

Abstract
Background
On the basis of the DREAMM‐2 study (ClinicalTrials.gov identifier NCT03525678), single‐agent belantamab mafodotin (belamaf) was approved for patients with relapsed or refractory multiple myeloma (RRMM) who received ≥4 prior therapies, including anti‐CD38 therapy. The authors investigated longer term efficacy and safety outcomes in DREAMM‐2 after 13 months of follow‐up among patients who received belamaf 2.5 mg/kg.
Methods
DREAMM‐2 is an ongoing, phase 2, open‐label, 2‐arm study investigating belamaf (2.5 or 3.4 mg/kg) in patients with RRMM who had disease progression after ≥3 lines of therapy and were refractory to immunomodulatory drugs and proteasome inhibitors and refractory and/or intolerant to an anti‐CD38 therapy. The primary outcome was the proportion of patients that achieved an overall response, assessed by an independent review committee.
Results
As of January 31, 2020, 10% of patients still received belamaf 2.5 mg/kg. Thirty‐one of 97 patients (32%; 97.5% confidence interval [CI], 21.7%‐43.6%) achieved an overall response, and 18 responders achieved a very good partial response or better. Median estimated duration of response, overall survival, and progression‐free survival were 11.0 months (95% CI, 4.2 months to not reached), 13.7 months (95% CI, 9.9 months to not reached), and 2.8 months (95% CI, 1.6‐3.6 months), respectively. Response and survival outcomes in patients who had high‐risk cytogenetics or renal impairment were consistent with outcomes in the overall population. Outcomes were poorer in patients with extramedullary disease. In patients who had a clinical response and prolonged dose delays (>63 days; mainly because of corneal events), 88% maintained or deepened responses during their first prolonged dose delay. Overall, there were no new safety signals during this follow‐up.
Conclusions
Extended follow‐up confirms sustained clinical activity without new safety signals with belamaf in this heavily pretreated patient population with RRMM.
Keywords: antibody‐drug conjugate, B‐cell maturation antigen, clinical activity, monoclonal antibody, multiple myeloma
Short abstract
Extended follow‐up of patients enrolled in the ongoing phase 2 DREAMM‐2 study confirms sustained clinical activity without new safety signals in patients with relapsed or refractory multiple myeloma who receive belantamab mafodotin 2.5 mg/kg every 3 weeks. These data show that belantamab mafodotin has the potential to shift the treatment paradigm in this heavily pretreated, anti‐CD38 monoclonal antibody–exposed patient population, which has a poor prognosis and few alternative treatment options.
Introduction
The treatment of patients with relapsed or refractory multiple myeloma (RRMM) who have received multiple therapies remains challenging. 1 , 2 , 3 , 4 The depth and duration of responses (DoRs) to subsequent therapies diminish over time as the disease inevitably becomes refractory to the 3 pillars of currently available therapy: proteasome inhibitors, immunomodulatory agents, and monoclonal antibodies (moAbs). 1 , 2 , 3 , 4 Patients who have disease that is refractory to anti‐CD38 moAbs have a particularly poor prognosis (median progression‐free survival [PFS], 3.4 months; median overall survival [OS], 9.3 months). 3 Prognosis is also poor in patients who have RRMM with various characteristics and/or disease features, as detailed below; all such patient groups would benefit from novel, effective therapies. Patients with high‐risk cytogenetic abnormalities have a poor prognosis and derive reduced benefit from standard‐of‐care therapies. 5 Renal impairment is commonly present at RRMM diagnosis, can develop during treatment (as a toxicity of some treatment classes), and frequently necessitates dose adjustment or avoidance of certain therapies. 6 , 7 Renal impairment is also highly prevalent in the elderly (the age group with the highest prevalence of RRMM) and is associated with poorer RRMM outcomes. 8 , 9 , 10 , 11 At baseline and relapse, patients with RRMM may present with extramedullary disease (EMD), characterized by the presence of malignant plasma cells outside the bone marrow; this again confers a poor prognosis. 12 There are currently no guidelines on the treatment of patients with EMD. 12
Single‐agent belantamab mafodotin (belamaf) (BLENREP; GSK2857916) is a first‐in‐class antibody‐drug conjugate that binds to B‐cell maturation antigen (BCMA), a cell‐membrane receptor expressed on all malignant plasma cells that is essential for their proliferation and survival. 13 , 14 Belamaf comprises an anti‐BCMA MoAb conjugated to microtubule‐disrupting agent monomethyl auristatin F (MMAF) (mafodotin). 13 Belamaf eliminates multiple myeloma (MM) cells by a multimodal mechanism of action, including delivering mafodotin to BCMA‐expressing MM cells, thereby inducing apoptosis accompanied by the release of markers associated with immunogenic cell death. 15 The MoAb component enhances immune effector cell‐dependent mechanisms of action, such as antibody‐dependent cellular cytotoxicity and phagocytosis. 13 , 15
In the DREAMM‐2 (ClinicalTrials.gov identifier NCT03525678) primary analysis at 6 months of follow‐up, single‐agent belamaf demonstrated deep and durable responses with a manageable safety profile in patients with RRMM who had received ≥3 prior lines of therapy, were refractory to an immunomodulatory agent and proteasome inhibitor, and were refractory and/or intolerant to an anti‐CD38 MoAb. 16
Keratopathy (including superficial punctuate keratopathy and/or microcyst‐like epithelial changes) was the most common adverse event (AE) in DREAMM‐2 (67 of 95 patients [71%] receiving belamaf 2.5 mg/kg). 16 These events were also the most common reason for dose reductions (22 of 95 patients; 23%) and dose delays (45 of 95 patients; 47%) but rarely led to permanent discontinuation of treatment (1 of 95 patients; 1%). 16 This is consistent with reports of other MMAF‐containing antibody‐drug conjugates and represents an effect of belamaf on the corneal epithelium (the outermost cell layer of the cornea). 17 , 18 Thrombocytopenia (33 of 95 patients; 35%) and anemia (23 of 95 patients; 24%) were the next most common AEs. 16
Single‐agent belamaf 2.5 mg/kg every 3 weeks was approved through an accelerated process in the United States 19 and the European Union 20 for patients with RRMM who have received ≥4 prior therapies, including a proteasome inhibitor, an immunomodulatory agent, and anti‐CD38 therapy. 21 , 22 The belamaf 2.5‐mg/kg dose was selected based on its efficacy and safety profile versus the 3.4‐mg/kg dose. 16 , 21
Here, we report updated efficacy and safety outcomes in patients who received belamaf 2.5 mg/kg in DREAMM‐2, with an additional 7 months of follow‐up (13‐month data cutoff). In addition, we report subanalyses of outcomes in patients with high‐risk cytogenetics, patients with renal impairment or EMD, and patients with prolonged dose delays.
Materials and Methods
Study Design and Patients
DREAMM‐2 (ClinicalTrials.gov identifier NCT03525678) is a phase 2, open‐label, 2‐arm, randomized study. 16 Eligible patients had progressed after ≥3 prior lines of therapy and were refractory to an immunomodulatory agent and a proteasome inhibitor and refractory and/or intolerant to an anti‐CD38 MoAb. Eligible patients had an estimated glomerular filtration rate (eGFR) ≥30 mL per minute per 1.73 m2. Full methodological details with inclusion criteria have been previously reported. 16
Randomization and Masking
Patients were randomly assigned (1:1) to receive belamaf 2.5 or 3.4 mg/kg. For this open‐label study, randomization was done centrally by using interactive response technology, with allocation and stratification based on the number of previous lines of therapy (≤4 vs >4 lines) and the presence or absence of high‐risk cytogenetic features. A centrally generated randomization schedule with permuted blocks (with a block size of 4) was used to conceal treatment allocation. Enrollment was done by study center staff who were not involved in the running of the clinical trial or in data collection.
Treatment, Procedures, and Assessments
Patients received single‐agent belamaf 2.5 or 3.4 mg/kg every 3 weeks by intravenous infusion (over ≥30 minutes) until disease progression or unacceptable toxicity occurred. 16 Dose modifications, including delays and reductions, were permitted for AEs or reasons not related to treatment (medical, surgical, or logistical). AEs were graded according to Common Terminology Criteria for Adverse Events (CTCAE), version 4.03. 23
In the DREAMM‐2 primary analysis, changes to the corneal epithelium observed on slit‐lamp examination (with or without symptoms or changes in visual acuity) were referred to as keratopathy, which is a general term for noninflammatory disease of the eye, and were reported according to CTCAE grade. 16 Corneal events were also graded using the protocol‐defined scale, which combined corneal examination findings with changes in best‐corrected visual acuity (BCVA). The corneal event grade according to this protocol‐defined scale was used to guide dose modifications. This protocol‐defined scale was later renamed the Keratopathy and Visual Acuity (KVA) scale (Table 1). 21 , 22 , 24 All keratopathy events were followed by an ophthalmologist or optometrist until full resolution or recovery to baseline or up to 1 year after the end of treatment, whichever came first.
TABLE 1.
Recommended Belantamab Mafodotin Dose Modifications Based on Eye Examination Findings According to the Keratopathy and Visual Acuity Scale a
| Grading Category: US/EU | Eye Examination Findings by KVA Scale | Recommended Dose Modifications |
|---|---|---|
| Grade 1/mild | Corneal examination finding(s) | Continue treatment at the current dose |
| Mild superficial keratopathy b | ||
| Change in BCVA c | ||
| Decline from baseline of 1 line on Snellen Visual Acuity | ||
| Grade 2/moderate | Corneal examination finding(s) | Withhold treatment until improvement in both corneal examination findings and change in BCVA to grade 1 (mild) or better and resume at the current dose; consider resuming at a reduced dose of 1.9 mg/kg |
| Moderate superficial keratopathy d | ||
| Change in BCVA | ||
| Decline from baseline of 2 or 3 lines on Snellen Visual Acuity and ≤20/200 | ||
| Grade 3/severe | Corneal examination finding(s) | Withhold treatment until improvement in both corneal examination findings and change in BCVA to grade ≥1 (mild) and resume at a reduced dose; for worsening symptoms that are unresponsive to appropriate management, consider discontinuation |
| Severe superficial keratopathy e | ||
| Change in BCVA | ||
| Decline from baseline by more than 3 lines on Snellen Visual Acuity and not worse than 20/200 f | ||
| Grade 4/severe | Corneal examination finding(s) | Consider permanent discontinuation of treatment; if continuing treatment, withhold treatment until improvement in both corneal examination findings and change in BCVA to grade ≥1 and resume at reduced a dose |
| Corneal epithelial defect g | ||
| Change in BCVA | ||
| Snellen Visual Acuity <20/200 f |
Abbreviations: BCVA, best‐corrected visual acuity; KVA scale, the Keratopathy and Visual Acuity scale; EU, European Union; US, United States.
DREAMM‐2 used a protocol‐defined scale that combined slit‐lamp examination findings with an assessment of BCVA. 24 This scale was renamed the KVA scale, as shown here, and is included in the current US and EU labels. This table provides a combined summary of these grades and dose‐modification guidelines in the current US and EU labels. 21 , 22 Recommendations unique to the EU label are italicized, whereas recommendations unique to the United States label are underlined; the EU label has the categories mild, moderate, and severe, with severe covering the KVA and US label grades 3 and 4. The severity category is defined by the most severely affected eye because both eyes may not be affected to the same degree. Prescribing physicians should follow the specific dose‐modification guidelines for corneal event management outlined in their local prescribing information.
This includes mild superficial keratopathy (documented worsening from baseline) with or without symptoms.
These are changes in visual acuity because of treatment‐related corneal findings.
This includes moderate superficial keratopathy with or without patchy microcyst‐like deposits, subepithelial haze (peripheral), or a new peripheral stromal opacity.
Severe superficial keratopathy with or without diffuse microcyst‐like deposits involving the central cornea, subepithelial haze (central), or a new central stromal opacity.
The EU label specifies only decline from baseline of more than 3 lines, without separation by Snellen Visual Acuity better or worse than 20/200.
Corneal epithelial defect such as corneal ulcers. The EU label states that these should be managed promptly and as clinically indicated by an eye care professional.
Outcomes and Analysis
The primary outcome was the proportion of patients achieving an overall response as assessed by an independent review committee, defined as the percentage of patients with a confirmed partial response (PR) or better (≥PR) (in accordance with the International Myeloma Working Group [IMWG] uniform response criteria for MM 25 ) when assessed every 3 weeks after cycle 1. 16 Other secondary end points have been previously reported and included PFS, DoR, and OS. The minimal residual disease (MRD) rate from centrally tested bone marrow samples, using next‐generation sequencing (clonoSEQ) (sensitivity, 1 × 10−5 cells) in patients who achieved a very good partial response (VGPR) or better (≥VGPR), was an exploratory end point.
The intention‐to‐treat population comprised all randomized patients regardless of treatment administration. 26 The safety population comprised all patients who received ≥1 belamaf dose, and the efficacy population comprised all patients who received ≥2 doses and completed ≥1 disease assessment after the second dose. For response‐rate analyses, patients with unknown or missing data were considered nonresponders.
Post hoc analyses were performed on the impact of dose delays in >3 treatment cycles (>63 days with cycles every 21 days) on clinical response and on outcomes in patients who had high‐risk cytogenetics (tested locally), renal impairment, or EMD. High‐risk cytogenetic markers, including t(4:14), t(14:16), 17p13del, or 1q21+, were determined using fluorescence in situ hybridization testing performed by a local laboratory (if available) or a central laboratory (if not available locally). Two definitions of high‐risk cytogenetics were used in this analysis: high‐risk according to IMWG criteria 27 (referred to here as HR‐IMWG), which includes patients who had t(4:14), t(14:16), or 17p13del; and high‐risk cytogenetics with 1q21+ (referred to here as HR‐cyto), which also includes patients with 1q21+. Neither 1q21 copy number nor the percentage 17p13 deletion was reported. Outcomes in these groups were compared with those in patients who had standard‐risk cytogenetics, defined as those without these cytogenetic features.
For renal impairment, patients were categorized by eGFR into 3 groups: normal (≥90 mL per minute per 1.73 m2), mildly impaired (≥60 to <90 mL per minute per 1.73 m2), or moderately impaired (≥30 to <60 mL per minute per 1.73 m2). Patients with severe renal impairment or missing data were not included in the analysis. EMD was confirmed based on imaging (computed tomography, magnetic resonance imaging, or positron emission tomography/computed tomography, according to local guidance) up to 30 days before the first treatment cycle. The type of EMD was not specified, and patients were classified according to the absence or presence of EMD. Imaging for EMD was conducted at baseline, every 12 weeks within the first 12 months, and thereafter when clinically indicated.
DREAMM‐2 was conducted in accordance with the Declaration of Helsinki and Good Clinical Practice guidelines after approval by ethics committees and institutional review boards at each study site. All patients provided written informed consent.
Statistical Methods
Statistical methods for DREAMM‐2 have been previously reported. 16 Briefly, demographics and safety data were analyzed using descriptive statistics. Overall response, DoR, duration of clinical benefit, and PFS were calculated with 2‐sided 95% exact confidence intervals (CIs).
Results
Demographics and baseline disease characteristics for patients enrolled in DREAMM‐2 have been reported. 16 Briefly, patients had heavily pretreated RRMM with a median of 7 (range, 3‐21) prior lines of treatment. Most patients (73 of 97 [75%] in the intention‐to‐treat population) had undergone prior autologous stem cell transplantation.
At the 13‐month data cutoff (January 31, 2020), 42 of 97 patients (43%) in the 2.5‐mg/kg group were ongoing in the study, and 10 (10%) of these patients were on treatment (Fig. 1). 16 , 28 Patients had received a median of 3 treatment cycles (range, 1‐17 treatment cycles). The median time on study treatment (not excluding any dose delay) was 2.1 months (range, 0.4‐4.0 months). Progressive disease was the primary reason for treatment discontinuation. The median time to treatment discontinuation was 2.1 months (95% CI, 2.1‐2.8 months). For patients who had data available (n = 37), the median time from study discontinuation to start of the next anticancer therapy was 1.4 months (range, 0.4‐4.8 months).
Figure 1.
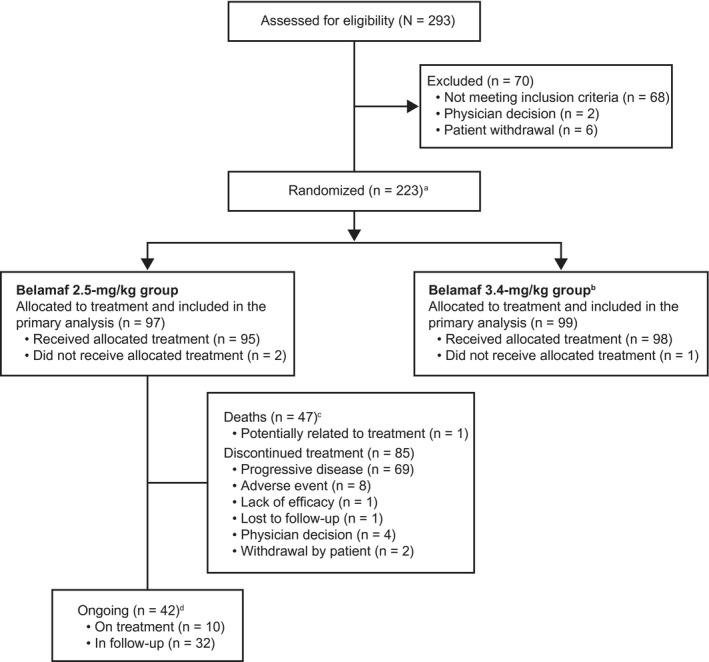
Patient disposition is illustrated. aTwo patients were re‐randomized and counted twice (once per each randomization). An additional independent group of 25 patients was randomized to receive a lyophilized configuration of belantamab mafodotin (belamaf) 3.4 mg/kg and underwent the same assessments and procedures as the main study. This group was analyzed separately from patients who were randomized to receive the frozen solution; as such, the results are not reported here. 28 bPatients who were allocated to receive belamaf 3.4 mg/kg were not included in this analysis and have been reported previously. 16 cPrimary causes of death in the safety population (n = 47) included the disease under study (n = 40), serious adverse event possibly related to study (n = 1; sepsis 16 ), other (n = 3), and unknown cause (n = 3). dA patient would be counted as ongoing with the study if no study conclusion record were included in the disposition data set.
Nineteen of 97 patients (20%) had normal renal function, whereas 48 of 97 (49%) had mild renal impairment, and 24 of 97 (25%) had moderate renal impairment (according to protocol definition). Patients with high‐risk cytogenetics were well represented using either the HR‐IMWG definition (26 of 97 patients; 27%) or the HR‐cyto definition (41 of 97 patients; 42%). Twenty‐two of 97 patients (23%) had EMD at baseline. Table 2 shows demographics, duration of follow‐up (for all patients: median, 12.4 months; range, 0.1‐17.9 months), and treatment details for these subgroups. 27
TABLE 2.
Patient Demographics and Baseline Disease Characteristics of the Overall (Intention‐to‐Treat) Population and Subgroups of Patients Enrolled to Receive Belantamab Mafodotin 2.5 mg/kg
| Variable | Overall, N = 97 | HR‐IMWG, N = 26 a | HR‐Cyto, N = 41 b | SR‐Cyto, N = 56 c | Normal Renal Function, N = 19 d | Mild Renal Impairment, N = 48 e | Moderate Renal Impairment, N = 24 f | Extramedullary Disease. N = 22 |
|---|---|---|---|---|---|---|---|---|
| Age: Median [range], y | 65.0 [60‐70] | 67.5 [47‐83] | 67.0 [42‐85] | 62.5 [39‐85] | 63.0 [39‐74] | 66.0 [40‐85] | 68.0 [45‐85] | 58.5 [40‐73] |
| Sex: No. (%) | ||||||||
| Women | 46 (47) | 10 (38) | 16 (39) | 30 (54) | 8 (42) | 26 (54) | 11 (46) | 9 (41) |
| Men | 51 (53) | 16 (62) | 25 (61) | 26 (46) | 11 (58) | 22 (46) | 13 (54) | 13 (59) |
| No. of prior therapies: Median [range] | 7 [3‐21] | 7 [3‐11] | 6 [3‐11] | 7 [3‐21] | 6 [4‐10] | 7 [3‐12] | 7 [3‐21] | 6 (3‐11) |
| ISS stage at screening: No. (%) | ||||||||
| I | 21 (22) | 4 (15) | 9 (22) | 13 (23) | 4 (21) | 14 (29) | 4 (17) | 3 (14) |
| II | 33 (34) | 14 (54) | 17 (41) | 16 (29) | 10 (53) | 15 (31) | 7 (29) | 6 (27) |
| III | 42 (43) | 8 (31) | 15 (37) | 27 (48) | 5 (26) | 19 (40) | 13 (54) | 13 (59) |
| HR‐Cyto markers: No. (%) g | ||||||||
| 17p13del | 16 (16) | 16 (62) | 16 (39) | 0 (0) | 1 (5) | 6 (13) | 8 (33) | 3 (14) |
| t(4;14) | 11 (11) | 11 (42) | 11 (27) | 0 (0) | 2 (11) | 5 (10) | 4 (17) | 0 (0) |
| t(14;16) | 7 (7) | 7 (27) | 7 (17) | 0 (0) | 0 (0) | 4 (8) | 2 (8) | 0 (0) |
| 1q21+ | 25 (26) | 10 (38) | 25 (61) | 0 (0) | 4 (21) | 11 (23) | 8 (33) | 3 (14) |
| Duration of follow‐up: Median [range], mo | 12.4 [0.1‐17.9] | 12.2 [0.9‐17.5] | 9.4 [0.5‐17.5] | 13.0 [0.1‐17.9] | 12.5 [0.7‐16.9] | 12.5 [1.0‐17.5] | 13.1 [0.5‐17.9] | 7.7 [0.1‐13.1] |
| No. of cycles: Median [range] | 3 [1‐11] | 3 [1‐17] | 3 [1‐17] | 3 [1‐16] h | 3 [1‐7] | 3 [1‐14] | 3 [1‐15] | 2 [0‐8] |
Abbreviations: HR‐cyto, high‐risk cytogenetics; HR‐IMWG, high‐risk cytogenetics according to International Myeloma Working Group criteria; ISS, International Staging System. SR‐Cyto, standard‐risk cytogenetics.
HR‐IMWG was defined as patients with any of t(4:14), t(14:16), or 17p13del from the overall population. 27
HR‐cyto was defined as patients with any of t(4:14), t(14:16), 17p13del, or 1q21+ from the overall population.
SR‐cyto was defined as patients with none of t(4:14), t(14:16), 17p13del, or 1q21+ from the overall population.
Normal renal function was defined as patients with an estimated glomerular filtration rate (eGFR) ≥90 mL per minute per 1.73 m2 from the overall population.
Mild renal impairment was defined as patients with an eGFR from ≥60 to <90 mL per minute 1.73 m2 from the overall population.
Moderate renal impairment was defined as patients with an eGFR from ≥30 to <60 mL per minute per 1.73 m2 from the overall population.
Patients could have >1 HR‐cyto marker.
Data were not available for 2 patients.
Efficacy
In this updated analysis, 31 of 97 patients (32%; 97.5% CI, 21.7%‐43.6%) achieved an overall response in the entire belamaf 2.5‐mg/kg group (Table 3). 25 , 27 Responses deepened over time in some patients: 12 responders who initially had a ≥PR subsequently deepened their response. Eighteen of 97 patients (19% [18 of 31 responders; 58%]) achieved a ≥VGPR, including a stringent complete response (sCR) or a complete response (CR) in 7 patients. A clinical benefit (minimal response [MR] or better [≥MR]) was achieved in 35 of 97 patients (36%). Among patients with a ≥VGPR who were tested for MRD status, 5 of 13 (38%) achieved MRD negativity at the 1 × 10−5 sensitivity level, including 2 of 2 patients (100%) with an sCR, 2 of 5 (40%) with a CR, and 1 of 6 (17%) with a VGPR.
TABLE 3.
Clinical Response in the Overall Population and Subgroups of Patients Enrolled to Receive Belantamab Mafodotin 2.5 mg/kg
| Independent Review Committee‐Assessed Best Response a | Overall, N = 97 | HR‐IMWG, N = 26 b | HR‐Cyto, N = 41 c | SR‐Cyto, N = 56 d | Normal Renal Function, N = 19 e | Mild Renal Impairment, N = 48 f | Moderate Renal Impairment, N = 24 g | Extramedullary Disease, N = 22 |
|---|---|---|---|---|---|---|---|---|
| ORR: No. (%) h | 31 (32) | 9 (35) | 12 (29) | 19 (34) | 7 (37) | 16 (33) | 8 (33) | 1 (5) |
| 97.5% or 95% CI i | 21.7‐43.6 | 17.2‐55.7 | 16.1‐45.5 | 21.8‐47.8 | 16.3‐61.6 | 20.4‐48.4 | 15.6‐55.3 | — |
| sCR | 2 (2) | 0 (0) | 1 (2) | 1 (2) | 1 (5) | 0 (0) | 1 (4) | 0 (0) |
| CR | 5 (5) | 2 (8) | 3 (7) | 2 (4) | 0 (0) | 2 (4) | 3 (13) | 0 (0) |
| VGPR | 11 (11) | 5 (19) | 5 (12) | 6 (11) | 1 (5) | 6 (13) | 4 (17) | 0 (0) |
| PR | 13 (13) | 2 (8) | 3 (7) | 10 (18) | 5 (26) | 8 (17) | 0 (0) | 0 (0) |
| MR | 4 (4) | 3 (12) | 3 (7) | 1 (2) | 2 (11) | 2 (4) | 0 (0) | 2 (9) |
| SD | 27 (28) | 5 (19) | 9 (22) | 18 (32) | 6 (32) | 13 (27) | 8 (33) | 8 (36) |
| CBR: No. (%) j | 35 (36) | 12 (46) | 15 (37) | 20 (36) | 9 (47) | 18 (38) | 8 (33) | 3 (14) |
| 95% CI | 26.6‐46.5 | 26.6‐66.6 | 22.1‐53.1 | 23.4‐49.6 | 24.4‐71.1 | 24.0‐52.6 | 15.6‐55.3 | 2.9‐34.9 |
Abbreviations: CBR, clinical benefit rate; CI, confidence interval; CR, complete response; HR‐cyto, high‐risk cytogenetics; HR‐IMWG, high‐risk cytogenetics according to International Myeloma Working Group criteria; MR, minimal response; ORR, overall response rate; PR, partial response; sCR, stringent complete response; SD, stable disease; SR‐Cyto, standard‐risk cytogenetics; VGPR, very good partial response.
Responses were assessed in the intention‐to‐treat population (including all randomly assigned patients) by an independent review committee according to the IMWG uniform criteria consensus recommendations. 25 Six patients (6%) were not evaluable for response and were treated as nonresponders.
HR‐IMWG was defined as patients with any of t(4:14), t(14:16), or 17p13del. 27
HR‐cyto was defined as patients with any of t(4:14), t(14:16), 17p13del, or 1q21+.
SR‐cyto was defined as patients with none of t(4:14), t(14:16), 17p13del, or 1q21+.
Normal renal function was defined as patients with an estimated glomerular filtration rate (eGFR) ≥90 mL per minute per 1.73 m2.
Mild renal impairment was defined as patients with an eGFR from ≥60 to <90 mL per minute per 1.73 m2.
Moderate renal impairment was defined as patients with an eGFR from ≥30 to <60 mL per minute per 1.73 m2.
The ORR was calculated as follows: sCR + CR + VGPR + PR.
The 97.5% CI is presented for the overall population, and the 95% CI is presented for all other groups.
The CBR was calculated as follows: sCR + CR + VGPR + PR + MR.
In the overall belamaf 2.5‐mg/kg group, the median DoR estimate was 11 months (95% CI, 4.2 months to not reached) (Fig. 2A), 25 and the estimated probability of maintaining a response at 12 months was 50% (95% CI, 29%‐68%). In patients with a ≥MR (n = 35), the estimated duration of clinical benefit was 11.7 months (95% CI, 4.2 months to not reached) (Fig. 2B). Correction added on 7 September 2021, after first online publication: This statement has been revised.
Figure 2.
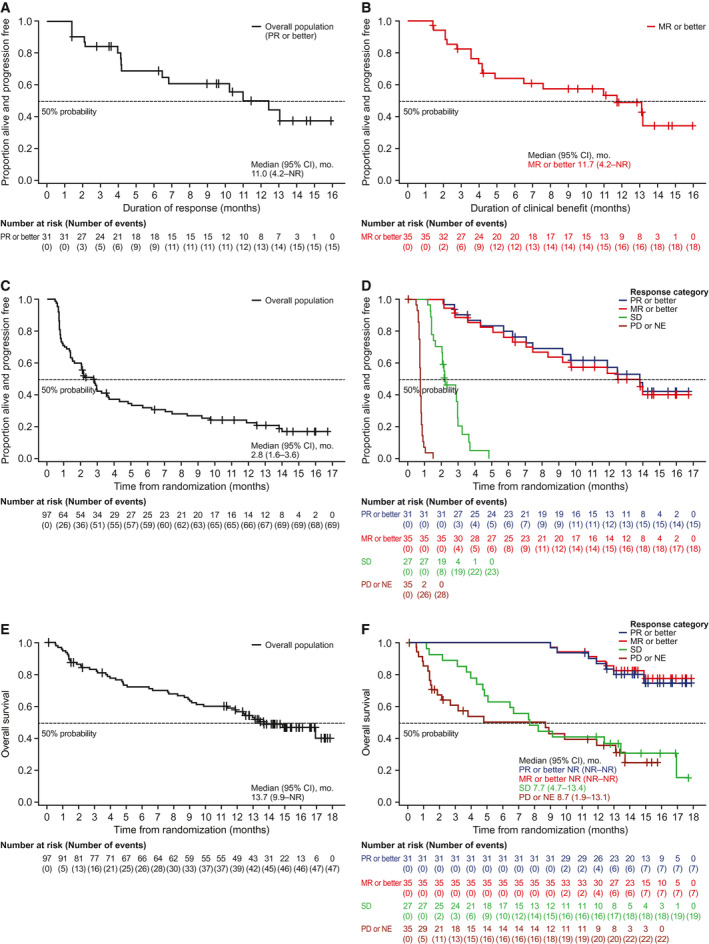
Duration of response in the overall population (PR or better) is illustrated in (A), by duration of clinical benefit (MR or better) in (B), progression‐free survival is illustrated in (C) the overall population and (D) by response category, and overall survival is illustrated in (E) the overall population and (F) by response category. Responses were assessed in the intention‐to‐treat population (including all randomly assigned patients) by an independent review committee according to the International Myeloma Working Group uniform criteria consensus recommendations. 25 Six patients (6%) were not evaluable (NE) for response and were treated as nonresponders. CI indicates confidence interval; MR, minimal response; NR, not reached; PD, progressive disease; PR, partial response; SD, stable disease. Correction added on 7 September 2021, after first online publication: Figure 2 and its legend have been corrected.
The median PFS was 2.8 months (95% CI, 1.6‐3.6 months) in the overall belamaf 2.5‐mg/kg group (Fig. 2C). In patients (n = 7) with an sCR or a CR, the median PFS was not reached (95% CI, 7.1 months to not reached). PFS by response category is illustrated in Figure 2D. Patients (n = 11) who achieved a VGPR had an estimated median PFS of 14 months (95% CI, 7.5 months to not reached). In patients (n = 13) with a PR, the median PFS estimate was 6.2 months (95% CI, 2.8 months to not reached). The median estimated OS was 13.7 months (95% CI, 9.9 months to not reached) (Fig. 2E), with an estimated 1‐year survival probability of 58% (95% CI, 47%‐67%). OS by response category is illustrated in Figure 2F. The estimated median OS was not reached in patients (n = 35) who had an ≥MR or in patients (n = 31) who had a ≥PR, with estimated 1‐year survival probabilities of 88% (95% CI, 72%‐95%) and 87% (95% CI, 69%‐95%), respectively.
Efficacy outcomes were similar in patients who had high‐risk or standard‐risk cytogenetics and were comparable to those of the overall population. The most common cytogenetic change (n = 7; 13%) in the standard‐risk group was t(11:14), which has been associated with favorable OS outcomes. 29 In our study, an overall response was achieved by 9 of 26 patients (35%; 95% CI, 17.2%‐55.7%) in the HR‐IMWG group, 12 of 41 (29%; 95% CI, 16.1%‐45.5%) in the HR‐cyto group, and 19 of 56 (34%; 95% CI, 21.8%‐47.8%) in the standard‐risk cytogenetic group (Table 3). The median estimated DoR was 10.3 months (95% CI, 1.4‐13.1 months) for both the HR‐IMWG and HR‐cyto groups and was not reached (95% CI, 4.2 months to not reached) in the standard‐risk cytogenetic group (see Supporting Fig. 1A). The median PFS was 3.3 months (95% CI, 0.9‐7.1 months) in the HR‐IMWG group, 2.1 months (95% CI, 0.8‐3.7 months) in the HR‐cyto group, and 2.9 months (95% CI, 1.6‐4.8 months) in the standard‐risk cytogenetic group. OS in these groups is presented in Supporting Figure 1B. The median OS estimate was 13.1 months (95% CI, 8.2 months to not reached) in the HR‐IMWG group, 9.9 months (95% CI, 4.3 months to not reached) in the HR‐cyto group, and 17 months (95% CI, 12.4 months to not reached) in the standard‐risk cytogenetic group.
Compared with patients who had normal renal function, the efficacy of belamaf was similar in patients who had mild and moderate renal impairment according to the protocol definition. A similar proportion of patients in all 3 groups achieved an overall response (Table 3). The median estimated DoR was 4.2 months (95% CI, 1.4 months to not reached) for patients with normal renal function, 12.5 months (95% CI, 2.2 months to not reached) for those with mild renal impairment, and 13.1 months (95% CI, 4.2 months to not reached) for those with moderate renal impairment. The median PFS was 3.0 months (95% CI, 1.3‐6.2 months) for patients with normal renal function, 2.2 months (95% CI, 2.0‐3.6 months) for those with mild renal impairment, and 3.7 months (95% CI, 1.0‐12.5 months) for those with moderate renal impairment. The median estimated OS was 14.9 months (95% CI, 7.7 months to not reached) for patients with normal renal function, 13.7 months (95% CI, 11.4 months to not reached) for those with mild renal impairment, and not reached (95% CI, 5.1 months to not reached) for those with moderate renal impairment.
In the subgroup of patients with EMD, 1 of 22 patients (5%) achieved an overall response (Table 3). The DoR in this patient was 1.4 months, and the PFS was 2.8 months. Eight of 22 patients (36%) had stable disease, and 2 of 22 patients (9%) were not evaluable for a response. In this subgroup, the median PFS was 1.1 months (range, 0‐8.3 months), and the median estimated OS was 13.4 months (range, 2.7 months to not reached). The median number of treatment cycles given to patients who had EMD was 2 (range, 0‐8 treatment cycles).
Safety
Overall, 93 of 95 patients (98%) receiving belamaf 2.5 mg/kg experienced at least 1 AE (Table 4), 23 , 24 with treatment‐related AEs occurring in 84 of 95 patients (88%). Fatal serious AEs (SAEs) occurred in 3 of 95 patients (3%). One of these events (sepsis) was considered treatment‐related. 16 Grade 3 and 4 AEs were reported in 79 of 95 patients (83%) (Table 4) and were treatment‐related in 54 of 95 patients (57%). The most common any grade and grade ≥3 AEs were keratopathy, thrombocytopenia, and anemia (Table 4).
TABLE 4.
Most Common Adverse Events (Occurring in ≥15%) and Grade ≥3 Adverse Events (Occurring in ≥5%) in the Overall Population a
| Event | Belamaf 2.5 mg/kg, N = 95: No. of Patients (%) | |
|---|---|---|
| Any Grade | Grade ≥3 | |
| Any event | 93 (98) | 80 (84) |
| Eye examination finding | ||
| Keratopathy b | 68 (72) | 44 (46) |
| Change in BCVA | 51 (54) | 29 (31) |
| Thrombocytopenia c | 36 (38) | 21 (22) |
| Anemia | 26 (27) | 20 (21) |
| Blurred vision d | 24 (25) | 4 (4) |
| Nausea | 24 (25) | 0 (0) |
| Pyrexia e | 22 (23) | 4 (4) |
| Aspartate aminotransferase increased | 20 (21) | 2 (2) |
| Infusion‐related reaction f | 20 (21) | 3 (3) |
| Fatigue | 15 (16) | 2 (2) |
| Neutropenia g | 14 (15) | 10 (11) |
| Dry eye h | 14 (15) | 1 (1) |
| Hypercalcemia | 14 (15) | 7 (7) |
| Lymphocyte count decreased | 13 (14) | 12 (13) |
| Pneumonia | 9 (9) | 6 (6) |
Abbreviations: BCVA, best‐corrected visual acuity; Belamaf, belantamab mafodotin.
Events are listed in order of decreasing frequency for any grade and are reported based on Common Terminology Criteria for Adverse Events, version 4.03 23 (except for keratopathy) in the safety population (including all patients who received ≥1 dose of trial treatment).
These were changes in the corneal epithelium observed on eye examination and were graded according to a protocol‐defined scale, which was renamed the Keratopathy and Visual Acuity scale (see Table 1). 24
Thrombocytopenia (considered an adverse event of special interest) includes the preferred terms thrombocytopenia and platelet count decreased.
Blurred vision includes the preferred terms vision blurred, diplopia, visual acuity reduced, and visual impairment.
Events occurring within 24 hours of infusion are included for individual adverse events (preferred terms) and are also counted within infusion‐related reactions.
Infusion‐related reactions (considered adverse events of special interest) include the preferred terms infusion‐related reaction, pyrexia, chills, diarrhea, nausea, asthenia, hypertension, lethargy, and tachycardia occurring within 24 hours of infusion.
Neutropenia includes neutropenia and neutrophil count decreased.
Dry eye includes the preferred terms dry eye, ocular discomfort, and eye pruritus.
Thrombocytopenia of any grade (including the preferred terms thrombocytopenia and decreased platelet count) was reported in 36 of 95 patients (38%). Grade ≥3 events occurred in 21 of 95 patients (22%). Infusion‐related reactions (IRRs), which included grouped terms pyrexia, chills, diarrhea, nausea, asthenia, hypertension, lethargy, and tachycardia, occurring within 24 hours of infusion were reported in 20 of 95 patients (21%) and were mostly mild (85% grade 1‐2).
Keratopathy comprising changes in the corneal epithelium observed on eye examination, with or without BCVA change from baseline or symptoms, occurred in 68 of 95 patients (72%) (Fig. 3). 24 The median time to the onset of the first keratopathy examination finding was 37 days (range, 19‐143 days), with 66 of 68 patients who had keratopathy experiencing their first finding by treatment cycle 4. As of this analysis and where data were available, 46 of 60 patients (77%) recovered (resolution or return to baseline) from their first keratopathy examination finding of grade ≥2 according to the KVA scale, and 29 of 60 patients (48%) recovered from their last event. The median time to recovery of the first examination finding was 86.5 days (range, 8‐358 days), and it was 81 days (range, 11‐232 days) for the last event. Among the 31 patients whose recovery had yet to be recorded from their last event, 9 were still receiving treatment, 5 were in follow‐up, and 17 did not complete follow‐up as of this analysis. Thirty‐seven of 44 patients (84%) who had grade 3 and 4 keratopathy examination findings were improving or had recovered at last follow‐up. One patient had reported corneal erosions of approximately 2 mm (without stromal involvement) bilaterally, and 2 patients had transient whorl‐like keratopathy identified by fluorescein staining. Corneal erosion and whorl‐like keratopathy resolved with dose delay.
Figure 3.
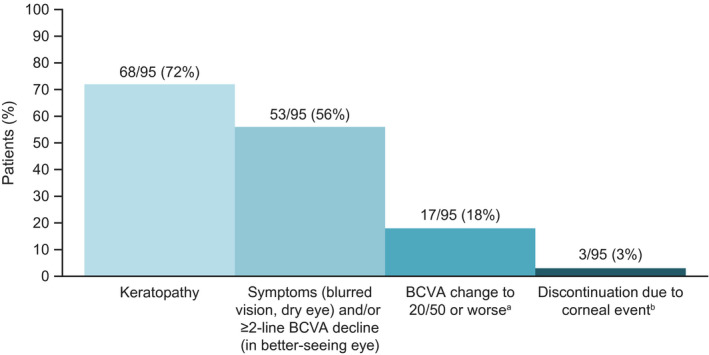
The frequency of corneal and vision‐related events in patients treated with belantamab mafodotin (belamaf) 2.5 mg/kg in the DREAMM‐2 trial (n = 95) is illustrated. aClinically meaningful best‐corrected visual acuity (BCVA) change represents a BCVA of Snellen Visual Acuity 20/50 or worse in the better seeing eye. bDiscontinuation included 1 patient with keratopathy, 1 patient with blurred vision, and 1 patient with reduced visual acuity.
Seventeen of 95 patients (18%) experienced a meaningful decline in BCVA (to a Snellen Visual Acuity of 20/50 or worse) in their better seeing eye at least once during or after the treatment period (Fig. 3). In patients with normal or near‐normal vision at baseline, change to a Snellen Visual Acuity score of 20/50 indicates a meaningful reduction in visual acuity and is used as a threshold for legal driving in many countries. 30 Fourteen of 17 patients (82%) recovered (BCVA improvement to better than a Snellen Visual Acuity of 20/50) while on treatment at the time of this follow‐up. 24 The median duration of these declines in BCVA was 21.5 days (range, 7‐64 days); therefore, most patients recovered after one 21‐day assessment interval. No permanent complete loss of vision (irreversible BCVA decline worse than a Snellen Visual Acuity of 20/200) has been reported to date from DREAMM‐2.
Blurred vision was reported by 24 of 95 patients (25%), and dry eye was reported by 14 of 95 patients (15%) (Table 4). 24 Grade 3 and 4 blurred vision (4 of 95 patients; 4%) or dry eye (1 of 95 patients; 1%) were less common. As of the latest follow‐up, 15 of 24 patients (63%) with blurred vision and 11 of 14 patients (79%) with dry eye had recovered.
The frequency of AEs, treatment‐related AEs, SAEs, and most common any‐grade and grade 3 and 4 events was similar in patients with high‐risk cytogenetics (including HR‐IMWG and HR‐cyto) and standard‐risk cytogenetics and was comparable to their frequency in the overall population (see Supporting Table 1). Similarly, patients who had mild or moderate renal impairment had a similar incidence of AEs, treatment‐related AEs, SAEs, and most common any‐grade and grade 3 and 4 events compared with those who had normal renal function (see Supporting Table 2). Few patients who had available postbaseline renal laboratory values developed signs of active renal conditions, with 1 of 45 patients (2%) who had mild renal impairment and 2 of 20 patients (10%) in the moderate renal impairment group showing an increased albumin creatinine ratio (≥500 mg/g). The incidence of keratopathy was similarly high in patient subgroups with high‐risk cytogenetics (HR‐IMWG, 17 of 26 patients [65%]; HR‐cyto, 25 of 41 patients [61%]) and renal impairment (mild renal impairment, 34 of 48 patients [71%]; moderate renal impairment, 15 of 24 patients [63%]). In patients with EMD, the frequency of AEs, including those that were treatment‐related or serious, was similar to the frequency in the overall population (see Supporting Table 3). The most common any‐grade and grade 3 and 4 events were also similar.
In the overall belamaf 2.5‐mg/kg dose group, dose delays and reductions because of AEs were common and occurred in 51 (54%) and 33 (35%) of 95 patients, respectively (see Supporting Table 4). Keratopathy was the most frequent reason for dose delays (45 of 95 patients; 47%) and dose reductions (24 of 95 patients; 25%). However, few (9 of 95 patients; 9%) permanently discontinued study treatment because of AEs, and only 1 of 95 patients (1%) discontinued for keratopathy; 2 additional patients discontinued for blurred vision and reduced visual acuity, respectively (Fig. 3).
A post hoc analysis to evaluate the outcomes of patients with a response who had prolonged dose delays (in which ≥3 treatment cycles were missed) was performed. Of the patients who had a response according to IMWG criteria in the 2.5‐mg/kg group, 16 of 31 patients (52%) had ≥1 prolonged dose delay. Fourteen of 16 patients (88%) continued to experience a clinical benefit during the first prolonged delay (see Supporting Fig. 2): 6 of 16 patients (38%) deepened their response during delay, 6 of 16 (38%) maintained the same response as of the last evaluable assessment, 2 of 16 (13%) had rising paraproteins during the delay but did not meet criteria for disease progression, and 2 of 16 (13%) developed disease progression. Among patients who had ≥2 prolonged dose delays, 4 of 5 (80%) maintained their response after the second prolonged dose delay. The 1 patient who experienced 3 prolonged dose delays maintained their response after the first and third delays and deepened their response after the second delay. Keratopathy was also the most frequent reason for dose delays in patients who had prolonged delays. In patients who had grade 3 and 4 keratopathy at the beginning of the first prolonged dose delay, 8 of 10 (80%) improved to grade ≤2 at the end of this delay. The 2 patients who had grade 3 keratopathy events at the end of the first prolonged delay for keratopathy are still in follow‐up as of this analysis. Belamaf was resumed at 1.92 mg/kg after a 105‐day delay and after a 68‐day delay, respectively; both patients had ongoing keratopathy events graded at 2 or 3 throughout follow‐up, but neither had a further prolonged dose delay.
Discussion
Clinically meaningful (overall responses achieved by 32% of patients) and deep (58% of responders with a ≥VGPR) responses with single‐agent belamaf 2.5 mg/kg were sustained at the 13‐month follow‐up in the DREAMM‐2 study. The median estimated OS was 13.7 months in this extended analysis, which is substantially longer than that reported in a similar population. 3 The median estimated DoR in the 2.5‐mg/kg group was 11 months. (Correction added on 7 September 2021, after first online publication: This statement has been revised.) In the overall population, the median PFS was 2.8 months; in patients who had an sCR or a CR, the median PFS was not reached. It is likely that the gap between PFS and OS is bridged by using salvage therapies, which were not explored here. Furthermore, in patients who had a ≥VGPR, the median PFS was 14 months. In patients with deep responses (≥VGPR) who were tested for MRD status, 5 of 13 (38%) achieved MRD negativity at this analysis. Follow‐up is continuing for long‐term responders.
No new safety signals were identified, although further information describing the epitheliopathy of the keratopathy was reported. Corneal erosions, which are considered an expression of the observed epitheliopathy, were observed in 1 patient, and whorl‐like keratopathy was observed in 2 patients. Additional follow‐up will be reported in future communications. The occurrence of AEs was comparable in subgroups of patients with high‐risk cytogenetics and renal impairment. As previously described, thrombocytopenia was common but was considered self‐limited; IRRs occurred early in treatment and were mainly grade 1 and 2. 16 The low rates of grade ≥3 hematologic AEs (thrombocytopenia, 21 of 95 patients [22%]; anemia, 20 of 95 patients [21%]; and neutropenia, 10 of 95 patients [11%]) and IRRs of any grade (20 of 95 patients; 21%), coupled with the short outpatient administration time and no mandatory requirement for premedication, make belamaf an attractive treatment option.
The efficacy of belamaf in patients who had high‐risk cytogenetics, a subgroup with a particularly high unmet need, was comparable to that in patients who had standard‐risk cytogenetics. Efficacy outcomes were similar in the HR‐IMWG (excluding 1q21+) and HR‐cyto (including 1q21+) groups (median DoR, 10.3 months for both). Although the DoR was not reached in patients who had standard‐risk cytogenetics, this is likely because of the low number of events at later timepoints. Patients with ≥3 copies of 1q21 have worse outcomes compared with those who lack this cytogenetic feature because 1q21 gain or amplification is associated with the overexpression of CKS1B, a gene involved in regulation of MM cell growth and survival. 5 , 31 In DREAMM‐2, 1q21 copy numbers were not reported, so further stratification and analysis based on 1q21 copy numbers are not possible.
Efficacy in patients who had mild or moderate renal impairment according to the protocol definition mirrored that in patients who had normal renal function, although the DoR was shorter in patients with normal renal function (4.2 months). We speculate that this may be due to the baseline demographic characteristics of these patients, although, because the number of patients included is small, any further analysis would not be sufficiently powered to determine causality. Outcomes from the DREAMM‐12 study (ClinicalTrials.gov identifier NCT04398745), which includes patients with severe renal impairment, will further inform the benefit:risk profile of single‐agent belamaf in patients with RRMM and renal impairment, a subgroup with limited treatment options.
In patients with EMD, there has not been strong evidence of efficacy to date with immunomodulatory agents, proteasome inhibitors, or moAbs. 12 In DREAMM‐2, patients who had EMD had poorer outcomes compared with the overall population, further highlighting the unmet need for novel agents and combinations in this subgroup of patients. Future studies are needed to determine whether responses to belamaf treatment vary depending on the EMD subtype.
No new safety signals were identified in longer term follow‐up with single‐agent belamaf, and the occurrence of AEs was comparable in subgroups of patients with high‐risk cytogenetics, renal impairment, and EMD. The increase in grade 3 and 4 keratopathy reported here relative to the primary analysis 16 predominantly reflects the difference in grading scale used to report keratopathy. Keratopathy was previously reported according to the grade for this event using the CTCAE version 4.03 scale. 16 Here, we report keratopathy grading according to a protocol‐defined scale (subsequently renamed the KVA scale), which combines corneal examination findings and BCVA changes and is consistent with the US and EU labeling. 21 , 22 In both the primary and follow‐up analyses, dose modifications were based on the protocol‐defined scale, which combined corneal examination findings and BCVA changes and thus are unaffected by this change in reporting.
Although keratopathy was frequently observed on eye examination, fewer patients experienced symptoms, most did not experience a clinically meaningful BCVA decline, and events rarely led to treatment discontinuation. At data cutoff, 77% of patients had recovered from their first keratopathy event, highlighting the reversibility of these events. Given the long half‐life of belamaf (14 days), continued follow‐up of patients for evidence of corneal event resolution is crucial because it takes 70 days (5 half‐lives) before the drug is eliminated; accordingly, the median time to resolution observed here was 86.5 days. Although complete resolution of all keratopathy events was not observed during follow‐up in this study, we might expect resolution with longer follow‐up; where patients have been lost to follow‐up, it is possible that their keratopathy resolved without this being recorded. Continued follow‐up of patients beyond this current data cutoff is ongoing. Additional studies are ongoing to determine the etiology and optimal management strategy for belamaf‐associated corneal events.
Changes in BCVA were manageable with dose modifications and resolved around the time of the next eye examination (conducted approximately every 21 days). No permanent complete loss of vision has been reported to date. Corneal events associated with belamaf may be adequately managed by close liaison with eye care professionals, according to the KVA scale guidelines (Table 1). 24 Prescribing physicians should follow the specific dose‐modification guidelines for corneal event management outlined in their local prescribing information.
Although dose modifications (delays or reductions) to manage AEs were common, there was minimal impact on patient responses to belamaf. A high proportion of patients with a clinical response who had prolonged dose interruptions continued to have clinical benefit after their first delay. Clinical responses were typically maintained in patients who had >1 prolonged dose delay. Rates of permanent treatment discontinuation were also low (9 of 95 patients; 9%), suggesting dose that modifications were effective at managing AEs. These data, along with outcomes from other studies using dose delays to manage corneal events associated with MMAF‐containing antibody‐drug conjugates, support the use of dose delays as a corneal event management strategy. 24 The effect of shorter dose delays on efficacy remains to be formally analyzed, but we anticipate it would be no greater than the effect of prolonged dose delays.
One limitation of the DREAMM‐2 study is that there is no comparator arm and thus no option to compare directly with other available treatment options. The ongoing, open‐label, phase 3, DREAMM‐3 study (ClinicalTrials.gov identifier NCT04162210) will compare belamaf versus standard‐of‐care pomalidomide plus dexamethasone therapy in patients with ≥2 prior lines of therapy. 32
The mechanism of action and manageable safety profile of belamaf offers the potential for combination with other agents; several studies are ongoing. The DREAMM‐5 platform study (ClinicalTrials.gov identifier NCT04126200) allows comparison of belamaf combined with a range of agents that have differing mechanisms of action and targets versus a shared belamaf monotherapy arm. 33 Limited expression of BCMA may predict response to belamaf and similar BCMA‐targeting drugs for the treatment of MM. In DREAMM‐5, combination therapy with belamaf and the γ‐secretase inhibitor nirogacestat is hypothesized to increase BCMA expression on MM cells. Therefore, results of DREAMM‐5 are expected to demonstrate a new therapeutic strategy for increasing the efficacy of MM drugs targeting BCMA.
The DREAMM‐4 (ClinicalTrials.gov identifier NCT03848845) and DREAMM‐6 (ClinicalTrials.gov identifier NCT03544281) trials investigating belamaf in combination with pembrolizumab and bortezomib plus dexamethasone, respectively, have shown clinical responses and an acceptable safety profile to date. 22 , 34 Studies of alternative dosing schedules and corneal event management strategies are underway to help mitigate belamaf‐associated corneal events.
Treatment options for RRMM are evolving, with the 2019 approval of selinexor (as part of a combination regimen) 35 and a recent confirmatory phase 3 study. 36 Single‐agent belamaf represents a further important new treatment option for patients with heavily pretreated RRMM, including those with high‐risk cytogenetics and renal impairment. The efficacy and safety data presented here demonstrate that belamaf has the potential to shift the treatment paradigm in this heavily pretreated, anti‐CD38 MoAb‐exposed patient population with a poor prognosis and few alternative treatment options.
Funding Support
This study was funded by GlaxoSmithKline. GlaxoSmithKline contributed to study design, implementation, data collection, interpretation, and analysis. Medical writing support was funded by GlaxoSmithKline.
Conflict of Interest Disclosures
Sagar Lonial reports grant funding and personal fees from Celgene and Takeda and personal fees from Novartis, Bristol‐Myers Squibb, GlaxoSmithKline, Amgen, Merck, and Janssen outside the submitted work. Hans C. Lee reports research funding and personal fees from Amgen, Celgene, Janssen, and Takeda; personal fees from Genentech, GlaxoSmithKline, and Sanofi; and research funding from Daiichi Sankyo and Regeneron outside the submitted work. Ashraf Badros reports consulting fees from Amgen outside the submitted work. Suzanne Trudel reports consulting fees from Celgene, Amgen, and GlaxoSmithKline; honoraria from Celgene, Janssen, Takeda, Sanofi, Karyopharm, and Amgen Canada; and research funding from Celgene, Janssen, Amgen, GlaxoSmithKline, and Genentech outside the submitted work. Ajay K. Nooka reports consulting fees from Amgen, Janssen Oncology, Celgene, Spectrum Pharmaceuticals, Bristol‐Myers Squibb, GlaxoSmithKline, Takeda, Oncopeptides, and Karyopharm Therapeutics; personal fees from GlaxoSmithKline; and research funding from Amgen, Janssen Oncology, and Takeda outside the submitted work. Ajai Chari reports consulting fees from Celgene, Novartis, Amgen, Janssen Oncology, Seattle Genetics, Bristol‐Myers Squibb, Karyopharm Therapeutics, Genzyme, Oncopeptides, Takeda, Antengene, GlaxoSmithKline, and Secura Bio; and research funding from Celgene, Novartis, Janssen, Pharmacyclics, Amgen, Seattle Genetics, and Takeda outside the submitted work. Natalie Callander reports research funding from Cellectar outside the submitted work. Douglas Sborov reports consulting fees, honoraria, and personal fees from Janssen outside the submitted work. Attaya Suvannasankha reports consulting fees from GlaxoSmithKline, Janssen, and Karyopharm Therapeutics; research funding from GlaxoSmithKline, Janssen, Bristol‐Myers Squibb, and Celgene; and personal fees from GlaxoSmithKline and Janssen outside the submitted work. Katja Weisel reports consulting fees/honoraria from Amgen, Adaptive, Bristol‐Myers Squibb, Celgene, Janssen, GlaxoSmithKline, Karyopharm, Takeda and Sanofi; and research funding from Amgen, Celgene, Sanofi, and Janssen outside the submitted work. Peter M. Voorhees reports personal fees from Adaptive Biotechnologies, Bristol‐Myers Squibb/Celgene, Janssen, Novartis, Oncopeptides, and TeneoBio outside the submitted work. Linsey Womersley is an employee of GlaxoSmithKline. January Baron, Trisha Piontek, Eric Lewis, and Joanna Opalinska are employees of and hold stock/shares in GlaxoSmithKline. Ira Gupta is an employee of and holds stock/shares in GlaxoSmithKline and Novartis. Adam D. Cohen reports grant funding from GlaxoSmithKline, Bristol‐Myers Squibb, and Novartis; personal fees from Janssen, Takeda, Oncopeptides, Kite Pharma, Genentech/Roche, AstraZeneca, and Seattle Genetics; and personal fees and other support from GlaxoSmithKline and Celgene outside the submitted work. Al‐Ola Abdallah made no disclosures.
Author Contributions
Sagar Lonial contributed to study conception/design and acquisition of data. Ashraf Badros, Ajai Chari, Adam D. Cohen, Ajay K. Nooka, Al‐Ola Abdallah, Attaya Suvannasankha, Douglas Sborov, Hans C. Lee, Katja Weisel, Natalie Callander, Peter M. Voorhees, and Suzanne Trudel contributed to acquisition of data. Eric Lewis, Ira Gupta, January Baron, Joanna Opalinska, and Trisha Piontek contributed to study design and data analysis and interpretation. Lynsey Womersley contributed to data analysis and interpretation. All authors had access to the study data and accept responsibility for submitting the article for publication. All authors were involved at each stage of article preparation and approved the final version.
Supporting information
Fig S1
Fig S2
Table S1‐4
Supplementary Material
Lonial S, Lee HC, Badros A, Trudel S, Nooka AK, Chari A, Abdallah A‐O, Callander N, Sborov D, Suvannasankha A, Weisel K, Voorhees PM, Womersley L, Baron J, Piontek T, Lewis E, Opalinska J, Gupta I, Cohen AD. Longer term outcomes with single‐agent belantamab mafodotin in patients with relapsed or refractory multiple myeloma: 13‐month follow‐up from the pivotal DREAMM‐2 study. Cancer. 2021. 10.1002/cncr.33809
We thank the ophthalmology and optometry colleagues who provided eye examinations to patients enrolled in the study and Eric Zhi, Julie Byrne, Jeff Jackson, Karrie Wang, Joe Kovach, Kaytlyn Nungesser, and Alessandra Tosolini. Medical writing support was provided by Sarah Hauze and Gemma Corr of Fishawack Indicia, part of Fishawack Health, and was funded by GlaxoSmithKline. Drug linker technology was licensed from Seagen, Inc (Bothell, Washington), and the monoclonal antibody was produced with POTELLIGENT technology licensed from BioWa (Princeton, New Jersey). Trademarks are owned by or licensed to the GSK group of companies.
Correction added on 7 September 2021, after first online publication: Minor text corrections have been made throughout the article.
Data Availability
Information about GlaxoSmithKline's data‐sharing commitments and access requests to anonymized individual participant data and associated documents can be requested for further research from ClinicalStudyDataRequest.com.
References
- 1. Chari A, Vogl DT, Gavriatopoulou M, et al. Oral selinexor‐dexamethasone for triple‐class refractory multiple myeloma. N Engl J Med. 2019;381:727‐738. doi: 10.1056/NEJMoa1903455 [DOI] [PubMed] [Google Scholar]
- 2. Verelst SGR, Blommestein HM, De Groot S, et al. Long‐term outcomes in patients with multiple myeloma: a retrospective analysis of the Dutch Population‐based HAematological Registry for Observational Studies (PHAROS). Hemasphere. 2018;2:e45. doi: 10.1097/hs9.0000000000000045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gandhi UH, Cornell RF, Lakshman A, et al. Outcomes of patients with multiple myeloma refractory to CD38‐targeted monoclonal antibody therapy. Leukemia. 2019;33:2266‐2275. doi: 10.1038/s41375-019-0435-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mikhael J. Treatment options for triple‐class refractory multiple myeloma. Clin Lymphoma Myeloma Leuk. 2020;20:1‐7. doi: 10.1016/j.clml.2019.09.621 [DOI] [PubMed] [Google Scholar]
- 5. Sonneveld P, Avet‐Loiseau H, Lonial S, et al. Treatment of multiple myeloma with high‐risk cytogenetics: a consensus of the International Myeloma Working Group. Blood. 2016;127:2955‐2962. doi: 10.1182/blood-2016-01-631200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Dimopoulos MA, Delimpasi S, Katodritou E, et al. Significant improvement in the survival of patients with multiple myeloma presenting with severe renal impairment after the introduction of novel agents. Ann Oncol. 2014;25:195‐200. doi: 10.1093/annonc/mdt483 [DOI] [PubMed] [Google Scholar]
- 7. Wanchoo R, Abudayyeh A, Doshi M, et al. Renal toxicities of novel agents used for treatment of multiple myeloma. Clin J Am Soc Nephrol. 2017;12:176‐189. doi: 10.2215/CJN.06100616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen J, Liu H, Li L, et al. Clinical features and treatment outcome of elderly multiple myeloma patients with impaired renal function. J Clin Lab Anal. 2019;33:e22888. doi: 10.1002/jcla.22888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fraz MA, Warraich FH, Warraich SU, et al. Special considerations for the treatment of multiple myeloma according to advanced age, comorbidities, frailty and organ dysfunction. Crit Rev Oncol Hematol. 2019;137:18‐26. doi: 10.1016/j.critrevonc.2019.02.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mallappallil M, Friedman EA, Delano BG, McFarlane SI, Salifu MO. Chronic kidney disease in the elderly: evaluation and management. Clin Pract (Lond). 2014;11:525‐535. doi: 10.2217/cpr.14.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Palumbo A, Bringhen S, Ludwig H, et al. Personalized therapy in multiple myeloma according to patient age and vulnerability: a report of the European Myeloma Network (EMN). Blood. 2011;118:4519‐4529. doi: 10.1182/blood-2011-06-358812 [DOI] [PubMed] [Google Scholar]
- 12. Jagosky MH, Usmani SZ. Extramedullary disease in multiple myeloma. Curr Hematol Malig Rep. 2020;15:62‐71. doi: 10.1007/s11899-020-00568-3 [DOI] [PubMed] [Google Scholar]
- 13. Tai YT, Mayes PA, Acharya C, et al. Novel anti–B‐cell maturation antigen antibody‐drug conjugate (GSK2857916) selectively induces killing of multiple myeloma. Blood. 2014;123:3128‐3138. doi: 10.1182/blood-2013-10-535088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tai YT, Acharya C, An G, et al. APRIL and BCMA promote human multiple myeloma growth and immunosuppression in the bone marrow microenvironment. Blood. 2016;127:3225‐3236. doi: 10.1182/blood-2016-01-691162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Montes De Oca R, Bhattacharya S, Vitali N, et al. PF558. The anti‐BCMA antibody‐drug conjugate GSK2857916 drives immunogenic cell death and immune‐mediated anti‐tumor responses, and in combination with an OX40 agonist potentiates in vivo activity. Hemasphere. 2019;3(S1):231. [Google Scholar]
- 16. Lonial S, Lee HC, Badros A, et al. Belantamab mafodotin for relapsed or refractory multiple myeloma (DREAMM‐2): a two‐arm, randomised, open‐label, phase 2 study. Lancet Oncol. 2020;21:207‐221. doi: 10.1016/S1470-2045(19)30788-0 [DOI] [PubMed] [Google Scholar]
- 17. Eghrari AO, Riazuddin SA, Gottsch JD. Overview of the cornea: structure, function, and development. Prog Mol Biol Transl Sci. 2015;134:7‐23. doi: 10.1016/bs.pmbts.2015.04.001 [DOI] [PubMed] [Google Scholar]
- 18. Eaton JS, Miller PE, Mannis MJ, Murphy CJ. Ocular adverse events associated with antibody‐drug conjugates in human clinical trials. J Ocul Pharmacol Ther. 2015;31:589‐604. doi: 10.1089/jop.2015.0064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. US Food and Drug Administration (FDA) . FDA granted accelerated approval to belantamab mafodotin‐blmf for multiple myeloma. Accessed December 14, 2020. https://www.fda.gov/drugs/drug‐approvals‐and‐databases/fda‐granted‐accelerated‐approval‐belantamab‐mafodotin‐blmf‐multiple‐myeloma
- 20. GlaxoSmithKline (GSK) . GSK announces European Medicines Agency (EMA) accepted marketing authorisation application for belantamab mafodotin for the treatment of relapsed or refractory multiple myeloma. Accessed December 14, 2020. https://www.gsk.com/en‐gb/media/press‐releases/gsk‐announces‐european‐medicines‐agency‐ema‐accepted‐marketing‐authorisation‐application‐for‐belantamab‐mafodotin‐for‐the‐treatment‐of‐relapsed‐or‐refractory‐multiple‐myeloma/
- 21. GlaxoSmithKline . BLENREP prescribing information. Accessed December 14, 2020. https://gsksource.com/pharma/content/dam/GlaxoSmithKline/US/en/Prescribing_Information/Blenrep/pdf/BLENREP‐PI‐MG.PDF
- 22. Nooka AK, Stockerl‐Goldstein K, Quach H, et al. DREAMM‐6: safety and tolerability of belantamab mafodotin in combination with bortezomib/dexamethasone in relapsed/refractory multiple myeloma (RRMM) [abstract]. J Clin Oncol. 2020;38(15 suppl):8502. [Google Scholar]
- 23. US Department of Health and Human Services, National Institutes of Health, National Cancer Institute . Common Terminology Criteria for Adverse Events (CTCAE). Version 4.03. Accessed March 3, 2021. https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03/CTCAE_4.03_2010‐06‐14_QuickReference_8.5x11.pdf
- 24. Farooq AV, Esposti SD, Popat R, et al. Corneal epithelial findings in patients with multiple myeloma treated with antibody‐drug conjugate belantamab mafodotin in the pivotal, randomized, DREAMM‐2 study. Ophthalmol Ther. 2020;9:889‐911. doi: 10.6084/m9.figshare.12326546.v1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol. 2014;15:e538‐e548. doi: 10.1016/S1470-2045(14)70442-5 [DOI] [PubMed] [Google Scholar]
- 26. Lonial S, Lee HC, Badros A, et al. Pivotal DREAMM‐2 study: single‐agent belantamab mafodotin (GSK2857916) in patients with relapsed/refractory multiple myeloma (RRMM) refractory to proteasome inhibitors (PIs), immunomodulatory agents, and refractory and/or intolerant to anti‐CD38 monoclonal antibodies (mAbs) [abstract]. J Clin Oncol. 2020;38(15 suppl):8536. [Google Scholar]
- 27. Rajkumar SV. Multiple myeloma: 2016 update on diagnosis, risk‐stratification, and management. Am J Hematol. 2016;91:719‐734. doi: 10.1002/ajh.24402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Richardson PG, Lee HC, Abdallah AO, et al. Single‐agent belantamab mafodotin for relapsed/refractory multiple myeloma: analysis of the lyophilised presentation cohort from the pivotal DREAMM‐2 study. Blood Cancer J. 2020;10:106. doi: 10.1038/s41408-020-00369-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Durie BG, Goldschmidt H, Mateos MV, et al. Outcomes of patients with t(11;14) multiple myeloma: an International Myeloma Working Group (IMWG) multicenter study [abstract]. J Clin Oncol. 2019;37(15 suppl):8015. doi: 10.1200/JCO.2019.37.15_suppl.8015 [DOI] [Google Scholar]
- 30. Bron AM, Viswanathan AC, Thelen U, et al. International vision requirements for driver licensing and disability pensions: using a milestone approach in characterization of progressive eye disease. Clin Ophthalmol. 2010;4:1361‐1369. doi: 10.2147/OPTH.S15359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhan F, Colla S, Wu X, et al. CKS1B, overexpressed in aggressive disease, regulates multiple myeloma growth and survival through SKP2‐ and p27Kip1‐dependent and ‐independent mechanisms. Blood. 2007;109:4995‐5001. doi: 10.1182/blood-2006-07-038703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Weisel K, Hopkins T, Fecteau D, et al. DREAMM‐3: a phase 3, open‐label, randomized study to evaluate the efficacy and safety of belantamab mafodotin (GSK2857916) monotherapy compared with pomalidomide plus low‐dose dexamethasone (pom/dex) in participants with relapsed/refractory multiple myeloma (RRMM) [abstract]. Blood. 2019;134(suppl 1):1900. [Google Scholar]
- 33. Richardson PG, Biswas S, Holkova B, et al. DREAMM‐5 platform trial: belantamab mafodotin in combination with novel agents in patients with relapsed/refractory multiple myeloma (RRMM) [abstract]. J Clin Oncol. 2020;38(15 suppl):TPS8552. [Google Scholar]
- 34. Nooka AK, Mateos Manteca MV, Bahlis N, et al. DREAMM‐4: evaluating safety and clinical activity of belantamab mafodotin in combination with pembrolizumab in patients with relapsed/refractory multiple myeloma (RRMM). Hemasphere. 2020;4(S1):433‐434. [Google Scholar]
- 35. US Food and Drug Administration (FDA) . FDA approves new treatment for refractory multiple myeloma [press release]. Accessed December 14, 2020. https://www.fda.gov/news‐events/press‐announcements/fda‐approves‐new‐treatment‐refractory‐multiple‐myeloma
- 36. Grosicki S, Simonova M, Spicka I, et al. Once‐per‐week selinexor, bortezomib, and dexamethasone versus twice‐per‐week bortezomib and dexamethasone in patients with multiple myeloma (BOSTON): a randomised, open‐label, phase 3 trial. Lancet. 2020;396:1563‐1573. doi: 10.1016/S0140-6736(20)32292-3 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Table S1‐4
Supplementary Material
Data Availability Statement
Information about GlaxoSmithKline's data‐sharing commitments and access requests to anonymized individual participant data and associated documents can be requested for further research from ClinicalStudyDataRequest.com.