 aa and no eGFP). A) Native PAGE analysis of the co‐purified constructs and analysis by Coomassie staining and in‐gel fluorescence. B) Quantitative assessment of the fluorescence intensities detected by in‐gel fluorescence using ImageJ. The analysis was done in triplicate. Error bars represent ± SEM. Key: amino acid (aa), encapsulin localization sequence (ELS).
Engineered protein tags, such as HaloTag, [20] are powerful tools to selectively and covalently label co‐transcribed proteins with for example, a fluorophore or a catalyst of interest or any small molecule that is tethered to a chloroalkane (CA) moiety (Figure S3). In order to install an organometallic catalyst inside StrepEncSM, we engineered a HaloTag construct carrying an ELS. Firstly, we validated successful uptake of HaloTag into StrepEncSM by co‐purification and native/denaturing PAGE analysis and subsequently we characterized the construct by TEM and DLS analysis (Figure S4B,D). Particles StrepEncSM{HaloTag} displayed an outer diameter of 21±2 nm (n=100) in our TEM experiments (Figure S4B). Again, negative staining indicated a filled cavity. Furthermore, analysis by DLS identified a mean hydrodynamic diameter of 28.7±0.1 nm (PDI=0.11±0.02, Figure S4D). Labeling of the protein tag with a commercially available fluorescent HaloTag substrate (CA‐AlexaFluor 660®, AF660) and in‐gel fluorescence analysis of StrepEncSM{HaloTag‐AF660} gave an estimate of 6.6±0.4 labeled cargo proteins per capsid by comparing to a standard curve of HaloTag‐AF660 only (Figure S4A,C). We further established a labeling efficiency within the capsule of 60 % at which all accessible binding sites were blocked (Figure S4E,F). The assembly and fluorescent labeling of StrepEncSM{HaloTag} now provided the unique opportunity to profoundly study the engineered system on the single‐molecule level using two independent techniques 1) a single‐molecule Förster resonance energy transfer (smFRET)‐based method [21] (Figure 3) shows that there are often more than two fluorescently labeled HaloTag proteins inside single StrepEncSM constructs; and 2) MINFLUX imaging [22] combined with a step‐wise off‐switching (Figure 4) even allowed counting them. To investigate StrepEncSM{HaloTag} using smFRET, we co‐labeled StrepEncSM{HaloTag} with two commercially available HaloTag ligands (CA‐JaneliaFluor 549®, JF549, and CA‐JaneliaFluor 646®, JF646) in a 1:1 ratio. The measurements were performed on a confocal microscope with pulsed interleaved excitation (PIE). [23] We used fluorescence correlation spectroscopy (FCS) to confirm that we have freely diffusing vesicles in solution (Figure S5A). Furthermore, we applied PIE‐FRET[ 21 , 23 ] and different burst search parameters to unveil at least three fluorophore binding sites per vesicle. Figure 3 A shows the result of an all photon burst search (APBS, see the Supporting Information for details). There a FRET population can be clearly seen at a FRET efficiency of around 0.85 and stoichiometry of around 0.5. In addition, a significant amount of data is found at higher stoichiometries, indicating at least a third dye in many vesicles [24] (for details see the experimental details in the Supporting Information, Figure S5). The presence of at least three binding sites was confirmed by applying a dual color burst search (DCBS) with the same data to select for the FRET‐species (for details, see the Supporting Information). Figure 3 B shows at least two FRET populations, which, are very unlikely to be caused by just two fluorophore positions (see SI for details). Unfortunately, we cannot extract the exact number of dyes in an encapsulin shell from the FRET efficiency vs. stoichiometry plots, likely because they are strongly affected by quenching, bleaching and homotransfer. Therefore, we used another method to count the number of dyes.
Figure 3.

Quantification of HaloTag labels inside StrepEncSM by single‐molecule fluorescence. A) smFRET measurements of StrepEncSM{HaloTag*JF549*JF646}. All photon burst search (APBS), time window 500 μs, threshold 50 photons, FRET‐2CDE filter: 0–10, DA rate: 15–20 kHz, DD rate: 2–20 kHz, burst duration 3–20 ms. B) Dual color burst search (DCBS), time window 500 μs, DD rate: 2–200 kHz, AA rate 5–50 kHz, DA rate: 20–150 kHz, FRET‐2CDE filter: 5–15, burst duration 2–7 ms, 2 Gaussian fits.
Figure 4.

MINFLUX measurements of StrepEncSM{HaloTag‐AF647}. A) Off‐switching count trace with visible off‐switching steps for a single StrepEncSM{HaloTag‐AF647} construct. B) Estimated number of fluorophores per StrepEncSM{HaloTag‐AF647} construct for all measurements (histogram, n=125) and median of the distribution (dashed line). C) 3D MINFLUX localizations of the single StrepEncSM{HaloTag‐AF647} construct. Each scatter dot represents an individual localization estimate for the position of a fluorescent AF647 molecule. The color‐coding describes when the localization was registered after starting the acquisition. Individual fluorescent molecules can deliver tens of localizations. We centered the localization with the origin by subtracting the mean position from all localizations. A sphere of 14 nm diameter (gray) encloses 94 % of localizations.
We employed a nanometer‐precise 3D single‐molecule nanoscopy method termed MINFLUX[ 22 , 25 ] to investigate our StrepEncSM{HaloTag} construct. StrepEncSM{HaloTag} was labeled with a HaloTag ligand (CA‐AlexaFluor 647, AF647, synthesized in house) and imaged the samples in a redox buffer system (for details see the Supporting Information, Figure S6A). In a first step, we transferred all fluorescent molecules to an off‐state by illumination with low‐power excitation light. This allowed us to count Alexa Fluor 647 molecules present in individual nanocompartments from the off‐switching steps (Figure 4 A), delivering an overall estimate of seven molecules per StrepEncSM{HaloTag‐AF647} (Figure 4 B). Subsequently, we localized individually photo‐activated molecules using an iterative MINFLUX approach (Figure 4 C). [22] By analyzing the standard deviation of all localizations that was slightly larger than our localization precision of about 2 nm, and comparing the result to simulations, we validated the presence of the molecules within the shell (shell inner diameter 14 nm, Supporting Information, Figure S6).
Following single‐molecule analysis of StrepEncSM{HaloTag}, we turned to the conversion of the construct as a nanoreactor. Importantly, the same protein assembly could be used, and only the CA as a substrate of HaloTag had to be attached to a different small molecule. As proof of concept of this enabling strategy, we chose to target a CA‐derivative of the water‐compatible and well‐studied ruthenium catalyst [CpRu(HQ)(allyl)]PF6 (Cp=η5‐cyclopentadienyl, HQ=8‐hydroxyquinolinate‐motif) [26] towards the interior of EncSM. Biocompatible metal‐promoted catalysis is facing significant complications as compared to classical transition metal catalysis in organic solvent, yet a growing body of research is available using Au, Pd, Ru or Cu species. Ru complexes present a good balance between reactivity and stability, when employed in biologically relevant media.[ 27 , 28 ] [CpRu(HQ)(allyl)]PF6 was firstly described by Meggers et al. [26] and is well suited for applications in aqueous systems, which is a pre‐requisite for our studies in self‐assembled protein nanocontainers. [CpRu(HQ)(allyl)]PF6 is, amongst other transformations, catalyzing the de‐N‐allylation of coumarin profluorophores (e.g. 1) to the corresponding coumarin dyes (2, Figure 5; Supporting Information, Scheme SS1), a transformation that can be easily followed in vitro and in cellulo by fluorescence spectroscopy.[ 26 , 29 ] For covalent labeling of HaloTag, [CpRu(HQ)(allyl)]PF6 was modified with a CA‐spacer (4), [30] yielding CA‐PEG3‐Ru (5, Figure 5; Scheme SS2).
Figure 5.

Uncaging reaction of 1 (125 μM, 1 equiv) to 2. Anaerobic labeling and washing steps with standard aerobic reaction conditions. Yields were determined by fluorescence intensity in three independent experiments (λ ex=339 nm, λ em=439 nm). Error bars represent ± SEM. Key: StrepEncSM{HaloTag‐PEG3‐Ru} (black square), HaloTag‐PEG3‐Ru (blue triangle), StrepEncSM{GFP} (green triangle), CA‐PEG3‐Ru (5) (turquoise diamond).
Initially, we monitored the activity of non‐encapsulated 5 in the deallylation reaction of 1 by fluorescence detection and LC‐MS analysis in three different buffer systems (Figures S7 and S8), applying biologically relevant conditions as described by Meggers et al. [26] CA‐PEG3‐Ru (5) catalysis provided product yields after 4 hours of 46 % and 49 %, respectively (0.1 equiv. cat., H2O/DMSO or potassium phosphate (KPi)) and diminished yields of 30 % in HEPES buffer containing 0.05 % Tween 20, our StrepEncSM storing buffer. Corresponding turnover numbers (TON=mol product per mol catalyst) are 4 (0.1 equiv cat., H2O/DMSO), 5 (0.1 equiv cat., KPi) and 3 (0.1 equiv cat., HEPES + 0.05 % Tween 20). The yields and TONs observed are low, yet in the range of previously reported values for Ru catalysts[ 5 , 26 , 28 , 29 , 31 ] applied in water‐based solvents under aerobic conditions. Additionally, the catalytic activity was also demonstrated by a linear correlation of the turnover frequency of 5 (Figure S9). Subsequently, we performed the reaction under anaerobic conditions and observed product yields after 4 hours of 88 % (0.1 equiv cat., HEPES + 0.05 % Tween 20) indicating that the presence of oxygen leads to catalyst inactivation (Figure S7A).
Subsequently, we labeled StrepEncSM{HaloTag} and HaloTag only, with CA‐PEG3‐Ru (5) in HEPES buffer (+ 0.05 % Tween 20, pH 7.4). Removal of the unreacted catalyst was achieved by extensive dialysis and subsequent washing of the constructs using Amicon® centrifugal filter devices. Integrity of the Enc constructs following labeling, purification and incubation (37 °C) was verified by native PAGE analysis (Figure S10). A slight degradation of HaloTag‐PEG3‐Ru was observed, while the Enc construct remained intact. ICP‐MS analysis of the constructs revealed a ruthenium content of 6.1±1.2 and 1.6±0.6 ruthenium molecules for StrepEncSM{HaloTag‐PEG3‐Ru} and HaloTag‐PEG3‐Ru, respectively. Due to the time consuming protocol, labeling and removal of unreacted catalyst was performed under anaerobic conditions, to limit oxygen driven degradation of 5. Next, we incubated each construct and CA‐PEG3‐Ru (5) only with profluorophore 1 and followed its deallylation reaction to 2 by measurements of fluorescence increase (Figure 5). Importantly, the CA‐PEG3‐Ru (5) catalyst and StrepEncSM{HaloTag‐PEG3‐Ru} deallylated 1 with yields of 41 % and 36 %, respectively, providing clear evidence for efficient transition‐metal‐mediated catalysis within the encapsulin structure.
In further control experiments, StrepEncSM{eGFP} and HaloTag‐PEG3‐Ru yielded 9 % and 25 % conversion, after 24 hours. The low yet observable catalytic activity of StrepEncSM{eGFP} is likely originating from impurities as a result of incomplete removal of the CA‐PEG3‐Ru (5) construct during the dialysis/washing steps, and non‐treated StrepEncSM{eGFP} is displaying no catalytic activity (Figure S11). Interestingly, significant loss of activity of the HaloTag‐PEG3‐Ru catalyst outside the nanocontainer was observed under anaerobic conditions, pointing towards beneficial effects of catalyst encapsulation, as the HaloTag attached catalyst regains its activity inside of StrepEncSM. Repeating the experiment under aerobic labeling and washing conditions validated our initial hypothesis of catalyst inactivation in the presence of oxygen (Figure S11). All constructs under investigation displayed ≥10‐fold lower product yields, except StrepEncSM{HaloTag‐PEG3‐Ru}, with a 2‐fold lower product yield, pointing towards a certain catalyst protection within the encapsulin environment. Protection of the construct from oxygen during labeling/washing steps and catalysis reaction, restored catalyst activity, and product yields of >75 % were observed for StrepEncSM{HaloTag‐PEG3‐Ru} and HaloTag‐PEG3‐Ru.
Furthermore, we have determined corresponding k cat and K m [32] values and found: K m=21.7±3.5 μM, 34.5±1.5 μM and 39.8±1.5 μM for CA‐PEG3‐Ru (5), HaloTag‐PEG3‐Ru and StrepEncSM{HaloTag‐PEG3‐Ru}, as well as k cat×102 [min−1]=8.6, 4.6 and 6.2, respectively (Figure S11). Interestingly, we find an increased Km value for encapsulin, suggesting that diffusion of the substrate across the shell is hindered, while k cat values are slightly higher for encapsulin, as compared to HaloTag bound catalyst, indicating that once the substrate entered the shell, the reaction might be faster for example, due to limited space and packed environment.
Modified Encs provide significant opportunities for intracellular applications, such as pro‐drug activation or synthetic biology approaches, and we have validated cellular uptake of StrepEncSM applying StrepEnc{eGFP} on mouse monocytes (J774A.1), a cell line that has been shown to internalize Enc particles (Figure S2D). [15] In order to investigate the applicability of StrepEncSM{HaloTag‐PEG3‐Ru} in cellulo, we incubated a monolayer of cells with the construct for 24 hours and, after washing the cells, subsequently applied profluorophore 1 to the cell supernatant (Figure 6). Fluorescence microscopy of live cells revealed that there is a considerable intracellular build‐up of blue fluorescence after 24 hours mainly located in round vesicles. Control experiments of cells treated with 1 or CA‐PEG3‐Ru (5) + 1 only, confirmed that such increase is a consequence of intracellular product formation. These results provide evidence that engineered Encs can be used to perform transition‐metal‐mediated catalysis in living cells.
Figure 6.

Fluorescence microscopy and bright‐field images of J774A.1 monocytes treated with StrepEncSM{HaloTag‐PEG3‐Ru} + 1, CA‐PEG3‐Ru (5) + 1 and 1 only (negative controls). Cells were seeded and treated with 1 μM StrepEncSM{HaloTag‐PEG3‐Ru} in DMEM overnight. Subsequently, 1 (20 μM) was applied to the cell supernatant. Following incubation and washing, images were acquired with a digital inverted microscope. Fluorescence microscopy images of monocytes treated with encapsulin show fluorescence from uncaging product 2 that is localized within distinct vesicles. Fluorescence microscopy images show no blue fluorescence in cells treated with 1 only. Key: Dulbecco's Modified Eagle Medium (DMEM).
In summary, we demonstrate that StrepEncSM nanocapsules can be engineered as hosts for transition metal catalysts inside cells using HaloTag in combination with a localization sequence.
Conclusion
We engineer and purify encapsulins equipped with the protein HaloTag as a modular platform for functional nanocompartments. By applying two independent single‐molecule techniques, smFRET and MINFLUX, we are able to delineate the number of labeled cargo proteins bound inside encapsulin. We then install a transition metal catalyst inside the capsule. Subsequently, we show that catalysis inside this capsule is possible and that encapsulation affects reaction yields. The deallylation of a profluorophore is taking place with lower yields with the HaloTag‐PEG3‐Ru construct outside of the capsule as compared to within encapsulin. We go on to demonstrate that the same reaction is also efficiently catalyzed inside a living cell line that takes up the engineered Enc and produces localized fluorescent vesicles. These results open up a plethora of potential applications for bioorthogonal catalysis inside confined space. Importantly, the HaloTag approach can be used to install a large variety of different catalysts and we envision the controlled placement of two different metal centers in one capsule to perform dual catalysis. We conclude that encapsulins are a highly modular platform, ideally suited to perform reactions that are new to biology under biologically relevant conditions.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information
Acknowledgements
We thank S. Ferlaino for measurement of NMR spectra and C. Warth for supporting ESI‐MS analysis. Furthermore, we thank Prof. A. Bechthold and Prof. M. Müller for their support. We thank Prof. D. Lamb and Prof. M. Schlierf for helpful discussions. J.T and T.H. acknowledge funding by the European Research Council through ERC grant agreement No. 681891. T.H. and A.W. acknowledge funding by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany's Excellence Strategy–EXC‐2193/1‐390951807. C.J.T and M.Z. acknowledge funding by the Deutsche Forschungsgemeinschaft grant RTG 2202 (transport across and into membranes). We furthermore thank all members of LIC for supporting fluorescence imaging efforts and image analysis, funded by DFG‐Funding INST 380/109‐1 FUGG (Zeiss LSM 880). Open Access funding enabled and organized by Projekt DEAL.
P. Lohner, M. Zmyslia, J. Thurn, J. K. Pape, R. Gerasimaitė, J. Keller-Findeisen, S. Groeer, B. Deuringer, R. Süss, A. Walther, S. W. Hell, G. Lukinavičius, T. Hugel, C. Jessen-Trefzer, Angew. Chem. Int. Ed. 2021, 60, 23835.
References
- 1.
- 1a. Hinzpeter F., Gerland U., Tostevin F., Biophys. J. 2017, 112, 767; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1b. Tsitkov S., Hess H., ACS Catal. 2019, 9, 2432. [Google Scholar]
- 2. Tracey J. C., Coronado M., Giessen T. W., Lau M. C. Y., Silver P. A., Ward B. B., Sci. Rep. 2019, 9, 20122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.
- 3a. Frey R., Mantri S., Rocca M., Hilvert D., J. Am. Chem. Soc. 2016, 138, 10072; [DOI] [PubMed] [Google Scholar]
- 3b. Moon H., Lee J., Kim H., Heo S., Min J., Kang S., Biomater. Res. 2014, 18, 21; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Ren H., Zhu S., Zheng G., Int. J. Mol. Sci. 2019, 20, 592; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Fiedler J. D., Brown S. D., Lau J. L., Finn M. G., Angew. Chem. Int. Ed. 2010, 49, 9648–9651; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 9842; [Google Scholar]
- 3e. Seebeck F. P., Woycechowsky K. J., Zhuang W., Rabe J. P., Hilvert D., J. Am. Chem. Soc. 2006, 128, 4516; [DOI] [PubMed] [Google Scholar]
- 3f. Goldsmith L. E., Pupols M., Kickhoefer V. A., Rome L. H., Monbouquette H. G., ACS Nano 2009, 3, 3175; [DOI] [PubMed] [Google Scholar]
- 3g. Fiedler J. D., Fishman M. R., Brown S. D., Lau J., Finn M. G., Biomacromolecules 2018, 19, 3945; [DOI] [PubMed] [Google Scholar]
- 3h. Minten I. J., Hendriks L. J. A., Nolte R. J. M., Cornelissen J. J. L. M., J. Am. Chem. Soc. 2009, 131, 17771. [DOI] [PubMed] [Google Scholar]
- 4. Giessen T. W., Curr. Opin. Chem. Biol. 2016, 34, 1. [DOI] [PubMed] [Google Scholar]
- 5. Hsu H.-T., Trantow B. M., Waymouth R. M., Wender P. A., Bioconjugate Chem. 2016, 27, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.
- 6a. Lagoutte P., Mignon C., Stadthagen G., Potisopon S., Donnat S., Mast J., Lugari A., Werle B., Vaccine 2018, 36, 3622; [DOI] [PubMed] [Google Scholar]
- 6b. Sigmund F., Massner C., Erdmann P., Stelzl A., Rolbieski H., Desai M., Bricault S., Wörner T. P., Snijder J., Geerlof A., et al., Nat. Commun. 2018, 9, 1990; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6c. Choi B., Moon H., Hong S. J., Shin C., Do Y., Ryu S., Kang S., ACS Nano 2016, 10, 7339; [DOI] [PubMed] [Google Scholar]
- 6d. Giessen T. W., Silver P. A., ACS Synth. Biol. 2016, 5, 1497. [DOI] [PubMed] [Google Scholar]
- 7. Giessen T. W., Silver P. A., Nat. Microbiol. 2017, 2, 17029. [DOI] [PubMed] [Google Scholar]
- 8.
- 8a. Giessen T. W., Orlando B. J., Verdegaal A. A., Chambers M. G., Gardener J., Bell D. C., Birrane G., Liao M., Silver P. A., eLife 2019, 8, e46070; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8b. Sigmund F., Pettinger S., Kube M., Schneider F., Schifferer M., Schneider S., Efremova M. V., Pujol-Martí J., Aichler M., Walch A., et al., ACS Nano 2019, 13, 8114. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Ross J., Lambert T., Piergentili C., He D., Waldron K. J., Logan Mackay C., Marles-Wright J., Clarke D. J., Chem. Commun. 2020, 56, 3417; [DOI] [PubMed] [Google Scholar]
- 9b. He D., Hughes S., Vanden-Hehir S., Georgiev A., Altenbach K., Tarrant E., Mackay C. L., Waldron K. J., Clarke D. J., Marles-Wright J., eLife 2016, 5, e18972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rurup W. F., Snijder J., Koay M. S., Heck A. J., Cornelissen J. J., J. Am. Chem. Soc. 2014, 136, 3828. [DOI] [PubMed] [Google Scholar]
- 11. Nichols R. J., Cassidy-Amstutz C., Chaijarasphong T., Savage D. F., Crit. Rev. Biochem. Mol. Biol. 2017, 52, 583. [DOI] [PubMed] [Google Scholar]
- 12. Cassidy-Amstutz C., Oltrogge L., Going C. C., Lee A., Teng P., Quintanilla D., East-Seletsky A., Williams E. R., Savage D. F., Biochemistry 2016, 55, 3461. [DOI] [PubMed] [Google Scholar]
- 13. Lau Y. H., Giessen T. W., Altenburg W. J., Silver P. A., Nat. Commun. 2018, 9, 1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rahmanpour R., Bugg T. D. H., FEBS J. 2013, 280, 2097. [DOI] [PubMed] [Google Scholar]
- 15. Putri R. M., Allende-Ballestero C., Luque D., Klem R., Rousou K.-A., Liu A., Traulsen C. H.-H., Rurup W. F., Koay M. S. T., Castón J. R., et al., ACS Nano 2017, 11, 12796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Tamura A., Fukutani Y., Takami T., Fujii M., Nakaguchi Y., Murakami Y., Noguchi K., Yohda M., Odaka M., Biotechnol. Bioeng. 2015, 112, 13. [DOI] [PubMed] [Google Scholar]
- 17. Contreras H., Joens M. S., McMath L. M., Le V. P., Tullius M. V., Kimmey J. M., Bionghi N., Horwitz M. A., Fitzpatrick J. A. J., Goulding C. W., J. Biol. Chem. 2014, 289, 18279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stetefeld J., McKenna S. A., Patel T. R., Biophys. Rev. 2016, 8, 409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tang Y., Mu A., Zhang Y., Zhou S., Wang W., Lai Y., Zhou X., Liu F., Yang X., Gong H., et al., Proc. Natl. Acad. Sci. USA 2021, 118, e2025658118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.
- 20a. England C. G., Luo H., Cai W., Bioconjugate Chem. 2015, 26, 975; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20b. Fischer S., Ward T. R., Liang A. D., ACS Catal. 2021, 11, 6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hellenkamp B., Schmid S., Doroshenko O., Opanasyuk O., Kühnemuth R., Rezaei Adariani S., Ambrose B., Aznauryan M., Barth A., Birkedal V., et al., Nat. Methods 2018, 15, 669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gwosch K. C., Pape J. K., Balzarotti F., Hoess P., Ellenberg J., Ries J., Hell S. W., Nat. Methods 2020, 17, 217. [DOI] [PubMed] [Google Scholar]
- 23. Müller B. K., Zaychikov E., Bräuchle C., Lamb D. C., Biophys. J. 2005, 89, 3508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Coullomb A., Bidan C. M., Qian C., Wehnekamp F., Oddou C., Albigès-Rizo C., Lamb D. C., Dupont A., Sci. Rep. 2020, 10, 6504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Balzarotti F., Eilers Y., Gwosch K. C., Gynnå A. H., Westphal V., Stefani F. D., Elf J., Hell S. W., Science 2017, 355, 606. [DOI] [PubMed] [Google Scholar]
- 26. Völker T., Meggers E., ChemBioChem 2017, 18, 1083. [DOI] [PubMed] [Google Scholar]
- 27.
- 27a. Yang M., Li J., Chen P. R., Chem. Soc. Rev. 2014, 43, 6511; [DOI] [PubMed] [Google Scholar]
- 27b. Vidal C., Tomás-Gamasa M., Destito P., López F., Mascareñas J. L., Nat. Commun. 2018, 9, 1913; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27c. Chankeshwara S. V., Indrigo E., Bradley M., Curr. Opin. Chem. Biol. 2014, 21, 128; [DOI] [PubMed] [Google Scholar]
- 27d. Ward T. R., Acc. Chem. Res. 2011, 44, 47. [DOI] [PubMed] [Google Scholar]
- 28. Eda S., Nasibullin I., Vong K., Kudo N., Yoshida M., Kurbangalieva A., Tanaka K., Nat. Catal. 2019, 2, 780. [Google Scholar]
- 29. Streu C., Meggers E., Angew. Chem. Int. Ed. 2006, 45, 5645; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 5773. [Google Scholar]
- 30.
- 30a. Clark S. A., Singh V., Mendoza D. V., Margolin W., Kool E. T., Bioconjugate Chem. 2016, 27, 2839; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30b. Liu Y., Miao K., Dunham N. P., Liu H., Fares M., Boal A. K., Li X., Zhang X., Biochemistry 2017, 56, 1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Okamoto Y., Kojima R., Schwizer F., Bartolami E., Heinisch T., Matile S., Fussenegger M., Ward T. R., Nat. Commun. 2018, 9, 1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cao-Milán R., He L. D., Shorkey S., Tonga G. Y., Wang L.-S., Zhang X., Uddin I., Das R., Sulak M., Rotello V. M., Mol. Syst. Des. Eng. 2017, 2, 624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supporting Information