

Abstract
Staphylococcus aureus have evolved into diverse lineages, known as clonal complexes (“CC”), which exhibit differences in the coding sequences of core virulence factors. Whether these alterations impact functionality is poorly understood. Here, we studied the highly polymorphic pore-forming toxin LukAB. We discovered that the LukAB toxin variants produced by S. aureus CC30 and CC45 kill human phagocytes regardless of whether CD11b, the previously established LukAB receptor, is present, and instead target the human hydrogen voltage-gated channel 1 (HVCN1). Biochemical studies identified the domain within human HVCN1 that drives LukAB species specificity, enabling the generation of humanized HVCN1 mice with enhanced susceptibility to CC30 LukAB and to bloodstream infection caused by CC30 S. aureus strains. Altogether, this work advances our understanding of an important S. aureus toxin and underscores the importance of considering genetic variation to characterizing virulence factors and understand the tug of war between pathogens and the host.
Keywords: Staphylococcus aureus, MRSA, LukAB, leukocidin, HVCN1, toxin receptor
Staphylococcus aureus is a Gram-positive bacterium responsible for a broad range of invasive diseases1. To escape clearance by the immune system, S. aureus employs a wide range of strategies including the production of potent bi-component pore-forming toxins known as leukocidins2,3. The leukocidins bind to specific cellular receptors to assemble into oligomeric pores that lead to cell lysis3. Amongst these toxins, LukAB (also known as LukGH4) is the most recently identified leukocidin4,5 and is the dominant toxin responsible for S. aureus-mediated phagocyte lysis in ex vivo infection models4–7. The tropism of LukAB towards human phagocytes is mediated by binding to the integrin component CD11b8. Although the lukAB locus is part of the core S. aureus genome, there is significant nucleotide and amino acid diversity at this locus, up to 18% amino acid diversity in the LukAB toxin as compared to up to 5% in other leukocidins9. The majority of prior research on LukAB, however, has focused on the prototype toxin produced by the community-acquired MRSA clone USA300, a member of clonal complex 8 (CC8)4–6,8,10–18. In the United States, isolates of the CC8 lineage cause a large proportion of both community-associated (CA) and health care-associated (HA) methicillin-resistant S. aureus (MRSA) infections11,19,20. However, other major lineages, such as CC1, CC5, CC15, CC22, CC30 and CC45, contribute significantly to infections both in the United States and globally21.
Here, we set out to characterize the activity of LukAB variants. Phylogenetic analyses indicated that the polymorphisms in lukAB loci are largely clonal complex-specific. Representative LukAB variants from the most diverse lineages exhibited comparable cytotoxic activities against primary human phagocytes. Nevertheless, two closely related variants produced by S. aureus belonging to the CC30 and CC45 lineages were found to kill cells lacking CD11b, the previously established LukAB receptor. By performing a genome-wide CRISPR-Cas9 screen, we identified the hydrogen voltage-gated channel 1 (HVCN1) as a critical cellular receptor for cell killing by CC30/CC45 LukAB. As with CD11b8,18, LukAB was found to kill mammalian cells containing human but not murine HVCN1. Mapping the LukAB-targeted domain within human HVCN1 enabled the generation of a humanized HVCN1 mouse (“hHVCN1”). Phagocytes from the hHVCN1 mice were susceptible to CC30 LukAB-mediated cytotoxicity. Compared to wildtype mice, the hHVCN1 mice were more susceptible to bacterial burdens following bloodstream infection with both community- and hospital-acquired CC30 S. aureus isolates. Taken together, these findings highlight how lineage-specific changes in the sequence of virulence factors influence the pathogenic potential of bacteria and identify HVCN1 as a human protein targeted by S. aureus.
RESULTS
Sequences of lukA and lukB cluster according to S. aureus clonal complexes
The lukAB locus is a part of the core genome of S. aureus, but it exhibits high allelic variability5,9. To define the extent of lukAB diversity across S. aureus, we analyzed the lukA and lukB sequences from 4,187 publicly available S. aureus genomes as well as the closely related species S. argenteus (previously known as CC75) and S. schweitzeri. Identical nucleotide sequences were collapsed into a single representative sequence, thereby reducing the number of unique lukA and lukB alleles to 94 and 83, respectively (Supplementary Table 1). Phylogenetic analysis revealed several distinct groups of toxin variants that reflect the overall phylogenetic structure of the Staphylococcal species (Figures 1A–1B)22. Major branches correspond to the S. aureus clonal complexes (CCs) 1/5/8/97, 10/395, 398, 30/45, as well as S. argenteus (CC75) and S. schweitzeri. Notably, toxins are highly similar within each group but exhibit long branch lengths between groups (Figures 1A–1B). However, comparisons between groups show strong levels of purifying selection, suggesting these differences occur disproportionally at synonymous sites and are not primarily due to positive selection. Specifically, whole-gene ratios of nonsynonymous divergence (dN) to synonymous divergence (dS) are in the range 0.07–0.12, significantly less than 1 (P<0.0001) (Extended Data Figure 1). These dN/dS values are similar to the mean across all orthologous loci in comparisons between S. aureus reference genomes (e.g., MW2 vs Mu50), which fall in the range 0.07 to 0.0823. Taken together, these findings suggest that the function of LukAB is critical for the fitness in all the groups studied, and that, with the possible exception of a few residues, the amino acid sequences of these proteins experience levels of functional constraint typical for S. aureus.
Figure 1. Clonal complex-specific LukAB variants exhibit different cytotoxic activities towards human leukocytes.
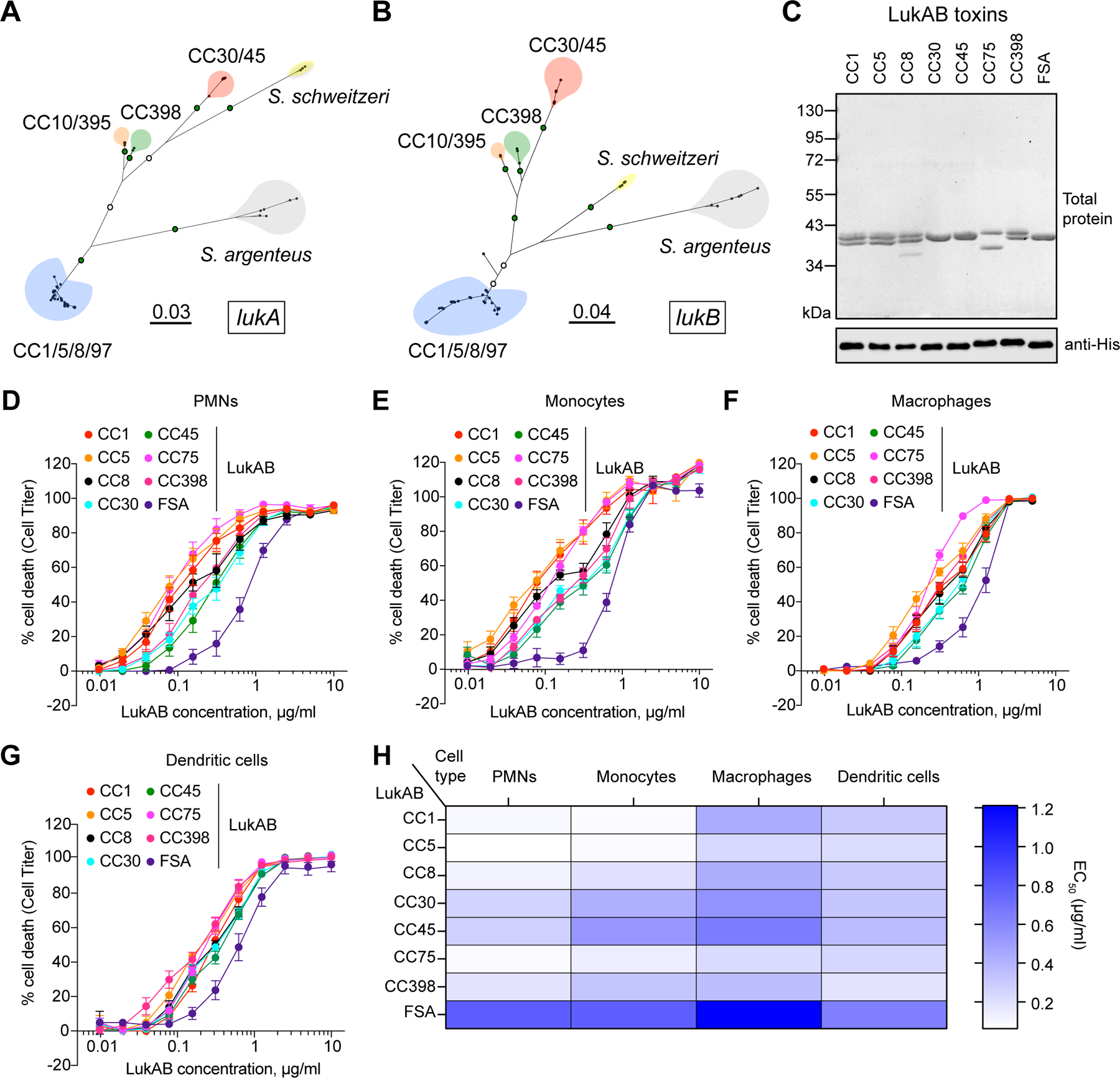
A-B: Unrooted maximum likelihood phylogeny for lukA (A) and lukB (B). Branch lengths are in substitutions/site. Key bootstrap values are depicted in green (>90%) or white (>70%) on branches. Major clades are colored blue (CC1/5/8/97), red (CC30/45), orange (CC10/395), green (CC398), yellow (S. schweitzeri, FSA), and grey (S. argenteus, CC75). C: Recombinant co-purified 6xHis-LukA and LukB proteins visualized by total protein staining (1 μg) and immunoblotting (100 ng) with anti-His antibody. One replicate of this experiment was performed. D-G: Intoxication of primary human PMNs (D), monocytes (E), macrophages (F), and dendritic cells (G) with indicated concentrations of LukAB toxins and cell viability measured with Cell Titer. Data were collected using cells isolated from six independent donors (PMNs), four donors (monocytes), five donors (monocyte-derived macrophages), four donors (monocyte-derived dendritic cells), and are represented as mean values ±SEM. H: Half-maximal lytic concentrations (EC50) of each toxin calculated from data presented in panels D-G. Also refer to Extended Data Figures 1–2 and Supplementary Tables 1–2.
LukAB variants kill primary human phagocytes
To evaluate the functional significance of the variations observed in LukAB, we cloned and purified LukAB from eight clonal complexes that encode the major lukAB alleles: CC1, CC5, CC8, CC30, CC45, S. argenteus (herein referred to as CC75), CC398, and S. schweitzeri (strain FSA-084, herein referred to as FSA) (Figure 1C, Extended Data Figure 2, Supplementary Table 2). We tested the cytotoxic activity of the LukAB variants towards primary human neutrophils (PMNs), monocytes, monocyte-derived macrophages and monocyte-derived dendritic cells. We found that all tested LukAB variants lysed human phagocytes in a dose-dependent manner (Figures 1D–1H). However, while cytotoxicity towards dendritic cells was similar across clonal complexes, we observed much higher variability in toxin activity towards PMNs and monocytes (Figures 1D–1H).
Unlike other variants, CC30 and CC45 LukAB are cytotoxic in the absence of CD11b
We sought to determine whether non-CC8 LukAB variants also rely on CD11b for cytotoxicity by evaluating their lytic activity towards control and CD11b-depleted human monocytic THP1 cells (Figure 2A). In cells transduced with scramble shRNA, the LukAB variants exhibited variable but overall comparable cytotoxicity (Figure 2B). Intoxication with CC30 and CC45 LukAB, two closely related LukAB variants that differ by only four amino acids, resulted in 100% cell death at toxin concentrations as low as 2.5 μg/ml (Figure 2B). As expected8, depletion of CD11b by shRNA (Figure 2A) abolished CC8 LukAB-mediated cytotoxicity. Depletion of CD11b (encoded by the ITGAM gene) also protected cells from lysis by the CC1, CC5, CC75, CC398, and FSA LukAB variants (Figure 2C). Unexpectedly, the CC30 and CC45 LukAB variants killed the THP1 cells depleted of CD11b (Figure 2C). Potencies of both toxins were, however, slightly reduced, as indicated by the increase in EC50 values (from 0.1368 μg/ml and 0.1835 μg/ml in control cells to 0.5425 μg/ml and 0.6604 μg/ml in CD11b-depleted cells for CC30 and CC45 LukAB, respectively). To ensure that the findings described above were not specific for THP1 cells, we also used the human promyelocytic leukemia cell line (HL60)8 differentiated into neutrophil-like cells (PMN-HL60). As with THP1 cells, CC30 and CC45 LukAB variants killed CD11b-depleted PMN-HL60 (Figure 2D). Furthermore, CD18 (encoded by the ITGB2 gene), which enables surface localization of all β2 integrins24,25, including CD11a, CD11b, CD11c, and CD11d, was also dispensable for the cytotoxicity of CC30 and CC45, but not CC8 LukAB (Figure 2D).
Figure 2. CC30 and CC45 LukAB kill cells lacking CD11b/CD18.
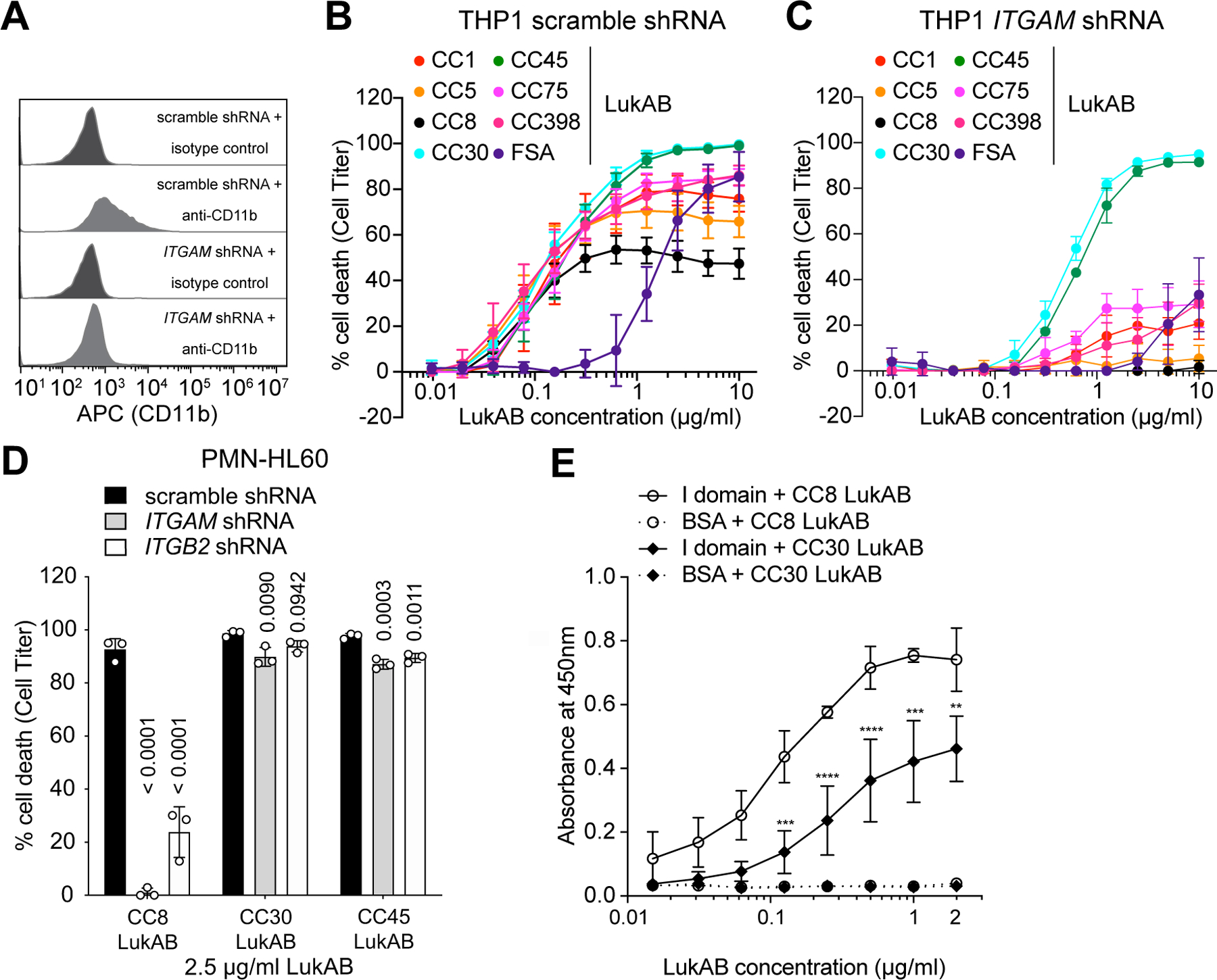
A: Flow cytometric analysis of surface CD11b levels of scramble shRNA and ITGAM shRNA (CD11b-depleted) expressing THP1 cells. Histograms are representative of two independent experiments. B-C: Intoxication of scramble shRNA (B) and ITGAM shRNA (C) expressing THP1 cells with indicated concentration of different LukAB variants. Data from three independent experiments are shown as mean values ±SD. D: Intoxication of scramble shRNA, ITGAM shRNA, and ITGB2 (CD18) shRNA expressing PMN-like HL60 cells with CC8, CC30, and CC45 LukAB (2.5 μg/ml). Data from three independent experiments are represented as mean values ±SD. B-D: Cell viability was measured with Cell Titer and statistical significance was determined by one-way ANOVA, numbers indicate P values. E: Binding of biotinylated CC8 and CC30 LukAB to the CD11b I-domain measured by ELISA. Data from three independent experiments are represented as mean values ±SD. Statistical significance was determined by two-way ANOVA (****, P ≤ 0.0001; ***, P ≤ 0.001; **, P ≤ 0.01; ns, not significant). Also refer to Extended Data Figures 3 and 6.
Consistent with the observed reduced dependence on CD11b for cytotoxicity, ELISA-based binding studies demonstrated that CC30 LukAB bound weaker to the CD11b I-domain compared to CC8 LukAB (Figure 2E). Furthermore, bio-layer interferometry revealed that CC30 LukAB exhibits lower binding affinity to the CD11b I-domain with an apparent Kd of 214 ± 3.28 nM, as opposed to CC8 LukAB with an apparent Kd of 9.41 ± 0.05 nM (Extended Data Figure 3). Altogether, these results indicate that unlike the other clonal complexes, CC30 and CC45 LukAB variants do not require CD11b to kill human phagocytes.
Identification of HVCN1 as the cellular target for the CC30 and CC45 LukAB toxins
To identify the cell surface target of CC30 and CC45 LukAB variants, we employed a genome-wide CRISPR-Cas9 approach26. We introduced two independent genome-wide subpools of single-guide RNAs (sgRNAs) together with Cas9 into CD11b-depleted THP1 cells and subjected the cells to two sequential rounds of CC30 LukAB intoxication. The sgRNAs from the surviving cells were then sequenced and the frequencies of sgRNAs in the surviving cell population compared to untreated cells were determined (Figure 3A). Among the recovered sgRNAs that target genes encoding cell surface proteins, sgRNAs for HVCN1, OGT, and ASGR1 were the most highly enriched in the toxin resistant cells (Figure 3B, Supplementary Table 3). Studies using individual knockout cell lines revealed that the cytotoxicity of CC30 LukAB was fully dependent on HVCN1, while remaining unaffected or only minimally affected by the ASGR1 and OGT depletion, respectively (Figure 3C). HVCN1 encodes the hydrogen voltage-gated channel 1, primarily found on leukocytes, where its major function is to balance charges across the membrane to ensure full NADPH oxidase activity and ROS production27–30. CRISPR-Cas9-mediated HVCN1 targeting (Figures 3D–3E) ablated the cytotoxicity of both CC30 and CC45 LukAB (Figure 3F), while the cytotoxicity of HlgAB, another bi-component pore-forming toxin that targets CCR2, CXCR1, and CXCR231, remained unaffected (Figure 3F). These data demonstrate that HVCN1 is required for CC30 and CC45 LukAB-mediated cell killing.
Figure 3. Identification of HVCN1 as a cellular target for the CC30 and CC45 LukAB variants.
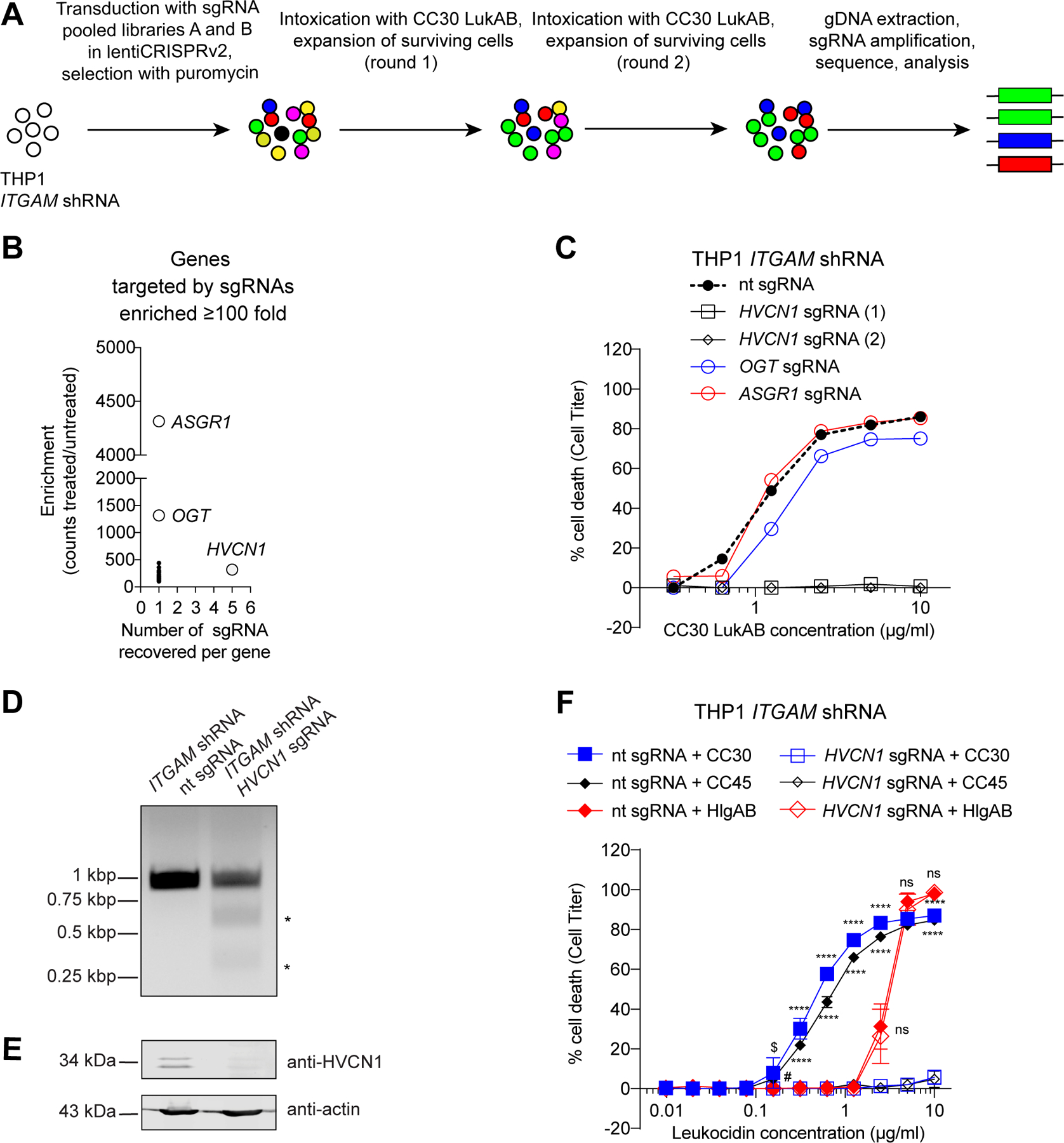
A: Schematic of the CC30 LukAB GeCKO screen in ITGAM shRNA THP1 cells. B: Enrichment of specific sgRNAs from the GeCKO library following two rounds of CC30 LukAB selection. Data are presented as the number of sgRNAs significantly enriched in the intoxicated sample versus the average fold enrichment as compared to untreated control. C: Intoxication of ITGAM shRNA THP1 cells transduced with lentiCRISPRv2 expressing sgRNAs targeting indicated genes with CC30 LukAB. Cell viability was measured with Cell Titer. Data are represented as the average of two independent experiments each performed in duplicate. D: Gel image of T7 Endonuclease I-treated HVCN1 PCR products confirming HVCN1 targeting by the sgRNA. HVCN1 was amplified from genomic DNA of ITGAM shRNA THP1 cells transduced with lentiCRISPRv2 expressing non-targeting (nt) sgRNA or HVCN1 sgRNA. Asterisks indicate T7 Endonuclease I cleavage bands. One replicate of this experiment was performed. E: Immunoblot of HVCN1 in ITGAM shRNA THP1 cells transduced with lentiCRISPRv2 expressing non-targeting (nt) sgRNA or HVCN1 sgRNA. Anti-actin immunoblot is shown below as a loading control. Representative image of three independent experiments is shown. F: Intoxication of ITGAM shRNA THP1 cells transduced with lentiCRISPRv2 expressing non-targeting (nt) sgRNA or HVCN1 sgRNA with indicated concentration of CC30 LukAB, CC45 LukAB, and HlgAB. Cell viability was measured with Cell Titer. Data from three independent experiments are represented as mean values ±SD. For each toxin, statistical significance was determined by two-way ANOVA (****, P ≤ 0.0001; $, P = 0.0021; #, P = 0.0072; ns, not significant, >0.9999). Also refer to Supplementary Table 3.
CC30 S. aureus targets human leukocytes in a LukAB- and HVCN1-dependent manner
To define the role of HVCN1-mediated LukAB cytotoxicity in the virulence of CC30 S. aureus, we generated an isogeneic lukAB deletion strain in a community-acquired CC30 S. aureus isolate from our collection (strain 62300D1) (Figures 4A–4B). Potent killing of primary human PMNs was observed with wildtype CC30 S. aureus, a phenotype significantly reduced when lukAB was deleted (Figure 4C). Similar to what we observed with purified toxins, CC30 S. aureus also killed CD11b-depleted THP1 cells in a LukAB-dependent manner (Figure 4D). In contrast, cytotoxicity of the wildtype CC30 S. aureus strain in HVCN1-deficient cells was significantly impaired and was indistinguishable from the cytotoxicity observed with the ΔlukAB strain (Figure 4D).
Figure 4. CC30 S. aureus kills leukocytes in a LukAB and HVCN1 dependent manner.
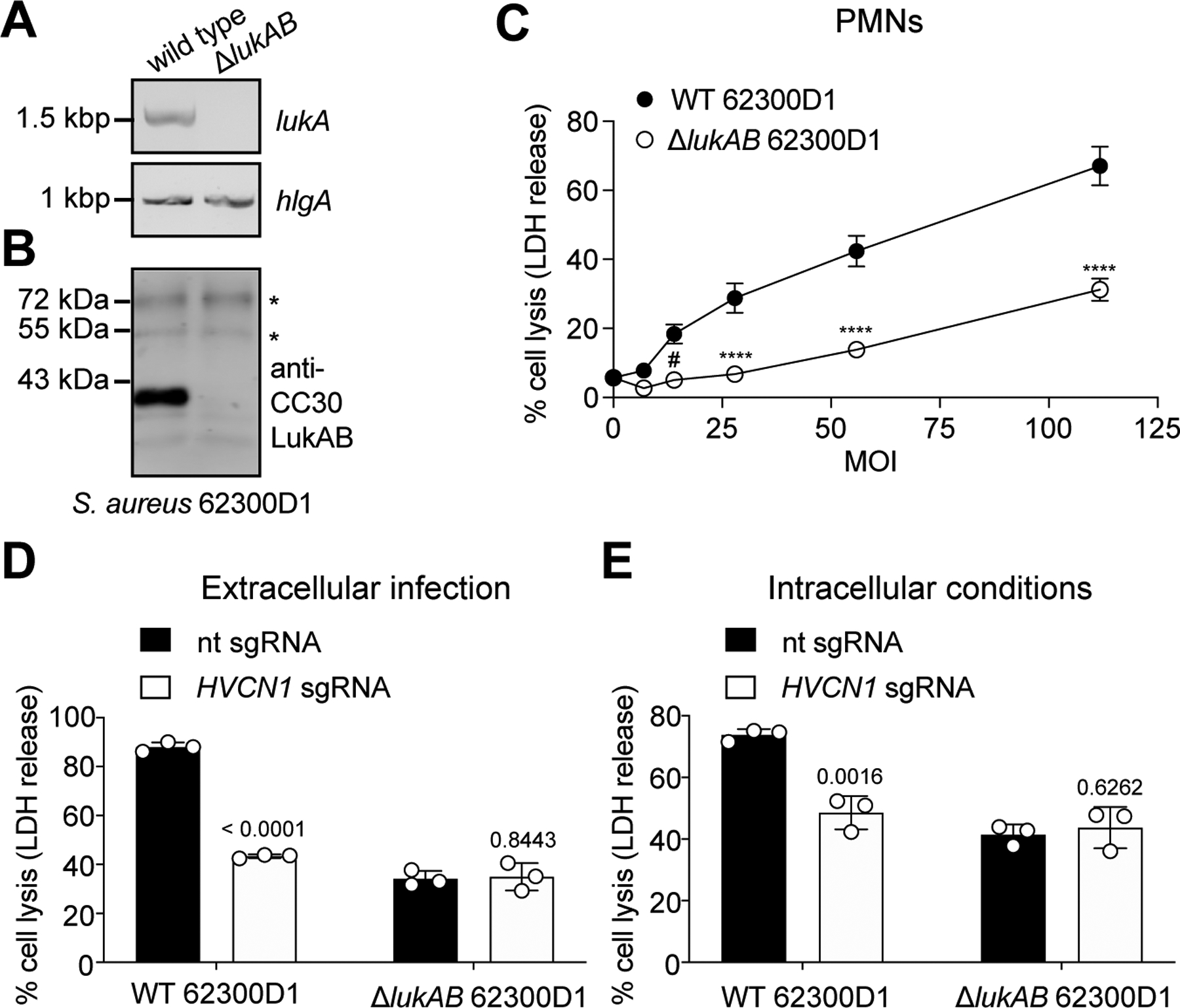
A-B: PCR targeting lukA and hlgA (A) and immunoblot of CC30 LukAB in supernatants of wild type and ΔlukAB CC30 S. aureus 62300D1 (B). Asterisks indicate non-specific bands that serve as loading controls. One replicate of this experiment was performed (A). Representative image of two independent experiments is shown (B). C: Viability of human PMNs following a 2-h infection with nonopsonized wild type (WT) or isogenic ΔlukAB CC30 S. aureus 62300D1 at the indicated multiplicity of infection (MOI). PMN lysis measured by LDH release. Data are from PMNs isolated from six independent donors represented as the mean values ±SEM. Statistical significance was determined by two-way ANOVA (****, P ≤ 0.0001; #, P = 0.0119). D-E: Viability of ITGAM shRNA THP1 cells transduced with lentiCRISPRv2 expressing non-targeting (nt) sgRNA or HVCN1 sgRNA and infected with nonopsonized (extracellular infection, D) or with opsonized (intracellular conditions, E) WT and ΔlukAB CC30 S. aureus 62300D1 for 2h (MOI=100). THP1 cell lysis was measured by LDH release. Data from three independent experiments are represented as the mean ±SD. Statistical significance was determined by t-test (two-tailed), numbers indicate P values.
LukAB mediates cell lysis not only by targeting the extracellular membrane but also when produced by phagocytosed bacteria6–8,13. Since HVCN1 is transported within phagosomes32,33, we next evaluated if LukAB produced by S. aureus upon phagocytosis targeted HVCN1 for cytotoxicity. CC30 S. aureus cells were opsonized with normal human serum, incubated with THP1 cells to promote bacterial uptake6 and the viability of the THP1 cells was evaluated. We observed that HVCN1-positive cells were killed by phagocytosed S. aureus in a LukAB- and HVCN1-dependent manner (Figure 4E). Therefore, CC30 S. aureus targets HVCN1 via LukAB to lyse phagocytes.
HVCN1 sensitizes mammalian cells to CC30 and CC45 LukAB cytotoxicity
To determine whether HVCN1 is sufficient to render mammalian cells susceptible to LukAB, HVCN1 was expressed in Chinese hamster ovary (CHO) cells, which lack endogenous CD11b and HVCN1. As shown in Figure 5A, control cells expressing firefly luciferase (Fluc) were not affected by CC30 and CC45 LukAB, whereas HVCN1 sensitized CHO cells to both toxins. We hypothesized that HVCN1 facilitates the interaction of LukAB with the plasma membrane of the target cells. Indeed, while only minimal interaction between CC30 and CC45 LukAB and control cells was observed, a dose-dependent interaction of the toxins with HVCN1-expressing cells was detected (Figure 5B). Moreover, unlabeled LukAB competed off binding of biotinylated toxin in a dose-dependent manner (Figure 5C). To further these studies, a pull-down assay was performed to evaluate the direct interaction between LukAB and HVCN1. Analysis of the elution of resin-immobilized HVCN1 revealed that HVCN1 interacts with LukAB, but not LukSF (another bi-component pore-forming leukocidin used as a negative control) (Figure 5D). The absence of LukAB in the elution fraction of the empty resin control (TBS) further confirmed that co-elution of LukAB with HVCN1 is not due to a non-specific LukAB-resin interaction (Figure 5D). Together, these data support the notion that HVCN1 is a receptor for LukAB.
Figure 5. Human HVCN1 mediates CC30 and CC45 LukAB binding and cytotoxicity.
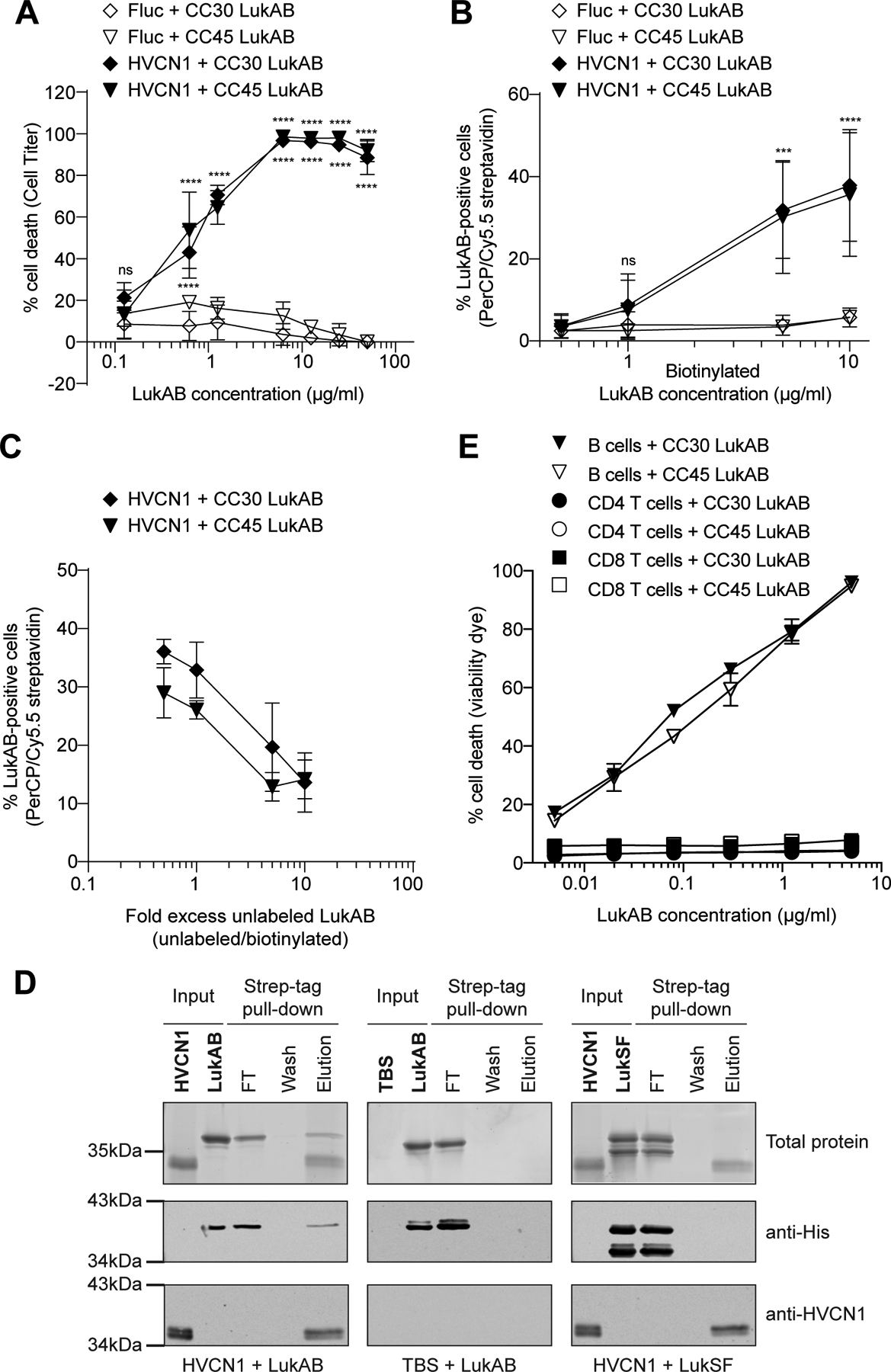
A: Intoxication of CHO cells expressing firefly luciferase (Fluc) or HVCN1 with CC30 and CC45 LukAB. Cell viability was measured with Cell Titer. Data from three independent experiments are represented as mean values ±SD. For each toxin, statistical significance was determined by two-way ANOVA (****, P ≤ 0.0001; ns, not significant). B: Binding of biotinylated CC30 and CC45 LukAB to CHO cells expressing Fluc or HVCN1. Binding was measured by PerCP/Cy5.5 streptavidin staining. Data from three independent experiments are represented as mean values ±SD. For each toxin, statistical significance was determined by two-way ANOVA (****, P ≤ 0.0001; ***, P ≤ 0.001; ns, not significant). C: Binding of biotinylated CC30 and CC45 LukAB (3 μg/ml) to CHO cells transduced with Fluc or HVCN1 in the presence of the indicated excess of unlabeled toxins. Binding was measured by PerCP/Cy5.5 streptavidin staining. Data from three independent experiments are represented as mean values ±SD. D: Pull-down of purified His-tagged LukAB or LukSF with Strep-tagged HVCN1. Input represents resin-bound ligand (HVCN1 or TBS control) and toxin binding partner (LukAB or LukSF). Flow-through (FT), wash, and elution lanes represent fractions from the pull-down after toxin binding (see Methods). Top panel is Sypro Ruby stained SDS-PAGE, middle panel is an immunoblot to detect the toxins, and bottom panel is an immunoblot to detect HVCN1. Representative images of two independent experiments are shown. E: Intoxication of primary human B cells, CD4-T and CD8-T cells with indicated concentrations of CC30 and CC45 LukAB. Membrane damage was detected using Fixable Viability Dye eFluor™ 450. Data from cells isolated from four different donors are represented as mean values ±SEM. Also refer to Extended Data Figure 4.
In addition to phagocytes, HVCN1 is also highly expressed in B cells while being minimally expressed in T cells (Extended Data Figure 4)34. This observation is consistent with the reported higher H+ currents in B cells as compared to T cells35. To evaluate if HVCN1 renders lymphocytes susceptible to LukAB, we purified primary B cells and CD4-positive and CD8-positive T cells from human peripheral blood mononuclear cells and exposed the cells to the toxins. In contrast to CD4 and CD8 T lymphocytes, which were resistant to both CC30 and CC45 LukAB toxins, human B lymphocytes were highly sensitive to LukAB-mediated membrane damage (Figure 5E). Therefore, HVCN1 broadens the repertoire of leukocytes that could be targeted by LukAB during infection.
CC30 and CC45 LukAB target human but not murine HVCN1
CC8 LukAB binds with high affinity to the human, but not murine, CD11b I-domain8,18,36. Treatment of murine peritoneal exudate cells (PECs) with CC8, CC30, and CC45 LukAB revealed that murine leukocytes are resistant to LukAB regardless of the toxin lineage (Figure 6A). In contrast, when the same cells were exposed to LukED, a bi-component leukocidin that targets both human and murine leukocytes37,38, extensive cell death was observed (Figure 6A). Since CC30 and CC45 LukAB are closely related and exhibited identical phenotypes with regard to HVCN1, we chose to focus on CC30 LukAB for the rest of the study.
Figure 6. LukAB targeting of HVCN1 promotes S. aureus pathogenesis.
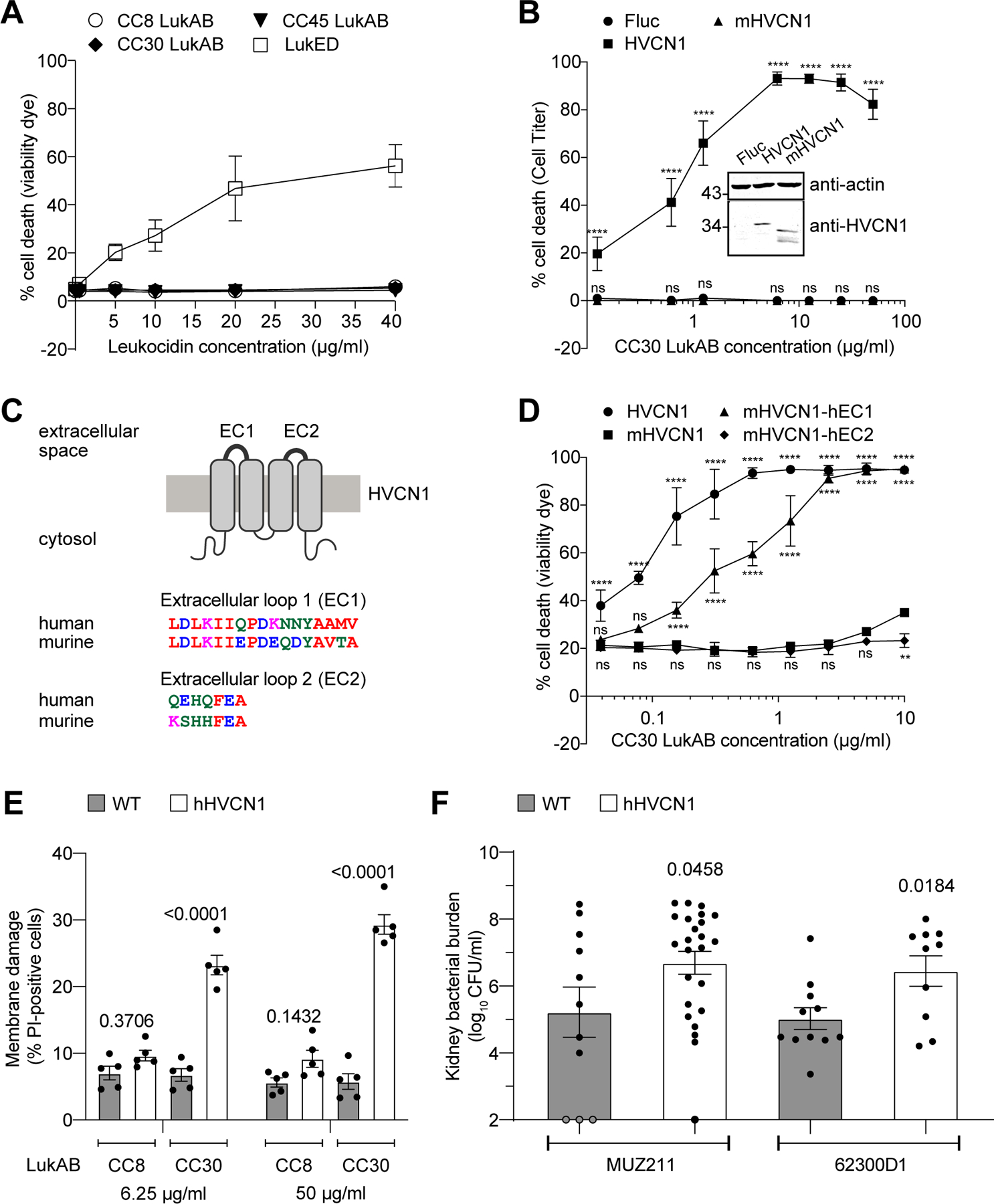
A: Intoxication of murine PECs with indicated concentrations of leukocidins. Membrane damage was detected using Fixable Viability Dye eFluor™ 450. Data are represented as the average of three independent experiments ± SEM B: (inset) Immunoblot of HVCN1 in CHO cells expressing firefly luciferase (Fluc), human (HVCN1) or murine (mHVCN1) HVCN1. Anti-actin immunoblot is shown above as a loading control. Representative images of four independent samples from one immunoblot are shown, see corresponding Source Data for full gel. Numbers on the left indicate migration of the corresponding molecular weight standards (in kDa). Target protein levels normalized by actin were obtained using ImageJ from four independent protein samples: HVCN1 = 0.310 ± 0.111, mHVCN1 = 0.333 ± 0.066 (mean ± SD), P = 0.742 as determined by unpaired t test. (main figure) Intoxication of Fluc, HVCN1, and mHVCN1 expressing CHO cells with indicated concentrations of CC30 LukAB. Cell viability was measured with Cell Titer. Data from three independent experiments are represented as mean values ±SD. Statistical significance was determined by two-way ANOVA (****, P ≤ 0.0001; ns, not significant). C: Schematic architecture of HVCN1 and the amino acid alignments of human and murine extracellular loops generated using Clustal Omega. D: Intoxication of Lenti-X 293T cells expressing C-terminal GFP-tagged human, murine, and chimeric HVCN1 proteins with indicated concentrations of CC30 LukAB. Membrane damage was detected using Fixable Viability Dye eFluor™ 450. Data from three independent experiments are represented as the mean values ±SD. Statistical significance was determined by two-way ANOVA (****, P ≤ 0.0001; **, P ≤ 0.01; ns, not significant). E: Intoxication of PECs from wild type (WT) and hHVCN1 mice with indicated LukAB. Membrane damage was detected using Propidium Iodide (PI) incorporation. Data from five mice per genotype over three independent experiments are represented as mean values ±SEM. Statistical significance was determined by two-way ANOVA, numbers indicate P values. F: CFUs in the kidneys of WT and homozygous hHVCN1 mice infected intravenously with MUZ211 (CFU obtained from 11 WT and 24 hHVCN1 mice) and 62300D1 (CFU obtained from 11 WT and 10 hHVCN1 mice). Data for each isolate are from mice infected over three independent experiments and is represented as mean values ±SEM. Statistical significance was determined by t-test (two-tailed), numbers indicate P values. Also refer to Extended Data Figures 5 and 7.
Human HVCN1 and its murine homolog (herein referred to as mHVCN1, also known as mVSOP) share 79.2% amino acid identity. To test whether the observed species specificity of LukAB could be explained by the interspecies differences in HVCN1, human and murine HVCN1-expressing CHO cells were exposed to CC30 LukAB. While both HVCN1 proteins were produced, only the human HVCN1 expressing cells were susceptible to LukAB (Figure 6B).
HVCN1 is comprised of 4 transmembrane segments with 2 extracellular loops (17 and 7 amino acid residues long, respectively) (Figure 6C). While human and murine HVCN1 share close to 80% overall sequence identity, the surface exposed regions are less than 60% identical between species (Figure 6C). To test if these differences affect LukAB targeting, we introduced each of the human extracellular loops into mHVCN1 and produced these chimeric proteins in 293T cells. The chimeric proteins were translationally fused to GFP via a C-terminal linker to ensure comparable protein levels, and LukAB-mediated cell death was assessed by flow cytometry. These experiments revealed that replacing the first, but not the second, extracellular loop of mHVCN1 with its human ortholog rendered the receptor compatible with CC30 LukAB-mediated killing (Figure 6D). Collectively, these data demonstrate that CC30 LukAB preferentially target human HVCN1, primarily via the first extracellular loop.
Humanizing HVCN1 renders murine leukocytes susceptible to CC30 LukAB
Based on the cytotoxicity data observed in 293T cells (Figure 6D), we hypothesized that humanizing the first extracellular loop of mHVCN1 in the mouse genome would result in a similar increase in susceptibility to the toxin. Using CRISPR-Cas9 gene editing39, we modified murine exon 4 to introduce the corresponding residues of the human HVCN1 extracellular loop one (Extended Data Figure 5). The humanized HVCN1 mice (hHVCN1) were generated in the C57BL/6J background and appeared to be normal in appearance, viability, and breeding ability.
To evaluate the susceptibility of hHVCN1 mice towards LukAB, we first exposed PECs from wild type and hHVCN1 mice to CC8 and CC30 LukAB. PECs from wild type mice were highly resistant to both toxins. In contrast, cells from the hHVCN1 mice were susceptible to CC30 LukAB-mediated membrane damage, while remaining resistant to CC8 LukAB (Figure 6E). Therefore, facilitating toxin-receptor interaction by humanizing the extracellular loop one of mHVCN1 allows for CC30 LukAB-mediated cytotoxicity.
hHVCN1 mice are more susceptible to CC30 S. aureus bloodstream infection
To rule out any unappreciated immunologic alterations in the hHVCN1 mice, we infected wild type and hHVCN1 mice with 1×107 colony forming units (CFU) of lukAB-deficient strain LAC13 and evaluated bacterial burdens 3 days post infection. No significant difference in bacterial burdens in the kidneys, liver, heart, spleen, and lungs was observed between the wild type and our gene-targeted animals (Extended Data Figure 5).
Next, wild type and hHVCN1 mice were infected intravenously with 5–10×107 CFU of CC30 S. aureus and bacterial burdens in organs examined 14 days post infection. To eliminate potential strain-specific effects, we tested two CC30 clinical isolates: a methicillin sensitive hospital-acquired bloodstream infection isolate MUZ211; and a community-acquired skin infection isolate 62300D140. Independent of the isolate, the hHVCN1 mice displayed increased bacterial burden in the kidneys as compared to wild type mice (Figure 6F). No significant difference was observed in the other tissues examined (Extended Data Figure 5). Thus, humanizing mHVCN1 increases the overall susceptibility of mice to renal infections with CC30 S. aureus.
DISCUSSION
Most studies characterizing LukAB and its role in S. aureus immune evasion have focused on the toxin variant produced by CC8 strains, although multiple other clonal complexes exist, including 11 major ones41. Here, we show that CC30 and CC45 LukAB, which differ only by four amino acids, have diverged significantly from the other LukAB proteins. We established a critical role for HVCN1 in the lytic activity of CC30/CC45 LukAB variants and determined that the first extracellular loop of HVCN1 was responsible for the human tropism exhibited by these toxins. Finally, we generated a humanized HVCN1 mouse to study CC30 LukAB, which revealed both increased susceptibility to CC30 LukAB cytotoxic activity in murine leukocytes ex vivo and decreased resistance to bloodstream infection with hospital- and community-acquired CC30 S. aureus in vivo.
Our bioinformatic and functional analyses of the LukAB-encoding alleles showed that while the overall toxin sequence and function are largely conserved, diverse clonal variants exist between clonal complexes but not within them. Along with our selection analyses, this pattern of diversity suggests both strong purifying selection for these toxins, but also potential inter-CC functional differences such as what we have uncovered herein. CC30 alone accounted for 21% of the MSSA strains causing disease worldwide between 1961 and 200441. Moreover, clinical studies have demonstrated that the CC30 lineage is associated with persistent bacteremia and increased risk for hematogenous complications (e.g., endocarditis, septic arthritis, vertebral osteomyelitis)42,43. Interestingly, many CC30 isolates harbor attenuating mutations in the global virulence regulator Agr (agrC), a stop codon in the alpha-toxin-encoding gene (hla), and lack the potent LukED toxin44. These alterations are believed to significantly reduce the cytotoxic potential of contemporary CC30 S. aureus45. It is tempting to speculate that the lack of alpha-toxin, LukED and reduced function of the Agr-master regulatory system could have provided a selective pressure for a LukAB variant that compensates for these toxins.
CC8 LukAB is highly cytotoxic towards human leukocytes but ~1,000-fold less potent towards murine immune cells12, a phenotype linked to the high binding affinity for human CD11b8,18. Identification of the human leukocidin receptors provides critical information that can be harnessed to generate transgenic mouse models where the human (or humanized) receptors are expressed in the appropriate leukocytes. Recently, the determinants of the human-specific CC8 LukAB-CD11b interaction were defined and used to generate a CD11b-humanized (hCD11b) mouse18. Interestingly, studies using these mice uncovered that during the initial phase of bloodstream infection, one to three days post-infection, LukAB contributes to USA300 bacterial burden only in the liver18. Using the hHVCN1 mice described here, we instead observed increased bacterial burden in the kidneys 14 days post-infection. This disparate tissue tropism could be due to differences in the activity of the LukAB toxins in vivo (e.g., CC8 LukAB vs CC30 LukAB) or to altered tropism by the S. aureus isolates (e.g., USA300 strain LAC vs. CC30 strains MUZ211 and 62300D1). Future studies utilizing congenic S. aureus strains are needed to clarify these possibilities. Ultimately, a combination of currently available humanized murine models (e.g., hCD11b18, hC5aR146 and the hHVCN1) might be needed to fully define the roles of LukAB and the other leukocidins in vivo.
In conclusion, the findings presented here emphasize the importance of considering the sequence diversity of S. aureus while attempting to characterize the role and function of virulence factors. Moreover, these data highlight that S. aureus lineages associated with human infections have evolved strategies to specifically target the human host. A better understanding of lineage-specific toxin activity and the interaction of S. aureus with the human host will aid in development of toxin inhibitors to prevent toxin-mediated immune depletion and combat infections caused by diverse S. aureus isolates.
METHODS
Ethics statement
Human blood samples were obtained as buffy coats from healthy, anonymous, consenting adult donors (New York Blood Center).
All experiments involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of NYU Langone Health and were performed according to guidelines from the National Institutes of Health (NIH), the Animal Welfare Act, and US Federal Law.
Bacterial strains
The following E. coli strains were used in cloning procedures: DH5a and Endura™ Competent Cells (Lucigen). E. coli IM30B47 was provided by Timothy J. Foster and used to passage plasmid DNA to enable direct transformation of CC30 S. aureus. E. coli T7 LysY/LacQ was used for Flag-tagged CD11b I domain expression. E. coli OverExpress C43(DE3) Chemically Competent Cells (Lucigen) were used for Strep-tagged HVCN1 expression. All E. coli strains were grown in Luria-Bertani (LB) broth.
S. aureus strains used in the study are listed in the Supplementary Table 4. For recombinant protein expression, S. aureus cultures were prepared in Tryptic Soy Broth (TSB). For PMN and THP1 infection studies, S. aureus strains were streaked to single colonies on tryptic soy agar (TSA) plates. Single colonies were inoculated in yeast-Casamino Acids (YC) broth for overnight culture and then subsequently subcultured 1:100 for 5 h in YC with 2% sodium pyruvate (Fisher Scientific) (YCP) at 37°C with shaking48. For in vivo infections, S. aureus strains were streaked to single colonies on tryptic soy agar (TSA) plates. Single colonies were inoculated in TSB broth for overnight culture and then subsequently subcultured 1:100 for 3 h in TSB at 37°C with shaking.
Oligonucleotides
All oligonucleotides used in the study are listed in Supplementary Table 5.
Antibodies
The following antibodies were used:
mouse anti-His tag (1:3000, CSI20563B, Cell Sciences), rabbit anti-HVCN1 (1:1000, OAPB01154, Aviva Systems Biology), rabbit anti-HVCN1 (1:200, AHC-001, Alomone Labs), rabbit anti-HVCN1 (1:1000, PA5–24964, Invitrogen, Thermo Fisher Scientific), mouse anti-β-Actin (1:1000, 8H10D10, Cell Signaling Technology), mouse PE/Cy7 anti-human CD19 (1:100, 302215, BioLegend), mouse FITC anti-human CD3 (1:100, 300406, BioLegend), mouse Alexa Fluor® 700 anti-human CD14 (1:100, 325614, BioLegend), mouse PE anti-human CD4 (1:100, 317410, BioLegend), mouse APC anti-human CD8a (1:50, 300912, BioLegend), mouse APC anti-human CD11b (1:100, 301310, BioLegend), mouse Alexa Fluor® 700 anti-mouse/human CD11b (1:300, 101222, BioLegend). Additionally, the following isotype control antibodies were used: PE/Cy7 Mouse IgG1, κ Isotype Ctrl Antibody (1:100, 400126, BioLegend), FITC Mouse IgG1, κ Isotype Ctrl Antibody (1:100, 400108, BioLegend), Alexa Fluor® 700 Mouse IgG1, κ Isotype Ctrl Antibody (1:100, 400143, BioLegend), PE Mouse IgG1, κ Isotype Ctrl Antibody (1:100, 400111, BioLegend), APC Mouse IgG1, κ Isotype Ctrl Antibody (1:100, 400120, BioLegend). The following secondary antibodies were used for immunoblotting: goat anti-Mouse IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 680 (1:25000, A-21057, Invitrogen), Goat anti-Rabbit IgG (H+L) Cross-Adsorbed Secondary Antibody, Alexa Fluor 680 (1:25000, A-21076, Invitrogen).
Anti-CC30 LukAB monoclonal antibody:
The anti-LukAB monoclonal antibodies were custom made at Envigo Inc. according to their approved standard operating procedures for mouse monoclonal hybridoma generation. Briefly, 5–100 μg of recombinant CC30 LukAB (rLukAB) were emulsified with Freud’s complete adjuvant for the primary immunization, followed by one boost of rLukAB emulsified with Freud’s incomplete adjuvant and 1–2 additional boosts of rLukAB emulsified with Titermax adjuvant (~ 1 month apart). The immunized mouse was sacrificed and splenocytes were removed and fused with NS01 myeloma cells to generate hybridomas. The monoclonal hybridoma cell lines were selected using enzyme-linked immunosorbent assay (ELISA). For ELISAs, Immulon 2HB flat bottom microtiter plates (Thermo Scientific) were coated with 1 μg/ml of antigen. The antigen-coated plates were blocked using 2% milk in saline containing 0.05% Tween-20 for 1 h at room temperature. 50 μl of serially diluted purified antibodies (2 μg/ml, 0.02 μg/ml, and 0.0002 μg/ml) or undiluted culture supernatants were added to the ELISA plate and incubated at room temperature for 1 h with rocking, followed by 3 washes of 100 μl/well PBST (1x PBS, 0.1% Tween-20, 0.01% NaN3). Then, 50 μl of goat anti-mouse HRP (1:1000; Bio-Rad) was added to each well and incubated at room temperature for 1 h with rocking, followed by 3 washes of 100 μl/well PBST. 50 μl of 3, 3’, 5, 5’- Tetramethylbenzidine (TMB) solution (Thermo Scientific) was added to each well and developed for 3 minutes, followed by addition of 50 μl of H2SO4. The plates were read at absorbance 450 nm on the Envision Multilabel Plate Reader (PerkinElmer). Anti-CC30 LukAB monoclonal antibody was used for immunoblotting at 1 μg/ml.
Bioinformatic analysis of LukAB diversity
LukA and LukB diversity screen:
Nucleotide sequences of lukA and lukB were used to query a nucleotide database of 4,187 of genomes using blastn. All genomes except 5 (C0673.fa, CO_98.fa, ST2282.fa, ST2283.fa, ST228.fa) had a hit to both lukA and lukB sequences (blastn, evalue cutoff=1×10−5). BLAST aligned portions were captured, and all identical nucleotide sequences were collapsed to a single representative sequence for further analysis using cd-hit. This reduced lukA to 94 and lukB to 83 representative sequences (see Supplementary Table 1).
Phylogenetic Analysis:
Gene alignments were done in MAFFT 7.271 (default settings) and optimized by aligning manually based on codons. Maximum likelihood phylogenies were constructed for lukA and lukB with RAxML v8.2.449 using the general time-reversible (GTR) substitution model50 accounting for among-site rate heterogeneity using the Γ distribution and four rate categories51 (GTRGAMMA model) with maximum parsimony random-addition starting trees. Node support was evaluated with 100 nonparametric bootstrap pseudoreplicates52.
Selection Analysis:
Sequences with mid-sequence STOP codons (n=188) were excluded from analysis. Sequences not evenly divisible by 3 were end-trimmed to form a complete codon set. Genes were translated and aligned at the amino acid level using the ClustalW algorithm with default settings as provided in MEGA7 version 7.0.2653, and this alignment was imposed on the nucleotide sequence. Whole-gene nonsynonymous (dN) and synonymous (dS) divergence were estimated for all pairs of sequences between each pair of clades using the between-group method of SNPGenie version 1.054 (https://github.com/chasewnelson/snpgenie). A Jukes-Cantor correction was used with the Nei-Gojobori (1986) method because synonymous p distances approached 0.555. Standard errors were determined using a bootstrap procedure with 10,000 replicates. Individual codons exhibiting potential positive selection were identified using the FUBAR method of HyPhy version 2.3.420171008beta(MP)56 (https://veg.github.io/hyphy-site/). All genes exhibited similar, significant levels of purifying selection (dN<dS; P<0.0001; Bonferroni correction). Two codons in lukA were identified as exhibiting positive selection with a posterior probability of >90%: lukA codons 50 and 148. All statistical operations were performed in R version 3.2.0.
Clade-defining sites:
Clade-defining sites were determined using EBT::aligned_fasta_group_diffs.pl (https://github.com/chasewnelson/ebt; accessed March 28, 2018). Specifically, sites were identified at which two or more clades differed in their major (i.e., consensus) nucleotides with frequencies of ≥75% in each clade, a criterion chosen to exclude singletons for clades with 2 members and doubletons for clades with 3 members. A clade was not considered to have a defining nucleotide at a site if its major nucleotide was present at a frequency <75% or the site contained ≤2 defined (i.e., non-gap, non-ambiguous) nucleotides. Among these sites, the nucleotide present in CC1/5/8 was unique for a nonsynonymous nucleotide difference for 17 sites in lukA and 8 sites in lukB. Sites undergoing episodic diversifying selection at internal branches were sought using the MEME approach in HyPhy57. MEME identified codon 37 in lukB (P=0.0020, LRT). No sites were identified in lukA.
Comparison of selected mature LukA and LukB:
Signal peptides and the location of cleavage sites in full-length LukA and LukB were predicted using SignalP 5.0 server58. Multiple protein alignments of mature (without signal peptide) LukA and LukB (listed in Supplementary Table 2) were performed using ClustalW alignment method algorithm with default settings as provided in MegAlign (DNAStar, Madison, WI). Further, MegAlign was used to generate phylogenetic trees and pairwise percent identity and divergence tables (presented in Extended Data Figure 2).
Engineering of mammalian cell lines
Tissue culture:
THP1 cells (ATCC TIB-202) and HL60 cells (ATCC CCL-240) were maintained in Roswell Park Memorial Institute medium 1640 (RPMI) medium (Cellgro) supplemented with 10% Foundation B™ Fetal Bovine Serum (FBS, 900–208, Gemini Bio-Products), penicillin (100 U/ml), and streptomycin (0.1 mg/ml) (1xP/S) at 37°C with 5% CO2. pLKO.1-blast transduced THP1 were maintained in full medium supplemented with 10 μg/ml blasticidin. LentiCRIPSRv2 transduced THP1 cells were additionally supplemented with 1.3 μg/ml puromycin.
HL60 cells were differentiated into polymorphonuclear (PMN)-HL60 cells by incubating with 1.5% (v/v) dimethyl sulfoxide (DMSO, Sigma Aldrich) for 72 h at an initial density of approximately 2.5 × 106 cells per ml. pLKO.1 puro transduced HL-60 cells were maintained in full medium supplemented with 2 μg/ml puromycin.
CHO-K1 Chinese hamster ovary cells (ATCC CCL-61) stably expressing the CD18 subunit (CD18 CHO-K1) (kindly provided by Radim Osicka59) were maintained in Ham’s F12 Nutrient Mixture medium with L-Glutamine (Gibco) supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (0.1 mg/ml) (1xP/S) and 300 μg/ml zeocin at 37°C with 5% CO2. Lentivirally transduced CHO-K1 cells were maintained in full medium supplemented with 5 μg/ml puromycin.
Lenti-X 293T cells (Takara Bio USA) were maintained in Dulbecco’s Modified Eagle’s Medium (DMEM) with L-Glutamine, 4.5 g/L Glucose and Sodium Pyruvate (Cellgro) supplemented with 10% FBS, penicillin (100 U/ml), and streptomycin (0.1 mg/ml).
Plasmids for protein expression in mammalian cells:
Plasmids used in this study are listed in the Supplementary Table 4. cDNA for human HVCN1 was obtained from the Ultimate ORF Clones (96-well plate) collection (Thermo Fisher Scientific) as a Gateway-compatible pENTR221 vector. cDNA for murine Hvcn1 (Clone ID: 4913027) was purchased from Dharmacon (Horizon Inspired Cell Technologies) in pCMV-SPORT6 vector and amplified by PCR with primers VJT1816 and VJT1817 containing Kozak sequence and attB sites (see Supplementary Table 5). PCR product was purified with the QIAquick PCR Purification Kit (Qiagen) and then recombined into a pDONR221 vector (Invitrogen, Thermo Fisher Scientific) using BP Clonase II Enzyme mix (Invitrogen, Thermo Fisher Scientific). BP reactions were transformed into E. coli Endura™ Competent Cells (Lucigen), and inserts verified by sequencing. pENTR221 clones were further recombined into pLenti CMV Puro DEST (w118–1) for generation of stable cell lines (a gift from Eric Campeau (Addgene plasmid #17452)60 using LR Clonase II Enzyme mix (Invitrogen, Thermo Fisher Scientific). LR reactions were transformed into E. coli Endura™ Competent Cells (Lucigen) and verified by sequencing. pLenti CMV Puro Fluc, encoding firefly luciferase and used as a control, was generated in a similar manner using pENTR221 Fluc (a gift of John Schoggins (UTSW)).
cDNA for chimeric Hvcn1 were codon optimized for synthesis and expression in human cells using Codon Optimization Tool (Integrated DNA Technologies) and synthesized as gBlocks Gene Fragments (Integrated DNA Technologies). Wild type human HVCN1 in pCMV6-AC-GFP, a mammalian vector with C-terminal GFP tag, was obtained from OriGene Technologies. Murine Hvcn1, and chimeric Hvcn1 genes were PCR amplified to add AsiSI and MluI restriction sites and cloned into pCMV6-AC-GFP using standard cloning procedures.
Generation of lentiviral shRNA expression vectors:
For shRNA-mediated knockdown of CD11b (ITGAM) and CD18 (ITGB2) in HL60 cells, pLKO.1 puro containing shRNA targeting CD18 (G8) and CD11b (C5) were used8. Knockdown of both ITGAM and ITGB2 in HL60 cells has been previously confirmed by flow cytometry8.
To allow compatibility with pLentiCRISPRv2 (with puromycin resistance) in THP1 cells, shRNA-mediated knockdown of ITGAM was achieved by using pLKO.1-blast encoding a previously validated ITGAM-targeting shRNA (C5, target sequence CGCAATGACCTTCCAAGAGAA, http://portals.broadinstitute.org/gpp/public/trans/details?transName=NM_000632.3). As a control, pLKO.1-blast-SCRAMBLE (a gift from Keith Mostov; Addgene plasmid # 26701) was used.
Generation of lentiviral pseudoparticles:
Lentiviral pseudoparticles were generated using Lenti-X 293T cells as previously described with XtremeGene 9 (Roche) as a transfection reagent61.
Generation of CD18 CHO-K1 stably expressing Fluc, HVCN1 and mHVCN1:
Lentiviral transduction was performed as previously described61. Briefly, CD18 CHO-K1 cells were seeded in 24-well tissue culture plates at a density of 7×104 cells per well and transduced the following day with 100 μl lentiviral pseudoparticles via spinoculation at 1000 × g for 45 min in medium containing 3% FBS, 20 mM HEPES, and 4 μg/ml polybrene. 6 h after spinoculation, pseudoparticle-containing media was removed and replaced with full cell culture medium. To generate stable cell lines, cells were selected with 5 μg/ml puromycin for 7 days 48h after transduction. HVCN1 and mHVCN1 protein expression was evaluated by Western blot. Cells were incubated in HEPES lysis buffer (1% Triton X-100, 20 mM HEPES, 137 mM NaCl, 2 mM EDTA, supplemented with 1x Halt™ Protease Inhibitor Cocktail (Thermo Scientific)) for 15 min on ice, homogenized by passing the sample 10 times through a 21G needle, and centrifuged at 14,000 rpm and 4°C for 15 min to separate from cellular debris. Total protein concentration was measured using Pierce™ BCA Protein Assay Kit (Thermo Scientific). 20 μg of total protein lysate was loaded per well with 1x Laemmli buffer. Proteins were separated by SDS-PAGE and blotted onto nitrocellulose membranes. After blocking in 5% milk in PBS-T for 1 h at room temperature, membranes were incubated with primary antibodies (anti-HVCN1 (1:1000, OAPB01154, Aviva Systems Biology), and anti-β-Actin (1:1000, 8H10D10, Cell Signaling Technology)) diluted in 5% milk in PBS-T for 16 h at 4°C. Membranes were then washed three times with PBS-T for 10 min, then incubated with secondary Alexa Fluor 680 conjugated antibodies diluted 1:25,000 in 5% milk in PBS-T for 1 h at room temperature. Membranes were imaged using Odyssey Infrared Imaging System (LI-COR). Images were analyzed and protein levels quantified using ImageJ 1.51s.
Generation of THP1 shRNA cell lines:
THP1 cell lines expressing scramble shRNA or ITGAM (CD11b)-targeting shRNA (C5) were generated as described previously8. Knockdown of CD11b by shRNA in THP1 cells was verified as described previously8. Briefly, cells were stained with anti–CD11b–APC (clone ICRF44, 301310, Biolegend) or APC-conjugated isotype control (clone MOPC-21, 400120, Biolegend) antibodies for 30 min on ice, then washed twice with 1× PBS+2% FBS + 0.05% sodium azide (FACS buffer) and resuspended in FACS buffer. Flow cytometry data were acquired using CytoFLEX Flow Cytometer (Beckman Coulter). Data were analyzed using FlowJo (version 10.7.1) software (Treestar).
LentiCRISPRv2 GeCKO transduction of THP1 cells:
ITGAM shRNA-Blast THP1 cells were used as the recipient for LentiCRISPRv2 GeCKO lentiviral transduction. Overall, our approach followed previously published protocols26,62. Briefly, two pooled human GeCKO v2 half-libraries (A and B; from Adgene) were independently electroporated into E. coli Endura™ Competent Cells (Lucigen), transformant bacterial cells were extracted as culture lawns, harvested, and processed using QIAGEN Plasmid Maxi Kit. Viral particles were generated as described above and used to transduce THP1 ITGAM shRNA-blast cells. Transduced cells were selected with 1.3 μg/ml puromycin.
Generation of THP1 cell lines expressing individual sgRNAs:
Following the GeCKO screen, THP1 cell lines expressing individual sgRNAs targeting most highly enriched hits were generated according to the previously published procedures63. For HVCN1, primer pairs VJT1759/VJT1760 and VJT1761/VJT1762 were used, for ASGR1 – VJT1767/VJT1768, and for OGT – VJT1769/VJT1770. Additionally, primer pair VJT1775/VJT1776 was used to clone non-targeting sgRNA (HGLibA_64423).
Nuclease assay to confirm HVCN1 targeting in THP1 cells:
To confirm HVCN1 targeting by the sgRNA, we performed the nuclease assay. Genomic DNA was extracted from 100’000 CD11b-depleted (ITGAM shRNA) THP1 cells transduced with non-targeting sgRNA or HVCN1-targeting sgRNA in 100 μL QuickExtract™ DNA Extraction Solution (QE09050, Lucigen) heated at 65 °C for 15 min, followed by 68 °C for 15 min and 98 °C for 10 min. Target region within HVCN1 was PCR-amplified using primers VJT2845 and VJT2846, and purified using MinElute PCR Purification Kit (Qiagen). 200ng of PCR products were annealed in the thermocycler in presence of 1xNEBuffer 2 (NEB) under the following conditions: denatured at 95 °C for 5 min and annealed at −2 °C per sec temperature ramp to 85 °C, followed by a −0.1 °C per sec ramp to 25 °C. T7 Endonuclease I (M0302S, NEB) was added to the annealed products. Reaction was incubated at 37 °C for 15 min and terminated with addition of EDTA, as per manufacturer’s instructions. DNA products from the assay were electrophoresed and imaged.
Western blot to confirm HVCN1 depletion in THP1 cells:
HVCN1 protein levels on CD11b-depleted THP1 cells transduced with non-targeting sgRNA or HVCN1-targeting sgRNA were evaluated by Western blot. Cells were incubated in HEPES lysis buffer (1% Triton X-100, 20 mM HEPES, 137 mM NaCl, 2 mM EDTA, supplemented with 1x Halt™ Protease Inhibitor Cocktail (Thermo Scientific)) for 15 min on ice, homogenized by passing the sample 10 times through a 21G needle, and centrifuged at 14,000 rpm and 4°C for 15 min to separate from cellular debris. Total protein concentration was measured using Pierce™ BCA Protein Assay Kit (Thermo Scientific). 150 μg of total protein lysate was loaded per well with 1x Laemmli buffer. Proteins were separated by SDS-PAGE and blotted onto nitrocellulose membranes. After blocking in 5% milk in PBS-T for 1 h at room temperature, membranes were incubated with primary antibodies ((anti-HVCN1 (1:200, AHC-001, Alomone Labs), and anti-β-Actin (1:1000, 715 8H10D10, Cell Signaling Technology)) diluted in 5% milk in PBS-T for 16 h at 4°C. Membranes were then washed three times with PBS-T for 10 min, then incubated with secondary Alexa Fluor 680 conjugated antibodies diluted 1:25,000 in 5% milk in PBS-T for 1 h at room temperature. Membranes were imaged using Odyssey Infrared Imaging System (LI-COR).
Ex vivo toxin exposure of mammalian cells
Generation of lukAB expression plasmids:
lukAB loci, encoding mature LukA and full length LukB, were PCR amplified using genomic DNA of the following S. aureus strains: MW2 (CC1) (primers VJT544 and VJT543), Mu50 (CC5) (primers VJT544 and VJT543), Clinical isolate # 52 (CC8) (primers VJT544 and VJT543), MRSA252 (CC30) (primers VJT1331 and VJT628), Clinical isolate # 33 (CC45) (primers VJT1331 and VJT1399). To obtain CC398 lukAB locus, the BVED 022 strain (CC398) was collected under the IRB protocol described before40,64 (primers VJT1331 and VJT1415). The S. argenteus (CC75) lukAB locus was synthesized by Genewiz using MSHR1132 genome as a template. The S. schweitzeri lukAB locus was synthesized by Genewiz using FSA084 genome as a template. lukAB loci were then cloned into the pOS1 plasmid containing the PlukAB-lukAs.s.-6xHis13. All plasmids were passaged through S. aureus strain RN4220 and then transformed into the toxinless S. aureus strain Newman ΔΔΔΔ for protein expression (see below).
For LukED, HlgAB, LukSF purification plasmids pOS1_PlukAB-lukAs.s.-6xHis-LukD and pOS1_PlukAB-lukAs.s.-6xHis-LukE, pOS1_PlukAB-lukAs.s.-6xHis-HlgA and pOS1_PlukAB-lukAs.s.-6xHis-HlgB, and pOS1_PlukAB-lukAs.s.-6xHis-LukS and pOS1_PlukAB-lukAs.s.-6xHis-LukF were used, respectively65.
Toxin purification:
LukAB, LukED, HlgAB, and LukSF toxins were purified as described previously15,65. All purified toxins were dialyzed into the protein storage buffer (10% glycerol/ 1xTBS). Buffer exchanged toxins were filtered through 0.2 μm filters to remove any debris and protein aggregates. The concentration of all purified toxins was measured using NanoDrop spectrophotometer (Thermo Scientific).
Isolation of primary human PMNs and PBMCs:
Primary human PMNs were isolated using a Ficoll-Paque method as described previously65. To isolate PBMCs, the fuzzy white layer resting on the Ficoll was collected and transferred to a new tube. RPMI medium was then added to bring the volume to 50 ml and tubes were centrifuged at 1500 rpm for 15 min. Then supernatant was removed and cells resuspended in 20–30 ml RPMI and centrifuged at 1500 rpm for 10 min. Finally, the supernatant was removed and cells resuspended in 20 ml RPMI/ 0.1% HSA/ 10 mM HEPES.
Isolation of human monocytes and generation of monocyte-derived macrophages and dendritic cells:
Fresh human PBMCs were isolated as described above. Cells were washed twice in 1xPBS, counted and resuspended in 950 ml ice cold MACS buffer (PBS/ 2 mM EDTA/ 0.5% BSA) per 100 × 106 cells. Next, 50 μl CD14 MicroBeads (Miltenyi Biotec) per 100 × 106 PBMCs were added, and cells were incubated for 20 min at 4°C. Following incubation, cells were washed in 25 ml MACS buffer, resuspended in 10 ml MACS buffer, passed through a 0.4 μm cell strainer, and divided into two 15-ml tubes per donor. Magnetic separation was then performed using the autoMACS Pro Separator following manufacturer’s instructions (Posselds program). CD14-positive selection cells (monocytes) were kept on ice, centrifuged at 1200 rpm for 5 min at 4°C to remove MACS buffer, resuspended in MDDC medium (RPMI/ 10% FBS/ 10 mM HEPES/ 1xP/S) at a concentration of ~1 × 106 cells / ml, and divided into 10 cm dishes (10–20 ml per dish). CD14-negative cells were kept on ice, centrifuged at 1200 rpm for 5 min at 4°C to remove MACS buffer, resuspended in PBMC freezing medium (RPMI/ 40% FBS/ 10% DMSO) at about 50 × 106 cells / ml, frozen overnight at −80°C, and stored in the gas phase in the liquid nitrogen tank until further use. To differentiate monocytes into macrophages, 0.05 μg/ml GM-CSF was added. To obtain monocyte-derived dendritic cells, IL-4 (2.82 × 102 IU/ml) and GM-CSF (1.08 × 102 IU/ml) were added. Plates were kept in the tissue culture incubator at 37°C with 5% CO2 and treatment was repeated 48 h later. 48 h after the second treatment, media containing loosely attached cells was collected. Remaining cells were detached by gently washing with cold 1xPBS twice, incubating for 5 min at 4°C, and washing twice with RPMI/ 10% FBS/ 10 mM HEPES/ 1xP/S.
Toxin exposure of human PMNs, monocytes, monocyte-derived macrophages, dendritic cells, THP1, and PMN-HL60 cells:
Cells of interest were counted and resuspended at 1.11×105 cells/ml. 1×105 cells per well in a total volume of 100 μl were incubated with the indicated concentrations of purified recombinant LukAB or HlgAB in flat-bottom tissue culture treated 96-well plates. PMNs, monocytes, monocyte-derived macrophages, dendritic cells, and PMN-HL60 cells were incubated with toxin for 1 h at 37°C, 5% CO2. THP1 cells were incubated for 2 h at 37°C, 5% CO2. Cell viability was measured with the metabolic dye Cell Titer by adding 10 μl of the CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS, Promega) to the wells, and incubating the cells for an additional 1 h at 37°C, 5% CO2 (1.5 h for THP1 cells). Absorbance at 492 nm was measured using EnVision Multimode Plate Reader (PerkinElmer).
Selection of CC30 LukAB resistant LentiCRISPRv2 THP1 cells:
LentiCRISPRv2 GeCKO transduced THP1 cells were exposed to 0.63 μg/ml CC30 LukAB in two sequential rounds. After the first round of toxin exposure, cells were allowed to recover for 10 days. After the second round, surviving cells were expanded, genomic DNA was extracted using a DNA Isolation Kit for Cells and Tissues (Roche), and sgRNA were amplified by nested PCR and sequenced as described previously26,62. As a control, LentiCRISPRv2 GeCKO transduced THP1 cells that were not exposed to LukAB were also kept in culture before isolating the DNA. Briefly, in the first round of PCR, primers VJT1627 and VJT1627 were used to anneal upstream and downstream of the sgRNA sequence. PCR product was then subjected to the second round of amplification with universal reverse primer VJT1667 in combination with forward primers VJT1663, VJT1664, VJT1665, and VJT1666 to add barcodes and stagger sequences for Illumina sequencing. Eight independent reactions were performed per sample to maintain diversity and prevent PCR bias. The PCR products containing sgRNAs were pooled and sequenced by NextSeq HighOutput (150 cycles) (Illumina). Reads for each sgRNA were normalized to counts per million total reads. Enrichment of each individual sgRNA was calculated compared to the unselected THP1 cells and expressed as fold enrichment. Data for sgRNAs enriched > 100-fold were plotted as the number of enriched sgRNAs per gene vs. average fold enrichment per gene.
Toxin exposure of CHO-K1 cells:
For toxin exposure, CHO-K1 cells generated as described above were detached from tissue culture treated plates using 0.05% Trypsin/ 0.53mM EDTA in HBSS (Corning, Fisher Scientific), resuspended in full medium, counted and resuspended at 1.11×105 cells/ml. 1×105 cells per well in a total volume of 100 μl were incubated with the indicated concentrations of purified recombinant LukAB in flat-bottom tissue culture treated 96-well plates for 1 h at 37°C, 5% CO2. Cell viability was measured with the metabolic dye Cell Titer by adding 10 μl of the CellTiter 96® AQueous One Solution Cell Proliferation Assay (MTS, Promega) to the wells, and incubating the cells for additional 1 h at 37°C, 5% CO2 (1.5 h for THP1 cells). Absorbance at 492 nm was measured using EnVision Multimode Plate Reader (PerkinElmer).
Isolation and toxin exposure of human B cells:
Cryopreserved human CD14-monocyte depleted PBMCs were thawed at 37°C and resuspended in 10 ml of RPMI/ 10% FBS. Cells were then treated with Benzonase® Nuclease (Millipore), incubated at room temperature for 10 min and passed through a 0.4 μm cell strainer into a fresh tube. Cells were centrifuged at 1500 rpm for 5 min and B lymphocytes isolated using the EasySep™ Human B Cell Isolation Kit (StemCell Technologies) following manufacturer’s instructions. 2×105 cells per well were intoxicated for 3 h at 37°C, 5% CO2 with the indicated concentrations of purified recombinant LukAB in tissue culture treated 96-well V-bottom plates. Cells were then washed twice in 100 μl 1xPBS and stained for 20 min on ice with the following antibodies: PE/Cy7 anti-human CD19 (1:100), APC anti-human CD11b (1:100), FITC anti-human CD3 (1:100), and Alexa Fluor® 700 anti-human CD14 (1:100) in 25 μl 1xPBS. Untreated cells were also stained with the corresponding isotype control antibodies. Following immunostaining, cells were washed twice in 100 μl 1xPBS, and stained with eBioscience™ Fixable Viability Dye eFluor™ 450 (Invitrogen™) at 1:1500 in 1xPBS. Finally, cells were washed twice in 100 μl 1xPBS and resuspended in 50 μl FACS/ Fix buffer (1x PBS/ 2% FBS / 2% PFA/ 0.05% (w/v) sodium azide). Flow cytometry data were acquired using CytoFLEX Flow Cytometer (Beckman Coulter). Data were analyzed using FlowJo (version 10.7.1) software (Treestar). Purity of 75–95% CD19-positive cells was achieved. B cell membrane damage was determined as percentage of PE/Cy7 (CD19)-positive cells stained with the eFluor™ 450 viability dye.
Isolation and toxin exposure of human T cells:
Cryopreserved human PBMCs were thawed at 37°C and resuspended in 10 ml of RPMI/ 10% FBS. CD4 or CD8 positive T lymphocytes were isolated using the EasySep™ Human CD4+ T Cell Isolation Kit (StemCell Technologies) or EasySep™ Human CD8+ T Cell Isolation Kit (StemCell Technologies), respectively, following manufacturer’s instructions. 2×105 cells per well were intoxicated for 3 h at 37°C, 5% CO2 with the indicated concentrations of purified recombinant LukAB in tissue culture treated 96-well V-bottom plates. Cells were then washed twice in 100 μl 1xPBS and stained for 20 min on ice with the following antibodies: PE anti-human CD4 (1:100), APC anti-human CD8a (1:50), and Alexa Fluor® 700 anti-mouse/human CD11b (1:300) in 25 μl 1xPBS. Untreated cells were also stained with the corresponding isotype control antibodies. Following immunostaining, cells were washed twice in 100 μl 1xPBS, and stained with eBioscience™ Fixable Viability Dye eFluor™ 450 (Invitrogen™) at 1:1500 in 1xPBS. Finally, cells were washed twice in 100 μl 1xPBS and resuspended in 50 μl FACS/ Fix buffer (1x PBS/ 2% FBS / 2% PFA/ 0.05% (w/v) sodium azide). Flow cytometry data were acquired using CytoFLEX Flow Cytometer (Beckman Coulter). Data were analyzed using FlowJo (version 10.7.1) software (Treestar). Purity of >93% and 75–95% was achieved of PE (CD4)-positive and APC (CD8)-positive T cells, respectively. Cell membrane damage was determined as percentage of CD4 or CD8 positive cells stained with the eFluor™ 450 viability dye.
Isolation of murine peritoneal exudate cells:
Eight- to 10-week-old WT or hHVCN1 C57BL/6 mice were injected intraperitoneally with 1 × 108 CFU of heat-killed S. aureus strain Newman 24 and 48 hours before harvest. Mice were euthanized with CO2, sprayed with 70% ethanol and mounted on the Styrofoam block on its back. Murine peritoneal exudate cells were collected as follows. 5 ml of RPMI/ 10% FBS was injected into the peritoneal cavity using a BD Angiocath I.V. catheter (18G 1.88IN, 381147) and the peritoneum was massaged to dislodge the cells. A maximum possible volume of cell-containing fluid was then collected. Cell were collected by centrifugation at 1500 rpm for 5 min, resuspended in 5–10 ml of RPMI/ 10% FBS and counted.
Ex vivo toxin exposure of freshly isolated murine cells analyzed by flow cytometry:
Approximately 2×105 cells isolated as described above were added per well to tissue culture treated 96-well V-bottom plates and incubated with the indicated concentration of purified recombinant CC8, CC30, or CC45 LukAB, or LukED. Cells were intoxicated for 2 h at 37°C, 5% CO2. To measure toxin-induced membrane damage, cells were washed twice in 100 μl 1xPBS, and stained with eBioscience™ Fixable Viability Dye eFluor™ 450 (Invitrogen™) at 1:1500 in 1xPBS for 20 min on ice. Finally, cells were washed twice in 100 μl 1xPBS and resuspended in 50 μl FACS/ Fix buffer (1x PBS/ 2% FBS / 2% PFA/ 0.05% (w/v) sodium azide). Flow cytometry data were acquired using CytoFLEX Flow Cytometer (Beckman Coulter). Data were analyzed using FlowJo (version 10.7.1) software (Treestar) and presented as percentage of cells stained with the eFluor™ 450 viability dye.
Ex vivo toxin exposure of freshly isolated murine cells analyzed by microscopy:
Approximately 1.5×105 cells isolated as described above were added per well to tissue culture treated 96-well black wall clear bottom plates and incubated with the indicated concentration of purified recombinant CC8 or CC30 LukAB. Cells were intoxicated for 3 h at 37°C, 5% CO2 followed by staining with Propidium Iodide Solution (1:500; G Biosciences, 786–1272) and NucBlue™ Live ReadyProbes™ Reagent (Hoechst 33342) (1:8; Thermo Fisher Scientific, R37605) for 20 min at room temperature. Stained cells were imaged using CellInsight CX7 High-Content Screening (HCS) Platform (Thermofisher) with the 4x objective acquiring 9 fields per well to cover the entire 96-well. Images were analyzed using HCS Studio software and toxin-induced membrane damage was quantified as a percentage of propidium iodide positive cells.
LukAB cell binding and competition assays:
For binding assays, 1×105 CD18 CHO-K1 cells59 expressing Fluc, HVCN1, or mHVCN1 were incubated on ice for 10 min with a titration of biotinylated CC30 and CC45 LukAB in a total volume of 100 μl of ice-cold full media. Following incubation, cells were washed twice with cold 1xPBS and stained with PerCP/Cy5.5 Streptavidin (405214, BioLegend) (diluted 1:200 in 1xPBS) on ice for 20 min. Cells were then washed twice with cold 1xPBS and resuspended in 50 μl FACS/ Fix buffer (1xPBS/ 2% FBS / 2% PFA/ 0.05% (w/v) sodium azide). Cell-bound toxins were detected by flow cytometry (CytoFLEX Flow Cytometer (Beckman Coulter)), data are shown as percentage cells stained with PerCP/Cy5.5 (streptavidin). For competition assays, 1×105 CD18 CHO-K1 cells expressing Fluc, HVCN1, or mHVCN1 were incubated on ice for 10 min with 3 μg/ml biotinylated CC30 or CC45 LukAB and a titration of unlabeled CC30 or CC45 LukAB in a total volume of 100 μl of ice-cold full media. LukAB binding was measured as described above.
Transient transfection and intoxication of Lenti-X 293T cells:
Lenti-X 293T cells were seeded in 6-well tissue culture plates at a density of 4.8×105 cells per well and transfected the following day with 400 ng pCMV6-AC-GFP plasmids encoding FLUC (Fluc), HVCN1 (human, HVCN1), Hvcn1 (murine, mHVCN1) or chimeric genes. For each transfection, 2 μl XtremeGene 9 (Roche) was combined with 400 ng total DNA in 50 μl Optimem (Gibco) and added to cells in 1 ml DMEM/ 3% FBS. Transfections were carried out for 6 h followed by a media change to 2 ml DMEM/ 10% FBS per well. Cells were harvested at 48 h post-transfection with Accumax (Innovative Cell Technologies) and counted using Countess™ II Automated Cell Counter (Thermo Fisher). Cells were intoxicated for 30 min with the indicated concentration of purified recombinant LukAB toxins in tissue culture treated 96-well V-bottom plates. Cells were then washed twice in 100 μl 1xPBS and stained for 20 min with the eBioscience™ Fixable Viability Dye eFluor™ 450 at 1:5000 in 1xPBS on ice. Finally, cells were washed twice with 100 μl 1xPBS and resuspended in 60 μl FACS/ Fix buffer (1x PBS/ 2% FBS / 2% PFA/ 0.05% (w/v) sodium azide). To ensure equal levels of protein expression between samples, during flow cytometry analysis cells were first gated on populations with equal GFP fluorescence intensity. Cell death was then determined as a percentage of these cells stained with the eFluor™ 450 viability dye.
Ex vivo S. aureus infection
Generation of ΔlukAB CC30 S. aureus strain:
An isogenic mutant lacking lukAB was constructed using pIMAY plasmid as described previously66. Briefly, 500 bp sequences flanking the lukAB locus were PCR amplified using VJT1878 and VJT1879 primers for the upstream fragment and VJT1880 and VJT1881 primers for the downstream fragment from MRSA252 genomic DNA. The gene encoding the kanamycin resistance marker aphA-3 was PCR amplified from the pBTK plasmid67 using VJT1882 and VJT1883 primers containing sequences overlapping with regions upstream and downstream of the CC30 lukAB locus, respectively. The PCR amplicons were then assembled using Splicing by Overlap Extension (SOE) PCR, digested with KpnI and SacI, ligated into previously digested and dephosphorylated pIMAY vector, and transformed into DH5alpha E. coli cells following standard cloning procedures. Correct insert was verified by sequencing, and plasmids transformed into CC30 S. aureus-compatible IM30B cells47. Deletion of the lukAB locus was achieved by allelic replacement as described previously66. Mutagenesis was confirmed by PCR using primers VJT1878 and VJT535 for lukAB (expected product size 1.5 kb) and primers VJT599 and VJT600 for hlgA as a positive control (expected product size 1 kb). Additionally, proteins secreted by wild type and isogenic mutant lacking lukAB following 5 h subculture in YCP medium were precipitated using trichloroacetic acid (TCA) as previously described13. Protein pellet was resuspended in 8M urea, incubated for 30 min at room temperature, mixed 1:1 with 2x TCA-SDS buffer (4X SDS-Laemmli buffer diluted 1:1 with 0.5 M Tris-HCl buffer pH 8.0 + 4% SDS), boiled for 10 min at 95°C and finally immunoblotted with anti-CC30 LukAB antibody.
Ex vivo human PMN and THP1 infection:
Killing of PMNs and THP1 cells by extracellular CC30 S. aureus was determined as follows. Briefly, the day before infection single colonies were inoculated from fresh tryptic soy agar (TSA) plates and grown overnight in YC media at 37°C with shaking. The next day, bacterial cultures were diluted 1:100 in YCP media and incubated for 5 h at 37°C with shaking. Following the subculture, bacteria were pelleted, washed once in 1xPBS, resuspended in 1xPBS and optical densities measured using a spectrophotometer (Genesys 20; Thermo Scientific). Bacteria were then distributed on flat-bottom 96-well tissue culture treated plates to achieve desired multiplicity of infection (MOI). PMNs or THP1 cells were added at 1×105 cells/well in 90 μl RPMI/ 0.1% HSA/ 10mM HEPES and incubated for 2 h at 37°C, 5% CO2. Cell lysis was evaluated using CytoTox-ONE™ Homogeneous Membrane Integrity Assay (Promega) according to the manufacturer’s protocol. MOIs were confirmed by serially diluting the input cultures and counting CFU on TSA plates.
Cell killing by opsonized CC30 S. aureus was measured as described previously6,13. Briefly, bacteria were cultured as described above and opsonized by resuspending in 20% normal human serum, prepared as described68 and incubated for 30 min at 37°C with shaking. THP1 cells were plated at 2×105 cells/well and infected with bacteria at MOI of 100. To promote phagocytosis, infections were synchronized by centrifuging the plates at 1500 rpm for 5 min, before incubating for 2 h at 37°C, 5% CO2. Cell lysis was evaluated using CytoTox-ONE™ Homogeneous Membrane Integrity Assay (Promega). MOIs were confirmed by serially diluting the input cultures and counting CFU on TSA plates.
LukAB interaction with the CD11b I domain in vitro
CD11b I domain purification:
Human Flag and 6xHis tagged CD11b I domain was purified as described previously8.
Protein biotinylation:
Proteins were biotinylated using EZ-Link™ NHS-PEG4-Biotin, No-Weigh™ Format (Thermo Scientific) according to the manufacturer’s instructions. Briefly, proteins were purified as described above and dialyzed overnight in 1xPBS. EZ-Link™ NHS-PEG4-Biotin reagent was added to the protein at a 1:1 molar ratio and incubated for 30 min at room temperature. Unreacted biotin reagent was removed by dialyzing overnight in 1xPBS. Labeled protein was then filter sterilized, aliquoted and stored at −80°C.
ELISA:
ELISA assay to measure CD11b I domain-LukAB interaction was performed as described recently18. As a loading control, plates were coated with a series of 1:2 dilutions of CC8 and CC30 LukAB starting at 1 μg/ml.
Bio-layer interferometry:
Kd values for CC8 and CC30 LukAB binding to CD11b I domain were determined by bio-layer interferometry using an Octet RED96 System (Pall ForteBio). Biotinylated CD11b I domain (10 μg/ml) was loaded onto streptavidin coated biosensors in kinetics buffer (1x PBS/ 1.5% Bovine serum albumin (BSA)/ 0.3% Tween-20) for 180 seconds. Association of CC8 and CC30 LukAB at 3 concentrations (125nM, 62.5nM, 31.3nM for CC8 LukAB, and 400nM, 200nM, 100nM for CC30 LukAB) was measured for 180 sec in kinetics buffer, followed by dissociation measurement for 180 sec in kinetics buffer. Experiments were performed at 30°C with shaking at 1000 rpm. To obtain Kd values, the best global fits of the experimental data were generated using a 1:1 binding model.
LukAB interaction with recombinant HVCN1-Strep in vitro
HVCN1-Strep purification:
Human HVCN1 gene (NM_032369) with a short linker and C-terminal Strep-tag (SA-WSHPQFEK) was codon optimized for expression in E. coli using publicly available gene optimization algorithm GeneArt (Thermo Fisher) and synthesized as gBlock Gene Fragment (Integrated DNA Technologies). The gene was then cloned into pET24a (+) vector using NdeI and BamHI restriction sites. The plasmid was transformed into OverExpress C43(DE3) chemically competent cells (Lucigen) for expression.
Overnight cultures of transformed C43 cells were subcultured 1:50 in LB supplemented with kanamycin (50 μg/ml) at 37°C shaking at 200 rpm and induced with 1mM IPTG at OD600~0.6–0.8. After induction, temperature was decreased to 30°C, and cells were grown for an additional 6 h. Cultures were pelleted at 5000 g, 15 min, 4°C. Cell pellets were resuspended in 50 mM Tris pH 8, 300 mM NaCl, 10% glycerol and frozen until lysis. Prior to lysis, cells were supplemented with Pierce Protease Inhibitor Mini Tablets (A32955). Cells were lysed by 4 passes through Emulsiflex-C3 (Avestin). Following lysis, cell debris was pelleted by centrifugation (15000g, 30 min, 4°C). Cleared lysate was ultracentrifuged (37000 rpm, 1 h 45 min, 4°C) to pellet the membrane fraction. Membrane was resuspended in 53 mM Tris pH 8, 320 mM NaCl, 10.6% glycerol, 20 mM FC-12 (Anatrace) and allowed to solubilize overnight by rocking at 4°C. The following day, solubilized membrane fraction was ultracentrifuged (37000 rpm, 1 h 30 min, 4°C) and the soluble fraction was incubated with StrepTactin XT Superflow High Capacity resin (IBA Lifesciences) by rocking for 1 h at 4°C. The resin was washed with wash buffer (150 mM Tris pH 8, 150 mM NaCl, 1 mM EDTA, 3 mM FC-12), and HVCN1-Strep was eluted with wash buffer supplemented with 50 mM biotin. All purification steps were performed at 4°C. FC-12 detergent was exchanged for amphipol by incubation of purified HVCN1 with an excess of amphipol (A8–35, Anatrace) at 4°C for 5 h, followed by overnight incubation with Bio-beads SM-2 (Bio-Rad). Excess amphipol was removed by gel filtration (Superdex 200 10/300 Increase, GE Healthcare) equilibrated in TBS (20mM Tris pH 7.4, 150mM NaCl).
CC30 LukAB purification for pull-down assay:
Overnight cultures of S. aureus Newman ΔΔΔΔ pOS1_PlukAB-lukAs.s.-6xHis-CC30 LukAB were subcultured 1:100 in TSB supplemented with chloramphenicol (5 μg/mL) and grown for 5 h. Cells were spun down (6000rpm, 15 min, 4°C) and the supernatant was filtered through a 0.22 μm filter. The supernatant was adjusted to pH 7.4, and supplemented with 400 mM NaCl. LukAB was purified using AKTA Pure FLPC (GE Healthcare). A HisTrap Excel column (GE Healthcare) was equilibrated in 20 mM Na2HPO4 pH 7.4, 500 mM NaCl. After sample load, the column was washed with 20 mM Na2HPO4 pH 7.4, 500 mM NaCl, 15 mM imidazole, and LukAB was eluted with a linear gradient of 20 mM Na2HPO4 pH 7.4, 500 mM NaCl, 500 mM Imidazole. Purified protein was exchanged into TBS (20 mM Tris pH 7.4, 150 mM NaCl) supplemented with 10% glycerol.
Pull-down assay:
StrepTactin XT Superflow High Capacity resin (IBA Lifesciences) was incubated with 50 μg of purified HVCN1-Strep (or TBS as a control) for 1 h at 4°C. All assay steps were performed at 4°C. Resin was loaded onto a column, washed with 20 mL wash buffer (150 mM Tris pH 8, 150 mM NaCl, 1 mM EDTA). 30 μg of LukAB or LukSF was added to the resin, and allowed to bind for 5 min. Excess toxin was washed with 20 mL wash buffer. HVCN1-Strep was eluted with 1.25 mL wash buffer supplemented with 50 mM biotin. 250 ng of input and 20 μL of pull-down output was separated by SDS-PAGE, and stained using Sypro Ruby (Invitrogen, Thermo Fisher Scientific) following the manufacturer’s Rapid Staining protocol. For Western blots, 125 ng of input and 20 μL of pull-down output proteins were separated by SDS-PAGE and blotted onto nitrocellulose membranes. After blocking in 5% milk in PBS-T for 1 h at room temperature, membranes were incubated with primary antibodies - anti-His (1:1000, Cell Sciences) or anti-HVCN1 (1:1000, PA5–24964, Invitrogen, Thermo Fisher Scientific), diluted in 5% milk in PBS-T for 1 h at room temperature. Membranes were then washed three times with PBS-T for 10 min, incubated with secondary Alexa Fluor680 conjugated antibodies diluted 1:25,000 in 5% milk in PBS-T for 1 h at room temperature. Membranes were washed three times with PBS-T for 10 min and imaged using Odyssey Infrared Imaging System (LI-COR).
In vivo S. aureus infection
Animal housing conditions:
Animals received PicoLab® Rodent Diet 20 (LabDiet) and acidified water, and were housed under normal lighting cycle conditions (12hours ON/12 hours OFF) and temperature 70°F (±2°F).
Generation of hHVCN1 mouse:
C57BL/6J fertilized zygotes were collected from superovulated C57BL/6J mice. Microinjection was performed with 45 μl of filtered injection mix consisting of 50 ng/μl guide RNA (Alt-R® CRISPR-Cas9 crRNA (IDT, guide sequence: GAAACAGACTGGGTGGCTAGGGG)) + Alt-R® CRISPR-Cas9 tracrRNA (IDT, 1072533), 80 ng/μl GeneArt™ Platinum™ Cas9 nuclease (Invitrogen), and 10 ng/ml template DNA (synthesized by Gene Universal) (Supplementary Table 5) in TE buffer. Guide RNA and DNA template were designed on Benchling.com. A NdeI restriction site was engineered into the DNA template to facilitate genotyping.
Injected zygotes were implanted into eight- to 9-week-old pseudopregnant CD-1 (Charles River Laboratory) females and the resulting pups were screened for alleles encoding the expected modifications in Hvcn1 exon 4. Genomic DNA was extracted from tail samples using DNeasy Blood & Tissue Kit (Qiagen) following manufacturer’s instructions. 100–150 ng genomic DNA was used to amplification with primers VJT2065 and VJT2069 or VJT2681 and VJT2683 (see Supplementary Table 5), PCR products were purified using MinElute PCR Purification Kit (Qiagen) and 200 ng digested with NdeI enzyme (NEB) overnight (Extended Data Figure 5B). Animals with PCR products cleaved by NdeI were identified and PCR product from those animals were sequenced. Two independent founder mice with the modified Hvcn1 exon 4 were backcrossed to WT C57BL/6J mice (JAX labs) and the progeny intercrossed to produce homozygous breeder pairs. Homozygous G1, G2, and G4 animals from both lines were used for downstream studies. Mice were bred in-house and maintained under specific pathogen-free conditions and used age-matched and sex-matched at eight – to −10 weeks of age. Representative gel of the genotyping strategy using genomic DNA isolated from wild type (WT), heterozygous (het), and homozygous (homo) hHVCN1 mice is presented in Figure 6F. DNA was isolated from tail samples of wild type (WT), heterozygous (het), and homozygous (homo) hHVCN1 mice, the region surrounding exon 4 was PCR amplified using primers VJT2065 (Fw) and VJT2069 (Rev), and samples were digested with NdeI.
Murine infections with S. aureus:
Eight- to 10-week-old WT or hHVCN1 homozygote C57BL/6 mice were infected retro-orbitally with 5–10×107 CFU of S. aureus (100-μl inoculum). Animals were randomly assigned to infection groups, investigators were not blinded. Prior to infection, mice were anesthetized intraperitoneally with 300 μl of Avertin (2,2,2-tribromoethanol dissolved in tert-amyl alcohol and diluted to a final concentration of 2.5% [vol/vol] in sterile saline). Over the course of infection mice were monitored three times a day for signs of morbidity (hunched posture, lack of movement, paralysis, and inability to acquire food or water) or ≥30% weight loss, at which time the animals were euthanized. 14 days post-infection, mice were euthanized with CO2. The livers, kidneys, lungs, spleens, and hearts were removed, homogenized in 1 ml sterile PBS using FastPrep-24™ Classic Instrument (MB Biomedicals), serially diluted, and plated on tryptic soy agar (TSA) plates for CFU counts. 3 independent experiments were performed per isolate.
Quantification And Statistical Analysis
All experiments were performed as three independent replicates, unless otherwise stated. Experiments with primary human cells were performed using cells obtained from at least 4 donors. For datasets where only two groups of samples were compared, two-tailed unpaired t-test was used to determine if difference between groups was statistically significant. To determine statistical significance in experiments with three or more groups of samples, one-way analysis of variance (ANOVA) was used. For dose-response experiments statistical significance was determined by two-way ANOVA. Data analysis was performed in GraphPad Prism 8 software.
Extended Data
Extended Data Fig. 1. Related to Figure 1.
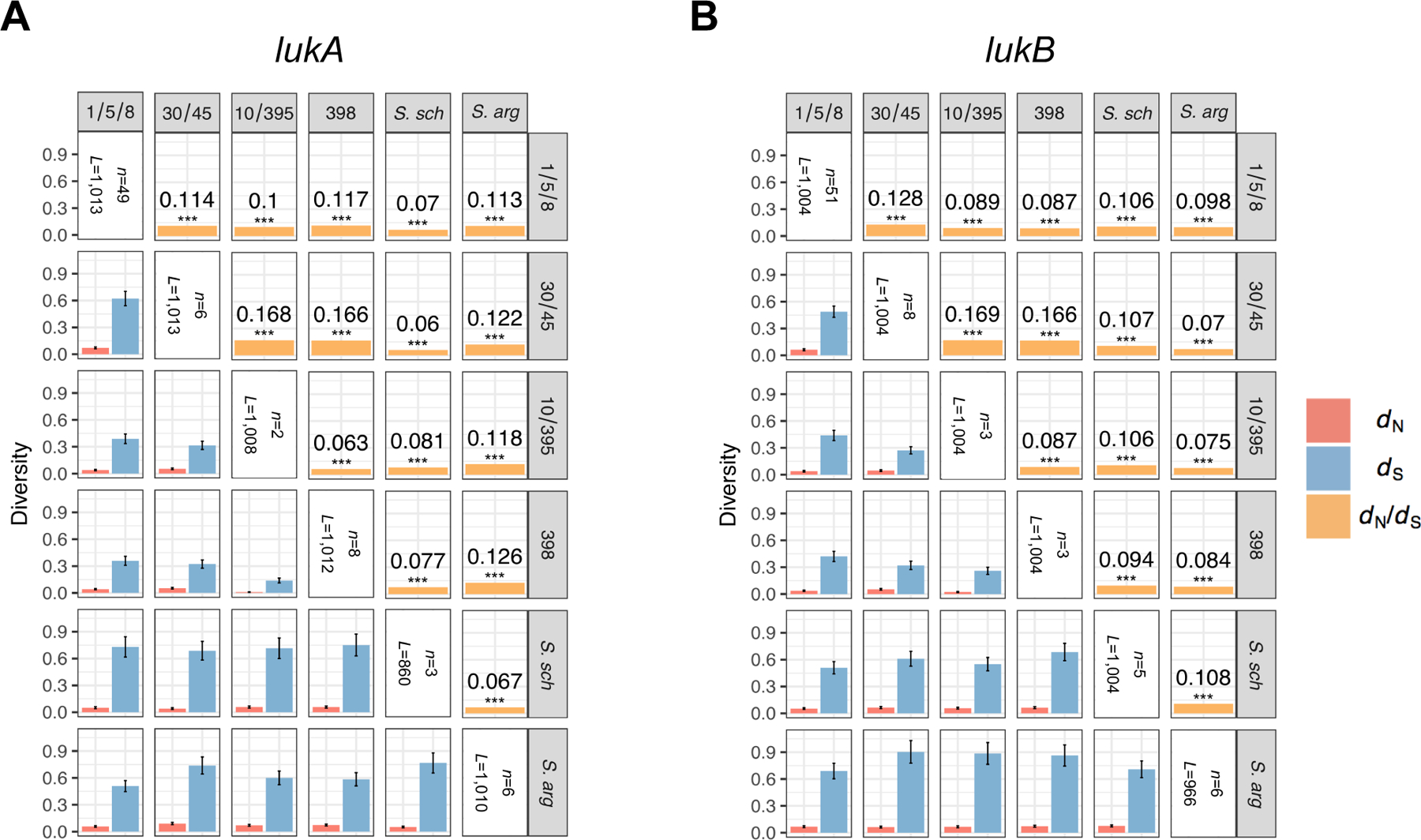
A-B: Mean between-clade dN and dS values are shown below the diagonal, after applying a Jukes-Cantor correction for multiple substitutions at the same sites. Error bars show the standard error of the mean (SEM), estimated using 10,000 bootstrap replicates (codon unit). Corresponding mean between-clade dN/dS ratios are shown above the diagonal, with significance evaluated using a Z-test of the null hypothesis that dN=dS (10,000 bootstrap replicates, codon unit). All P-values are <1.82×10−6 (see corresponding Figure Source Data) such that asterisks *** indicate a significance level of 0.0001 after Bonferroni correction. Mean between-clade estimates were derived by comparing each member of one clade to each member of a second clade for all pairs of clades. For lukA, the numbers of taxa (n) and independent aligned codon positions (L) for each clade are n=49 and L=1013 (clade 1/5/8); 6 and 1013 (30/45); 2 and 1008 (10/395); 8 and 1012 (398); 3 and 860 (S. schweitzeri, labeled as S. sch.); and 6 and 1010 (S. argenteus, labeled as S. arg.). For lukB, the numbers of taxa (n) and independent aligned codon positions (L) for each clade are n=51 and L=1004 (clade 1/5/8); 8 and 1004 (30/45); 3 and 1004 (10/395); 3 and 1004 (398); 5 and 1004 (S. schweitzeri); and 6 and 966 (S. argenteus). The number of aligned codon positions (L) are given after eliminating positions with ≤2 defined (non-gap, non-ambiguous) codons in either clade, separately for each between-clade comparison.
Extended Data Fig. 2. Related to Figure 1.
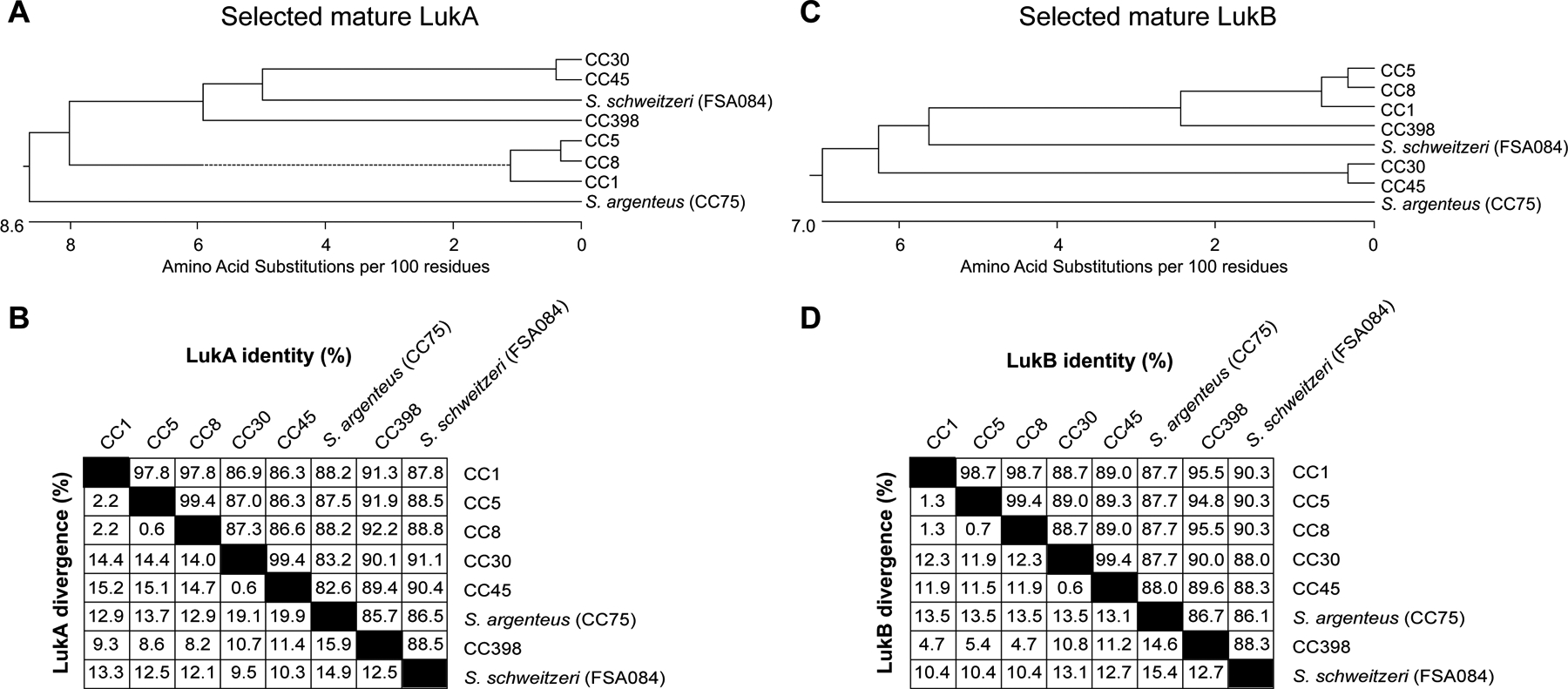
A, C: Phylogenetic tree based on amino acid sequences of mature LukA (A) and LukB (C) produced by S. aureus belonging to the indicated clonal complexes (CCs). The branch length in the tree is proportional to the number of amino acid substitutions per 100 residues. B, D: Percent identity and divergence of mature LukA (B) and LukB (D) proteins produced by S. aureus belonging to the indicated CCs.
Extended Data Fig. 3. Related to Figure 2.
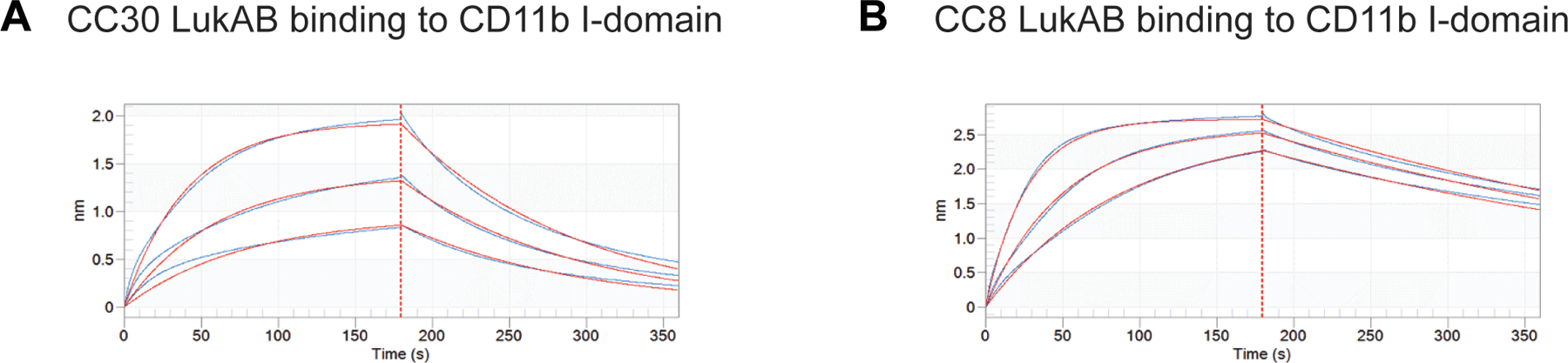
A, B: Binding curves of CC30 (A) and CC8 (B) LukAB toxins to CD11b I-domain. The association and dissociation kinetics of LukAB with the I-domain coated sensor are represented in blue. Toxin concentrations are 400nM, 200nM, and 100nM for CC30 LukAB, or 125nM, 62.5nM, and 31.3nM for CC8 LukAB. Red curves show the best global fit using a 1:1 binding model.
Extended Data Fig. 4. Related to Figure 5.
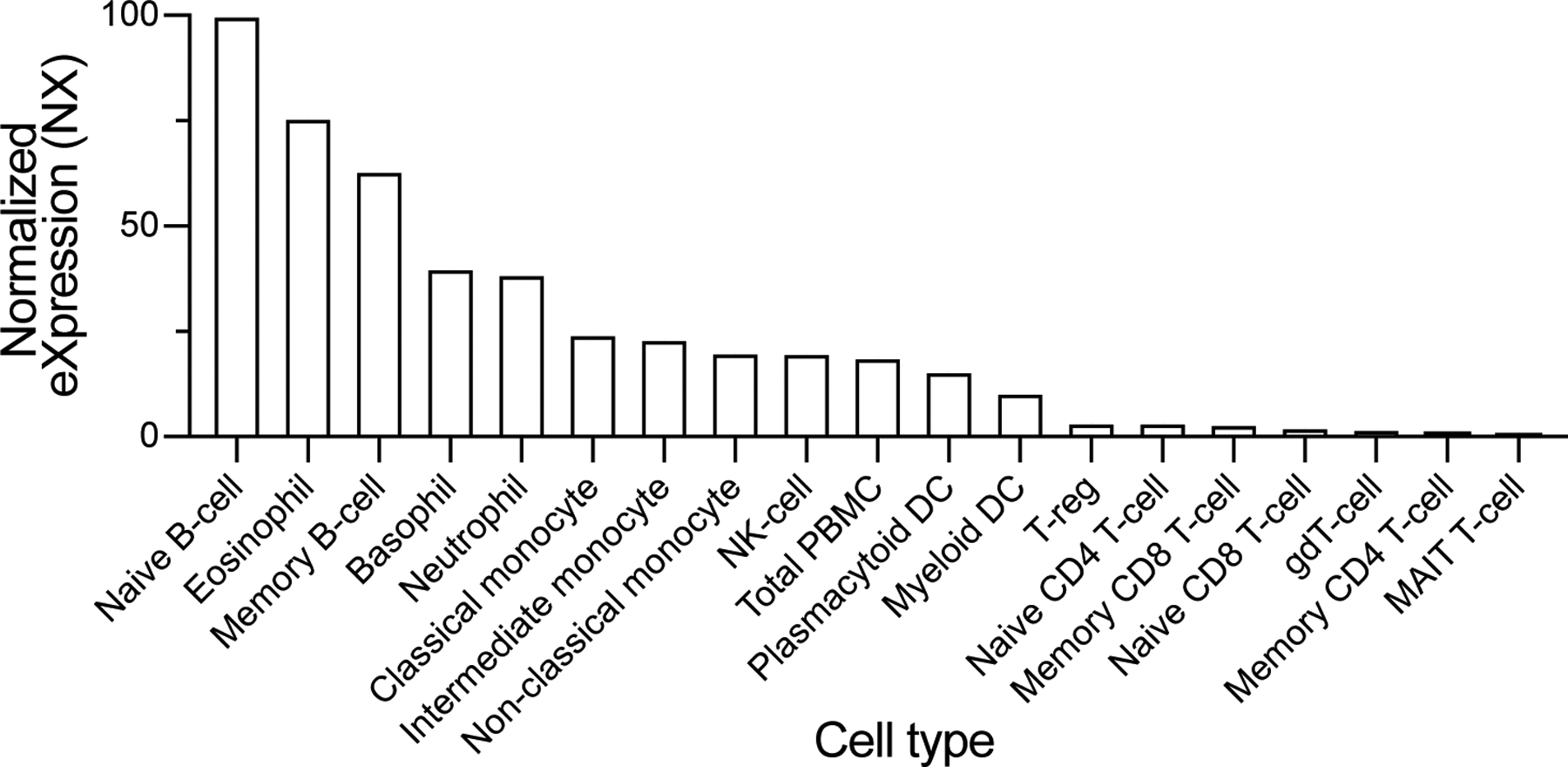
Consensus human blood cell type expression of HVCN1 derived from RNA-seq data from internally generated Human Protein Atlas (HPA) data1. Transcript expression values are presented as Normalized eXpression (NX), resulting from the internal normalization pipeline for 18 blood cell types and total peripheral blood mononuclear cells (PBMC). Data is available at v20.proteinatlas.org/ENSG00000122986-HVCN1/blood, Human Protein Atlas available from www.proteinatlas.org34
Extended Data Fig. 5. Related to Figure 6.
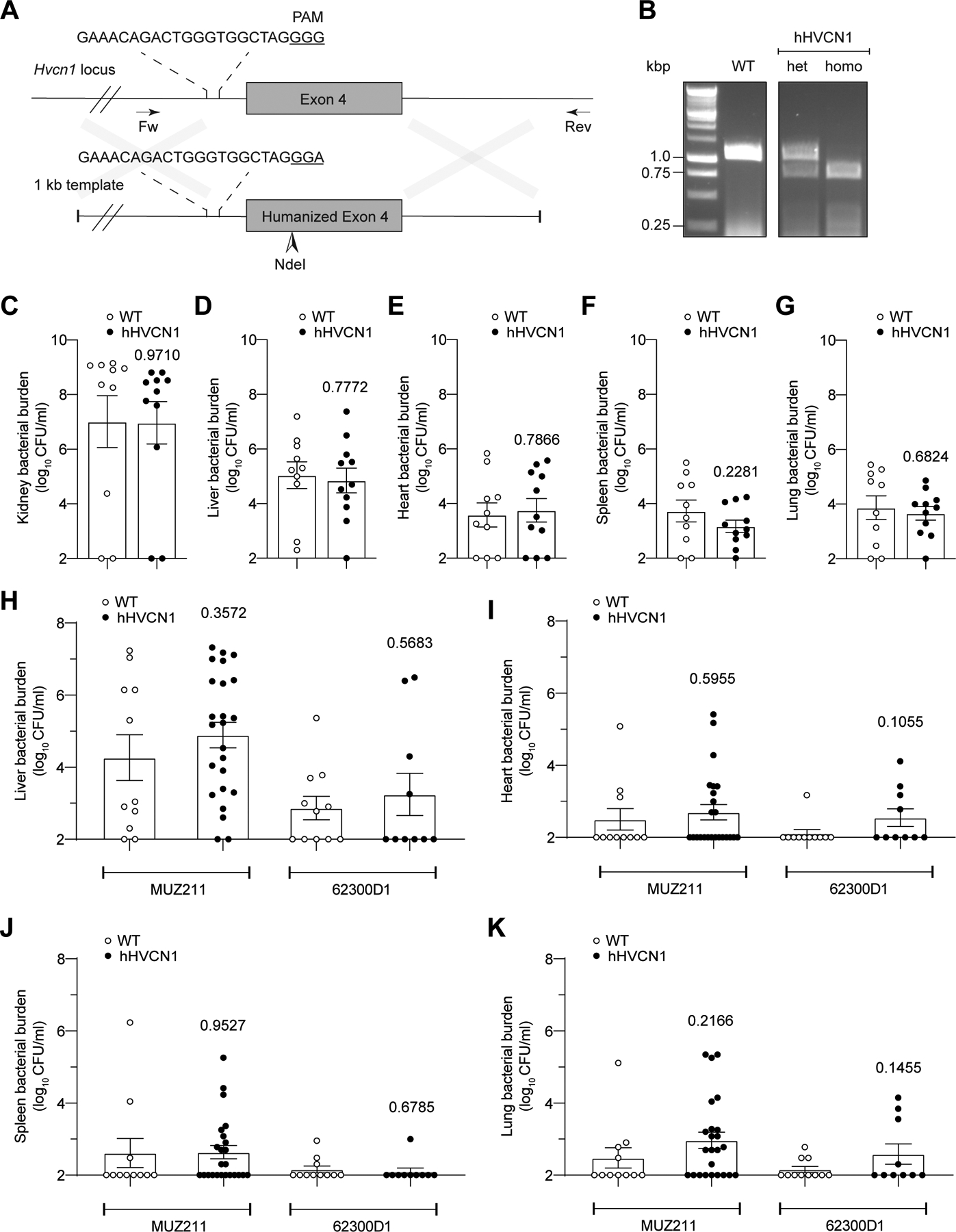
A: Schematic representation of murine Hvcn1 locus and DNA template used to humanize exon 4. B: Genotyping strategy using genomic DNA isolated from wild type (WT), heterozygous (het), and homozygous (homo) hHVCN1 mice using primers VJT2065 and VJT2069. Images are representative of multiple independent experiments as routinely performed for hHVCN1 mouse genotyping. C-G: CFUs in the kidneys (C), livers (D), hearts (E), spleens (F), and lungs (G) collected from WT and hHVCN1 mice infected intravenously with 1×107 CFU of lukAB-deficient USA300 strain LAC. Data from 11 WT and 10 hHVCN1 mice are represented as mean values ±SEM. Statistical significance was determined by t-test (two-tailed), numbers above bars indicate P values. H-K: CFUs in the livers (H), hearts (I), spleens (J), and lungs (K) collected from WT and hHVCN1 mice infected intravenously with 5–10×107 CFU CC30 S. aureus MUZ211 (CFU obtained from 11 WT and 24 hHVCN1 mice) and 62300D1 (CFU obtained from 11 WT and 10 hHVCN1 mice). Data for each isolate are from mice infected over three independent experiments and is represented as mean values ±SEM. Statistical significance was determined by t-test (two-tailed), numbers above bars indicate P values.
Extended Data Fig. 6. Flow cytometry gating (part 1).
A: Flow cytometry gating scheme utilized to measure surface CD11b levels in scramble shRNA (top) and ITGAM shRNA (bottom) expressing THP1 cell (Figure 2A) using APC-conjugated anti-CD11b antibody.
B: Flow cytometry gating scheme utilized to measure binding of biotinylated LukAB (CC30 LukAB is shown as an example) to CHO cells expressing Fluc (top) or HVCN1 (bottom) using PerCP/Cy5.5-conjugated streptavidin staining (Figures 5B–5C).
C: Flow cytometry gating scheme utilized to measure membrane damage in B cells following treatment with PBS control (top) and LukAB (CC30 LukAB is shown as an example, bottom) using Fixable Viability Dye eFluor™ 450 (Figure 5E).
D-E: Flow cytometry gating scheme utilized to measure membrane damage in CD4-positive (D) and CD8-positive (E) T cells following treatment with PBS control (top) and LukAB (CC30 LukAB is shown as an example, bottom) using Fixable Viability Dye eFluor™ 450 (Figure 5E).
Extended Data Fig. 7. Flow cytometry gating (part 2).
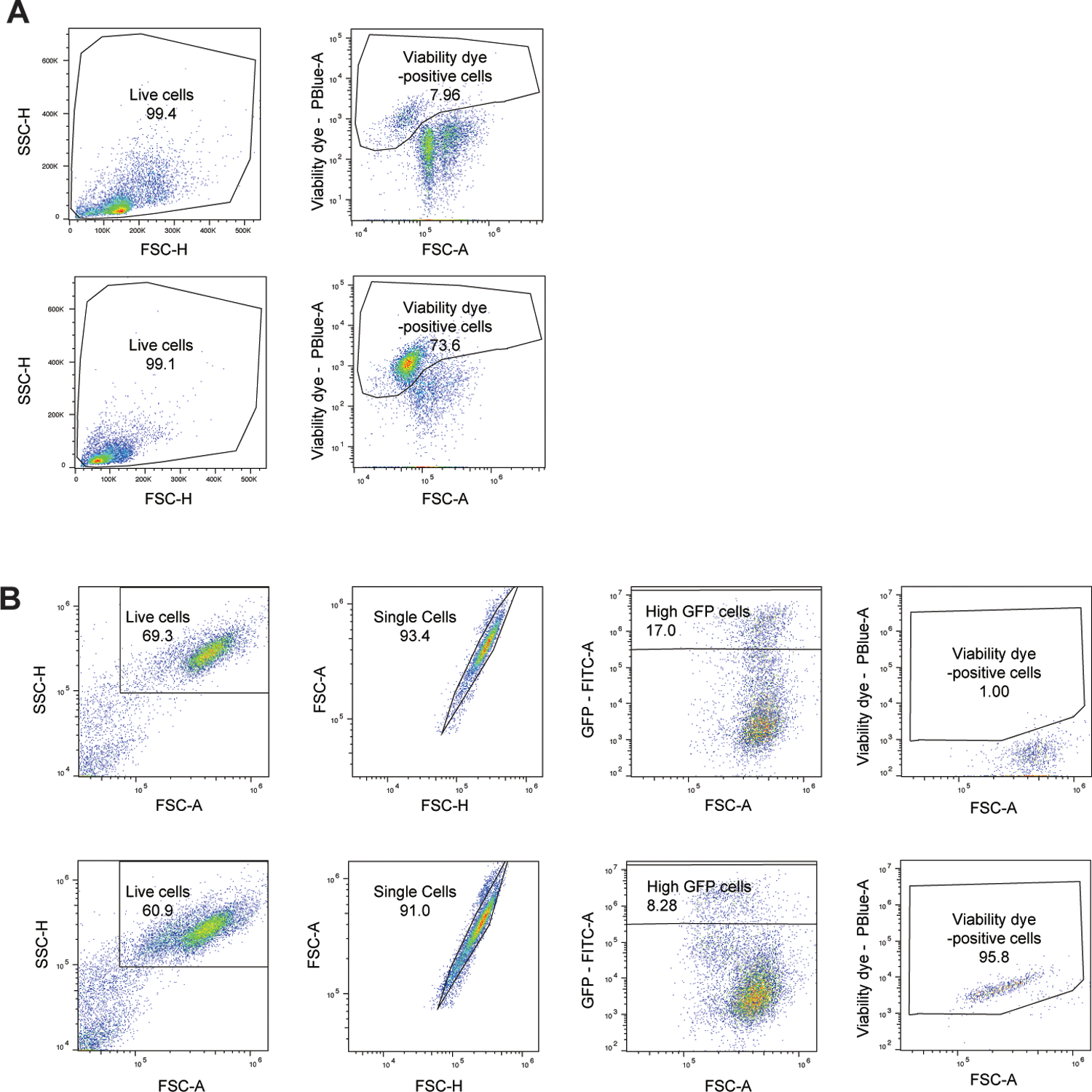
A: Flow cytometry gating scheme utilized to measure membrane damage in PECs after treatment with PBS control (top) and leukocidins (LukED is shown as an example, bottom) using Fixable Viability Dye eFluor™ 450 (Figure 6A).
B: Flow cytometry gating scheme utilized to measure membrane damage in Lenti-X 293T cells expressing C-terminal GFP-tagged wildtype HVCN1 and chimeric proteins (human HVCN1 is shown as an example) following treatment with PBS control (top) and CC30 LukAB (bottom) using Fixable Viability Dye eFluor™ 450 (Figure 6D).
Supplementary Material
Acknowledgements
We are thankful to members of the Torres and Planet labs for insightful discussions and comments on the manuscript, and to Hannah Behrens, Bianca Dunn, Michael Abrams, and Rebecca Plessel for help with reagents, and Ze Chen for help with data analysis. We would like to thank Tenzin Lhakhang (NYU Grossman School of Medicine’s Applied Bioinformatics Laboratories) for help with the bioinformatic analysis of the CRISPR/Cas9 screen and the NYU Grossman School of Medicine’s Genome Technology Center for helping with the Next generation sequencing. Lastly, we would like to acknowledge Eric Campeau, Paul Kaufman, Charles Rice, Neal Alto, John Schoggins, Keith Mostov, Tim Foster, Radim Osicka, and Feng Zhang for providing reagents.
This work was supported in part by the National Institute of Health-National Institute of Allergy and Infectious Diseases award numbers R01 AI099394, R01 AI105129, R01 AI121244 and contract HHSN272201400019C (V.J.T.), R01 AI137336 and R01 AI140754 (B.S. and V.J.T.), T32 AI007180 (D.B.A.J., E.E.Z. and K.T.), and F32 AI122486 (D.B.A.J.). S.S.P. was supported by the Jan Vilcek/David Goldfarb Fellowship Endowment Funds and Bernard B. Levine Program for Postdoctoral Research Fellowships in Immunology, K.M.B. was supported by the Vilcek MSTP Scholars award and T32 GM007308, and C.W.N. was supported by a Gerstner Scholars Fellowship from the Gerstner Family Foundation at the American Museum of Natural History, and a Postdoctoral Research Fellowship from Academia Sinica. P.J.P and A.M.M. were supported by R01 AI137526. The Genome Technology Center and the Flow cytometry technologies are partially supported by the Cancer Center Support Grant P30 CA016087 from the National Institutes of Health/National Cancer Institute at the Laura and Isaac Perlmutter Cancer Center. V.J.T. is a Burroughs Wellcome Fund Investigator in the pathogenesis of infectious diseases.
COMPETING INTERESTS STATEMENT
V.J.T. is an inventor on patents and patent applications filed by New York University, which are currently under commercial license to Janssen Biotech Inc. Janssen Biotech Inc. provides research funding and other payments associated with the licensing agreement. All other authors declare no conflict of interest.
Footnotes
Code availability
Any code generated during this study is available upon request with no restrictions.
Contact For Reagent And Resource Sharing
Further information and requests for reagents should be directed to the Lead Contact, Victor J. Torres (Victor.Torres@nyulangone.org). S. aureus strains and cells could be obtained through an MTA. Mice could also be obtained through an MTA and a licensing fee.
Data availability
The data generated during this study are available upon request with no restrictions.
References
- 1.Tong SY, Davis JS, Eichenberger E, Holland TL & Fowler VG Jr. Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin Microbiol Rev 28, 603–661, doi: 10.1128/CMR.00134-14 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Alonzo F 3rd & Torres VJ The Bicomponent Pore-Forming Leucocidins of Staphylococcus aureus. Microbiology and molecular biology reviews: MMBR 78, 199–230, doi: 10.1128/MMBR.00055-13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Spaan AN, van Strijp JAG & Torres VJ Leukocidins: staphylococcal bi-component pore-forming toxins find their receptors. Nat Rev Microbiol 15, 435–447, doi: 10.1038/nrmicro.2017.27 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ventura CL et al. Identification of a novel Staphylococcus aureus two-component leukotoxin using cell surface proteomics. PLoS One 5, e11634, doi: 10.1371/journal.pone.0011634 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dumont AL et al. Characterization of a new cytotoxin that contributes to Staphylococcus aureus pathogenesis. Mol Microbiol 79, 814–825, doi: 10.1111/j.1365-2958.2010.07490.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Melehani JH, James DB, DuMont AL, Torres VJ & Duncan JA Staphylococcus aureus Leukocidin A/B (LukAB) Kills Human Monocytes via Host NLRP3 and ASC when Extracellular, but Not Intracellular. PLoS Pathog 11, e1004970, doi: 10.1371/journal.ppat.1004970 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Berends ETM et al. Staphylococcus aureus Impairs the Function of and Kills Human Dendritic Cells via the LukAB Toxin. MBio 10, doi: 10.1128/mBio.01918-18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.DuMont AL et al. Staphylococcus aureus LukAB cytotoxin kills human neutrophils by targeting the CD11b subunit of the integrin Mac-1. Proceedings of the National Academy of Sciences of the United States of America 110, 10794–10799, doi: 10.1073/pnas.1305121110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Badarau A, Trstenjak N & Nagy E Structure and Function of the Two-Component Cytotoxins of Staphylococcus aureus - Learnings for Designing Novel Therapeutics. Adv Exp Med Biol 966, 15–35, doi: 10.1007/5584_2016_200 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Planet PJ Life After USA300: The Rise and Fall of a Superbug. J Infect Dis 215, S71–S77, doi: 10.1093/infdis/jiw444 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carrel M, Perencevich EN & David MZ USA300 Methicillin-Resistant Staphylococcus aureus, United States, 2000–2013. Emerg Infect Dis 21, 1973–1980, doi: 10.3201/eid2111.150452 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Malachowa N et al. Staphylococcus aureus leukotoxin GH promotes inflammation. J Infect Dis 206, 1185–1193, doi: 10.1093/infdis/jis495 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.DuMont AL et al. Staphylococcus aureus elaborates leukocidin AB to mediate escape from within human neutrophils. Infection and immunity 81, 1830–1841, doi: 10.1128/IAI.00095-13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Malachowa N, Kobayashi SD, Freedman B, Dorward DW & DeLeo FR Staphylococcus aureus leukotoxin GH promotes formation of neutrophil extracellular traps. Journal of immunology 191, 6022–6029, doi: 10.4049/jimmunol.1301821 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DuMont AL et al. Identification of a crucial residue required for Staphylococcus aureus LukAB cytotoxicity and receptor recognition. Infection and immunity 82, 1268–1276, doi: 10.1128/IAI.01444-13 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Badarau A et al. Structure-function analysis of heterodimer formation, oligomerization, and receptor binding of the Staphylococcus aureus bi-component toxin LukGH. The Journal of biological chemistry 290, 142–156, doi: 10.1074/jbc.M114.598110 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Badarau A et al. Context matters: The importance of dimerization-induced conformation of the LukGH leukocidin of Staphylococcus aureus for the generation of neutralizing antibodies. MAbs 8, 1347–1360, doi: 10.1080/19420862.2016.1215791 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boguslawski KM et al. Exploiting species specificity to understand the tropism of a human-specific toxin. Sci Adv 6, eaax7515, doi: 10.1126/sciadv.aax7515 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bal AM et al. Genomic insights into the emergence and spread of international clones of healthcare-, community- and livestock-associated meticillin-resistant Staphylococcus aureus: Blurring of the traditional definitions. J Glob Antimicrob Resist 6, 95–101, doi: 10.1016/j.jgar.2016.04.004 (2016). [DOI] [PubMed] [Google Scholar]
- 20.Diekema DJ et al. Continued emergence of USA300 methicillin-resistant Staphylococcus aureus in the United States: results from a nationwide surveillance study. Infect Control Hosp Epidemiol 35, 285–292, doi: 10.1086/675283 (2014). [DOI] [PubMed] [Google Scholar]
- 21.Lakhundi S & Zhang K Methicillin-Resistant Staphylococcus aureus: Molecular Characterization, Evolution, and Epidemiology. Clin Microbiol Rev 31, doi: 10.1128/CMR.00020-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Planet PJ et al. Architecture of a Species: Phylogenomics of Staphylococcus aureus. Trends Microbiol 25, 153–166, doi: 10.1016/j.tim.2016.09.009 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Hughes AL & Friedman R Nucleotide substitution and recombination at orthologous loci in Staphylococcus aureus. J Bacteriol 187, 2698–2704, doi: 10.1128/JB.187.8.2698-2704.2005 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Springer TA, Thompson WS, Miller LJ, Schmalstieg FC & Anderson DC Inherited deficiency of the Mac-1, LFA-1, p150,95 glycoprotein family and its molecular basis. The Journal of experimental medicine 160, 1901–1918 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.McKillop WM, Barrett JW, Pasternak SH, Chan BM & Dekaban GA The extracellular domain of CD11d regulates its cell surface expression. J Leukoc Biol 86, 851–862, doi: 10.1189/jlb.0309150 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Shalem O et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87, doi: 10.1126/science.1247005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DeCoursey TE, Morgan D & Cherny VV The voltage dependence of NADPH oxidase reveals why phagocytes need proton channels. Nature 422, 531–534, doi: 10.1038/nature01523 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Morgan D et al. Voltage-gated proton channels maintain pH in human neutrophils during phagocytosis. Proceedings of the National Academy of Sciences of the United States of America 106, 18022–18027, doi: 10.1073/pnas.0905565106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramsey IS, Ruchti E, Kaczmarek JS & Clapham DE Hv1 proton channels are required for high-level NADPH oxidase-dependent superoxide production during the phagocyte respiratory burst. Proceedings of the National Academy of Sciences of the United States of America 106, 7642–7647, doi: 10.1073/pnas.0902761106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeCoursey TE Voltage-gated proton channels find their dream job managing the respiratory burst in phagocytes. Physiology (Bethesda) 25, 27–40, doi: 10.1152/physiol.00039.2009 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spaan AN et al. The staphylococcal toxins gamma-haemolysin AB and CB differentially target phagocytes by employing specific chemokine receptors. Nature communications 5, 5438, doi: 10.1038/ncomms6438 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Okochi Y, Sasaki M, Iwasaki H & Okamura Y Voltage-gated proton channel is expressed on phagosomes. Biochem Biophys Res Commun 382, 274–279, doi: 10.1016/j.bbrc.2009.03.036 (2009). [DOI] [PubMed] [Google Scholar]
- 33.El Chemaly A, Nunes P, Jimaja W, Castelbou C & Demaurex N Hv1 proton channels differentially regulate the pH of neutrophil and macrophage phagosomes by sustaining the production of phagosomal ROS that inhibit the delivery of vacuolar ATPases. J Leukoc Biol 95, 827–839, doi: 10.1189/jlb.0513251 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Uhlen M et al. A genome-wide transcriptomic analysis of protein-coding genes in human blood cells. Science 366, doi: 10.1126/science.aax9198 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Schilling T, Gratopp A, DeCoursey TE & Eder C Voltage-activated proton currents in human lymphocytes. J Physiol 545, 93–105, doi: 10.1113/jphysiol.2002.028878 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Trstenjak N et al. Molecular mechanism of leukocidin GH-integrin CD11b/CD18 recognition and species specificity. Proceedings of the National Academy of Sciences of the United States of America 117, 317–327, doi: 10.1073/pnas.1913690116 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alonzo F 3rd et al. Staphylococcus aureus leucocidin ED contributes to systemic infection by targeting neutrophils and promoting bacterial growth in vivo. Mol Microbiol 83, 423–435, doi: 10.1111/j.1365-2958.2011.07942.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Alonzo F 3rd et al. CCR5 is a receptor for Staphylococcus aureus leukotoxin ED. Nature 493, 51–55, doi: 10.1038/nature11724 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang H et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell 153, 910–918, doi: 10.1016/j.cell.2013.04.025 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pelzek AJ et al. Human Memory B Cells Targeting Staphylococcus aureus Exotoxins Are Prevalent with Skin and Soft Tissue Infection. mBio 9, doi: 10.1128/mBio.02125-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chambers HF & Deleo FR Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat Rev Microbiol 7, 629–641, doi: 10.1038/nrmicro2200 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nienaber JJ et al. Methicillin-susceptible Staphylococcus aureus endocarditis isolates are associated with clonal complex 30 genotype and a distinct repertoire of enterotoxins and adhesins. J Infect Dis 204, 704–713, doi: 10.1093/infdis/jir389 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fowler VG Jr. et al. Potential associations between hematogenous complications and bacterial genotype in Staphylococcus aureus infection. J Infect Dis 196, 738–747, doi: 10.1086/520088 (2007). [DOI] [PubMed] [Google Scholar]
- 44.Holden MT et al. Complete genomes of two clinical Staphylococcus aureus strains: evidence for the rapid evolution of virulence and drug resistance. Proceedings of the National Academy of Sciences of the United States of America 101, 9786–9791, doi: 10.1073/pnas.0402521101 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeLeo FR et al. Molecular differentiation of historic phage-type 80/81 and contemporary epidemic Staphylococcus aureus. Proceedings of the National Academy of Sciences of the United States of America 108, 18091–18096, doi: 10.1073/pnas.1111084108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tromp AT et al. Human CD45 is an F-component-specific receptor for the staphylococcal toxin Panton-Valentine leukocidin. Nat Microbiol 3, 708–717, doi: 10.1038/s41564-018-0159-x (2018). [DOI] [PubMed] [Google Scholar]
- 47.Monk IR, Tree JJ, Howden BP, Stinear TP & Foster TJ Complete Bypass of Restriction Systems for Major Staphylococcus aureus Lineages. MBio 6, e00308–00315, doi: 10.1128/mBio.00308-15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harper L et al. Staphylococcus aureus Responds to the Central Metabolite Pyruvate To Regulate Virulence. MBio 9, doi: 10.1128/mBio.02272-17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stamatakis A RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313, doi: 10.1093/bioinformatics/btu033 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lanave C, Preparata G, Saccone C & Serio G A new method for calculating evolutionary substitution rates. Journal of molecular evolution 20, 86–93 (1984). [DOI] [PubMed] [Google Scholar]
- 51.Yang Z Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. Journal of molecular evolution 39, 306–314 (1994). [DOI] [PubMed] [Google Scholar]
- 52.Felsenstein J Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39, 783–791 (1985). [DOI] [PubMed] [Google Scholar]
- 53.Kumar S, Stecher G & Tamura K MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol Biol Evol 33, 1870–1874, doi: 10.1093/molbev/msw054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Nelson CW, Moncla LH & Hughes AL SNPGenie: estimating evolutionary parameters to detect natural selection using pooled next-generation sequencing data. Bioinformatics 31, 3709–3711, doi: 10.1093/bioinformatics/btv449 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nei M & Kumar S Molecular Evolution and Phylogenetics. (Oxford University Press, New York, 2000). [Google Scholar]
- 56.Kosakovsky Pond SL & Frost SD Not so different after all: a comparison of methods for detecting amino acid sites under selection. Mol Biol Evol 22, 1208–1222, doi: 10.1093/molbev/msi105 (2005). [DOI] [PubMed] [Google Scholar]
- 57.Murrell B et al. Detecting individual sites subject to episodic diversifying selection. PLoS Genet 8, e1002764, doi: 10.1371/journal.pgen.1002764 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Almagro Armenteros JJ et al. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nat Biotechnol 37, 420–423, doi: 10.1038/s41587-019-0036-z (2019). [DOI] [PubMed] [Google Scholar]
- 59.Osicka R et al. Bordetella adenylate cyclase toxin is a unique ligand of the integrin complement receptor 3. Elife 4, e10766, doi: 10.7554/eLife.10766 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Campeau E et al. A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS One 4, e6529, doi: 10.1371/journal.pone.0006529 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schoggins JW et al. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472, 481–485, doi: 10.1038/nature09907 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Virreira Winter S, Zychlinsky A & Bardoel BW Genome-wide CRISPR screen reveals novel host factors required for Staphylococcus aureus alpha-hemolysin-mediated toxicity. Sci Rep 6, 24242, doi: 10.1038/srep24242 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sanjana NE, Shalem O & Zhang F Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11, 783–784, doi: 10.1038/nmeth.3047 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Radke EE et al. Hierarchy of human IgG recognition within the Staphylococcus aureus immunome. Sci Rep 8, 13296, doi: 10.1038/s41598-018-31424-3 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Reyes-Robles T, Lubkin A, Alonzo F 3rd, Lacy DB & Torres VJ Exploiting dominant-negative toxins to combat Staphylococcus aureus pathogenesis. EMBO Rep 17, 780, doi: 10.15252/embr.201670010 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Monk IR, Shah IM, Xu M, Tan MW & Foster TJ Transforming the untransformable: application of direct transformation to manipulate genetically Staphylococcus aureus and Staphylococcus epidermidis. MBio 3, doi: 10.1128/mBio.00277-11 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fuller JR et al. Identification of a lactate-quinone oxidoreductase in Staphylococcus aureus that is essential for virulence. Front Cell Infect Microbiol 1, 19, doi: 10.3389/fcimb.2011.00019 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Berends ET et al. Distinct localization of the complement C5b-9 complex on Gram-positive bacteria. Cellular microbiology 15, 1955–1968, doi: 10.1111/cmi.12170 (2013). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated during this study are available upon request with no restrictions.