

Abstract
Lignin is an abundant natural feedstock that offers great potential as a renewable substitute for fossil‐based resources. Its polyaromatic structure and unique properties have attracted significant research efforts. The advantages of an enzymatic over chemical or thermal approach to construct or deconstruct lignins are that it operates in mild conditions, requires less energy, and usually uses non‐toxic chemicals. Laccase is a widely investigated oxidative enzyme that can catalyze the polymerization and depolymerization of lignin. Its dual nature causes a challenge in controlling the overall direction of lignin‐laccase catalysis. In this Review, the factors that affect laccase‐catalyzed lignin polymerization were summarized, evaluated, and compared to identify key features that favor lignin polymerization. In addition, a critical assessment of the conditions that enable production of novel lignin hybrids via laccase‐catalyzed grafting was presented. To assess the industrial relevance of laccase‐assisted lignin valorization, patented applications were surveyed and industrial challenges and opportunities were analyzed. Finally, our perspective in realizing the full potential of laccase in building lignin‐based materials for advanced applications was deduced from analysis of the limitations governing laccase‐assisted lignin polymerization and grafting.
Keywords: enzyme catalysis, grafting, laccase, lignin, polymerization
Laccases as builders: Similar to their role in the formation of lignin in nature, laccases are capable of building advanced lignin‐based materials, given the optimum laboratory conditions. This Review evaluates the factors that affect laccase‐catalyzed lignin polymerization and grafting reactions. Patented applications of lignin‐based materials produced via laccase catalysis are highlighted and potential routes towards unlocking the full capacity of laccase in lignin valorization are proposed.
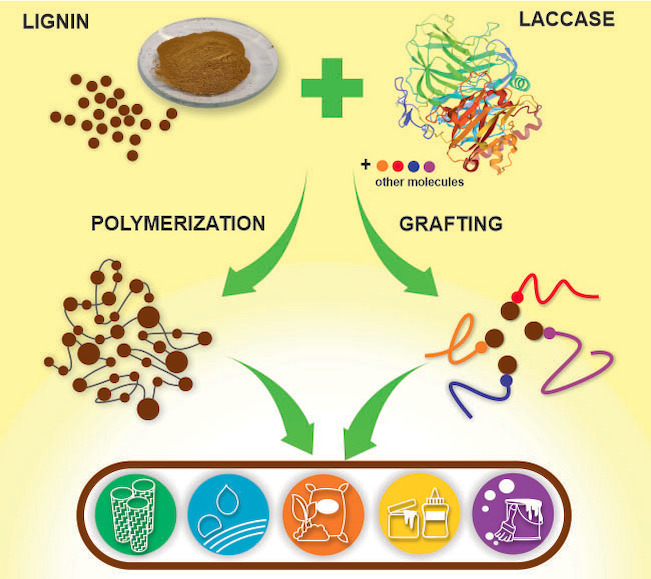
1. Introduction
The production of fossil‐based materials has long been associated with processes that negatively affect the environment, especially accelerating climate change through carbon dioxide emissions. In addition to their environmental impact, the depletion of non‐renewable fossil resources prompted a global call for greener and sustainable substitutes. This gave rise to the increasing research interest in valorizing bio‐based materials as alternatives for fossil resources in the past decades. Among these bio‐based materials, lignin attracts significant attention because of its abundance in plant biomass, polyaromatic structure, and unique properties.
Lignin is an enormous bio‐based feedstock. As one of the major constituents of the plant cell walls, lignin is the second most abundant biopolymer on earth, next to cellulose and hemicelluloses. It is a renewable material biosynthesized in plants with an estimated annual production of 5–36×108 tons globally. [1] Industrially, lignin is generated as a side‐stream product traditionally from pulp and paper industries, and recently also from cellulosic bioethanol production. At present, pulp and paper industries worldwide generate about 60 million tons of lignin each year. [2] By 2030, an additional 225 million tons of lignin annually is estimated to come from processing 750 million tons of biomass for cellulosic bioethanol production in the US alone. [3]
Lignin is a complex, highly heterogeneous aromatic biopolymer. It has a three‐dimensional, amorphous structure, constructed from the oxidative coupling of plant phenolic monomers (monolignols). Because of the randomness of linkage generation among monomers, the resulting lignin macromolecules are highly variable and exhibit different physicochemical features. [4] Lignin has various functional groups such as methoxy, phenolic hydroxy, alcoholic hydroxy, and carbonyl groups, which are in various proportions and have a profound impact on its properties. [5]
The complex and variable structure of lignin determine its various unique functions and characteristics. In plants, lignin provides structural support, water‐conductive properties, and defense against pathogens. [6] In soil, lignin is a precursor for the formation of humus, which is the recalcitrant organic matter that affects soil fertility. [7] Biodegradation of lignin by microbes such as fungi and bacteria generates modified organic compounds that accumulate in soil organic matter and aid in the formation of humus and humic substances. [8] Thus, lignin acts as a carbon storage pool both in the biosphere (in plants) as well as in the soil (as organic matter). In line with these unique functions of lignin in nature, industrial lignins isolated from biomass through various pulping methods also exhibit exceptional properties. Lignin is known to exhibit UV‐shielding, antimicrobial, antioxidant, hydrophobic, amphiphilic, emulsifying, and excellent binding properties. [9]
Despite the promising potential lignin could play in a bio‐based economy, its utilization is still limited to low‐value applications. Lignin is highly available, but a large proportion of it remains untapped and is only burned for energy recovery at the pulping facility in plant biomass. [10] Lignin is the largest reservoir of renewable aromatic biopolymers, but its conversion to high‐value aromatic chemicals is still far from industrial scale. One of the biggest drawbacks toward unlocking the full potential of lignin is its heterogeneity in structure, composition, and properties, which all vary depending on the source and mode of isolation.
Exhaustive efforts to valorize lignin led to various approaches that take advantage of lignin's unique features while overcoming its heterogeneity. Traditionally, effort has been put on depolymerization of lignin, that is, degradation and fragmentation of natural or technical lignins to smaller‐molecular‐weight soluble compounds and subunits. [11] This has been the aim in delignification processes in wood pulping and papermaking. Another route of lignin valorization is by utilizing the exceptional characteristics of lignin as a polymeric material itself or coupled with other molecules. In this approach, lignin is either further polymerized or grafted with other molecules to produce lignin‐based materials with properties tailored for specific applications. [12] Various methods based on chemical, [13] thermal, [14] and enzymatic techniques [15] were explored with the aim of constructing novel lignin‐based polymeric materials. Among these methods, enzymatic lignin valorization received great attention because it has environmentally more acceptable processing conditions. Enzyme‐catalyzed processes operate under mild conditions, require less energy input, and are performed most often in the absence of toxic solvents. [16] Enzymatic valorization of lignin adapts the concept of how enzymes in nature catalyze the biosynthesis and biodegradation of lignin. Plant, fungal, and microbial enzymes involved in these natural processes were harnessed and investigated for their capabilities to catalyze lignin polymerization or degradation under controlled laboratory conditions, with the aim of finding novel applications for lignin. [17]
Laccase is one of the oxidative enzymes that offers vast possibilities for lignin valorization via polymerization or grafting reactions. Laccases are phenol‐oxidizing metalloenzymes widely distributed in nature. [18] They act on diverse substrates, which can be further expanded by using laccase‐mediator systems, require only oxygen as co‐substrate, and release water as the first product.[ 5 , 16a , 16b ] These characteristics and their ability to act on phenolic and polymeric aromatic compounds make laccases enzymes of high interest for lignin valorization. As biocatalysts, laccases can either polymerize or depolymerize lignin and phenols, depending on the substrate, mediator, and radical reaction conditions.[ 16b , 19 ]
In this Review, we present a comprehensive summary of the applications of laccases in building lignin‐based materials via polymerization or grafting with other molecules. First, we present a general overview of lignin structure and the properties and sources of different types of lignins. Next, a brief summary about laccases and their mode of action towards lignin‐based materials is introduced. We then evaluate and analyze various studies that utilized laccase for lignin polymerization in order to identify key features that enable successful construction of lignin‐based materials via laccase‐assisted polymerization. Numerous reports have proven the potential of laccase‐mediated polymerization of various types of lignin, but this is the first time that an overall evaluation of the factors that direct the reaction towards polymerization is formulated in a Review. Advanced insights into these factors help future researchers in identifying crucial elements that must be considered when building lignin‐based materials via laccase catalysis.
Apart from polymerization, we highlighted the application of laccase in constructing lignin‐based materials via grafting of other molecules to lignin. Previous Reviews on this aspect have been more general, covering various types of enzymes [15a] or different types of substrates, including small molecules and polymers other than lignin.[ 15c , 16a , 16b ] Here, we focused only on the building of new materials with lignin as a starting material and laccase as the enzyme. Our aim is to highlight the potential of laccases in building novel materials from lignin via polymerization or grafting. In order to assess the potential of laccases in lignin valorization in an industrial setting, we looked for patented processes that utilized laccase in producing new lignin‐based materials. Finally, we present our perspectives on how to further expand the application of laccases for lignin valorization and how to overcome a few obstacles in the development of applicable processes.
2. Structure of Native and Technical Lignins
2.1. Formation and chemical structure of native lignin in plants
The formation of the aromatic biopolymer lignin (i. e., lignification) takes place at the final stages of biosynthesis of lignin through radical coupling of monolignols, which are first derived from phenylalanine. For a more detailed description of plant lignin biosynthesis, there are many review articles and book chapters available. [20] There are three types of monolignols, which are structurally p‐hydroxycinnamyl alcohols differing in the amount of methoxy substituents attached to the aromatic ring, adjacent to the aromatic hydroxy group. The monolignols (Figure 1a) are p‐coumaryl alcohol (no methoxy groups), coniferyl alcohol (one methoxy group in position 3 of the monolignol aromatic ring), and sinapyl alcohol (two methoxy groups at positions 3 and 5 of the aromatic ring). By definition, the position 1 of the aromatic ring of monolignol is the carbon bearing the side‐chain moiety, and its opposite (position 4) carbon possesses the phenolic hydroxy group.
Figure 1.

Structure of lignin: (a) monolignols involved in biosynthesis of different types of lignins and that differ in the amount of methoxyl groups present in the aromatic ring and (b) model structure of softwood/gymnosperm lignin comprising of G‐units (adapted from Ref. [20f]).
During biosynthesis, the dehydrogenation of lignin starts with one‐electron oxidation by laccases and/or peroxidases and loss of a proton. Laccases use oxygen (O2) and peroxidases use hydrogen peroxide (H2O2) for their oxidative power. In the resulting resonance‐stabilized radical the unpaired electron is delocalized throughout the aromatic ring and conjugated double bond (Figure 1b).[ 20a , 20c , 20d , 20e , 20f ] The monolignols primarily attach to the growing polymer (i. e., endwise polymerization), or they may couple more rarely through dimerization. The resulting phenylpropanoid units of lignin are called p‐hydroxyphenyl (H), guaiacyl (G), and syringyl (S) units. The proportions of the different lignin units are dependent on the plant species and specific tissue. Softwood (gymnosperm) lignin consists mainly of G units and some H units. Hardwood (angiosperm/dicot) lignin contains G and S units and minor amount of H units. Grass (monocot) lignin also contains G and S units, but a higher level of H units compared to hardwood lignin.[ 20a , 20c , 20f ]
The electron density of the delocalized monolignol radical or the end of the growing lignin polymer is concentrated on the β‐carbon and, consequently, the main linkage patterns of plant cell wall lignin involve coupling through this site. The main structural pattern of lignin is β‐aryl ether (β‐O‐4’) linkage comprising a share of around 80, 60, and 50 % for grass, hardwood, and softwood lignins, respectively.[ 20e , 20f ] Lignins also have other types of common linkages (Figure 1b), which are phenylcoumaran (β‐5’), resinol (β‐β’), dibenzodioxocin (5’‐5’’/α‐O‐4’/β‐O‐4’’), diaryl ether (4‐O‐5’), and spirodienone (β‐1’/α‐O‐α’). According to present knowledge, the biphenyl (5‐5’) linkage in native lignin is often associated with the dibenzodioxocin structure. [20e]
Softwood is composed mainly of G units, which offers the possibility for branching through position 5 of the monolignol. Therefore, softwood lignin has been considered to be more branched and polymerized in comparison to hardwood lignin. [10] However, there have been recent studies and discussions about the degree of linearity and/or branching of native softwood lignin.[ 20f , 21 ]
2.2. Properties of various types of technical and isolated lignins
Lignocellulosic plant biomass consists of cellulose fibrils surrounded by lignin and hemicelluloses.[ 20b , 22 ] The share of the different biopolymers in wood are: cellulose 33–51 %, lignin 21–32 %, and hemicelluloses 17–31 %. [20b] Isolation of lignin in a completely detached and native form is not possible because of the layered structure of the lignocellulose matrix in the plant cell walls, in which lignin and hemicelluloses are partially covalently linked to each other. [23] The situation is even more complex considering that, for the most optimal use of materials in future biorefineries, the isolation of all fractions of plant biomass in high yield would be desirable.
On a laboratory scale, milled wood lignin (MWL) is considered to be the most representative lignin when compared to native lignin. This well‐known assumption is supported by the more recently developed analytical and computational methods opening the structural arrangement of intact plant cell walls.[ 20b , 22b , 24 ] The MWL method includes ball milling of the plant material, which through degradation of the cell wall enables extraction of part of the lignin using dioxane‐water as the solvent.
Kraft lignin is the main technical lignin produced by the pulp and paper industry. Globally, the annual production of kraft pulp is around 130 million tons. [20e] During kraft pulping process, wood chips are processed with an alkaline aqueous solution of pulping chemicals (NaOH and Na2S) at elevated temperatures of 140–170 °C. [25] The main reaction resulting from the kraft process is the cleavage of β‐aryl ether linkages of lignin resulting in alkali‐soluble fragments. Dissolved lignin is one of the components in black liquor, from which the pulping chemicals are recovered and recycled back to the pulping process. [26] Part of the lignin in the black liquor is required as energy for the process and, consequently, burned for electricity and heating. [20e] However, the production of lignin is in excess of the energy required for the process. Kraft lignin can be isolated by precipitation with an acid, which protonates the phenolate anionic groups to their phenolic forms resulting in aggregation of lignin. This phenomenon has led to the development of the LignoBoost process, where CO2 is used as the acid, increasing the throughput of the pulp mill by enhancing recovery of chemicals, and similarly increasing the amount of available kraft lignin in the market.[ 26 , 27 ] The chemical structure of kraft lignin has been studied intensively; earlier in order to understand the chemistry of the kraft process, and more recently in order to use the material efficiently considering the globally reducing oil resources. Isolated kraft lignin contains many interunit linkages compared to native lignin. However, because of the degradation reactions, the amount of different structural patterns is significantly lower. [28] Kraft lignin is also considered more condensed than native lignin, at least because of accumulating 5‐5’ and 4‐O‐5’ structures. [28b] The phenolic hydroxy content of dissolved kraft lignin is much higher compared to the wood lignin. [25a] Similarly, as a result of the fragmentation, the molar masses of kraft lignins are lower compared to wood lignin, however, there is a high variation of the reported results (M w=200–20000 Da), which depend on the source of lignin, isolation method, and analysis method. [10] In addition to aromatic structures, kraft lignin also contains fatty acids originating from wood extractives and sulfur bound to the lignin during pulping reactions.[ 20e , 28d ]
Lignosulfonates are produced by the sulfite pulping process, which was a more common pulping process in the beginning of 20th century when it was patented, before being replaced almost completely by the kraft pulping process.[ 20b , 29 ] The global production of lignosulfonates is around a million tons per year. [29] In the process, sulfite or bisulfite is present in the pulping liquor, depending on the pH of the solution, which, is dependent on the base used (Ca2+, Mg2+, Na+, or NH4 +).[ 20b , 26 ] The temperature of the process is also dependent on the pH, varying from 125–140 °C for acidic conditions to 160–180 °C for neutral conditions. [20b] During the process, lignin is sulfonylated, for example from the α‐position of the β‐aryl ether structure, enhancing the solubility of lignin. Therefore, in contrast to kraft lignin, which is soluble at alkaline pH, lignosulfonates are soluble over a wide pH range. [26] In addition to the sulfonylation reaction, cleavage and condensation reactions take place, while the route of the reactions is dependent on pH. In comparison with other technical lignins, the phenolic content of lignosulfonates is lower and their molar masses are higher. [30] According to a recently developed method based on multiangle laser light scattering (MALLS), the range of M w for different types of lignosulfonates is in the range of 15000–60000 Da. [31] In addition to the sulfite processing, lignosulfonates can be prepared using kraft lignin as the starting material. [20b]
Organosolv lignin is a process in which organic solvents are used for pulping to produce sulfur‐free lignin.[ 20e , 26 ] The organic solvent used in combination with water can be, for example, methanol, ethanol, dioxane, tetrahydrofurane, acetone, or ethylene glycol. In the Alcell process wood chips were treated with aqueous ethanol at 180–200 °C for 30–90 min. Organosolv liquor can also include an acid catalyst, such as HCl, oxalic acid, acetic acid, or formic acid, which at the temperature of 140–190 °C induces degradation of both hemicelluloses and lignin. In acidic conditions, the organosolv process is based on the hydrolysis of β‐aryl ether structure. [20e] Organosolv process without added acid catalyst usually involves the removal of acetyl groups of hemicelluloses, which decreases the pH of the solution to a level prompting the hydrolysis of some of the β‐aryl ether structures (i. e., auto‐catalyzed system). Delignification in non‐catalyzed conditions is not so clear and assumingly induced by the degradation of α‐O‐4’ structures. According to present knowledge, however, the α‐O‐4’ structures are rare, and most often associated with phenylcoumaran or dibenzodioxocin structures. As a result of the degradation of β‐aryl ether structures in acidic or auto‐catalyzed conditions, the amount of phenolic hydroxy groups is increased. [32] The molar masses of organosolv preparations are in general lower compared to MWL samples from the same biomass (M w=2000–9000). [10] However, the molar mass is dependent on the source of the lignin and the severity of the treatment, including parameters such as time and the amount of organic solvent.
In addition to these most common technical lignins, there is a constant development of different isolation methods. The treatment of biomass can include, for example, enzymatic hydrolysis, treatment with hot water or dilute acids, alkaline treatment, and treatment with ionic liquids. Furthermore, in developing new types of biorefineries, attention will be paid to isolation methods that will not severely modify the structure of lignin, that is, “lignin‐first” biorefineries. [33]
3. Laccase and Mechanism of Action on Lignin
Laccases (EC 1.10.3.2, benzenediol: oxygen oxidoreductase) belong to the multicopper oxidase (MCO) protein superfamily, which is classified to CAZy family AA1 (www.cazy.org). [34] The protein folding structure of laccase contains four copper atoms, which differ in their coordination and spectroscopic properties.[ 34 , 35 ] The primary site of oxidation of the reducing substrate (like a phenolic compound) is the type‐1 copper center (T1), where copper is typically coordinated by a single cysteine and two histidine residues. T1 copper is responsible for the blue color of the enzyme. Trinuclear type T2/T3 copper center includes one type‐2 (T2) and two type‐3 (T3 and T3’) coppers, which are coordinated by conserved histidines. Electrons from T1 center are transferred to T2/T3 coppers, where the reduction of oxygen to water takes place.
Laccases are found in plants, fungi, insects, and bacteria with varying roles.[ 18b , 35 , 36 ] In plants the known roles of laccases include lignification, wound healing, and polymerization of seed coat. [37] Filamentous fungi, especially in the phyla Ascomycota and Basidiomycota, express several laccase isoenzymes encoded by multigene families.[ 35 , 38 ] Secretion of laccases with peroxidases and other enzymes is especially common in plant biomass degrading fungi and bacteria active in deadwood, plant litter, and organic matter containing environments like forest, plain, and agricultural soils. [39] The polymerizing activity of laccases is probably also related to detoxification of phenolic compounds in the soil environments through incorporating them into the humic substances and soil mineral layers, and secretion of laccases to protection against fungicides.[ 36 , 38d , 39 , 40 ] Bacterial laccases have specific functions, for example, in formation of bacterial endospores, like in Bacillus subtilis.[ 18c , 41 ] One of the best‐known roles of insect laccases is taking part in the cuticle sclerotization, that is, hardening of cuticle comprising of chitin and proteins.[ 18c , 42 ] Sclerotization involves oxidation of catecholamines to quinones and quinone methides, which react further to form cross‐links with the amino acid residues in the proteins. [43]
In contrast to many other enzymes, laccases have a wide substrate range, including, for example, various types of phenols and polyphenols (including lignin), polyamines, aryl diamines, and some inorganic ions.[ 16b , 16c , 18b , 35 , 36 ] The substrate range of laccases can be even wider by the use of so‐called mediators, which are small molecules that act as intermediate electron shuttles.[ 5 , 44 ] The mediators are first oxidized by laccases, and the resulting oxidized mediators can react further with some molecules, which are not accessible otherwise by these enzymes. Some of the laccase mediators are small phenols, including, for example, vanillin and syringaldehyde, so‐called natural mediators, which might be present in isolated lignin preparations and in technical lignins. [45] The reactions leading to attack and oxidation of lignin non‐phenolic units require a laccase‐mediator system.[ 16b , 16c , 46 ]
During the catalytic cycle of laccases, four reducing substrate molecules are oxidized by one‐electron transfer, and finally the four electrons are transferred to reduce oxygen to two water molecules. For a thermodynamically spontaneous reaction, the redox potential of the reducing substrate molecule must be equal or fall below the redox potential of the oxidized (resting) enzyme. Redox values of T1 Cu sites in fungal laccases vary within the range of 420–790 mV versus normal hydrogen electrode (NHE). [35] Oxidation of the substrate at the T1 site is the rate‐determining step of the laccase catalytic cycle. [34]
The rate‐determining step in the laccase‐catalyzed oxidation of a phenolic substrate is the removal of one electron followed by a fast removal of a proton, resulting in formation of a resonance‐stabilized phenoxy radical.[ 16b , 34 , 36 ] The radical reactions following the laccase‐catalyzed oxidation are mainly dependent on possibilities provided by the structure of the reacting molecules. Because of the complex structure of lignin, the reaction mechanisms on a molecular level have been mainly studied using smaller model compounds representing structurally versatile lignins. The reactions of different lignin model compounds with and without the presence of a mediator have been reviewed.[ 46 , 47 ] Studies performed with laccases and lignin model compounds showed that phenolic, dimeric β‐1’ and β‐O‐4’ model compounds treated with laccases resulted in cleavage of Cα−Cβ bonds or aryl−Cα bonds, and oxidation of Cα−OH to Cα=O. [48] Another type of transformation found in laccase treatments of 2,6‐dimethoxyphenol and vanillyl alcohol, methoxylated phenol monomeric lignin model compounds, is demethylation.[ 38c , 49 ] Regarding the polymerization (or coupling) reactions, the formation of lignin dehydropolymer from monolignol is well known. With monomeric guaiacyl‐type model compounds it has been shown that laccases can induce the formation of dimeric 5‐5’ and 4‐O‐5’ compounds, as well as Cα‐oxidation.[ 49a , 50 ] A further reaction of a dimeric 5‐5’ model compound results in formation of dibenzodioxepin‐type tetrameric structures as well as Cα‐oxidation. [50b] With sulfonylated derivatives of the same phenolic monomer precursors, coupled 5‐5’ and 4‐O‐5’ dimers were produced since the Cα‐position was occupied by a sulfonyl group preventing oxidation. [51] One interesting aspect of the laccase‐catalyzed reactions of lignin model compounds is the effect of reaction conditions on the product distribution. The product distribution is affected by, for example, the pH and dosage of the enzyme, which may be explained by the thermodynamic and kinetic factors.[ 38a , 50a , 52 ]
In addition to model compound studies, there are some reports about the polymerization of different lignins: MWL, kraft lignin, and sulfonylated kraft lignin. [53] Although the detailed reaction pathways could not be explained in detail at a molecular level, some chemical transformations were identified. In general, during laccase‐catalyzed modification of lignins, the amount of carbonyl groups were increased (e. g., indicating Cα‐oxidation), the changes observed for the aromatic protons indicated formation of condensed structures through 5‐position, and in a couple of reports demethylation was also observed.[ 32 , 53b , 53c ] The content of phenolic hydroxy groups in treated lignins was also determined in all studies, but the results were varying a lot, for an unknown reason, which would benefit from a more systematic investigation. However, the change in phenolic content during polymerization was explained accordingly: during laccase treatment, a decrease in phenolic content can be associated with coupling through the phenolic group; unchanged level indicates the reaction is taking place in side‐chains, and increased level means cleavage of bonds to create new phenolic hydroxy groups. In order to achieve polymerization of lignin, optimization of the reaction conditions is essential, as pH, temperature, and time have impact on the molar mass of the laccase‐oxidized lignin.[ 53b , 53c ] This result may reflect the varying product distributions observed in model compound studies mentioned at the end of the previous paragraph. The lack of thorough knowledge regarding the laccase‐catalyzed polymerization of lignin is evident, as after decades of research it was only just very recently reported that H2O2 is formed during the laccase‐catalyzed oxidation of lignin, a phenomenon that is not observed during oxidation of smaller compounds. [54] The formation of radicals during laccase‐catalyzed oxidation of lignin can be quantified by electron paramagnetic resonance (EPR) spectroscopy, which has shown that different laccases have differing rates of oxidation towards organosolv lignin. [55] Furthermore, the EPR studies have shown that the radical decay reactions, which take place after oxidation, are indeed independent of the enzyme. However, the EPR technique does not enable identification of the formed products and the reaction pathways, which are yet to be identified.
4. Factors Affecting Laccase‐Catalyzed Lignin Polymerization
Laccases play a dual role, as they both build and degrade lignin in nature.[ 11a , 20d , 56 ] This dual nature of laccases causes a challenge in directing the overall reaction of laccase‐lignin catalysis to either polymerization or depolymerization. Achieving successful polymerization of lignin during laccase catalysis requires systematic control of various factors. In the following sections, the different factors that affect laccase‐catalyzed lignin polymerization are discussed in detail, and the studies cited herein are summarized in Table 1 and Figure 2.
Table 1.
List of studies cited in the text and their major findings on laccase‐catalyzed lignin polymerization arranged according to publication date.
|
Substrates |
Laccase origin |
Highlighted findings |
Ref. |
|---|---|---|---|
|
lignosulfonates |
Trametes hirsuta |
low‐molecular‐weight lignin fraction favored polymerization |
[63] |
|
flax soda, mild acidolysis spruce, eucalyptus organosolv lignins |
Trametes hirsuta |
lowest degree of polymerization in spruce mild acidolysis lignin attributed to low solubility in the reaction mixture |
[59] |
|
spruce alkali, steam‐exploded wheat straw alkali lignins |
Mycelia Sterillia YY‐5 (MS‐Lac) from Rhus chinensis |
molecular weight increased for both lignins; MS‐Lac more effective in wheat straw alkali lignin than in spruce alkali lignin |
[62a] |
|
various types of lignosulfonates |
Trametes villosa, Myceliophthora thermophila |
high redox TviL gave higher increase in molecular weight than the low redox MtL; polymerization improved at higher initial lignin concentration and at higher enzyme activity for MtL; degree of sulfonation influenced the extent of polymerization |
[65] |
|
lignosulfonates |
Trametes villosa, Trametes hirsuta |
Tvil more effective than ThL in increasing the molecular weight; use of mediator improved polymerization |
[66] |
|
birch organosolv, mixed hardwood organosolv, steam‐exploded softwood and hardwood lignins |
Trametes hirsuta, Melanocarpus albomyces, Thielavia arenaria TaLcc and TaLcc2 |
highest polymerization observed at pH 6 where the lignins are better dissolved; higher polymerization in hardwood than in SW lignin; TaLcc1 and 2 were less effective than MaL and ThL |
[60] |
|
birch organosolv, spruce organosolv, hardwood and softwood kraft lignins |
Melanocarpus albomyces, Streptomyces ipomoea |
low initial molecular weight of lignin and high phenolic hydroxy groups favored polymerization; efficiency of SiL improved and made similar to MaL with the use of mediator |
[62b] |
|
eucalyptus kraft lignin |
Myceliophthora thermophila |
organic solvent‐based fractionation yielded fractions with varying reactivity towards laccases; decrease in syringyl/guiacyl ratio seemed to favor polymerization; low‐molecular‐weight mediators had no effect on polymerization |
[61a] |
|
mixed hardwood organosolv, wheat straw soda, mixed sarkanda grass/wheat straw soda, Indulin AT softwood kraft, alkali‐ pretreated wheat straw lignins |
Trametes versicolor |
fractionation enabled the isolation of low‐molecular‐weight lignin fractions favorable for polymerization; highest increase in molecular in alkali wheat straw lignin |
[61b] |
|
enzyme saccharified, lignosulfonate, steam‐exploded pine, stea‐exploded eucalyptus, karft lignins |
Trametes hirsuta |
polymerization correlated well with low molecular weight and high amount of monomeric phenolics in the starting lignin |
[64] |
|
hardwood organosolv, softwood Indulin AT kraft lignins and softwood lignosulfonates |
Trametes villosa, Myceliophthora thermophila |
highest increase in molecular weight observed in lignosulfonates for both laccases; oxygen supply had no clear correlation with increase in molecular weight but reduced the reaction time |
[67] |
|
lignosulfonates |
Trametes villosa, Aspergillus oryzea |
polymerization improved with the use of mediators |
[68] |
|
lignosulfonates |
Myceliophthora thermophila |
polymerization was favored with continuous oxygen supply over use of mediators |
[69] |
|
lignosulfonates and kraft lignin |
Myceliophthora thermophila |
lignosulfonates polymerized more than kraft lignin using immobilized MtL; increasing MtL dosage improved polymerization only for lignosulfonates |
[70] |
|
hardwood organosolv lignin and softwood lignosulfonates |
Trametes versicolor |
lignosulfonates with higher number of phenolic hydroxy groups per molecule had higher degree of polymerization than the organosolv lignin |
[71] |
|
lignosulfonates |
Trametes hirsuta |
low degree of polymerization at pH 3, 4, and 8; extensive polymerization at pH 6 and 7; increase in temperature increased the rate of oxidation |
[72] |
|
hardwood lignosulfonates |
Myceliophthora thermophila |
increasing enzyme dosage increased the extent of polymerization |
[73] |
Figure 2.
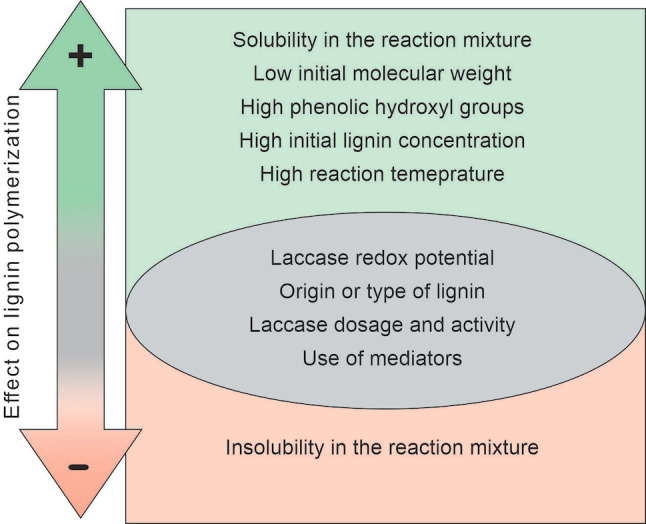
Factors affecting the extent of laccase‐catalyzed lignin polymerization summarized from references cited in the text. Factors in the green region generally favor polymerization and the factor in the orange region generally hinders polymerization. Factors in the central grey region are not generalized to favor or inhibit polymerization and depending on the reaction conditions can yield variable results.
4.1. Solubility
Lignin has poor solubility in water and in common organic solvents, and this is one of the limitations to overcome in valorizing it. [57] Similarly, this limitation in solubility is reflected in laccase‐lignin catalysis as early studies of this system have been broadly investigated but limited to water‐soluble lignin, modified lignin, or lignin model compounds. [58] Whether the final aim is to polymerize or depolymerize lignin, the solubility of the starting lignin in the reaction medium is an important factor to consider for successful laccase catalysis. For example, Mattinen et al. [59] studied the oxidative polymerization of three different technical lignins by a high‐redox‐potential Trametes hirsuta laccase (ThL). Their findings showed that the lowest degree of polymerization was in spruce mild acidolysis lignin, which was insoluble in the aqueous medium buffered at pH 5. Similarly, van de Pas et al. [60] found that extensive polymerization was favorable at pH 6, where the lignin was in dissolved state. In contrast, at pH 5, where the lignin was in a dispersed state, extensive oxidation of the lignin surfaces was favored.
Efforts made to address this limitation in solubility include fractionating lignin to small‐molecular‐weight fractions using organic solvents[ 60 , 61 ] or performing the reaction at alkaline pH, where lignin is more soluble, by using alkali‐stable laccases. [62]
4.2. Molecular weight and phenolic groups
The molecular weight of lignin that varies widely among lignin types is one of the factors affecting the recalcitrance of biomass. [10] It is not surprising that this property has a profound effect on the reactivity of lignin towards laccases.
The effect of initial molecular weight of lignin on laccase catalysis was clearly demonstrated in the work of Leonowicz et al., [63] who investigated the behavior of fractionated spruce lignosulfonates with Trametes versicolor laccase (TvrL). It was found that the low‐molecular‐weight fraction (1 kDa) polymerized upon incubation with TvrL, whereas depolymerisation was observed in the high‐molecular‐weight fraction (97 kDa). Several other studies also observed a higher extent of polymerization in lower‐molecular‐weight lignin than in higher‐molecular‐weight polymers.[ 60 , 64 ] A common observation linked to this finding is treatments that decrease the molecular weight of lignin increase the reactivity, making lignin a more effective substrate in these reactions. [64]
The functional groups that were associated with the increased reactivity of lignin with decreasing molecular weight were the phenolic hydroxy groups. Thus, in addition to low initial molecular weight, the amount of phenolic hydroxy groups in lignin was found to correlate well with the extent of polymerization. Moya et al. [62b] investigated the polymerization of various types of lignin with Melanocarpus albomyces laccase (MaL) at alkaline pH. They found that the highest increase in molecular weight was in the softwood kraft lignin, which had the lowest molecular weight and the highest proportion of phenolic hydroxy groups among the investigated lignins, which included softwood and hardwood organosolv and hardwood kraft lignins. The effect of phenolic groups was further supported by West et al., [64] who studied the reactivity of different types of industrial lignins (kraft, lignosulfonate, steam exploded, and enzyme saccharified) with ThL at pH 5.5. Their findings showed that molecular weight and the amount of free monomeric phenolic compounds affected the reactivity of lignin more than the solubility. The lignosulfonates, despite being the most soluble at pH 5.5, exhibited the smallest increase in molecular weight after ThL treatment. It appeared that higher reactivity and the extent of polymerization correlated well with lower molecular weight and higher amounts of monomeric phenolic compounds in the lignin substrate. [64]
The number of phenolic hydroxy groups, however, seemed to have a more pronounced effect on the extent of polymerization than having a low initial molecular weight. This was shown in the work of Gillgren et al., [71] who compared the extent of polymerization of a softwood lignosulfonate (M w=4.4–61 kDa) with a hardwood organosolv lignin (M w=1.9–5 kDa) during incubation with TvrL. The softwood lignosulfonates, which had a higher molecular weight and higher number of phenolic hydroxy groups per lignin macromolecule (6.5 vs. 1.4), showed exponential growth in molecular weight with time, while that of the hardwood organosolv lignin had a linear growth. It has to be noted that in the study the number of phenolic hydroxy groups per molecule was calculated from the experimental values of number‐average molecular weight and amount of phenolic hydroxy groups per g of lignin. Even though there was no significant difference in the number of moles of phenolic hydroxy groups per g of the lignin (1.8 vs. 1.4), a clear difference was observed when those values were converted to per macromolecule basis. [71] The high number of phenolic hydroxy groups per molecule translated to a higher number of active groups that were able to participate in crosslinking reactions, either intra‐ or intermolecularly. [71]
4.3. Type of laccase
Different types of laccases, depending on the origin and mode of isolation, display wide variation in properties such as pH and optimal temperature, redox potential, and substrate specificity.[ 18b , 38a ] These differences affect the reactivity of laccases with lignin and limit the reaction conditions, particularly the pH and temperature, during incubation.
The redox potential is often the property of laccase that is most correlated to the outcome of lignin‐laccase catalysis. This comes as no surprise because the redox potential of laccase plays a significant role in the overall kinetics and energetics of the rate of electron transfer during the oxidation of the substrate. [19b] The redox potential of laccase varies widely, with fungal laccases often having higher redox potential than laccases of plant or bacterial origin.[ 18b , 19b ]
In the performance between the high‐redox Trametes villosa laccase (TviL) and the low‐redox Myceliophthora thermophila laccase (MtL) during incubation with different types of lignosulfonates, it was found that TviL yielded a higher increase in molecular weight after 24 h of incubation. [65] Accordingly, TviL, with its higher redox potential was capable of oxidizing not only the phenolic subunits of lignin but also the non‐phenolic subunits. On the other hand, the findings of van de Pas et al. [60] in their investigation on the reactivity of four fungal laccases with various types of lignin showed a different result. Even though the high‐redox‐potential TviL displayed the highest degree of oxidation among the laccases studied, it was the low redox potential MaL that caused the highest degree of polymerization. Furthermore, for both the low‐ (MaL) and the high‐redox‐potential (ThL) laccases, polymerization was generally higher in the hardwood than in the softwood lignin. It was interesting to note from this study that the other two laccases from Thielavia arenaria have a similar redox potential as the MaL but were less effective than MaL and ThL; which is a clear manifestation that the behavior of laccase enzyme in polymerizing lignin cannot be generalized based on redox potential alone. Another study that demonstrated the differing reactivity of different laccases even with almost similar redox potential is the comparison of the polymerization of lignosulfonates using ThL and TviL. [66] Both are high‐redox‐potential laccases, but TviL displayed five times higher efficiency than ThL in increasing the molecular weight of lignosulfonates after 17 h of incubation.
Apart from redox potential, the pH optimum of the laccase is an essential factor to consider in laccase‐lignin reactions as it limits the pH range of the reaction mixture. Most of the fungal laccases have an optimal pH in mildly acidic range (pH 4–6), [38d] which is not the optimum for lignin dissolution. Thus, laccases that are stable under alkaline conditions have been of recent interest to overcome the insolubility of lignin in acidic reaction mixtures. An example is the extracellular bacterial laccase produced by Streptomyces ipomoea (SiL), which is known for its pH insensitivity and salt‐resistant characteristics. SiL was used to polymerize various types of lignin at pH 7–10, and the efficiency of polymerization was compared to the fungal laccase MaL. [62b] It was shown that, for both enzymes, polymerization occurred in the alkaline pH range for various types of lignin, but the efficiency of SiL was less than that of MaL.
4.4. Reaction conditions
Reaction conditions play a crucial role in directing the overall outcome of laccase incubation with lignin towards polymerization. The wide variability in the properties of lignin coupled to the varying performance of the laccase at various conditions clearly indicates the need to find the optimum reaction conditions, which would favor the target outcome. Among the parameters that have been shown to affect the extent of polymerization of lignin during laccase catalysis were the pH, enzyme dosage and activity, initial lignin concentration, and temperature.
The pH of the reaction mixture is an important parameter affecting both the activity of laccases and the properties of lignin, in particular its solubility. A similar finding to that observed by van de Pas et al. [60] showed a higher degree of polymerization of lignosulfonates with ThL incubations at pH 6 and 7 than at pH 3–5. [72] At pH 8, where the lignosulfonates were also completely soluble, significant polymerization did not occur. Though not mentioned, the possible reason could have been the loss of activity at pH 8 of ThL, which has a pH optimum in the acidic range.
The effect of increasing the enzyme dosage and activity on lignin polymerization vary. Magina et al. [73] varied the MtL dosage (42–500 U g−1 of lignosulfonates) and found an increasing degree of polymerization with increasing MtL dosage. An 11‐fold increase in molecular weight of the lignosulfonates was attained at the highest dosage within 90 min of reaction time at the optimized conditions (40 °C and pH 4.5). On the other hand, the findings of Areskogh et al. [65] suggested that the increase in molecular weight with increasing enzyme activity was enzyme specific. In their study, the increase in the molecular weight of various types of lignosulfonates with increasing enzyme activity was only significant with the low‐redox MtL but not with the high‐redox TviL. Not only was the effect of increasing the dosage enzyme specific, but also substrate specific. In the polymerization of lignosulfonates and kraft lignin during incubation with immobilized MtL, doubling the enzyme activity increased the extent of polymerization of lignosulfonates but not of the kraft lignin. [70]
Increasing the lignin concentration in the reaction mixture seemed favorable for polymerization as the number of lignin substrate molecules that can participate in the crosslinking increased. In the work of Areskogh et al., [65] increasing the initial concentration (1–100 g L−1) of the water‐soluble lignosulfonates improved polymerization, regardless of the type of laccase enzyme used. However, it should be noted that increasing the concentration of other types of lignin to as high as 100 g L−1, especially in a slightly acidic aqueous system, is possibly not feasible because of the poor water solubility of lignin in general. [57b]
The reaction temperature is expected to affect laccase‐catalyzed lignin polymerization because temperature influences both the catalytic activity of the laccase [38d] and the solubility of lignin. [74] Bacterial laccases are often more thermally stable than fungal laccases, which have a catalytic activity often dropping rapidly at temperatures above 60 °C.[ 38a , 38d ] Despite the importance of thermotolerant and thermostable laccases in industrial applications,[ 38a , 75 ] most of the laccase‐catalyzed lignin polymerization reactions cited herein were performed at ambient conditions or at a constant temperature slightly above ambient. Legras‐Lecarpentier et al., [72] however, investigated the effect of increasing the reaction temperature (from 23 up to 60 °C) on the ThL‐catalyzed polymerization of lignosulfonates. It revealed that the rate of oxidation rapidly increased with increasing temperature, and the highest molecular weight was observed at 60 °C within 90 min.
4.5. Use of mediators
The oxidation of non‐phenolic groups, which typically have higher redox potential than the laccases, by using mediators during lignin‐laccase catalysis could promote either bond cleavage or coupling reaction, depending on the type of mediators, laccases, lignin, and reaction conditions.[ 5 , 44 ]
Although most of the efforts on laccase‐catalyzed lignin polymerization proved to be feasible in the absence of mediators, a number of studies investigated the effect of laccase‐mediator systems in lignin polymerization. Nugroho Prasetyo et al. [66] observed extensive polymerization without degradation of the aromatic backbone of lignosulfonates upon incubation with ThL or TviL and 1‐hydroxybenzotriazole (HBT) as the mediator. In particular, the incubation in TviL‐HBT systems led to 572 % increase in molecular weight and improved the dispersion property of the lignosulfonates. Detailed chemical characterization revealed new ether and C−C, aryl–aryl, or aryl–alkyl linkages, which were deduced to be formed from condensation reactions of non‐phenolic units in the lignosulfonates. Similarly, Euring et al. [68] observed enhanced polymerization of lignosulfonates with TviL and caffeic acid (CA) or vanillyl alcohol (CA) as mediator. The molecular weight was doubled after 30 min of incubation in the TviL‐CA system, which was found to be more efficient than the TviL‐VA system. Investigation of the electrochemical properties of CA and VA in the presence of TviL by cyclic voltammetry showed that the radicals formed by CA were possibly more stable than those of VA.
In another study, the efficiency of the bacterial laccase, SiL, in polymerizing spruce organosolv lignin at pH 10 was found to double in the presence of acetosyringone as mediator. [62b] The findings revealed that a similar performance as that of MaL could be achieved by the acetosyringone‐SiL system in polymerizing the spruce organosolv lignin with the advantage of performing the reaction at highly alkaline conditions, which is favorable for lignin dissolution. On the other hand, Gouveia et al. [61a] reported that the addition of low‐molecular‐weight additives (violuric acid, syringaldehyde, pyrocatechol, guaiacol, vanillin, and polyphenon 60) had no significant effect on the polymerization of kraft lignin with MtL. Even though an increase in molecular weight was observed in the laccase‐mediator‐treated lignin, none of the additives surpassed the polymerizing efficiency of MtL alone. It has to be noted that in many other reports, depolymerization was the dominant outcome of lignin incubation in laccase‐mediator systems.[ 5 , 49a , 58 , 76 ] However, the aim of those studies was to deconstruct lignin structure into small aromatic molecules, which is not covered by this Review.
4.6. Other factors: oxygen supply, pre‐treatment, degree of sulfonation
One of the advantages of laccase‐catalyzed reaction is that the co‐substrate is only oxygen, and it is not surprising that the effect of oxygen supply during incubation with laccase is a factor to consider. Ortner et al. [67] investigated the effect of external oxygen supply on the laccase‐catalyzed polymerization of various types of lignin. It appeared that the modes of supplying oxygen, which include shaking, aeration, or oxygenation (supplying pure oxygen), had no clear correlation to the increase in molecular weight of the lignin. Supplying oxygen to the system, however, ensured complete oxidation (indicated by a decrease in fluorescence intensity to almost zero) and shortened the laccase‐lignin incubation time. A follow‐up of their work investigated the effect of oxygen supply and addition of mediators to MtL‐catalyzed polymerization of lignosulfonates. [69] It was shown that continuous oxygen supply was more important than the addition of mediators in polymerizing the lignosulfonates. Apparently, the oxidation of non‐phenolic moieties seemed to counteract polymerization, that is, an increase in the concentration of the mediator resulted in a decrease in lignin molecular weight.
Pre‐treatment processes like fractionation in order to lessen the heterogeneity of lignin proved to be an effective means of improving lignin polymerization with laccase. When Gouveia et al. [61a] fractionated eucalyptus kraft lignin, acid‐based fractionation yielded lignin fractions showing only slight differences in reactivity with laccases whereas the organic‐solvent fractionation yielded significant differences. The organic‐solvent fractionation led to isolates of different molecular weight ranges, phenolic content, and S/G ratios. Unlike previous reports, finding correlation of phenolic content with molecular weight increase, their work showed that the phenolic content of the fractions had no direct correlation with the extent of polymerization. Interestingly, the decrease in S/G ratios was found to favor polymerization.
Fractionation not only lessened the heterogeneity of the lignin, but also enabled the isolation of low‐molecular‐weight soluble fractions as demonstrated in the work of Fitigău et al. [61b] Various types of lignins were fractionated in 50 : 50 acetone/water mixture and the isolated fractions were treated with TviL. Despite the slight decrease in the activity of TviL in the presence of acetone, polymerization still occurred and was higher in wheat straw lignin than in hardwood lignin.
The degree of sulfonation in lignosulfonates seemed to affect the extent of polymerization during laccase treatment. The study of Areskogh et al. [65] on the laccase‐catalyzed polymerization of four different types of commercial lignosulfonates showed that the lignosulfonates with the lowest degree of sulfonation exhibited the lowest degree of polymerization. From the previous work of the same group based on lignin model compounds, it was deduced that sulfonation of the α‐carbon prevented unproductive radical coupling, thereby promoting polymerization. [51] A separate study of Hilgers et al. [78] using model compounds further indicated that lignin sulfonation drives the overall outcome of laccase catalysis with lignin and mediator systems towards polymerization.
5. Laccase‐Catalyzed Grafting to Lignin
Grafting refers to the formation of a covalent bond between a macromolecule and a small molecule.[ 15c , 16a ] In polymer chemistry, the macromolecule is the main polymer chain where small molecules covalently bond as side chains. There are three modes how the small molecules can be grafted to the main polymer chains: grafting‐to, grafting‐from, and grafting‐through.[ 12d , 15c ] In the grafting‐to approach, the small molecule or a small polymer with a reactive chemical group covalently binds to a compatible reactive group present in the main polymer chain. In the grafting‐from method, a small molecule, which can be a monomer, binds and polymerizes to the initiating sites present in the main polymer backbone. Lastly, in the grafting‐through method, two types of monomers are simultaneously polymerized, one monomer creating the main polymer chain, and the other monomer, forming the side chains. [15c]
In the context of lignin derivatization, grafting is the binding of small molecules to lignin, which acts as the main polymer chain, referred also as the polymer core. Because lignin lacks a well‐defined chemical structure and has an irregular three‐dimensional networked structure, grafting‐to and grafting‐from approaches are reported more than grafting‐through. [12d] Grafting molecules to lignin has been a promising approach to produce new lignin hybrids with tunable properties, which depend on the chemical characteristics, and proportion and distribution, of the small molecules on the lignin core. Grafting small molecules to lignin, similar to other polymers, requires the use of catalysts, and laccases have shown great potential to catalyze grafting reactions in lignin. The following sections highlight the various laccase‐mediated grafting in lignin, and the summary is presented in Table 2. The reaction medium used during the synthesis was especially identified as it affects both the reactivity of lignin and the activity of laccases.
Table 2.
Molecules grafted to lignin via laccase‐assisted reactions.
|
Grafted compounds |
Laccase origin |
Substrate |
Reaction medium |
Potential application |
Ref. |
|
|---|---|---|---|---|---|---|
|
acrylic monomers |
|
Trametes versicolor |
organosolv lignin, Indulin AT lignosulfonates |
aq. dioxane |
engineering plastics |
[79,81–83] |
|
phenolic compounds |
vanillic acid |
Trametes versicolor |
organosolv lignin, Indulin AT |
aq. dioxane |
engineering plastics |
[79] |
|
water‐soluble phenols |
Trametes villosa |
|
dioxane |
– |
[84] |
|
|
plant‐based phenols (gallic acid, tannic acid) |
Myceliophtora thermophila |
soda lignin, kraft lignin |
aq. buffer (pH 8.5) |
adhesive |
[85] |
|
|
nitrogen‐containing compounds |
isocyanates (4,4′‐methyl‐ enediphenyl diisocyanate) |
Trametes versicolor |
organosolv lignin, Indulin AT |
aq. dioxane |
engineering plastics |
[79] |
|
N−OH‐type mediators |
Trametes versicolor, Pleurotus ostreatus |
cellulase‐treated wheat straw enzyme lignin, beech organosolv lignin |
aq. buffer (pH 4.8) |
alternative route for lignin modification |
[86] |
|
|
glucosamine, tripeptide |
Trametes versicolor |
various types of hardwood, softwood, and grass lignin |
50 : 50 acetone/water |
lignin‐based macro‐ molecular structures |
[87] |
|
|
polyethylimine, chitosan, soy protein |
Galerina sp. |
kraft lignin |
aq. alkali |
adhesive |
[88] |
|
|
inorganic silanes |
|
Trametes hirsuta |
softwood and hardwood lignosulfonates, kraft lignin |
aq. buffer (pH 4.5) |
adhesive |
[89] |
|
cellulose |
|
not specified |
lignosulfonates |
aq. buffer (pH 5.5) |
– |
[90] |
|
thiols [tris(2‐mercap‐ toethyl)amine] |
|
Trametes villosa |
kraft lignin |
aq. dioxane |
lignin‐based thermoplastic |
[91] |
The work of Milstein et al. [79] is among the pioneering works that demonstrated the potential of laccase to catalyze the grafting of lignin with small molecular weight compounds. In particular, the reaction between acrylic monomers and lignin, a reaction initially performed using radical initiators such as hydroperoxides and chloride ions [80] proved to be feasible by laccase catalysis. The incubation of TvrL in dioxane/water (7 : 3) with an organosolv lignin and acrylic monomer resulted in non‐homogeneous, nitrogen‐containing products. The acetone‐insoluble fraction of the product was hard and sparingly soluble in water and in dioxane/water (7 : 3). It was deduced that lignin provided the active free‐radical sites for the polymerization of acrylic monomers because polymerization was not detected without lignin. A series of works investigating the mechanism and kinetics of the said reaction followed from the same group because of the potential in producing plastics with biodegradable properties. The first follow‐up work investigated the effect of organic peroxides in the incubation of TvrL with lignin and acrylic monomers. [81] It was found that both the enzymes and the peroxides were required to achieve significant product yield and sufficient incorporation of acrylamide into the lignin. It was proposed that the phenoxy radicals generated by laccase alone are not reactive enough to initiate a side‐chain polymerization of acrylamide. The peroxy radicals initiated the polymerization of acrylamide and the radical end of the polymerized acrylamide combined with the phenoxy radicals generated by laccases in lignin, terminating the reaction. Further studies confirmed the grafting reaction and revealed the fibril, crystal‐like morphology of the lignin–acrylamide copolymer. [82] The polymerization of acrylamide with other types of lignin such as lignosulfonates, the effect of different initiators (tert‐butylhydroperoxide and Fenton reagent), and the underlying mechanism were also subsequently studied. [83]
The capacity of laccase to act on phenolic substrates certainly attracted investigations of grafting phenolic derivatives to lignin. The laccase‐catalyzed copolymerization of vanillic acid (VA) with lignin was demonstrated by incubating radio‐labeled [14COOH]VA with lignin and laccase in aqueous dioxane. The study showed that the carboxyl group of the aromatic VA participated in forming linkages with the lignin as revealed by the recovered radioactivity of VA in the acid‐insoluble copolymer. [79]
In another study, Lund and Ragauskas [84] investigated systematically the laccase‐mediated grafting of water‐soluble phenol derivatives to kraft lignin. The phenols had either carboxylic or sulfonic acid groups, which produced water‐soluble homopolymers upon enzymatic oxidation. The removal of the water‐soluble homopolymers in the lignin‐laccase incubation was easily achieved by extensive washing with water, eliminating the effect of adsorbed phenolics when determining the amount of grafted phenols by conductometric titration and quantitative 1H and 31P NMR spectroscopy. It was shown that only phenols having electron‐donating groups, such as methoxy on guaiacol sulfonate and the methylene group on 4‐hydroxyphenylacetic acid, were significantly incorporated into the lignin. Accordingly, electron‐donating substituents decreased the reduction potential of phenols and stabilized the generated phenoxy radicals, promoting bond formation with lignin, rather than the dimerization or oligomerization of the phenols. Interestingly, the incorporation of water‐soluble phenols, in particular the guaiacol sulfonate, greatly altered the solubility of lignin by producing a lignin derivative highly soluble in acidic aqueous medium of pH 2.4. [84]
Plant‐based phenolic compounds such as gallic acid (GA) and tannic acid (TA) were grafted to various types of lignin via laccase oxidation during the preparation of lignin‐based adhesive for amine‐containing wool carpets. [85] The rationale of grafting GA or TA was to increase the phenolic groups to be oxidized to quinone structures during laccase treatment. The quinone structures, which are reactive to the amino groups in the wool, were expected to improve the bonding strength between the wool and lignin‐based adhesive. The grafting of GA or TA was performed by pre‐activating the lignin with acetosyringone with the action of laccase, followed by the addition of GA or TA. Instead of spectroscopic methods, cyclic voltammetry was used to deduce the grafting of GA or TA to lignin. The electrochemical behavior of the adhesive precursors (lignin, acetosyringone, TA, and GA) was studied methodically and revealed that the plant‐based phenolic compounds were able to autopolymerize and copolymerize between themselves and with the lignin during enzymatic reaction. The lignin‐based adhesive proved to be comparable with synthetic latex‐based adhesives in terms of bonding strength. [85]
Nitrogen‐containing compounds such as isocyanates, N−OH‐type mediators, amines, and amides are grafted to lignin via laccase catalysis. The grafting of 4,4’‐methylenediphenyl diisocyanate (MDI) to organosolv lignin was achieved via laccase‐catalysis in aqueous dioxane. The recovered lignin–MDI copolymer showed fibrillar structures resembling the structure of the hard domains of linear‐segmented polyurethanes obtained from the reaction of polyethers or polyesters with symmetrical diisocyanates and short‐chain dialdehydes. [79] It was assumed that the various types of hydroxy groups in lignin enabled the donation of hydrogen atom to isocyanate and possibly formed lignin‐based polyurethanes during laccase incubation.
The coupling of N−OH‐type mediators to lignin is a novel application of laccase‐mediator system and offers an alternative route of functionalizing lignin. While mediators are often used as intermediate electron shuttles during lignin‐laccase treatment, the study of Munk et al. [86] revealed that some mediators, when oxidized, could covalently bond to lignin and do not return to their original reduced form. It was demonstrated that among the investigated artificial mediators HBT, N‐hydroxyacetanilide (HPI), 2,20‐azino‐bis(3‐ethylbenzthiazoline‐6‐sulfonate) (ABTS), and 2,2,6,6‐tetramethylpiperidin‐1‐yloxy, only the N−OH types (HBT and HPI) were able to graft to lignin. It was proposed that the N‐oxyl radicals formed by the mediators upon oxidation by laccase could covalently bond by radical‐radical coupling to (1) phenoxyl radicals on a laccase‐activated lignin subunit, (2) non‐phenolic moieties of lignin that were activated into radicals by the N‐oxyl radicals, and (3) double bonds (aliphatic alkenes) in lignin, with the copper in laccase acting as a catalyst. It was found that the grafting efficiency was higher with HPI than with HBT, which was attributed possibly to the differing stability of the radicals formed by the mediators during laccase oxidation. The study also showed that the differing reactivity of the lignin substrates (wheat straw lignin vs. beech organosolv lignin) affected the extent of coupling, while the type of laccase showed no significant difference in grafting efficiency.
Another example of N‐containing compounds grafted to lignin was the laccase‐catalyzed coupling of hydrophilic amines and amides. The grafting of glucosamine and a tyrosine‐containing tripeptide to various types of lignin in acetone/water mixture was demonstrated in the work of Fitigău et al. [87] According to the proposed mechanism in the study, the quinonoid structures generated in lignin as intermediates during laccase catalysis can react further with nucleophiles (the amino groups in glucosamine) via Michael addition. For the incorporation of a tyrosine‐containing tripeptide, the tyrosine end acted as a mediator, which upon oxidation with laccase formed tyrosyl radicals that can couple with the phenoxyl radical of lignin. From the various types of lignin tested as the starting material, it was found that not all lignin could be grafted with the chosen hydrophilic compounds at the reaction conditions used. Only the organosolv hardwood lignin and the alkali‐pretreated wheat straw lignin were able to form coupling reactions with the glucosamine and the tripeptide, as evidenced by structural and molecular weight changes confirmed by various spectroscopic techniques. [87]
Not only small amine‐containing compounds, but also amine‐containing polymers, can be enzymatically grafted to lignin. Hardwood kraft lignin dissolved in aqueous alkali was grafted with polyethyleneimine, chitosan, or soy protein in the presence of laccase with the aim of formulating eco‐friendly adhesives. [88] The soy protein–lignin formulation, where the lignin was treated with laccase and subsequently reduced by NaBH4, yielded a binding strength 50 % higher than that of the commercial polyurethane adhesive. The lignin‐containing adhesive exhibited good water‐resistance properties and could be suitable for binding paper or cardboards.
Inorganic compounds, such as silanes, could also be crosslinked to lignin using laccases and producing hybrid polymers with organic and inorganic constituents. In the study of Prasetyo et al., [89] a lignin‐siloxane hybrid was produced by mixing laccase‐oxidized lignosulfonate salts or kraft lignin with siloxane precursors (silanes) via a sol‐gel process. To confirm the contribution of laccase in the hybrid formation, pure lignin monomers as model compounds were reacted with silanes that do not form polymers upon hydrolysis, and the reaction product was analyzed using 29Si NMR spectroscopy. It was revealed that without pre‐oxidation with laccase, coupling between the lignin monomers and siloxane did not occur. It was deduced that the presence of laccase increased the oxygen‐containing groups in lignin, which increased their interaction with siloxanes. The produced interpenetrating polymer network of lignin and siloxane was used as an adhesive between two strips of paper and was found to increase the tensile strength of the bound papers. [89]
Laccase‐catalyzed co‐polymerization of lignin with carbohydrates, particularly cellulose, is a reaction not easily achieved in laboratory conditions. However, Hüttermann et al. [90] showed that the enzymatic activation of 20 % (w/w) lignin in water with high concentration of laccase and intensive aeration unexpectedly led to a sudden increase in the reactivity of lignin with cellulose. According to their findings, the intensive condition for activation yielded highly reactive phenoxyl radicals that can react with nucleophiles such as cellulose or starch. [90]
Thiol‐containing compounds are another class of nucleophiles that can be grafted to lignin producing lignin derivatives with thermoplastic properties. [91] In the study, a kraft lignin dissolved in aqueous dioxane was treated with methylhydroquinone in the presence of laccase, which was then followed by the addition of a trithiol, tris(2‐mercaptoethyl)amine. In contrast to the work of Fitigău et al., [87] where the site for nucleophilic addition was proposed to be generated as quinonoid structures by the lignin during laccase catalysis, in this thiol addition, the para‐quinone structures were laccase‐generated from the precursor, methyl hydroquinone, and served as sites for the coupling of thiols. The formation of covalent linkage between each individual component was analyzed using various NMR techniques (1H, 13C, 13C distortionless enhancement by polarization transfer‐135, and 1H‐13C heteronuclear multiple bond correlation). Although the original aim was to produce discrete lignin‐core hyperbranched copolymers (LCHCs) by grafting the thiols only on the surface of the lignin, it was revealed that crosslinking between LCHCs occurred and resulted in a network of lignin copolymers. Nevertheless, characterization of the copolymers showed that the crosslinked LCHCs had a potential as lignin‐based thermoplastics with good thermal stability and a moderate glass transition temperature. [91]
6. Patented Applications
Laccases are promising green tools for enhancing the utilization of technical lignin in industrial processes and expand the incorporation of lignin‐based products commercially (Figure 3). Indeed, numerous patents have highlighted the potential of laccase for lignin valorization.[ 15a , 92 ] The following sections present a Review of relevant patents on novel lignin‐based materials engineered via laccases. The current challenges and opportunities for lignin valorization by laccase in the industry are also discussed. More information on the patents cited in the text and details of the type of technical lignin, laccase origin, grafted compounds, and target applications are provided in Table 3.
Figure 3.

Examples of patented products derived from laccase‐assisted lignin valorization.
Table 3.
Summary of patents cited in the text with type of technical lignin, laccase origin, grafted compounds, and target applications.
|
Substrates |
Laccase origin |
Grafted compounds |
Target applications |
Ref. |
|---|---|---|---|---|
|
lignin sulfonate, kraft lignin, alkaline lignin, organosolv lignin, acetosolv lignin, ASAM lignin |
not specified |
– |
binding agents |
[93] |
|
alkali lignin |
not specified |
– |
binding agents |
[94] |
|
lignosulfonate, kraft lignin, alkaline lignin and/or organosolv lignin |
not specified |
– |
binding agents |
[95] |
|
lignosulfonate or soluble lignin in pulp itself (if in enough amount) |
not specified |
– |
trapping mineral oil in packaging |
[96] |
|
lignin sulfonate |
Trametes villosa or Stachybotrys chartorum |
– |
paints |
[97] |
|
lignosulfonate, lignosulfonic acid |
Trametes villosa or Stachybotrys chartorum |
– |
coating |
[98] |
|
lignin sulfonate, kraft lignin, organosolv lignin, acetosolv lignin, ASAM lignin |
not specified |
– |
thermosets |
[99] |
|
lignin sulfonate and kraft lignin |
not specified |
– |
fiber‐reinforced thermosetting composites, thermosetting plastics, waterproof paper, and cardboard |
[100] |
|
kraft lignin |
Trametes versicolor |
– |
carbon fiber |
[101] |
|
lignosulfonates and/or derived lignosulfonates |
Trametes villosa |
– |
wastewater treatment |
[102] |
|
lignosulfonate |
Myceliophthora thermophila |
– |
delivery systems for fertilizers/pesticides with water storage capacity |
[103] |
|
water‐soluble lignosulfonic acid and its salt (i. e., sodium lignosulfonate and calcium lignosulfonate) |
Myceliophthora thermophile, Trametes villosa, Rhizoctonia praticola, Scytalidium thermophilium, Coriolus hirsitus, among others |
– |
deodorant |
[104] |
|
organosolv lignin |
not specified |
glucose, vanillic acid, sorbit, and acrylamide |
production of polymers from lignin with target functional groups |
[105] |
|
residual lignin from aqueous lignocellulosic extracts |
Trametes villosa |
hemicelluloses |
preparation of a new class of technical lignin for novel materials manufacture (e. g., films) |
[106] |
|
kraft, organosolv, soda, and acid lignin and lignin from enzymatic degradation of cellulose |
Trametes versicolor |
low‐molar‐mass organic compounds functionalized by secondary amino, hydroxy, or phenyl groups |
mineral binding |
[107] |
|
alkali lignin (e. g., wheat straw alkali lignin, bamboo pulp alkali lignin, or bagasse alkali lignin) |
Rhus lacquer (Rhus vernicifera) or fungal (Trametes versicolor, Coriolus versicolor, Agaricus bisporus, Pleurotus ostreatus) |
epichlorohydrin, sulfonating agent, and an aldehyde compound |
concrete and coal water slurry dispersing agents |
[108] |
|
industrial lignin |
not specified |
wheat straw fiber |
formaldehyde‐free straw fiber board |
[109] |
|
industrial lignin such as alkali lignin and lignosulphonates |
Aspergillus sp. |
pulp fibers |
paper with improved strength |
[110] |
6.1. Patented applications of laccase‐polymerized lignin
Initially, the use of laccases for the valorization of technical lignin mimicked nature, where the laccase‐polymerized lignin acts as a natural glue that binds the components in the plant cell wall. Bioinspired by such mechanisms, lignin‐based binding agents for wooden composites were developed by laccase‐assisted lignin polymerization from various types of technical lignin. [93] Similarly, Hatakka et al. disclosed the capacity of fungal laccases to catalyze the polymerization of alkali lignin by radical coupling. [94] Inventors reported that the polymerized lignin, exhibiting a gel‐like appearance at room temperature, had great potential as a binder for paper and boards.
With the consolidation of the methodology to produce lignin‐based binders, improvement in the technology was oriented towards enhanced lignin reactivity. An elegant combination of enzymatic strategies, including an accelerated lignin polymerization by laccases, was disclosed for the production of binding agents from lignosulfonate, kraft lignin, alkaline lignin, and/or organosolv lignin. [95] These inventors applied a pretreatment using proteolytic enzyme that released new reaction centers in the lignin in a position previously bound to proteins. This technology enhanced the laccase reactivity and made the lignin activation stronger, resulting in an increased polymerization rate and gain of 167 % in lignin molar mass.
The increasing hydrophobicity is another consequence of technical lignin polymerization. Nolsen and Miller disclosed the use of laccase to increase hydrophobicity in paper food packaging and minimize or suppress mineral oil migration from packaging to food. [96] The intensification of hydrophobic regions increases the tendency of mineral oil to be trapped and/or bound in the polymerized lignin by fixation and/or hydrophobic interaction. In this invention, only soluble lignin works as substrate, which can be the soluble lignin remaining in the pulp or lignosulfonates supplemented before, during, and/or after the laccase addition.
Lignin‐based paints and coatings can also be produced via laccase catalysis. Lignosulfonates have proven to be suitable substrates for the production of paints through an environmentally safe laccase‐ABTS method in which dye or pigment is supplemented during the oxidative reaction. [97] Paints obtained from this invention can be applied to protect and beautify lignin‐containing surfaces, such as wooden materials. Later, the same substrate was also used for the manufacture of coating, also using a similar laccase‐ABTS polymerization technique. [98] Notably, the coating agent obtained was applicable to non‐lignocellulosic surfaces. Although lignosulfonate is the preferred substrate in both technologies, kraft and organosolv lignins are also listed as alternative substrates.[ 97 , 98 ] This indicated that, despite variation in reactivity of technical lignin, laccase treatments are quite comprehensive in terms of action.
The manufacture of thermosets from various types of technical lignin proved to be feasible via laccase catalysis. Mai et al. [99] disclosed that lignin can be polymerized by laccase, producing highly active intermediates suitable for thermoset preparation. According to the inventors, the technology was pioneered for the manufacture of thermosets from renewable sources. Later, Hüttermann et al. [100] also demonstrated that the aforementioned lignin substrates are suitable lignin‐based precursors for material manufacturing of fiber‐reinforced thermosetting composites, thermosetting plastics, waterproof paper, and cardboard after laccase treatment. [100]
Laccase‐polymerized lignin also served as precursor for carbon fiber production. For example, Yuan and Li [101] disclosed a technology to produce carbon fibers from kraft lignin treated by laccase‐HBT. The laccase‐assisted process releases lignin precursors with high uniformity, increased frequency of either uncondensed (e. g., β‐O‐4) or condensed linkages (e. g., β‐5), optimized according to the need, and concentrated in intermolecular hydrogen bonds. Lignin precursors with target structures are then subjected to thermostabilization or carbonization for carbon fiber production.
Another trending field for lignin application is in environmental treatment solutions or bioremediation. For example, a gel formed through laccase polymerization and cross‐linking of lignosulfonates was previously hypothesized as having potential to treat wastewater. [102]
In the field of agriculture, laccase‐polymerized lignin is a novel and green material for preparing specialized delivery systems for fertilizers or pesticides. Nyanhongo et al. [103] disclosed the preparation of a biodegradable lignosulfonate polymer with water storage capacity produced through lignosulfonate polymerization by laccase. [103] The resultant lignosulfonate polymer is water insoluble, can store water, and can be also used for the continuous release of water‐soluble compounds, such as fertilizers, crop defensives, and other agrochemicals.
An interesting application of laccase‐treated lignosulfonate is for removing unpleasant odors. [104] The inventors prepared a deodorant by combining sodium or calcium lignosulfonates with laccase in equal ratio, and both liquid or powdery compositions were obtained. The produced deodorant had satisfactory deodorizing capacity for unpleasant odors and potential use in food, feed, oral care, sanitary, and pet products. The mechanism for removing odors was described as an oxidative process of phenolic compounds catalyzed by laccase.
6.2. Patented applications of grafted lignin produced via laccase catalysis
Various types of compounds, from small molecules to fiber matrix (Figure 4) grafted to lignin via laccase catalysis were given patent approval, indicating the industrial significance of such inventions.
Figure 4.

Examples of patented applications of laccase‐aided grafting of lignin with known compounds and materials.
Hüttermann and Milstein [105] demonstrated the co‐polymerization of glucose, vanillic acid, sorbit, and acrylamide with organosolv lignin by laccase oxidation. The inventors disclosed that the lignin co‐polymers are produced in a simple manner that does not require an oxygen‐free atmosphere.
Laccase can also catalyze the formation of a new class of technical lignin. This is the case of the method developed to coupling hemicelluloses and aromatic moieties, such as residual lignin in lignocellulosic extracts, by laccases. [106] Although the invention aimed for increasing the molecular weight of wood mannans and xylans, the methodology was adapted to produce a new type of technical lignin called Ecohelix. [111] The technology yields a new technical lignin with specific properties, including high sulfonates and aliphatic OH groups, suitable molecular weight and thermal stability, and capacity to be used in the development of novel materials, such as films. [111]
The solubility of lignin is a parameter usually limiting its reactivity during enzymatic processes as mentioned earlier. A recent invention by Schober et al. [107] has dealt with this limitation and disclosed a technique to produce water‐soluble lignin from water‐insoluble lignin (e. g., kraft, organosolv, soda, and acid lignin, and lignin from enzymatic degradation of cellulose) by coupling it to low‐molar‐mass organic compounds functionalized by secondary amino, hydroxy, or phenyl groups. [107] The laccase‐ABTS method applied for this conversion occurs under mild condition, without toxic components, and results in the formation of lignin with an exceptional dispersant capacity for mineral binding applications.
Amphiphilic behavior can also be created by laccase catalysis. Xueqing et al. [108] demonstrated the production of a modified sulfonated soda lignin amphiphilic polymer by a laccase‐catalyzed cross‐linking of alkali lignin with epichlorohydrin, sulfonating agent, and an aldehyde compound (e. g., formaldehyde, acetaldehyde, oxalic dialdehyde, propionic aldehyde, butyraldehyde, isobutyric aldehyde, and glutaraldehyde). [108] The resultant lignin amphiphilic polymer had improved reactivity compared to alkali lignin, in addition to higher molecular weight and sulfonation degree. Such amphiphilic polymer is suggested to be used as concrete and coal water slurry dispersing agents in substitution of petroleum‐based amphiphilic polymers.
The capacity of laccase for engineering industrial lignin into more sustainable and less toxic compounds was demonstrated by Lei et al. [109] by using laccase to crosslink industrial lignin and wheat straw fiber and manufacture a formaldehyde‐free straw fiber board. [109] The simplicity of operations and the valorization of agricultural and industrial wastes are additional benefits of this invention.
An undeniable advantage of the inventions using laccase for modifying technical lignin is their natural insertion in the models of circular bioeconomy. A great example is the technique for improving paper strength disclosed by Na et al. [110] in which alkali lignin or lignosulphonates were grafted to pulp fibers by laccase. The incorporation of lignin rich in phenolic hydroxy groups improved the paper strength. Accordingly, laccase promotes the formation of phenolic oxygen free radicals and their consequent crosslinking, which increases the bonding strength between fibers. A further aspect of this invention is that, in addition to functionalizing the strength of unbleached kraft pulp, the technique also opens the possibility to create fibrous binding force in paper from wastes, such as old corrugated containers, allowing their efficient re‐utilization in industry. [110]
6.3. Industrial challenges and opportunities
Laccase has a great potential for integrating biotechnological processes in various industries, such as lignocellulosic biorefinery (including in the traditional pulp and paper industry), chemical, biomedical, and pharmaceutical industries.[ 92a , 92c ] Despite this, information on the current use of laccase for valorizing lignins on industrial scale is not easily found, likely due to the confidentiality that protects such processing conditions. On the other hand, some practical and technical aspects available in the literature published on laboratory‐scale experiments help to identify the challenges and opportunities regarding the current industrial use of laccases.
Nowadays, laccases are commercially available from a number of market suppliers. Despite this, the availability of stocks and the cost of laccases still raise certain concerns when their industrial use is discussed, especially when high‐purity enzyme is required.[ 92a , 112 ] In this sense, the invention of economic, safe, and efficient methods for producing microbial laccases is highly opportune. [113] Novel technologies can also benefit the development of biotechnical methods for enzyme production to increase laccase purity and stability.[ 92a , 114 ] In line with this, other technical aspects such as the usual low tolerance of laccases towards alkaline pH and high temperature might restrict their activity and stability in certain industrial conditions and also limit the efficient solubilization of lignin.[ 38a , 114 ] Regarding laccase limitations, an alternative would be using thermostable laccases that are expressed by microbes living in harsh environments, or may be more easily produced and engineered by genetic modification. [75]
High degree of polymerization of the lignin substrate and the low redox potential of laccase enzymes usually requires their combination with mediators for efficiently enabling substrate oxidations.[ 45 , 49a , 58 ] However, certain mediators, especially the synthetic ones, are expensive, toxic, and might cause a certain degree of laccase deactivation.[ 5 , 16b ] Obviously, prompting lignin valorization by treatment with laccases should also consider production of efficient and compatible mediators. This is the case of the natural lignin‐derivative phenolic mediator compounds recovered as by‐products from industrial side‐streams, which has the advantage of not deactivating the enzymes.[ 45 , 115 ]
These industrial challenges related to the use of laccase in lignin valorization can be overcome by fostering close cooperation between industrial sectors, including those responsible for the production and modification of technical lignins, with enzyme producers and biorefineries yielding natural mediator compounds and precursors for grafting. Obviously, this depends on a clear definition of the properties and attributes of the laccase enzymes. With renewable substrates like plant biomass and lignin, inventions of new applications and processes will promote sustainability in industrial production and transition towards a bio‐based economy.
7. Summary and Outlook
Laccase is indeed a promising tool to construct lignin‐based materials via either polymerization or grafting of other molecules. Various types of technical lignin have shown to be suitable substrates for laccase‐assisted valorization. Similarly, a number of different laccases are also available for this task. Fundamental understanding of the factors affecting laccase‐catalyzed lignin polymerization is essential, and being able to identify, compare, and evaluate in detail all these factors provides a guide for future work intending to dwell on this topic. Not only are these factors essential for producing polymerized lignin but also for grafting reactions, because during laccase‐catalyzed grafting, lignin self‐polymerization must be efficiently controlled while coupling reaction with the intended molecules is promoted.
The property of lignin that proved essential in laccase‐catalysis was solubility in the reaction medium. In both polymerization and grafting, dissolution of the starting lignin material seemed to be the pre‐requisite. The grafting reactions, in most cases, used toxic organic solvents such as dioxane, which forfeits the principle of a fully green bio‐based technology using laccase and lignin. Instead of aiming to dissolve lignin in the reaction medium, one promising approach is to use lignin nanoparticles, a form of lignin that can be homogeneously dispersed in an aqueous system even at slightly acidic conditions. [116] The action of laccase on lignin nanoparticles is still relatively unexplored despite the exponential interest arising from these particles recently. [116] Using lignin nanoparticles could possibly pave the way out of using toxic organic solvents during laccase treatment.
One of the gaps worth considering in this laccase‐lignin catalysis involves characterization of the final material. The characterization of laccase‐catalyzed lignin‐based materials are mostly limited to increase in molecular weight for polymerization and elucidation of the formation of covalent bonding between lignin and the grafted molecule. While all these characterizations are important in establishing the success of the laccase‐lignin reaction, the changes in morphology and surface properties of the starting lignin are also worth exploring in the future, especially if lignin nanoparticles are to be used as starting materials.
The use of lignin nanoparticles may also improve the synthesis of lignin core hybrids, produced by grafting other molecules only on the surface of the lignin. So far, most of the lignin‐grafted copolymers produced via laccase catalysis were based on random and uncontrolled co‐polymerization reactions. A more controlled laccase‐assisted synthesis of lignin core hybrids could very well be feasible with lignin nanoparticles dispersed in an aqueous system or at interfaces of emulsion systems.
To bridge the gap between laboratory and industrial scale, it would be beneficial to include in future studies more of the effect of temperature variation, as most of the previous works were based on reaction at ambient conditions. While room temperature is favorable and convenient in laboratory scale, elevated temperatures (40–60 °C) are more common and durable in industrial processes. On this aspect, thermostable laccases offer the advantage of performing the reaction at elevated temperature, which increases the solubility of lignin and reaction kinetics. With all the advances resulting from exhaustive research efforts on thermostable laccases in the past decades,[ 38a , 75 ] it is imperative that the utilization of thermostable laccases in building lignin‐based materials be further explored.
Patented applications realized so far are remarkable, but there are definitely more advanced applications that can be derived from lignin. With various advanced applications being developed using lignin nanoparticles including applications in controlled drug delivery systems, surface interfacial stabilizers, energy storage devices, shape‐memory materials, and sensors,[ 9b , 117 ] using lignin nanoparticles in laccase catalysis could then possibly realize the use of laccases in building highly advanced, functional, and smart lignin‐based materials.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Melissa B. Agustin spent her early career as a chemistry instructor in the Philippines before completing her DSc in Agricultural Science at Kyoto University, Japan, in 2017. She joined the University of Helsinki as a post‐doctoral researcher in 2018, and since then her research interest expanded from chemical modification of nanocelluloses to lignin and its valorization. She is currently working on the utilization of laccase in building lignocellulose‐based adsorbents in a post‐doc project funded by the Academy of Finland.

Biographical Information
Danila M. Carvalho is postdoctoral researcher in the Food Materials Science Research group at the Department of Food and Nutrition, University of Helsinki. In her research, she utilizes chemical, physical, thermal, and enzymatic treatments for the extraction and fractionation of biopolymers from lignocellulosic biomass. Using a combination of classical and advanced analytical technique, she assesses the chemical structure of biopolymers, which is of paramount importance for designing novel materials. Her research interests include the valorization of biopolymers (native or derivative) in sustainable and functional applications.

Biographical Information
Maarit Lahtinen received her Ph.D. in Organic Chemistry at University of Helsinki, Department of Chemistry in 2013. Her Ph.D. work focused on studying laccase‐catalyzed reactions of synthetic lignin model compounds. After that, she continued working in the academy participating in research projects collaborating with industrial partners. She joined the Food Materials Science group at University of Helsinki, Department of Food and Nutrition in 2017. She has an ongoing research interest in lignin and laccases, while expanding to new areas, such as hemicelluloses, hydrocolloids, and emulsion systems.

Acknowledgements
The authors acknowledge Troy Faithfull for his help editing the manuscript and Nelson M. Panajon for drawing the graphical and TOC abstracts. The following are acknowledged for providing funding: the European Commission through the European Research Council PARTIFACE (Grant No. 863808) for DMC and KSM, the Academy of Finland PickPollutants PostDoc Project (Grant No. 330617) for MBA and AromaFung project (Grant No. 297847) for KH, and the Jane and Aatos Erkko Foundation GOOD project for ML.
M. B. Agustin, D. M. de Carvalho, M. H. Lahtinen, K. Hilden, T. Lundell, K. S. Mikkonen, ChemSusChem 2021, 14, 4615.
Contributor Information
Dr. Melissa B. Agustin, Email: melissa.agustin@helsinki.fi.
Dr. Danila Morais de Carvalho, Email: danila.moraisdecarvalho@helsinki.fi.
Dr. Maarit H. Lahtinen, Email: maarit.lahtinen@helsinki.fi.
References
- 1. Gellerstedt G., Henriksson G. in Monomers, Polymers and Composites from Renewable Resources (Eds.: Belgacem M. N., Gandini A.), Elsevier Inc., Oxford, 2008, pp. 201. [Google Scholar]
- 2. Nagy M., Kosa M., Theliander H., Ragauskas A. J., Green Chem. 2010, 12, 31. [Google Scholar]
- 3.J. E. Holladay, J. F. White, J. J. Bozell, D. Johnson, Vol. II, US Department of Energy, 2007, Top-Value Added Chemicals from Lignin..
- 4.
- 4a. Ralph J., Lundquist K., Brunow G., Lu F., Kim H., Schatz P. F., Marita J. M., Hatfield R. D., Ralph S. A., Christensen J. H., Boerjan W., Phytochem. Rev. 2004, 3, 29; [Google Scholar]
- 4b. Mottiar Y., Vanholme R., Boerjan W., Ralph J., Mansfield S. D., Curr. Opin. Biotechnol. 2016, 37, 190. [DOI] [PubMed] [Google Scholar]
- 5. Christopher L. P., Yao B., Ji Y., Front. Energy Res. 2014, 2, 1. [Google Scholar]
- 6.
- 6a. Behling R., Valange S., Chatel G., Green Chem. 2016, 18, 1839; [Google Scholar]
- 6b. Tobimatsu Y., Schuetz M., Curr. Opin. Biotechnol. 2019, 56, 75. [DOI] [PubMed] [Google Scholar]
- 7. Hubbe M. A., Nazhad M., Sánchez C., BioResources 2010, 5, 2808. [Google Scholar]
- 8. Gul S., Yanni S. F., Whalen J. K. in Lignin: Structural Analysis, Applications in Biomaterials and Ecological Significance (Ed.: Lu F.), Nova Science Publishers, Inc, New York, USA, 2014, pp. 375. [Google Scholar]
- 9.
- 9a. Collins M. N., Nechifor M., Tanasă F., Zănoagă M., McLoughlin A., Stróżyk M. A., Culebras M., Teacă C. A., Int. J. Biol. Macromol. 2019, 131, 828; [DOI] [PubMed] [Google Scholar]
- 9b. Sipponen M. H., Henn A., Penttilä P., Österberg M., Chem. Eng. J. 2020, 393, 124711. [Google Scholar]
- 10. Tolbert A., Akinosho H., Khunsupat R., Naskar A. K., Ragauskas A. J., Biofuels Bioprod. Biorefin. 2014, 8, 836. [Google Scholar]
- 11.
- 11a. Kirk T. K., Gifford O., Drive P., Farrell R. L., Annu. Rev. Microbiol. 1987, 41, 465; [DOI] [PubMed] [Google Scholar]
- 11b. Bugg T. D., Rahmanpour R., Curr. Opin. Chem. Biol. 2015, 29, 10. [DOI] [PubMed] [Google Scholar]
- 12.
- 12a. Wang Y. Y., Meng X., Pu Y., Ragauskas A. J., Polymer 2020, 12; [Google Scholar]
- 12b. Ganewatta M. S., Lokupitiya H. N., Tang C., Polymer 2019, 11; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12c. Matsushita Y., J. Wood Sci. 2015, 61, 230; [Google Scholar]
- 12d. Liu H., Chung H., J. Polym. Sci. Part A 2017, 55, 3515; [Google Scholar]
- 12e. Cannatelli M. D., Ragauskas A. J., Appl. Microbiol. Biotechnol. 2016, 100, 8685. [DOI] [PubMed] [Google Scholar]
- 13.
- 13a. Figueiredo P., Lintinen K., Hirvonen J. T., Kostiainen M. A., Santos H. A., Prog. Mater. Sci. 2018, 93, 233; [Google Scholar]
- 13b. Laurichesse S., Avérous L., Prog. Polym. Sci. 2014, 39, 1266. [Google Scholar]
- 14.
- 14a. Nguyen N. A., Meek K. M., Bowland C. C., Naskar A. K., Polymer 2019, 160, 210; [Google Scholar]
- 14b. Zhang Y., Wu J.-Q., Li H., Yuan T.-Q., Wang Y.-Y., Sun R.-C., ACS Sustainable Chem. Eng. 2017, 5, 7269; [Google Scholar]
- 14c. Sun Q., Khunsupat R., Akato K., Tao J., Labbé N., Gallego N. C., Bozell J. J., Rials T. G., Tuskan G. A., Tschaplinski T. J., Naskar A. K., Pu Y., Ragauskas A. J., Green Chem. 2016, 18, 5015. [Google Scholar]
- 15.
- 15a. Sena-Martins G., Almeida-Vara E., Duarte J. C., Ind. Crops Prod. 2008, 27, 189; [Google Scholar]
- 15b. Hämäläinen V., Grönroos T., Suonpää A., Heikkilä M. W., Romein B., Ihalainen P., Malandra S., Birikh K. R., Front. Bioeng. Biotechnol. 2018, 6, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15c. Slagman S., Zuilhof H., Franssen M. C. R., ChemBioChem 2018, 19, 288; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15d. Cannatelli M. D., Ragauskas A. J., J. Biotechnol. Biomater. 2015, 5, 1. [Google Scholar]
- 16.
- 16a. Kudanga T., Nyanhongo G. S., Guebitz G. M., Burton S., Enzyme Microb. Technol. 2011, 48, 195; [DOI] [PubMed] [Google Scholar]
- 16b. Bassanini I., Ferrandi E. E., Riva S., Monti D., Catalysts 2021, 11; [Google Scholar]
- 16c. Riva S., Trends Biotechnol. 2006, 24, 219. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Lee S., Kang M., Bae J. H., Sohn J. H., Sung B. H., Front. Bioeng. Biotechnol. 2019, 7, 1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Bugg T. D. H., Williamson J. J., Rashid G. M. M., Curr. Opin. Chem. Biol. 2020, 55, 26; [DOI] [PubMed] [Google Scholar]
- 17c. Li C., Chen C., Wu X., Tsang C. W., Mou J., Yan J., Liu Y., Lin C. S. K., Bioresour. Technol. 2019, 291, 121898. . [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Matsumoto Y., Hattori M., Arch. Insect Biochem. Physiol. 2019, 102, 1; [DOI] [PubMed] [Google Scholar]
- 18b. Dwivedi U. N., Singh P., Pandey V. P., Kumar A., J. Mol. Catal. B 2011, 68, 117; [Google Scholar]
- 18c. Janusz G., Pawlik A., Swiderska-Burek U., Polak J., Sulej J., Jarosz-Wilkolazka A., Paszczynski A., Int. J. Mol. Sci. 2020, 21, 966.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.
- 19a. Widsten P., Kandelbauer A., Enzyme Microb. Technol. 2008, 42, 293; [Google Scholar]
- 19b. Cannatelli M. D., Ragauskas A. J., Chem. Rec. 2017, 17, 122. [DOI] [PubMed] [Google Scholar]
- 20.
- 20a.D. Fengel, G. Wegener, Wood: Chemistry, ultrastructure, Reactions, De Gruyter, Inc., Berlin/Boston, Germany, 1983;
- 20b.E. Sjöström, Wood Chemistry: Fundamentals and Applications, Academic Press, Inc., San Diego, California, 1993;
- 20c.J. Ralph, G. Brunow, W. Boerjan in Encyclopedia of Life Sciences, Nature Publishing Group, London, 2007;
- 20d. Vanholme R., Demedts B., Morreel K., Ralph J., Boerjan W., Plant Physiol. 2010, 153, 895; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20e.R. Rinaldi, R. Jastrzebski, M. T. Clough, J. Ralph, M. Kennema, P. C. A. Bruijnincx, B. M. Weckhuysen, Angew. Chem. Int. Ed. Engl. 2016, 55, 8164; [DOI] [PMC free article] [PubMed]
- 20f. Ralph J., Lapierre C., Boerjan W., Curr. Opin. Biotechnol. 2019, 56, 240. [DOI] [PubMed] [Google Scholar]
- 21. Balakshin M., Capanema E. A., Zhu X., Sulaeva I., Potthast A., Rosenau T., Rojas O. J., Green Chem. 2020, 22, 3985. [Google Scholar]
- 22.
- 22a. Doherty W. O. S., Mousavioun P., Fellows C. M., Ind. Crops Prod. 2011, 33, 259; [Google Scholar]
- 22b. Smith M. D. in Understanding Lignocellulose: Synergistic Computational and Analytic Methods, Vol. 1338, American Chemical Society, 2019, pp. 1. [Google Scholar]
- 23. Giummarella N., Pu Y., Ragauskas A. J., Lawoko M., Green Chem. 2019, 21, 1573. [Google Scholar]
- 24. Mansfield S. D., Kim H., Lu F., Ralph J., Nat. Protoc. 2012, 7, 1579. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Chakar F. S., Ragauskas A. J., Ind. Crops Prod. 2004, 20, 131; [Google Scholar]
- 25b. Brännvall E., BioResources 2017, 12, 2081.. [Google Scholar]
- 26. Li T., Takkellapati S., Biofuels Bioprod. Biorefin. 2018, 12, 756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Tomani P., Cellul. Chem. Technol. 2010, 44, 53. [Google Scholar]
- 28.
- 28a. Liitiä T. M., Maunu S. L., Hortling B., Toikka M., Kilpeläinen I., J. Agric. Food Chem. 2003, 51, 2136; [DOI] [PubMed] [Google Scholar]
- 28b. Balakshin M. Y., Capanema E. A., Chen, Gracz H. S., J. Agric. Food Chem. 2003, 51, 6116; [DOI] [PubMed] [Google Scholar]
- 28c. Crestini C., Lange H., Sette M., Argyropoulos D. S., Green Chem. 2017, 19, 4104; [Google Scholar]
- 28d. Lancefield C. S., Wienk H. L. J., Boelens R., Weckhuysen B. M., Bruijnincx P. C. A., Chem. Sci. 2018, 9, 6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.P. Bajpai, Biermann's Handbook of Pulp and Paper : Volume 1: Raw Material and Pulp Making : Volume 1: Raw Material and Pulp Making, Elsevier, San Diego, USA, 2018.
- 30. Mansouri N.-E. E., Salvadó J., Ind. Crops Prod. 2006, 24, 8. [Google Scholar]
- 31. Sulaeva I., Vejdovszky P., Henniges U., Mahler A. K., Rosenau T., Potthast A., ACS Sustainable Chem. Eng. 2019, 7, 216. [Google Scholar]
- 32.J.-L. Wen, B.-L. Xue, S.-L. Sun, R.-C. Sun, J. Chem. Technol. Biotechnol. 2013, 88, 1663.
- 33. Renders T., Van den Bosch S., Koelewijn S. F., Schutyser W., Sels B. F., Energy Environ. Sci. 2017, 10, 1551. [Google Scholar]
- 34. Solomon E. I., Sundaram U. M., Machonkin T. E., Chem. Rev. 1996, 96, 2563. [DOI] [PubMed] [Google Scholar]
- 35. Giardina P., Faraco V., Pezzella C., Piscitelli A., Vanhulle S., Sannia G., Cell. Mol. Life Sci. 2010, 67, 369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Thurston C. F., Microbiology 1994, 140, 8. [Google Scholar]
- 37.
- 37a. Gavnholt B., Larsen K., Physiol. Plant. 2002, 116, 273; [Google Scholar]
- 37b. Wang X., Zhuo C., Xiao X., Wang X., Docampo-Palacios M., Chen F., Dixon R. A., Plant Cell 2020, 32, 3825; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37c. Pourcel L., Routaboul J. M., Kerhoas L., Caboche M., Lepiniec L., Debeaujon I., Plant Cell 2005, 17, 2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.
- 38a. Hilden K., Hakala T. K., Lundell T., Biotechnol. Lett. 2009, 31, 1117; [DOI] [PubMed] [Google Scholar]
- 38b. Lundell T. K., Mäkelä M. R., Hildén K., J. Basic Microbiol. 2010, 50, 5; [DOI] [PubMed] [Google Scholar]
- 38c. Hatakka A., Hammel K. E., in Mycota, Vol. 10: Industrial applications, Springer, 2011, pp. 319; [Google Scholar]
- 38d. Baldrian P., FEMS Microbiol. Rev. 2006, 30, 215. [DOI] [PubMed] [Google Scholar]
- 39. Lundell T. K., Mäkelä M. R., de Vries R. P., Hildén K. S., Adv. Bot. Res. 2014, 70, 329. [Google Scholar]
- 40. Claus H., Arch. Microbiol. 2003, 179, 145. [DOI] [PubMed] [Google Scholar]
- 41. Bugg T. D. H., Ahmad M., Hardiman E. M., Singh R., Curr. Opin. Biotechnol. 2011, 22, 394. [DOI] [PubMed] [Google Scholar]
- 42. Dittmer N. T., Kanost M. R., Insect Biochem. Mol. Biol. 2010, 40, 179. [DOI] [PubMed] [Google Scholar]
- 43. Suderman R. J., Dittmer N. T., Kanost M. R., Kramer K. J., Insect Biochem. Mol. Biol. 2006, 36, 353. [DOI] [PubMed] [Google Scholar]
- 44. Bourbonnais R., Paice M. G., FEBS Lett. 1990, 267, 4. [DOI] [PubMed] [Google Scholar]
- 45. Cañas A. I., Camarero S., Biotechnol. Adv. 2010, 28, 694. [DOI] [PubMed] [Google Scholar]
- 46. Munk L., Sitarz A. K., Kalyani D. C., Mikkelsen J. D., Meyer A. S., Biotechnol. Adv. 2015, 33, 13. [DOI] [PubMed] [Google Scholar]
- 47. Zakzeski J., Bruijnincx P. C. A., Jongerius A. L., Weckhuysen B. M., Chem. Rev. 2010, 110, 3552. [DOI] [PubMed] [Google Scholar]
- 48.
- 48a. Kirk T. K., Harkin J. M., Cowling E. B., Biochim. Biophys. Acta 1968, 165, 145; [DOI] [PubMed] [Google Scholar]
- 48b. Kawai S., Umezawa T., Shimada M., Higuchi T., Koide K., Nishida T., Morohoshi N., Haraguchi T., Mokuzai Gakkaishi. 1987, 33, 792.; [Google Scholar]
- 48c. Kawai S., Umezawa T., Higuchi T., Arch. Biochem. Biophys. 1988, 262, 99. [DOI] [PubMed] [Google Scholar]
- 49.
- 49a. Crestini C., Jurasek L., Argyropoulos D. S., Chem. Eur. J. 2003, 9, 5371; [DOI] [PubMed] [Google Scholar]
- 49b. Kallio J., Auer S., Jänis J., Andberg M., Kruus K., Rouvinen J., Koivula A., Hakulinen N., J. Mol. Biol. 2009, 392, 895. [DOI] [PubMed] [Google Scholar]
- 50.
- 50a.D. Areskogh, J. Li, P. Nousiainen, G. Gellerstedt, J. Sipilä, G. J. H. Henriksson, Holzforschung 2010, 64, 21;
- 50b. Lahtinen M., Kruus K., Heinonen P., Sipila J., J. Agric. Food Chem. 2009, 57, 8357. [DOI] [PubMed] [Google Scholar]
- 51. Areskogh D., Li J., Gellerstedt G., Nousiainen P., Henriksson G., Sipilä J., Ind. Biotechnol. 2010, 6, 50. [Google Scholar]
- 52.
- 52a. Leonowicz A., Edgehill R. U., Bollag J. M., Arch. Microbiol. 1984, 137, 89; [Google Scholar]
- 52b. Lahtinen M., Heinonen P., Oivanen M., Karhunen P., Kruus K., Sipila J., Org. Biomol. Chem. 2013, 11, 5454. [DOI] [PubMed] [Google Scholar]
- 53.
- 53a. Grönqvist S., Viikari L., Niku-Paavola M. L., Orlandi M., Canevali C., Buchert J., Appl. Microbiol. Biotechnol. 2005, 67, 489; [DOI] [PubMed] [Google Scholar]
- 53b. Gouveia S., Fernández-Costas C., Sanromán M. A., Moldes D., Bioresour. Technol. 2013, 131, 288; [DOI] [PubMed] [Google Scholar]
- 53c. Ai M.-Q., Wang F.-F., Huang F., J. Microbiol. Biotechnol. 2015, 25, 1361; [DOI] [PubMed] [Google Scholar]
- 53d. Sun Y., Qiu X., Liu Y., Biomass Bioenergy 2013, 55, 198. [Google Scholar]
- 54. Perna V., Meyer A. S., Holck J., Eltis L. D., Eijsink V. G. H., Wittrup Agger J., ACS Sustainable Chem. Eng. 2020, 8, 831. [Google Scholar]
- 55. Perna V., Agger J. W., Andersen M. L., Holck J., Meyer A. S., ACS Sustainable Chem. Eng. 2019, 7, 10425. [Google Scholar]
- 56.
- 56a. Ralph J., Brunow G., Harris P. J., Dixon R. A., Schatz P. F., Boerjan W. in Recent Advances in Polyphenol Research, Vol. 1 (Eds.: Daayf F., Lattanzio V.), Wiley-Blackwell Publishing, Oxford, 2008, pp. 36; [Google Scholar]
- 56b. Hatakka A. in Biopolymers. Biology, Chemistry, Biotechnology, Applications. Vol 1. Lignin, Humic Substances and Coal (Eds.: Hofrichter M., Steinbuchel A.), Wiley-VCH, Weinheim, 2001, 129; [Google Scholar]
- 56c. Singh Arora D., Kumar Sharma R., Appl. Biochem. Biotechnol. 2010, 160, 1760. [DOI] [PubMed] [Google Scholar]
- 57.
- 57a. Xu A., Guo X., Zhang Y., Li Z., Wang J., Green Chem. 2017, 19, 4067; [Google Scholar]
- 57b. Sun J., Dutta T., Parthasarathi R., Kim K. H., Tolic N., Chu R. K., Isern N. G., Cort J. R., Simmons B. A., Singh S., Green Chem. 2016, 18, 6012. [Google Scholar]
- 58. Dillies J., Vivien C., Chevalier M., Rulence A., Chataigne G., Flahaut C., Senez V., Froidevaux R., Biotechnol. Appl. Biochem. 2020, 67, 774. [DOI] [PubMed] [Google Scholar]
- 59. Mattinen M.-L., Suortti T., Gosselink R., Argyropoulos D. S., Evtuguin D., Suurnäkki A., Jong E. D.., Tamminena T., BioResources 2008, 3, 549. [Google Scholar]
- 60. van de Pas D., Hickson A., Donaldson L., Lloyd-Jones G., Tamminen T., Fernyhough A., Mattinen M. L., BioResources 2011, 6, 1105. [Google Scholar]
- 61.
- 61a. Gouveia S., Fernandez-Costas C., Sanroman M. A., Moldes D., Bioresour. Technol. 2012, 121, 131; [DOI] [PubMed] [Google Scholar]
- 61b. Fitigău I. F., Peter F., Boeriu C. G., Acta Biochim. Pol. 2013, 60, 817. [PubMed] [Google Scholar]
- 62.
- 62a. Qiu W., Chen H., Bioresour. Technol. 2008, 99, 5480;18096384 [Google Scholar]
- 62b. Moya R., Saastamoinen P., Hernández M., Suurnäkki A., Arias E., Mattinen M. L., Bioresour. Technol. 2011, 102, 10006. [DOI] [PubMed] [Google Scholar]
- 63. Leonowicz A., Szklarz G., Wojtaś-Wasilewska M., Phytochemistry 1985, 24, 393. [Google Scholar]
- 64. West M. A., Hickson A. C., Mattinen M.-L., Lloyd-Jones G., BioResources 2014, 9, 14. [Google Scholar]
- 65. Areskogh D., Li J., Gellerstedt G., Henriksson G., Biomacromolecules 2010, 11, 904. [DOI] [PubMed] [Google Scholar]
- 66. Nugroho Prasetyo E., Kudanga T., Ostergaard L., Rencoret J., Gutierrez A., del Rio J. C., Ignacio Santos J., Nieto L., Jimenez-Barbero J., Martinez A. T., Li J., Gellerstedt G., Lepifre S., Silva C., Kim S. Y., Cavaco-Paulo A., Seljebakken Klausen B., Lutnaes B. F., Nyanhongo G. S., Guebitz G. M., Bioresour. Technol. 2010, 101, 5054. [DOI] [PubMed] [Google Scholar]
- 67. Ortner A., Huber D., Haske-Cornelius O., Weber H. K., Hofer K., Bauer W., Nyanhongo G. S., Guebitz G. M., Process Biochem. 2015, 50, 1277. [Google Scholar]
- 68. Euring M., Kirsch A., Schneider P., Kharazipour A., J. Mater. Sci. Res. 2016, 5. [Google Scholar]
- 69. Huber D., Ortner A., Daxbacher A., Nyanhongo G. S., Bauer W., Guebitz G. M., ACS Sustainable Chem. Eng. 2016, 4, 5303. [Google Scholar]
- 70. Huber D., Pellis A., Daxbacher A., Nyanhongo G. S., Guebitz G. M., Polymer 2016, 8, 280.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Gillgren T., Hedenström M., Jönsson L. J., Int. J. Biol. Macromol. 2017, 105, 438. [DOI] [PubMed] [Google Scholar]
- 72. Legras-Lecarpentier D., Stadler K., Weiss R., Guebitz G. M., Nyanhongo G. S., ACS Sustainable Chem. Eng. 2019, 7, 12621.. [Google Scholar]
- 73. Magina S., Barros-Timmons A., Evtuguin D. V., Holzforschung 2020, 74, 589. [Google Scholar]
- 74. Evstigneyev E. I., Shevchenko S. M., Wood Sci. Technol. 2018, 53, 7. [Google Scholar]
- 75. Liu Y., Luo G., Ngo H. H., Guo W., Zhang S., Bioresour. Technol. 2020, 298, 122511. [DOI] [PubMed] [Google Scholar]
- 76. Bourbonnais R., Paice M. G., Reid I. D., Lanthier P., Yaguchi M., Appl. Environ. Microbiol. 1995, 61, 1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Areskogh D., Li J., Gellerstedt G., Henriksson G., Ind. Crops Prod. 2010, 32, 458. [Google Scholar]
- 78. Hilgers R., Twentyman-Jones M., van Dam A., Gruppen H., Zuilhof H., Kabel M. A., Vincken J.-P., Catal. Sci. Technol. 2019, 9, 1535. [Google Scholar]
- 79. Milstein O., Hüttermann A., Fründ R., Lüdemann H. D., Appl. Microbiol. Biotechnol. 1994, 40, 760. [Google Scholar]
- 80. Meister J. J., Patil D. R., Channell H., J. Appl. Polym. Sci. 1984, 50, 398. [Google Scholar]
- 81. Mai C., Milstein O., Hüttermann A., Appl. Microbiol. Biotechnol. 1999, 51, 527. [DOI] [PubMed] [Google Scholar]
- 82. Mai C., Milstein O., Huttermann A., J. Biotechnol. 2000, 79, 10. [DOI] [PubMed] [Google Scholar]
- 83.
- 83a. Mai C., Schormann W., Hüttermann A., Enzyme Microb. Technol. 2001, 28, 460; [DOI] [PubMed] [Google Scholar]
- 83b. Mai C., Schormann W., Hüttermann A., Kappl R., Hüttermann J., Enzyme Microb. Technol. 2002, 30, 66. [Google Scholar]
- 84. Lund M., Ragauskas A. J., Appl. Microbiol. Biotechnol. 2001, 55, 699. [DOI] [PubMed] [Google Scholar]
- 85. Aracri E., Díaz Blanco C., Tzanov T., Green Chem. 2014, 16, 2597. [Google Scholar]
- 86. Munk L., Punt A. M., Kabel M. A., Meyer A. S., RSC Adv. 2017, 7, 3358. [Google Scholar]
- 87. Fitigău I. F., Boeriu C. G., Peter F., Macromol. Symp. 2015, 352, 78. [Google Scholar]
- 88. Ibrahim V., Mamo G., Gustafsson P.-J., Hatti-Kaul R., Ind. Crops Prod. 2013, 45, 343. [Google Scholar]
- 89. Prasetyo E. N., Kudanga T., Fischer R., Eichinger R., Nyanhongo G. S., Guebitz G. M., Biotechnol. J. 2012, 7, 284. [DOI] [PubMed] [Google Scholar]
- 90.A. Hüttermann, A. Majcherczyk, A. Braun-Lüllemann, C. Mai, M. Fastenrath, A. Kharazipour, J. Hüttermann, A. H. Hüttermann, Naturwissenschaften 2000, 87, 539. [DOI] [PubMed]
- 91. Cannatelli M. D., Ragauskas A. J., Appl. Microbiol. Biotechnol. 2017, 101, 6343. [DOI] [PubMed] [Google Scholar]
- 92.
- 92a. Mayolo-Deloisa K., González-González M., Rito-Palomares M., Front. Bioeng. Biotechnol. 2020, 8, 222; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92b. Zerva A., Simić S., Topakas E., Nikodinovic-Runic J., Catalysts 2019, 9, 1023; [Google Scholar]
- 92c. Moreno A. D., Ibarra D., Eugenio M. E., Tomás-Pejó E., J. Chem. Technol. Biotechnol. 2020, 95, 481. [Google Scholar]
- 93.A. Hüttermann, K. Nonninger, A. Kharazipour, (Pfleiderer Unternehmensverwaltung GmbH and Co KG), WO1998031729 A1, 1997.
- 94.A. Hatakka, K. Nyyssoenen-Hiekka, A. Temmes, (Metsä-Serla Oy), FI94261B, 1993.
- 95.E. Srebotnik, T. Ters, K. Fackler, K. Messner, O. Ertl, (Annikki GMBH), US009206292B2, 2015.
- 96.A. Nolsen, R. Miller, (Buckman Laboratories International Inc.), US009464386B2, 2016.
- 97.R. Bolle, W. Aehle, (Genencor International, Inc), 2000.
- 98.R. Bolle, W. Aehle, (Genencor International, Inc), US006217942B1, 2001.
- 99.C. Mai, A. Majcherczyk, A. Braun-Luellemann, J. Hüttermann, A. Hüttermann, (Evonik Stockhausen GmbH), 1997.
- 100.A. Hüttermann, A. Majcherczyk, C. Mai, A. Braun-Lullemann, M. Fastenrath, S. Noetzold, (Evonik Stockhausen GmbH), US20010009955 A1, 2001.
- 101.J. S. Yuan, Q. Li, (Texas A&M University System), US20200407884 A1, 2019.
- 102.M. B. Andersen, B. Yde, (Novo Nordisk A/S), WO1995023232 A1, 1995.
- 103.G. S. Nyanhongo, G. Guebitz, A. Ortner, S. Bischof, (Universität fur Bodenkultur Wien), WO2020152314 A1, 2020.
- 104.T. Hiramoto, Y. Mishima, T. Yamamoto, T. Hansen, K. Abe, (Novozymes AS and Takasago International Corp), US20060239939 A1, 2006.
- 105.A. Hüttermann, O. Milstein, (Hüttermann Aloy, Evonik Stockhausen GmbH), US5608040 A, 1994.
- 106.G. Henriksson, D. Areskogh, P. Oinonen, (Ecohelix AB), US009243078B2, 2016.
- 107.I. Schober, M. Richter, T. Heck, D. Jankowska, (Sika Technology AG, EMPA), US010202495B2, 2019.
- 108.Q. Xueqing, Y. Dongjie, Z. Haifeng, L. Hongming, Z. Mingsong, S. Yong, O. Xinping, CN102199268 A, 2011.
- 109.W. Lei, X. Yimin, D. Na, L. Yi, L. Jin, Y. Haitao, Y. Lan, W. Peng, F. Jianyun, CN102817286 A, 2012.
- 110.L. Na, Q. Menghua, X. Qinghua, F. Yingjuan, CN102296474 A, 2011.
- 111. Abbadessa A., Oinonen P., Henriksson G., BioResources 2018, 13, 7606. [Google Scholar]
- 112.
- 112a. Couto S. R., Herrera J. L. T., Biotechnol. Adv. 2006, 24, 500;16716556 [Google Scholar]
- 112b. Osma J. F., Toca-Herrera J. L., Rodríguez-Couto S., J. Environ. Manage. 2011, 92, 2907. [DOI] [PubMed] [Google Scholar]
- 113.Z. Ding, C. Guo, Z. Liting, B. Lu, Q. Wang, L. Peng, J. Lu, Z. Gu, G. Shi, (Jiangnan University), US10626380B2, 2020.
- 114. Agrawal K., Chaturvedi V., Verma P., Bioresour. Bioprocess. 2018, 5, 4.. [Google Scholar]
- 115. Gutiérrez A., Rencoret J., Ibarra D., Molina S., Camarero S., Romero J., Del Río J. C., Martínez Á. T., Environ. Sci. Technol. 2007, 41, 4124. [DOI] [PubMed] [Google Scholar]
- 116. Österberg M., Rojas O. J., Sipponen M. H., Mattos B. D., Green Chem. 2020, 22, 2712. [Google Scholar]
- 117.
- 117a. Moreno A., Sipponen M. H., Mater. Horiz. 2020, 7, 2237; [Google Scholar]
- 117b. Wu X., Jiang J., Wang C., Liu J., Pu Y., Ragauskas A., Li S., Yang B., Biofuels Bioprod. Biorefin. 2020, 14, 650. [Google Scholar]