

Abstract
Genetic diversity underpins species conservation and management goals, and ultimately determines a species’ ability to adapt. Using freshwater environmental DNA (eDNA) samples, we examined mitochondrial genetic diversity using multigene metabarcode sequence data from four Oncorhynchus species across 16 sites in Oregon and northern California. Our multigene metabarcode panel included targets commonly used in population genetic NADH dehydrogenase 2 (ND2), phylogenetic cytochrome c oxidase subunit 1 (COI) and eDNA (12S ribosomal DNA) screening. The ND2 locus showed the greatest within‐species haplotype diversity for all species, followed by COI and then 12S rDNA for all species except Oncorhynchus kisutch. Sequences recovered for O. clarkii clarkii were either identical to, or one mutation different from, previously characterized haplotypes (95.3% and 4.5% of reads, respectively). The greatest diversity in O. c. clarkii was among coastal watersheds, and subsets of this diversity were shared with fish in inland watersheds. However, coastal streams and the Umpqua River watershed appear to harbour unique haplotypes. Sequences from O. mykiss revealed a disjunction between the Willamette watershed and southern watersheds suggesting divergent histories. We also identified similarities between populations in the northern Deschutes and southern Klamath watersheds, consistent with previously hypothesized connections between the two via inland basins. Oncorhynchus kisutch was only identified in coastal streams and the Klamath River watershed, with most diversity concentrated in the coastal Coquille watershed. Oncorhynchus tshawytscha was only observed at one site, but contained multiple haplotypes at each locus. The characterization of genetic diversity at multiple loci expands the knowledge gained from eDNA sampling and provides crucial information for conservation actions and genetic management.
Keywords: aquatic community, biodiversity, environmental DNA, genetic diversity, metabarcoding, Oncorhynchus
1. INTRODUCTION
Observing, quantifying, and mapping the genetic diversity present within and among populations can provide insights into the basis of adaptation to environmental extremes, susceptibility to diseases, barriers to migration and reproduction, and paths of speciation, all of which are influenced by selection and are under genetic control (e.g., Gershoni et al., 2009). Mitochondrial DNA (mtDNA) variation is a common focus of intraspecific genetic surveys in animals due to the central relevance of mitochondrial variation in bioenergetics (specifically oxidative phosphorylation; e.g., Dingley et al., 2014; Hill, 2016), and the simplicity of modelling uniparental transmission of nonrecombining haploid genomes across the landscape and evolutionary time (Ballard & Rand, 2005). Mitochondrial DNA‐based estimates of genetic diversity provide a rich resource for biodiversity assessments, and they are frequently used to inform and even guide conservation and management programmes.
The coordination of mitochondrial and nuclear genomes has consequences on fitness (Healy & Burton, 2020), and it influences the phyletic patterns of sequence evolution, as mitonuclear coadaptation leads to rapid sequence divergence and the acquisition of species‐specific DNA motifs (Hill, 2016). This coordinated mutational process creates phylogenetic “gaps” that are the basis of “DNA barcoding”, a method of DNA‐based detection that is widely applied for species evaluation and biodiversity assessments, and underlies nearly all indirect genetic detection methods, such as environmental DNA (eDNA) analysis. Mitochondrial DNA‐based eDNA analysis has become a nearly universal tool for observing and monitoring the presence and absence of species in a wide variety of ecosystems (e.g., aquatic, terrestrial; Ogram et al., 1987; Rees et al., 2014; Valentin et al., 2020) and environmental media (water: Valentini et al., 2016; Venter et al., 2004; soil: Ogram et al., 1987; Schmidt et al., 2013; air: Be et al., 2015; Johnson, Cox, et al., 2019; even snow: Franklin et al., 2019; Kinoshita et al., 2019). The most commonly‐used methods for detecting eDNA use quantitative PCR (qPCR) methods that exploit these phylogenetic gaps, as taxon‐specific primers and probes are designed to anneal to predefined regions that are unique to target taxa, and differ from nontarget species. These assays are quantitative, producing a signal that is proportional to the starting amount of target DNA in the sample, allowing target species to be scored as present or absent, and occasionally ranked for abundance (Levi et al., 2019). Despite its many advantages (e.g., simplicity, low cost; Sigsgaard et al., 2015), qPCR provides no information on genetic diversity, leaving questions about the magnitude, apportionment, and relevance of mitochondrial diversity unanswered (but see Uchii et al., 2016).
Newer sequence‐based “metabarcoding” (Taberlet et al., 2012) detection methods share many of the same properties as qPCR, but they enrich DNA sequences that are nearly‐universal by PCR amplification, and then taxonomically classify each sequence using the wealth of accumulated mitochondrial DNA barcoding information that relates DNA sequence to taxon identity. This approach provides information on presence and absence (like qPCR), but also allows for verification of taxon identity, as well as genetic variation for single or multiple species, in one assay (Valentini et al., 2016). Recent eDNA metabarcoding studies have used DNA sequencing to identify species and characterize within‐species sequence variation (Baker et al., 2018; Elbrecht et al., 2018; Parsons et al., 2018; Sigsgaard et al., 2016; Stat et al., 2017). Sigsgaard et al. (2016) showed that mitochondrial haplotypes of whale shark (Rhincodon typus) obtained from eDNA metabarcoding of ocean water matched those obtained from tissue samples, and that haplotype frequencies were similar between methods. Tsuji, Maruyama, et al. (2020) also showed that haplotype identities and frequencies were highly similar when estimated from eDNA analysis and individually sampled Ayu fish (Plecoglossus altivelis altivelis). Using a high degree of replication (20 filter replicates; 15 PCR amplifications per filter), these authors showed that eDNA sampling with modest replication (e.g., three independent filters) has the same haplotype detection efficiency as traditional sampling methods based on a large sample of specimens (e.g., 70–90 fish). More recent studies (for review, see Sigsgaard et al., 2020) use eDNA to infer intraspecific diversity from multiple species simultaneously, based on metabarcode data and mitochondrial regions targeting large clades (Elbrecht et al., 2018; Stat et al., 2017; Zizka et al., 2020), or narrowly targeted for detection of closely‐related species (Marshall & Stepien, 2019; Stepien et al., 2019; Tsuji et al., 2020).
In this analysis, we build on these studies by characterizing intraspecific genetic diversity at a regional scale from multiple taxonomic and mitogenomic targets, focusing on multiple species of Pacific salmon and trout. Few taxonomic groups provide a better illustration of the role of genetics in the management of a nondomesticated species than North American Pacific salmon and trout (Oncorhynchus spp.). This group of species inhabit a complex network of streams, rivers, lakes, and marine environments, and show dynamic population relationships that can change as a function of geological, hydrological, and human forces across the landscape (Eaton et al., 2018; Minckley et al., 1986). Oncorhynchus populations are among the most intensively managed nondomesticated species in North America, with widespread hatchery‐raised stocks adding additional complexity on top of native populations, and they are the focus of continuous monitoring of genetic diversity across management units (Johnson, Johnson, et al., 2019). Genetic sampling of Pacific salmon and trout has traditionally required laborious sampling of individuals caught in the field or in hatcheries, from commercial marine fishing (for anadromous individuals), or from fish found in lakes and streams, although the latter requires angling, electrofishing, or the use of weirs (Johnson et al., 2007). However, certain information can only be gained from stream‐sampled individuals, such as the distribution of genetic diversity across the landscape or on a local scale, taxonomic identification at a local scale, the genetic influence of introduced or transplanted stocks or the presence of non‐native alleles, or information about nonanadromous life stages and phenotypes. The emergence of eDNA as a monitoring tool may be able to augment traditional studies with useful genetic diversity data in addition to species presence/absence observations. Sampling eDNA may require fewer resources than capturing individuals, allowing eDNA to enable increased geographic sampling, observe a different subset of the population than that sampled via captured individuals, and perform continued observations over time, in between periodic tissue sampling (Lim et al., 2016; Mächler et al., 2014; Sigsgaard et al., 2015; Smart et al., 2016).
Here, we obtain eDNA from multiple mitochondrial loci within Oncorhynchus clarkii clarkii (Coastal Cutthroat Trout), O. mykiss (Rainbow Trout, steelhead), O. kisutch (Coho Salmon), and O. tshawytscha (Chinook Salmon), identify its species of origin, and characterize the genetic diversity within each species across multiple watersheds in western Oregon and northwestern California. Across most of the focal watersheds, O. mykiss, O. kisutch, and O. tshawytscha have undergone major declines in their population numbers and distributions which has led to their listing as threatened species under the U.S. Endangered Species Act (Crozier et al., 2019), and O. clarkii clarkii have been precluded from listing in their current assessment owing to their broad distribution in watersheds from headwaters to river mouths, even though numbers in some places are in decline (Budy et al., 2019).
2. MATERIALS AND METHODS
Sixteen sites were sampled from streams or mainstem rivers in western Oregon and northwestern California, in September or October 2017, including three watersheds from coastal streams originating west of the Oregon Coast Range (n = 3), and five interior watersheds: Deschutes (n = 2) and Willamette (n = 5) rivers (both drain to the Columbia River), and Umpqua (n = 3), Rogue (n = 2), and Klamath (n = 1) rivers that drain into the Pacific Ocean (Figure 1, Table S1). Collection sites are a subset of locations surveyed for the Aquatic and Riparian Effectiveness Monitoring Program (AREMP) of the Northwest Forest Plan (Miller et al., 2017).
FIGURE 1.
Study region in western North America. White squares indicate sampling sites
Sample collection, DNA extraction, target amplification, sequencing, read processing, and read classification followed the methods of Hauck et al. (2019). For each of six replicates at a site, 3 L of stream water was filtered through a 0.45 µm nitrocellulose filter using a peristaltic pump. Sites were entered downstream from the point of sample collection, and a 50% bleach solution, followed by triple rinsing with deionized water, was used to decontaminate equipment. Filters were stored and transported on ice until being placed at –20°C within 6 h of collection. DNA was extracted from filters using a MoBio Power Water extraction kit (Qiagen), and extracted DNA was cleaned and concentrated using the Zymoclean Large fragment DNA Recovery Kit (Zymo Research). Target DNA concentration was adjusted to 12 ng/µl for subsequent amplification on a Fluidigm Access Array (Fluidigm) for 38 samples. DNA was limiting in the remaining 54 samples, so DNA was diluted to one of four lower target concentrations: 9 ng/µl (n = 9), 6 ng/µl (n = 23), 3 ng/µl (n = 10), and <2 ng/µl (n = 12).
Three mitochondrial gene regions were targeted using primers tailored to multiple Oncorhynchus species to mitigate possible PCR bias from a single priming sequence (Table 1). These include cytochrome c oxidase subunit 1 (COI), NADH dehydrogenase 2 (ND2), and the small subunit ribosomal 12S rRNA gene (12S). ND2 and COI were each targeted with two sets of primers spanning the same locus coordinates, but containing SNPs in the priming region identified from reference sequences within and between species. To identify taxonomic “barcode gaps” (Hebert et al., 2004) a sliding window analysis was conducted in SPIDER version 1.5.0 (Brown et al., 2012). Windows showing maximum intertaxon divergence were targeted for primer development. The ND2 target includes one of these windows, however the COI target does not because COI barcode gaps were not compatible with other primer design requirements. Gene targets for other taxa were also included on the Fluidigm array, including other teleosts, amphibians, mollusks, arthropods, and fungal and oomycete pathogens (Table S2; also see Hauck et al., 2019); these were not included in the current analysis. Positive and negative control samples were prepared as in Hauck et al. (2019), with positive controls containing amplicons for target loci at a concentration of 2 million molecules of each amplicon per reaction (2 × 106 molecules/31.35 nl), and were then supplemented with genomic DNA from Ginkgo biloba L. and Pinus lambertiana Dougl. (gymnosperms not targeted by primers in this study) to bring the final DNA concentration to 15 ng/µl. Negative controls contained 15 ng/µl duplex DNA from bacteriophage lambda (New England Biolabs). Samples were submitted on two 48‐sample plates, with a positive and negative control present on each plate.
TABLE 1.
ND2, COI, and 12S metabarcoding primers
| Target gene | Primers | F primer sequence | F primer length | F primer GC% | R primer sequence | R primer length | R primer GC% | Insert length |
|---|---|---|---|---|---|---|---|---|
| 12S | 63F+64R | GTCAGGTCGAGGTGTAGCGC | 20 | 65.0 | CATGTTACGACTTGCCTCCCCT | 22 | 54.5 | 198 |
| ND2.Set1 | 84F+85R | ACCGCCGCAGCAATGATYCT | 20 | 57.9 | ATTTTACGTAGYTGGGTTTGGTTAAGYCC | 29 | 40.7 | 292 |
| ND2.Set2 | 82F+197R | CAACMGCCGCAGCAATAATCCT | 22 | 52.4 | ATTTTACGCAGTTGGGTTTGATTAAGCCC | 29 | 41.4 | 292 |
| COI.Set1 | 102F+104R | ACCACTTTCTTTGACCCGGCAG | 22 | 54.5 | GGTGGCAGATGTRAAGTAGGCACG | 24 | 56.5 | 233 |
| COI.Set2 | 102F+105R | ACCACTTTCTTTGACCCGGCAG | 22 | 54.5 | GGTRGCAGATGTAAAGTARGCACG | 24 | 50.0 | 233 |
Primer sequence excludes the Fluidigm amplification sequence appended to the 5′ end. Insert length does not include the primer lengths.
Target amplification was performed by the University of Illinois at Urbana‐Champaign Roy J. Carver Biotechnology Center using a Fluidigm 48.48 Access Array, FastStart High Fidelity PCR System dNTPack (Roche), and standard 2‐step Fluidigm cycling parameters (35 cycles for individual primers, 14 cycles for sample indices and Illumina control sequences) with a modified annealing temperature of 58°C. Samples were uniquely tagged during amplification with 10 basepair (bp) indices. Bovine serum albumin (BSA) was added at 0.2 µg/µl final volume to mitigate PCR inhibition. Amplified targets were then sequenced on an Illumina MiSeq with 250 bp paired‐end reads.
Sequenced reads were trimmed of Illumina adapters and trimmed reads <35 bp were removed using Trimmomatic 0.33 (Bolger et al., 2014). Reads were demultiplexed by sample and primer, trimmed on the 3′ end where sequence quality fell below Phred = 20, and those <60 bp were dropped using dbcAmplicons version 0.9.1 (Settles & Gerritsen, 2014). Overlapping read pairs were joined using dbcAmplicons. Joined reads >15 bp shorter than the expected length for that locus were removed using Trimmomatic in order to exclude spurious sequences, while retaining sequences from taxa that may have deletions relative to the expected locus length.
Following read processing, reads were classified to taxon of origin using the program centrifuge version 1.0.4‐beta (Kim et al., 2016) and a database containing target gene regions for all eukaryotes present in NCBI, as described in Hauck et al. (2019). Sequences were allowed to match to multiple equal‐scoring taxa, and then classified as the lowest taxonomic unit encompassing those taxa. Sequences classified below the species level were elevated to species. Sequences classified as O. mykiss, O. clarkii, O. kisutch, or O. tshawytscha were extracted, and found to be of equal length within a locus for each species, with no evidence of indel‐containing haplotypes (data not shown). These sequences were filtered to retain only those exactly matching the expected length of the target regions. Quality score summary statistics were generated from these sequences using FastQC version 0.11.9 (Andrews, 2019).
False haplotypes of the ND2 and COI loci were removed for each species using a denoising process modeled after Turon et al. (2020). Haplotypes with total abundance <8 were removed. Remaining haplotypes were screened for the presence of intraspecific chimeras, and denoised at a range of stringencies using the program unoise2 (Edgar, 2016): the stringency parameter (alpha) was tested at a range from 1 to 10, and the optimal value was selected by analysing the ratio of mean entropy at the second codon position to that at the third position, choosing a value near the inflection point intended to optimize the reduction of sequencing error while retaining true rare haplotypes. This process could not be applied to the 12S locus due to the lack of codons, so 12S haplotypes were denoised using the same alpha values applied to ND2 and COI.
Phylogenetic trees were constructed from denoised haplotypes for each locus. Maximum‐likelihood (ML) phylogenies were constructed with the online tool phyml 3.0 (Guindon et al., 2010), with the optimal models of nucleotide substitution, rate variation among sites, and equilibrium frequencies inferred using the Akaike information criterion as implemented by SMS version 1.8.4 (Lefort et al., 2017). The same method was used to construct a phylogeny including the O. clarkii clarkii ND2 haplotypes recovered from this study together with other O. clarkii subspecies haplotypes from Loxterman and Keeley (2012). Haplotypes falling outside of their respective species or subspecies clade, or on unusually long branches, were manually examined for chimeric sequence regions, and were removed if confirmed.
For each species and locus, haplotype sequences and counts were summed across replicates at a site, dropping replicates with a single read, and used to calculate diversity indices and pairwise F ST values among sites and watersheds. Haplotype diversity (Hd; Nei & Tajima, 1981, equation 6), nucleotide diversity (π; Nei & Li, 1979, equation 22), and segregating sites (S) were calculated in r version 3.6.3 (R Core Team, 2020), using the packages ape 5.3 (Paradis & Schliep, 2018) and pegas 0.12 (Paradis, 2010). Haplotype counts by site were used to construct rarefaction curves in r using the vegan 2.5–6 package (Oksanen et al., 2019) and F ST values were calculated using arlequin 3.5.2.2 (Excoffier & Lischer, 2010). F ST p‐values are equal to the proportion of 1023 permutations of haplotypes among populations that produced F ST values greater than the observed value; permutations were performed with Arlequin, with a significance threshold of 0.05. Haplotype frequencies were mapped using the r package scatterpie 0.1.4 (Yu, 2019) using shapefile data from the package usaboundaries 0.3.1 (Mullen & Bratt, 2018) and the StreamNet Project (2012). All haplotypes were compared against the NCBI “nt” database (downloaded 2019‐11‐26) using blastn version 2.10.0+ (Zhang et al., 2000).
3. RESULTS
3.1. Sequencing results and gene characteristics
The MiSeq run produced >13 M total read pairs, with 10.8 M being assigned to a sample and primer of origin. Of those assigned to field samples from this project (e.g., excluding controls), the 12S primer produced 1366 K read pairs with 829 K meeting all filtering criteria and being assigned to one of the Oncorhynchus species (829/1366 Oncorhynchus/total); ND2 primer set 1 produced 148/213, ND2 set 2 produced 102/154, COI set 1 produced 99/125, and COI set 2 produced 621/823. The 12S primers produced 49 K Oncorhynchus pairs that could not be assigned to species, and the ND2 and COI primers each produced <20. Negative controls produced <4 reads for all taxon × primer combinations evaluated for this study (two primers not evaluated here produced read counts of 959 and 7659 in negative controls). At all positions in the COI and 12S loci, >90% of reads had Phred quality scores of 38; every position in the ND2 locus had >90% of reads with quality ≥27 (Figure S1).
An alpha value of 4 was selected for haplotype denoising as a stringency level providing robust removal of spurious sequence variants while retaining rare haplotypes (Figure 2). The denoising process reduced ND2 from 2462 raw sequence variants to 76, COI was reduced from 1704 to 124, and 12S was reduced from 1180 to 19. Following denoising, over 1.4 million sequences informed haplotype counts (Table 2).
FIGURE 2.
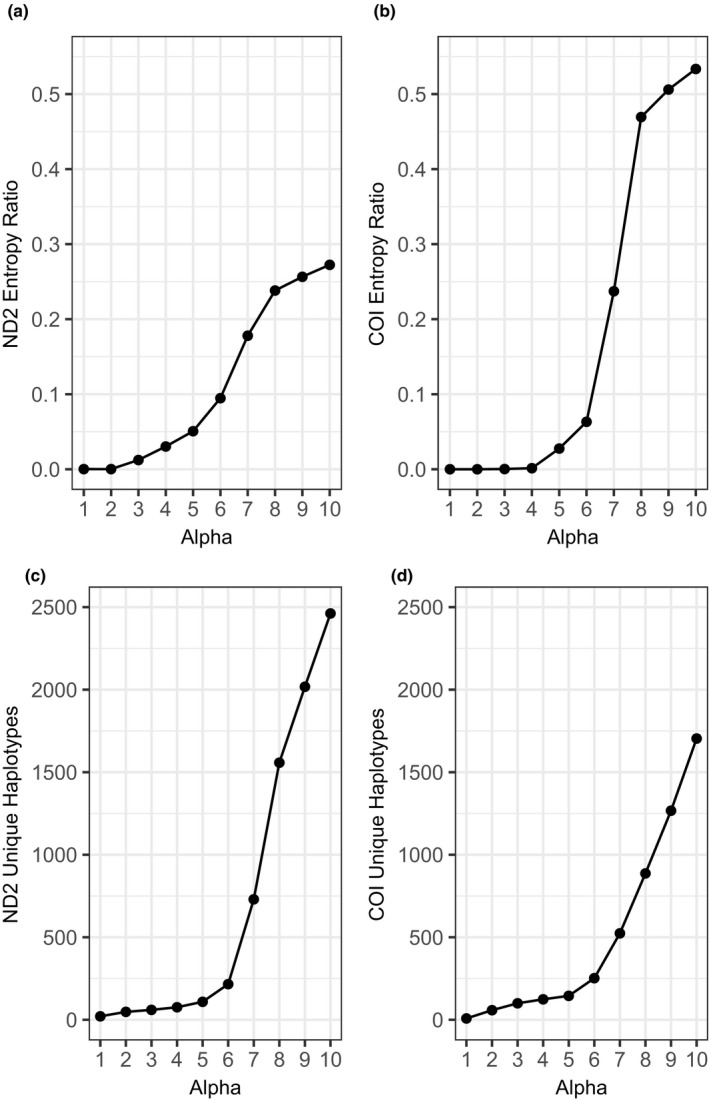
The effect of strict (low alpha) or relaxed (high alpha) filtering on the entropy ratio and the number of unique haplotypes retained for NADH dehydrogenase 2 (ND2) (a, c) and cytochrome c oxidase subunit 1 (COI) (b, d). An alpha value of 4 was selected as the inflection point representing the trade‐off of minimizing the entropy ratio while retaining true haplotypes
TABLE 2.
Diversity statistics by Oncorhynchus species and mitochondrial locus
| Reads | Streams | h | S | Hd | π | |
|---|---|---|---|---|---|---|
| ND2 (292 bp) | ||||||
| O. clarkii clarkii | 86,423 | 11 | 13 | 16 | 0.738 | 0.0048 |
| O. mykiss | 75,587 | 10 | 28 | 45 | 0.719 | 0.0046 |
| O. kisutch | 6409 | 3 | 16 | 14 | 0.701 | 0.0035 |
| O. tshawytscha | 1562 | 1 | 11 | 24 | 0.305 | 0.0043 |
| COI (233 bp) | ||||||
| O. clarkii clarkii | 244,802 | 16 | 25 | 31 | 0.013 | 0.0002 |
| O. mykiss | 295,205 | 16 | 52 | 46 | 0.555 | 0.0030 |
| O. kisutch | 24,545 | 4 | 15 | 16 | 0.189 | 0.0011 |
| O. tshawytscha | 38,499 | 8 | 9 | 9 | 0.227 | 0.0012 |
| 12S (198 bp) | ||||||
| O. clarkii clarkii | 301,893 | 16 | 2 | 3 | 0.001 | <0.0001 |
| O. mykiss | 327,470 | 16 | 8 | 14 | 0.257 | 0.0014 |
| O. kisutch | 24,244 | 5 | 5 | 5 | 0.243 | 0.0013 |
| O. tshawytscha | 51,558 | 13 | 4 | 2 | 0.116 | 0.0006 |
Reads, number of sequence reads assigned to each species and locus; Streams, number of sampling locations where reads were observed, out of 16; h, number of unique haplotypes; S, number of segregating sites; Hd, haplotype diversity; π, nucleotide diversity.
A ML phylogeny of ND2 haplotypes formed two clades under the GTR+G+I model corresponding to O. tshawytscha + O. kisutch and O. mykiss + O. c. clarkii (Figure S2). Within these, O. kisutch was monophyletic within a paraphyletic O. tshawytscha, and O. c. clarkii was monophyletic within a paraphyletic O. mykiss. A ML phylogeny of O. clarkii clarkii haplotypes together with O. clarkii haplotypes from Loxterman and Keeley (2012) formed an O. c. clarkii clade within an O. clarkii clade, under the HKY85+G model (Figure S3). Four haplotypes on a long branch within O. c. clarkii, and four haplotypes appearing to split from O. clarkii prior to the divergence of O. clarkii subspecies were manually examined as potential chimeric sequences and removed (Figure S3, ellipses).
A ML phylogeny of COI haplotypes under the model formed two clades, but with unclear root placement (Figure S4). The two clades had similar topologies, each with nearly monophyletic O. c. clarkii, O. kisutch, and O. tshawytscha haplotypes within paraphyletic O. mykiss haplotypes. The O. c. clarkii, O. kisutch, and O. tshawytscha haplotypes in one half of the tree, and O. kisutch sequences within an otherwise O. c. clarkii clade, were manually inspected as potential chimeric sequences and removed (Figure S4, ellipses).
A ML analysis of 12S haplotypes, under the GTR+I model, produced a tree with unclear root placement, and no clades corresponding to species (Figure S5). All haplotypes were retained in further analyses.
COI had the greatest number of haplotypes (h) within O. c. clarkii, O. mykiss, and O. tshawytscha, followed by ND2 and 12S, although O. kisutch had more ND2 haplotypes than COI (Table 2). The ND2 locus had the greatest values of Hd and π across all four species (Table 2). COI had greater values of Hd and π than 12S in O. c. clarkii, O. mykiss, and O. tshawytscha, but lower values than 12S for both measures in O. kisutch. Rarefaction curves suggest all or most haplotypes present at each site were observed (Figure S6).
3.2. Species characteristics
3.2.1. Oncorhynchus clarkii clarkii
Overall, 13 ND2 haplotypes were observed in O. c. clarkii. Of these, the five most abundant, representing 95.3% of reads, were also observed in a previous study of O. clarkii ND2 diversity (Figure S3; Table S3; Loxterman & Keeley, 2012). The next four most abundant haplotypes, representing 4.5% of reads, differed from those observed by Loxterman and Keeley (2012) by one SNP.
Oncorhynchus clarkii clarkii of coastal streams, particularly tributaries on the Nestucca and Alsea rivers, displayed the greatest Hd at the ND2 locus among the sites sampled (Hd = 0.73 and 0.71, respectively; Table 3), and shared haplotypes with every other watershed (Figure 3a). The coastal Alsea watershed contained one nearly‐private haplotype, which was observed in two replicates at this site, and at no other site with >10 reads. The Willamette and Umpqua watersheds each held sites with moderately high diversity (Hd > 0.5), but did not share any haplotypes. The Rogue River had low diversity (Hd = 0.08), sharing haplotypes with sites in the Willamette River and coastal streams, but not sharing haplotypes with the Umpqua River (Figure 3a).
TABLE 3.
Diversity statistics of Oncorhynchus clarkii clarkii by site and mitochondrial locus
| Watershed | Site | ND2 | COI | 12S | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reads a | h | Hd | π | Reads a | h | Hd | π | Reads a | h | Hd | π | ||
| Deschutes | Badger Crk | 0 | — | — | — | 30 | 2 | 0.0667 | 0.0011 | 126 | 1 | 0 | 0 |
| Canyon Crk | 430 | 5 | 0.0231 | 0.0001 | 3249 | 11 | 0.2190 | 0.0060 | 28 | 1 | 0 | 0 | |
| Willamette | Cabin Crk | 5 | 2 | 0.4000 | 0.0041 | 12,479 | 1 | 0 | 0 | 22,375 | 1 | 0 | 0 |
| Humbug Crk | 974 | 2 | 0.0102 | <0.0001 | 2 | 1 | 0 | 0 | 8809 | 1 | 0 | 0 | |
| Owl Crk | 1100 | 3 | 0.0036 | <0.0001 | 981 | 6 | 0.0885 | 0.0013 | 271 | 1 | 0 | 0 | |
| Quartz Crk | 9195 | 6 | 0.5745 | 0.0056 | 22,752 | 6 | 0.0026 | <0.0001 | 22,694 | 1 | 0 | 0 | |
| Hills Crk | 13,085 | 5 | 0.4600 | 0.0046 | 54,444 | 3 | 0.0001 | <0.0001 | 56,423 | 2 | 0.0003 | <0.0001 | |
| Coastal b | Clarence Crk (Nestucca) | 13,852 | 9 | 0.7286 | 0.0050 | 40,257 | 12 | 0.0527 | 0.0008 | 52,157 | 2 | 0.0059 | <0.0001 |
| Eckman Crk (Alsea) | 16,161 | 7 | 0.7071 | 0.0053 | 14,037 | 1 | 0 | 0 | 7033 | 1 | 0 | 0 | |
| Elk Crk (Coquille) | 2793 | 4 | 0.5357 | 0.0039 | 11,669 | 6 | 0.0113 | 0.0002 | 5706 | 2 | 0.0042 | <0.0001 | |
| Umpqua | Bear Crk | 0 | — | — | — | 3 | 1 | 0 | 0 | 9 | 1 | 0 | 0 |
| Stouts Crk | 0 | — | — | — | 19 | 1 | 0 | 0 | 24 | 1 | 0 | 0 | |
| Yellow Crk | 2788 | 5 | 0.5717 | 0.0039 | 9553 | 1 | 0 | 0 | 3976 | 1 | 0 | 0 | |
| Rogue | Mill Crk | 26,034 | 8 | 0.0792 | 0.0005 | 74,682 | 1 | 0 | 0 | 122,150 | 1 | 0 | 0 |
| Lone Tree Crk | 0 | — | — | — | 5 | 1 | 0 | 0 | 15 | 1 | 0 | 0 | |
| Klamath | Indian Crk | 0 | — | — | — | 24 | 1 | 0 | 0 | 91 | 1 | 0 | 0 |
Reads, number of sequence reads assigned to each site and locus.; h, number of unique haplotypes; Hd, haplotype diversity; π , nucleotide diversity
Read counts may differ from Table 2 because singleton counts were dropped.
Coastal streams occupy separate watersheds originating west of the Oregon Coast Range, indicated next to the site names.
FIGURE 3.
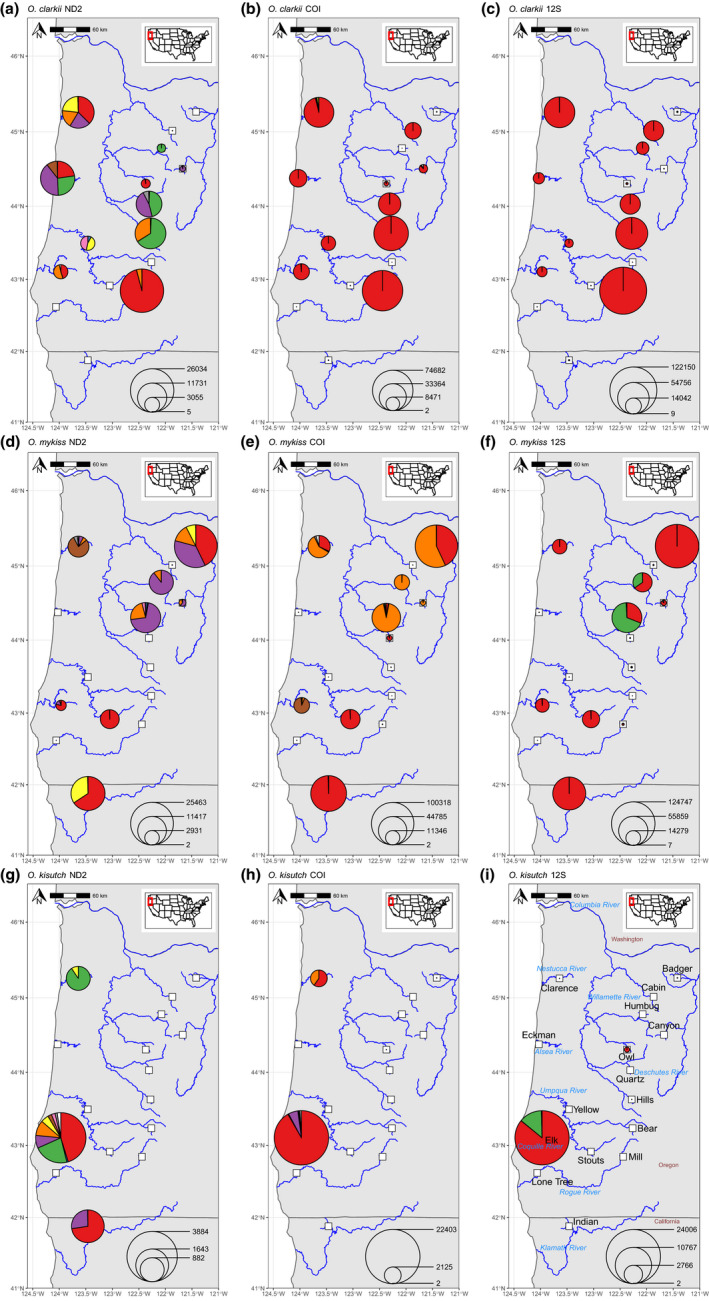
Mitochondrial haplotypes observed by species and locus. White squares indicate sampling locations. Coloured pies represent reads from that location assigned to each species and locus, with pie size proportional to the number of reads observed (summed across replicates), given by the scale in the lower right. Coloured slices represent unique haplotypes for each species and locus, with white slices representing a combination of low frequency haplotypes. Note that colours are independent across panels (e.g., red represents a different haplotype for each panel)
In O. c. clarkii, haplotype differentiation measured by F ST at ND2 was 0.476, and values ranged between 0.020 and 0.994 in pairwise comparisons (Figure 4a). F ST was significantly greater than zero between each pair of sites with >50 O. c. clarkii reads. Mill Creek, in the Rogue watershed, had the highest mean F ST value against other sites (0.679); although F ST between Mill Creek and Owl Creek, in the Willamette watershed, was the lowest among all comparisons (0.020). Eckman Creek, in the coastal Alsea watershed, had the lowest mean F ST value against other sites (0.273).
FIGURE 4.
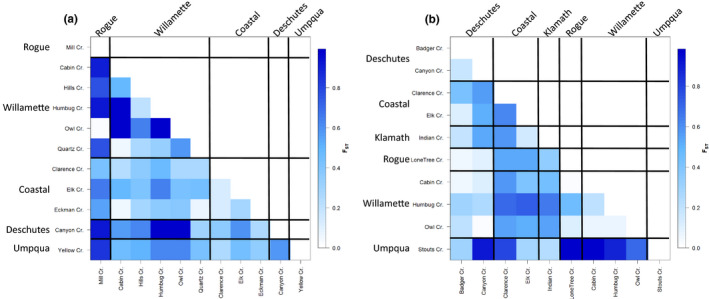
Pairwise F ST values between populations of (a) Oncorhynchus clarkii clarkii and (b) O. mykiss. Heavy lines delineate populations within the same watershed (or a member of coastal watersheds)
Both the 12S and COI loci were dominated by single haplotypes within O. c. clarkii across all sampled sites (99.9% and 99.3% overall frequency, respectively). Only one other 12S haplotype was observed (Figure 3c). Twenty‐four minor COI haplotypes were observed, but together they never made up more than 12% of the reads at any site (Figure 3b). No O. clarkii COI or 12S haplotypes observed here were present in NCBI (Table S3).
3.2.2. Oncorhynchus mykiss
Twenty‐eight O. mykiss ND2 haplotypes were observed, with the five most abundant representing 98.1% of reads (Figure 3d), and three of these also present in NCBI (Table S3). The most abundant haplotype (27,678 reads; Figure 3d, red) was primarily observed in southern Oregon and California in the Umpqua, Coquille, and Klamath watersheds, but also with a large proportion at Badger Creek, a tributary of the White River in the Deschutes watershed. The next haplotype was nearly as abundant overall (26,441 reads; Figure 3d, purple), but almost entirely found within the Willamette and Deschutes watersheds, with only a small proportion observed from the coastal Coquille and Nestucca watersheds. This pattern is also seen in the third most abundant haplotype (7852 reads; Figure 3d, orange). The fourth haplotype (735 reads; Figure 3d, yellow) was observed almost exclusively in the Klamath watershed and Badger Creek, while the fifth was a nearly private haplotype from the coastal Nestucca watershed (4860 reads; Figure 3d, brown). Badger Creek held the highest Hd, 0.6616, among O. mykiss sites with >100 reads (Table 4).
TABLE 4.
Diversity statistics for Oncorhynchus mykiss by site and mitochondrial locus
| Watershed | Site | ND2 | COI | 12S | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reads a | h | Hd | π | Reads a | h | Hd | π | Reads a | h | Hd | π | ||
| Deschutes | Badger Crk | 25,463 | 6 | 0.6616 | 0.0038 | 100,316 | 5 | 0.4918 | 0.0021 | 124,745 | 4 | 0.0016 | <0.0001 |
| Canyon Crk | 681 | 10 | 0.5166 | 0.0045 | 1446 | 6 | 0.0990 | 0.0015 | 1980 | 3 | 0.2035 | 0.0049 | |
| Willamette | Cabin Crk | 7 | 2 | 0.4762 | 0.0033 | 8 | 4 | 0.7857 | 0.0120 | 110 | 3 | 0.0882 | 0.0006 |
| Humbug Crk | 8129 | 5 | 0.1904 | 0.0007 | 13,049 | 4 | 0.0137 | <0.0001 | 24,150 | 4 | 0.4545 | 0.0023 | |
| Owl Crk | 12,636 | 16 | 0.4563 | 0.0024 | 45,602 | 24 | 0.1213 | 0.0014 | 58,599 | 5 | 0.4399 | 0.0024 | |
| Quartz Crk | 0 | — | — | — | 1472 | 13 | 0.0952 | 0.0015 | 71 | 2 | 0.0821 | 0.0004 | |
| Hills Crk | 0 | — | — | — | 20 | 3 | 0.5105 | 0.0070 | 191 | 2 | 0.0807 | 0.0004 | |
| Coastal b | Clarence Crk (Nestucca) | 6114 | 19 | 0.3778 | 0.0032 | 27,512 | 25 | 0.5436 | 0.0050 | 13,962 | 4 | 0.0033 | <0.0001 |
| Eckman Crk (Alsea) | 0 | — | — | — | 7 | 2 | 0.5714 | 0.0025 | 23 | 2 | 0.1660 | 0.0008 | |
| Elk Crk (Coquille) | 1488 | 6 | 0.3565 | 0.0021 | 13,813 | 11 | 0.1442 | 0.0013 | 12,286 | 5 | 0.0165 | 0.0002 | |
| Umpqua | Bear Crk | 0 | — | — | — | 2 | 2 | 1.0000 | 0.0043 | 7 | 1 | 0 | 0 |
| Stouts Crk | 4970 | 3 | 0.0120 | <0.0001 | 20,854 | 4 | 0.0178 | <0.0001 | 19,469 | 4 | 0.0060 | <0.0001 | |
| Yellow Crk | 0 | — | — | — | 3 | 2 | 0.6667 | 0.0029 | 12 | 1 | 0 | 0 | |
| Rogue | Mill Crk | 0 | — | — | — | 31 | 3 | 0.5462 | 0.0025 | 365 | 2 | 0.0430 | 0.0002 |
| Lone Tree Crk | 2 | 2 | 1.0000 | 0.0103 | 10 | 2 | 0.5556 | 0.0024 | 8 | 2 | 0.2500 | 0.0013 | |
| Klamath | Indian Crk | 16,092 | 5 | 0.4532 | 0.0016 | 71,047 | 4 | 0.0093 | <0.0001 | 71,487 | 4 | 0.0010 | <0.0001 |
Reads, number of sequence reads assigned to each site and locus; h, number of unique haplotypes; Hd, haplotype diversity; π, nucleotide diversity.
Read counts may differ from Table 2 because singleton counts were dropped.
Coastal streams occupy separate watersheds originating west of the Oregon Coast Range, indicated next to the site names.
In O. mykiss, the mean F ST among sites at the ND2 locus was 0.411, and it ranged from 0.021 to 0.984 in pairwise comparisons (Figure 4b). F ST was significantly greater than zero between each pair of sites with >50 O. mykiss reads. Stouts Creek, in the Umpqua watershed, had the highest mean F ST value against other sites (0.674); Badger Creek, in the Deschutes watershed had the lowest mean F ST value (0.202).
Three haplotypes represented 98.5% of the O. mykiss COI reads, with the remainder represented by 49 other haplotypes (Figure 3e). The two most abundant haplotypes were present in NCBI (Table S3). Similar to the pattern in the ND2 data, the most abundant COI haplotype was primarily observed in the Umpqua, Klamath, and Deschutes watersheds, but was also seen in the coastal Nestucca watershed and, to a lesser extent, in the Willamette watershed (Figure 3e, red). The second most abundant COI haplotype was observed primarily at the same sites as the second most abundant ND2 locus: the Deschutes, Willamette, Nestucca, and Coquille watersheds (Figure 3e, orange). The third COI haplotype was observed almost exclusively in the Coquille watershed, with only trace observations at other sites (<0.8% of the COI reads at a site).
The mean F ST among sites with >50 reads at the COI locus was 0.553, ranging from 0.002 to 0.990. However, the distribution of pairwise F ST had values clustered near 0 and 1: among 36 pairwise comparisons, eight had F ST values <0.2 and 15 had values >0.8.
Eight O. mykiss 12S haplotypes were observed, with the two most abundant representing 99.7% of reads. The most abundant haplotype was observed at every site (Figure 3f, red), and was also present in NCBI (Table S3). The second haplotype was restricted almost entirely to the Willamette watershed (<0.6% or <10 reads at all other sites).
3.2.3. Oncorhynchus kisutch
Oncorhynchus kisutch was observed with the ND2 marker in the Nestucca, Coquille, and Klamath watersheds (Figure 3g). Sixteen haplotypes were observed, with five producing >100 reads and representing 94.2% of reads. Hd was highest in the Coquille watershed, which contained all observed O. kisutch ND2 haplotypes (Table 5, Figure 3g). Only two haplotypes were observed with more than two reads in the Klamath watershed (haplotypes 1 and 3 in ranked abundance) and the Nestucca watershed (haplotypes 2 and 5). The Coquille watershed had a pairwise F ST with the Nestucca and Klamath watersheds of 0.357 and 0.120, respectively. The pairwise F ST between the Nestucca and Klamath watersheds was 0.691. All pairwise F ST values were significantly greater than zero.
TABLE 5.
Diversity statistics for Oncorhynchus kisutch by site and mitochondrial locus
| Watershed | Site | ND2 | COI | 12S | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reads a | h | Hd | π | Reads a | h | Hd | π | Reads a | h | Hd | π | ||
| Deschutes | Badger Crk | 0 | — | — | — | 2 | 1 | 0 | 0 | 5 | 2 | 0.4000 | 0.0020 |
| Canyon Crk | 0 | — | — | — | 0 | — | — | — | 0 | — | — | — | |
| Willamette | Cabin Crk | 0 | — | — | — | 0 | — | — | — | 0 | — | — | — |
| Humbug Crk | 0 | — | — | — | 0 | — | — | — | 0 | — | — | — | |
| Owl Crk | 0 | — | — | — | 2 | 1 | 0 | 0 | 218 | 1 | 0 | 0 | |
| Quartz Crk | 0 | — | — | — | 0 | — | — | — | 0 | — | — | — | |
| Hills Crk | 0 | — | — | — | 0 | — | — | — | 2 | 1 | 0 | 0 | |
| Coastal b | Clarence Crk (Nestucca) | 882 | 2 | 0.1707 | 0.0006 | 2122 | 3 | 0.4816 | 0.0021 | 2 | 1 | 0 | 0 |
| Eckman Crk (Alsea) | 0 | — | — | — | 0 | — | — | — | 0 | — | — | — | |
| Elk Crk (Coquille) | 3884 | 16 | 0.7258 | 0.0040 | 22,152 | 15 | 0.1369 | 0.0009 | 24,006 | 5 | 0.2445 | 0.0013 | |
| Umpqua | Bear Crk | 0 | — | — | — | 0 | — | — | — | 0 | — | — | — |
| Stouts Crk | 0 | — | — | — | 0 | — | — | — | 0 | — | — | — | |
| Yellow Crk | 0 | — | — | — | 0 | — | — | — | 0 | — | — | — | |
| Rogue | Mill Crk | 0 | — | — | — | 0 | — | — | — | 0 | — | — | — |
| Lone Tree Crk | 0 | — | — | — | 0 | — | — | — | 0 | — | — | — | |
| Klamath | Indian Crk | 1,643 | 6 | 0.4000 | 0.0014 | 0 | — | — | — | 0 | — | — | — |
Reads, number of sequence reads assigned to each site and locus.; h, number of unique haplotypes; Hd, haplotype diversity; π, nucleotide diversity
Read counts may differ from Table 2 because singleton counts were dropped.
Coastal streams occupy separate watersheds originating west of the Oregon Coast Range, indicated next to the site names.
Oncorhynchus kisutch was observed with the COI locus in the Nestucca and Coquille watersheds, but not in the Klamath watershed (Figure 3h). Fifteen haplotypes were observed, with four producing >100 reads and representing 99.2% of reads. All haplotypes were observed in the Coquille watershed, but only the first and third most abundant haplotypes were observed in the Nestucca watershed. Pairwise F ST between the two locations was significantly greater than zero, at 0.450.
Oncorhynchus kisutch was observed with the 12S locus in the Coquille watershed (24,006 reads) and at Owl Creek in the Willamette watershed (218 reads), but at no other site with >10 reads (Figure 3i). Five 12S haplotypes were observed, with the two most abundant representing 99.5% of reads. Only the most abundant O. kisutch 12S haplotype was observed at Owl Creek, but this haplotype differs by one SNP from the most abundant O. tshawytscha haplotype, which was highly abundant at this site.
One haplotype from each O. kisutch locus was represented in NCBI (Table S3).
3.2.4. Oncorhynchus tshawytscha
Oncorhynchus tshawytscha was observed by all three loci in Owl Creek, a tributary in the Willamette watershed. No other site showed ≥50 reads for any locus, except in the Coquille watershed, with 50 reads at the 12S locus, which we believe is due to sequencing error from O. kisutch 12S reads at this site (Table 6). At Owl Creek, 11 ND2 haplotypes were observed (six at a frequency >1%), along with nine COI (4 > 1%) and four 12S (3 > 1%). The most abundant ND2, COI, and 12S haplotypes represented 83.1%, 87.6%, and 93.9% of total reads for each locus, respectively (Figure S7). Two COI and one 12S haplotype were represented in NCBI (Table S3).
TABLE 6.
Diversity statistics for Oncorhynchus tshawytscha by site and mitochondrial locus
| Watershed | Site | ND2 | COI | 12S | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Reads a | h | Hd | π | Reads a | h | Hd | π | Reads a | h | Hd | π | ||
| Deschutes | Badger Crk | 0 | — | — | — | 2 | 1 | 0 | 0 | 13 | 1 | 0 | 0 |
| Canyon Crk | 0 | — | — | — | 0 | — | — | — | 6 | 2 | 0.3333 | 0.0017 | |
| Willamette | Cabin Crk | 0 | — | — | — | 0 | — | — | — | 5 | 2 | 0.4000 | 0.0020 |
| Humbug Crk | 0 | — | — | — | 2 | 1 | 0 | 0 | 6 | 1 | 0 | 0 | |
| Owl Crk | 1562 | 11 | 0.3045 | 0.0043 | 37,413 | 9 | 0.2272 | 0.0012 | 51,399 | 4 | 0.1157 | 0.0006 | |
| Quartz Crk | 0 | — | — | — | 6 | 1 | 0 | 0 | 15 | 1 | 0 | 0 | |
| Hills Crk | 0 | — | — | — | 3 | 3 | 1.0000 | 0.0086 | 7 | 1 | 0 | 0 | |
| Coastal b | Clarence Crk (Nestucca) | 0 | — | — | — | 0 | — | — | — | 11 | 2 | 0.4364 | 0.0022 |
| Eckman Crk (Alsea) | 0 | — | — | — | 0 | — | — | — | 0 | — | — | — | |
| Elk Crk (Coquille) | 0 | — | — | — | 2 | 1 | 0 | 0 | 50 | 1 | 0 | 0 | |
| Umpqua | Bear Crk | 0 | — | — | — | 0 | — | — | — | 0 | — | — | — |
| Stouts Crk | 0 | — | — | — | 0 | — | — | — | 2 | 1 | 0 | 0 | |
| Yellow Crk | 0 | — | — | — | 0 | — | — | — | 0 | — | — | — | |
| Rogue | Mill Crk | 0 | — | — | — | 6 | 1 | 0 | 0 | 11 | 1 | 0 | 0 |
| Lone Tree Crk | 0 | — | — | — | 0 | — | — | — | 3 | 1 | 0 | 0 | |
| Klamath | Indian Crk | 0 | — | — | — | 3 | 2 | 0.6667 | 0.0029 | 10 | 2 | 0.2000 | 0.0010 |
Reads, number of sequence reads assigned to each site and locus.; h, number of unique haplotypes; Hd, haplotype diversity; π, nucleotide diversity
Read counts may differ from Table 2 because singleton counts were dropped.
Coastal streams occupy separate watersheds originating west of the Oregon Coast Range, indicated next to the site names.
4. DISCUSSION
Here, we show that eDNA metabarcoding can effectively characterize in‐stream genetic diversity from Pacific salmon and trout. We used eDNA metabarcoding in conjunction with microfluidic processing to characterize the genetic diversity of four species of Oncorhynchus in watersheds of the Pacific Northwest at multiple mitochondrial loci. We show that eDNA samples collected at a regional scale can provide estimates of genetic similarities and differences among populations. This includes uncovering geographically localized (potentially private) haplotypes, which in some cases may be evidence of long‐term isolation or possibly local adaptation (Sjöstrand et al., 2014), warranting consideration by managers as a conservation priority and a source of genetic diversity. When applied over time and at a regional scale, this approach enables the continued biomonitoring of diversity dynamics among multiple in‐stream species or populations, and supplements traditional studies of captured individuals by tracking changes in genetic diversity for heavily‐studied species and possibly providing novel diversity data for understudied species or populations.
4.1. Gene characteristics
The three mitochondrial loci we surveyed had differing levels of intraspecific diversity. COI held the greatest number of haplotypes (h), followed by ND2 and 12S, except in O. kisutch which had more ND2 haplotypes than COI. Despite having fewer unique haplotypes, the greatest haplotype (Hd) and nucleotide (π) diversity was seen in ND2, followed by COI then 12S in O. c. clarkii, O. mykiss, and O. tshawytscha, while O. kisutch had greater diversity at the 12S locus than COI. These observations agree with prior studies showing ND2 to be less conserved, making it more suited for intraspecific investigations, but possibly less useful as a barcoding region (Loxterman & Keeley, 2012; Wilcox et al., 2015). COI and 12S, on the other hand, show greater conservation within species‐‐in several cases having a single allele fixed or nearly fixed within a population‐‐but are able to distinguish among these salmonids, hence, increasing their utility as barcoding regions (Freeland, 2016; Shaw et al., 2016; Valentini et al., 2016).
The discrepancy between COI holding the highest values of h and ND2 holding the highest values of Hd and π may relate to the sensitivity of these measures to false haplotypes. The inclusion of false haplotypes will inflate h, even if those sequences are at very low abundance, while rare haplotypes will have little effect on Hd and π. The ability to distinguish true rare haplotypes from false haplotypes requires further refinement (see below), and as such we caution that h may currently be an unreliable measure for eDNA metabarcoding.
4.2. Species characteristics
4.2.1. Oncorhynchus clarkii clarkii
Observations of O. c. clarkii haplotypes reveal comparatively high diversity at the ND2 locus, but low intraspecific diversity at COI and 12S, with most populations being fixed for single COI and 12S haplotypes. The number of O. c. clarkii ND2 haplotypes we detected is broadly congruent with a previous study of O. c. clarkii intraspecific diversity by Loxterman and Keeley (2012). We observed 13 O. c. clarkii ND2 haplotypes in our study, while Loxterman and Keeley (2012) observed 23 ND2 haplotypes in a sample of 83 O. c. clarkii individuals collected from Oregon to southern Alaska. Fewer haplotypes are expected to be recovered in our study since the segment of the ND2 locus observed by Loxterman and Keeley (2012) was larger and contained polymorphisms flanking the segment used here. While the geographic range sampled by Loxterman and Keeley (2012) is broader than our sampling scheme, covering much of the range of O. c. clarkii (Penaluna et al., 2016), most of the O. c. clarkii diversity they observed was found in Oregon, suggesting that Oregon is a hot spot for O. c. clarkii diversity. Loxterman and Keeley (2012) identified three O. c. clarkii clades, all of which occurred in Oregon, and one of which was exclusive to the state. We recovered sequences identical to haplotypes found in all three of these clades.
We observed the highest O. c. clarkii ND2 diversity in coastal watersheds, specifically the Nestucca and Alsea rivers. In Oregon, a study using microsatellite analysis similarly showed that coastal streams had greater O. c. clarkii genetic diversity (mean alleles = 47) than streams originating in the Cascades (mean alleles = 30) (Gresswell et al., 2006). O. c. clarkii exhibit a diversity of life histories, including anadromous, lake, fluvial, and stream‐living populations. Several life‐history expressions often co‐occur leading to individuals with different life histories simultaneously using the same habitat, including rivers, tributaries, lakes, estuaries, and the nearshore ocean. Anadromous O. c. clarkii individuals can migrate a great distance from their spawning areas to the sea, returning to their natal streams with high site fidelity and straying among watersheds by relatively short distances (<15 km), although some migrants may travel farther (Goetz et al., 2013; Losee et al., 2017, 2018). Other work using microsatellite analysis shows evidence that O. c. clarkii are genetically structured at the level of individual streams in Washington (Wenburg et al., 1998) and Oregon (Wofford et al., 2005). Despite this general pattern of high geographic structure, the frequency of long‐range migrants may be sufficiently high, and barriers to upstream travel sufficiently strong, to produce a pattern of downstream migration that over time effectively mixes alleles from upstream tributaries together in small coastal watersheds, reducing genetic structure at these sites.
An ND2 haplotype was observed in multiple replicates from the Alsea River (1653 reads, Figure 3a) that was not seen anywhere else with >10 reads. This suggests that trout in coastal streams may harbour endemic variation, and exhibit reduced migration to neighbouring watersheds. However, further study with expanded sampling is needed to confirm that this haplotype is restricted to coastal watersheds, and if so, whether it is an artefact of historical demographic patterns, or possibly maintained by local adaptation. Such private or highly restricted haplotypes may be especially useful to managers for population monitoring and brood‐stock analysis.
The Umpqua watershed has a unique suite of ND2 haplotypes, two of which are not observed anywhere else, and the third only observed in the coastal Nestucca watershed. Loxterman and Keeley (2012) observe haplotypes from the Umpqua watershed belonging to their O. c. clarkii “Clade B,” members of which were observed in coastal streams from southern Oregon to southern British Columbia and Vancouver Island. However, the Umpqua River watershed may indeed harbour unique ND2 haplotypes, as it is otherwise recognized for its unique freshwater fish assemblage, consisting of more native and endemic fishes than any other Oregon river that drains to the Pacific Ocean (excluding the Columbia River; Markle, 2019). As with the private allele from the Alsea watershed, more widespread sampling is needed to better estimate the geographic extent of these alleles. Nevertheless, the collection of rare alleles for Coastal Cutthroat Trout in the Umpqua watershed is striking, and warrants further examination from dedicated studies.
4.2.2. Oncorhynchus mykiss
Observations of O. mykiss haplotypes reveal high genetic diversity at the ND2 locus similar to that observed in O. c. clarkii, however O. mykiss has higher diversity than O. c. clarkii at the COI and 12S loci, which contain three and two major alleles, respectively. As with O. c. clarkii, our sampling probably includes individuals with varying life history phenotypes, such as winter‐run and summer‐run steelhead, and nonanadromous Rainbow Trout. Sampling of eDNA from sites that sustain individuals from multiple life histories may be contributing to the increase in genetic diversity relative to O. c. clarkii, and although O. mykiss of different life histories from the same watershed are more closely related to each other than to fish from other watersheds, winter‐ and summer‐run steelhead have been shown to be genetically distinct (Arciniega et al., 2016). It has also been hypothesized that O. mykiss has undergone various periods of isolation and convergence following its divergence from O. clarkii, leading to greater overall mixing and fewer distinct lineages than other Pacific trout lineages (Behnke 2002, 2007).
The most abundant ND2 and COI haplotypes of the Willamette watershed are not observed in the Umpqua or Klamath watersheds, and are only a minor component of the Coquille. The 12S locus is represented mainly by a single haplotype across the range of O. mykiss, except in the Willamette and upper Deschutes watersheds, where an alternative allele is observed with high abundance. These observations support a disjunction between southern watersheds (Umpqua and Klamath) and the Willamette watershed, mirroring prior studies that found strong divergence among O. mykiss lineages associated with the upper Sacramento, Klamath, and Columbia River regions (Behnke, 1992; Currens et al., 2009).
The disjunction between the Willamette and southern watersheds does not extend to samples from Badger Creek, a tributary to the White River in the Deschutes watershed, which consistently shares haplotypes with the southern watersheds. This unusual pattern may be a reflection of ancient continuity with the fauna of the Klamath watershed, via changes in the hydrological connections of the intervening Fort Rock Basin, followed by later isolation of the White River from the main Deschutes due to waterfall formation (Currens et al., 1990, 2009). The Fort Rock Basin, which currently does not drain to the sea, borders both the Deschutes and Klamath watersheds, and contains populations of O. mykiss with morphological and genetic similarities to those of the White River (Currens et al., 1990, 2009). Geological evidence indicates the basin was previously drained by the Deschutes (Allison, 1979), while biogeographic patterns from other species indicate previous connections to Klamath fauna (Currens et al., 2009; Minckley et al., 1986).
Samples from a second site in the Deschutes watershed, Canyon Creek, exhibit allele frequencies at all three loci similar to those from the Willamette watershed. O. mykiss from these watersheds may represent different lineages and are sometimes considered different subspecies, including O. mykiss irideus (Coastal Rainbow Trout) in the Willamette and O. mykiss gairdneri (Columbia River redband Rainbow Trout) in the Deschutes. However, previous studies have reflected the genetic similarities seen here (Brunelli et al., 2010). Brunelli et al. (2010) suggest there is sex‐biased dispersal in O. mykiss, with females dispersing from their natal streams more successfully than males, mitigating geographic structuring in mitochondrial loci that is otherwise seen in the male‐inherited Y‐chromosome. Moreover, the Willamette River contains a mix of native and hatchery‐introduced steelhead, including an introduced summer run, which is phenologically similar to steelhead runs in the Deschutes (Van Doornik et al., 2015).
Private haplotypes were observed in O. mykiss from coastal watersheds at both the ND2 and COI loci, suggesting either a higher frequency of generally rare haplotypes, or the potential for local adaptation. As with O. c. clarkii, further study is needed to confirm the geographic distribution of these alleles and their importance to the genetic diversity of this species.
4.2.3. Oncorhynchus kisutch and O. tshawytscha
eDNA signal from O. kisutch was observed in three watersheds: the Nestucca, Coquille, and Klamath; O. tshawytscha was observed at Owl Creek in the Willamette watershed, although both species are known to occupy broader ranges. O. kisutch are not naturally found in the upper Willamette River basin above Willamette Falls (although hatchery runs are artificially stocked there), but they occur naturally in the Rogue, Umpqua, and Alsea watersheds, and Cabin Creek (a tributary of the Clackamas River in the lower Willamette watershed). Although O. tshawytscha occur throughout the sampled range, except in Badger Creek in the Deschutes watershed, in these streams they are generally found lower in watersheds, in mainstem rivers and estuaries. Both species migrate as juveniles and adults, and hence, may be highly localized within watersheds when present (Groot & Margolis, 1991). Although juveniles of both species can remain in streams year‐round, our sampling in the summer may have occurred at sites that are not juvenile habitat or are above adult migration, or were sampled before adults had arrived (e.g., upriver migration timing of adults, Flitcroft et al., 2016).
Despite being observed in only three watersheds, O. kisutch exhibits comparatively high levels of genetic diversity. Four COI haplotypes and two 12S haplotypes made up >99% of reads for those loci, and five ND2 haplotypes were observed with >100 reads each. This level of diversity agrees with previous studies indicating higher amounts of mitochondrial genetic diversity harboured in the southern portion of the O. kisutch range in Washington, Oregon, and California relative to watersheds farther north (Smith et al., 2001). The high number of ND2 haplotypes within the Coquille watershed, and the observation of shared haplotypes between the Coquille watershed and the Klamath and Nestucca watersheds agrees with previous studies demonstrating higher within‐population genetic variation than between populations (Ford et al., 2004; Olsen et al., 2003). In contrast, the Nestucca and Coquille O. kisutch populations belong to a different evolutionarily significant unit than the Klamath population (Lawson et al., 2007), and the Klamath population has been shown to belong to a lineage distinct from other California populations (Gilbert‐Horvath et al., 2016). The pattern of haplotypes observed here may imply migration from the Coquille watershed to the Klamath and Nestucca watersheds, and the Coquille watershed has been classified as a “functionally independent population” by Lawson et al. (2007), meaning migration from other populations does not substantially affect it and it is a net donor to small populations (Lawson et al., 2007, p. 10). However, the Nestucca population received the same classification, and although the Klamath population was not assessed, it, too, would probably be classified the same way based on its large population (Lawson et al., 2007).
Multiple haplotypes of each locus were seen in O. tshawytscha at the single site where it was observed in the Willamette watershed. With six, four, and three haplotypes at a frequency >1% for the ND2, COI, and 12S loci, the level of allelic richness is comparable to previous studies of O. tshawytscha mitochondrial diversity. For example, four haplotypes of the D‐loop/trn‐F/12S locus were observed from 24 fish from the Willamette River, with a total of nine overlapping with fish from the broader Columbia watershed (Martin et al., 2010).
4.3. Caveats
The sequence processing implemented here produced haplotypes that were largely identical to (95.3% in O. clarkii) or differing by one SNP from (4.5%) previously characterized haplotypes (e.g., Loxterman & Keeley, 2012). The haplotype denoising process assumes that the main source of erroneous haplotypes is from random single nucleotide errors introduced through the sequencing process. The denoising process includes an abundance threshold based on the similarity of a query sequence to a more abundant “true” sequence (Edgar, 2016), while the stringency threshold relies on the positions of SNPs within a sequence, with SNPs occurring at first or second codon positions thought more likely to be caused by error (Turon et al., 2020).
As studies of this type advance, it may be found that alternative methods of error removal are more effective generally or in certain cases. The use of a single abundance threshold, as opposed to the dynamic threshold employed by denoising, may be found to be sufficient for certain uses, but could risk eliminating true rare haplotypes that differ by only one or two SNPs from an abundant haplotype. Alternatively, haplotypes that only appear in a single replicate may be eliminated, as it would be unlikely for an identical error to occur in multiple replicates. (On the Fluidigm platform used here, we think this would be most appropriate for technical replicates from the same filter and DNA extraction, as we have seen individual biological replicates miss certain targets that are otherwise moderately to highly abundant, leading us to sum replicates in this case.) Finally, careful use of mock communities, known tissue samples, or spiked‐in DNA may allow for an error rate to be estimated for the procedure overall or for certain steps, guiding the selection of a denoising alpha value or flat abundance threshold. However, even with robust filtering, some false haplotypes are likely to be retained, as will false haplotypes produced by other mechanisms (e.g., nuclear DNA of mitochondrial origin, sequence chimeras).
By constructing a phylogenetic tree of COI haplotypes from the four Oncorhynchus species, we were able to manually examine and identify multiple haplotypes probably representing chimeras. The resulting unrooted tree (Figure S4) had topologies for the four species recapitulated in two areas of the tree. Close inspection of these haplotypes revealed that haplotypes in one of these areas were probably chimeras between O. mykiss and the other species. Similarly, constructing a phylogenetic tree of O. c. clarkii ND2 haplotypes together with those obtained by Loxterman and Keeley (2012) identified four haplotypes from this study that were sister to all other O. clarkii haplotypes, and four haplotypes that were in the O. c. clarkii clade but on an unusually long branch (Figure S3). Inspection of these sequences also revealed that they were probably chimeras. Suspected chimeric sequences were rare overall, representing 2.73% of O. tshawytscha COI reads, and 0.13% of O. c. clarkii ND2 reads. Our chimera filtering, which screened for intraspecific chimeras during the denoising process, and screened for interspecific chimeras using the phylogenetic approach outlined above, probably overlooked some chimeric sequences that otherwise appear to have proper phylogenetic placement.
In rare instances, depending on the genetic locus targeted, species may only be distinguishable at a single SNP, increasing the risk of classification error and false positive detections. We saw this at the 12S locus with O. kisutch and O. tshawytscha: the most abundant haplotypes for these species differ by one SNP, signal from O. tshawytscha was high at Owl Creek in the Willamette watershed (48,286 reads), and O. kisutch signal at this site was low (218 reads; Figure 3i), probably originating from sequencing error. The reciprocal error probably produced O. tshawytscha signal in the Coquille watershed (50 reads), where O. kisutch was highly abundant (20,606 reads). Different species may also carry identical haplotypes, especially at more conservative loci, which may explain the greater number of ambiguous 12S sequences found here relative to ND2 and COI. Careful targeting of genetic loci and construction of haplotype networks can reduce this likelihood and warn users when this type of error occurs.
The estimation of species abundance from metabarcoding sequence abundance remains a challenge for the field, with multiple mechanisms decoupling the correlation between biomass and sequence count, such as variable eDNA shedding rates and PCR bias (Elbrecht & Leese, 2015); although some studies have had success in this area (Levi et al., 2019; Thomsen et al., 2016). In contrast, the correlation of intraspecific haplotype frequencies with metabarcoding sequence frequencies is reported as robust, with Sigsgaard et al. (2016), Parsons et al. (2018) and Tsuji, Shibata, et al. (2020) all reporting strong correlations between mitochondrial haplotypes from eDNA and the relative frequencies of those haplotypes in whale shark (Rhincodon typus), harbour porpoise (Phocoena phocoena), and Ayu (Plecoglossus altivelis altivelis) populations, respectively. This may be due to lower variation in eDNA shedding rates among conspecific individuals than between individuals of different species, and reduced primer and PCR bias among sequences that are more similar within a species than between species. In this study, we used multiple primers to target the same locus, reducing the likelihood of consistent bias against any specific haplotype. To date, mitochondrial haplotype variation has only been examined for one taxon/gene combination (O. clarkii/ND2; Loxterman & Keeley, 2012) in this geographic region, so it's difficult to know how accurately our eDNA‐based diversity estimates reflect true values. Coordinating this type of eDNA‐based genetic analysis at sampling locations that make direct counts for species and permit individual specimen genotyping (e.g., fish hatcheries; monitoring weirs) would provide direct evaluation of the accuracy of the method, as well as produce reference haplotypes that can guide the denoising step (Tsuji, Shibata, et al., 2020). This information would provide high confidence in eDNA methods to provide the full spectrum of biodiversity data inferred from mitochondrial genomes, including species presence, absence, and measures of genetic diversity and population connectivity.
A larger challenge with the use of eDNA to monitor Oncorhynchus genetic diversity may owe to the widespread stocking of hatchery‐raised fish. Signals of isolation‐by‐distance in historical populations of O. mykiss have been obscured by long‐term stocking practices in California (Pearse et al., 2011), but eDNA can also be affected by contemporary stocking. Hatchery populations of Oncorhynchus may contain haplotype frequencies different from those of wild populations (Nielsen et al., 1994). Unless hatchery haplotypes are known to be distinct from wild haplotypes, eDNA will not be able to distinguish among them. If isolation of wild haplotype frequencies is needed, then frequencies obtained from eDNA will need to be corrected, using both knowledge of stocking histories and hatchery genetic profiles (records that may be hard to come by) or from frequencies obtained from a sample of captured individuals (which can be distinguished as wild or hatchery‐raised by an intact or clipped adipose fin).
Environmental DNA assays, including those presented here, generally target mitochondrial loci, particularly when surveying animals (reviewed by Sigsgaard et al., 2020). The maternal inheritance of mitochondria seen in most species may create phylogeographic patterns different from those found in biparentally inherited loci. This includes Oncorhynchus species, which may exhibit sex‐biased dispersal (Brunelli et al., 2010; Kendall et al., 2014; McMillan et al., 2007). Effective assays for single‐copy nuclear loci in eDNA should mitigate this bias, but require further development (Pinfield et al., 2019; Sigsgaard et al., 2020).
Finally, hybridization may also mislead species detection efforts and population genetic inferences, especially those based on uniparentally inherited loci. Relevant to this study, hybridization is known to occur between O. clarkii and O. mykiss, particularly in anthropogenically disturbed or hatchery‐stocked watersheds (Heath et al., 2010), and can affect migration timing and within‐watershed spawning location (Corsi et al., 2013). We cannot rule out the possibility that haplotypes assigned to a species actually originated from hybrid individuals. However, if mitochondrial introgression was widespread throughout the region we would expect identical sequences, assigned to multiple species, to populate NCBI and cause our observed sequences to be assigned ambiguously to Oncorhynchus sp. The low number of ambiguous sequences we observed, particularly in ND2 and COI, suggests our results are not strongly influenced by hybrid individuals.
5. CONCLUSIONS
The use of multilocus microfluidic eDNA metabarcoding in streams can effectively sample multiple haplotypes from multiple species, shown here for three mitochondrial loci in members of Oncorhynchus from Oregon and northern California, and produce estimates of genetic diversity and population structure broadly in agreement with prior studies. Although erroneous haplotypes will be introduced to the raw data through PCR and sequencing error, adequate filtering can effectively mitigate this noise, so that most recovered sequences match previously characterized haplotypes (95.3%) or differ by one SNP (4.5%). The ability to monitor multiple genetic loci helps mitigate presence/absence false positives in single loci with low interspecific divergence, and provides a more complete assessment of genetic diversity by incorporating loci with varying levels of sequence evolution. The adoption of these techniques as a biomonitoring tool can augment genetic studies from captured individuals, allowing for increased sampling density both geographically and temporally. As human activities continue to impact aquatic habitats and their populations, continued genetic monitoring of multiple aquatic species, not only Oncorhynchus, is necessary to assess the direction and extent of those impacts and provide critical information to managers tasked with maintaining healthy ecosystems.
AUTHOR CONTRIBUTIONS
B.E.P., R.C., and K.W. conceived the initial study. B.E.P. designed, implemented, and coordinated the field portion of the study. B.E.P., R.C., K.W., and L.L.H. designed the laboratory portion of the study. L.L.H., and L.J.L. implemented the laboratory portion of the study and R.C., and L.L.H. supervised laboratory analyses. K.W. developed computational pipelines and led data analysis and interpretation, with assistance from all authors. K.W. drafted the manuscript, and all authors contributed to writing and revisions.
Supporting information
Supplementary Material
Table S1
Table S2
Table S3
ACKNOWLEDGEMENTS
We thank Chris Hirsch, Mark Raggon, Dave Hockman‐Wert, and AREMP field crews for partnering on this work. Mark Raggon also helped BEP select field sites. Stacy Crumley performed DNA extractions. We thank Dr Mark Band and the University of Illinois at Urbana‐Champaign Roy J. Carver Biotechnology Center for DNA amplification, sequencing, and consultation. Computational infrastructure was supported and maintained by the Oregon State University Center for Genome Research and Biocomputing. Bruce Hansen collected stocking information for focal species in these basins. We thank Dr Benjamin Sibbett, Dr. Naiara Rodriguez‐Ezpeleta, Dr Owen S. Wangensteen and one anonymous reviewer for their valuable comments.
Funding information
Financial support was provided by the US Forest Service Pacific Northwest Research Station, the Bureau of Land Management, and NCASI.
This article has been contributed to by US Government employees and their work is in the public domain in the USA
DATA AVAILABILITY STATEMENT
Raw sequence reads are archived under NCBI Sequence Read Archive BioProject PRJNA641714. Oncorhynchus sequence reads, as well as scripts and commands for the denoising process, haplotype analyses, and haplotype mapping are archived in the Oregon State University institutional repository ScholarsArchive@OSU (Weitemier, 2020; https://doi.org/10.7267/9wg8‐f298). Table S3 provides haplotype sequences and a key between haplotype names and haplotype colors in Figure 3 and Figure S7.
REFERENCES
- Allison, I. S. (1979). Pluvial Fort Rock Lake, Lake County, Oregon. Oregon Department of Geology and Mineral Industries, Special Paper, 7, 1–72. [Google Scholar]
- Andrews, S. (2019). FastQC ‐ A quality control application for FastQ files (Version 0.11.9). http://www.bioinformatics.babraham.ac.uk/projects/fastqc/.
- Arciniega, M. , Clemento, A. J. , Miller, M. R. , Peterson, M. , Garza, J. C. , & Pearse, D. E. (2016). Parallel evolution of the summer steelhead ecotype in multiple populations from Oregon and Northern California. Conservation Genetics, 17(1), 165–175. 10.1007/s10592-015-0769-2 [DOI] [Google Scholar]
- Baker, C. S. , Steel, D. , Nieukirk, S. , & Klinck, H. (2018). Environmental DNA (eDNA) From the wake of the whales: Droplet digital PCR for detection and species identification. Frontiers in Marine Science, 5, 133. 10.3389/fmars.2018.00133 [DOI] [Google Scholar]
- Ballard, J. W. O. , & Rand, D. M. (2005). The population biology of mitochondrial DNA and its phylogenetic implications. Annual Review of Ecology, Evolution, and Systematics, 36(1), 621–642. 10.1146/annurev.ecolsys.36.091704.175513 [DOI] [Google Scholar]
- Be, N. A. , Thissen, J. B. , Fofanov, V. Y. , Allen, J. E. , Rojas, M. , Golovko, G. , Fofanov, Y. , Koshinsky, H. , & Jaing, C. J. (2015). Metagenomic analysis of the airborne environment in urban spaces. Environmental Microbiology, 69, 346–355. 10.1007/s00248-014-0517-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behnke, R. J. (1992). Native Trout of Western North America. American Fisheries Society. [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30(15), 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, S. D. J. , Collins, R. A. , Boyer, S. , Lefort, M.‐C. , Malumbres‐Olarte, J. , Vink, C. J. , & Cruickshank, R. H. (2012). SPIDER: An R package for the analysis of species identity and evolution, with particular reference to DNA barcoding. Molecular Ecology Resources, 12(3), 562–565. 10.1111/j.1755-0998.2011.03108.x [DOI] [PubMed] [Google Scholar]
- Brunelli, J. P. , Steele, C. A. , & Thorgaard, G. H. (2010). Deep divergence and apparent sex‐biased dispersal revealed by a Y‐linked marker in rainbow trout. Molecular Phylogenetics and Evolution, 56(3), 983–990. 10.1016/j.ympev.2010.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budy, P. , Rogers, K. B. , Kanno, Y. , Penaluna, B. E. , Hitt, N. P. , Thiede, G. P. , & DeRito, J. (2019). Distribution and status of trout and char in North America. In Kersher J. L., Williams J. E., Gresswell R. E., & Lobon‐Cervia J. (Eds.), Trout and char of the world (pp. 193–250). American Fisheries Society. [Google Scholar]
- Corsi, M. P. , Eby, L. A. , & Barfoot, C. A. (2013). Hybridization with rainbow trout alters life history traits of native westslope cutthroat trout. Canadian Journal of Fisheries and Aquatic Sciences, 70(6), 895–904. 10.1139/cjfas-2012-0312 [DOI] [Google Scholar]
- Crozier, L. G. , McClure, M. M. , Beechie, T. , Bograd, S. J. , Boughton, D. A. , Carr, M. , Cooney, T. D. , Dunham, J. B. , Greene, C. M. , Haltuch, M. A. , Hazen, E. L. , Holzer, D. M. , Huff, D. D. , Johnson, R. C. , Jordan, C. E. , Kaplan, I. C. , Lindley, S. T. , Mantua, N. J. , Moyle, P. B. , … Willis‐Norton, E. (2019). Climate vulnerability assessment for Pacific salmon and steelhead in the California Current Large Marine Ecosystem. PLoS One, 14(7), e0217711. 10.1371/journal.pone.0217711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currens, K. P. , Schreck, C. B. , & Li, H. W. (1990). Allozyme and morphological divergence of Rainbow Trout (Oncorhynchus mykiss) above and below waterfalls in the Deschutes River, Oregon. Copeia, 1990(3), 730–746. 10.2307/1446439 [DOI] [Google Scholar]
- Currens, K. P. , Schreck, C. B. , & Li, H. W. (2009). Evolutionary ecology of Redband Trout. Transactions of the American Fisheries Society, 138(4), 797–817. 10.1577/T08-120.1 [DOI] [Google Scholar]
- Dingley, S. D. , Polyak, E. , Ostrovsky, J. , Srinivasan, S. , Lee, I. , Rosenfeld, A. B. , Tsukikawa, M. , Xiao, R. , Selak, M. A. , Coon, J. J. , Hebert, A. S. , Grimsrud, P. A. , Kwon, Y. J. , Pagliarini, D. J. , Gai, X. , Schurr, T. G. , Hüttemann, M. , Nakamaru‐Ogiso, E. , & Falk, M. J. (2014). Mitochondrial DNA variant in COX1 subunit significantly alters energy metabolism of geographically divergent wild isolates in Caenorhabditis elegans . Journal of Molecular Biology, 426(11), 2199–2216. 10.1016/j.jmb.2014.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eaton, K. R. , Loxterman, J. L. , & Keeley, E. R. (2018). Connections and containers: Using genetic data to understand how watershed evolution and human activities influence cutthroat trout biogeography. PLoS One, 13(8), e0202043. 10.1371/journal.pone.0202043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R. C. (2016). UNOISE2: Improved error‐correction for Illumina 16S and ITS amplicon sequencing. BioRxiv, 081257. 10.1101/081257 [DOI] [Google Scholar]
- Elbrecht, V. , & Leese, F. (2015). Can DNA‐based ecosystem assessments quantify species abundance? Testing primer bias and biomass—Sequence relationships with an innovative metabarcoding protocol. PLoS One, 10(7), e0130324. 10.1371/journal.pone.0130324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elbrecht, V. , Vamos, E. E. , Steinke, D. , & Leese, F. (2018). Estimating intraspecific genetic diversity from community DNA metabarcoding data. PeerJ, 6, e4644. 10.7717/peerj.4644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Excoffier, L. , & Lischer, H. E. L. (2010). Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10(3), 564–567. [DOI] [PubMed] [Google Scholar]
- Flitcroft, R. L. , Lewis, S. L. , Arismendi, I. , LovellFord, R. , Santelmann, M. V. , Safeeq, M. , & Grant, G. (2016). Linking hydroclimate to fish phenology and habitat use with ichthyographs. PLoS One, 11(12), e0168831. 10.1371/journal.pone.0168831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford, M. J. , Teel, D. , Van Doornik, D. M. , Kuligowski, D. , & Lawson, P. W. (2004). Genetic population structure of central Oregon coast Coho Salmon (Oncorhynchus kisutch). Conservation Genetics, 5(6), 797–812. 10.1007/s10592-004-1983-5 [DOI] [Google Scholar]
- Franklin, T. W. , McKelvey, K. S. , Golding, J. D. , Mason, D. H. , Dysthe, J. C. , Pilgrim, K. L. , Squires, J. R. , Aubry, K. B. , Long, R. A. , Greaves, S. E. , Raley, C. M. , Jackson, S. , MacKay, P. , Lisbon, J. , Sauder, J. D. , Pruss, M. T. , Heffington, D. , & Schwartz, M. K. (2019). Using environmental DNA methods to improve winter surveys for rare carnivores: DNA from snow and improved noninvasive techniques. Biological Conservation, 229, 50–58. 10.1016/j.biocon.2018.11.006 [DOI] [Google Scholar]
- Freeland, J. R. (2016). The importance of molecular markers and primer design when characterizing biodiversity from environmental DNA. Genome, 60(4), 358–374. 10.1139/gen-2016-0100 [DOI] [PubMed] [Google Scholar]
- Gershoni, M. , Templeton, A. R. , & Mishmar, D. (2009). Mitochondrial bioenergetics as a major motive force of speciation. BioEssays, 31(6), 642–650. 10.1002/bies.200800139 [DOI] [PubMed] [Google Scholar]
- Gilbert‐Horvath, E. A. , Pipal, K. A. , Spence, B. C. , Williams, T. H. , & Garza, J. C. (2016). Hierarchical phylogeographic structure of Coho Salmon in California. Transactions of the American Fisheries Society, 145(5), 1122–1138. 10.1080/00028487.2016.1201003 [DOI] [Google Scholar]
- Goetz, F. A. , Baker, B. , Buehrens, T. , & Quinn, T. P. (2013). Diversity of movements by individual anadromous Coastal Cutthroat Trout Oncorhynchus clarkii clarkii . Journal of Fish Biology, 83(5), 1161–1182. 10.1111/jfb.12209 [DOI] [PubMed] [Google Scholar]
- Gresswell, R. E. , Torgersen, C. E. , Bateman, D. S. , Guy, T. J. , Hendricks, S. R. , & Wofford, J. E. B. (2006). A spatially explicit approach for evaluating relationships among Coastal Cutthroat Trout, habitat, and disturbance in small Oregon streams. American Fisheries Society Symposium, 48, 457–471. [Google Scholar]
- Groot, C. , & Margolis, L. (Eds.) (1991). Pacific salmon life histories. UBC Press. [Google Scholar]
- Guindon, S. , Dufayard, J.‐F. , Lefort, V. , Anisimova, M. , Hordijk, W. , & Gascuel, O. (2010). New algorithms and methods to estimate maximum‐likelihood phylogenies: Assessing the performance of PhyML 3.0. Systematic Biology, 59(3), 307–321. 10.1093/sysbio/syq010 [DOI] [PubMed] [Google Scholar]
- Hauck, L. L. , Weitemier, K. A. , Penaluna, B. E. , Garcia, T. S. , & Cronn, R. (2019). Casting a broader net: Using microfluidic metagenomics to capture aquatic biodiversity data from diverse taxonomic targets. Environmental DNA, 1(3), 251–267. 10.1002/edn3.26 [DOI] [Google Scholar]
- Healy, T. M. , & Burton, R. S. (2020). Strong selective effects of mitochondrial DNA on the nuclear genome. Proceedings of the National Academy of Sciences, 117(12), 6616. 10.1073/pnas.1910141117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heath, D. , Bettles, C. M. , & Roff, D. (2010). Environmental factors associated with reproductive barrier breakdown in sympatric trout populations on Vancouver Island. Evolutionary Applications, 3(1), 77–90. 10.1111/j.1752-4571.2009.00100.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert, P. D. N. , Stoeckle, M. Y. , Zemlak, T. S. , & Francis, C. M. (2004). Identification of birds through DNA barcodes. PLOS Biology, 2(10), e312. 10.1371/journal.pbio.0020312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, G. E. (2016). Mitonuclear coevolution as the genesis of speciation and the mitochondrial DNA barcode gap. Ecology and Evolution, 6(16), 5831–5842. 10.1002/ece3.2338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson, B. M. , Johnson, M. S. , & Thorgaard, G. H. (2019). Salmon genetics and management in the Columbia River Basin. Northwest Science, 92(sp5), 346–363. 10.3955/046.092.0505 [DOI] [Google Scholar]
- Johnson, D. H. , Shrier, B. M. , O’Neal, J. S. , Knutzen, J. A. , Augerot, X. , O’Neil, T. A. , & Pearsons, T. N. (2007). Salmonid field protocols handbook. Techniques for assessing status and trends in salmon and trout populations. American Fisheries Society. [Google Scholar]
- Johnson, M. D. , Cox, R. D. , & Barnes, M. A. (2019). Analyzing airborne environmental DNA: A comparison of extraction methods, primer type, and trap type on the ability to detect airborne eDNA from terrestrial plant communities. Environmental DNA, 1(2), 176–185. 10.1002/edn3.19 [DOI] [Google Scholar]
- Kendall, N. W. , McMillan, J. R. , Sloat, M. R. , Buehrens, T. W. , Quinn, T. P. , Pess, G. R. , & Zabel, R. W. (2014). Anadromy and residency in steelhead and Rainbow Trout (Oncorhynchus mykiss): A review of the processes and patterns. Canadian Journal of Fisheries and Aquatic Sciences, 72(3), 319–342. 10.1139/cjfas-2014-0192 [DOI] [Google Scholar]
- Kim, D. , Song, L. , Breitwieser, F. P. , & Salzberg, S. L. (2016). Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Research, 26(12), 1721–1729. 10.1101/gr.210641.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinoshita, G. , Yonezawa, S. , Murakami, S. , & Isagi, Y. (2019). Environmental DNA collected from snow tracks is useful for identification of mammalian species. Zoological Science, 36(3), 198–207. 10.2108/zs180172 [DOI] [PubMed] [Google Scholar]
- Lawson, P. W. , Bjorkstedt, E. P. , Chilcote, M. W. , Huntington, C. W. , Mills, J. S. , Moore, K. M. S. , Weitkamp, L. A. (2007). Identification of historical populations of Coho salmon (Oncorhynchus kisutch) in the Oregon coast evolutionary significant unit (NOAA Technical Memorandum No. NMFS‐NWFSC‐79; p. 129). United States Department of Commerce.
- Lefort, V. , Longueville, J.‐E. , & Gascuel, O. (2017). SMS: Smart model selection in PhyML. Molecular Biology and Evolution, 34(9), 2422–2424. 10.1093/molbev/msx149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi, T. , Allen, J. M. , Bell, D. , Joyce, J. , Russell, J. R. , Tallmon, D. A. , Vulstek, S. C. , Yang, C. , & Yu, D. W. (2019). Environmental DNA for the enumeration and management of Pacific salmon. Molecular Ecology Resources, 19(3), 597–608. 10.1111/1755-0998.12987 [DOI] [PubMed] [Google Scholar]
- Lim, N. K. M. , Tay, Y. C. , Srivathsan, A. , Tan, J. W. T. , Kwik, J. T. B. , Baloğlu, B. , Meier, R. , & Yeo, D. C. J. (2016). Next‐generation freshwater bioassessment: eDNA metabarcoding with a conserved metazoan primer reveals species‐rich and reservoir‐specific communities. Royal Society Open Science, 3(11), 160635. 10.1098/rsos.160635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Losee, J. P. , Claiborne, A. M. , Dionne, P. E. , Faulkner, H. S. , & Seamons, T. R. (2018). Size, age, growth and site fidelity of anadromous cutthroat trout Oncorhynchus clarkii clarkii in the Salish Sea. Journal of Fish Biology, 93(5), 978–987. 10.1111/jfb.13824 [DOI] [PubMed] [Google Scholar]
- Losee, J. P. , Seamons, T. R. , & Jauquet, J. (2017). Migration patterns of anadromous Cutthroat Trout in South Puget Sound: A fisheries management perspective. Fisheries Research, 187, 218–225. 10.1016/j.fishres.2016.12.006 [DOI] [Google Scholar]
- Loxterman, J. L. , & Keeley, E. R. (2012). Watershed boundaries and geographic isolation: Patterns of diversification in cutthroat trout from western North America. BMC Evolutionary Biology, 12(1), 38. 10.1186/1471-2148-12-38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mächler, E. , Deiner, K. , Steinmann, P. , & Altermatt, F. (2014). Utility of environmental DNA for monitoring rare and indicator macroinvertebrate species. Freshwater Science, 33(4), 1174–1183. 10.1086/678128 [DOI] [Google Scholar]
- Markle, D. F. (2019). Drainage evolution and freshwater fish zoogeography in coastal Oregon and Washington. Northwestern Naturalist, 100(2), 71–89. 10.1898/NWN-18-18 [DOI] [Google Scholar]
- Marshall, N. T. , & Stepien, C. A. (2019). Invasion genetics from eDNA and thousands of larvae: A targeted metabarcoding assay that distinguishes species and population variation of zebra and quagga mussels. Ecology and Evolution, 9(6), 3515–3538. 10.1002/ece3.4985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin, K. E. , Steele, C. A. , Brunelli, J. P. , & Thorgaard, G. H. (2010). Mitochondrial variation and biogeographic history of Chinook Salmon. Transactions of the American Fisheries Society, 139(3), 792–802. 10.1577/T09-080.1 [DOI] [Google Scholar]
- McMillan, J. R. , Katz, S. L. , & Pess, G. R. (2007). Observational evidence of spatial and temporal structure in a sympatric anadromous (winter steelhead) and resident Rainbow Trout mating system on the Olympic Peninsula, Washington. Transactions of the American Fisheries Society, 136(3), 736–748. 10.1577/T06-016.1 [DOI] [Google Scholar]
- Miller, S. A. , Gordon, S. N. , Eldred, P. , Beloin, R. M. , Wilcox, S. , Raggon, M. , Muldoon, A. (2017). Northwest Forest Plan—the first 20 years (1994–2013): Watershed condition status and trends (General Technical Report No. PNW‐GTR‐932; p. 74). Portland, OR: United States Department of Agriculture, Forest Service, Pacific Northwest Research Station.
- Minckley, W. L. , Hendrickson, D. A. , & Bond, C. E. (1986). Geography of western North American freshwater fishes: Description and relations to intracontinental tectonism. In Hocutt C. H., & Wiley E. O. (Eds.), Zoogeography of Western North American freshwater fishes (pp. 519–613). John Wiley and Sons. [Google Scholar]
- Mullen, L. A. , & Bratt, J. (2018). USAboundaries: Historical and contemporary boundaries of the United States of America. The Journal of Open Source Software, 3(34), 314. 10.21105/joss.00314 [DOI] [Google Scholar]
- Nei, M. , & Li, W.‐H. (1979). Mathematical model for studying genetic variation in terms of restriction endonucleases. Proceedings of the National Academy of Sciences, 76(10), 5269–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M. , & Tajima, F. (1981). DNA polymorphism detectable by restriction enonucleases. Genetics, 97(1), 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, J. L. , Gan, C. , & Thomas, W. K. (1994). Differences in genetic diversity for mitochondrial DNA between hatchery and wild populations of Oncorhynchus . Canadian Journal of Fisheries and Aquatic Sciences, 51(S1), 290–297. 10.1139/f94-316 [DOI] [Google Scholar]
- Ogram, A. , Sayler, G. S. , & Barkay, T. (1987). The extraction and purification of microbial DNA from sediments. Journal of Microbiological Methods, 7(2), 57–66. 10.1016/0167-7012(87)90025-X [DOI] [Google Scholar]
- Oksanen, J. , Blanchet, F. G. , Friendly, M. , Kindt, R. , Legendre, P. , McGlinn, D. , Wagner, H. (2019). vegan: Community Ecology Package (Version 2.5‐6). https://CRAN.R‐project.org/package=vegan.
- Olsen, J. B. , Miller, S. J. , Spearman, W. J. , & Wenburg, J. K. (2003). Patterns of intra‐ and inter‐population genetic diversity in Alaskan Coho Salmon: Implications for conservation. Conservation Genetics, 4(5), 557–569. 10.1023/A:1025684104113 [DOI] [Google Scholar]
- Paradis, E. (2010). pegas: An R package for population genetics with an integrated–modular approach. Bioinformatics, 26(3), 419–420. 10.1093/bioinformatics/btp696 [DOI] [PubMed] [Google Scholar]
- Paradis, E. , & Schliep, K. (2018). ape 5.0: An environment for modern phylogenetics and evolutionary analyses in R. Bioinformatics, 35(3), 526–528. 10.1093/bioinformatics/bty633 [DOI] [PubMed] [Google Scholar]
- Parsons, K. M. , Everett, M. , Dahlheim, M. , & Park, L. (2018). Water, water everywhere: Environmental DNA can unlock population structure in elusive marine species. Royal Society Open Science, 5(8), 180537. 10.1098/rsos.180537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearse, D. E. , Martinez, E. , & Garza, J. C. (2011). Disruption of historical patterns of isolation by distance in coastal steelhead. Conservation Genetics, 12(3), 691–700. 10.1007/s10592-010-0175-8 [DOI] [Google Scholar]
- Penaluna, B. E. , Abadía‐Cardoso, A. , Dunham, J. B. , García‐Dé León, F. J. , Gresswell, R. E. , Luna, A. R. , Taylor, E. B. , Shepard, B. B. , Al‐Chokhachy, R. , Muhlfeld, C. C. , Bestgen, K. R. , Rogers, K. , Escalante, M. A. , Keeley, E. R. , Temple, G. M. , Williams, J. E. , Matthews, K. R. , Pierce, R. , Mayden, R. L. , … Fausch, K. D. (2016). Conservation of native Pacific trout diversity in western North America. Fisheries, 41(6), 286–300. 10.1080/03632415.2016.1175888 [DOI] [Google Scholar]
- Pinfield, R. , Dillane, E. , Runge, A. K. W. , Evans, A. , Mirimin, L. , Niemann, J. , & Foote, A. D. (2019). False‐negative detections from environmental DNA collected in the presence of large numbers of killer whales (Orcinus orca). Environmental DNA, 1(4), 316–328. 10.1002/edn3.32 [DOI] [Google Scholar]
- R Core Team (2020). R: A language and environment for statistical computing. R Foundation for Statistical Computing. http://www.R‐project.org/. [Google Scholar]
- Rees, H. C. , Maddison, B. C. , Middleditch, D. J. , Patmore, J. R. M. , & Gough, K. C. (2014). REVIEW: The detection of aquatic animal species using environmental DNA – A review of eDNA as a survey tool in ecology. Journal of Applied Ecology, 51(5), 1450–1459. 10.1111/1365-2664.12306 [DOI] [Google Scholar]
- Schmidt, P.‐A. , Bálint, M. , Greshake, B. , Bandow, C. , Römbke, J. , & Schmitt, I. (2013). Illumina metabarcoding of a soil fungal community. Soil Biology and Biochemistry, 65, 128–132. 10.1016/j.soilbio.2013.05.014 [DOI] [Google Scholar]
- Settles, M. , & Gerritsen, A. (2014). dbcAmplicons (Version 0.8.6). https://github.com/msettles/dbcAmplicons.
- Shaw, J. L. A. , Clarke, L. J. , Wedderburn, S. D. , Barnes, T. C. , Weyrich, L. S. , & Cooper, A. (2016). Comparison of environmental DNA metabarcoding and conventional fish survey methods in a river system. Biological Conservation, 197, 131–138. 10.1016/j.biocon.2016.03.010. [DOI] [Google Scholar]
- Sigsgaard, E. E. , Carl, H. , Møller, P. R. , & Thomsen, P. F. (2015). Monitoring the near‐extinct European weather loach in Denmark based on environmental DNA from water samples. Biological Conservation, 183, 46–52. 10.1016/j.biocon.2014.11.023. [DOI] [Google Scholar]
- Sigsgaard, E. E. , Jensen, M. R. , Winkelmann, I. E. , Møller, P. R. , Hansen, M. M. , & Thomsen, P. F. (2020). Population‐level inferences from environmental DNA—Current status and future perspectives. Evolutionary Applications, 13(2), 245–262. 10.1111/eva.12882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigsgaard, E. E. , Nielsen, I. B. , Bach, S. S. , Lorenzen, E. D. , Robinson, D. P. , Knudsen, S. W. , Pedersen, M. W. , Jaidah, M. A. , Orlando, L. , Willerslev, E. , Møller, P. R. , & Thomsen, P. F. (2016). Population characteristics of a large Whale Shark aggregation inferred from seawater environmental DNA. Nature Ecology & Evolution, 1(1), 0004. 10.1038/s41559-016-0004. [DOI] [PubMed] [Google Scholar]
- Sjöstrand, A. E. , Sjödin, P. , & Jakobsson, M. (2014). Private haplotypes can reveal local adaptation. BMC Genetics, 15(1), 61. 10.1186/1471-2156-15-61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart, A. S. , Weeks, A. R. , van Rooyen, A. R. , Moore, A. , McCarthy, M. A. , & Tingley, R. (2016). Assessing the cost‐efficiency of environmental DNA sampling. Methods in Ecology and Evolution, 7(11), 1291–1298. 10.1111/2041-210X.12598 [DOI] [Google Scholar]
- Smith, C. T. , Nelson, R. J. , Wood, C. C. , & Koop, B. F. (2001). Glacial biogeography of North American Coho Salmon (Oncorhynchus kisutch). Molecular Ecology, 10(12), 2775–2785. 10.1046/j.1365-294X.2001.t01-1-01405.x [DOI] [PubMed] [Google Scholar]
- Stat, M. , Huggett, M. J. , Bernasconi, R. , DiBattista, J. D. , Berry, T. E. , Newman, S. J. , Harvey, E. S. , & Bunce, M. (2017). Ecosystem biomonitoring with eDNA: Metabarcoding across the tree of life in a tropical marine environment. Scientific Reports, 7(1), 12240. 10.1038/s41598-017-12501-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stepien, C. A. , Snyder, M. R. , & Elz, A. E. (2019). Invasion genetics of the silver carp Hypophthalmichthys molitrix across North America: Differentiation of fronts, introgression, and eDNA metabarcode detection. PLoS One, 14(3), e0203012. 10.1371/journal.pone.0203012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- StreamNet Project . (2012). StreamNet Mixed Scale Hydrography (MSHv3.1) ‐ updated September, 2012 [Vector digital data]. Portland, OR: StreamNet Project, Pacific States Marine Fisheries Commission. http://www.streamnet.org/gisdata/map_data_base/MSHv31_93.zip [Google Scholar]
- Taberlet, P. , Coissac, E. , Hajibabaei, M. , & Rieseberg, L. H. (2012). Environmental DNA. Molecular Ecology, 21(8), 1789–1793. 10.1111/j.1365-294X.2012.05542.x [DOI] [PubMed] [Google Scholar]
- Thomsen, P. F. , Møller, P. R. , Sigsgaard, E. E. , Knudsen, S. W. , Jørgensen, O. A. , & Willerslev, E. (2016). Environmental DNA from seawater samples correlate with trawl catches of subarctic, deepwater fishes. PLoS One, 11(11), e0165252. 10.1371/journal.pone.0165252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuji, S. , Maruyama, A. , Miya, M. , Ushio, M. , Sato, H. , Minamoto, T. , & Yamanaka, H. (2020). Environmental DNA analysis shows high potential as a tool for estimating intraspecific genetic diversity in a wild fish population. Molecular Ecology Resources, 20(5), 1248–1258. 10.1111/1755-0998.13165 [DOI] [PubMed] [Google Scholar]
- Tsuji, S. , Shibata, N. , Sawada, H. , & Ushio, M. (2020). Quantitative evaluation of intraspecific genetic diversity in a natural fish population using environmental DNA analysis. Molecular Ecology Resources, 20(5), 1323–1332. 10.1111/1755-0998.13200 [DOI] [PubMed] [Google Scholar]
- Turon, X. , Antich, A. , Palacín, C. , Præbel, K. , & Wangensteen, O. S. (2020). From metabarcoding to metaphylogeography: Separating the wheat from the chaff. Ecological Applications, 30(2), e02036. 10.1002/eap.2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchii, K. , Doi, H. , & Minamoto, T. (2016). A novel environmental DNA approach to quantify the cryptic invasion of non‐native genotypes. Molecular Ecology Resources, 16(2), 415–422. 10.1111/1755-0998.12460 [DOI] [PubMed] [Google Scholar]
- Valentin, R. E. , Fonseca, D. M. , Gable, S. , Kyle, K. E. , Hamilton, G. C. , Nielsen, A. L. , & Lockwood, J. L. (2020). Moving eDNA surveys onto land: Strategies for active eDNA aggregation to detect invasive forest insects. Molecular Ecology Resources, 20(3), 746–755. 10.1111/1755-0998.13151 [DOI] [PubMed] [Google Scholar]
- Valentini, A. , Taberlet, P. , Miaud, C. , Civade, R. , Herder, J. , Thomsen, P. F. , Bellemain, E. , Besnard, A. , Coissac, E. , Boyer, F. , Gaboriaud, C. , Jean, P. , Poulet, N. , Roset, N. , Copp, G. H. , Geniez, P. , Pont, D. , Argillier, C. , Baudoin, J.‐M. , … Dejean, T. (2016). Next‐generation monitoring of aquatic biodiversity using environmental DNA metabarcoding. Molecular Ecology, 25(4), 929–942. 10.1111/mec.13428 [DOI] [PubMed] [Google Scholar]
- Van Doornik, D. M. , Hess, M. A. , Johnson, M. A. , Teel, D. J. , Friesen, T. A. , & Myers, J. M. (2015). Genetic population structure of Willamette River steelhead and the influence of introduced stocks. Transactions of the American Fisheries Society, 144(1), 150–162. 10.1080/00028487.2014.982178 [DOI] [Google Scholar]
- Venter, J. C. , Remington, K. , Heidelberg, J. F. , Halpern, A. L. , Rusch, D. , Eisen, J. A. , & Smith, H. O. (2004). Environmental genome shotgun sequencing of the Sargasso Sea. Science, 304(5667), 66–74. 10.1126/science.1093857 [DOI] [PubMed] [Google Scholar]
- Weitemier, K. A. (2020). Supplemental data to “Estimating the Genetic Diversity of Pacific salmon and trout using Multi‐gene eDNA Metabarcoding”. Oregon State University, 10.7267/9wg8-f298 [DOI] [PMC free article] [PubMed]
- Wenburg, J. K. , Bentzen, P. , & Foote, C. J. (1998). Microsatellite analysis of genetic population structure in an endangered salmonid: The Coastal Cutthroat Trout (Oncorhynchus clarki clarki). Molecular Ecology, 7(6), 733–749. 10.1046/j.1365-294x.1998.00386.x [DOI] [PubMed] [Google Scholar]
- Wilcox, T. M. , Carim, K. J. , McKelvey, K. S. , Young, M. K. , & Schwartz, M. K. (2015). The dual challenges of generality and specificity when developing environmental DNA markers for species and subspecies of Oncorhynchus . PLoS One, 10(11), e0142008. 10.1371/journal.pone.0142008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wofford, J. E. B. , Gresswell, R. E. , & Banks, M. A. (2005). Influence of barriers to movement on within‐watershed genetic variation of Coastal Cutthroat Trout. Ecological Applications, 15(2), 628–637. 10.1890/04-0095 [DOI] [Google Scholar]
- Yu, G. (2019). scatterpie: Scatter Pie Plot (Version 0.1.4). https://CRAN.R‐project.org/package=scatterpie.
- Zhang, Z. , Schwartz, S. , Wagner, L. , & Miller, W. (2000). A Greedy Algorithm for aligning DNA sequences. Journal of Computational Biology, 7(1–2), 203–214. 10.1089/10665270050081478 [DOI] [PubMed] [Google Scholar]
- Zizka, V. M. A. , Weiss, M. , & Leese, F. (2020). Can metabarcoding resolve intraspecific genetic diversity changes to environmental stressors? A test case using river macrozoobenthos. Metabarcoding and Metagenomics, 4, e51925. 10.3897/mbmg.4.51925 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Weitemier, K. A. (2020). Supplemental data to “Estimating the Genetic Diversity of Pacific salmon and trout using Multi‐gene eDNA Metabarcoding”. Oregon State University, 10.7267/9wg8-f298 [DOI] [PMC free article] [PubMed]
Supplementary Materials
Supplementary Material
Table S1
Table S2
Table S3
Data Availability Statement
Raw sequence reads are archived under NCBI Sequence Read Archive BioProject PRJNA641714. Oncorhynchus sequence reads, as well as scripts and commands for the denoising process, haplotype analyses, and haplotype mapping are archived in the Oregon State University institutional repository ScholarsArchive@OSU (Weitemier, 2020; https://doi.org/10.7267/9wg8‐f298). Table S3 provides haplotype sequences and a key between haplotype names and haplotype colors in Figure 3 and Figure S7.