

Abstract
Aims
To evaluate pharmacokinetic equivalence and preliminary safety of the adalimumab biosimilar CT‐P17 administered via autoinjector (CT‐P17 AI) or prefilled syringe (CT‐P17 PFS) in healthy subjects.
Methods
This phase I, open‐label study (ClinicalTrials.gov: NCT04295356) randomised subjects (1:1) to receive a single 40‐mg (100 mg/mL) dose of CT‐P17 AI or CT‐P17 PFS. Primary endpoint was pharmacokinetic equivalence of CT‐P17 AI to CT‐P17 PFS for: area under the concentration–time curve from time zero to infinity (AUC0–inf); area under the concentration–time curve from time zero to the last quantifiable concentration (AUC0–last); maximum serum concentration (Cmax). Equivalence was determined if the 90% confidence interval for the geometric least‐squares mean ratio was within the 80–125% equivalence margin. Additional pharmacokinetic endpoints, safety and immunogenicity were evaluated.
Results
Of 193 subjects who were randomised (98 CT‐P17 AI; 95 CT‐P17 PFS), 180 received study drug. Pharmacokinetic equivalence was demonstrated: 90% confidence intervals were within the 80–125% equivalence margin (AUC0–inf: 93.98–114.29; AUC0–last: 91.09–121.86; Cmax: 94.08–111.90). Mean serum CT‐P17 concentrations, secondary pharmacokinetic parameters and numbers of subjects with antidrug antibodies (ADAs) or neutralising ADAs were comparable between groups. AUC0–inf, AUC0–last and Cmax were numerically lower for ADA‐positive than for ADA‐negative subjects (both groups); pharmacokinetic equivalence was also demonstrated among ADA‐positive subjects. CT‐P17 AI and CT‐P17 PFS were well tolerated, with comparable overall safety profiles.
Conclusions
CT‐P17 AI and CT‐P17 PFS were pharmacokinetically equivalent. Overall safety and immunogenicity were comparable between the 2 delivery devices.
Keywords: biologicals, drug delivery, monoclonal antibodies, pharmacokinetics, phase I

1. What is already known about this subject
There are several approved biosimilars of reference adalimumab (50 mg/mL).
CT‐P17 is a biosimilar of reference adalimumab with a citrate free, high‐concentration (100 mg/mL) formulation.
Autoinjector and prefilled syringe devices for CT‐P17 administration have not been previously evaluated; reports suggest patients with rheumatic disease prefer the convenience of autoinjectors.
What this study adds
Single doses of CT‐P17 administered to healthy subjects by autoinjector or prefilled syringe were pharmacokinetically equivalent, with comparable overall safety and immunogenicity.
Findings support use of CT‐P17, developed with a 100 mg/mL citrate‐free formulation, as a treatment option offering flexibility through its administration by either autoinjector or prefilled syringe.
1. INTRODUCTION
Reference adalimumab (Humira; AbbVie, North Chicago, IL, USA) is a tumour necrosis factor (TNF)‐targeting human monoclonal antibody approved by the US Food and Drug Administration (FDA; 2002) and European Medicines Agency (EMA; 2003) for indications including rheumatoid arthritis (RA). 1 , 2 Adalimumab has contributed to the revolutionary effect of biologics on immune‐mediated inflammatory disease treatment 3 and, with other TNF inhibitors, is important in treatment guidelines. 4 , 5 , 6 Since 2016, several adalimumab biosimilars have been approved by the FDA and EMA. 7 , 8 , 9 Subsequently, adalimumab biosimilars have been adopted rapidly in European countries. 10 However, despite FDA approval, no adalimumab biosimilar has yet been marketed in the USA due to patent disputes 11
CT‐P17 is the first adalimumab biosimilar with a high‐concentration formulation, approved by the EMA in February 2021 for the same indications as reference adalimumab. 2 , 12 , 13 , 14 , 15 Aligned with the devices available for reference adalimumab self‐administration, prefilled syringe (PFS) and autoinjector (AI) options are in development for CT‐P17. 1 , 2 AI devices represent favourable treatment options for patients with rheumatic disease, who prefer AI over PFS administration. 16 , 17 , 18 , 19 , 20 In addition, high levels of usability have been demonstrated in patients with RA despite hand disability. 21 , 22 , 23 To improve convenience, the device for CT‐P17 administration by AI (CT‐P17 AI) was developed as a single‐use, disposable prefilled pen, containing the same formulation as the PFS for CT‐P17 (CT‐P17 PFS).
This phase I, randomised, open‐label study compared the pharmacokinetics (PK), safety and immunogenicity of CT‐P17 in healthy subjects following subcutaneous administration of a single dose of CT‐P17 AI (comparator) or, as used in prior CT‐P17 clinical studies, CT‐P17 PFS (reference). The study was designed to fulfil the requirement to demonstrate PK similarity between the 2 delivery methods, per FDA recommendations for developing drug products for RA treatment. 24
2. SUBJECTS AND METHODS
2.1. Study design and procedures
This phase I, randomised, open‐label, 2‐arm, parallel‐group study was conducted at 2 US PPD Development sites (Austin, TX; Las Vegas, NV). Subjects were randomised (1:1) to receive CT‐P17 AI or CT‐P17 PFS (day 1) followed by PK, safety and immunogenicity assessment over the 10‐week study period (until end‐of‐study visit [day 71]). The randomisation code was generated by the contract research organisation before the study and provided to the study centre pharmacist, with randomisation numbers assigned on day −1. Randomisation was balanced by permuted blocks and stratified by day −1 body weight (≥80 kg vs. <80 kg), sex (male vs. female) and study centre.
Subjects received a single 40‐mg dose of CT‐P17 (100 mg/mL) via AI or PFS to the lower abdomen (excluding the 5 cm surrounding the navel) on day 1. CT‐P17 AI and CT‐P17 PFS were both administered using 29‐gauge, 1.25‐cm needles. The single‐use CT‐P17 AI was placed over the injection site at a 90° angle and pressed firmly against the skin. Subjects fasted for ≥8 hours before and 4 hours after study drug administration; ingestion of water was permitted as desired, excluding from 1 hour before to 1 hour after study drug administration. Although not required for PK evaluation of this subcutaneously administered drug, the fasting state ensured uniformity.
The study was conducted according to the principles of the Declaration of Helsinki and the International Council for Harmonisation Good Clinical Practice guideline. All national, state and local laws and regulations were followed. Written approval of the study protocol and informed consent form was obtained from the institutional review board (for both study sites), and written informed consent obtained from each subject, before study initiation. The study was registered retrospectively with ClinicalTrials.gov (NCT04295356).
2.2. Subjects
The Supplementary Methods detail full eligibility criteria. Briefly, subjects were healthy male or female adults aged 18–55 years with body mass index of 18.0–29.9 kg/m2. Key exclusion criteria were history of infection with hepatitis B virus (except past resolved infection), hepatitis C virus, human immunodeficiency virus, or syphilis; history of opportunistic infection, tuberculosis (latent or active), or malignancy; and biologic treatment ≤6 months before day 1.
2.3. Study endpoints
The primary objective was to demonstrate PK equivalence of CT‐P17 AI vs. CT‐P17 PFS in terms of area under the concentration–time curve from time zero to infinity (AUC0–inf), area under the concentration–time curve from time zero to the last quantifiable concentration (AUC0–last) and maximum serum concentration (Cmax). Secondary PK endpoints were similarity of CT‐P17 AI vs. CT‐P17 PFS in terms of percentage of the area extrapolated for calculation of AUC0–inf (%AUCextrap), time to Cmax (Tmax), terminal elimination half‐life (t½), terminal elimination rate constant (λz), apparent total body clearance (CL/F) and apparent volume of distribution (Vz/F) during the terminal phase after nonintravenous administration. Additional secondary endpoints were safety and immunogenicity.
2.4. Study assessments
Blood samples for PK assessment were collected at all study visits from day 1 (Table S1). Serum adalimumab concentrations were determined using an electrochemiluminescent method (Meso Scale Discovery, Rockville, MD, USA). Streptavidin‐coated plates were coated with biotinylated TNF‐α and the chemiluminescent signal was measured in relative light units using a SECTOR plate reader (Meso Scale Discovery). The lower limit of quantification was 100 ng/mL. Concentrations below the lower limit of quantification were set to zero before study drug administration, then set to missing thereafter. Measurable concentrations after consecutive concentrations below the lower limit of quantification during the terminal phase were also set to missing. PK parameters were calculated by noncompartmental method using Phoenix WinNonlin version 8.0 (Certara, Princeton, NJ, USA).
Safety evaluations included adverse events (AEs), clinical monitoring for tuberculosis, prior/concomitant medications and clinical laboratory tests (Table S1). AEs of special interest were injection‐site reactions (ISRs; assessed 30 ± 10 minutes after study drug administration), hypersensitivity/allergic reactions (assessed by additional electrocardiogram and vital sign monitoring [days 1 and 2]), infections and malignancies. Local site pain was evaluated by 100‐mm visual analogue scale within 15 minutes postdose.
For immunogenicity assessment (Table S1), antidrug antibodies (ADAs) were detected using a validated electrochemiluminescent bridging assay with acid dissociation (Meso Scale Discovery). ADA‐positive samples underwent further analysis to confirm the specificity of binding and to quantify ADA titre; assay sensitivity was 0.588 ng/mL. Samples with detectable specific anti‐adalimumab antibodies were tested for neutralising antibodies (NAbs). A validated electrochemiluminescent assay with affinity capture elution (Meso Scale Discovery) was used to measure neutralising activity against adalimumab in human serum. Biotinylated drug and Sulfo‐Tag‐labelled target were used for detection of NAbs; the resulting electrochemiluminescent signal was inversely proportional to the amount of NAbs. This method was developed and validated in accordance with EMA and FDA guidance. 25 , 26 Intrarun, interday and inter‐run precision were below 13% and assay sensitivity was 113.4 ng/mL.
2.5. Statistical analysis
The log‐transformed primary endpoints (AUC0–inf, AUC0–last and Cmax) were analysed by analysis of covariance (ANCOVA) with treatment as a fixed effect. Stratification factors were sex, study centre and day −1 body weight. The difference in least‐squares mean (LSM) between CT‐P17 AI and CT‐P17 PFS and associated 90% confidence interval (CI) were back‐transformed to provide the ratio of geometric LSM and corresponding 90% CI. PK equivalence (AUC0–inf, AUC0–last and Cmax) was concluded if the 90% CI was within the 80–125% equivalence margin. A sample size of 162 subjects (81 per treatment group) provided ≥90% power to show PK equivalence of CT‐P17 AI and CT‐P17 PFS, given the primary endpoint equivalence margin, expected geometric mean ratio of 1.0, and coefficient of variation of 45% (assumed based on historical PK data in healthy individuals). Considering a 10% dropout rate, randomisation of approximately 180 subjects (90 per group) was required.
Analysis populations are described in the Supplementary Methods. PK parameters were analysed in the PK population; analyses were also conducted separately for ADA‐positive (≥1 positive post‐treatment ADA result) and ADA‐negative (all negative post‐treatment ADA results) subjects. A posthoc analysis assessed the relationship between ADA titre and primary PK endpoints by Pearson correlation coefficients (using log2‐transformed ADA titres) in each group. P‐values for correlation coefficients, and for the difference in correlation coefficients between groups, were calculated using Fisher's z‐transformation. A posthoc analysis was conducted to compare local site pain scores between groups using 2‐sample t‐tests. Statistical analyses were performed using SAS version 9.4 (SAS Institute, Cary, NC, USA). Missing data were not imputed.
3. RESULTS
3.1. Subject disposition
Subjects were randomised between 21 June 2019 and 14 August 2019; the last subject's last visit was on 15 November 2019. Overall, 193 subjects were randomised (CT‐P17 AI: 98; CT‐P17 PFS: 95; Figure 1). Thirteen (6.7%) subjects discontinued before study drug administration (CT‐P17 AI: 5 [5.1%]; CT‐P17 PFS: 8 [8.4%]), mostly by investigator decision considering out‐of‐range laboratory values or vital signs (CT‐P17 AI: 5 [5.1%]; CT‐P17 PFS: 4 [4.2%]). Overall, 175 (90.7%) subjects completed the study. Five (2.6%) subjects discontinued after study drug administration due to loss to follow‐up (n = 3, all in the CT‐P17 PFS group), protocol deviation (n = 1; CT‐P17 PFS group) and investigator decision (n = 1; CT‐P17 AI group).
FIGURE 1.
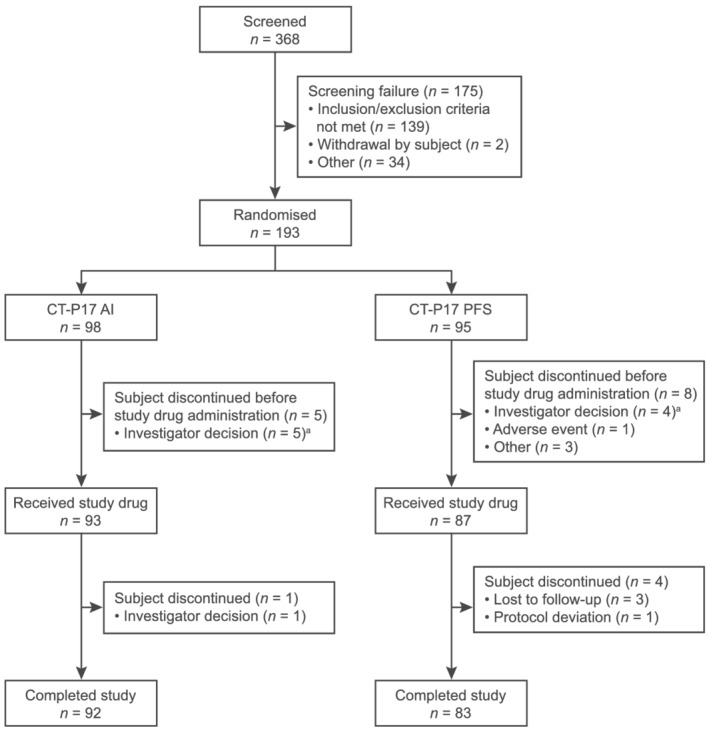
Subject flow diagram. a Due to out‐of‐range laboratory values or vital signs. AI = autoinjector; PFS = prefilled syringe
Subject demographics and baseline characteristics were similar between groups, with 50.0% male and 55.6% white, overall (Table 1). At day −1, 65 (69.9%) and 62 (71.3%) subjects in the CT‐P17 AI and CT‐P17 PFS groups, respectively, weighed <80 kg.
TABLE 1.
Baseline demographics and disease characteristics (safety population)
| Characteristic | CT‐P17 AI (n = 93) | CT‐P17 PFS (n = 87) |
|---|---|---|
| Age (y), median (range) | 34 (18–55) | 36 (18–55) |
| Male, n (%) | 48 (51.6) | 42 (48.3) |
| Ethnicity, n (%) | ||
| Hispanic or Latino | 32 (34.4) | 25 (28.7) |
| Not Hispanic or Latino | 61 (65.6) | 62 (71.3) |
| Race, n (%) a | ||
| American Indian or Alaska native | 1 (1.1) | 0 |
| Asian | 3 (3.2) | 1 (1.1) |
| Black or African American | 34 (36.6) | 39 (44.8) |
| White | 53 (57.0) | 47 (54.0) |
| Multiracial | 2 (2.2) | 0 |
| Screening height (cm), median (range) | 168.80 (146.0–186.6) | 169.50 (151.2–197.6) |
| Screening weight (kg), median (range) | 70.60 (48.2–101.6) | 73.00 (47.4–106.3) |
| Screening BMI (kg/m2), median (range) | 25.40 (19.0–29.9) | 26.00 (18.5–29.9) |
| Day −1 weight category, n (%) | ||
| Weight <80 kg | 65 (69.9) | 62 (71.3) |
| Weight ≥80 kg | 28 (30.1) | 25 (28.7) |
AI = autoinjector; BMI = body mass index; PFS = prefilled syringe.
Subjects from multiple races were counted only in the multiracial category.
3.2. PK results
Mean serum CT‐P17 concentrations observed until 71 days postdose were comparable between groups (Figure 2). Mean peak and total systemic exposure of CT‐P17 (assessed by AUC0–inf, AUC0–last and Cmax) were equivalent for CT‐P17 AI and CT‐P17 PFS: in each case, the 90% CIs for the geometric LSM ratios for CT‐P17 AI vs. CT‐P17 PFS determined by ANCOVA were within the predefined 80–125% equivalence margin (Table 2). Median Tmax occurred at 132 hours for both groups (Table 3). Mean secondary PK parameters (t½, λz, CL/F, Vz/F and %AUCextrap) were comparable between groups.
FIGURE 2.
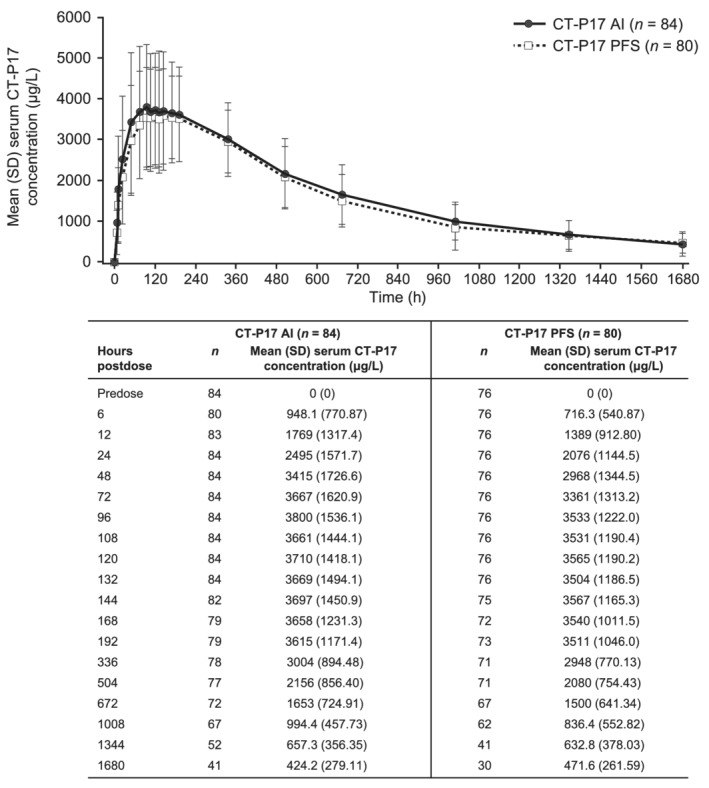
Mean (SD) serum concentrations of CT‐P17 for CT‐P17 AI and CT‐P17 PFS (PK population a). a In the CT‐P17 AI and CT‐P17 PFS groups, 6 and 7 subjects, respectively, were excluded due to absence of ≥3 time points following Cmax. Three subjects (CT‐P17 AI) were excluded due to major protocol deviations (whole volume of study drug was not administered successfully [n = 2] and dosing with morphine in a previous clinical study [n = 1]). AI = autoinjector; Cmax = maximum serum concentration; PFS = prefilled syringe; PK = pharmacokinetic; SD = standard deviation
TABLE 2.
Statistical analysis of the primary PK endpoints (PK population a )
| Parameter (units) | Treatment | n | gLSM | Ratio of gLSMs (90% CI) | P‐value |
|---|---|---|---|---|---|
| Cmax (μg/mL) | CT‐P17 AI | 84 | 3.801 | 102.60 (94.08–111.90) | .6244 |
| CT‐P17 PFS | 76 | 3.705 | |||
| AUC0–inf (h·μg/mL) | CT‐P17 AI | 69 b | 2606.4 | 103.64 (93.98–114.29) | .5459 |
| CT‐P17 PFS | 63 b | 2514.8 | |||
| AUC0–last (h·μg/mL) | CT‐P17 AI | 84 | 2110.7 | 105.36 (91.09–121.86) | .5537 |
| CT‐P17 PFS | 76 | 2003.4 |
AI = autoinjector; AUC0–inf = area under the concentration–time curve from time zero to infinity; AUC0–last = area under the concentration–time curve from time zero to the last quantifiable concentration; %AUCextrap = percentage of the area extrapolated for calculation of area under the concentration–time curve from time zero to infinity; CI = confidence interval; Cmax = maximum serum concentration; gLSM = geometric least‐squares mean; PFS = prefilled syringe; PK = pharmacokinetic.
CT‐P17 AI (n = 84); CT‐P17 PFS (n = 80). Four subjects in the CT‐P17 PFS group who were included in the PK population discontinued the study early and were not included in the summary statistics.
AUC0–inf PK parameter values were excluded from the summary statistics after not meeting ≥1 of the following: terminal elimination rate constant was calculated with an adjusted correlation coefficient r2 of ≥0.85 and/or a %AUCextrap ≤ 20%.
TABLE 3.
PK parameters for CT‐P17 AI and CT‐P17 PFS (PK population a )
| Parameter (units) | CT‐P17 AI (n = 84) | CT‐P17 PFS (n = 80) |
|---|---|---|
| Tmax (h) | ||
| n | 84 | 76 |
| Median (range) | 132.000 (24.00–504.18) | 132.000 (48.00–505.97) |
| t½ (h) | ||
| n | 69 b | 63 b |
| Mean (SD) | 369.0 (139.76) | 355.4 (141.80) |
| λz (1/h) | ||
| n | 69 b | 63 b |
| Mean (SD) | 0.002228 (0.0011047) | 0.002313 (0.0010012) |
| CL/F (L/h) | ||
| n | 69 b | 63 b |
| Mean (SD) | 0.01630 (0.0074049) | 0.01690 (0.0059949) |
| Vz/F (L) | ||
| n | 69 b | 63 b |
| Mean (SD) | 7.885 (2.7536) | 7.898 (2.7145) |
| %AUCextrap (%) | ||
| n | 69 b | 63 b |
| Mean (SD) | 7.274 (4.5181) | 7.104 (4.5240) |
λz = terminal elimination rate constant; %AUCextrap = percentage of the area extrapolated for calculation of area under the concentration–time curve from time zero to infinity; AI = autoinjector; AUC0–inf = area under the concentration–time curve from time zero to infinity; CL/F = apparent total body clearance; PFS = prefilled syringe; PK = pharmacokinetic; SD = standard deviation; t½ = terminal elimination half‐life; Tmax = time to the maximum serum concentration; Vz/F = apparent volume of distribution during the terminal phase after nonintravenous administration.
Four subjects in the CT‐P17 PFS group who were included in the PK population discontinued the study early and were not included in the summary statistics.
AUC0–inf PK parameter values were excluded from the summary statistics after not meeting ≥1 of the following: terminal elimination rate constant was calculated with an adjusted correlation coefficient r2 of ≥0.85 and/or a %AUCextrap ≤ 20%.
3.3. Immunogenicity
Overall, 10 (10.8%) and 5 (5.7%) subjects in the CT‐P17 AI and CT‐P17 PFS groups, respectively, were ADA‐positive at baseline. No subject had a positive NAb result at baseline. Overall, 91 (97.8%)/81 (87.1%) and 85 (97.7%)/75 (86.2%) subjects in the CT‐P17 AI and CT‐P17 PFS groups, respectively, had ≥1 post‐treatment ADA/NAb‐positive result (Table 4).
TABLE 4.
Immunogenicity results for CT‐P17 AI and CT‐P17 PFS (safety population)
| Subjects, n (%) | CT‐P17 AI (n = 93) | CT‐P17 PFS (n = 87) |
|---|---|---|
| ≥1 ADA‐positive result after study drug administration | 91 (97.8) | 85 (97.7) |
| ≥1 NAb‐positive result after study drug administration | 81 (87.1) | 75 (86.2) |
| Day 1 | ||
| ADA‐positive | 10 (10.8) | 5 (5.7) |
| NAb‐positive | 0 | 0 |
| ADA‐negative | 83 (89.2) | 82 (94.3) |
| Day 15 | ||
| ADA‐positive | 62 (66.7) | 59 (67.8) |
| NAb‐positive | 21 (22.6) | 25 (28.7) |
| ADA‐negative | 30 (32.3) | 27 (31.0) |
| Day 29 | ||
| ADA‐positive | 78 (83.9) | 73 (83.9) |
| NAb‐positive | 39 (41.9) | 44 (50.6) |
| ADA‐negative | 10 (10.8) | 11 (12.6) |
| Day 57 | ||
| ADA‐positive | 82 (88.2) | 73 (83.9) |
| NAb‐positive | 65 (69.9) | 60 (69.0) |
| ADA‐negative | 6 (6.5) | 7 (8.0) |
| End of study | ||
| ADA‐positive | 87 (93.5) | 78 (89.7) |
| NAb‐positive | 73 (78.5) | 68 (78.2) |
| ADA‐negative | 4 (4.3) | 6 (6.9) |
ADA = antidrug antibody; AI = autoinjector; NAb = neutralising antibody; PFS = prefilled syringe.
3.4. PK findings by ADA status
Mean serum concentrations and mean peak and total systemic exposure parameters (AUC0–inf, AUC0–last and Cmax) were lower for ADA‐positive vs. ADA‐negative subjects in both groups (P ≥ .05, except for AUC0‐inf [P = .0246]). ANCOVA showed that positive ADA status had no impact on the equivalence of CT‐P17 AI and CT‐P17 PFS (Table S2). Due to low numbers, ANCOVA could not be conducted for ADA‐negative subjects. Mean secondary PK parameters were comparable between CT‐P17 AI and CT‐P17 PFS in either ADA‐positive or ADA‐negative subgroups.
In addition, primary PK endpoints were analysed by ADA titre. In both treatment groups, there was a negative correlation between AUC0‐inf or AUC0‐last and ADA titre (Pearson r ranged from −0.6956 to −0.4582; Figure S1). There were no significant differences between treatment groups for all primary PK endpoints (P > .05).
3.5. Safety
The whole volume of study drug was successfully received by 91 (97.8%) and 87 (100.0%) subjects in the CT‐P17 AI and CT‐P17 PFS groups, respectively; device malfunction occurred for 2 (2.2%) subjects in the CT‐P17 AI group.
Overall, 56 (60.2%) and 45 (51.7%) subjects reported at least 1 treatment‐emergent AE (TEAE) in the CT‐P17 AI and CT‐P17 PFS groups, respectively (Table 5). All TEAEs recovered by study end, except for grade 1 TEAEs in 3 subjects (electrocardiogram T‐wave abnormal and dental caries [study drug‐unrelated]; alanine aminotransferase increased [study drug‐related]). Two subjects receiving CT‐P17 AI reported treatment‐emergent serious AEs (TESAEs): viral meningitis and rhabdomyolysis. Both were grade 3, study drug‐related TESAEs; subjects recovered and completed the study. There were no grade 4 TEAEs or deaths; no TEAEs led to discontinuation.
TABLE 5.
Adverse events (safety population)
| Subjects, n (%) | CT‐P17 AI (n = 93) | CT‐P17 PFS (n = 87) |
|---|---|---|
| Subjects with ≥1 TEAE a | 56 (60.2) | 45 (51.7) |
| Study drug‐related | 47 (50.5) | 38 (43.7) |
| Subjects with ≥1 TESAE | 2 (2.2) | 0 |
| Subjects with ≥1 TEAE due to hypersensitivity/allergic reactions b | 3 (3.2) | 1 (1.1) |
| Subjects with ≥1 TEAE due to ISR c | 8 (8.6) | 6 (6.9) |
| Subjects with ≥1 TEAE due to infection | 10 (10.8) | 6 (6.9) |
| Study drug‐related | 7 (7.5) d | 2 (2.3) e |
| Study drug‐unrelated | 5 (5.4) f | 4 (4.6) g |
AI = autoinjector; ISR = injection‐site reaction; PFS = prefilled syringe; TEAE = treatment‐emergent adverse event; TESAE = treatment‐emergent serious adverse event.
No subjects experienced TEAEs leading to study drug discontinuation or death or TEAEs due to malignancy.
All TEAEs classified as hypersensitivity/allergic reactions were considered study drug‐related and were grade 1–2 in intensity.
All TEAEs classified as ISR were considered study drug‐related and were grade 1 in intensity.
Grade 1 upper respiratory tract infection (n = 3), grade 3 viral meningitis (n = 1), grade 2 subcutaneous abscess (n = 1), grade 2 urinary tract infection (n = 1), grade 1 pyuria (n = 1).
Grade 2 urinary tract infection (n = 2).
Grade 2 urinary tract infection (n = 2), grade 2 upper respiratory tract infection (n = 1), grade 1 nasopharyngitis (n = 1), grade 1 vaginal infection (n = 1).
Grade 2 upper respiratory tract infection (n = 1), grade 1 upper respiratory tract infection (n = 1), grade 1 cystitis (n = 1), grade 1 vulvovaginal mycotic infection (n = 1).
TEAEs by System Organ Class (SOC) are displayed in Table S3. For most SOCs, incidence of TEAEs was comparable between groups. TEAEs in the musculoskeletal and connective‐tissue disorders SOC occurred in 14 (15.1%) and 3 (3.4%) subjects in the CT‐P17 AI and CT‐P17 PFS groups, respectively. All were grade 1, nonserious events except for the grade 3 TESAE of rhabdomyolysis, and all subjects recovered. Musculoskeletal pain (including the Preferred Term of musculoskeletal chest pain) was experienced by 7 (7.5%) and 2 (2.3%) subjects in the CT‐P17 AI and CT‐P17 PFS groups, respectively, while, correspondingly, 4 (4.3%) and 1 (1.1%) subjects reported back pain. In addition, 4 (4.3%) and 2 (2.2%) subjects (both CT‐P17 AI group) reported arthralgia and myalgia, respectively. One (1.1%) subject (CT‐P17 AI group) reported a grade 3 TESAE of rhabdomyolysis following strenuous activity; the investigator confirmed there was no significant change in renal or cardiac function. This subject recovered in 8 days after receiving hydration; they completed the study.
Overall, the most frequently reported TEAEs were headache and ISR (Table S4). Increased blood creatine phosphokinase (CPK) was reported in 9 (9.7%) and 4 (4.6%) subjects in the CT‐P17 AI and CT‐P17 PFS groups, respectively. This included 3 subjects with CPK levels >10× the upper limit of normal (ULN) in the CT‐P17 AI group: 1 subject with a TEAE of rhabdomyolysis had a CPK level of 16 652 IU/L, 1 subject had a CPK level of 11 620 IU/L that was reported as TEAE of increased blood CPK and 1 subject had a CPK level of 4519 IU/L that was captured from laboratory testing but not reported as a TEAE. Ten subjects (CT‐P17 AI: 6 [6.5%]; CT‐P17 PFS: 4 [4.6%]) had CPK results 2.0–5.1× the ULN. CPK elevations returned to normal at the next regular visit (within 14 days) in 8 subjects, including the 2 who had reported the highest CPK values. Of the 5 remaining subjects who developed CPK elevation, CPK decreased to 1.5× the ULN within 14 days in 3 subjects and within 28 days and 42 days in the remaining 2. All 13 subjects completed the study, and other than for the subject with rhabdomyolysis, no relevant clinical AEs were reported. There was no concordance between the subjects who reported musculoskeletal or connective‐tissue disorders and those with blood CPK elevations.
Three (3.2%) and 1 (1.1%) subjects experienced TEAEs of hypersensitivity/allergic reaction in the CT‐P17 AI and CT‐P17 PFS groups, respectively (Table 5). Pruritus was the most common sign and symptom, reported by 2 (1.1%) subjects receiving CT‐P17 AI only. During hypersensitivity monitoring, there were some clinically notable low and high vital sign results; however, none were reported as TEAEs and there were no marked differences between groups. Eight (8.6%) and 6 (6.9%) subjects experienced TEAEs classified as ISRs in the CT‐P17 AI and CT‐P17 PFS groups, respectively (Table 5). Injection‐site erythema was the most common sign and symptom (CT‐P17 AI: 5 [5.4%] subjects; CT‐P17 PFS: 2 [2.3%]). Ten (10.8%) and 6 (6.9%) subjects experienced TEAEs classified as infection in the CT‐P17 AI and CT‐P17 PFS groups, respectively, with these being deemed study drug‐related for 7 (7.5%) and 2 (2.3%) subjects, correspondingly. Study drug‐related infections experienced by at least 1 subject were upper respiratory tract infection (CT‐P17 AI group: 3 [3.2%] subjects) and urinary tract infection (CT‐P17 AI: 1 [1.1%]; CT‐P17 PFS: 2 [2.3%]). No subject had any signs or symptoms indicative of tuberculosis during the study. There were no TEAEs of malignancy.
No more than 1 subject in each group had abnormal, clinically significant postbaseline haematology laboratory parameters. There were no notable differences between groups. In both groups, the most frequently reported grade 3 laboratory parameter was decreased neutrophil count (CT‐P17 AI: 2 [2.2%] subjects; CT‐P17 PFS: 3 [3.4%]). Neutropenia (neutrophil count <1.0 × 109/L) occurred in 5 subjects. Neutrophil counts returned to above this level when evaluation was repeated on the same day (2 subjects) or at the next routine visit within 5 or 14 days (2 subjects); this was also the case for 1 subject who had 2 episodes of neutropenia. No infections were associated with these transient episodes of neutropenia, which were considered not to be clinically significant by the investigator.
Following study drug administration, mean (standard deviation) local site pain score by 100‐mm visual analogue scale was 0.8 (2.55) mm in the CT‐P17 AI group and 1.2 (7.33) mm in the CT‐P17 PFS group. Local site pain scores did not differ (P > .05) between groups.
4. DISCUSSION
PK equivalence was established for CT‐P17 AI and CT‐P17 PFS regarding mean peak and total systemic exposure, assessed by AUC0–inf, AUC0–last and Cmax, following administration of a single subcutaneous dose in healthy individuals. Secondary PK parameters (%AUCextrap, Tmax, Vz/F, λz, t½ and CL/F) were comparable between groups. CT‐P17 AI and CT‐P17 PFS were well tolerated, with comparable overall safety, although a higher frequency of TEAEs in the musculoskeletal and connective‐tissue disorders SOC was reported for subjects in the CT‐P17 AI group. Immunogenicity profiles were comparable between groups, with lower mean peak and total exposure parameters reported for ADA‐positive than for ADA‐negative subjects in both groups.
Comparability of PK parameters between CT‐P17 AI and CT‐P17 PFS is consistent with previous reports for other adalimumab biosimilars. Equivalence 27 , 28 or similarity 29 , 30 of mean peak and total systemic exposure, in terms of parameters including AUC0–inf, AUC0–last and Cmax, has been demonstrated following the administration of single doses of adalimumab biosimilars (40 mg/0.8 mL) via AI or PFS in healthy volunteers. Regarding the secondary PK endpoints evaluated in this study, median Tmax (132 hours in both groups) was similar to that in 2 studies that evaluated the adalimumab biosimilar BI 695501 (108–132 and 144–168 hours). 29 In addition, the comparability of t½ 28 and CL/F 27 , 29 between groups aligns with previous reports for adalimumab biosimilars administered by AI or PFS.
In this study, a slightly greater proportion of subjects in the CT‐P17 AI group experienced TEAEs than in the CT‐P17 PFS group, consistent with the occurrence of TEAEs when the adalimumab biosimilars FKB327 28 or SB5 27 were administered to healthy individuals. However, when patients with RA were treated with SB5, overall safety was comparable for AI and PFS administration. 16 As in previous studies comparing administration of adalimumab biosimilars by both AI and PFS, headache and ISRs were among the most common TEAEs in this study. 27 , 28 , 30
Compared with subjects in the CT‐P17 PFS group, a greater number of subjects in this study who received CT‐P17 by AI reported musculoskeletal pain, ISRs and transient blood CPK increases; such AEs are listed as very common or common in the reference adalimumab summary of product characteristics. 2 TEAEs in the musculoskeletal and connective‐tissue disorders SOC were reported more frequently in subjects receiving CT‐P17 by AI than PFS. Musculoskeletal pain was the most frequently reported event in this SOC. All but 1 event were nonserious TEAEs of grade 1 intensity and all subjects recovered without sequalae. The proportion of subjects reporting musculoskeletal and connective‐tissue disorders in the CT‐P17 AI group is aligned with those reported in the VOLTAIRE‐AI and VOLTAIRE‐TAI studies of BI 695501 administered by AI (11.1 and 20.0%). 29 As was also observed for BI 695501, a slightly greater proportion of subjects receiving CT‐P17 by AI reported ISRs than did those receiving CT‐P17 by PFS. 29 Three subjects who reported musculoskeletal pain also reported ISRs, although these events are unlikely to have been related since the location of the reported pain did not correspond to the injection site or they were not experienced concurrently. No subject reported musculoskeletal‐related TEAEs or ISR TEAEs in addition to CPK elevation, other than 1 subject in the CT‐P17 AI group who reported a grade 3 TESAE of rhabdomyolysis, which is listed as an uncommon AE in the summary of product characteristics for reference adalimumab 2 ; however, this subject recovered and completed the study. Transient neutropenia (neutrophil count <1.0 × 109/L) occurred in 5 subjects (CT‐P17 AI: 2 [2.2%]; CT‐P17 PFS: 3 [3.4%]), but neutrophil counts returned to >1.0 × 109/L when retested on the same day or at the next routine visit, without correlation with infection. Taken together, these safety findings are consistent with those previously reported for reference adalimumab and adalimumab biosimilars. Local site pain scores were minimal and did not differ between groups, consistent with the comparability of injection‐site pain for SB5 administered by AI or PFS to patients with RA. 16
The proportion of ADA‐positive subjects at baseline (CT‐P17 AI: 10.8%; CT‐P17 PFS: 5.7%) falls within the 2–11% false‐positive rate that might be expected for such an assay. 31 , 32 No subject was NAb‐positive at baseline. During the study, the proportions of both ADA‐positive and NAb‐positive subjects were comparable between groups. More than 97% of subjects in each group had ADA‐positive status, reflecting the high sensitivity of the electrochemiluminescent detection method. Of those who were ADA‐positive, most had positive ADA results at consecutive visits (until end of study). Similarly high rates of ADA positivity have been reported in studies of adalimumab biosimilars administered to healthy subjects by AI or PFS, 28 and comparable immunogenicity of PFS‐administered reference adalimumab and adalimumab biosimilars has also been observed. 33 , 34 We also analysed PK parameters by ADA status. ANCOVA findings demonstrated that mean peak and total exposure parameters were lower for ADA‐positive than for ADA‐negative subjects in both CT‐P17 groups, in line with previous reports for reference adalimumab and adalimumab biosimilars. 30 , 35 However, because of the small number of ADA‐negative subjects, these subgroup analyses should be interpreted with caution. Primary PK parameters were also analysed by ADA titre: PK parameters tended to be lower for subjects with higher titres, except for Cmax, for which there was no correlation.
Other studies in the CT‐P17 clinical development programme evaluated biosimilarity of CT‐P17 and reference adalimumab administered by PFS. 14 , 15 Patients with rheumatic disease have indicated a preference for AI over PFS administration of medications 16 , 17 , 18 , 19 , 20 ; therefore, the availability of an AI device option for CT‐P17 administration could benefit patients. In accordance with FDA recommendations for the development of drug–device combinations for RA treatment, 24 this study compared the PK profile for a single 40‐mg dose of CT‐P17 AI and CT‐P17 PFS. The findings from this study establish the PK equivalence of CT‐P17 AI and CT‐P17 PFS. Human factor studies (unpublished data on file, Celltrion, Inc., Incheon, Republic of Korea) have established that CT‐P17 AI is safe and effective for the intended users, uses and use environments, and a study in the CT‐P17 clinical development programme 36 confirmed the usability of CT‐P17 AI in patients with RA (unpublished data on file, Celltrion, Inc.).
Limitations of this study include that it was not powered to formally assess safety, so there could be numerical differences in safety findings between groups that may not reflect true differences. Enrolment of both male and female subjects might increase variability in PK findings beyond that explained by differences in body weight, since drug PK profiles can differ between males and females, 37 including those reported for reference adalimumab. 38 , 39
As an adalimumab biosimilar with a concentration of 100 mg/mL, replicating the high‐concentration formulation of reference adalimumab, CT‐P17 offers potential benefits for patients. Patients with plaque psoriasis, inflammatory bowel disease, or adult uveitis who are initiating adalimumab would require fewer injections than if they were treated with low‐concentration reference or biosimilar adalimumab. Although reports vary, citrate‐buffered vs. citrate‐free drug formulations have been associated with increased injection‐site pain on subcutaneous administration, possibly due to greater tissue pH changes with citrate‐containing formulations. 40 , 41 Indeed, reduced discomfort during injection has been observed with citrate‐free preparations of reference adalimumab and the adalimumab biosimilar ABP 501 in RA, FKB327 in a pooled analysis of patients with RA and healthy subjects, ABP 501 in plaque psoriasis, and reference adalimumab in inflammatory bowel disease. 42 , 43 , 44 , 45 In addition to the citrate‐free buffer composition, the smaller needle gauge (29‐ vs. 27‐gauge) and lower injection volume compared with the low‐concentration reference adalimumab formulation 46 may contribute to the minimal local site pain reported in this study, since these are 3 major product‐related factors that influence injection‐site pain. 40 , 41
In summary, this study demonstrated equivalence of PK parameters for CT‐P17 AI and CT‐P17 PFS in healthy subjects. Overall safety and immunogenicity profiles were comparable for the 2 administration methods. These results suggest that CT‐P17 administered via AI could be an alternative option for adalimumab treatment.
COMPETING INTERESTS
Jonathan Kay has received consultancies, speaker fees and/or honoraria from AbbVie, Alvotech Suisse AG, Boehringer Ingelheim GmbH, Celltrion Healthcare Co. Ltd, Mylan Inc., Merck & Co., Inc., Novartis AG, Samsung Bioepis, Sandoz and UCB. Edward Keystone has received speaker honoraria and/or consultancies from AbbVie, Amgen, Celltrion, Gilead, Janssen, Lilly, Merck, Pfizer, Roche, Samsung Bioepis, Sandoz and Sanofi. Sang Joon Lee, Sung Hyun Kim, Yun Ju Bae and Eun Jin Choi are employed by Celltrion, Inc. Daniel E. Furst has received grant/research support from Corbus, CSL Behring, Galapagos, Gilead, GlaxoSmithKline, Kadmon, PICORI, Pfizer and Talaris, and consultancies from AbbVie, Amgen, Boehringer Ingelheim, Corbus, CSL Behring, Galapagos, Gilead, Novartis, Pfizer, Roche/Genentech and Talaris. Antonia Davidson and Darin Brimhall have no conflicts of interest to report.
CONTRIBUTORS
All authors made substantial contributions to study conception and design, acquisition of data, or analysis and interpretation of data; were involved in drafting of the article or revising it critically for important intellectual content; and gave their final approval of the version to be published.
PRIOR PRESENTATION
The manuscript has not been previously published and the manuscript is not under consideration elsewhere. Selected results from this study were presented as a poster at the American College of Rheumatology (ACR) Convergence 2020 (5–9 November 2020; virtual).
Supporting information
TABLE S1 Study assessment time points
TABLE S2 Primary serum PK parameters for CT‐P17 AI and CT‐P17 PFS for ADA‐positive subjects a (PK population b)
FIGURE S1 Primary PK endpoints by ADA titre (PK population).
TABLE S3 TEAEs by SOC for CT‐P17 AI and CT‐P17 PFS (safety population)
TABLE S4 TEAEs reported by ≥3% of subjects in either treatment group (safety population)
ACKNOWLEDGEMENTS
We thank all study investigators, staff and individuals who contributed to this study. Medical writing support, including development of a draft outline and subsequent drafts in consultation with the authors, collating author comments, copyediting, fact checking and referencing, was provided by Beatrice Tyrrell, DPhil, at Aspire Scientific Limited (Bollington, UK). Funding for medical writing support for this article was provided by Celltrion, Inc. (Incheon, Republic of Korea). This study was funded by Celltrion, Inc. (Incheon, Republic of Korea).
Davidson A, Brimhall D, Kay J, et al. Randomised, phase I pharmacokinetic study of adalimumab biosimilar CT‐P17 (40 mg/0.4 mL) by autoinjector and prefilled syringe in healthy subjects. Br J Clin Pharmacol. 2021;87(11):4323-4333. 10.1111/bcp.14850
The authors confirm that the Principal Investigators for this paper were Antonia Davidson and Darin Brimhall and that they had direct clinical responsibility for patients.
Summary for Twitter/Facebook: Pharmacokinetic equivalence demonstrated for single‐dose administration of CT‐P17 by autoinjector or prefilled syringe in healthy adults.
Funding information Celltrion, Inc.
DATA AVAILABILITY STATEMENT
Available data and methodological information for this ongoing study are included in this article and accompanying supplementary materials.
REFERENCES
- 1. US Food and Drug Administration . Humira Prescribing Information 2021. https://www.accessdata.fda.gov/drugsatfda_docs/label/2021/125057s417lbl.pdf. Accessed March 16, 2021.
- 2. European Medicines Agency . Humira Summary of Product Characteristics 2021. https://www.ema.europa.eu/en/documents/product-information/humira-epar-product-information_en.pdf. Accessed March 16, 2021.
- 3. Bellinvia S, Cummings JRF, Ardern‐Jones MR, Edwards CJ. Adalimumab biosimilars in Europe: an overview of the clinical evidence. BioDrugs. 2019;33(3):241‐253. [DOI] [PubMed] [Google Scholar]
- 4. Singh JA, Saag KG, Bridges SL Jr, et al. 2015 American College of Rheumatology guideline for the treatment of rheumatoid arthritis. Arthritis Rheumatol. 2016;68(1):1‐25. [DOI] [PubMed] [Google Scholar]
- 5. Smolen JS, Landewé R, Bijlsma J, et al. EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease‐modifying antirheumatic drugs: 2016 update. Ann Rheum Dis. 2017;76(6):960‐977. [DOI] [PubMed] [Google Scholar]
- 6. Lau CS, Chia F, Dans L, et al. 2018 update of the APLAR recommendations for treatment of rheumatoid arthritis. Int J Rheum Dis. 2019;22(3):357‐375. [DOI] [PubMed] [Google Scholar]
- 7. US Food and Drug Administration . Scientific considerations in demonstrating biosimilarity to a reference product: guidance for industry 2015. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/scientific-considerations-demonstrating-biosimilarity-reference-product. Accessed March 16, 2021.
- 8. European Medicines Agency . Guideline on similar biological medicinal products containing biotechnology‐derived proteins as active substance: nonclinical and clinical issues 2014. https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐similar‐biological‐medicinal‐products‐containing‐biotechnology‐derived‐proteins‐active_en‐2.pdf. Accessed March 16, 2021.
- 9. Generics and Biosimilars Initiative . Biosimilars of adalimumab 2021. http://www.gabionline.net/Biosimilars/General/Biosimilars-of-adalimumab. Accessed March 16, 2021.
- 10. IQVIA . The impact of biosimilar competition in Europe 2019. https://ec.europa.eu/docsroom/documents/38461/attachments/1/translations/en/renditions/native. Accessed March 16, 2021.
- 11. Jensen TB, Kim SC, Jimenez‐Solem E, Bartels D, Christensen HR, Andersen JT. Shift from adalimumab originator to biosimilars in Denmark. JAMA Intern Med. 2020;180(6):902‐903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. European Medicines Agency . Yuflyma https://www.ema.europa.eu/en/medicines/human/EPAR/yuflyma. Accessed March 16, 2021.
- 13. European Medicines Agency . Yuflyma Summary of Product Characteristics 2021. https://www.ema.europa.eu/en/documents/product-information/yuflyma-epar-product-information_en.pdf. Accessed March 16, 2021.
- 14. Yu KS, Jang IJ, Lim HS, et al. Pharmacokinetic equivalence of CT‐P17 to high‐concentration (100 mg/ml) reference adalimumab: A randomized phase I study in healthy subjects. Clin Transl Sci. 2021;1‐12. 10.1111/cts.12967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kay J, Jaworski J, Wojciechowski R, et al. Efficacy and safety of biosimilar CT‐P17 versus reference adalimumab in subjects with rheumatoid arthritis: 24‐week results from a randomized study. Arthritis Res Ther. 2021;23(1):51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ghil J, Zielińska A, Lee Y. Usability and safety of SB5 (an adalimumab biosimilar) prefilled syringe and autoinjector in patients with rheumatoid arthritis. Curr Med Res Opin. 2019;35(3):497‐502. [DOI] [PubMed] [Google Scholar]
- 17. Kivitz A, Cohen S, Dowd JE, et al. Clinical assessment of pain, tolerability, and preference of an autoinjection pen versus a prefilled syringe for patient self‐administration of the fully human, monoclonal antibody adalimumab: the TOUCH trial. Clin Ther. 2006;28(10):1619‐1629. [DOI] [PubMed] [Google Scholar]
- 18. Rho YH, Rychlewska‐Hańczewska A, Śliwowska B, Kim TH. Usability of prefilled syringe and autoinjector for SB4 (an etanercept biosimilar) in patients with rheumatoid arthritis. Adv Ther. 2019;36(9):2287‐2295. [DOI] [PubMed] [Google Scholar]
- 19. Borrás‐Blasco J, Gracia‐Pérez A, Rosique‐Robles JD, Casterá MD‐E, Abad FJ. Acceptability of switching adalimumab from a prefilled syringe to an autoinjection pen. Expert Opin Biol Ther. 2010;10(3):301‐307. [DOI] [PubMed] [Google Scholar]
- 20. Saraux A, Hudry C, Zinovieva E, Herman‐Demars H, Self‐I Investigators group . Use of auto‐injector for methotrexate subcutaneous self‐injections: high satisfaction level and good compliance in SELF‐I study, a randomized, open‐label, parallel group study. Rheumatol Ther. 2019;6(1):47‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xiao X, Li W, Clawson C, Karvani D, Sondag P, Hahn JK. Evaluation of performance, acceptance, and compliance of an auto‐injector in healthy and rheumatoid arthritic subjects measured by a motion capture system. Patient Prefer Adherence. 2018;12:515‐526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schwarzenbach F, Dao Trong M, Grange L, et al. Results of a human factors experiment of the usability and patient acceptance of a new autoinjector in patients with rheumatoid arthritis. Patient Prefer Adherence. 2014;8:199‐209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cohen S, Klimiuk PA, Krahnke T, Assudani D. Successful administration of BI 695501, an adalimumab biosimilar, using an autoinjector (AI): results from a Phase II open‐label clinical study (VOLTAIRE®‐RL). Expert Opin Drug Deliv. 2018;15(6):545‐548. [DOI] [PubMed] [Google Scholar]
- 24. US Food and Drug Administration . Rheumatoid arthritis: developing drug products for treatment ‐ Draft guidance 2013. https://www.fda.gov/regulatory‐information/search‐fda‐guidance‐documents/rheumatoid‐arthritis‐developing‐drug‐products‐treatment. Accessed March 16, 2021.
- 25. European Medicines Agency . Guideline on immunogenicity assessment of therapeutic proteins 2017. https://www.ema.europa.eu/en/documents/scientific‐guideline/guideline‐immunogenicity‐assessment‐therapeutic‐proteins‐revision‐1_en.pdf. Accessed March 16, 2021.
- 26. US Food and Drug Administration . Immunogenicity testing of therapeutic protein products — developing and validating assays for anti‐drug antibody detection. Guidance for industry 2019. https://www.fda.gov/media/119788/download. Accessed March 16, 2021.
- 27. Shin D, Lee Y, Jeong D, Ellis‐Pegler R. Comparative pharmacokinetics of an adalimumab biosimilar SB5 administered via autoinjector or prefilled syringe in healthy subjects. Drug des Devel Ther. 2018;12:3799‐3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bush J, Kawakami K, Muniz R. A phase 1, randomized, open‐label, single‐dose study to assess the relative bioavailability of a subcutaneous dose of FKB327 when administered using a prefilled syringe, a prefilled auto‐injector, or a vial with disposable syringe in healthy subjects. BMC Pharmacol Toxicol. 2019;20(1):87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ramael S, Van Hoorick B, Tiessen R, et al. Similar pharmacokinetics of the adalimumab (Humira®) biosimilar BI 695501 whether administered via subcutaneous autoinjector or prefilled syringe (VOLTAIRE®‐AI and VOLTAIRE®‐TAI): phase 1, randomized, open‐label, parallel‐group trials. Rheumatol Ther. 2018;5(2):403‐421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. von Richter O, Lemke L, Haliduola H, et al. GP2017, an adalimumab biosimilar: pharmacokinetic similarity to its reference medicine and pharmacokinetics comparison of different administration methods. Expert Opin Biol Ther. 2019;19(10):1075‐1083. [DOI] [PubMed] [Google Scholar]
- 31. Amaravadi L, Song A, Myler H, et al. 2015 White Paper on recent issues in bioanalysis: focus on new technologies and biomarkers (Part 3‐‐LBA, biomarkers and immunogenicity). Bioanalysis. 2015;7(24):3107‐3124. [DOI] [PubMed] [Google Scholar]
- 32. Shankar G, Devanarayan V, Amaravadi L, et al. Recommendations for the validation of immunoassays used for detection of host antibodies against biotechnology products. J Pharm Biomed Anal. 2008;48(5):1267‐1281. [DOI] [PubMed] [Google Scholar]
- 33. Shin D, Lee Y, Kim H, Körnicke T, Fuhr R. A randomized phase I comparative pharmacokinetic study comparing SB5 with reference adalimumab in healthy volunteers. J Clin Pharm Ther. 2017;42(6):672‐678. [DOI] [PubMed] [Google Scholar]
- 34. Wynne C, Altendorfer M, Sonderegger I, et al. Bioequivalence, safety and immunogenicity of BI 695501, an adalimumab biosimilar candidate, compared with the reference biologic in a randomized, double‐blind, active comparator phase I clinical study (VOLTAIRE®‐PK) in healthy subjects. Expert Opin Investig Drugs. 2016;25(12):1361‐1370. [DOI] [PubMed] [Google Scholar]
- 35. Kaur P, Chow V, Zhang N, Moxness M, Kaliyaperumal A, Markus R. A randomised, single‐blind, single‐dose, three‐arm, parallel‐group study in healthy subjects to demonstrate pharmacokinetic equivalence of ABP 501 and adalimumab. Ann Rheum Dis. 2017;76(3):526‐533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. ClinicalTrials.gov . A study to evaluate usability of subcutaneous auto‐injector of CT‐P17 in patients with active rheumatoid arthritis [NCT04171414] 2019. https://clinicaltrials.gov/ct2/show/NCT04171414. Accessed March 16, 2021.
- 37. Soldin OP, Mattison DR. Sex differences in pharmacokinetics and pharmacodynamics. Clin Pharmacokinet. 2009;48(3):143‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ternant D, Ducourau E, Fuzibet P, et al. Pharmacokinetics and concentration‐effect relationship of adalimumab in rheumatoid arthritis. Br J Clin Pharmacol. 2015;79(2):286‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. US Food and Drug Administration . Humira Clinical Pharmacology and Biopharmaceutics Review(s) 2002. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2002/BLA_125057_S000_HUMIRA_BIOPHARMR.PDF. Accessed March 16, 2021.
- 40. St Clair‐Jones A, Prignano F, Goncalves J, Paul M, Sewerin P. Understanding and minimising injection‐site pain following subcutaneous administration of biologics: a narrative review. Rheumatol Ther. 2020;7(4):741‐757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Srivastava A, Brophy G, Agarkhed M. Approaches to alleviating subcutaneous injection‐site pain for citrate formulations. Pharm Tech. 2020;44:32‐37. [Google Scholar]
- 42. Nash P, Vanhoof J, Hall S, et al. Randomized crossover comparison of injection site pain with 40 mg/0.4 or 0.8 mL formulations of adalimumab in patients with rheumatoid arthritis. Rheumatol Ther. 2016;3(2):257‐270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Markus R, McBride HJ, Ramchandani M, et al. A review of the totality of evidence supporting the development of the first adalimumab biosimilar ABP 501. Adv Ther. 2019;36(8):1833‐1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gely C, Marín L, Gordillo J, et al. Impact of pain associated with the subcutaneous administration of adalimumab. Gastroenterol Hepatol. 2020;43(1):9‐13. [DOI] [PubMed] [Google Scholar]
- 45. Alten R, Keller H, Boyce M, Yonemura T, Ito T, Genovese MC. Systematic analysis of injection‐site pain and reactions caused by subcutaneous administration of the adalimumab biosimilar FKB327 versus the adalimumab reference product via different delivery methods. GaBI J. 2020;9(3):108‐116. [Google Scholar]
- 46. US Food and Drug Administration . Humira Prescribing Information 2017. https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125057s402lbl.pdf. Accessed March 18, 2021.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 Study assessment time points
TABLE S2 Primary serum PK parameters for CT‐P17 AI and CT‐P17 PFS for ADA‐positive subjects a (PK population b)
FIGURE S1 Primary PK endpoints by ADA titre (PK population).
TABLE S3 TEAEs by SOC for CT‐P17 AI and CT‐P17 PFS (safety population)
TABLE S4 TEAEs reported by ≥3% of subjects in either treatment group (safety population)
Data Availability Statement
Available data and methodological information for this ongoing study are included in this article and accompanying supplementary materials.