

The anthracycline drug doxorubicin is an effective anticancer drugs with both DNA‐ and chromatin‐damaging activity. While the chromatin‐damaging activity constitutes major anticancer efficacy of doxorubicin, combination with the DNA‐damaging activity plagues the drug with long‐term toxicities such as cardiotoxicity, therapy‐related malignancies and gonadotoxicity. Therefore, developing DNA damage‐free anthracyclines is a promising direction for novel treatment options with limited side effects, which has been shown to be possible.
Keywords: aclarubicin, anthracyclines, cancer, cardiotoxicity, chromatin damage, DNA damage, doxorubicin, histone eviction, therapy‐related tumours, topoisomerase II
Abstract
The anthracycline drug doxorubicin is among the most used—and useful—chemotherapeutics. While doxorubicin is highly effective in the treatment of various hematopoietic malignancies and solid tumours, its application is limited by severe adverse effects, including irreversible cardiotoxicity, therapy‐related malignancies and gonadotoxicity. This continues to motivate investigation into the mechanisms of anthracycline activities and toxicities, with the aim to overcome the latter without sacrificing the former. It has long been appreciated that doxorubicin causes DNA double‐strand breaks due to poisoning topoisomerase II. More recently, it became clear that doxorubicin also leads to chromatin damage achieved through eviction of histones from select sites in the genome. Evaluation of these activities in various anthracycline analogues has revealed that chromatin damage makes a major contribution to the efficacy of anthracycline drugs. Furthermore, the DNA‐damaging effect conspires with chromatin damage to cause a number of adverse effects. Structure–activity relationships within the anthracycline family offer opportunities for chemical separation of these activities towards development of effective analogues with limited adverse effects. In this review, we elaborate on our current understanding of the different activities of doxorubicin and their contributions to drug efficacy and side effects. We then offer our perspective on how the activities of this old anticancer drug can be amended in new ways to benefit cancer patients, by providing effective treatment with improved quality of life.
Abbreviations
- AML
acute myeloid leukaemia
- APL
acute promyelocytic leukaemia
- DAMPs
damage‐associated molecular patterns
- DSBs
double‐strand breaks
- H2O2
hydrogen peroxide
- ICD
immunogenic cell death
- diMe‐doxorubicin
N,N‐dimethyl‐doxorubicin
superoxide anion
- ROS
reactive oxygen species
- t‐AML
therapy‐related acute myeloid leukaemia
- t‐APL
therapy‐related acute promyelocytic leukaemia
- t‐MNs
therapy‐related malignant neoplasms
- Topo II
topoisomerase II
- Topo IIα
topoisomerase II alpha
- Topo IIβ
topoisomerase II beta
Introduction
Doxorubicin, also known as adriamycin, is a member of the anthracycline anticancer drug family (Fig. 1). The first anthracycline drug, daunorubicin, was isolated from a soil sample found in Italy in 1960 [1, 2]. Daunorubicin is a pigmented antibiotic produced by the actinobacterium strain Streptomyces peucetius [2]. Soon, it was discovered that daunorubicin displayed anticancer activity in mice, which spurred its clinical use for the treatment of leukaemia, lymphoma and solid tumours in the late 1960s [3, 4]. In 1969, a daunorubicin homologue, doxorubicin, was isolated from a culture of chemically mutated S. peucetius [5]. Doxorubicin showed an even broader anticancer activity than daunorubicin, especially against solid tumours [6, 7]. However, quickly a major side effect of both otherwise highly potent anticancer drugs was noted—cardiotoxicity [8]. Cardiotoxicity incited by anthracyclines develops in a dose‐dependent manner and can be lethal [9, 10]. As a result, treatment has to be stopped once the maximal tolerated cumulative dose is reached, while patients with poor heart function are excluded from chemo regimens containing anthracyclines. In addition to treatment‐limiting cardiotoxicity, therapy‐related malignancies and gonadotoxicity are also associated with anthracycline treatment [9, 11]. With latest improvements in cancer therapy, the emphasis in cancer management has changed from ‘cure at any cost’ to giving quality of life after treatment more consideration. In this light, quests to understand and alleviate the side effects incurred by anthracyclines have been revived. Here, we provide an overview of the mechanisms of action and toxicity of anthracycline drugs and discuss different attempts that have been made to improve them. This is followed by our perspective on how to detoxify doxorubicin for effective anticancer treatment with limited adverse effects.
Fig. 1.
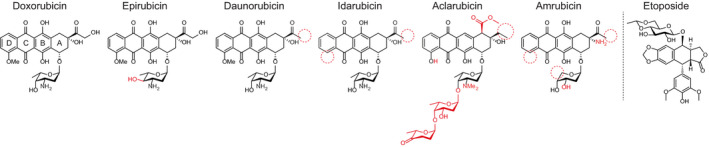
Structures of different anthracycline drugs and the structurally unrelated Topo II poison etoposide. Aglycon rings are numbered in doxorubicin. Structural differences compared with doxorubicin are indicated in red.
Mechanisms of action of anthracycline drugs
Topoisomerase II poison
The classical mechanism of action by which anthracyclines function is inhibition or poisoning of Topoisomerase II (Topo II) [12]. This enzyme plays a critical role in chromosome condensation, decatenation of intertwined DNA strands and relaxation of tension in the DNA strand in front of the replication fork [13, 14]. Topo II acts by introducing a transient double‐strand break (DSB) in one DNA strand (the G‐segment), allowing another DNA strand (the T‐segment) to pass through and subsequently closing the initial break by re‐ligation of the two DNA ends (Fig. 2) [13, 14, 15, 16, 17, 18]. Most anthracyclines (e.g. doxorubicin, epirubicin, daunorubicin, idarubicin, and amrubicin) intercalate into DNA and poison Topo II in its catalytic step following initial break induction by forming Topo II‐DNA complexes. These anthracyclines interfere at the interface of Topo II‐DNA with their sugar moieties and the cyclohexane ring A [19]. In essence, the interfacial positioning makes these anthracyclines act as molecular doorstops and prevent Topo II from re‐ligating the broken strand, which ultimately results in enzyme‐mediated DNA damage in the form of DSB [12, 20, 21]. Although the protein structure of a Topo II‐DNA‐doxorubicin complex is not available (reason will be discussed in the latter part), the door‐stopping act of doxorubicin can be deduced from the structure of a counterpart complex with the nonanthracycline Topo II poison, etoposide [22, 23, 24]. As a consequence of DSBs, DNA damage response (DDR) and TP53 pathways are activated, which lead to cell cycle arrest and cell death [25]. Some anthracyclines interrupt Topo II at other steps of the catalytic cycle, such as preventing the enzyme binding to the DNA (e.g. aclarubicin) or inhibiting ATP binding [13]. Topo II is essential for the survival of rapidly dividing cells, such as cancer cells that are more sensitive to DNA breaks than normal quiescent cells; hence, anthracyclines create a chemotherapeutic window by hijacking the essential enzyme function in cells [26]. For the same reason, anthracyclines also cause side effects, such as hair loss, bone marrow suppression and gastrointestinal complications.
Fig. 2.
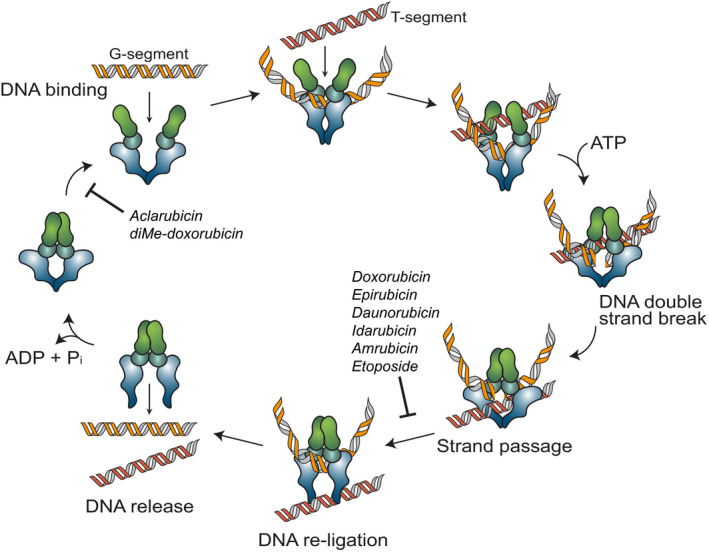
Schematic representation of the Topo II poisoning mechanism of anthracyclines. To entangle DNA or to remove DNA supercoils Topo II binds to DNA, introduce a transient DSB in one of the DNA strands (the G‐segment), allowing the second DNA strand (the T‐segment) to pass through. After re‐ligation of the G‐segment, the Topo II is released from DNA [15, 16, 17, 18]. The majority of Topo II poisons, including most anthracyclines (doxorubicin, daunorubicin, epirubicin, idarubicin and amrubicin) and etoposide, stabilize the Topo II complex after it has introduced the DNA DSB and prevent the DNA break from being resealed [13, 26]. Anthracycline variants aclarubicin and diMe‐doxorubicin inhibit the enzymatic activity by preventing Topo II from loading onto the DNA [13, 44]. Figure is inspired by [13].
DNA intercalation
Anthracyclines intercalate into the DNA helix with their anthraquinone moiety. While rings B and C of the tetracycline moiety overlap with adjacent DNA base pairs, and ring D passes through the intercalation site, the sugar moiety is pointed into the minor groove, which may compete for space with histones [19, 27]. In addition to stabilizing the Topo II‐DNA complex, DNA intercalation of anthracyclines has additional effects, such as inhibiting DNA and RNA synthesis [28, 29].
Oxidative stress
The quinone moiety in ring C of anthracyclines can be transformed into a semiquinone by a number of oxidoreductases, including cytochrome P450 reductases, xanthine oxidase and NADH dehydrogenase (complex I) of the mitochondrial electron transport chain [30, 31]. Subsequently, this semiquinone quickly regenerates and thereby converts oxygen into reactive oxygen species (ROS), such as superoxide anion () and hydrogen peroxide (H2O2), or oxidize the bond between the sugar and the aglycon resulting in reductive deglycosylation. Eventually , and H2O2 are converted into more reactive hydroxyl radicals (·OH) via the iron‐catalysed Haber‐Weiss reaction [32, 33]. In addition, anthracyclines can also mediate ROS production by directly interfering with iron metabolism. They can increase cellular levels of iron by interacting with iron regulatory proteins (IRP1 and/or IRP2) or accelerate the release of iron from ferritin, which then further amplifies iron‐mediated oxidative stress [34, 35, 36]. The excessive ROS production can lead to lipid oxidation, genomic and mitochondrial DNA damage, which are toxic to cells. Nevertheless, the contribution of ROS formation to the anticancer activity of anthracyclines is still unclear and heavily discussed. It is worth noting that excessive ROS production is often observed when cells were exposed to anthracycline doses that are much higher than clinical relevant concentrations. Yet, at physiological concentrations, significant ROS formation was observed at late time points after drug removal, indicating this might be a secondary effect of anthracycline treatment rather than a direct mode of action [37]. Notwithstanding, it cannot be excluded that ROS formation may reinforce other mechanisms of anthracyclines.
Chromatin damage
To organize two metres of DNA in the nucleus of a single cell, DNA is compacted at several levels. One level of organization is the formation of nucleosomes, where a segment of 146 base pairs of DNA is wrapped around eight histone proteins [38]. As mentioned above, when an anthracycline intercalates into DNA, the sugar moiety emanates into the DNA minor groove and competes with histones for space, resulting in the collapse of nucleosomes. As a result, histones are evicted from chromatin (Fig. 3) [27, 39]. In vitro experiments with reconstituted single nucleosomes showed that doxorubicin causes nucleosome dissociation in an ATP‐, transcription‐ and histone chaperone‐independent manner, which may explain why the structure of Topo II‐DNA‐doxorubicin complex is not available [27]. Moreover, the doxorubicin metabolite doxorubicinone, which lacks the sugar moiety of doxorubicin, was not able to dissociate nucleosomes under the same condition, suggesting a critical contribution of the sugar moiety to histone eviction [27, 40]. These data indicate that histone eviction induced by anthracyclines is a drug intrinsic process, which is cooperatively mediated by DNA intercalation of the anthraquinone group and nucleosome destabilization by the sugar moiety. This unique activity is not observed for other DNA intercalators (e.g. ethidium bromide [27]) or other chemotherapeutics (e.g. amsacrine or proflavin, data not published).
Fig. 3.
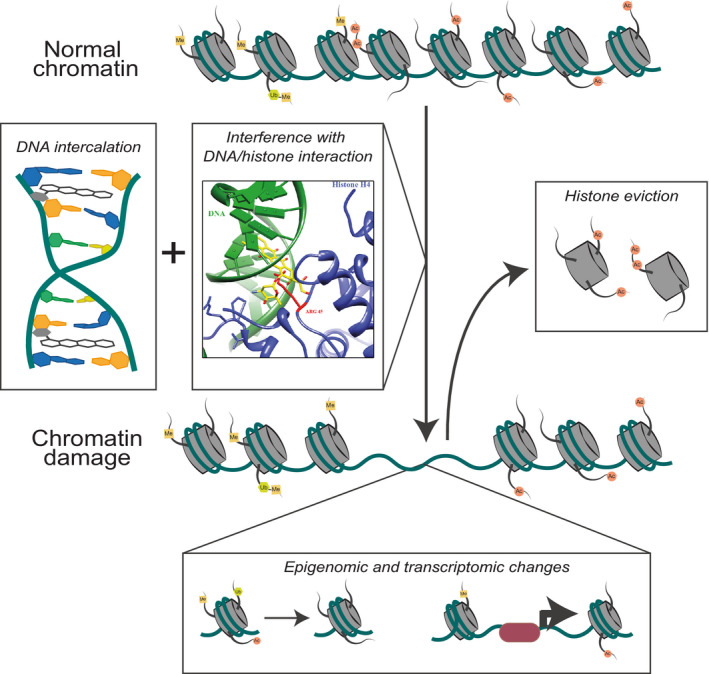
Schematic representation of chromatin damage induced by doxorubicin. Besides DNA intercalation by its anthraquinone group, doxorubicin's sugar moiety destabilizes nucleosome by competing for space with histones. Histone eviction caused by doxorubicin is shown to be ATP‐, transcription‐, and histone chaperone‐independent [27]. Histone eviction results in epigenetic and transcriptomic alterations and DSB repair attenuation, collectively referred to as chromatin damage. Part of the figure is reproduced from [27].
The dynamic structure of chromatin is essential for many nuclear processes, including transcription and replication. Therefore, the assembly, spatial organizing and compactization of chromatin are tightly regulated by various histone chaperones, ATP‐dependent chromatin remodelling complexes and histone‐modifying enzymes [41, 42]. Being the building blocks of chromatin, histones are directly involved in the regulation of these processes via different epigenetic modifications. Upon eviction, these modified histones are replaced by new/nascent ones with less or different epigenetic marks. This results in DDR delay, epigenetic and transcriptomic alterations, collectively termed as chromatin damage [43]. With the aid of next‐generation sequencing, unbiased (epi)genomic analysis revealed that each anthracycline evicts histones at select (epi)genomic regions [27, 43]. More specifically, doxorubicin evicts histones at open genomic regions marked by H3K36me3; while aclarubicin, whose sugar moiety is different from doxorubicin, induces histone eviction in a wider range, including compacted chromatin regions decorated by H3K27me3. As a matter of fact, anthracyclines could therefore be considered as epigenetic modifiers with defined (epi)genomic selectivity.
How histone eviction exactly causes cell death remains unclear, but it is likely to play a major contribution to the anticancer activity of the anthracycline drugs. This is illustrated by the anthracycline drugs aclarubicin and N,N‐dimethyl‐doxorubicin (diMe‐doxorubicin), which induce histone eviction without generating DNA damage [44]. Aclarubicin is prescribed mainly for the treatment of acute myeloid leukaemia (AML), showing similar efficacy as doxorubicin [27, 39, 44]. While aclarubicin was once used worldwide, it is currently only used in Japan and China. The specific reason behind this is not clear, and there are no clinical data that can explain the halt of usage. On the other hand, the doxorubicin analogue diMe‐doxorubicin was first reported in the 1980s [45]. It exhibited similar anticancer activity compared with doxorubicin in tissue culture experiments and in mice [27, 39, 44]. Further, its pharmacokinetics was tested in mice and rabbits [46], but no further follow‐up was reported. Surprisingly, it was recently shown by our laboratory that diMe‐doxorubicin only induces chromatin damage but no DSB, suggesting that chromatin damage rather that DNA breaks may be the dominant cytotoxic mechanism [44]. This is further substantiated by the anthracycline variant amrubicin, which only induces DSBs. Amrubicin is much less effective than doxorubicin, aclarubicin and diMe‐doxorubicin in killing cancer cells and thus did not enter clinic. Taken together, this implies that chromatin damage rather than DSB formation constitutes the major anticancer activity of anthracyclines.
Immune modulation
Besides the direct effect on eliminating tumour cells, anthracyclines can also promote antitumour immunity. During cell death, cell contents can be released into the tumour microenvironment, including tumour antigens and danger signals (also known as damage‐associated molecular patterns, DAMPs) [47]. These DAMPs can initiate inflammatory response, recruit immune cells and facilitate recognition of tumour cells. This process is known as immunogenic cell death (ICD) [48, 49, 50]. It has been shown that anthracyclines such as doxorubicin can induce ICD and thereby elicit a dendritic cell‐mediated tumour‐specific CD8+ T‐cell response in a colon carcinoma mouse model [51]. Moreover, doxorubicin was reported to selectively deplete myeloid‐derived suppressor cells from the tumour microenvironment, which relieved the immunosuppressive impact of these cells in a murine breast cancer model [52]. Recently, it is observed that the C‐type lectin receptor Clec2d is activated by binding histones to induce inflammation and tissue damage responses [53]. So it would be interesting to test whether histones can be externalized by doxorubicin, detected by the Clec2d receptor and cause an inflammation response. The immune stimulatory activity of doxorubicin, in the context of immune checkpoint blockade, was confirmed in a multi‐arm noncomparative phase II trial. Treatment of triple negative breast cancer patients with doxorubicin followed by PD1 blockade resulted in an overall response rate of 35%, compared with 17% for PD1 blockade alone [54]. Although this finding needs to be confirmed in larger cohorts, it suggests that the immune modulating function of anthracyclines may have a synergistic role in the overall anticancer activity in patients.
Anthracycline‐associated severe side effects and preventive solutions
Although doxorubicin has been a cornerstone in cancer treatment for nearly five decades, its use is plagued with severe and treatment‐limiting side effects. Next to common generally acute and reversible chemo‐related adverse effects, such as nausea, vomiting, diarrhoea and bone marrow suppression, anthracycline treatment is associated with long‐term side effects, namely cardiotoxicity, therapy‐related malignancies and gonadotoxicity. These long‐term adverse effects severely impact the quality of life of cancer survivors, which limit the further application of anthracyclines. Therefore, extensive research has been performed to understand and reduce the anthracycline‐induced long‐term side effects.
Cardiotoxicity
The most treatment‐limiting and therefore probably the best studied side effect of anthracyclines is cardiotoxicity. Anthracycline‐induced cardiotoxicity presents as cardiomyopathy, ventricular dysfunction, pericarditis‐myocarditis syndrome or arrhythmias and is dose‐dependent and irreversible [10, 55, 56]. As a result, doxorubicin treatment is limited to a cumulative dose of 450–550 mg·m−2 [9, 10]. Besides cumulative dose, the risk of cardiotoxicity is also associated with treatment schedule, age extremes, and combinations with other drugs or radiotherapy in the heart region [57, 58]. Currently, there is no management or medication to relieve anthracycline‐induced cardiotoxicity, and the only option for patients with severe symptoms is a heart transplantation. Therefore, doxorubicin is excluded from treating patients with a poor heart function, usually old patients. Thus, alleviating cardiotoxicity would greatly improve cancer treatment with anthracyclines.
Multiple mechanisms have been proposed, including mitochondrial dysfunction and/or lipid peroxidation as a result of ROS formation, targeting Topo II beta (Topo IIβ) in cardiomyocytes, and effects on calcium homeostasis [59, 60, 61, 62]. To reduce anthracycline‐induced cardiotoxicity, several attempts to manipulate these pathways have been made. In the following sections, we will discuss these in detail and propose a possible solution based on recent data.
ROS alleviation
The most intensely studied mechanism of anthracycline‐induced cardiotoxicity is ROS production through interference with redox cycling and mitochondrial function [63]. To meet the high demand of ATP supply, cardiomyocytes have a greater density of mitochondria compared with other tissues, which could explain why the heart is more affected by anthracycline‐induced ROS production than other tissues [59, 64]. Green et al. [65] showed that doxorubicin‐induced mitochondrial dysfunction coincided with the production of ROS and cytochrome C release, which in turn activated Caspase‐3 and initiate apoptosis in H9C2 cardiac cells. It was reported that pretreatment with the free radical scavenger tocopherol reduced the cardiotoxicity of doxorubicin in a lymphoma mouse model, without affecting its antitumour efficacy [60]. Although similar results were observed in an AML animal model, the cardiac protective effects of radical quenchers in clinical trials were disappointing [66, 67].
Similar to ROS scavengers, most iron‐chelating agents can reduce ROS formation and alleviate doxorubicin‐induced cardiotoxicity in preclinical models. However, such benefits were not observed in patients [68, 69]. The iron chelator dexrazoxane is an exceptional case. It was reported to reduce anthracycline‐induced cardiotoxicity in some clinical studies, albeit not in all [70, 71]. However, this reduced toxicity is likely mediated by mechanisms different from ROS quenching, since other iron chelators are not cardiac protective [72]. Several alternative mechanisms of dexrazoxane function have been proposed, including inhibition of both apoptosis and necroptosis of cardiomyocytes [73] and antagonizing doxorubicin‐induced DNA damage by interfering with Topo IIβ [74].
Although it is convincingly shown that anthracyclines can induce ROS formation in in vitro studies, the discrepancy between the effectivity of ROS scavengers and iron chelators in preclinical studies and patients challenges the contribution of ROS production in anthracycline‐induced heart damage. Using appropriate preclinical cardiotoxicity models and treatment with anthracyclines at clinical relevant concentrations and schedules may help clarifying this issue.
Precluding from targeting topoisomerase IIβ in cardiomyocytes
In human, Topo II enzymes are expressed in two isoforms, Topo IIα and Topo IIβ [75]. Although these two isoforms are encoded by different genes, they share substantial amino acid sequence identity and exhibit almost identical enzymological properties [76]. Notwithstanding their similarities, the expression patterns of Topo IIα and Topo IIβ are different. Topo IIα is mainly expressed in proliferating cells, and almost absent in quiescent and differentiated tissues. Topo IIα is associated with replication forks and stays bound to chromosomes during mitosis, which makes its expression essential for proliferation. On the contrary, Topo IIβ expression is independent of proliferation status and is high in most cell types [76]. In line with this notion, adult mammalian cardiomyocytes express Topo IIβ, but no detectable Topo IIα. Zhang et al. [61] reported that targeting Topo IIβ in cardiomyocytes by doxorubicin is important for the initiation of cardiotoxicity. It was shown that mice with cardiomyocytes‐selective conditional Topo IIβ knockout (Topo IIβ+/Δ and Topo IIβΔ/Δ) were not susceptible to the cardiac impairment caused by doxorubicin as observed in Topo IIβ+/+ mice. Further, Lyu et al. [74] reported that dexrazoxane reduced doxorubicin‐induced DNA damage in cardiomyocytes in vitro by rapid proteasomal degradation of Topo IIβ. These studies indicate that the DSBs mediated by Topo IIβ poisoning is a major cause of doxorubicin‐induced cardiotoxicity. Nevertheless, DSB cannot be the only reason, since the structurally nonrelated Topo II poison etoposide does not cause cardiotoxicity. From a clinical point of view, it suggests that Topo IIα‐specific anthracycline would prevent cardiotoxicity in patients and that Topo IIβ expression could be used as a prognostic marker for cardiotoxicity. Unfortunately, no genuine Topo IIα‐ or Topo IIβ‐specific drugs are available in clinic at present.
Novel delivery strategies to reduce anthracycline‐induced toxicity
Due to the unsatisfactory effects of ROS scavengers and iron chelators in the clinic, tumour‐specific drug delivery systems were introduced in 1990s to reduce doxorubicin‐induced toxicities. These delivery strategies included nanoparticle encapsulated liposomal doxorubicin (LD) and pegylated LD (PLD). LD and PLD both show prolonged serum half‐life and a smaller volume of distribution compared with conventional doxorubicin [77]. LD and PLD can extravasate into the tumour via gaps in the micro vessels, whereas other tissues are much less permeable through tight junctions. Therefore, the long serum circulation of LD and PLD results in more specific tumour accumulation. Various animal models, as well as clinical trials, showed that these particles significantly decreased cardiotoxicity compared with conventional doxorubicin, without compromising antitumour efficacy [78, 79, 80]. Therefore, both LD and PLD are approved by the FDA for treating AIDS‐related Kaposi's sarcoma, multiple myeloma, breast and ovarian cancer, but their clinical application is limited by drug leakage and higher costs.
Separating chromatin damage from DNA damage
With the aim to identify more effective anthracyclines with fewer side effects, thousands of doxorubicin analogues, either isolated from natural sources, produced by mutant enzymes or prepared by organic (semi)synthesis, have been evaluated in the past decades. However, only few variant drugs showed reduced cardiotoxicity without loss of anticancer activity. One such analogue which entered the clinic is epirubicin. In a meta‐analysis, epirubicin treatment showed significantly less cardiotoxicity compared with doxorubicin (OR, 0.39, 95% confidence interval: 0.20–0.78, P = 0.008) and subclinical cardiotoxicity (OR, 0.30, 0.16–0.57, P < 0.001) without compromising antitumour efficacy [81]. Therefore, epirubicin can be used at higher cumulative dose (900–1000 mg·m−2) compared with doxorubicin (450–550 mg·m−2). Although epirubicin can be used at higher cumulative dose, its application is still limited by cardiotoxicity.
The key question for the development of analogues with reduced toxicity is whether these toxic effects and anticancer activities are mediated by the same mechanism(s), which determines whether it is theoretically feasible to eliminate the cardiotoxicity of anthracycline without compromising its therapeutic efficacy. Recent work of our group provides some insight. We observed that aclarubicin, as well as the doxorubicin analogue diMe‐doxorubicin, showed strongly reduced cardiotoxicity in various mouse models and human‐induced pluripotent stem cells‐derived cardiomyocyte microtissues, without compromising anticancer activity [44]. N,N‐dimethylation of the amino sugar eliminated the DNA‐damaging capacity of these compounds, while retaining effective histone eviction activity (Fig. 4). On the other hand, etoposide and amrubicin, with only DNA‐damaging activity, are also not cardiotoxic in mouse models and patients, but display much lower anticancer activity. These observations indicate that the combination of DNA with chromatin damage, as for doxorubicin and other clinically used anthracyclines, is responsible for the cardiotoxicity of these drugs [44]. Therefore, variants with only chromatin‐damaging activity would be a promising direction for the development of next‐generation anthracyclines. Furthermore, the identification of the structure–activity relationship of the sugar moiety and cardiotoxicity provides a new strategy for anthracycline development.
Fig. 4.
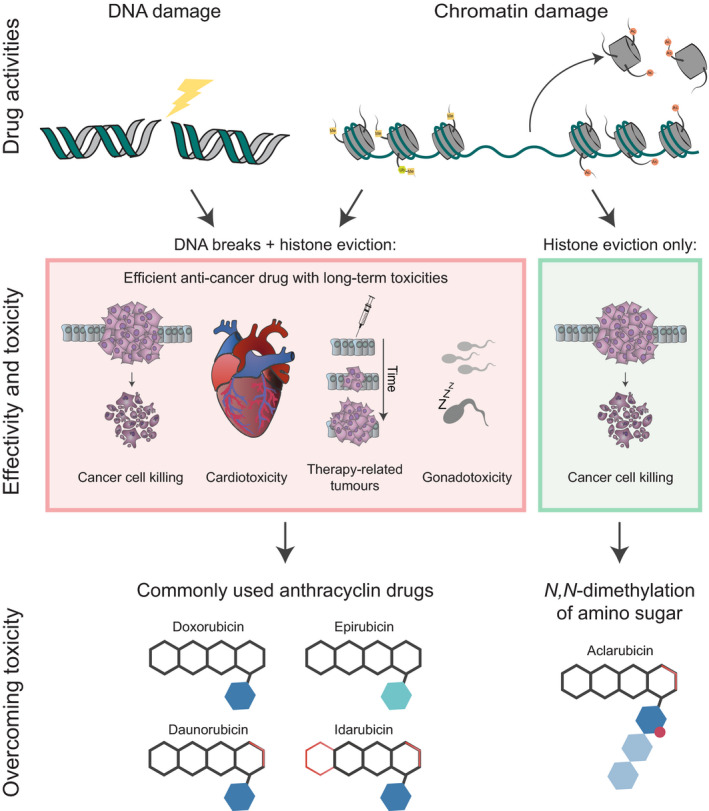
Schematic overview of the activities and toxicities of the clinically used anthracyclines and their underlying mechanisms. Most commonly used anthracyclines, including doxorubicin, epirubicin, daunorubicin and idarubicin, possess both DNA‐ and chromatin‐damaging activities. As a consequence, these drugs are associated with cardiotoxicity, therapy‐related malignancies and gonadotoxicity. N,N‐dimethylation of the sugar moiety, as for aclarubicin (and diMe‐doxorubicin), results in anthracycline variants with only chromatin‐damaging activity, which are effective anticancer drugs with limited toxicities.
Therapy‐related malignant neoplasms
Attributing to the increased survival of cancer patients which modern anticancer therapy has made possible, the long‐term side effects, such as tumorigenicity, have become an issue. Currently, 17–19% of all new primary malignancies occur in cancer survivors [82, 83]. Among all the long‐term adverse effects caused by chemotherapy, therapy‐related malignant neoplasms (t‐MNs) are one of the most deleterious, because of substantial morbidity and considerable mortality. Soon after discovery, anthracyclines (excluding aclarubicin hereafter in this section) has been found to cause transformation and mutagenesis in vitro and tumorigenic in vivo [84, 85, 86, 87, 88, 89, 90], and anthracycline exposure is associated with increased risks of t‐MNs in cancer survivors. The t‐MNs most often ascribed to anthracyclines are AML [91, 92, 93], sarcoma [94, 95, 96] and female breast cancer [96, 97]. Thyroid cancer [98] and acute promyelocytic leukaemia (APL) [99, 100] have also been linked to antecedent anthracycline treatment.
The anthracycline therapy‐related AMLs (t‐AMLs) frequently exhibit balanced chromosomal translocations at 11q23 (involving MLL1 gene) or 21q22 (involving AML1/RUNX1/CBFA2 gene), however occurring at unique breakpoints than de novo AML with the same cytogenetics [101, 102, 103, 104]. In contrast to alkylating agent‐associated t‐AMLs, these leukaemias are rarely preceded by a myelodysplastic phase [105]. They develop with a shorter latency, often within 1–3 years after the initial anthracycline‐based chemotherapy and, in some cases, within 1 year [106]. Due to unfavourable, complex or monosomal karyotypes, these t‐AMLs often present as aggressive diseases and are associated with poor prognosis compared with de novo AML [106, 107]. Anthracyclines are also involved in the development of therapy‐related APL (t‐APL) featured with balanced translocation of t(15;17) [99], which results in a double dominant‐negative fusion protein, PML‐RARα [100]. Anthracycline‐associated t‐APL also arises after a short latency period, usually without a preleukaemic phase [99, 100]. After a peak at 2 years following primary anthracycline treatment, the incidence of t‐APL quickly decreases with time. Although the chromosomal breakpoints induced by anthracyclines are distinct from those observed in de novo t(15;17) APL, the clinical outcomes of t‐APL and de novo APL are similar after all‐trans retinoic acid‐ and anthracycline‐based treatments, for which the 5‐year survival rate is about 80% [99, 100]. Anthracycline‐associated solid tumours typically occur >10 years after exposure and in a dose‐dependent manner [94, 95, 96, 97, 98]. There is not much known about the genetic alterations of anthracycline‐related solid tumours, though a strong dose–response correlation with doxorubicin was found in survivors of Li‐Fraumeni syndrome‐associated cancer types compared with other childhood cancer survivors [96]. Furthermore, we recently reported that doxorubicin single drug treatment induced breast cancer development in Trp53+/− female mice, indicating the direct contribution of doxorubicin treatment to tumour development [44].
Besides the tumorigenicity of anthracyclines, cancer survivors may be especially susceptible to developing t‐MNs due to a variety of other risk factors. These include genetic predisposition (such as the abovementioned Li‐Fraumeni syndrome), carcinogenic exposures in common (such as tobacco use or alcohol abuse), host effects (age, gender, immunodeficiency or obesity) and combination therapy with other mutagenic chemotherapeutics (alkylating agents, etoposide or radiotherapy) [82, 83, 95, 96, 97]. Therefore, the exact mechanisms how anthracyclines contribute to t‐MN development remain unclear. One option follows reports showing that leukaemia‐associated translocation t(8;21) can be detected in hematopoietic cells of healthy individuals with no overt leukaemia [108, 109], and anthracycline‐related t(8;21) t‐AMLs were found to be positive for JAK2 V617F mutation [110], which suggests that t‐AML is the consequence of a series of genetic alterations. Anthracyclines may facilitate the complete transformation of preleukaemic cells by introducing additional mutations. On the other hand, anthracyclines can cause chromosomal translocations through an indirect mechanism mediated by apoptotic nucleases [111, 112, 113]. Nevertheless, accumulating evidence suggests that anthracyclines play a direct role in causing t‐MN associated genetic aberrations. Anthracyclines generate DSBs by hijacking Topo II, particularly at breakpoint hotspot regions of leukaemic translocations [103]. Unfaithful repair by error‐prone DNA repair pathways can then result in mutagenesis or chromosomal translocations [114]. Through a similar mechanism of action, the structurally unrelated Topo II poison etoposide was also found to be associated with t‐MNs of similar karyotypes in a dose‐dependent manner, albeit less potent than anthracyclines [95, 115, 116]. The inferior potency of etoposide in transformation is also observed in a Trp53+/− mouse model treated with single agents of comparable dose and schedule, which excluded the influence of genetic predisposition of host and concurrent anticancer therapies [44]. This tumorigenic difference can be explained by the strongly delayed DNA repair of anthracycline due to eviction of histone variant H2AX [27].
H2AX is an important histone variant for DNA damage repair, which is phosphorylated at DNA damage sites and responsible for repair machinery recruitment. Eviction of H2AX by doxorubicin greatly attenuates DNA damage repair and consequently results in enhanced cell death and more transformation compared with etoposide [27, 43]. In line with this hypothesis, the same Trp53+/− mouse experiment and in vitro data showed that aclarubicin and diMe‐doxorubicin without DNA‐damaging activity are not tumorigenic [44, 117, 118]. Collectively, DNA damage induced by Topo II poisons is a main cause of t‐MNs.
As mentioned above, anthracyclines evict histones with different epigenomic selectivity. It is interesting to notice that t(11q23) AML with MLL1 translocation is also associated with epigenetic changes, since MLL1 is an H3K4 methyltransferase [119]. The C‐terminal SET domain of MLL1, which is responsible for methylating H3K4, is missing in the fusion oncoprotein of 5′‐MLL1–partner‐3′ rearrangement. Epigenetic profiling after MLL1 deletion or with MLL1 fusion proteins revealed reduced H3K4 methylation at promotor region of target genes [120, 121]. Considering the selectivity of doxorubicin for H3K4me3 at active promotors, this coincidence may provide another explanation for the development of t(11q23) AML and its resistance to doxorubicin‐based regimens [122, 123]. As a result, anthracyclines with different histone eviction profiles, such as aclarubicin and diMe‐doxorubicin, could provide alternative treatment options for doxorubicin‐resistant AMLs, and vice versa [45, 124, 125, 126, 127, 128].
Due to limited understanding of the mechanisms of action, t‐MN was previously considered as the original sin of anthracycline treatment because of resulted DNA damage. Hence, hope was laid on early detection of t‐MNs by intense follow‐up screening in susceptible cancer survivors or restraint of high cumulative dose of anthracyclines. However, the discovery of histone eviction activity of anthracyclines not only offers a new anticancer mechanism, but also provides a strategy to prevent t‐MNs, which is experimentally illustrated by aclarubicin [44, 117]. The recent understanding on the structure–activity relationship of anthracyclines makes it possible to eliminate the DNA‐damaging activity of anthracycline and related toxicities, while remaining their anticancer efficacy.
Gonadotoxicity
Owing to its mechanisms, doxorubicin also targets healthy tissues with high proliferating rates, such as myeloid and lymphoid tissues, gastrointestinal mucosa and gonads. Since the survival rates of cancer patients improved spectacularly in the last two decades, the number of cancer survivors suffering from doxorubicin‐induced gonadotoxicity also strongly increased [129]. Gonadotoxicity not only causes psychosocial distress, but also increases the risk of subsequent complications, such as osteoporosis, infertility and cardiovascular disease [130]. Gonadal damage caused by doxorubicin treatments happens to patients at all stages of life. Although many of the cancer survivors could regain gonadal functions in a few months or years after doxorubicin treatment [131], they may have a shortened reproductive lifespan or late effects on pregnancy than the age‐matched normal population [132, 133, 134]. Currently, cryopreservation of gametes or embryos is the only option to preserver fertility in patients receiving doxorubicin‐containing therapy. However, this approach is only applicable to patients in a reproductive age and can be problematic in adolescent patients. For patients who have not yet commenced puberty, there is no clinically approved method for fertility preservation at present [135], despite that previous doxorubicin treatment during prepubertal period can lead to severe injury of the adult fertility [136].
Several classes of compounds have been proposed to protect gonads from doxorubicin insult in mouse models, including hormone agonists [137], anti‐oxidants [138, 139], proteasome inhibitors [140], tyrosine kinase and DDR inhibitors [141]. Before validating these drugs in a patient cohort, it is more important to test whether these inhibitors alleviate the gonadotoxicity without compromising the anticancer activity of doxorubicin in vivo. Nevertheless, development of active anthracycline variants with limited gonadotoxicity would be a preferable strategy, if possible. The depletion of follicular reserve in females and depletion of spermatogenesis in males caused by doxorubicin treatment can be attributed to the DSBs generated by the drug and subsequent cell death of germ cells [134, 142, 143, 144]. Besides direct germ cell destruction, doxorubicin also causes DSBs in somatic cells, vasculature and apoptosis of the stromal compartments in gonads [136, 143, 145, 146, 147]. The latter then further impairs the development of fertile germ cells. Similar effects were also observed for the nonanthracycline Topo II poison etoposide, which also causes DSBs and destruction of gonads [148, 149]. These data suggest that the DNA‐damaging activity of doxorubicin plays an important role in mediating gonadotoxicity. This observation is further strengthened by our recent study showing that aclarubicin and diMe‐doxorubicin, both lacking DNA‐damaging activity but with comparable antitumour capacity as doxorubicin, did not cause apoptosis of developing follicles in female mice [44]. However, diMe‐doxorubicin, with different histone eviction profile than aclarubicin and doxorubicin (unpublished results), still induced depletion of spermatogenesis in male mice, albeit at a lower degree than doxorubicin.
Oxidative stress has also been proposed as a mechanism of doxorubicin‐induced gonadotoxicity [150]. However, some work using spermatogonia and immature Sertoli cell lines has shown no increase of ROS formation before the onset of cytotoxicity [151]. In line with this observation, co‐administration of anti‐oxidants showed no protective effect on doxorubicin‐induced testicular toxicity in vivo [139, 152]. Collectively, these data suggest that DNA‐damaging activity of doxorubicin is a major cause for gonadotoxicity, especially in females, with perhaps some contribution of specific histone eviction in the case of diMe‐doxorubicin in male gonadotoxicity.
Perspectives
Since the discovery of daunorubicin and doxorubicin in the 1960s, a search for less toxic yet effective alternatives to doxorubicin was initiated in the 1980s. Out of thousands of anthracycline variants tested, only a few entered the clinic, most notably epirubicin, idarubicin and aclarubicin. One reason for this limited number of successful compounds might be the lack of consensus on the mechanism of action of anthracyclines for their anticancer activity and toxicities. Furthermore, whether the severe toxicities of these drugs are intimately connected with their anticancer activity has been a lingering topic in the field. For a long time, DSB induction was considered as the main anticancer activity of anthracyclines. While only recently, a second activity –chromatin damage as a result of histone eviction– was proposed [27, 39]. Chromatin damage is not only a novel activity of anthracyclines, but also a new anticancer mechanism, which is not found in other types of chemotherapeutics. The ground‐breaking discovery of chromatin damage is granted by modern molecular technologies, such as time‐lapse confocal imaging, photoactivation and various next‐generation sequencing techniques. Hence, it is still meaningful to re‐investigate old drugs with modern technology. This may yield new mechanisms of action that can be explored to arrive at active and detoxified doxorubicin and other drug variants. Additionally, this resulted in the rediscovery of an anthracycline variant, aclarubicin, as a less toxic but very active drug in (relapsed) AML treatment.
While the potential cardiotoxicity‐low/free anthracyclines need to be tested in clinic, some improvements of current anthracycline‐containing chemotherapy regimen should be considered. For instance, it would be debatable to combine anthracyclines with etoposide in the same treatment regimen concerning the contribution of DNA‐damaging activity to multiple toxicities, although this is frequently used in AML treatment. Likewise, specific anthracycline variant should be carefully selected for children cancer patients or patients with predisposal genetic disorder to avoid toxicities. The new mechanism, histone eviction with certain (epi)genomic selectivity, indicates that anthracyclines are in fact also epigenetic drugs. Preliminary data showed that diffuse large B‐cell lymphoma cells with elevated levels of H3K27me3 were more susceptible to aclarubicin than daunorubicin [43], indicating anthracycline variant selection can be personalized for cancer treatment based on their histone eviction profiles.
The recent understanding on anthracycline anticancer activity and toxicities suggests that anthracycline development should focus on depleting DNA‐damaging activity from chromatin‐damaging activity. Such drugs should allow effective anthracycline‐based therapies devoid of the major treatment‐limiting adverse effects: cardiotoxicity, therapy‐related malignancies and gonadotoxicity. This would especially benefit cancer patients with a poor heart function, which are currently excluded from anthracycline‐based chemotherapy. In addition, drug variants lacking these side effects could be used in more intense and/or longer therapy and could be used for relapsed patients with a history of anthracyclines‐based therapies.
In conclusion, despite the long history of anthracyclines, the novel discovery of chromatin damage as the major antitumour activity and its collective contribution with DNA‐damaging activity to toxicities, allows the development of potentially new treatment strategies to improve cancer therapy and the quality of life of cancer survivors.
Conflict of interest
JN is a shareholder in NIHM that aims to produce aclarubicin for clinical use.
Author contributions
SvdZ and XQ conceived the manuscript and constructed the text under supervision of JN.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1111/febs.15583.
Acknowledgements
We thank I. Berlin for critical reading of the manuscript and L. Hamoen for input on the figures. This work was supported by ERC Adv. Grant and KWF grants to JN and the Institute for Chemical Immunology, an NWO Gravitation project funded by the Ministry of Education, Culture and Science of the Netherlands to JN.
Sabina Y. van der Zanden and Xiaohang Qiao contributed equally to this work.
References
- 1. Camerino B & Palamidessi G(1960) Derivati della parazina II. Sulfonamdopir (in Italian). Gazz Chim Ital 90, 1802–1815. [Google Scholar]
- 2. Di Marco A, Cassinelli G & Arcamone F (1981) The discovery of daunorubicin. Cancer Treat Rep 65 (Suppl 4), 3–8. [PubMed] [Google Scholar]
- 3. Tan C, Tasaka H, Yu KP, Murphy ML & Karnofsky DA(1967) Daunomycin, an antitumor antibiotic, in the treatment of neoplastic disease. Cancer 20, 333–353. [DOI] [PubMed] [Google Scholar]
- 4.drugs.com, (2000). Drugs.com/daunorubicin. https://www.drugs.com/pro/daunorubicin.html, Accessed April 8th 2020.
- 5. Arcamone F, Cassinelli G, Fantini G, Grein A, Orezzi P, Pol C & Spalla C (1969) Adriamycin, 14‐hydroxydaunomycin, a new antitumor antibiotic from S. peucetius var. caesius. Biotechnol Bioeng 11, 1101–1110. [DOI] [PubMed] [Google Scholar]
- 6. Ho AD(1996)New anthracyclines–a comparative analysis of efficacy and toxicity. In Acute Leukemias V Experimental Approaches and Management of Refractory Disease (Hiddemann W, Büchner T, Wörmann B, Ritter J, Creutzig U, Plunkett W & Keating M, eds), pp. 591–594. Springer Berlin Heidelberg, Berlin Heidelberg. [Google Scholar]
- 7.drugs.com, (2000). Drugs.com/doxorubicin. https://www.drugs.com/pro/doxorubicin.html. Accessed April 8th 2020.
- 8. Tan C, Tasaka H, Yu KP, Murphy ML & Karnofsky DA (1967) Daunomycin an antitumor antibiotic in treatmentt of neoplastic disease ‐ clinical evaluation with special reference to childhood leukemia. Cancer 20, 333. [DOI] [PubMed] [Google Scholar]
- 9. Lotrionte M, Biondi‐Zoccai G, Abbate A, Lanzetta G, D'Ascenzo F, Malavasi V, Peruzzi M, Frati G & Palazzoni G (2013) Review and meta‐analysis of incidence and clinical predictors of anthracycline cardiotoxicity. Am J Cardiol 112, 1980–1984. [DOI] [PubMed] [Google Scholar]
- 10. Jones RL, Swanton C & Ewer MS (2006) Anthracycline cardiotoxicity. Expert Opin Drug Saf 5, 791–809. [DOI] [PubMed] [Google Scholar]
- 11. Mistry AR, Felix CA, Whitmarsh RJ, Mason A, Reiter A, Cassinat B, Parry A, Walz C, Wiemels JL, Segal MR et al. (2005) DNA topoisomerase II in therapy‐related acute promyelocytic leukemia. N Engl J Med 352, 1529–1538. [DOI] [PubMed] [Google Scholar]
- 12. Tewey KM, Rowe TC, Yang L, Halligan BD & Liu LF (1984) Adriamycin‐induced DNA damage mediated by mammalian DNA topoisomerase‐Ii. Science 226, 466–468. [DOI] [PubMed] [Google Scholar]
- 13. Nitiss JL (2009) Targeting DNA topoisomerase II in cancer chemotherapy. Nat Rev Cancer 9, 338–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nitiss JL (2009) DNA topoisomerase II and its growing repertoire of biological functions. Nat Rev Cancer 9, 327–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Roca J & Wang JC (1992) The Capture of a DNA double helix by an Atp‐dependent protein clamp ‐ a key step in DNA transport by type‐Ii DNA topoisomerases. Cell 71, 833–840. [DOI] [PubMed] [Google Scholar]
- 16. Roca J & Wang JC (1994) DNA transport by a type‐Ii DNA topoisomerase ‐ evidence in favor of a 2‐gate mechanism. Cell 77, 609–616. [DOI] [PubMed] [Google Scholar]
- 17. Pommier Y, Sung YL, Huang SYN & Nitiss JL (2016) Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat Rev Mol Cell Biol 17, 703–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Dong KC & Berger JM (2007) Structural basis for gate‐DNA recognition and bending by type IIA topoisomerases. Nature 450, 1201–1205. [DOI] [PubMed] [Google Scholar]
- 19. Wang AHJ, Ughetto G, Quigley GJ & Rich A (1987) Interactions between an anthracycline antibiotic and DNA ‐ molecular‐structure of daunomycin complexed to D(Cpgptpapcpg) at 1.2‐a resolution. Biochemistry‐Us 26, 1152–1163. [DOI] [PubMed] [Google Scholar]
- 20. Pommier Y, Schwartz RE, Zwelling LA & Kohn KW (1985) Effects of DNA intercalating agents on topoisomerase‐Ii induced DNA strand cleavage in isolated mammalian‐cell nuclei. Biochemistry‐Us 24, 6406–6410. [DOI] [PubMed] [Google Scholar]
- 21. Tewey KM, Chen GL, Nelson EM & Liu LF (1984) Intercalative antitumor drugs interfere with the breakage‐reunion reaction of mammalian DNA topoisomerase‐Ii. J Biol Chem 259, 9182–9187. [PubMed] [Google Scholar]
- 22. Wu CC, Li TK, Farh L, Lin LY, Lin TS, Yu YJ, Yen TJ, Chiang CW & Chan NL (2011) Structural basis of type II topoisomerase inhibition by the anticancer drug etoposide. Science 333, 459–462. [DOI] [PubMed] [Google Scholar]
- 23. Wilstermann AM, Bender RP, Godfrey M, Choi S, Anklin C, Berkowitz DB, Osheroff N & Graves DE (2007) Topoisomerase II‐drug interaction domains: identification of substituents on etoposide that interact with the enzyme. Biochemistry‐Us 46, 8217–8225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bender RP, Jablonksy MJ, Shadid M, Romaine I, Dunlap N, Anklin C, Graves DE & Osheroff N (2008) Substituents on etoposide that interact with human topoisomerase II alpha in the binary enzyme‐drug complex: Contributions to etoposide binding and activity. Biochemistry‐Us 47, 4501–4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Perego P, Corna E, De Cesare M, Gatti L, Polizzi D, Pratesi G, Supino R & Zunino F (2001) Role of apoptosis and apoptosis‐related genes in cellular response and antitumor efficacy of anthracyclines. Curr Med Chem 8, 31–37. [DOI] [PubMed] [Google Scholar]
- 26. Liu LF (1989) DNA topoisomerase poisons as antitumor drugs. Annu Rev Biochem 58, 351–375. [DOI] [PubMed] [Google Scholar]
- 27. Pang B, Qiao X, Janssen L, Velds A, Groothuis T, Kerkhoven R, Nieuwland M, Ovaa H, Rottenberg S, van Tellingen O et al. (2013) Drug‐induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat Commun 4, 1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Di Marco A, Silvestrini R, Di Marco S & Dasdia T (1965) Inhibiting effect of the new cytotoxic antibiotic daunomycin on nucleic acids and mitotic activity of HeLa cells. J Cell Biol 27, 545–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Munger C, Ellis A, Woods K, Randolph J, Yanovich S & Gewirtz D (1988) Evidence for inhibition of growth related to compromised DNA synthesis in the interaction of daunorubicin with H‐35 rat hepatoma. Cancer Res 48, 2404–2411. [PubMed] [Google Scholar]
- 30. Davies KJ & Doroshow JH (1986) Redox cycling of anthracyclines by cardiac mitochondria. I. Anthracycline radical formation by NADH dehydrogenase. J Biol Chem 261, 3060–3067. [PubMed] [Google Scholar]
- 31. Doroshow JH & Davies KJ (1986) Redox cycling of anthracyclines by cardiac mitochondria. II. Formation of superoxide anion, hydrogen peroxide, and hydroxyl radical. J Biol Chem 261, 3068–3074. [PubMed] [Google Scholar]
- 32. Licata S, Saponiero A, Mordente A & Minotti G (2000) Doxorubicin metabolism and toxicity in human myocardium: role of cytoplasmic deglycosidation and carbonyl reduction. Chem Res Toxicol 13, 414–420. [DOI] [PubMed] [Google Scholar]
- 33. Minotti G, Cairo G & Monti E (1999) Role of iron in anthracycline cardiotoxicity: new tunes for an old song? FASEB J 13, 199–212. [PubMed] [Google Scholar]
- 34. Gammella E, Maccarinelli F, Buratti P, Recalcati S & Cairo G (2014) The role of iron in anthracycline cardiotoxicity. Frontiers in Pharmacology 5, 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Demant EJF (1984) Transfer of ferritin‐bound iron to adriamycin. FEBS Lett 176, 97–100. [DOI] [PubMed] [Google Scholar]
- 36. Hasinoff BB & Kala SV (1993) The removal of metal‐ions from transferrin, ferritin and ceruloplasmin by the cardioprotective agent Icrf‐187 [(+)‐1,2‐Bis(3,5‐dioxopiperazinyl‐1‐Yl)Propane] and its hydrolysis product Adr‐925. Agents Actions 39, 72–81. [DOI] [PubMed] [Google Scholar]
- 37. Minotti G, Menna P, Salvatorelli E, Cairo G & Gianni L (2004) Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol Rev 56, 185–229. [DOI] [PubMed] [Google Scholar]
- 38. Luger K, Mader AW, Richmond RK, Sargent DF & Richmond TJ (1997) Crystal structure of the nucleosome core particle at 2.8 angstrom resolution. Nature 389, 251–260. [DOI] [PubMed] [Google Scholar]
- 39. Yang F, Kemp CJ & Henikoff S (2013) Doxorubicin enhances nucleosome turnover around promoters. Curr Biol 23, 782–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jawad B, Poudel L, Podgornik R, Steinmetz NF & Ching WY (2019) Molecular mechanism and binding free energy of doxorubicin intercalation in DNA. Phys Chem Chem Phys 21, 3877–3893. [DOI] [PubMed] [Google Scholar]
- 41. Talbert PB & Henikoff S (2017) Histone variants on the move: substrates for chromatin dynamics. Nat Rev Mol Cell Biol 18, 115–126. [DOI] [PubMed] [Google Scholar]
- 42. Alabert C, Jasencakova Z & Groth A (2017) Chromatin replication and histone dynamics. Adv Exp Med Biol 1042, 311–333. [DOI] [PubMed] [Google Scholar]
- 43. Pang B, de Jong J, Qiao X, Wessels LF & Neefjes J (2015) Chemical profiling of the genome with anti‐cancer drugs defines target specificities. Nat Chem Biol 11, 472–480. [DOI] [PubMed] [Google Scholar]
- 44. Qiao X, van der Zanden SY, Wander DPA, Borras DM, Song JY, Li X, van Duikeren S, van Gils N, Rutten A, van Herwaarden T et al. (2020) Uncoupling DNA damage from chromatin damage to detoxify doxorubicin. Proc Natl Acad Sci USA 117, 15182–15192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schaefer A, Westendorf J, Lingelbach K, Schmidt CA, Mihalache DL, Reymann A & Marquardt H (1993) Decreased resistance to N, N‐dimethylated anthracyclines in multidrug‐resistant Friend erythroleukemia cells. Cancer Chemother Pharmacol 31, 301–307. [DOI] [PubMed] [Google Scholar]
- 46. Egorin MJ, Clawson RE, Ross LA, Chou FTE, Andrews PA & Bachur NR (1980) Disposition and metabolism of N, N‐dimethyldaunorubicin and N, N‐dimethyladriamycin in rabbits and mice. Drug Metab Dispos. 8, 353–362. [PubMed] [Google Scholar]
- 47. Galluzzi L, Buque A, Kepp O, Zitvogel L & Kroemer G (2015) Immunological effects of conventional chemotherapy and targeted anticancer agents. Cancer Cell 28, 690–714. [DOI] [PubMed] [Google Scholar]
- 48. Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N et al. (2007) Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 13, 54–61. [DOI] [PubMed] [Google Scholar]
- 49. Martins I, Wang Y, Michaud M, Ma Y, Sukkurwala AQ, Shen S, Kepp O, Metivier D, Galluzzi L, Perfettini JL et al. (2014) Molecular mechanisms of ATP secretion during immunogenic cell death. Cell Death Differ 21, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Yamazaki T, Hannani D, Poirier‐Colame V, Ladoire S, Locher C, Sistigu A, Prada N, Adjemian S, Catani JP, Freudenberg M et al. (2014) Defective immunogenic cell death of HMGB1‐deficient tumors: compensatory therapy with TLR4 agonists. Cell Death Differ 21, 69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Casares N, Pequignot MO, Tesniere A, Ghiringhelli F, Roux S, Chaput N, Schmitt E, Hamai A, Hervas‐Stubbs S, Obeid M et al. (2005) Caspase‐dependent immunogenicity of doxorubicin‐induced tumor cell death. J Exp Med 202, 1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Alizadeh D, Trad M, Hanke NT, Larmonier CB, Janikashvili N, Bonnotte B, Katsanis E & Larmonier N (2014) Doxorubicin eliminates myeloid‐derived suppressor cells and enhances the efficacy of adoptive T‐cell transfer in breast cancer. Cancer Res 74, 104–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lai JJ, Cruz FM & Rock KL (2020) Immune sensing of cell death through recognition of histone sequences by C‐type lectin‐receptor‐2d causes inflammation and tissue injury. Immunity 52, 123–135.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Voorwerk L, Slagter M, Horlings HM, Sikorska K, van de Vijver KK, de Maaker M, Nederlof I, Kluin RJC, Warren S, Ong S et al. (2019) Immune induction strategies in metastatic triple‐negative breast cancer to enhance the sensitivity to PD‐1 blockade: the TONIC trial. Nat Med 25, 920–928. [DOI] [PubMed] [Google Scholar]
- 55. Lefrak EA, Pitha J, Rosenheim S & Gottlieb JA (1973) A clinicopathologic analysis of adriamycin cardiotoxicity. Cancer 32, 302–314. [DOI] [PubMed] [Google Scholar]
- 56. Shan K, Lincoff AM & Young JB (1996) Anthracycline‐induced cardiotoxicity. Ann Intern Med 125, 47–58. [DOI] [PubMed] [Google Scholar]
- 57. Swain SM, Whaley FS & Ewer MS (2003) Congestive heart failure in patients treated with doxorubicin: a retrospective analysis of three trials. Cancer 97, 2869–2879. [DOI] [PubMed] [Google Scholar]
- 58. Von Hoff DD, Layard MW, Basa P, Davis HL Jr, Von Hoff AL, Rozencweig M & Muggia FM (1979) Risk factors for doxorubicin‐induced congestive heart failure. Ann Intern Med 91, 710–717. [DOI] [PubMed] [Google Scholar]
- 59. Berthiaume JM & Wallace KB (2007) Adriamycin‐induced oxidative mitochondrial cardiotoxicity. Cell Biol Toxicol 23, 15–25. [DOI] [PubMed] [Google Scholar]
- 60. Myers CE, McGuire WP, Liss RH, Ifrim I, Grotzinger K & Young RC (1977) Adriamycin: the role of lipid peroxidation in cardiac toxicity and tumor response. Science 197, 165–167. [DOI] [PubMed] [Google Scholar]
- 61. Zhang S, Liu XB, Bawa‐Khalfe T, Lu LS, Lyu YL, Liu LF & Yeh ETH (2012) Identification of the molecular basis of doxorubicin‐induced cardiotoxicity. Nat Med 18, 1639–1642. [DOI] [PubMed] [Google Scholar]
- 62. Wallace KB (2007) Adriamycin‐induced interference with cardiac mitochondrial calcium homeostasis. Cardiovasc Toxicol 7, 101–107. [DOI] [PubMed] [Google Scholar]
- 63. Mei SB, Hong L, Cai XY, Xiao B, Zhang P & Shao L (2019) Oxidative stress injury in doxorubicin‐induced cardiotoxicity. Toxicol Lett 307, 41–48. [DOI] [PubMed] [Google Scholar]
- 64. Barth E, Stammler G, Speiser B & Schaper J (1992) Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J Mol Cell Cardiol 24, 669–681. [DOI] [PubMed] [Google Scholar]
- 65. Green PS & Leeuwenburgh C (2002) Mitochondrial dysfunction is an early indicator of doxorubicin‐induced apoptosis. Biochim Biophys Acta 1588, 94–101. [DOI] [PubMed] [Google Scholar]
- 66. Sonneveld P (1978) Effect of alpha‐tocopherol on the cardiotoxicity of adriamycin in the rat. Cancer Treat Rep 62, 1033–1036. [PubMed] [Google Scholar]
- 67. Legha SS, Wang YM, Mackay B, Ewer M, Hortobagyi GN, Benjamin RS & Ali MK (1982) Clinical and pharmacologic investigation of the effects of alpha‐tocopherol on adriamycin cardiotoxicity. Ann Ny Acad Sci 393, 411–418. [DOI] [PubMed] [Google Scholar]
- 68. Dresdale AR, Barr LH, Bonow RO, Mathisen DJ, Myers CE, Schwartz DE, Dangelo T & Rosenberg SA (1982) Prospective randomized study of the role of N‐acetyl cysteine in reversing doxorubicin‐induced cardiomyopathy. Am J Clin Oncol 5, 657–663. [DOI] [PubMed] [Google Scholar]
- 69. Ladas EJ, Jacobson JS, Kennedy DD, Teel K, Fleischauer A & Kelly KM (2004) Antioxidants and cancer therapy: a systematic review. J Clin Oncol 22, 517–528. [DOI] [PubMed] [Google Scholar]
- 70. Speyer JL, Green MD, Sanger J, Zeleniuchjacquotte A, Kramer E, Rey M, Wernz JC, Blum RH, Hochster H, Meyers M et al. (1990) A prospective randomized trial of Icrf‐187 for prevention of cumulative doxorubicin‐induced cardiac toxicity in women with breast‐cancer. Cancer Treat Rev 17, 161–163. [DOI] [PubMed] [Google Scholar]
- 71. Lipshultz SE, Scully RE, Lipsitz SR, Sallan SE, Silverman LB, Miller TL, Borry EV, Asselin BL, Athale U, Clavell LA et al. (2010) Assessment of dexrazoxane as a cardioprotectant in doxorubicin‐treated children with high‐risk acute lymphoblastic leukaemia: long‐term follow‐up of a prospective, randomised, multicentre trial. Lancet Oncol 11, 950–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hasinoff BB, Patel D & Wu X (2003) The oral iron chelator ICL670A (deferasirox) does not protect myocytes against doxorubicin. Free Radical Biol Med 35, 1469–1479. [DOI] [PubMed] [Google Scholar]
- 73. Yu XX, Ruan Y, Huang XQ, Dou L, Lan M, Cui J, Chen BD, Gong H, Wang Q, Yan MJ et al. (2020) Dexrazoxane ameliorates doxorubicin‐induced cardiotoxicity by inhibiting both apoptosis and necroptosis in cardiomyocytes. Biochem Bioph Res Commun 523, 140–146. [DOI] [PubMed] [Google Scholar]
- 74. Lyu YL, Kerrigan JE, Lin CP, Azarova AM, Tsai YC, Ban Y & Liu LF (2007) Topoisomerase II beta‐mediated DNA double‐strand breaks: implications in doxorubicin cardiotoxicity and prevention by dexrazoxane. Cancer Res 67, 8839–8846. [DOI] [PubMed] [Google Scholar]
- 75. Drake FH, Zimmerman JP, Mccabe FL, Bartus HF, Per SR, Sullivan DM, Ross WE, Mattern MR, Johnson RK, Crooke ST et al. (1987) Purification of topoisomerase‐Ii from amsacrine‐resistant p388 leukemia‐cells ‐ evidence for 2 forms of the enzyme. J Biol Chem 262, 16739–16747. [PubMed] [Google Scholar]
- 76. Austin CA & Marsh KL (1998) Eukaryotic DNA topoisomerase II beta. BioEssays 20, 215–226. [DOI] [PubMed] [Google Scholar]
- 77. Gabizon A, Shmeeda H & Barenholz Y (2003) Pharmacokinetics of pegylated liposomal doxorubicin ‐ Review of animal and human studies. Clin Pharmacokinet 42, 419–436. [DOI] [PubMed] [Google Scholar]
- 78. Vaage J, Barberaguillem E, Abra R, Huang A & Working P (1994) Tissue distribution and therapeutic effect of intravenous free or encapsulated liposomal doxorubicin on human prostate carcinoma xenografts. Cancer 73, 1478–1484. [DOI] [PubMed] [Google Scholar]
- 79. Cabanes A, Tzemach D, Goren D, Horowitz AT & Gabizon A (1998) Comparative study of the antitumor activity of free doxorubicin and polyethylene glycol‐coated liposomal doxorubicin in a mouse lymphoma model. Clin Cancer Res 4, 499–505. [PubMed] [Google Scholar]
- 80. Harris L, Batist G, Belt R, Rovira D, Navari R, Azarnia N, Welles L, Winer E; TLC D‐99 Study Group (2002) Liposome‐encapsulated doxorubicin compared with conventional doxorubicin in a randomized multicenter trial as first‐line therapy of metastatic breast carcinoma. Cancer 94, 25–36. [DOI] [PubMed] [Google Scholar]
- 81. Smith LA, Cornelius VR, Plummer CJ, Levitt G, Verrill M, Canney P & Jones A (2010) Cardiotoxicity of anthracycline agents for the treatment of cancer: systematic review and meta‐analysis of randomised controlled trials. BMC Cancer 10, 337. 10.1186/1471-2407-10-337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Travis LB, Demark Wahnefried W, Allan JM, Wood ME & Ng AK (2013) Aetiology, genetics and prevention of secondary neoplasms in adult cancer survivors. Nat Rev Clin Oncol 10, 289–301. [DOI] [PubMed] [Google Scholar]
- 83. Morton LM, Swerdlow AJ, Schaapveld M, Ramadan S, Hodgson DC, Radford J & van Leeuwen FE (2014) Current knowledge and future research directions in treatment‐related second primary malignancies. EJC Suppl 12, 5–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Bertazzoli C, Chieli T & Solcia E (1971) Different incidence of breast carcinomas or fibroadenomas in daunomycin or adriamycin treated rats. Experientia 27, 1209–1210. [DOI] [PubMed] [Google Scholar]
- 85. Sternberg SS, Philips FS & Cronin AP (1972) Renal tumors and other lesions in rats following a single intravenous injection of daunomycin. Cancer Res 32, 1029–1036. [PubMed] [Google Scholar]
- 86. Price PJ, Suk WA, Skeen PC, Chirigos MA & Huebner RJ (1975) Transforming potential of the anticancer drug adriamycin. Science 187, 1200–1201. [DOI] [PubMed] [Google Scholar]
- 87. Philips FS, Gilladoga A, Marquardt H, Stemberg SS & Vidal PR (1975) Some observations on the toxicity of Adriamycin (NSC‐123127).Cancer Chemother. Rep. Part III, 6. 177‐181 [Google Scholar]
- 88. Marquardt H, Philips FS & Sternberg SS (1976) Tumorigenicity in vivo and induction of malignant transformation and mutagenesis in cell cultures by adriamycin and daunomycin. Cancer Res 36, 2065–2069. [PubMed] [Google Scholar]
- 89. Solcia E, Ballerini L, Bellini O, Sala L & Bertazzoli C (1978) Mammary tumors induced in rats by adriamycin and daunomycin. Cancer Res 38, 1444–1446. [PubMed] [Google Scholar]
- 90. Overall evaluations of carcinogenicity: an updating of IARC monographs volumes 1 to 42. IARC Monogr Eval Carcinog Risks Hum Suppl 7, 1–440. [PubMed] [Google Scholar]
- 91. Diamandidou E, Buzdar AU, Smith TL, Frye D, Witjaksono M & Hortobagyi GN (1996) Treatment‐related leukemia in breast cancer patients treated with fluorouracil‐doxorubicin‐cyclophosphamide combination adjuvant chemotherapy: the University of Texas M.D. Anderson Cancer Center experience. J Clin Oncol 14, 2722–2730. [DOI] [PubMed] [Google Scholar]
- 92. Praga C, Bergh J, Bliss J, Bonneterre J, Cesana B, Coombes RC, Fargeot P, Folin A, Fumoleau P, Giuliani R et al. (2005) Risk of acute myeloid leukemia and myelodysplastic syndrome in trials of adjuvant epirubicin for early breast cancer: correlation with doses of epirubicin and cyclophosphamide. J Clin Oncol 23, 4179–4191. [DOI] [PubMed] [Google Scholar]
- 93. Le Deley MC, Suzan F, Cutuli B, Delaloge S, Shamsaldin A, Linassier C, Clisant S, de Vathaire F, Fenaux P & Hill C (2007) Anthracyclines, mitoxantrone, radiotherapy, and granulocyte colony‐stimulating factor: risk factors for leukemia and myelodysplastic syndrome after breast cancer. J Clin Oncol 25, 292–300. [DOI] [PubMed] [Google Scholar]
- 94. Henderson TO, Whitton J, Stovall M, Mertens AC, Mitby P, Friedman D, Strong LC, Hammond S, Neglia JP, Meadows AT et al. (2007) Secondary sarcomas in childhood cancer survivors: a report from the Childhood Cancer Survivor Study. J Natl Cancer Inst 99, 300–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Henderson TO, Rajaraman P, Stovall M, Constine LS, Olive A, Smith SA, Mertens A, Meadows A, Neglia JP, Hammond S et al. (2012) Risk factors associated with secondary sarcomas in childhood cancer survivors: a report from the childhood cancer survivor study. Int J Radiat Oncol Biol Phys 84, 224–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Teepen JC, van Leeuwen FE, Tissing WJ, van Dulmen‐den Broeder E, van den Heuvel‐Eibrink MM, van der Pal HJ, Loonen JJ, Bresters D, Versluys B, Neggers S et al. (2017) Long‐term risk of subsequent malignant neoplasms after treatment of childhood cancer in the DCOG LATER Study Cohort: role of chemotherapy. J Clin Oncol 35, 2288–2298. [DOI] [PubMed] [Google Scholar]
- 97. van Leeuwen FE & Ronckers CM (2016) Anthracyclines and alkylating agents: new risk factors for breast cancer in childhood cancer survivors? J Clin Oncol 34, 891–894. [DOI] [PubMed] [Google Scholar]
- 98. Veiga LH, Bhatti P, Ronckers CM, Sigurdson AJ, Stovall M, Smith SA, Weathers R, Leisenring W, Mertens AC, Hammond S et al. (2012) Chemotherapy and thyroid cancer risk: a report from the childhood cancer survivor study. Cancer Epidemiol Biomarkers Prev 21, 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Beaumont M, Sanz M, Carli PM, Maloisel F, Thomas X, Detourmignies L, Guerci A, Gratecos N, Rayon C, San Miguel J et al. (2003) Therapy‐related acute promyelocytic leukemia. J Clin Oncol 21, 2123–2137. [DOI] [PubMed] [Google Scholar]
- 100. Rashidi A & Fisher SI (2013) Therapy‐related acute promyelocytic leukemia: a systematic review. Med Oncol 30, 625. [DOI] [PubMed] [Google Scholar]
- 101. Pedersen‐Bjergaard J & Philip P (1991) Balanced translocations involving chromosome bands 11q23 and 21q22 are highly characteristic of myelodysplasia and leukemia following therapy with cytostatic agents targeting at DNA‐topoisomerase II. Blood 78, 1147–1148. [PubMed] [Google Scholar]
- 102. Larson RA, Le Beau MM, Ratain MJ & Rowley JD (1992) Balanced translocations involving chromosome bands 11q23 and 21q22 in therapy‐related leukemia. Blood 79, 1892–1893. [PubMed] [Google Scholar]
- 103. Felix CA, Kolaris CP & Osheroff N (2006) Topoisomerase II and the etiology of chromosomal translocations. DNA Repair (Amst) 5, 1093–1108. [DOI] [PubMed] [Google Scholar]
- 104. Cowell IG & Austin CA (2012) Mechanism of generation of therapy related leukemia in response to anti‐topoisomerase II agents. Int J Environ Res Public Health 9, 2075–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Pendleton M, Lindsey RH Jr, Felix CA, Grimwade D & Osheroff N (2014) Topoisomerase II and leukemia. Ann N Y Acad Sci 1310, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Park SH, Chi HS, Cho YU, Jang S & Park CJ (2013) Evaluation of prognostic factors in patients with therapy‐related acute myeloid leukemia. Blood Res 48, 185–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Kayser S, Dohner K, Krauter J, Kohne CH, Horst HA, Held G, von Lilienfeld‐Toal M, Wilhelm S, Kundgen A, Gotze K et al. (2011) The impact of therapy‐related acute myeloid leukemia (AML) on outcome in 2853 adult patients with newly diagnosed AML. Blood 117, 2137–2145. [DOI] [PubMed] [Google Scholar]
- 108. Basecke J, Cepek L, Mannhalter C, Krauter J, Hildenhagen S, Brittinger G, Trumper L & Griesinger F (2002) Transcription of AML1/ETO in bone marrow and cord blood of individuals without acute myelogenous leukemia. Blood 100, 2267–2268. [DOI] [PubMed] [Google Scholar]
- 109. Mori H, Colman SM, Xiao Z, Ford AM, Healy LE, Donaldson C, Hows JM, Navarrete C & Greaves M (2002) Chromosome translocations and covert leukemic clones are generated during normal fetal development. Proc Natl Acad Sci USA 99, 8242–8247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Schnittger S, Bacher U, Kern W, Haferlach C & Haferlach T (2007) JAK2 seems to be a typical cooperating mutation in therapy‐related t(8;21)/ AML1‐ETO‐positive AML. Leukemia 21, 183–184. [DOI] [PubMed] [Google Scholar]
- 111. Stanulla M, Wang J, Chervinsky DS, Thandla S & Aplan PD (1997) DNA cleavage within the MLL breakpoint cluster region is a specific event which occurs as part of higher‐order chromatin fragmentation during the initial stages of apoptosis. Mol Cell Biol 17, 4070–4079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Betti CJ, Villalobos MJ, Diaz MO & Vaughan AT (2001) Apoptotic triggers initiate translocations within the MLL gene involving the nonhomologous end joining repair system. Cancer Res 61, 4550–4555. [PubMed] [Google Scholar]
- 113. Betti CJ, Villalobos MJ, Diaz MO & Vaughan AT (2003) Apoptotic stimuli initiate MLL‐AF9 translocations that are transcribed in cells capable of division. Cancer Res 63, 1377–13781. [PubMed] [Google Scholar]
- 114. Mays AN, Osheroff N, Xiao Y, Wiemels JL, Felix CA, Byl JA, Saravanamuttu K, Peniket A, Corser R, Chang C et al. (2010) Evidence for direct involvement of epirubicin in the formation of chromosomal translocations in t(15;17) therapy‐related acute promyelocytic leukemia. Blood 115, 326–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Kudo K, Yoshida H, Kiyoi H, Numata S, Horibe K & Naoe T (1998) Etoposide‐related acute promyelocytic leukemia. Leukemia 12, 1171–1175. [DOI] [PubMed] [Google Scholar]
- 116. Le Deley MC, Leblanc T, Shamsaldin A, Raquin MA, Lacour B, Sommelet D, Chompret A, Cayuela JM, Bayle C, Bernheim A et al. (2003) Risk of secondary leukemia after a solid tumor in childhood according to the dose of epipodophyllotoxins and anthracyclines: a case‐control study by the Societe Francaise d'Oncologie Pediatrique. J Clin Oncol 21, 1074–1081. [DOI] [PubMed] [Google Scholar]
- 117. Westendorf J, Marquardt H, Ketkar MB, Mohr U & Marquardt H (1983) Tumorigenicity in vivo and induction of mutagenesis and DNA repair in vitro by aclacinomycin A and marcellomycin: structure‐activity relationship and predictive value of short‐term tests. Cancer Res 43, 5248–5251. [PubMed] [Google Scholar]
- 118. Westendorf J, Marquardt H & Marquardt H (1984) Structure‐activity relationship of anthracycline‐induced genotoxicity in vitro . Cancer Res 44, 5599–565604. [PubMed] [Google Scholar]
- 119. Del Rizzo PA & Trievel RC (2011) Substrate and product specificities of SET domain methyltransferases. Epigenetics 6, 1059–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Jakovcevski M, Ruan H, Shen EY, Dincer A, Javidfar B, Ma Q, Peter CJ, Cheung I, Mitchell AC, Jiang Y et al. (2015) Neuronal Kmt2a/Mll1 histone methyltransferase is essential for prefrontal synaptic plasticity and working memory. J Neurosci 35, 5097–5108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Xu J, Li L, Xiong J, denDekker A, Ye A, Karatas H, Liu L, Wang H, Qin ZS, Wang S et al. (2016) MLL1 and MLL1 fusion proteins have distinct functions in regulating leukemic transcription program. Cell Discov 2, 16008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Zuber J, Radtke I, Pardee TS, Zhao Z, Rappaport AR, Luo W, McCurrach ME, Yang MM, Dolan ME, Kogan SC et al. (2009) Mouse models of human AML accurately predict chemotherapy response. Genes Dev 23, 877–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Estey E & Dohner H (2006) Acute myeloid leukaemia. Lancet 368, 1894–1907. [DOI] [PubMed] [Google Scholar]
- 124. Erttmann R, Munchmeyer M, Looft G & Winkler K (1991) Conserved cytostatic activity of aclarubicin in a doxorubicin selected Friend leukaemia cell line with multifactorial multidrug resistance. Eur J Cancer 27, 1064. [DOI] [PubMed] [Google Scholar]
- 125. Natale RB, Cody RL, Simon MS & Wheeler RH (1993) An in vivo and in vitro trial of aclarubicin in metastatic breast cancer: a novel approach to the study of analogs. Cancer Chemother Pharmacol 31, 485–488. [DOI] [PubMed] [Google Scholar]
- 126. Lehne G, De Angelis P, Clausen OP & Rugstad HE (1996) Human hepatoma cells rich in P‐glycoprotein are sensitive to aclarubicin and resistant to three other anthracyclines. Br J Cancer 74, 1719–1729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Liu L, Qu Q, Jiao W, Zhang Y, Li X, Ding C & Wu D (2015) Increasing aclarubicin dose in low‐dose cytarabine and aclarubicin in combination with granulocyte colony‐stimulating factor (CAG regimen) is efficacious as salvage chemotherapy for relapsed/refractory mixed‐phenotype acute leukemia. Leuk Res 39, 805–811. [DOI] [PubMed] [Google Scholar]
- 128. Qu Q, Liu L, Zhang Y, Li X & Wu D (2015) Increasing aclarubicin dosage of the conventional CAG (low‐dose cytarabine and aclarubicin in combination with granulocyte colony‐stimulating factor) regimen is more efficacious as a salvage therapy than CAG for relapsed/refractory acute myeloid leukemia. Leuk Res 39, 1353–1359. [DOI] [PubMed] [Google Scholar]
- 129. Delessard M, Saulnier J, Rives A, Dumont L, Rondanino C & Rives N (2020) Exposure to chemotherapy during childhood or adulthood and consequences on spermatogenesis and male fertility. Int J Mol Sci 21 4, 1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. European Society for Human Reproduction and Embryology (ESHRE) Guideline Group on POI , Webber L, Davies M, Anderson R, Bartlett J, Braat D, Cartwright B, Cifkova R, de Muinck Keizer‐Schrama S , Hogervorst E et al. (2016) ESHRE Guideline: management of women with premature ovarian insufficiency. Hum Reprod 31, 926–937. [DOI] [PubMed] [Google Scholar]
- 131. Viviani S, Santoro A, Ragni G, Bonfante V, Bestetti O & Bonadonna G (1985) Gonadal toxicity after combination chemotherapy for Hodgkin's disease. Comparative results of MOPP vs ABVD. Eur J Cancer Clin Oncol 21, 601–605. [DOI] [PubMed] [Google Scholar]
- 132. Byrne J, Fears TR, Gail MH, Pee D, Connelly RR, Austin DF, Holmes GF, Holmes FF, Latourette HB, Meigs JW et al. (1992) Early menopause in long‐term survivors of cancer during adolescence. Am J Obstet Gynecol 166, 788–793. [DOI] [PubMed] [Google Scholar]
- 133. Green DM, Whitton JA, Stovall M, Mertens AC, Donaldson SS, Ruymann FB, Pendergrass TW & Robison LL (2002) Pregnancy outcome of female survivors of childhood cancer: a report from the Childhood Cancer Survivor Study. Am J Obstet Gynecol 187, 1070–1080. [DOI] [PubMed] [Google Scholar]
- 134. Ben‐Aharon I, Bar‐Joseph H, Tzarfaty G, Kuchinsky L, Rizel S, Stemmer SM & Shalgi R (2010) Doxorubicin‐induced ovarian toxicity. Reprod Biol Endocrinol 8, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Anderson RA, Mitchell RT, Kelsey TW, Spears N, Telfer EE & Wallace WH (2015) Cancer treatment and gonadal function: experimental and established strategies for fertility preservation in children and young adults. Lancet Diabetes Endocrinol 3, 556–567. [DOI] [PubMed] [Google Scholar]
- 136. Brilhante O, Stumpp T & Miraglia SM (2011) Long‐term testicular toxicity caused by doxorubicin treatment during pre‐pubertal phase. Int J Med Sci 3, 52–60. [Google Scholar]
- 137. Manabe F, Takeshima H & Akaza H (1997) Protecting spermatogenesis from damage induced by doxorubicin using the luteinizing hormone‐releasing hormone agonist leuprorelin: an image analysis study of a rat experimental model. Cancer 79, 1014–1021. [DOI] [PubMed] [Google Scholar]
- 138. Kropp J, Roti Roti EC, Ringelstetter A, Khatib H, Abbott DH & Salih SM (2015) Dexrazoxane diminishes doxorubicin‐induced acute ovarian damage and preserves ovarian function and fecundity in mice. PLoS One 10, e0142588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Levi M, Tzabari M, Savion N, Stemmer SM, Shalgi R & Ben‐Aharon I (2015) Dexrazoxane exacerbates doxorubicin‐induced testicular toxicity. Reproduction 150, 357–366. [DOI] [PubMed] [Google Scholar]
- 140. Roti Roti EC, Ringelstetter AK, Kropp J, Abbott DH & Salih SM (2014) Bortezomib prevents acute doxorubicin ovarian insult and follicle demise, improving the fertility window and pup birth weight in mice. PLoS One 9, e108174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Tuppi M, Kehrloesser S, Coutandin DW, Rossi V, Luh LM, Strubel A, Hotte K, Hoffmeister M, Schafer B, De Oliveira T et al. (2018) Oocyte DNA damage quality control requires consecutive interplay of CHK2 and CK1 to activate p63. Nat Struct Mol Biol 25, 261–269. [DOI] [PubMed] [Google Scholar]
- 142. Perez GI, Knudson CM, Leykin L, Korsmeyer SJ & Tilly JL (1997) Apoptosis‐associated signaling pathways are required for chemotherapy‐mediated female germ cell destruction. Nat Med 3, 1228–1232. [DOI] [PubMed] [Google Scholar]
- 143. Roti Roti EC, Leisman SK, Abbott DH & Salih SM (2012) Acute doxorubicin insult in the mouse ovary is cell‐ and follicle‐type dependent. PLoS One 7, e42293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Beaud H, van Pelt A & Delbes G (2017) Doxorubicin and vincristine affect undifferentiated rat spermatogonia. Reproduction 153, 725–735. [DOI] [PubMed] [Google Scholar]
- 145. Marcello MF, Nuciforo G, Romeo R, Di Dino G, Russo I, Russo A, Palumbo G & Schiliro G (1990) Structural and ultrastructural study of the ovary in childhood leukemia after successful treatment. Cancer 66, 2099–2104. [DOI] [PubMed] [Google Scholar]
- 146. Kobayashi H, Urashima M, Hoshi Y, Uchiyama H, Fujisawa K, Akatsuka J, Maekawa K & Hurusato M (1996) Testicular morphological changes in children with acute lymphoblastic leukemia following chemotherapy. Acta Paediatr Jpn 38, 640–643. [DOI] [PubMed] [Google Scholar]
- 147. Brilhante O, Okada FK, Sasso‐Cerri E, Stumpp T & Miraglia SM (2012) Late morfofunctional alterations of the Sertoli cell caused by doxorubicin administered to prepubertal rats. Reprod Biol Endocrinol 10, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Stumpp T, Sasso‐Cerri E, Freymuller E & Miraglia SM (2004) Apoptosis and testicular alterations in albino rats treated with etoposide during the prepubertal phase. Anat Rec A Discov Mol Cell Evol Biol 279, 611–622. [DOI] [PubMed] [Google Scholar]
- 149. Ortiz RJ, Lizama C, Codelia VA & Moreno RD (2009) A molecular evaluation of germ cell death induced by etoposide in pubertal rat testes. Mol Hum Reprod 15, 363–371. [DOI] [PubMed] [Google Scholar]
- 150. Bar‐Joseph H, Ben‐Aharon I, Rizel S, Stemmer SM, Tzabari M & Shalgi R (2010) Doxorubicin‐induced apoptosis in germinal vesicle (GV) oocytes. Reprod Toxicol 30, 566–572. [DOI] [PubMed] [Google Scholar]
- 151. Tremblay AR & Delbes G (2018) In vitro study of doxorubicin‐induced oxidative stress in spermatogonia and immature Sertoli cells. Toxicol Appl Pharmacol 348, 32–42. [DOI] [PubMed] [Google Scholar]
- 152. Hou M, Chrysis D, Nurmio M, Parvinen M, Eksborg S, Soder O & Jahnukainen K (2005) Doxorubicin induces apoptosis in germ line stem cells in the immature rat testis and amifostine cannot protect against this cytotoxicity. Cancer Res 65, 9999–10005. [DOI] [PubMed] [Google Scholar]