

Abstract
The effects of ER stress on protein secretion by cardiac myocytes are not well understood. In this study, the ER stressor thapsigargin (TG), which depletes ER calcium, induced death of cultured neonatal rat ventricular myocytes (NRVMs) in high media volume but fostered protection in low media volume. In contrast, another ER stressor, tunicamycin (TM), a protein glycosylation inhibitor, induced NRVM death in all media volumes, suggesting that protective proteins were secreted in response to TG but not TM. Proteomic analyses of TG- and TM-conditioned media showed that the secretion of most proteins was inhibited by TG and TM; however, secretion of several ER-resident proteins, including GRP78 was increased by TG but not TM. Simulated ischemia, which decreases ER/SR calcium also increased secretion of these proteins. Mechanistically, secreted GRP78 was shown to enhance survival of NRVMs by collaborating with a cell-surface protein, CRIPTO, to activate protective AKT signaling and to inhibit death-promoting SMAD2 signaling. Thus, proteins secreted during ER stress mediated by ER calcium depletion can enhance cardiac myocyte viability.
Keywords: ER stress, Proteostasis, Cardiac myocyte death, Cardioprotection, Heart failure, Cardiokine
1. Introduction
Secreted proteins are important regulators of cellular function outside the heart, as well as within the heart. For example, proteins secreted from cardiac myocytes can function through paracrine and autocrine mechanisms to augment viability of myocytes and other cell types in the heart. The ER-dependent, or classical, protein secretion pathway is a major route for the secretion of such proteins by cells in the heart (Fig. 1A) [1,2]. Conditions that affect the environment in the ER of cardiac myocytes can decrease protein-folding efficiency; subsequent accumulation of misfolded proteins can be toxic, contributing to a condition of ER stress, which could affect protein secretion. The acute response to ER stress involves a global reduction in protein synthesis, including that in the ER, which relieves demands on the ER protein-folding machinery and potentially decreases secretory protein trafficking (Fig. 1B). ER stress results in the activation of several signaling pathways, i.e. the ER stress response, which genetically reprograms cells in order to fortify ER protein-folding and reduce the stress of misfolded protein accumulation (Fig. 1C). However, if the acute adaptive ER stress response is not sufficient to restore ER proteinfolding, chronic ER stress affects cellular programming in ways that lead to cell death, which is considered maladaptive.
Fig. 1.

Hypothetical Effects of ER Stress on Classical Secretion and Effect of Media Volume and Cell Density on Cardiac Myocyte Viability- Panel A- Proteins secreted by the classical secretory pathway are synthesized and folded in the endoplasmic reticulum (ER), then routed to the Golgi before they are secreted, after which they exert their functions through autocrine, paracrine, and endocrine signaling. Panel B- Hypothesis that acute ER stress impairs ER protein-folding and decreases secretion. Panel C- Hypothesis that in response to ER stress an adaptive response is initiated to reduce misfolded protein accumulation and restore secretion via the classical secretory pathway. Panel D- Diagram describing the hypothesis that secreted proteins could enhance cardiac myocyte viability by autocrine/paracrine-mediated protection. Panel E- Diagram depicting the effects of low (top) or high (bottom) media volumes on auto/paracrine-mediated protection. Panel F- Neonatal rat ventricular myocytes (NRVM) were plated in DMEM/F-12 media containing 10% fetal bovine serum (FBS) at respective densities of 2.5 (lanes 1 and 2), 5.0 (3,4), or 10.0 (5,6) × 105 cells/cm2 in 24-well culture dishes. After 24 h, media were replaced with DMEM/F-12 containing 2% FBS. After another 24 h, media were replaced with either a low volume (L = 0.25 ml; bars 1, 3, 5 in Panel G) or a high volume (H = 2.0 ml; bars 2, 4, 6 in Panel G) of serum-free DMEM/F-12 containing BSA at 1 mg/ml (minimal media). After 48 h, media samples from NRVMs were subjected to SDS-PAGE followed by silver staining. Panel G- Culture viabilities from NRVMs plated as described in Panel F were determined using an MTT assay. Shown are the mean optical density (O.D.) values ± S.E.M. * and #, p ≤ .05 different from paired control.
Recent evidence suggest that ER stress is involved in the development and progression of heart disease, including cardiac hypertrophy, ischemic heart disease, and heart failure [3–5]. However, little is known about the effects of ER stress in the heart on the secretion of proteins that could influence cardiac myocyte viability. Accordingly, this study was undertaken to examine the effects of ER stress on the secretion of proteins from cardiac myocytes, and to determine whether changes in protein secretion during ER stress affect cardiac myocyte viability.
2. Materials and methods
2.1. Cell culture neonatal rat ventricular myocytes
Neonatal rat ventricular myocytes (NRVM) were isolated by enzymatic digestion of 1–4 day–old neonatal rat hearts and purified by Percoll density gradient centrifugation, as described [6]. Cardiac myocytes were cultured at various densities, as described in the figure legends. NRVM-conditioned medium for proteomic analyses was prepared essentially as previously described [7], except that myocytes were purified by Percoll density gradient centrifugation before plating. Details concerning the generation of NRVM conditioned medium for proteomics can be found in the legend to Supplementary Table 1.
2.2. NRVM plating and culturing paradigm
Since the culture cell density and media volume affected myocyte viability in response to some treatments, with the exception of Fig. 1F and G, in most experiments NRVM plating and culturing paradigms were carefully scaled to well surface area so as to maintain a relatively constant cell density and relative media volume. Details are provided in the relevant figure legends. In general, the culturing protocol was as follows:
| Culture well plate size | NRVM plating density | Low volume (1×) | High volume (8×) |
|---|---|---|---|
| 6-well plate | 1 × 106/well | 1 ml | 8 ml |
| 12-well plate | 4 × 105/well | 0.5 ml | 4 ml |
| 24-well plate | 2.5 × 105/well | 0.25 ml | 2 ml |
| 48-well plate | 1.25 × 105/well | 0.125 ml | 1 ml |
2.3. Cell culture adult mouse ventricular myocytes
Adult mouse ventricular myocytes (AMVM) were isolated essentially as described [8,9]. AMVM were cultured in maintaining media consisting of 99 ml MEM supplemented with 1 ml 100× insulin-transferrin-selenium, 0.1 mg/ml bovine serum albumin (BSA), 10 ml 1 M HEPES solution, 1 ml 100× pen/strep-glutamine. Media did not contain blebbistatin.
2.4. Conditioned and treatment media
For all experiments not related to simulated ischemia, AMVM were cultured and treated in maintaining medium. Unless otherwise stated, NRVM were treated in either serum-free DMEM/F-12 supplemented with 1 mg/ml BSA, 1× pen/strep-glutamine (minimal media) or serumand BSA-free DMEM/F-12, 1× pen/strep-glutamine containing either no additions (Con), or combinations of 10 μg/ml TM, 2 μM TG, 5 μg/ml BFA, 2 μg/ml recombinant GRP78 (Prospec-Tany TechnoGene Ltd.; Cat# hsp-037), 10 μg/ml ALK4-Fe L75A (generously provided by Dr. Peter Gray), and 20 μM Z-VAD-FMK (R&D Systems; FMK001). Media samples were then collected for immunoblotting or proteomics analyses (see below).
2.5. Simulated ischemia
NRVM were plated at a density of either 2.5 or 4.0 × 105 cells, while AMVM were plated at 5.0 × 105 cells per 12-well culture dish for simulated ischemia or chemical ER stress experiments, respectively. NRVM cultures were maintained in DMEM/F-12 medium containing 10% FBS for 18–24 h, after which they were changed to DMEM/F-12 medium containing 2% FBS and maintained for 48 h. NRVM or AMVM culture media was then changed to glucose-free DMEM (Thermo Fisher; Cat# A1443001) medium containing 2% dialyzed FBS (Thermo Fisher; Cat# 26400044) and placed in a hypoxic chamber at < 0.1% oxygen for various times. Cultures were maintained in low or high media volumes of 0.5 or 5 ml, respectively. Control cultures were maintained in atmospheric O2, 5% CO2 incubator at 37 °C in DMEM medium containing 2% dialyzed FBS and 17.5 mM glucose ± BFA at 5 μg/ml during the ischemia.
2.6. MTT assay
The MTT assay was performed using the Cell Proliferation Kit I (Roche Diagnostics #11465007001).
2.7. Propidium iodide/hoechst cell staining
NRVM cultures were washed one- to two times with DMEM/F-12 and, in the case of simulated ischemia, they were washed with glucose-free media DMEM/F-12 after which 0.5 ml of media were added to each culture. Two μl of propidium iodide (2 μg/ml final concentration) and 2 μl of Hoechst 33342 (2 μg/ml final concentration) were added to each culture, which were then rocked for at least 5 min at 37 °C. Fluorescent images were captured and the red (PI-positive dead) and blue (Hoechstpositive total) cells were counted in at least 4–5 fields/well.
2.8. Calcein AM blue cell staining
AMVM cultures were treated with 2.5 μl of calcein AM stock (1 mg/ml in DMSO) and incubated for ~5 min at 37 °C. Phase and fluorescence images were obtained using a 10× objective on an Olympus X70 epifluorescence microscope. Cells that were calcein AM-positive and rod-shaped and showed no evidence of membrane blebs were counted as viable. Rod-shaped morphology is consistent with a highly organized internal cytoskeletal structure, which is missing in adult cardiac myocytes undergoing apoptosis or necrosis [10].
2.9. Immunocytofluorescence
NRVM were plated at a density of 1.1 × 105 per chamber on Falcon 4-Chamber Glass Slides (Ref #354104; Surface Area 1.8 cm2) in DMEM/F-12 containing 10% FBS for 24 h, after which the medium was replaced with DMEM/F-12 containing 2% FBS for an additional 48 h. The medium was then replaced with either 0.25 ml (low volume) or 2.5 ml (high volume) of minimal media, i.e. serum-free DMEM/F-12 containing bovine serum albumin (1 mg/ml) then treated ± TG (2 μM) or TM (10 μg/ml) for 8 h or 24 h for activated caspase staining or GRP78 staining, respectively. Cultures were then washed and fixed with 4% paraformaldehyde/0.5% Triton-X in PBS for 15 min. Slides were then treated with Superblock (Thermo Fisher; Cat #37515) blocking buffer for 60 min after which primary antibodies were added (see below) for 16 h at 4 °C in the dark. Primary antibodies used for immunocytofluorescence were as follows: α-actinin, Sigma Aldrich, Cat #A7811, 1:200; GRP78 (C-20) Santa Cruz Biotechnology, Cat #SC-1051 Lot #F0415, 1:30; cleaved caspase-3, Cell Signaling Technology, Cat #D175 Lot #37, 1:500. Slides were then washed four times with PBS and then treated with the appropriate secondary antibodies, as follows: Cy3 anti-mouse, FITC anti-goat, FITC anti-rabbit, all purchased from Jackson ImmunoResearch Laboratories and all used at 1:100 for 90-min at 25 °C in the dark. Slides were visualized on a Zeiss 710 laser-scanning confocal microscope using a C-Apochromat 40× objective with 1.20 W Korr M27 specifications.
2.10. XBP1 splicing
XBP1 splicing was assessed by RT-PCR, essentially as previously described [11].
2.11. Immunoblotting
Media or cell extract samples were combined with the appropriately concentrated form of Laemmli sample buffer and then boiled for 7 min at 95° C before SDS-PAGE, followed by transfer to PVDF membranes. The membranes were probed with the following antibodies diluted in 10 ml 5% non-fat milk: calreticulin (Upstate Cell Signaling Solutions; #06–661 Lot #29700; 1:2000, or Abcam Inc., ab2907; 1:5000); GRP94 (mAb 119G10) (ENZO Life Sciences; ADI-SPA-850; 1:1000); GRP78 (C20) (Santa Cruz Biotechnology, Inc.; SC1051; 1:1000), GAPDH (RDI, TRK5G4; 1:15,000), or KDEL (ENZO Life Sciences; ADI-SPA-827; 1:8000), pAKT (Ser473, Cell Signaling; 9271 s; 1:1000), AKT (Cell Signaling; 9272 s; 1:1000), pSMAD2 (Ser465/467, Cell Signaling; 3108S; 1:1000), SMAD2/3 (Cell Signaling; 8685S; 1:1000), ANF (Peninsula Laboratories; T-4013; 1:2000).
2.12. Proteomics
See the legend to Supplementary Table 1.
2.13. Quantitative real time PCR
Quantitative real time PCR (qRT-PCR) was carried out as previously described [8] using the primers listed in the table below.
| Gene (rat) | Forward primer | Reverse primer |
|---|---|---|
| Atf4 set 1 | CTCTCTGTACGCTGTTCCTTTC | CTCGGTCATGTTGTAGGGATTT |
| Atf4 set 2 | AGAATGGCTGGCTATGGATG | GAAGAGGCTGCAAGAATGTAAAG |
| Chop/Gadd153 | CCAAAATAACAGCCGGAACCT | GCCATGACTGTAATTGGACCG |
| Cripto | CCAGGGCTAGAACATTTGAGT | CCTCCTGGCTGTCATTCTTT |
| Crt | AGCAGTTCTTGGACGGAGATG | TGTTGGATTCGACCCAGC |
| Gapdh | CCTGGCCAAGGTCATCCAT | GTCATGAGCCCTTCCACGAT |
| Grp94 | AAGCATCTGATTACCTTGAATTGGAT | CTCCTCCACAGTCTCAGTCTTGCT |
| Grp78 | CACGTCCAACCCGGAGAA | ATTCCAAGTGCGTCCGATG |
| Hrd1 | TTTCCGCTCTTTGCCATTAGG | TTCCATGTTGCGGATGGCTCT |
| Manf | TGCAAAGGCTGTGCAGAGAAG | ATGAACTGCTGTTTCCCTCCG |
| Pdia6 | GGTGAAATTGGCAGCCGTA | CAGGAGACTCGCCTTTCTGAA |
| Puma | ATGGCGGACGACCTCAAC | AGTCCCATGAAGAGATTGTAC |
| XBP1 | CAGCAAGTGGGGATTTGGAA | ATCCCCAAGCGTGCCTTAAC |
|
| ||
| Gene (mouse) | Forward primer | Reverse primer |
|
| ||
| Crt | GAGGACGAAGAAGATGAGAAGG | GATGAGGGCTGAAGGAGAATTA |
| Gapdh | ATGTTCCAGTATGACTCCACTCACG | GAAGACACCAGTAGACTCCACGACA |
| Grp94 | GGGAGGTCACCTTCAAGTCG | CTCGAGGTGCAGATGTGGG |
| Grp78 | TTCTGCCATGGTTCTCACTAAA | TGTTCTTCTCTCCCTCTCTCTT |
2.14. Small interfering RNA (siRNA) transfection
Transfection of siRNA into NRVM was achieved using HiPerfect Transfection Reagent (Qiagen, Valencia, CA) following the vendor’s protocol. The sequence of siRNA targeting rat GRP78 was 5-AGUGUU GGAAGAUUCUGA-3 (Thermo Fisher; Silencer Select siRNA, Cat# 4390771) or rat cripto was 5-GGACCAAGACCUCAAGGUA-3 (Thermo Fisher; Silencer Select siRNA, Cat# 34390771). A non-targeting sequence (Thermo Fisher; Cat# 12935300) was used as a control siRNA.
2.15. Statistics
Cell culture experiments were performed at least 3 times with n = at least three cultures for each treatment. All multiple group comparisons were performed using a one-way analysis of variance (ANOVA) with a Newman–Keuls (sometimes called Student–Newman–Keuls or SNK) post hoc analysis. The Newman–Keuls analysis is based on the “Studentized range” or “Student’s q”. The q statistic is used to compare each mean with every other mean in a pairwise fashion. The Newman–Keuls test is often used whenever a significant difference between three or more sample means has been revealed by an analysis of variance [12]. Paired analyses were done with Student’s t-test. Unless otherwise described in the figure legends, data are represented as mean with all error bars indicating ± s.e.m. * or # P ≤ .05 different from paired control or from all other values.
3. Results
3.1. Effect of media volume on cardiac myocyte viability
For most of the studies here we used primary neonatal rat ventricular myocytes (NRVMs), which are a well-established model for examining cardiac myocyte function and are often used in studies of factors that influence cardiac myocyte viability [13–15]. To determine whether NRVMs secrete proteins that can enhance viability, cultures were maintained in either high or low volumes of media. We reasoned that in low media volumes, secreted proteins would be more concentrated than in high media volumes and, as a result, myocyte viability would be improved if cultures were maintained in low media volumes (Fig. 1D, E). When conditioned media samples were examined, we found that the concentration of proteins was higher in low media volumes than high volumes, and that the quantity of protein in the media increased as a function of increasing NRVM plating densities (Fig. 1F). Moreover, myocyte viability was greater in low media volume than high media volume, and this volume-dependent enhancement of viability was more pronounced at high plating densities (Fig. 1G). These results suggest that NRVM secrete proteins that act through auto/paracrine mechanisms to improve myocyte viability.
3.2. Effects of ER stress on myocyte viability
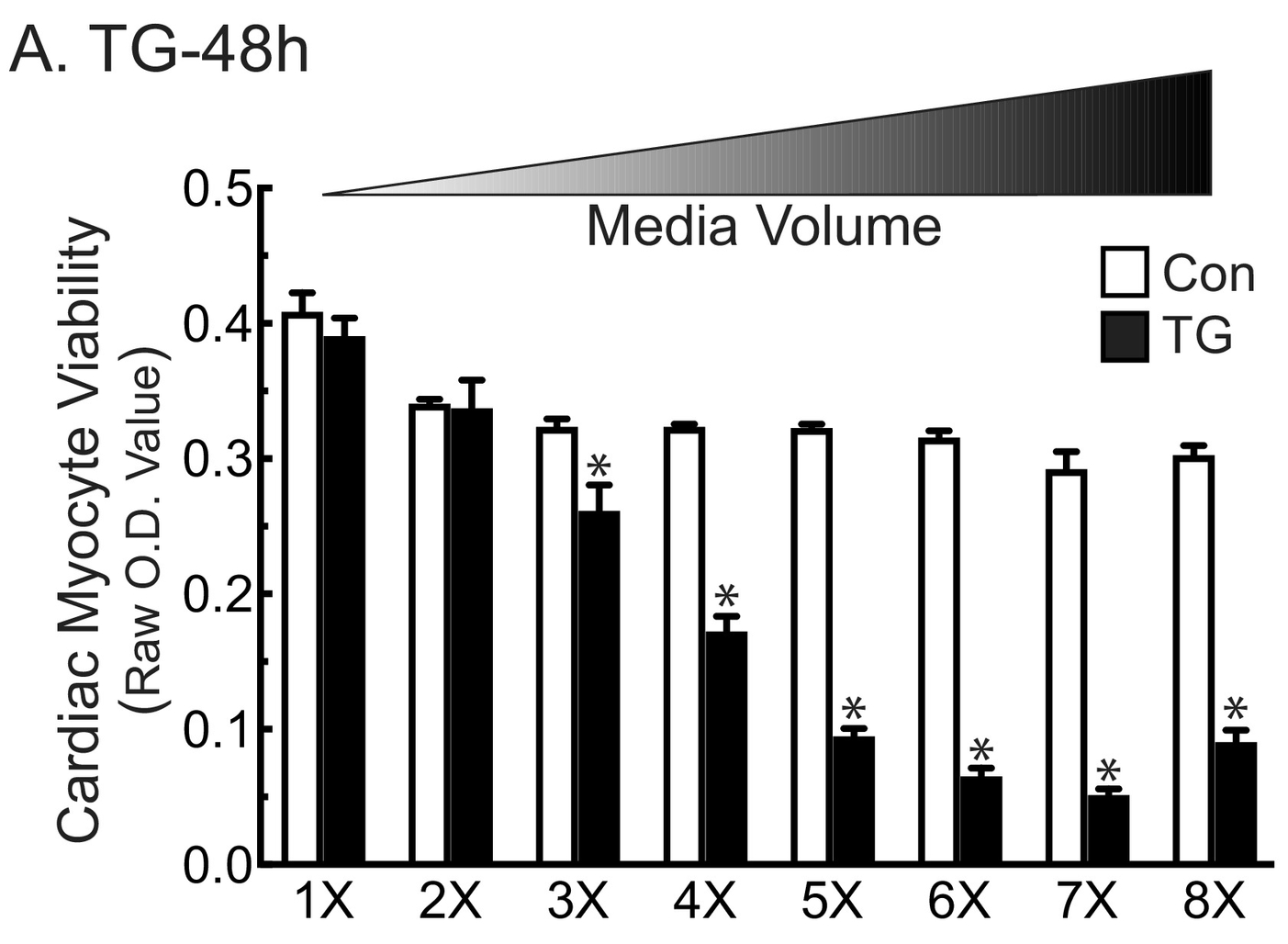
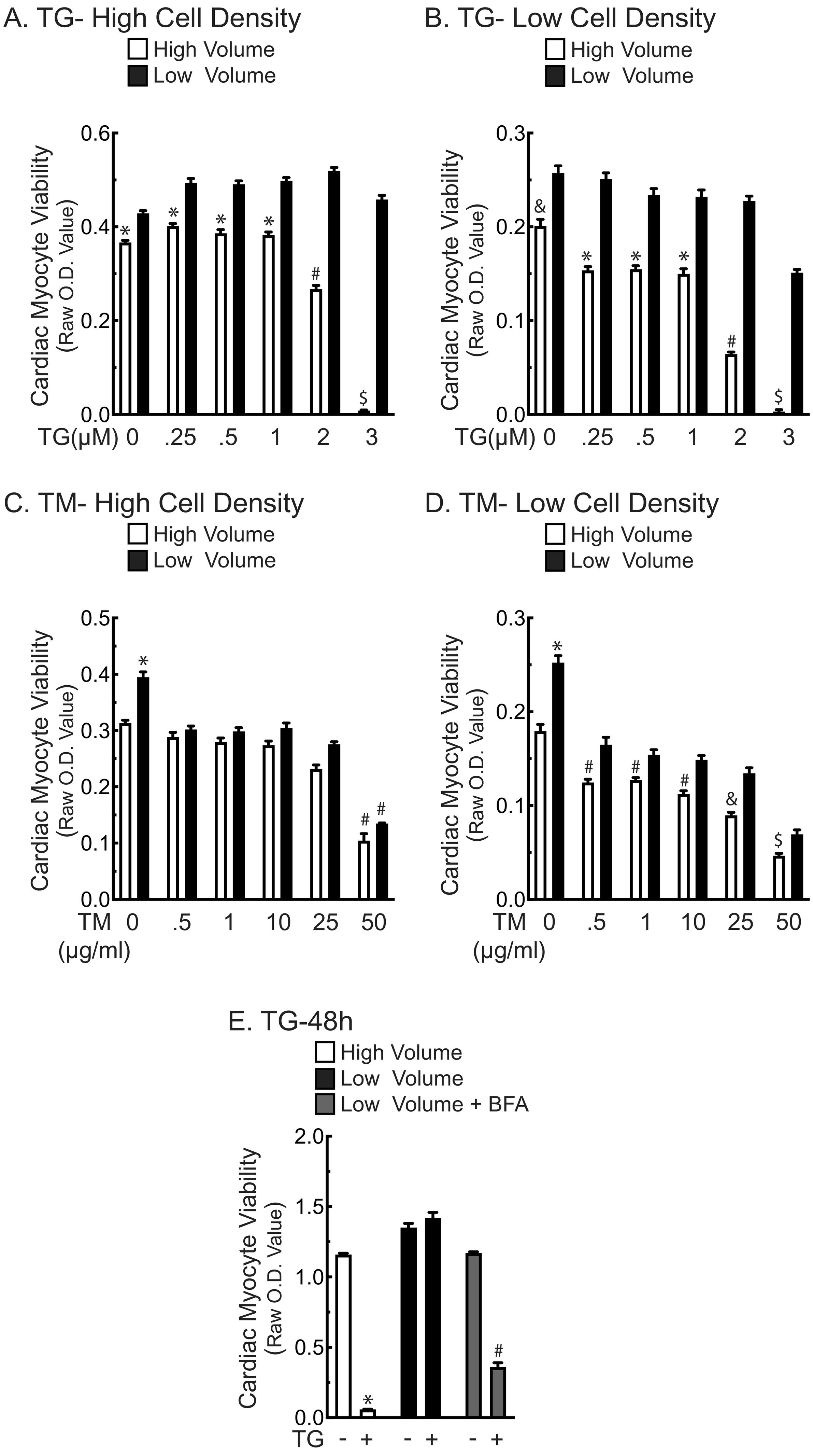
We then explored the effects of ER stress on the viability of NRVM in either high or low media volumes using two chemicals that induce ER stress and can lead to cell death; tunicamycin (TM), which induces ER stress by decreasing ER protein glycosylation, and thapsigargin (TG), which induces ER stress by decreasing ER calcium. In the absence of TM, myocytes maintained in low media volume (low) exhibited greater viability than those in high media volumes (high) (Fig. 2A –TM). Also as expected, TM decreased viability to the same extent, regardless of the media volume (Fig. 2A +TM). In contrast, myocytes treated with TG in high volume (TG/high) exhibited low viability, while those treated with TG in low volume (TG/low) were unaffected (Fig. 2B). This loss of viability caused by TG increased gradually as a function of increasing media volumes (Supplementary Fig. 1). Moreover, TG-induced myocyte death in high media volume was less pronounced at higher cell densities (Supplementary Fig. 2A and B, white bars), while in low volumes, TG actually increased myocyte viability but only at high cell density (Supplementary Fig. 2A and B, black bars). However, TM decreased cell viability at high cell density, regardless of media volume (Supplementary Fig. 2C), and at low density there was a modest but significant increase in viability in low volume (Supplementary Fig. 2D). Moreover, these data allowed us to determine the LD50 for each treatment. Therefore, for the rest of the paper we chose to use the LD50 of the low density and high volume for TM and TG. These findings suggest involvement of secreted proteins in the volume-dependent protective effects of TG. Consistent with this, the ability of TG/low to preserve myocyte viability was inhibited by brefeldin A (Supplementary Fig. 2E), which is known to block protein secretion via the ER-dependent secretory pathway [16]. Taken together, these results are consistent with the hypothesis that TG, but not TM, increases the secretion of a protein, or proteins, through the ER-dependent classical secretory pathway, and that these proteins can protect cardiac myocytes from ER stress-induced death.
Fig. 2.

ER Calcium Depletion Uniquely Alters the Cardiac Myocyte Secretome- Panel A- NRVMs were plated at 2.5 × 105 cells/cm2 in 24-well culture dishes, then treated in high (1 ml) or low (0.125 ml) minimal media volumes ± TM (50 μg/ml). After 48 h, culture viabilities were determined by MTT assay. *, # p ≤ .05 different from all other values, as determined by ANOVA followed by Newman Keul’s post hoc analysis. Panel B- NRVMs were plated as in Panel A, except treated ± TG (3 μM). *, # p ≤ .05 different from all other values, as determined by ANOVA followed by Newman Keul’s post hoc analysis. Panel C– Heatmap summary of proteomics findings identified in fresh, unconditioned media (Mdm), media from NRVMs without TG or TM (Con), and media from NRVMs treated with TG or TM. The unit of the scale is not LogFC or FC. The scale shows the z-score of the data that went into the heatmap. In order to generate the heatmap, the original data is scaled. In essence, the scaling computes the z-score, i.e. the input data is transformed in such a way that the mean is zero and the variance is one. This can be seen from the heatmap scale indicator that has its center at 0. Panel D- NRVMs were plated at 1 × 106 cells/6-well culture dish, then treated ± TG (2 μm) or TM (10 μg/ml) in 1 ml serum- and BSA-free media for 24 h. media samples were analyzed directly by immunoblotting for the ER stress-inducible proteins shown using antibodies specific for GRP94, GRP78, CRT, ANF, and GAPDH, as shown. Panel E- Cell extracts from the cultures described in Panel D were examined by immunoblotting. Panel F- Diagram of the hypothetical benefits of proteins secreted in response to TG.
3.3. TG increases the secretion of a select group of ER proteins
Since it appeared as though the protective factors were secreted via the ER protein secretory pathway in response to TG but not TM, we undertook proteomics analyses to identify proteins that are secreted from NRVM in response to TG but not TM. Using immunocytofluorescence (ICF) we showed that essentially all of the cells in the NRVM cultures expressed α-actinin, when assessed with an antibody specific for skeletal and cardiac muscle actinins, and that treatment with TM or TG robustly increased expression of the ER stress-inducible, ER-luminal protein, GRP78 (Supplementary Fig. 3A). This validated the vitality and purity of the cultures, indicating that proteins identified in the media must be secreted from cardiac myocytes, and are not released from other cell types, or as a result of cell death.
Proteomic analysis and mass spectrometry identified 104 different proteins in naïve (fresh), unconditioned media (DMEM/F-12 1% fetal bovine serum; Mdm) (Supplementary Table 1, columns G and T). To distinguish the proteins secreted by NRVMs from the proteins that were present in unconditioned media alone (Mdm), only proteins not found in Mdm were further examined; proteins found in Mdm came from fetal bovine serum and not from cells because this media was never conditioned with cells. Analysis of media conditioned by untreated cells (Con) identified 76 proteins that were either not found in Mdm, or were drastically increased in the conditioned media (CM) compared to Mdm (Supplementary Table 1, columns K and U). When the published characteristics of these 76 proteins were examined, it was found that 39 were known secreted proteins (Supplementary Table 1, column V). These known secreted proteins in Con CM are summarized in Fig. 2C (Control). Further analysis of the proteomics results showed that the secretion of a majority of these proteins was decreased upon treatment with TM (33 decreased, 4 increased and 2 unchanged) or TG (30 decreased, 5 increased and 4 unchanged) (Fig. 2C [TM, TG]; Supplementary Table 1, columns X and W), an expected result, since ER stress decreases trafficking through the ER-dependent secretory pathway. Remarkably, there were three ER-resident proteins that were actually found in increased levels in media from only the TG-treated cells compared to Control (Fig. 2C [TG yellow]); these proteins were identified as 78 kDa glucose-regulated protein (GRP78), calreticulin, and endoplasmin (GRP94). Supplementary Table 2 shows the peptide sequences and other identification characteristics for proteins listed in Supplementary Table 1 as identified by mass spectrometry.
3.4. Targeted antibody-dependent quantification of proteins
To further demonstrate the TG-dependent secretion of the ER-resident proteins, control-, TG-, and TM-conditioned media were immunoblotted for GRP94, GRP78, and CRT. We found that the levels of each of these proteins were increased but only in the TG-conditioned media (Fig. 2D), consistent with the mass spectrometric evidence. GAPDH detection in conditioned medium was used to discriminate between active secretion and passive release due to cell death. GAPDH would be passively released from cells if the cells were dying. GAPDH was not found in conditioned media, suggesting that secretion was not passive following cell death, but rather actively from living cells. Secretion of atrial natriuretic factor (ANF) was not increased by TG or TM, suggesting that not all secreted proteins exhibit the same characteristics. In contrast, TG and TM both increased cellular levels of GRP94, GRP78 and CRT, as expected, since they are known ER stress-inducible proteins (Fig. 2E). Thus, the appearance of GRP94, GRP78,. and CRT in the media is not the result of increased cellular levels of these proteins, since TG and TM both increased cellular levels of each of them, but only TG increased media levels of these proteins (Fig. 2F).
3.5. Simulated ischemia
To examine the effects of a pathophysiologically relevant treatment that, like TG, is known to dysregulate contractile calcium in cardiac myocytes and to activate ER stress, we assessed the effect of simulated ischemia (sI). As expected, sI upregulated GRP78 in cardiac myocytes in either high or low media volumes, as determined by ICF, qRT-PCR, and immunoblotting (Fig. 3A and Supplementary Fig. 3B and C). Consistent with previous studies by us and others [11,17,18], simulated ischemia activated the ER stress response. Simulated ischemia also resulted in secretion of GRP78, GRP94, and CRT, similar to TG (Supplementary Fig. 3D). The ICF results also suggested fewer myocytes, thus, more myocyte death in NRVM subjected to sI/high compared to sI/low. This was examined in more depth by quantitative live/dead staining of NRVM, which showed that sI/high resulted in considerably more death sI/low (Fig. 3B, white and black bars). Moreover, BFA increased death of NRVM in sI/low (Fig. 3B, gray bars), consistent with a role for proteins secreted via the ER-dependent secretory pathway. To examine the broader relevance of the volume-dependent effects of TG and sI on myocyte viability, similar experiments were performed using adult mouse ventricular myocytes (AMVM). The results of these experiments were similar to NRVM, demonstrating that while both TG and sI upregulated GRP78, GRP94, and CRT (Supplementary Fig. 3E and F), the volume-dependent, BFA-sensitive protective effects of TG and sI, as measured by calcein AM staining, were also observed in adult ventricular myocytes (Fig. 3C and D).
Fig. 3.

Low Volume Protects Cardiac Myocytes from Death during Simulated Ischemia- Panel A- NRVMs plated at 1.1 × 105 cells/chamber on 4-chamber glass slides were treated for 24 h in high (2.5 ml) or low (0.25 ml) media volume with or without simulated ischemia (sI). Cultures were then immunostained for α-actinin (red), GRP78 (green), and stained for DNA (TOPRO-3; blue). Panel B- NRVMs were plated at 4 × 105 cells/12-well culture dish and subjected to sI for 48 h in high or low media volume ± BFA (5 μg/ml), then analyzed for total and dead cells by Hoechst and propidium iodide staining. Shown are the mean values of cell death ± S.E.M., expressed as the percentage of dead cells. *, #, p ≤ .05 different from all other values, as determined by ANOVA followed by Newman Keul’s post hoc analysis. Panels C– Freshly isolated adult mouse ventricular myocytes (AMVM) were plated at 5 × 105 cells/12-well culture dish, then treated for 8 h in high or low maintaining media (see Methods) volume ± TG (2 μm) and BFA (5 μg/ml). AMVMs were scored for viability by quantifying the number of calcein AM-positive, rod-shaped myocytes, as described in the Methods. Shown are the mean values of viable cells ± S.E.M. *, p ≤ .05 different from all other values, as determined by ANOVA followed by Newman Keul’s post hoc analysis. Panel D- AMVMs were plated at 5 × 105 cells/12-well culture dish subjected to sI for 8 h in high or low volume glucose-free DMEM medium containing 2% dialyzed FBS ± BFA (5 μg/ml), and then scored for viability by quantifying the number of calcein AM-positive, rod-shaped myocytes, as described in the Methods. Shown are the mean values of viable cells ± S.E.M. *, p ≤ .05 different from all other values, as determined by ANOVA followed by Newman Keul’s post hoc analysis.
3.6. TG-mediated ER stress is not dependent on media volume
We considered it possible that TG/high might induce more maladaptive ER stress than TG/low. Accordingly, we compared the effects of TG/high and TG/low on ER stress using XBP1 splicing as a highly sensitive measure of ER protein misfolding (Fig. 4A, B) and using ER stress target genes as downstream effectors of the ER stress response. TG/high and TG/low induced XBP1 mRNA splicing to similar extents (Fig. 4C, D), and had similar effects on induction of numerous adaptive and maladaptive ER stress response genes (Fig. 4E). Thus, the media volume-dependent beneficial effects of TG on myocyte viability are not due to media volume-dependent differences in intracellular aspects of ER stress signaling, and were instead due to extracellular aspects, such as increased media levels of GRP78, CRT, or GRP94.
Fig. 4.

Effects of Media Volume and TG on Xbp1 mRNA Splicing and ER Stress Gene Induction- Panel A- Xbp1 splicing is a sensitive measure of protein misfolding in the ER. Panel B- Xbp1 mRNA splicing process and assay. In the absence of ER stress, the Xbp1 mRNA encodes a 261 AA protein that is not a transcription factor. However, upon ER stress, the endoribonuclease activity of IRE-1 is activated, resulting in the removal of a 26-nucleotide intron (pink) and generation of Xbp1 mRNA spliced, which encodes a 376 AA form of Xbp1 that is an active transcription factor. Use of the primers that are shown result in a 290 nucleotide PCR product for the unspliced form of the Xbp1 mRNA and a 264 nucleotide PCR product for the spliced Xbp1 mRNA. Panel C- NRVMs were plated at 1 × 106 cells/6-well culture dish and treated in high (8 ml) or low (1 ml) serum-free DMEM/F-12 containing 1 mg/ml BSA (minimal media) ± TG (2 μM) for the times shown, then extracts were analyzed by PCR for Xbp1 mRNA splicing. Panel D- Quantification of the Xbp1 splicing gel shown in Panel B. *, p ≤ .05 different from untreated control, as determined by ANOVA followed by Newman Keul’s post hoc analysis. Panel E- Gene expression was determined by qRT-PCR using RNA from NRVMs plated at 1 × 106 cells/6-well culture dish and treated for 8 h ± TG (2 μM) in high (8 ml) or low (1 ml) minimal media volumes, as described in Panel B. *, p ≤ .05 different from untreated control, as determined by ANOVA followed by Newman Keul’s post hoc analysis. Panels F, G- NRVMs were plated at 4 × 105 cells/well on 12-well plates. Sixteen hours after plating, cultures were treated for 24 h ± TG (2 μM) in minimal media in low (0.4 ml) or high (2 ml) volume, then examined by SDS-PAGE immunoblotting.
3.7. TG activates the GRP78/CRIPTO signaling pathway
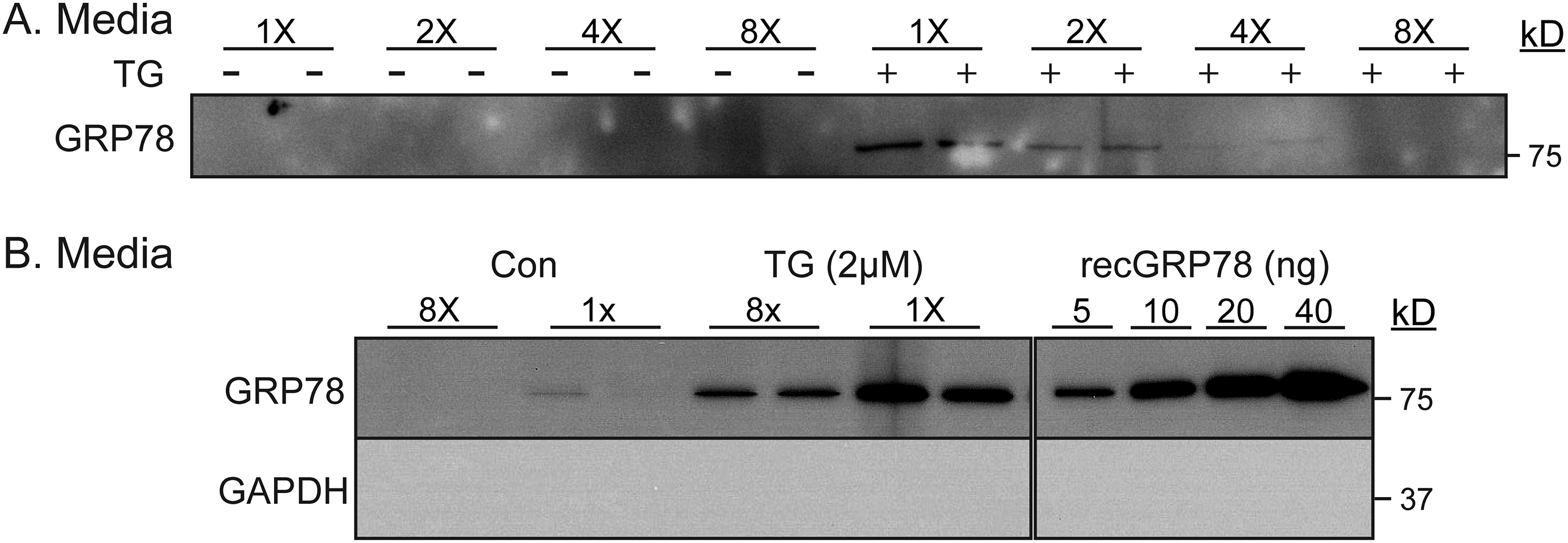
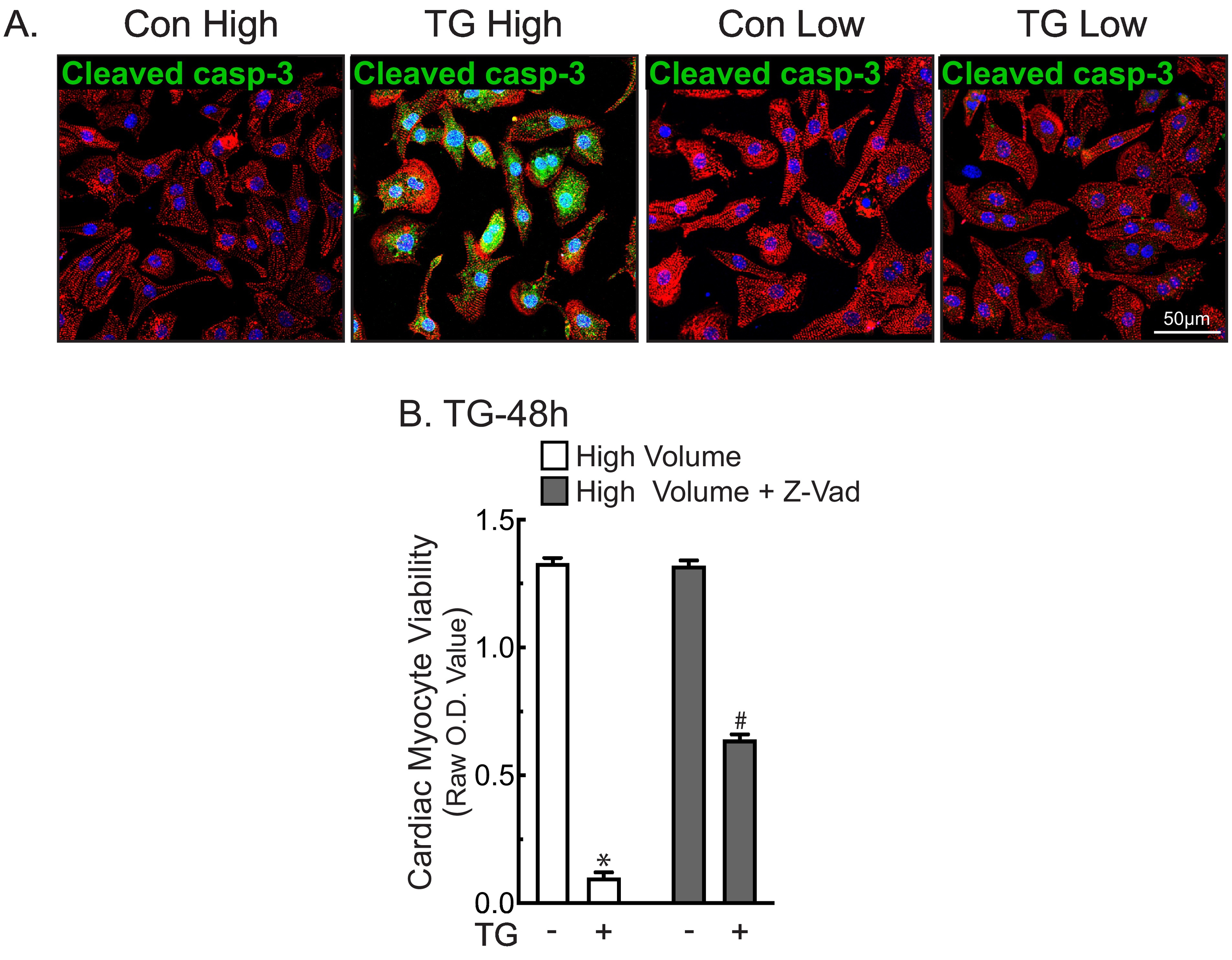
Recent studies have shown that cell-surface GRP78 can act as a binding partner of CRIPTO in cancer cells [19,20]. In those studies, it was shown that when cell-surface GRP78 binds to CRIPTO, AKT is activated, providing protection from apoptosis, while SMAD2 is inactivated, which inhibits SMAD2-associated apoptosis. Accordingly, we focused mechanistic studies on GRP78, asking whether it contributes to protection in TG/low and, if so, whether it does so by signaling through CRIPTO. We showed that TG/low, which increased media levels of GRP78, as expected (Fig. 4F), activated AKT, as shown by increased phosphorylation of AKT on serine 473, while SMAD2 remained inactive, as shown by undetectable phosphorylation of SMAD2 (Fig. 4G). These findings are consistent with the pro-survival effects of TG/low. In contrast, TG/high did not activate AKT but did activate SMAD2 (Fig. 4G), consistent with the pro-death effects of TG/high. Moreover, while the apoptosis mediator, caspase-3, was activated in TG/high, it was not activated in TG/low (Supplementary Fig. 4A). Additionally, NRVM death in TG/high was decreased by the apoptosis inhibitor, Z-VAD (Supplementary Fig. 4B). Thus, TG/low induces NRVM death by apoptosis, which is consistent with the hypothesis that it is secreted GRP78 binding to CRIPTO in TG/low that protects cardiac myocytes from apoptosis. Moreover, by immunoblotting for secreted GRP78 in increasing cell culture volumes, we found a volume-dependent decrease in GRP78 concentrations with increasing media volume, suggesting that effects of secreted GRP78 are seen only in low volume because the protein is more concentrated (Supplementary Fig. 5A and B).
3.8. TG activates protective CRIPTO signaling via secreted GRP78
To dive deeper into the mechanism by which secreted GRP78 protects cardiac myocytes, cellular GRP78 was knocked down using siRNA and CRIPTO/AKT/SMAD2 signaling was examined. As expected, treatment of NRVM with siRNA to GRP78 decreased cellular and secreted GRP78 in cultures treated with TG/low (Fig. 5A, B). Moreover, the pro-survival signaling profile of AKT and SMAD2 seen with TG/low volume were both lost upon GRP78 knockdown (Fig. 5B). Importantly, survival of NRVM in TG/low and simulated ischemia was also decreased upon knockdown of GRP78 (Fig. 5C and Supplementary Fig. 6A). To probe the dependence of NRVM survival on secreted GRP78, a neutralizing antibody to GRP78 was added to culture media and compared to the same amount of non-immune antibody, was also shown to reduce the survival of NRVM (Fig. 5D). Anti-GRP78 also reduced myocyte viability at baseline. Similarly, in Fig. 2B we found that there is a basal decrease in viability in high volume compared to low volume at baseline. In a complementing experiment, it was shown that adding recombinant GRP78 to cultures in high media volume protected NRVM against TG-induced death (Fig. 5E, red). These findings are consistent with the hypothesis that secreted GRP78 can protect NRVM in a CRIPTO-dependent manner.
Fig. 5.

Effects of GRP78 on Cardiac Myocyte Viability and AKT/SMAD2 Signaling- Panels A, B- NRVMs were plated at 4 × 105 cells/well on 12-well plates. Sixteen hours after plating, NRVM culture medium was replaced with DMEM/F-12 supplemented with 0.5% FBS without antibiotics, 120 nM siRNA, and 1.25 μl HiPerfect / 1 μl Grp78 or control siRNA. After 16 h the culture medium was replaced with DMEM/F-12 with BSA (1 mg/ml) for 48 h, after which cultures were treated for 24 h ± TG (2 μM) in low (0.4 ml) or high (2 ml) volume. Cell extracts and media were examined by immunoblotting. Panel C– NRVMs were plated at 1.25 × 105 cells/well on 48-well plates. Sixteen hours after plating, cultures were transfected with siRNA as described in Panel A. Forty-eight hours after transfection, cultures were treated without or with TG (2 μM) in serum- and BSA-free DMEM/F-12 in low (0.125 ml) or high (0.75 ml) volume. After 48 h, culture viabilities were determined by MTT assay. Shown are the mean optical density (O.D.) values ± S.E.M. *, #, p ≤ .05 different from all other values. Panel D- NRVMs were plated at 1.25 × 105 cells/well on 48-well plates. Sixteen hours after plating, media were then replaced with either high (0.75 ml) or low (0.125 ml) volume of serum- and BSA-free DMEM/F-12 ± TG (2 μM), ± 4 μg/ml anti-GRP78 or 4 μg/ml non-immune goat IgG. After 48 h, cell viability was determined by MTT assay; n = 4 cultures/treatment. *, #, $, p ≤ .05 different from all other values, as determined by ANOVA followed by Newman Keul’s post hoc analysis. Panel E- NRVMs were plated as described in Panel C. Media were then replaced with either high volume (0.75 ml) or low volume (0.125 ml) of serum- and BSA-free DMEM/F-12 without or with TG (2 μM), in combination with recombinant GRP78 (2 μg/ml) or bovine serum albumin (2 μg/ml). After 48 h, cell viability was determined by MTT assay; n = 4 cultures/treatment. After 48 h, culture viabilities were determined by MTT assay. *, #, p ≤ .05 different from all other values, as determined by ANOVA followed by Newman Keul’s post hoc analysis.
3.9. CRIPTO is required for TG-mediated protection by secreted GRP78
To examine the CRIPTO hypothesis, NRVM were treated with siRNA to CRIPTO. Treatment with TG/low induced GRP78 secretion (Fig. 6A). However, siRNA to CRIPTO completely blocked AKT activation and enhanced SMAD2 phosphorylation in response to TG/low (Fig. 6B), suggesting that CRIPTO is necessary for TG/low-mediated protection of NRVM. CRIPTO knockdown was validated by qRT-PCR (Fig. 6C). Moreover, CRIPTO knockdown significantly impaired the ability of TG to protect NRVM in low media volume (Fig. 6D). Finally, the CRIPTO-specific antagonist, ALK4L75A [21], mimicked the effect of CRIPTO knockdown, impairing the ability of TG to protect NRVM in low media volume (Fig. 6E). ALK4L75A had no effect on secreted or cellular GRP78 levels (Supplementary Fig. 6B), but significantly decreased AKT activation and increasing SMAD2 activation (Supplementary Fig. 6C). Thus, TG/low-mediated protection requires CRIPTO signaling to AKT and SMAD2.
Fig. 6.

Effects of CRIPTO on Cardiac Myocyte Viability and AKT/SMAD2 Signaling - Panels A, B- NRVMs were plated at 4.0 × 105 cells/12-well plates. Sixteen hours later, cells were transfected with (120 nM) siRNA to CRIPTO or control siRNA, then incubated for 16 h, after which the culture medium was replaced with DMEM/F-12 supplemented with BSA (1 mg/ml) for an additional 48 h, after which cultures were treated for 24 h with or without TG in low (0.4 ml) or high (2 ml) volume. Cell extracts and media were examined by immunoblotting. Panel C–NRVMs were plated and treated as described in Panel A, B. Cripto mRNA levels were determined by qRT-PCR. Shown are the mean values of rat Cripto/Gapdh mRNA, expressed as fold-of-control ± S.E.M. *, p ≤ .05 different from control. Panel D- NRVMs were plated at 1.25 × 105 cells/48-well plates. Sixteen hours after plating, cultures were transfected with siRNA as described in Panel A. Forty-eight hours later, cultures were treated ± TG (2 μM) in serum- and BSA-free DMEM/F-12 in low (0.125 ml) or high (0.75 ml) volume media. After 48 h, culture viabilities were determined by MTT assay. Shown are the mean optical density (O.D.) values ± S.E.M. *, p ≤ .05 different from all other values. Panel E- NRVMs were plated and maintained as in Panel D, except they were treated ± 10 μg/ml ALK4L75A-Fc. Shown are the mean optical density (O.D.) values ± S.E.M. *, p ≤ .05 different from all other values.
4. Discussion
In this study the effects of ER stress on the secretion of proteins that can influence cell viability were examined. Proteomic analysis of the secretome of cardiac myocytes showed that the movement of most proteins through the classical secretory pathway was inhibited by both TM and TG (Fig. 2D). But in contrast to TM, TG selectively increased the secretion of several proteins considered to be permanent ER-residents, i.e. GRP94, GRP78, and CRT. Also, in contrast to TM, TG was cytoprotective in low media volumes and cytotoxic in high media volumes, while TM cytotoxicity occurred similarly at all volumes examined. We extended these studies mechanistically to show that one of the proteins secreted in response to TG, GRP78, protects cardiac myocytes by activating the CRIPTO/AKT/SMAD2 signaling pathway. This is the first demonstration in any cell type that secreted GRP78 can protect through this signaling pathway.
Together, these results support the hypothesis that by reducing ER calcium, TG stimulates the release of proteins from the ER that can exhibit concentration-dependent cytoprotection (Fig. 7), while TM, which does not reduce ER calcium, does not increase secretion of protective proteins. Additionally, when cultured cardiac myocytes were subjected to simulated ischemia, which mimics the circulatory defects seen in cardiovascular disease and is known to cause ER/SR calcium depletion and ER stress [11,17], the low media volume-dependent cytoprotective effects of TG were replicated. Thus, the volume-dependent protective effects seen with TG were also observed during a pathophysiologically relevant maneuver that also activates ER/SR calcium depletion and ER stress. How ER/SR calcium depletion inhibits the secretion of some proteins, while enhancing the secretion of other proteins, and how proteins secreted under these conditions exert their functions remain to be determined. However, several previous studies shed some light on these unknowns.
Fig. 7.

Summary of the Effects on the Classical Secretory Pathway of TG- or Simulated Ischemia-induced ER Stress. - Part A- In cardiac myocytes, ER/SR-resident proteins are depicted as red, and secreted proteins as black within the ER lumen. Under control conditions, ER/SR-resident proteins are not secreted while secreted proteins are properly folded, routed through the Golgi, and eventually released into the media. Part B- Upon treatment with TG or sI, most ER luminal proteins that should be secreted misfold, do not traffic through the Golgi, and are therefore not secreted. However, in contrast to TM, treatment with TG or sI stimulates the secretion of a group of ER/SR resident proteins that are not secreted under other conditions; in this study these proteins were identified as GRP94, GRP78 and CRT. When these three proteins are secreted, they act in an autocrine/paracrine manner to protect cells from the maladaptive effects of ER stress.
We previously examined how ER/SR calcium depletion increased the secretion of the ER stress-inducible, ER-resident, mesencephalic astrocyte-derived neurotrophic factor (MANF) from cultured cardiac myocytes and HeLa cells [22]. In that study, we showed that MANF interacts directly with GRP78, which is a calcium-binding chaperone that requires calcium to interact with its client proteins. Moreover, we showed that depletion of ER calcium with TG or ischemia disrupted the calcium-dependent interaction between GRP78 and MANF, leading to the release of a portion of MANF from the ER lumen, after which it was secreted. We also showed that secreted MANF protects cardiac myocytes in culture and the heart, in vivo, from ischemia/reperfusion damage. Thus, it seems quite possible that like the GRP78-MANF complex, other protein complexes in the ER may depend on calcium, and that when ER calcium is reduced, for example by TG or ischemia, the retention of these proteins in the ER is reduced, which leads to their secretion. Moreover, it is reasonable to postulate the after their secretion, these proteins might have functions different from those in the ER, such as the protection from maladaptive ER stress that was observed in this study. It should be noted that MANF was not observed in the secretome analyses in this study, even though we had previously shown that TG and not TM stimulates MANF secretion. These findings suggest that, perhaps for technical reasons, such as the efficiency of tryptic peptide generation, extraction, and detection, some proteins, such as MANF might not be detected in our proteomics analysis, leaving open the possibility that there are other proteins in addition to GRP94, GRP78, and CRT that are secreted upon ER/SR calcium depletion. Therefore, it is likely that our finding of three ER-resident proteins in the TG secretome is an underestimate of the numbers of such proteins that are secreted in response to TG.
Here, we demonstrated one possible mechanism by which secreted GRP78 can protect cardiac myocytes. This is the first time secreted GRP78 has been shown to protect any cell type by activating the CRIPTO/AKT/SMAD2 signaling pathway. Our findings complement and add to several previous reports showing that some ER-resident proteins can traffic to the cell surface of tumor, immune, and several other cell types [23,24], mostly in cancer and stem cells. These studies suggest that when they reach the cell surface ER-resident chaperones might illicit protective signaling responses; however, the exact roles for cell surface ER-resident proteins are not yet known [25]. In terms of protective signaling, our findings are in agreement with several studies that have shown that cell-surface GRP78 can activate PI3K/AKT, ERK, and SMAD pathways [20,25], which can have pro-survival effects in a variety of cell types, including cardiac myocytes [26]. However, our results differ in that we found that secreted GRP78 can exert protective effects through this same signaling pathway.
It is also possible that there are other mechanisms responsible for protection by secreted ER-resident proteins. For example, one possible mechanism is that secreted ER-resident proteins might maintain the proper folding and function of other extracellular proteins, such as extracellular matrix proteins, autocrine and paracrine proteins, and cell-surface receptors. Support for such a mechanism was seen in studies of the ER-resident, ER stress protein, ERdj3, which is an HSP family member; it was shown that ERdj3 can bind to misfolded proteins in the extracellular space where it acts as a chaperone to decrease the proteotoxicity of misfolded extracellular proteins [27]. Moreover, in our study, conditioned media experiments have not demonstrated significant beneficial effects, indicating that the protective effects of GRP78 could not be transferred from one culture to another. While it is unclear why the conditioned media were not beneficial, it could be due to the labile nature of GRP78 after its secretion. For example, is it known that in the ER, GRP78 exists is in a large multi-protein complex with other chaperones, e.g. GRP94, PDI, ERp72, GRP170/ORP150, UGGT, CaBP1 (calcium binding protein), cyclophilin B and SDF2-L1, and that this complex forms a network for processing unfolded proteins in the ER [28]. It is feasible that this complex, or one like it, may also be secreted and provide the benefit we observe, yet it may be unstable, such that it has lost its effectiveness by the time conditioned medium is tested. Likewise, in our study we found that recombinant GRP78 conferred partial protection, perhaps due in part to the above reason, as well as the possibility that GRP78 may harbor a number of posttranslational modifications in the cells from which it is secreted [29], that were missing in the recombinant GRP78.
It is important to mention that in the study here, TG stimulated protein secretion from viable, healthy cardiac myocytes, and was not a result of release of intracellular contents upon cell death. Thus, while ER/SR calcium depletion decreased the secretion of some proteins, it facilitated the secretion of other proteins. This finding indicates that during calcium ER stress, the movement of proteins through the ER protein secretion pathway is not uniform. In fact, our results show that the calcium ER stress secretome is unique, such that during this stress, some proteins, i.e. those constituting the TG secretome (Fig. 7), are handled differently than all other proteins in the ER/Golgi pathway, so that they can circumvent the trafficking roadblock that inhibits the release of classically secreted proteins. Determining how proteins comprising the TG secretome are able to negotiate this roadblock, and examining the functions of these proteins, once they are secreted, will be required to better understand how and why ER calcium depletion inhibits the secretion of some proteins, while enhancing the secretion of other proteins.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.yjmcc.2020.04.012.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Dr. Peter Gray for kindly providing ALK4L75A-Fc.
Sources of funding
This work was supported by grants and fellowships to E.A.B. from the American Heart Association, United States; SDSU (17PRE33670796), the Inamori Foundation, and the ARCS Foundation Inc., San Diego Chapter, grants from the National Institutes of Health to E.A.B. (1F31HL140850), C.C.G. (R01HL135893, R01HL141463, P01HL085577, and R01HL149931), Institutional support RVO:68081715 to M.S., grants from the NIH to J.V.E. (R01HL132075, P01HL112730, and P01HL10026), and DZHK (German Centre for Cardiovascular Research) Excellence Grants to T.J. and S.D. E.A.B. is a Rees-Stealy Research Foundation Phillips Gausewitz, M.D., Scholar of the SDSU Heart Institute.
Abbreviations:
- BFA
Brefeldin A
- CM
Conditioned medium
- CRT
Calreticulin, CALR
- ER
Endoplasmic reticulum
- GRP78
78 kDa glucose-regulated protein, HSPA5
- GRP94
94 kDa glucose-regulated protein, HSP90b1, endoplasmin
- ICF
Immunocytoflourescence
- LDH
Lactate dehydrogenase
- MANF
Mesencephalic astrocyte-derived neurotrophic factor, ARMET
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- NRVM
Neonatal rat ventricular myocytes
- sI
Simulated ischemia
- SMAD
Mothers against decapentaplegic homolog
- SR
Sarcoplasmic reticulum
- TG
Thapsigargin
- TM
Tunicamcyin
Footnotes
Disclosures
None.
Declaration of Competing Interest
The authors declare that they have no conflict of interest.
References
- [1].Doroudgar S, Glembotski CC, The cardiokine story unfolds: ischemic stress-induced protein secretion in the heart, Trends Mol. Med. 17 (2011) 207–214, 10.1016/j.molmed.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Rabouille C, Malhotra V, Nickel W, Diversity in unconventional protein secretion, J. Cell Sci. 125 (2012) 5251–5255, 10.1242/jcs.103630. [DOI] [PubMed] [Google Scholar]
- [3].Wang S, Binder P, Fang Q, Wang Z, Xiao W, Liu W, Wang X, Endoplasmic reticulum stress in the heart: insights into mechanisms and drug targets, Br. J. Pharmacol. 175 (2018) 1293–1304, 10.1111/bph.13888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Henning RH, Brundel BJJM, Proteostasis in cardiac health and disease, Nat. Rev. Cardiol. 14 (2017) 637, 10.1038/nrcardio.2017.89. [DOI] [PubMed] [Google Scholar]
- [5].Arrieta A, Blackwood EA, Glembotski CC, Wiseman RL, Haynes CM (Eds.), ER Protein Quality Control and the Unfolded Protein Response in the Heart BT Coordinating Organismal Physiology Through the Unfolded Protein Response, Springer International Publishing, Cham, 2018, pp. 193–213, , 10.1007/82_2017_54. [DOI] [Google Scholar]
- [6].Doroudgar S, Völkers M, Thuerauf DJ, Khan M, Mohsin S, Respress JL, Wang W, Gude NA, Müller OJ, Wehrens XHT, Sussman MA, Glembotski CC, Hrd1 and ER-associated protein degradation, ERAD, are critical elements of the adaptive ER stress response in cardiac myocytes, Circ. Res. 117 (2015) 536–546, 10.1161/CIRCRESAHA.115.306993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Stastna M, Chimenti I, Marban E, Van Eyk JE, Identification and functionality of proteomes secreted by rat cardiac stem cells and neonatal cardiomyocytes, Proteomics. 10 (2010) 245–253, 10.1002/pmic.200900515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].O’Connell TD, Rodrigo MC, Simpson PC, Isolation and culture of adult mouse cardiac myocytes, Methods Mol. Biol. 357 (2007) 271–296, 10.1385/1-59745-214-9:271. [DOI] [PubMed] [Google Scholar]
- [9].Pinz I, Zhu M, Mende U, Ingwall JS, An improved isolation procedure for adult mouse cardiomyocytes, Cell Biochem. Biophys. 61 (2011) 93–101, 10.1007/s12013-011-9165-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kang PM, Armin H, Hiroki A, Anny U, Seigo I, Morphological and molecular characterization of adult cardiomyocyte apoptosis during hypoxia and reoxygenation, Circ. Res. 87 (2000) 118–125, 10.1161/01.RES.87.2.118. [DOI] [PubMed] [Google Scholar]
- [11].Thuerauf DJ, Marcinko M, Gude N, Rubio M, Sussman MA, Glembotski CC, Activation of the unfolded protein response in infarcted mouse heart and hypoxic cultured cardiac myocytes, Circ. Res. 99 (2006) 275–282, 10.1161/01.RES.0000233317.70421.03. [DOI] [PubMed] [Google Scholar]
- [12].Abdi H, Williams LJ, Encyclopedia of Research Design, (2010), 10.4135/9781412961288. [DOI]
- [13].Peter AK, Bjerke MA, Leinwand LA, Biology of the cardiac myocyte in heart disease, Mol. Biol. Cell 27 (2016) 2149–2160, 10.1091/mbc.E1601-0038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Louch WE, Sheehan KA, Wolska BM, Methods in cardiomyocyte isolation, culture, and gene transfer, J. Mol. Cell. Cardiol. 51 (2011) 288–298, 10.1016/j.yjmcc.2011.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Glembotski CC, Classic studies of cultured cardiac myocyte hypertrophy: interview with a transformer, Circ. Res. 113 (2013) 1112–1116, 10.1161/CIRCRESAHA.113.302490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Misumi Y, Misumi Y, Miki K, Takatsuki A, Tamura G, Ikehara Y, Novel blockade by brefeldin a of intracellular transport of secretory proteins in cultured rat hepatocytes, J. Biol. Chem. 261 (1986) 11398–11403 http://www.jbc.org/content/261/24/11398.abstract. [PubMed] [Google Scholar]
- [17].Doroudgar S, Thuerauf DJ, Marcinko MC, Belmont PJ, Glembotski CC, Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response, J. Biol. Chem. 284 (2009), 10.1074/jbc.M109.018036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yang W, Paschen W, Unfolded protein response in brain ischemia: a timely update, J. Cereb. Blood Flow Metab. 36 (2016) 2044–2050, 10.1177/0271678X16674488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shani G, Fischer WH, Justice NJ, Kelber JA, Vale W, Gray PC, GRP78 and Cripto form a complex at the cell surface and collaborate to inhibit transforming growth factor beta signaling and enhance cell growth, Mol. Cell. Biol. 28 (2008) 666–677, 10.1128/MCB.01716-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kelber JA, Panopoulos AD, Shani G, Booker EC, Belmonte JC, Vale WW, Gray PC, Blockade of Cripto binding to cell surface GRP78 inhibits oncogenic Cripto signaling via MAPK/PI3K and Smad2/3 pathways, Oncogene. 28 (2009) 2324–2336, 10.1038/onc.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Spike BT, Kelber JA, Booker E, Kalathur M, Rodewald R, Lipianskaya J, La J, He M, Wright T, Klemke R, Wahl GM, Gray PC, CRIPTO/GRP78 signaling maintains fetal and adult mammary stem cells ex vivo, Stem Cell Rep. 2 (2014) 427–439, 10.1016/j.stemcr.2014.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Glembotski CC, Thuerauf DJ, Huang C, Vekich JA, Gottlieb RA, Doroudgar S, Mesencephalic astrocyte-derived neurotrophic factor protects the heart from ischemic damage and is selectively secreted upon sarco/endoplasmic reticulum calcium depletion, J. Biol. Chem. 287 (2012) 25893–25904, 10.1074/jbc.M112.356345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Calderwood SK, Mambula SS, Gray PJ Jr., J.R. Theriault, Extracellular heat shock proteins in cell signaling, FEBS Lett. 581 (2007) 3689–3694, 10.1016/j.febslet.2007.04.044. [DOI] [PubMed] [Google Scholar]
- [24].Delpino A, Castelli M, The 78 kDa glucose-regulated protein (GRP78/BIP) is expressed on the cell membrane, is released into cell culture medium and is also present in human peripheral circulation, Biosci. Rep. 22 (2002) 407–420, 10.1023/A:1020966008615. [DOI] [PubMed] [Google Scholar]
- [25].Zhang Y, Liu R, Ni M, Gill P, Lee ASS, Cell surface relocalization of the endoplasmic reticulum chaperone and unfolded protein response regulator GRP78/BiP, J. Biol. Chem. 285 (2010) 15065–15075, 10.1074/jbc.M109.087445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bi X, Zhang G, Wang X, Nguyen C, May HI, Li X, Al-Hashimi AA, Austin RC, Gillette TG, Fu G, Wang ZV, Hill JA, Endoplasmic reticulum chaperone GRP78 protects heart from ischemia/reperfusion injury through Akt activation, Circ. Res. 122 (2018) 1545–1554, 10.1161/CIRCRESAHA.117.312641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Genereux JC, Qu S, Zhou M, Ryno LM, Wang S, Shoulders MD, Kaufman RJ, Lasmezas CI, Kelly JW, Wiseman RL, Lasmezas CI, Kelly JW, Wiseman RL, Unfolded protein response-induced ERdj3 secretion links ER stress to extracellular proteostasis, EMBO J. 34 (2014) 4–19, 10.15252/embj.201488896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Meunier L, Usherwood Y-K, Chung KT, Hendershot LM, A subset of chaperones and folding enzymes form multiprotein complexes in endoplasmic reticulum to bind nascent proteins, Mol. Biol. Cell 13 (2002) 4456–4469, 10.1091/mbc.e02-05-0311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang J, Lee J, Liem D, Ping P, HSPA5 gene encoding Hsp70 chaperone BiP in the endoplasmic reticulum, Gene. 618 (2017) 14–23, 10.1016/j.gene.2017.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.