

Abstract
Background
Obesity is a risk factor for the development of asthma. However, pharmacologic therapeutic strategies that specifically target obese asthmatics have not been identified. We hypothesize that glucagon‐like peptide‐1 receptor agonist (GLP‐1RA) treatment inhibits aeroallergen‐induced early innate airway inflammation in a mouse model of asthma in the setting of obesity.
Methods
SWR (lean) and TALLYHO (obese) mice were challenged intranasally with Alternaria alternata extract (Alt‐Ext) or PBS for 4 consecutive days concurrent with GLP‐1RA or vehicle treatment.
Results
TALLYHO mice had greater Alt‐Ext‐induced airway neutrophilia and lung protein expression of IL‐5, IL‐13, CCL11, CXCL1, and CXCL5, in addition to ICAM‐1 expression on lung epithelial cells compared with SWR mice, and all endpoints were reduced by GLP‐1RA treatment. Alt‐Ext significantly increased BALF IL‐33 in both TALLYHO and SWR mice compared to PBS challenge, but there was no difference in the BALF IL‐33 levels between these two strains. However, TALLYHO, but not SWR, mice had significantly higher airway TSLP in BALF following Alt‐Ext challenge compared to PBS, and BALF TSLP was significantly greater in TALLYHO mice compared to SWR mice following airway Alt‐Ext challenge. GLP‐1RA treatment significantly decreased the Alt‐Ext‐induced TSLP and IL‐33 release in TALLYHO mice. While TSLP or ST2 inhibition with a neutralizing antibody decreased airway eosinophils, they did not reduce airway neutrophils in TALLYHO mice.
Conclusions
These results suggest that GLP‐1RA treatment may be a novel pharmacologic therapeutic strategy for obese persons with asthma by inhibiting aeroallergen‐induced neutrophilia, a feature not seen with either TSLP or ST2 inhibition.
Keywords: glucagon‐like peptide‐1 receptor (GLP‐1R), group 2 innate lymphoid cells (ILC2), liraglutide, neutrophilia, obese asthma
Polygenic obese mice have larger level of Altrernaria extract‐induced TSLP, the number of lung ILC2, neutrophils in the airway, and ICAM‐1 on lung epithelial cells compared with lean mice. GLP‐1RA decreases the Altrernaria extract‐induced IL‐33 and TSLP release, type‐2 inflammation mediated by ILC2, eosinophilia and neutrophilia in the airway, and airway responsiveness in polygenic obese mice. Our findings indicate the potential for a new pharmacological therapeutic strategy to patients with asthma in the setting of obesity.
Abbreviations: ICAM‐1, intercellular adhesion molecule 1; ILC2, group 2 innate lymphoid cells; GLP‐1RA, glucagon‐like peptide‐1 agonist
Abbreviations
- Ab
antibody
- Alt‐Ext
Alternaria alternata extract
- AR
airway responsiveness
- BALF
bronchoalveolar lavage fluid
- ECs
epithelial cells
- GLP‐1
glucagon‐like peptide‐1
- GLP‐1RA
glucagon‐like peptide‐1 receptor agonist
- ICAM‐1
intercellular adhesion molecule 1
- ILC2
group 2 innate lymphoid cells
- Lin
lineage
- MFI
mean fluorescence intensity
- MPO
myeloperoxidase
- T2D
type 2 diabetes
- TSLP
thymic stromal lymphopoietin
- VCAM‐1
vascular cell adhesion molecule 1
1. INTRODUCTION
Obesity is a risk factor for numerous health problems, including type 2 diabetes (T2D), heart disease, and some types of cancer. 1 In addition, obesity is associated with an increased risk of asthma and increased asthma severity. 2 , 3 Asthma prevalence was significantly higher in obese adults (11.1%) compared with normal weight adults (7.1%). 4 Likewise, obesity prevalence was higher in individuals with asthma. 5 Further, the prevalence of obesity was higher in patients with difficult‐to‐control than well‐controlled asthma. 6 Some clinical studies reported that obese asthmatics, whether children or adults, were less responsive to corticosteroid treatment. 7 , 8 The percentage of sputum neutrophils or the number of blood neutrophils was elevated in obese patients with asthma compared with nonobese patients with asthma. 9 , 10 Although a cluster analytical approach has been used to determine the contribution of obesity and related factors to asthma phenotypes, 11 , 12 an integrated phenotype has yet to be determined due to the heterogeneity of obese asthma patients. Previous studies revealed that weight loss in obese asthmatic patients, including that which occurs with surgical procedures, improves asthma control. 13 , 14 However, current asthma treatment guidelines do not differentiate pharmacotherapeutic strategies for obese persons with asthma, 15 , 16 and the differences in asthma pathophysiology between the obese and nonobese suggest that this may be needed.
A recent focus for treatment of T2D and/or obesity is glucagon‐like peptide‐1 (GLP‐1) and its receptor (GLP‐1R). GLP‐1 is a peptide hormone synthesized and released by enteroendocrine L‐cells in the ileum and large intestine following nutrient intake. GLP‐1 has a role in glycemic control by inducing glucose‐dependent insulin secretion from β‐cells and inhibiting glucagon release from α‐cells in the pancreas. 17 , 18 Further, GLP‐1 induces weight loss. 19 GLP‐1R agonists (GLP‐1RA), such as liraglutide and semaglutide, are approved by the Food and Drug Administration (FDA) for the treatment of both obesity and T2D. 20 , 21 Several animal studies reported that GLP‐1RA had anti‐inflammatory effects in pulmonary diseases. For instance, liraglutide treatment decreased bleomycin‐induced fibrosis in mice and was associated with a reduction of DNA binding activity of nuclear‐factor kappa B in lung tissue. 22 Further, liraglutide treatment decreased ovalbumin (OVA) sensitization and challenge‐induced allergic pulmonary inflammation mediated by adaptive immune responses in lean mice. 23 In addition, our group reported that treatment with the GLP‐1RA liraglutide significantly decreased aeroallergen‐induced innate allergic inflammation associated with the reduction of IL‐33 release and activation of lung group 2 innate lymphoid cells (ILC2) in BALB/c lean mice. 24 Obesity in persons with asthma is associated with increased sputum neutrophilia, a marker of poor response to increased inhaled corticosteroids compared to lean persons with asthma. 25 Murine models of obesity support that IL‐33 mediates airway neutrophilia, 26 but to the best of our knowledge, the effect of IL‐33 or signaling through its receptor, ST2, has not been examined in allergen challenge models of obesity. Therefore, we hypothesized that treatment with the GLP‐1RA liraglutide decreases aeroallergen‐induced innate allergic inflammation in a mouse model of obesity, as well as in lean mice. To test this hypothesis, we used a polygenic mouse model of obesity characterized by hyperglycemia, hyperinsulinemia, and hyperlipidemia (TALLYHO mice), and the corresponding lean genetic control (SWR mice). We chose to use an innate model of allergen‐induced inflammation to specifically investigate the effect of GLP‐1RA signaling on IL‐33‐ and Thymic stromal lymphopoietin (TSLP)‐induced airway inflammatory responses in the setting of obesity. GLP‐1RA treatment, but neither anti‐TSLP nor anti‐ST2 therapy, inhibits aeroallergen‐driven innate immune system‐induced neutrophilia in obese mice and suggests that this strategy may be a novel therapeutic approach for the treatment of asthma in obese persons. This report is the first to determine the impact of GLP‐1RA treatment on aeroallergen‐induced innate immune responses in the setting of obesity.
2. METHODS
2.1. Mice
Nine‐ to twelve‐week old female TALLYHO/JngJ (TALLYHO) mice and SWR/J (SWR) mice were obtained from Jackson Laboratories (Bar Harbor, ME). Animal experiments were approved by the Institutional Animal Care and Use Committee at Vanderbilt University and were conducted according to the guidelines for the Care and Use of Laboratory Animals prepared by the Institute of Laboratory Animal Resources, National Research Council.
2.2. GLP‐1RA treatment and Alt‐Ext‐challenge in mouse model
The GLP‐1RA, liraglutide (Novo Nordisk Inc., Bagsvaerd, Denmark) or an equivalent volume of vehicle (0.1% bovine serum albumin (BSA)/phosphate‐buffered saline (PBS)) were administered subcutaneously twice per day from day −2 to day 0, or to day 3. The dose of GLP‐1RA was 0.05 mg/kg on day −2, 0.1 mg/kg on day −1, and 0.2 mg/kg on days 0–3 or an equivalent volume of vehicle (0.1% BSA/PBS). From day 0 to day 3, either 5 µg (protein amount) of Alternaria alternata extract (Alt‐Ext) (Stallergenes Greer, Lenoir, NC) in 80 µl of PBS or 80 µl of PBS as vehicle were administered intranasally to mice anesthetized with ketamine/xylazine. In the first protocol, the mice were euthanized and bronchoalveolar lavage fluid (BALF) was harvested 1 h or 12 h after the first challenge of Alt‐Ext or PBS for the detection of IL‐33 or TSLP, respectively. In the second protocol, whole lungs and the BALF were harvested 12 h after the last Alt‐Ext‐or PBS‐challenge (day 4) to evaluate cell differentials, as well as lung cytokine and chemokine expression, 24 h after the last challenge to enumerate lung ILC2 and ILC3 by flow cytometry, or 48 h after the last challenge to evaluate airway responsiveness (AR) and mucus.
2.3. Statistical analysis
All data were analyzed with GraphPad Prism 8 (GraphPad Software, La Jolla, CA). Statistical significance was assessed by one‐way analysis of variance (ANOVA) with Bonferroni‐multiple pairs comparisons test, or Mann‐Whitney test for two group comparison. Values of P < .05 were considered significant between two groups.
Additional detail on the methods is available in an Appendix S1.
3. RESULTS
3.1. Obese phenotype in TALLYHO mice
TALLYHO mice were 1.7‐fold heavier than SWR mice, and this difference was statistically significant (Figure 1A,B). Serum glucose levels were significantly increased in TALLYHO mice compared with SWR mice, both at baseline and after having received glucose, revealing obesity‐associated defects in glucose homeostasis (Figure 1C). Another obesity biomarker, the serum leptin level, was significantly higher in TALLYHO mice than SWR mice (Figure 1D). These results indicate that the TALLYHO mouse model replicates the key metabolic components of obesity and metabolic syndrome.
FIGURE 1.
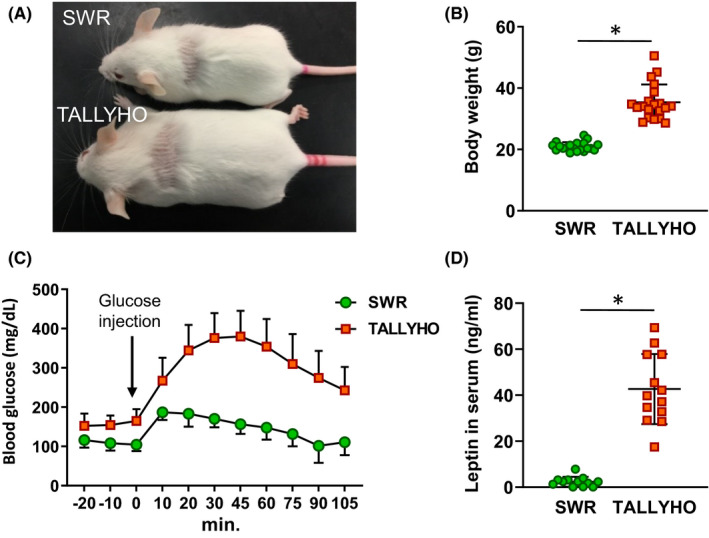
Physiological differences between SWR and TALLYHO mice. A, Representative picture of SWR and TALLYHO mice. B, Difference in body weight between SWR and TALLYHO mice. Age matched mice were used (n = 20). C, Comparison of glucose tolerance in SWR and TALLYHO mice. Blood glucose levels were measured before and after an equivalent amount of glucose that was administered by intraperitoneal injection (n = 6). D, Leptin concentrations in serum (n = 12). The values are the mean ± SD. *P < .05
3.2. GLP‐1RA treatment suppresses Alt‐Ext‐induced IL‐33 and TSLP in BALF from TALLYHO mice
IL‐33 and TSLP are key cytokines that induce activation of group 2 innate lymphoid cells (ILC2), as well as adaptive immune cells, and are critical contributors to allergic airway inflammation. 27 Thus, we determined the time course of IL‐33 and TSLP appearance in the BALF after a single Alt‐Ext‐challenge. IL‐33 protein peaked 1 h after the Alt‐Ext‐challenge in both SWR and TALLYHO mice (Figure S1). TSLP protein peaked 6 h after Alt‐Ext‐challenge in both SWR and TALLYHO mice, but TSLP release remained at near peak levels at 12 h only in TALLYHO mice (Figure S1). Therefore, we measured IL‐33 1 h after allergen challenge and TSLP 12 h after allergen challenge to determine the effect of GLP‐1RA treatment on IL‐33 and TSLP protein release (Figure 2A). Alt‐Ext‐challenge significantly increased IL‐33 in BALF in both SWR and TALLYHO mice compared with naïve mice in these strains (Figure 2B). GLP‐1RA treatment significantly decreased Alt‐Ext‐induced IL‐33 in both SWR and TALLYHO mice compared with vehicle treatment (Figure 2B). Alt‐Ext‐challenge significantly increased TSLP in the BALF of TALLYHO mice, but not in SWR mice (Figure 2C). GLP‐1RA treatment significantly decreased the Alt‐Ext‐induced TSLP in TALLYHO mice compared with the vehicle‐treated mice, while there was no effect of GLP‐1RA treatment on TSLP in SWR mice (Figure 2C). Taken together, these results indicate that GLP‐1RA treatment suppressed acute IL‐33 and TSLP release after a single Alt‐Ext‐challenge in TALLYHO mice.
FIGURE 2.
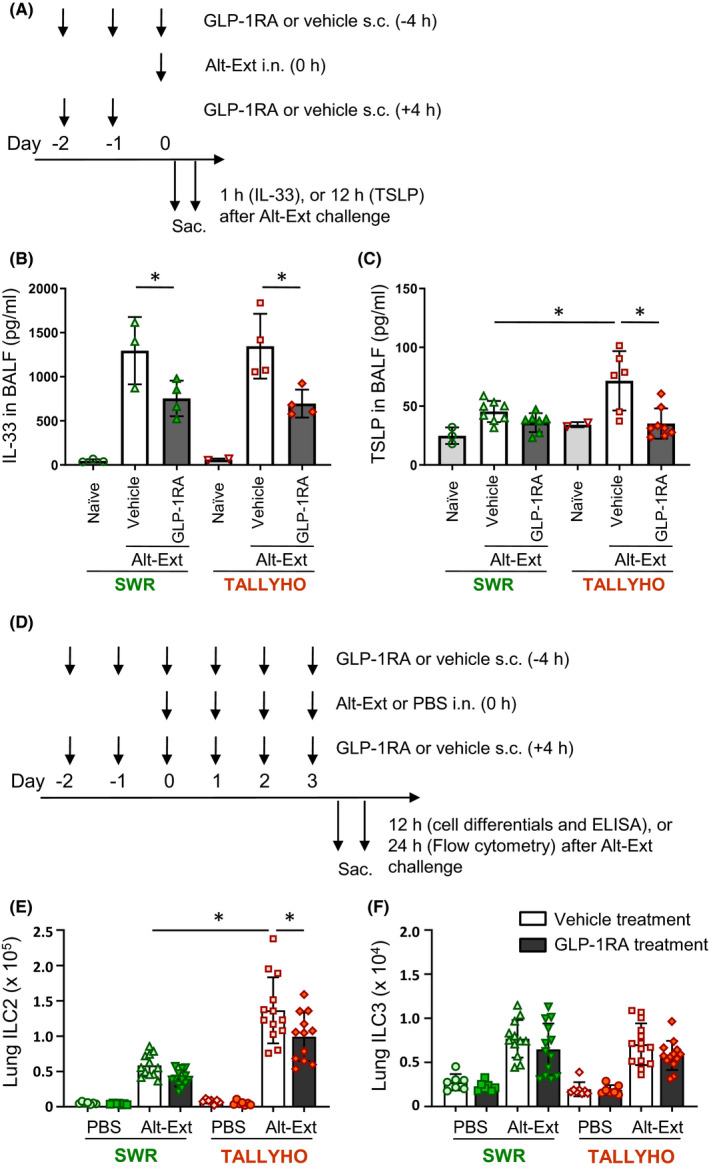
Experimental protocol. A, GLP‐1RA or its vehicle was administered subcutaneously on day −2 and −1, and then 4 h before Alt‐Ext‐challenge on day 0. The dose of GLP‐1RA was 0.05 mg/kg on day −2, 0.1 mg/kg on day −1, and 0.2 mg/kg on day 0. The BALF was harvested 1 h or 12 h after the Alt‐Ext‐challenge on day 0 to measure IL‐33 or TSLP protein, respectively. B, The protein level of IL‐33 in the BALF 1 h after Alt‐Ext challenge (n = 3–4). C, The protein level of TSLP in the BALF 12 h after Alt‐Ext challenge (n = 3–8). D, SWR and TALLYHO mice were challenged with Alt‐Ext or PBS intranasally from day 0 to day 3. GLP‐1RA or its vehicle was administered subcutaneously on day −2 and −1, and then every 4 h before and after Alt‐Ext‐challenge on day 0–3. The dose of GLP‐1RA was 0.05 mg/kg on day −2, 0.1 mg/kg on day −1, and 0.2 mg/kg on day 0–3. The mice were sacrificed 12 h, or 24 h after the last Alt‐Ext‐ or PBS‐challenge to evaluate cell differentials and cytokine/chemokine expression by ELISA, and lung ILC2/ILC3 or ICAM‐1/VCAM‐1 by flow cytometry, respectively. E,F, Flow cytometric analysis of the number of lung ILC2 and lung ILC3 (n = 7–13). The results shown are combined from 4 independent experiments. The values are the mean ± SD. *P < .05
3.3. GLP1‐RA treatment suppresses the number of mouse lung ILC2, but not ILC3 in response to airway Alt‐Ext‐challenge
We previously reported that GLP‐1RA treatment significantly decreased ILC2 activation that was associated with a reduction of Alt‐Ext‐induced IL‐33 release in lean BALB/c mice. 24 In addition, a previous study showed that ILC3 expressing IL‐17 was enhanced in high‐fat diet‐induced obese mice. 28 , 29 Therefore, we counted the number of total lung ILC2 and ILC3 in SWR and TALLYHO mice after 4 consecutive days of Alt‐Ext‐challenge with or without GLP‐1RA treatment. The lungs for flow cytometry were harvested 24 h after the last Alt‐Ext‐challenge (Figure 2D). The gating strategy of lung ILC2 and ILC3 is shown in Figure S2. ILC2 were identified as lineage (lin)− CD3− CD4− CD45+ ICOS+ ST2+ GATA3+ cells, and ILC3 were identified as lin− CD3− CD4− CD45+ ICOS+ ST2− GATA3−RORγt+ cells. Alt‐Ext‐challenge significantly increased the number of lung ILC2 compared with PBS‐challenge in both SWR and TALLYHO mice. The number of Alt‐Ext‐induced lung ILC2 was higher in TALLYHO mice than SWR mice (Figure 2E). GLP‐1RA treatment significantly decreased the number of Alt‐Ext‐induced lung ILC2 in TALLYHO mice compared with vehicle treatment (Figure 2E). Although the number of lung ILC3 was increased by Alt‐Ext‐challenge in both SWR and TALLYHO mice compared with PBS‐challenged groups, there was no difference in the number of Alt‐Ext‐induced ILC3 between SWR and TALLYHO mice. GLP‐1RA treatment did not decrease Alt‐Ext‐induced lung ILC3 (Figure 2F). Taken together, TALLYHO mice had a higher number of Alt‐Ext‐induced ILC2 compared with SWR mice, and GLP1‐RA treatment had an inhibitory effect on lung ILC2 proliferation in TALLYHO mice.
3.4. GLP1‐RA treatment suppresses Alt‐Ext‐induced cytokine and chemokine expression in the lung
Next, we measured the protein levels of inflammatory cytokines and chemokines in lung homogenates. Our preliminary time‐course test indicated that the BALF expression of the proinflammatory chemokine KC (CXCL1) peaked at 3 h and was no longer elevated 24 h after a single Alt‐Ext‐challenge in TALLYHO mice compared with SWR mice (Figure S1); however, IL‐5 peaked 24 h after Alt‐Ext‐challenge (Figure S1). Thus, an optimal timepoint to detect various cytokines and chemokines in the BALF and lungs was 12 h after the last Alt‐Ext‐challenge. Alt‐Ext‐challenge significantly increased the protein expression of lung IL‐1β, IL‐5, IL‐13, IL‐17, eotaxin (CCL11), eotaxin‐2 (CCL24), KC, and LIX (CXCL5) in both SWR and TALLYHO mice compared with PBS‐challenged groups (Figure 3). Alt‐Ext‐induced lung protein levels of IL‐5, IL‐13, eotaxin, KC, and LIX were higher in TALLYHO mice compared with SWR mice. In contrast, lung IL‐17 and eotaxin‐2 were higher in SWR mice compared with TALLYHO mice. Although lung IL‐17 in TALLYHO mice was not decreased by GLP1‐RA treatment, we found a statistically significant decrease in the other Alt‐Ext‐induced cytokines and chemokines in both SWR and TALLYHO mice treated with GLP1‐RA compared with vehicle‐treated mice (Figure 3). Thus, obese TALLYHO mice highly expressed neutrophil chemoattractants such as KC and LIX, as well as type 2 cytokines such as IL‐5 and IL‐13. GLP1‐RA treatment decreased almost all Alt‐Ext‐induced proinflammatory cytokines and chemokines in both obese and lean mice.
FIGURE 3.
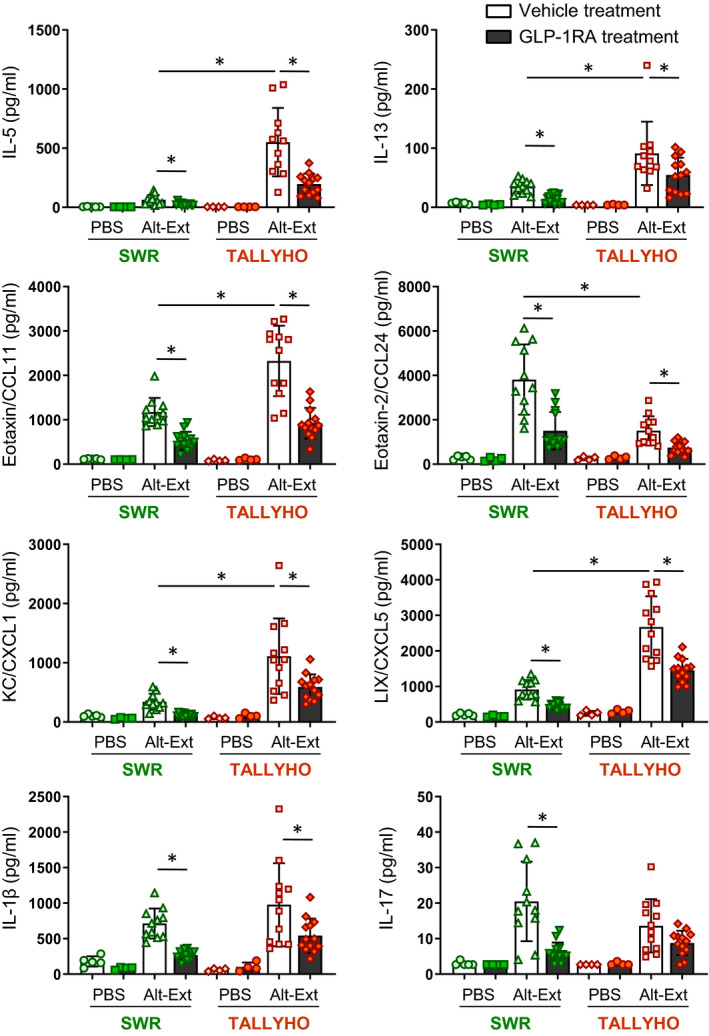
Effects of GLP1‐RA treatment on Alt‐Ext‐induced expressions of cytokines and chemokines in the lung. Lung homogenates were prepared to measure the protein expression of IL‐1β, IL‐5, IL‐13, IL‐17, eotaxin (CCL11), eotaxin‐2 (CCL24), KC (CXCL1), and LIX (CXCL5) by ELISA (n = 4–13). The results are combined with 3 independent experiments, and the all results and shown as mean ± SD. Veh = vehicle. PBS = phosphate‐buffered saline. *P < .05
3.5. GLP1‐RA treatment suppresses neutrophil and eosinophil recruitment in response to airway Alt‐Ext‐challenge
To determine the effect of obesity and GLP1‐RA treatment on the airway inflammatory phenotype, we enumerated the cell populations in BALF after 4 consecutive days of Alt‐Ext‐challenge with or without GLP1‐RA treatment. Alt‐Ext‐challenge significantly increased the number of eosinophils, lymphocytes, and neutrophils compared with PBS‐challenged groups in both SWR and TALLYHO mice (Figure 4A‐C). In contrast, the number of macrophages was significantly increased in SWR mice, but not in TALLYHO mice (Figure 4D). While the number of Alt‐Ext‐induced neutrophils and lymphocytes was significantly higher in TALLYHO mice than SWR mice, there was no difference in the number of eosinophils between SWR and TALLYHO mice (Figure 4B,C). GLP1‐RA treatment significantly decreased the number of Alt‐Ext‐induced eosinophils and neutrophils in both SWR and TALLYHO mice, and it also diminished the number of Alt‐Ext‐induced lymphocytes in TALLYHO mice (Figure 4A,B). To determine activation of the accumulated neutrophils in the lung, we measured myeloperoxidase (MPO) in BALF. The concentration of MPO was significantly increased by Alt‐Ext‐challenge, and the level of MPO was higher in TALLYHO mice than SWR mice (Figure 4E). GLP1‐RA treatment significantly decreased the Alt‐Ext‐induced MPO concentration in both SWR and TALLYHO mice (Figure 4E). Therefore, TALLYHO mice had neutrophil‐dominant inflammation compared with SWR mice, and GLP1‐RA treatment had an anti‐inflammatory effect for both eosinophilia and neutrophilia in Alt‐Ext‐induced airway inflammation. In addition, we measured total IgE and IgG in sera to evaluate immunoglobin‐mediated allergic responses. The IgE and IgG were higher in SWR mice compared to TALLYHO mice; however, there were no differences in serum IgE and IgG between PBS and Alt‐Ext‐challenge with or without GLP‐1RA treatment (Figure S3).
FIGURE 4.
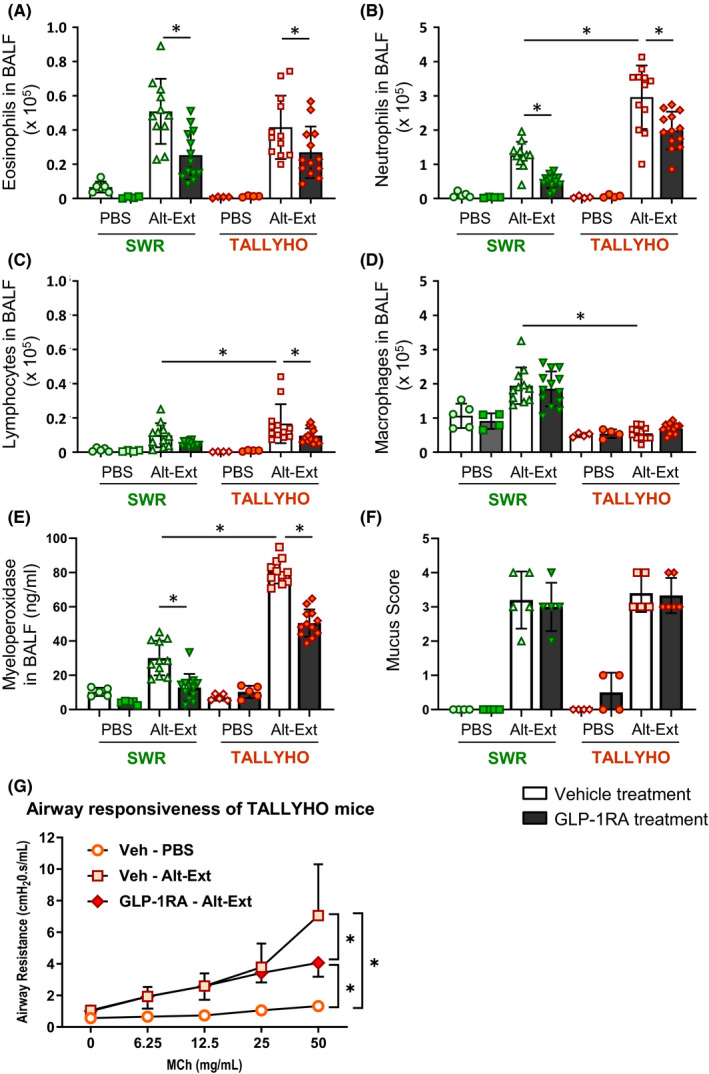
Effects of GLP1‐RA treatment on Alt‐Ext‐induced immune cell accumulation in the airway. A‐D, BALF were harvested 12 h after the last Alt‐Ext‐ or PBS‐challenge to evaluate cell differentials, macrophages, neutrophils, eosinophils, and lymphocytes in BALF (n = 4–13). E, The protein of myeloperoxidase in BALF was measured by ELISA. The results are combined with 3 independent experiments. F, Lungs were harvested 48 h after the last Alt‐Ext‐ or PBS‐challenge to evaluate mucus score by PAS staining (n = 4–5). G, Airway resistance to increasing dose of methacholine challenge was tested 48 h after the last Alt‐Ext‐ or PBS‐challenge in TALLYHO mice (n = 3–4). The all results and shown as mean ± SD. Veh = vehicle. PBS = phosphate‐buffered saline. *P < .05
3.6. GLP1‐RA treatment decreases Alt‐Ext‐induced airway responsiveness, but not mucus production
We found that GLP‐1RA treatment decreased the protein level of type2 cytokines, including IL‐13 in the lung. Since IL‐13 is a central mediator of airway responsiveness and mucus, we tested whether GLP‐1RA treatment decreases mucus in the airway following Alt‐Ext‐challenge. PBS challenge did not induce airway mucus. Alt‐Ext‐challenge increased airway mucus in both SWR and TALLYHO mice. However, there was no difference in the mucus score between SWR and TALLYHO mice. GLP‐1RA treatment did not decrease the Alt‐Ext‐induced mucus compared with vehicle treatment (Figure 4F).
Further, we tested whether GLP‐1RA treatment decreased AR in Alt‐Ext‐challenged TALLYHO mice. Alt‐Ext‐challenge significantly increased methacholine‐induced AR compared with PBS challenge. GLP‐1RA treatment significantly decreased the Alt‐Ext‐induced AR compared with vehicle treatment (Figure 4G).
3.7. GLP1‐RA treatment suppresses Alt‐Ext‐induced ICAM‐1, but not VCAM‐1 expression in the lung endothelial and epithelial cells
Our findings revealed that Alt‐Ext‐induced neutrophil chemokines and the number of neutrophils were significantly decreased by GLP‐1RA treatment. Further, we tested whether GLP‐1RA treatment decreases the cell adhesion molecules ICAM‐1 (Intercellular Adhesion Molecule 1) and VCAM‐1 (Vascular Cell Adhesion Molecule 1) on lung endothelial cells and epithelial cells. The gating strategies of lung endothelial cells and epithelial cells are shown in Figure S4. The lung endothelial cells were identified as CD45− CD31+ CD146+ EpCAM− cells, and the lung epithelial cells were identified as CD45− CD146− EpCAM+ cells.
Histograms of ICAM‐1 and VCAM‐1 expression on those cells are shown in Figure S4. The MFIs (Mean Fluorescence Intensity) of ICAM‐1 and VCAM‐1 were calculated in the range of positive expression in the histogram (Figure S5). Alt‐Ext‐challenge significantly increased the MFI of ICAM‐1 and VCAM‐1 compared with PBS‐challenge on both endothelial and epithelial cells (Figure 5). The MFI of ICAM‐1 on lung epithelial cells was higher in TALLYHO mice than SWR mice (Figure 5). GLP‐1RA treatment significantly decreased the Alt‐Ext‐induced ICAM‐1, but not VCAM‐1, on lung endothelial and epithelial cells (Figure 5). Taken together, epithelial cells from TALLYHO mice had higher MFI of ICAM‐1 expression compared with SWR mice, and GLP1‐RA treatment significantly reduced Alt‐Ext‐induced ICAM‐1 expression on lung endothelial and epithelial cells.
FIGURE 5.
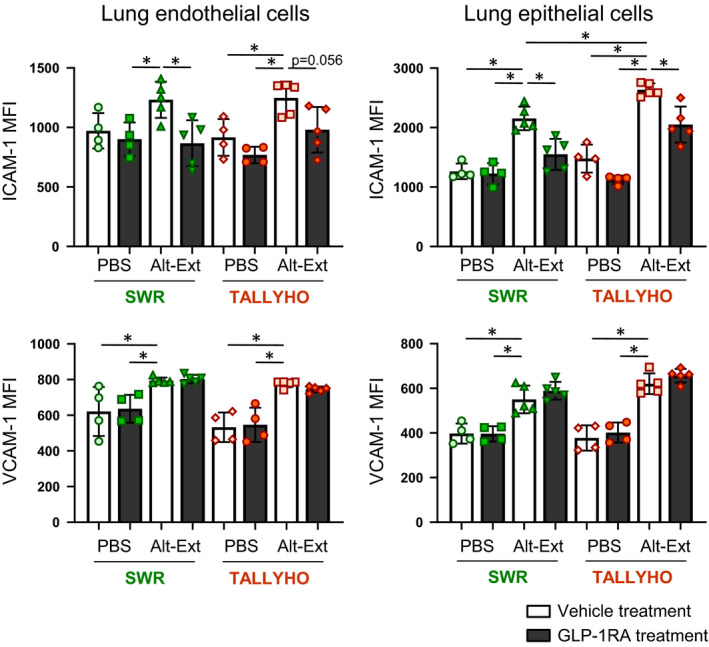
Effects of GLP1‐RA treatment on Alt‐Ext‐induced expressions of ICAM‐1(Intercellular Adhesion Molecule 1) and VCAM‐1(Vascular Cells Adhesion Molecule 1) in the lung endothelial and epithelial cells. The MFI (Mean Fluorescence Intensity) was calculated in positive expression area of ICAM‐1 and VCAM‐1 compared to respective isotype control staining. (n = 4–5). The results shown are combined from 2 independent experiments. The values are the mean ± SD. *P < .05
3.8. Neutralization of endogenous TSLP or ST2 did not reduce Alt‐Ext‐induced airway neutrophils in obese mice
We found that TSLP release after Alt‐Ext‐challenge was significantly greater in the BALF of TALLYHO mice than SWR mice (Figure 2C). To determine whether Alt‐Ext‐induced TSLP leads to the increased neutrophilia in TALLYHO mice, we performed anti‐TSLP antibody (ab) treatment prior to Alt‐Ext‐challenge. TALLYHO mice were treated with anti‐TSLP ab or isotype ab 1 h and 24 h prior to the first Alt‐Ext‐challenge to neutralize the Alt‐Ext‐induced endogenous TSLP. Twelve hours after the 4th Alt‐Ext‐challenge, BALF and lungs were harvested (Figure 6A). Anti‐TSLP ab treatment significantly decreased the number of total BALF cells, macrophages, eosinophils, and lymphocytes, but not neutrophils compared with isotype ab treatment (Figure 6B). Further, anti‐TSLP ab treatment significantly decreased Alt‐Ext‐induced levels of IL‐5, IL‐13, eotaxin, and eotaxin‐2, but not KC and LIX in lung homogenates compared with isotype ab treatment (Figure 6C‐H).
FIGURE 6.
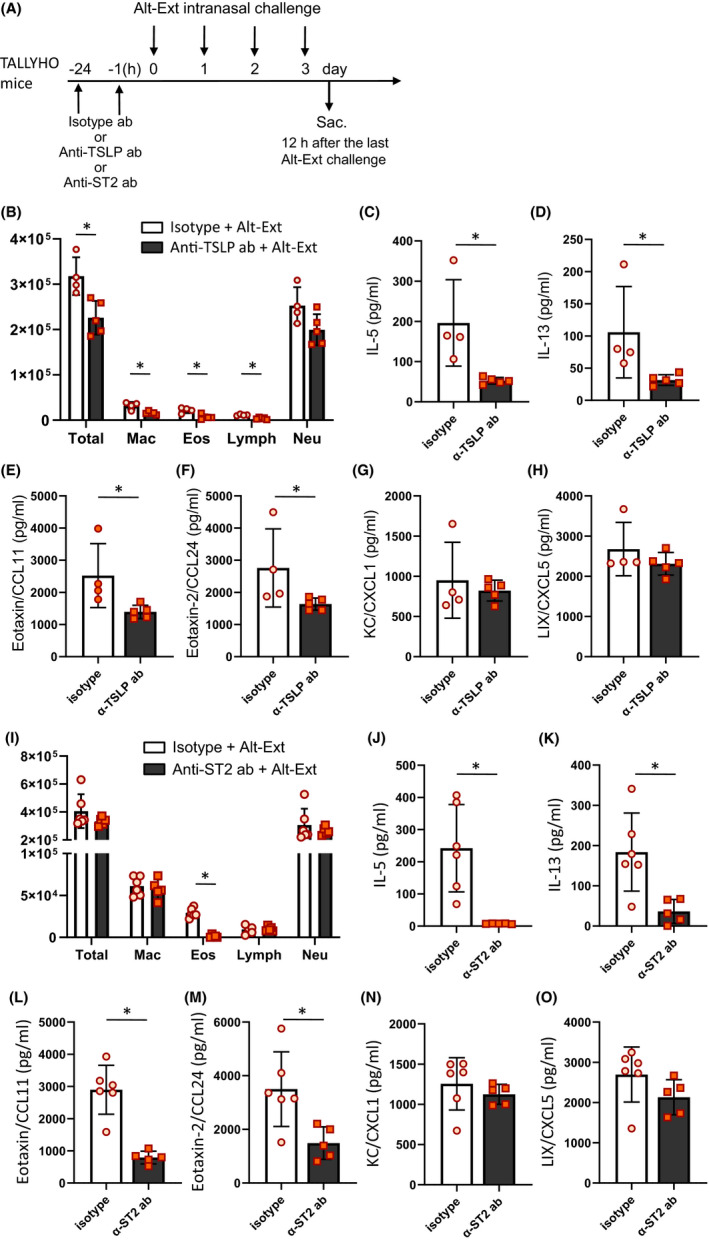
Endogenous TSLP or ST2 neutralization decreased Alt‐Ext‐induced type2 airway inflammation, but not neutrophilia. A, Anti‐TSLP antibody (200 μg/mouse), anti‐ST2 antibody (10 mg/kg), or the each isotype antibody was administered intraperitoneally 1 h and 24 h prior to first Alt‐Ext‐challenge. BALF and lung were harvested 12 h after the 4th Alt‐Ext‐challenge. B,I, Cell differentials in BALF. C‐H,J‐O, The protein level of IL‐5, IL‐13, eotaxin (CCL11), eotaxin‐2 (CCL24), KC (CXCL1), and LIX (CXCL5) in the lung homogenates. The data are shown as mean ± SD. (n = 4–6). *P < .05
In addition, we tested whether inhibition of ST2 signaling decreased airway neutrophilia in TALLYHO mice challenged with Alt‐Ext. The dose of anti‐ST2 antibody was the same as described a previous study. 26 BALF and lungs were harvested 12 h after 4th Alt‐Ext‐challenge (Figure 6A). Anti‐ST2 ab treatment significantly decreased the number of eosinophils, but not macrophages, lymphocytes, and neutrophils compared with isotype ab treatment (Figure 6I). Further, anti‐ST2 ab treatment significantly decreased Alt‐Ext‐induced levels of IL‐5, IL‐13, eotaxin, and eotaxin‐2, but not KC and LIX in lung homogenates compared with isotype ab treatment (Figure 6J‐O).
Thus, neutralization of endogenous TSLP or ST2 suppressed Alt‐Ext‐induced innate type 2 inflammation, but not neutrophilic inflammation in this obese animal model and these results strongly suggest that TSLP and IL‐33 are not responsible directly for Alt‐Ext‐induced innate neutrophilic inflammation.
4. DISCUSSION
We sought to determine whether obesity drives differences in Alt‐Ext‐induced allergic phenotypes and whether these are modified by treatment with a GLP‐1RA. We specifically chose a model of allergic airway inflammation driven by the alarmins IL‐33 and TSLP, as the contribution of this pathway has not been fully defined. Thus, we tested whether GLP1‐RA treatment decreases Alt‐Ext‐induced lung inflammation in the TALLYHO mouse obesity model, relative to the lean SWR control. The obese mice had significantly greater numbers of lung ILC2 and neutrophilia during Alt‐Ext‐induced innate allergic inflammation than lean mice. This confirms findings of other investigators who have shown that there is increased airway neutrophilia in mouse models of allergic airway inflammation in the setting of obesity compared to lean mice. 26 , 30 , 31 , 32 , 33 GLP1‐RA treatment had an inhibitory effect on the aeroallergen‐induced IL‐33 and TSLP release into the airway; lung ILC2 numbers; lung eotaxin, IL‐5 and IL‐13; AR to methacholine inhalation; and ICAM‐1 expression on lung endothelial and epithelial cells in TALLYHO mice. Importantly, and unexpectedly, GLP‐1RA treatment had an inhibitory effect on allergen‐induced neutrophilia and the production of the neutrophil chemoattractants, KC and LIX, differentiating the effect of GLP‐1RA from that of TSLP or ST2 inhibition.
So far, several different types of mouse models have been used for research studies of obesity in airway inflammation. 34 , 35 For instance, monogenic obese mice, ob/ob or db/db, have a deficiency of leptin or leptin receptor, respectively. 36 However, leptin, a hormone released from adipocytes, has multiple effects on immune responses, as well as on glycemic control. 37 , 38 , 39 , 40 Cpefat mice have a spontaneous mutation at the locus of carboxypeptidase E, but carboxypeptidase E is expressed in various tissues or cells including mast cells. 41 These monogenetic depletions may directly alter immune responses. The genetic predisposition for typical human obesity is polygenic. A strength of our study is the use of the TALLYHO polygenic obese mouse that replicates components of the metabolic syndrome. This mouse strain develops obesity and hyperinsulinemia at 6–8 weeks of age by regular chow diet in both male and female, 42 and we have confirmed the presence of these metabolic disorders. Since TALLYHO mice that were fed regular chow diet had an initial high weight gain within a few weeks of weaning, 42 , 43 this mouse strain was ideal to investigate allergen responsiveness to mimic asthma patients with obesity. To the best of our knowledge, this is the first report the use of TALLYHO mice in an airway allergen challenge model.
Alt‐Ext‐challenged TALLYHO mouse had significantly greater TSLP release in BALF compared with SWR mice. This higher level of TSLP may be one factor that enhances lung ILC2 activation with IL‐5 and IL‐13 production in TALLYHO mice compared with SWR mice, because TSLP signaling acts synergistically with IL‐33 to activate ILC2. 43 , 44 GLP‐1RA treatment of TALLYHO mice significantly decreased IL‐33 and TSLP release after the first Alt‐Ext challenge; the number of lung ILC2; and the production of IL‐5 and IL‐13 after 4 consecutive days of Alt‐Ext challenge in TALLYHO mice. These results are consistent with our previous report that GLP‐1RA treatment significantly decreased Alt‐Ext‐induced lung ILC2 expressing IL‐5 and IL‐13 mediated by reducing of IL‐33 release in lean mice. Interestingly, the number of eosinophils was not different between obese TALLYHO and lean controls in the current study. Eotaxin and eotaxin‐2, as well as IL‐5, are chemoattractants of eosinophils. 45 , 46 The expression level of eotaxin was higher in SWR than in TALLYHO mice. Conversely, eotaxin‐2 was higher in TALLYHO than in SWR mice. Therefore, the net effect of the changes in these chemokines seems to have been to increase eosinophils in TALLYHO and SWR mice to the same level.
In contrast, TALLYHO mice showed higher levels of airway neutrophils and expression of neutrophil chemoattractants, KC and LIX, compared with SWR mice after 4 consecutive days of Alt‐Ext challenge. These results are consistent with the human late onset, non‐type 2 obese asthma phenotype. 10 , 11 , 25 Previous studies using diet‐induced obese (DIO) mouse models reported that high‐fat diet fed mice had a greater number of lung ILC3 expressing IL‐17 compared with chow or low‐fat diet fed mice. 28 , 29 However, we determined that there was no difference in the number of ROR‐γt+ ILC3 cells between SWR and TALLYHO mice, and IL‐17 was not detected in PBS‐challenged mice. Although there was no difference in Alt‐Ext induced IL‐1β and IL‐17 in lungs between SWR and TALLYHO mice, Alt‐Ext‐induced lung KC and LIX were increased in TALLYHO mice to a greater extent than SWR mice. These results suggest that ILC3 expressing IL‐17 is not mechanistically involved in this model of aeroallergen‐induced acute neutrophilia in obesity.
There were no differences in the total IgE and IgG between PBS and Alt‐Ext challenge in either the SWR mice or the TALLYHO mice. Our previous study reported that 4 consecutive days of Alt‐Ext‐challenge induced type 2 inflammation paralleled by increases in lung ILC2, but not by either CD4 T cell influx or IgE induction in lean BALB/c mice. 47 Identical to our previous result, SWR and TALLYHO mice had no increase of total IgE and IgG after 4 consecutive days of Alt‐Ext‐challenge, revealing that this is a model of innate, and not adaptive immunity to aeroallergen challenge. While there was no difference in Alt‐Ext‐induced airway mucus between vehicle and GLP‐1RA treatment, AR was significantly decreased by GLP‐1RA treatment in TALLYHO mice. These results suggest that GLP‐1RA treatment reduces AR with neutrophil‐dominant inflammation in the setting of obesity independent of airway remodeling by mucus.
Furthermore, the obesity‐associated increased neutrophilia was not caused by an increase in airway TSLP level because anti‐TSLP ab treatment had no effect on KC and LIX in the lung, and airway neutrophilia in TALLYHO mice, whereas it did reduce IL‐5 and IL‐13, in addition to airway eosinophilia. The reduction in type 2 inflammation markers in the early innate model of allergen‐induced inflammation is consistent with our previous report using lean BALB/c mice. 44 To the best of our knowledge, TSLP antagonism has not been specifically investigated in either obese patients with asthma or in an animal model of allergic airway inflammation. Tezepelumab, an anti‐TSLP ab treatment, decreased allergen‐induced late phase reactions in subjects with allergic asthma 48 and decreased clinically significant asthma exacerbations in the patients with uncontrolled asthma. 49 However, the effect of anti‐TSLP ab treatment in the subset of obese subjects with asthma was not reported in either of these studies. Our results suggest that TSLP may not be responsible for the increase in innate neutrophilic inflammation that preferentially occurs in obese subjects with asthma, and by extension that TSLP targeted therapies might lack efficacy in obesity‐associated asthma. However, a clinical trial antagonizing TSLP in obese persons with asthma would have to be performed to determine whether this hypothesis is correct in human disease. Regardless, our study is the first to examine the role of TSLP antagonism in the obese mouse model of allergen‐induced innate immune responses.
Others have reported that subcutaneous injection of GLP‐1RA decreased intratracheal instillation of LPS‐induced lung injury score; the number of neutrophils and macrophages in the BALF; and mRNA expression of KC, MIP‐2 (CXCL2), and IL‐6. 50 In addition, liraglutide inhibited the TNF‐α‐induced ICAM‐1 on human endothelial cell line in vitro 51 and mouse endothelial cells in vivo. 52 These reports are consistent with our results that GLP‐1RA treatment decreased acute Alt‐Ext‐induced KC, LIX, ICAM‐1, and consequent neutrophil accumulation into the airway.
Previous studies reported that IL‐33 drove airway neutrophilic responses to ozone in obese mice. 26 , 53 In contrast, our results showed that anti‐ST2 ab treatment did not change airway neutrophilic responses to Alt‐Ext challenge, but significantly decreased eosinophilic responses including IL‐5, IL‐13, CCL11, and CCL24 expressions. However, this previous study reported that anti‐ST2 ab treatment or ST2 deficiency increased ozone‐induced neutrophil number in the BALF. 54 Thus, the effect of ST2 neutralization on pulmonary neutrophilia is dependent on the experimental design. Further studies are needed to fully define the effects of ST2 signaling on neutrophilic inflammation.
In data not shown, we attempted in vitro studies to define the mechanism by which GLP‐1RA regulates allergen‐induced proinflammatory cytokine and chemokine production in airway epithelial cells, since these cells are a major source of IL‐33 in the Alt‐Ext model. 24 We were deterred by the fact that there are no cell lines that express both GLP‐1 and IL‐33. GLP‐1R expression, in particularly, is notoriously difficult to detect at “functional” levels in cell lines for unclear reasons. This prevented us from dissecting signaling mechanisms by which GLP‐1R activation inhibits either IL‐33 production or release. The primary cell line currently used for in vitro studies of IL‐33 is HBE‐33 cells, and it was genetically engineered to express IL‐33 55 ; however, it does not express GLP‐1R. We also attempted to use primary human airway epithelial cells to study the interaction of GLP‐1RA signaling and IL‐33 release; however, the dose of Alt‐Ext needed to induce measurable IL‐33 release was toxic to the cells, preventing detailed molecular pharmacology studies. Regardless, our in vivo results clearly show that GLP‐1RA signaling inhibits allergen‐induced IL‐33 release in the airway in the setting of obesity, a finding that is likely highly clinically relevant.
Currently, non‐pharmacologic therapies such as dietary interventions and exercise are recommended for obese asthma patients as a result of the multiple immune phenotypes that obese asthma patients exhibit and because these patients have reduced therapeutic responses to standard asthma medications. 7 , 10 , 29 , 56 , 57 Our findings suggest that GLP‐1RA may fill an important unmet medical need and that this therapy will benefit different endotypes of obese asthma, including type 2 and non‐type 2 endotypes. The GLP‐1RA used here, liraglutide, is FDA‐approved for weight loss in settings of euglycemic obesity and T2D. 58 Our findings indicate the potential for a new therapeutic strategy in patients with asthma in the setting of obesity, by which GLP‐1RA treatment inhibits aeroallergen‐induced eosinophilic and neutrophilic airway inflammation.
CONFLICT OF INTEREST
The authors have no financial conflicts of interest.
AUTHOR CONTRIBUTIONS
ST designed and performed experiments, analyzed data, and wrote the manuscript. KDN and RSP designed and supervised this study and edited the manuscript. KLB scored the histopathology and provided the photomicrographs. RLP, DCN, and KNC provided scientific input into the study design, the interpretation, or the results, and edited the manuscript.
Supporting information
Fig S1‐S5
Appendix S1
ACKNOWLEDGMENTS
We thank Dr. Helen Bettencourt (Genentech, Inc. South San Francisco, CA) for the gift of anti‐ST2 antibody. This study was supported by Vanderbilt American Heart Association Strategically Focused Research Network Center award; Department of Veterans Affairs 2I01BX000624; and NIH AI 095227‐02, AI 111820, AI124456, AI 145265, AI 145397, and HL 090664‐04.
Kevin D. Niswender and R. Stokes Peebles contributed equally to this work.
Contributor Information
Shinji Toki, Email: shinji.toki@vumc.org.
Kevin D. Niswender, Email: kevin.niswender@vumc.org.
REFERENCES
- 1. Pi‐Sunyer X. The medical risks of obesity. Postgrad Med. 2015;121:21‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Peters U, Dixon AE, Forno E. Obesity and asthma. J Allergy Clin Immunol. 2018;141:1169‐1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Marko M, Pawliczak R. Obesity and asthma: risk, control and treatment. Postepy Dermatol Alergol. 2018;35:563‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Akinbami LJ, Fryar CD. Asthma Prevalence by Weight Status Among Adults: United States, 2001–2014. NCHS data brief, no 239. Hyattsville, MD: National Center for Health Statistics; 2016. https://www.cdc.gov/nchs/products/databriefs/db239.htm. Accessed November 14, 2020. [PubMed] [Google Scholar]
- 5. Novelli F, Bacci E, Latorre M, et al. Comorbidities are associated with different features of severe asthma. Clin Mol Allergy. 2018;16:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hekking P‐P, Amelink M, Wener RR, Bouvy ML, Bel EH. Comorbidities in difficult‐to‐control asthma. J Allergy Clin Immunol Pract. 2018;6:108‐113. [DOI] [PubMed] [Google Scholar]
- 7. Pradeepan S, Garrison G, Dixon AE. Obesity in asthma: approaches to treatment. Curr Allergy Asthma Rep. 2013;13:434‐442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Vijayakanthi N, Greally JM, Rastogi D. Pediatric obesity‐related asthma: the role of metabolic dysregulation. Pediatrics. 2016;137:e20150812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sutherland ER, Goleva E, King TS, et al. Cluster analysis of obesity and asthma phenotypes. PLoS One. 2012;7:e36631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Telenga ED, Tideman SW, Kerstjens HAM, et al. Obesity in asthma: more neutrophilic inflammation as a possible explanation for a reduced treatment response. Allergy. 2012;67:1060‐1068. [DOI] [PubMed] [Google Scholar]
- 11. Moore WC, Meyers DA, Wenzel SE, et al. Identification of asthma phenotypes using cluster analysis in the severe asthma research program. Am J Respir Crit Care Med. 2010;181:315‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Carr TF, Kraft M. Use of biomarkers to identify phenotypes and endotypes of severe asthma. Ann Allergy Asthma Immunol. 2018;121:414‐420. [DOI] [PubMed] [Google Scholar]
- 13. Dixon AE, Pratley RE, Forgione PM, et al. Effects of obesity and bariatric surgery on airway hyperresponsiveness, asthma control, and inflammation. J Allergy Clin Immunol. 2011;128:508‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Juel CT, Ali Z, Nilas L, Ulrik CS. Asthma and obesity: does weight loss improve asthma control? A systematic review. J Asthma Allergy. 2012;5:21‐26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mohanan S, Tapp H, McWilliams A, Dulin M. Obesity and asthma: pathophysiology and implications for diagnosis and management in primary care. ExpBiol Med. 2014;239:1531‐1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. National Heart, Lung, and Blood Institute, Guidelines for the Diagnosis and Management of Asthma (EPR‐3) NIH Publication No. 07‐4051 Originally Printed July 1997 Revised June 2002, August 2007. https://www.nhlbi.nih.gov/health‐topics/guidelines‐for‐diagnosis‐management‐of‐asthma. Accessed November 14, 2020.
- 17. Vilsboll T, Toft‐Nielsen MB, Krarup T, Madsbad S, Dinesen B, Holst JJ. Evaluation of beta‐cell secretory capacity using glucagon‐like peptide 1. Diabetes Care. 2000;23:807‐812. [DOI] [PubMed] [Google Scholar]
- 18. De Marinis YZ, Salehi A, Ward CE, et al. GLP‐1 inhibits and adrenaline stimulates glucagon release by differential modulation of N‐ and L‐type Ca2+ channel‐dependent exocytosis. Cell Metab. 2010;11:543‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Spreckley E, Murphy KG. The L‐cell in nutritional sensing and the regulation of appetite. Front Nutr. 2015;2:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Egan AG, Blind E, Dunder K, et al. Pancreatic safety of incretin‐based drugs–FDA and EMA assessment. N Engl J Med. 2014;370:794‐797. [DOI] [PubMed] [Google Scholar]
- 21. Hinnen D. Glucagon‐like peptide 1 receptor agonists for type 2 diabetes. Diabetes Spectr. 2017;30:202‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gou SI, Zhu T, Wang W, Xiao M, Wang X‐C, Chen Z‐H. Chen Z‐h. Glucagon like peptide‐1 attenuates bleomycin‐induced pulmonary fibrosis, involving the inactivation of NF‐κB in mice. Int Immunopharmacol. 2014;22:498‐504. [DOI] [PubMed] [Google Scholar]
- 23. Zhu T, Wu X‐L, Zhang W, Xiao M. Glucagon Like Peptide‐1 (GLP‐1) modulates OVA‐induced airway inflammation and mucus secretion involving a protein kinase a (PKA)‐dependent nuclear factor‐κB (NF‐κB) signaling pathway in mice. Int J Mol Sci. 2015;16:20195‐20211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Toki S, Goleniewska K, Reiss S, et al. Glucagon‐like peptide 1 signaling inhibits allergen‐induced lung IL‐33 release and reduces group 2 innate lymphoid cell cytokine production in vivo. J Allergy Clin Immunol. 2018;142:1515‐1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Peerboom S, Graff S, Seidel L, et al. Predictors of a good response to inhaled corticosteroids in obesity‐associated asthma. Biochem Pharmacol. 2020;179:113994. [DOI] [PubMed] [Google Scholar]
- 26. Mathews JA, Krishnamoorthy N, Kasahara DI, et al. IL‐33 drives augmented responses to ozone in obese mice. Environ Health Perspect. 2017;125:246‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roan F, Obata‐Ninomiya K, Ziegler SF. Epithelial cell‐derived cytokines: more than just signaling the alarm. J Clin Invest. 2019;129:1441‐1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim HY, Lee HJ, Chang Y‐J, et al. Interleukin‐17–producing innate lymphoid cells and the NLRP3 inflammasome facilitate obesity‐associated airway hyperreactivity. Nat Med. 2013;20:54‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Everaere L, Ait‐Yahia S, Molendi‐Coste O, et al. Innate lymphoid cells contribute to allergic airway disease exacerbation by obesity. J Allergy Clin Immunol. 2016;38:1309‐1318. [DOI] [PubMed] [Google Scholar]
- 30. Yanagisawa R, Koike E, Ichinose T, Takano H. Obese mice are resistant to eosinophilic airway inflammation induced by diesel exhaust particles. J Appl Toxicol. 2014;34:688‐694. [DOI] [PubMed] [Google Scholar]
- 31. Dahm PH, Richards JB, Karmouty‐Quintana H, et al. Effect of antigen sensitization and challenge on oscillatory mechanics of the lung and pulmonary inflammation in obese carboxypeptidase E‐deficient mice. Am J Physiol Regul Integr Comp Physiol. 2014;307:R621‐633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Silva FMC, Oliveira EE, Gouveia ACC, et al. Obesity promotes prolonged ovalbumin‐induced airway inflammation modulating T helper type 1 (Th1), Th2 and Th17 immune responses in BALB/c mice. Clin Exp Immunol. 2017;189:47‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tashiro H, Takahashi K, Sadamatsu H, et al. Saturated fatty acid increases lung macrophages and augments house dust mite‐induced airway inflammation in mice fed with high‐fat diet. Inflammation. 2017;40:1072‐1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kleinert M, Clemmensen C, Hofmann SM, et al. Animal models of obesity and diabetes mellitus. Nat Rev Endocrinol. 2018;14:140‐162. [DOI] [PubMed] [Google Scholar]
- 35. Dixon AE, Poynter ME. Mechanisms of asthma in obesity. pleiotropic aspects of obesity produce distinct asthma phenotypes. Am J Respir Cell Mol Biol. 2016;54:601‐608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Coleman DL. A historical perspective on leptin. Nat Med. 2010;16:1097‐1099. [DOI] [PubMed] [Google Scholar]
- 37. Tong K‐M, Shieh D‐C, Chen C‐P, et al. Leptin induces IL‐8 expression via leptin receptor, IRS‐1, PI3K, Akt cascade and promotion of NF‐κB/p300 binding in human synovial fibroblasts. Cell Signal. 2008;20:1478‐1488. [DOI] [PubMed] [Google Scholar]
- 38. Souza‐Almeida G, D’Avila H, Almeida PE, et al. Leptin mediates in vivo neutrophil migration: involvement of tumor necrosis factor‐alpha and CXCL1. Front Immunol. 2018;9:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Batra A, Okur B, Glauben R, et al. Leptin: a critical regulator of CD4+T‐cell polarization in vitro and in vivo. Endocrinology. 2010;151:56‐62. [DOI] [PubMed] [Google Scholar]
- 40. Zheng H, Zhang X, Castillo EF, Luo Y, Liu M, Yang XO. Leptin enhances TH2 and ILC2 responses in allergic airway disease. J Biol Chem. 2016;291:22043‐22052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cawley NX, Wetsel WC, Murthy SR, Park JJ, Pacak K, Loh YP. New roles of carboxypeptidase E in endocrine and neural function and cancer. Endocr Rev. 2012;33:216‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kim JH, Stewart TP, Soltani‐Bejnood M, et al. Phenotypic characterization of polygenic type 2 diabetes in TALLYHO/JngJ mice. J Endocrinol. 2006;191:437‐446. [DOI] [PubMed] [Google Scholar]
- 43. Leiter EH, Strobel M, O'Neill A, Schultz D, Schile A, Reifsnyder PC. Comparison of two new mouse models of polygenic type 2 diabetes at the jackson laboratory, NONcNZO10Lt/J and TALLYHO/JngJ. J Diabetes Res. 2013;2013:165327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Toki S, Goleniewska K, Zhang J, et al. TSLP and IL‐33 reciprocally promote each other's lung protein expression and ILC2 receptor expression to enhance innate type‐2 airway inflammation. Allergy. 2020;75:1606‐1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mould AW, Matthaei KI, Young IG, Foster PS. Relationship between interleukin‐5 and eotaxin in regulating blood and tissue eosinophilia in mice. J Clin Invest. 1997;99:1064‐1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Forssmann U, Uguccioni M, Loetscher P, et al. Eotaxin‐2, a novel CC chemokine that is selective for the chemokine receptor CCR3, and acts like eotaxin on human eosinophil and basophil leukocytes. J Exp Med. 1997;185:2171‐2176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhou W, Toki S, Zhang J, et al. Prostaglandin I2 signaling and inhibition of group 2 innate lymphoid cell responses. Am J Respir Crit Care Med. 2016;193:31‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gauvreau GM, O'Byrne PM, Boulet L‐P, et al. Effects of an anti‐TSLP antibody on allergen‐induced asthmatic responses. N Engl J Med. 2014;370:2102‐2110. [DOI] [PubMed] [Google Scholar]
- 49. Corren J, Parnes JR, Wang L, et al. Tezepelumab in adults with uncontrolled asthma. N Engl J Med. 2017;377:936‐946. [DOI] [PubMed] [Google Scholar]
- 50. Xu J, Wei G, Wang J, et al. Glucagon‐like peptide‐1 receptor activation alleviates lipopolysaccharide‐induced acute lung injury in mice via maintenance of endothelial barrier function. Lab Invest. 2019;99:577‐587. [DOI] [PubMed] [Google Scholar]
- 51. Di Tomo P, Lanuti P, Di Pietro N, et al. Liraglutide mitigates TNF‐α induced pro‐atherogenic changes and microvesicle release in HUVEC from diabetic women. Diabetes Metab Res Rev. 2017;33:e2925. [DOI] [PubMed] [Google Scholar]
- 52. Gaspari T, Liu H, Welungoda I, et al. A GLP‐1 receptor agonist liraglutide inhibits endothelial cell dysfunction and vascular adhesion molecule expression in an ApoE‐/‐ mouse model. Diab Vasc Dis Res. 2011;8:117‐124. [DOI] [PubMed] [Google Scholar]
- 53. Kasahara DI, Shore SA. IL‐33, diet‐induced obesity, and pulmonary responses to ozone. Respir Res. 2020;21:98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Michaudel C, Mackowiak C, Maillet I, et al. Ozone exposure induces respiratory barrier biphasic injury and inflammation controlled by IL‐33. J Allergy Clin Immunol. 2018;142:942‐958. [DOI] [PubMed] [Google Scholar]
- 55. Uchida M, Anderson EL, Squillace DL, et al. Oxidative stress serves as a key checkpoint for IL‐33 release by airway epithelium. Allergy. 2017;72:1521‐1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Forno E, Lescher R, Strunk R, Weiss S, Fuhlbrigge A, Celedon JC. Decreased response to inhaled steroids in overweight and obese asthmatic children. J Allergy Clin Immunol. 2011;127:741‐749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ather JL, Chung M, Hoyt LR, et al. Weight loss decreases inherent and allergic methacholine hyperresponsiveness in mouse models of diet‐induced obese asthma. Am J Respir Cell Mol Biol. 2016;55:176‐187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. van Bloemendaal L, Ten Kulve JS, la Fleur SE, Ijzerman RG, Diamant M. Effects of glucagon‐like peptide 1 on appetite and body weight: focus on the CNS. J Endocrinol. 2014;221:T1‐16. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S5
Appendix S1