

Abstract
Background:
REFINE was an exploratory, dose- and frequency-blinded, prospective, randomized, dose-ranging study in relapsing–remitting multiple sclerosis (RRMS) patients.
Objective:
To examine the efficacy, safety, and tolerability of natalizumab administered via various regimens in RRMS patients.
Methods:
Clinically stable RRMS patients previously treated with 300 mg natalizumab intravenously for ⩾12 months were randomized to one of six natalizumab regimens over 60 weeks: 300 mg administered intravenously or subcutaneously every 4 weeks (Q4W), 300 mg intravenously or subcutaneously every 12 weeks (Q12W), or 150 mg intravenously or subcutaneously Q12W. The primary endpoint was the mean cumulative number of combined unique active magnetic resonance imaging (MRI) lesions at week 60.
Results:
In total, 290 patients were enrolled. All Q12W dosing arms were associated with increased clinical and MRI disease activity and closed early; ⩾39.5% of patients in each Q12W arm met rescue criteria. In the 300 mg intravenous and subcutaneous Q4 W arms, the mean cumulative number of combined unique active MRI lesions was 0.23 and 0.02, respectively; annualized relapse rates were 0.07 and 0.08, respectively; and trough natalizumab serum levels and α4-integrin saturation were comparable.
Conclusion:
Natalizumab 300 mg subcutaneous Q4W was comparable to 300 mg intravenous Q4W dosing with respect to efficacy, pharmacokinetics/pharmacodynamics, and safety.
Keywords: Dosing interval, MRI, multiple sclerosis, natalizumab, neurology, relapses
Introduction
In patients with relapsing–remitting multiple sclerosis (RRMS), the currently approved natalizumab regimen of 300 mg administered intravenously (IV) every 4 weeks (Q4W) 1 significantly reduces disease activity on magnetic resonance imaging (MRI), clinical relapse rate, and confirmed disability worsening relative to placebo over 2 years of treatment. 2 In addition, the long-term safety profile and effectiveness of natalizumab administered IV have been demonstrated in clinical practice settings.3,4
An alternative administration route may, however, benefit patients on natalizumab. IV infusion requires visits to an infusion center, which may not be readily accessible to all patients with multiple sclerosis (MS). Subcutaneous (SC) administration could provide a more convenient ambulatory treatment setting. Patients with poor venous access could also benefit from SC administration. Evidence from other medical conditions indicates that many patients prefer SC over IV administration based on convenience5,6 and potential time savings. 7
In the phase 1 DELIVER study, SC and IV administration of natalizumab Q4W yielded similar serum trough concentrations and pharmacodynamic (PD) effects after repeated dosing and similar efficacy, safety, and immunogenicity. 8 In addition, dosing less than Q4W has been associated with significantly lower risk of progressive multifocal leukoencephalopathy (PML). 9 At the time of the design and planning of REFINE, observational studies and reports assessing natalizumab dosing interruption suggested that disease activity remained suppressed for ⩾3 months after the last dose.10–13 However, none of these studies prospectively addressed the effect of natalizumab interruption on disease reactivation using alternative dosing regimens in a controlled, blinded manner. REFINE examined the efficacy, safety, and tolerability of natalizumab administered using different doses, intervals, and routes in RRMS patients who were clinically stable while on approved natalizumab dosing (300 mg IV Q4W).
Methods
Study design and participants
REFINE (clinicaltrials.gov, NCT01405820) was an exploratory, dose- and frequency-blinded, prospective, randomized, dose-ranging phase 2 study. The 72-week study included 60 weeks of randomized, dose- and frequency-blinded treatment followed by 12 weeks of open-label treatment with natalizumab (300 mg IV Q4W; Figure 1).
Figure 1.

Study design.
EDSS: Expanded Disability Status Scale; IV: intravenous; PD: pharmacodynamics; PK: pharmacokinetics; Q4W: every 4 weeks; Q12W: every 12 weeks; SC: subcutaneous.
Enrollment in REFINE required a documented diagnosis of RRMS (Table 1). JC virus (JCV) seropositivity was not an exclusion criterion. Patients permanently discontinued study treatment if they developed PML, became pregnant, withdrew consent, or if medical issues necessitated unblinding or treatment discontinuation.
Table 1.
REFINE inclusion and exclusion criteria.
| Inclusion criteria |
|---|
| A documented diagnosis of RRMS |
| Aged 18–55 years, inclusive, at the time of informed consent |
| Free of MS relapse, as determined by the enrolling investigator, for 12 months prior to randomization |
| Received at least 11 doses of natalizumab during the 12 months prior to randomization, with no missed doses in the 3 months prior to randomization |
| Experienced ⩾2 documented clinical relapses or 1 relapse and ⩾1 Gd+ lesion on MRI unrelated to the relapse in the 12 months before initiating natalizumab approved dosing |
| Exclusion criteria |
| Known history of human immunodeficiency virus, hepatitis C (test for hepatitis C virus antibody), or hepatitis B virus (test for hepatitis B surface antigen and/or hepatitis B core antibody) |
| Positive for anti-natalizumab antibodies at screening visit |
| MRI positive for Gd+ lesions at study entry |
| Patients for whom MRI was contraindicated, e.g., had a pacemaker or other contraindicated implanted metal devices, had suffered or were at risk for side effects from gadolinium or had claustrophobia that could not be medically managed |
| History of any clinically significant disease (as determined by the investigator) that would preclude participation in a clinical study |
| History of malignant disease, including solid tumors and hematologic malignancies (with the exception of basal cell and squamous cell carcinomas of the skin that should have been completely excised and were considered cured) |
| History of transplantation or any anti-rejection therapy |
| History of severe allergic or anaphylactic reactions or known hypersensitivity to any drug |
| A clinically significant infectious illness (e.g., cellulitis, abscess, pneumonia, septicemia) within 30 days prior to screening, or PML or other opportunistic infections at any time |
| Signs or symptoms suggestive of any serious infection, based on medical history, physical examination or laboratory testing, as determined by the Investigator |
| Prior treatment with total lymphoid irradiation |
| Prior treatment with cladribine, mitoxantrone, fingolimod, T-cell or T-cell receptor vaccination, cyclophosphamide, cyclosporine, azathioprine, methotrexate, mycophenolate mofetil, or any therapeutic monoclonal antibody other than natalizumab within 24 months prior to randomization |
| Prior treatment with IV immunoglobulin, plasmapheresis or cytapheresis within 12 months prior to randomization |
| Treatment with IV or oral corticosteroids (topical corticosteroids were acceptable) or related products within 3 months prior to randomization |
| Female patients who considered becoming pregnant while in the study |
| Female patients who were pregnant or breastfeeding |
| History of drug or alcohol abuse within 2 years prior to entry per investigator judgment |
Gd+: gadolinium enhancing; IV: intravenous; MS: multiple sclerosis; PML: progressive multifocal leukoencephalopathy; RRMS: relapsing-remitting multiple sclerosis.
An ethics committee approved the study at each site. All patients provided written informed consent.
Interventions
Patients received natalizumab or placebo by infusion or injection at study visits Q4W. Patients were randomized equally to one of six natalizumab dosing regimens: 300 mg IV Q4W, 300 mg SC Q4W, 300 mg IV every 12 weeks (Q12W), 300 mg SC Q12W, 150 mg IV Q12W, or 150 mg SC Q12W. Patients who received active treatment Q12W received matching placebo at the week 4 and 8 study visits.
Study treatment was blinded for 60 weeks, after which patients received open-label natalizumab (300 mg IV) at weeks 60, 64, and 68. Patients, the study sponsor and study staff (except for the site pharmacist, who was unblinded to dispense treatment) were blinded to the study dose and frequency but not the administration route.
Patients could receive rescue treatment with high-dose corticosteroids or open-label natalizumab if they met any of the rescue criteria (Table 2). Those rescued with open-label natalizumab were advanced to the 60-week visit, received natalizumab 300 mg IV at that time and Q4W through week 68, and could remain in the study through week 72.
Table 2.
Rescue criteria.
| Rescue criteria |
|---|
| 1 new Gd+ lesion >0.8 cm3 or ⩾2 Gd+ lesions of any size |
| 1 new or newly enlarging T2 lesion >0.8 cm3 or ⩾2 new or newly enlarging T2 lesions of any size, compared with a previous scan performed as part of the study |
| A protocol-defined relapse, defined as new or recurrent (occurring ⩾30 days apart) neurological symptoms not associated with fever or infection, with a minimum duration of 24 hours and either an increase of ⩾1 grade in ⩾2 functional scales of the EDSS, an increase of ⩾2 grades in 1 functional scale of the EDSS, or a confirmed increase of ⩾1.5, ⩾1.0, or ⩾0.5 in EDSS score from a baseline score of 0.0, 1.0–5.5, or ⩾6.0, respectively, confirmed ⩾12 weeks after the initial increase |
| A confirmed increase of ⩾1.5, ⩾1.0, or ⩾0.5 in EDSS score from a baseline score of 0.0, 1.0–5.5, or ⩾6.0, respectively, confirmed ⩾12 weeks after the initial increase |
EDSS: Expanded Disability Status Scale; Gd+: gadolinium enhancing.
Patients who received only corticosteroids as rescue therapy could continue in the randomized treatment period.
Endpoints and assessments
The primary endpoint was the cumulative number of combined unique active lesions on MRI (scans at screening (week –4) and weeks 12, 24, 36, 48, and 60; Figure 1), calculated as the sum of new gadolinium-enhancing lesions and new or newly enlarging T2 hyperintense lesions that were non-enhancing on post-gadolinium T1-weighted scans. All scheduled MRI scans were evaluated at a central MRI reading center. Site staff involved with the study remained blinded to study scans.
Exploratory clinical efficacy endpoints included annualized relapse rate (ARR) at week 60, the proportion of patients who relapsed by week 60, the proportion of patients with 12-week confirmed disability worsening (defined as a ⩾1.5-point, ⩾1.0-point, or ⩾0.5-point increase from a baseline Expanded Disability Status Scale (EDSS) score of 0.0, 1.0–5.5, or ⩾6.0, respectively, sustained for 12 weeks ± 5 days) at week 60 and the change in EDSS score from baseline through week 60.
Data on pharmacokinetics (PK) and PD, clinical outcomes, and safety were collected and assessed at screening and/or baseline (week 0) and at weeks 12, 24, 36, 48, and 60.
PK of serum natalizumab concentrations were assessed using an enzyme-linked immunosorbent assay. PD were monitored using α4-integrin saturation and CD49d expression (corresponding to α4-integrin) by flow cytometry.
Safety and drug viability were monitored by adverse events (AEs), serious AEs (SAEs), physical examination findings, changes in clinical laboratory values, testing for the presence of anti-JCV antibodies and immunogenicity testing for anti-natalizumab antibodies.
Patients who tested positive for anti-natalizumab antibodies at two or more time points separated by ⩾42 days were considered persistently positive, while those with one positive test result but a negative result on retest were considered transiently positive. Once patients met the persistently positive criteria, they could not subsequently be classified as transiently positive.
Laboratory assessments were conducted at screening and week 60, and AEs were monitored on an ongoing basis. Safety across the six treatment arms was followed by a data and safety monitoring committee (DSMC), which could recommend closure of individual treatment arms in the event of safety concerns, including increased disease activity.
Statistical analyses
The study was planned to enroll 50 patients per treatment arm, which allowed for a 10% dropout rate and was expected to provide approximately 80% power to detect a trend toward 50% loss of efficacy with an alternative regimen compared with the standard regimen at a one-sided 0.1 level of significance. The study was not powered to test the noninferiority of alternative natalizumab dosing regimens.
Efficacy analyses were conducted using the modified intention-to-treat (mITT) population, which included all randomized patients who received ⩾1 dose of study treatment and had ⩾1 efficacy assessment. The unadjusted ARR was calculated as the total number of protocol-defined relapses experienced in each group divided by the duration of the randomized treatment period in days and multiplied by 365.25. The proportions of patients who met MRI rescue criteria, experienced a relapse, or had EDSS disability progression were estimated using the Kaplan–Meier method based on time to first relapse.
Efficacy endpoints were summarized using summary statistics for continuous or count variables and frequency distributions for categorical variables. A sensitivity analysis of EDSS scores excluded scores assessed within 29 days of a protocol-defined relapse. The duration of follow-up was insufficient to draw conclusions about clinical efficacy; thus, these endpoints were considered exploratory, and no formal comparisons between treatment arms were made.
Post hoc analyses were conducted in the mITT population to statistically compare the natalizumab 300 mg IV and SC Q4W arms with respect to the number and proportion of patients with combined unique active lesions since baseline. To account for the imbalance in mean T2 lesion volume at baseline between treatment arms, the number of combined unique active lesions since baseline was derived using a negative binomial regression with treatment arm and baseline T2 lesion volume as covariates and the log-transformed study duration as the offset variable. The proportion of patients with combined unique active lesions since baseline was analyzed using a logistic regression with treatment arm and baseline T2 lesion volume as covariates. Missing values were not imputed for the efficacy assessments.
The PK, PD, and safety populations included all patients who received ⩾1 dose of study treatment and had ⩾1 postbaseline assessment of PK, PD, or safety parameters.
Results
Study population
Study enrollment began in December 2010; the study was completed in October 2014. The study enrolled 290 patients from five countries (Belgium, Germany, Spain, France, and Italy; Figure 2). Of these patients, 289 received ⩾1 dose of study treatment and had ⩾1 postbaseline assessment and were thus included in the PK, PD, and safety populations. The mITT population included 277 patients who had ⩾1 efficacy assessment. Eleven patients (3.8%) were excluded from the mITT population because they received incorrect study treatment; one additional patient was excluded due to a diagnosis of PML. Baseline demographics and disease characteristics were comparable across the six treatment arms (Table 3).
Figure 2.

CONSORT flow diagram.
IV: intravenous; Q4W: every 4 weeks; Q12W: every 12 weeks; SC: subcutaneous.
aOne patient randomized to this cohort received no doses of study medication.
Table 3.
Patient baseline demographics and disease characteristics.
| Characteristic | Natalizumab 300 mg IV Q4W (n = 54) | Natalizumab 300 mg SC Q4W (n = 45) | Natalizumab 300 mg IV Q12W (n = 52) | Natalizumab 300 mg SC Q12W (n = 54) | Natalizumab 150 mg IV Q12W (n = 47) | Natalizumab 150 mg SC Q12W (n = 38) | Total (N = 290) |
|---|---|---|---|---|---|---|---|
| Age, mean (SD) | 38.4 (7.84) | 36.3 (8.92) | 38.7 (8.43) | 38.7 (7.85) | 38.7 (8.61) | 36.0 (9.03) | 37.9 (8.41) |
| Sex, female, n (%) | 39 (72.2) | 29 (64.4) | 37 (71.2) | 41 (75.9) | 34 (72.3) | 24 (63.2) | 204 (70.3) |
| Body weight, kg | |||||||
| n | 53 | 45 | 51 | 53 | 47 | 37 | 286 |
| Mean (SD) | 69.99 (16.579) | 70.82 (14.809) | 69.30 (12.629) | 68.37 (13.747) | 65.87 (12.885) | 69.28 (19.110) | 68.93 (14.894) |
| EDSS score, median (range) | 3.0 (0.0–6.5) | 2.5 (0.0–6.0) | 2.5 (0.0–6.0) | 2.5 (0.0–6.5) | 2.5 (0.0–6.0) | 2.5 (0.0–6.0) | 2.5 (0.0–6.5) |
| Time since MS diagnosis, y, mean (SD) | 9.7 (5.20) | 9.0 (6.08) | 9.5 (5.51) | 9.8 (6.08) | 9.6 (6.63) | 8.9 (4.66) | 9.5 (5.71) |
| Time since initiation of natalizumab, y, mean (SD) | 3.2 (1.50) | 2.7 (1.34) | 3.1 (1.46) | 2.9 (1.35) | 2.9 (1.22) | 2.8 (1.39) | 3.0 (1.38) |
| No. of natalizumab infusions prior to randomization, mean (SD) | 36.2 (15.14) | 31.0 (16.01) | 37.0 (15.53) | 34.7 (16.43) | 35.8 (15.74) | 34.4 (15.22) | 34.9 (15.68) |
| No. of Gd+ lesions, mean (SD) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) |
| T2 lesion volume, cm3, mean (SD) a | 11.3 (11.3) | 8.7 (8.4) | 10.9 (12.2) | 13.5 (13.7) | 11.0 (10.4) | 12.2 (10.4) | 11.3 (11.3) |
| Non-Gd+ T1 lesion volume, cm3, mean (SD) a | 4.7 (6.2) | 3.3 (4.9) | 4.0 (5.4) | 5.4 (7.6) | 4.3 (5.6) | 4.9 (5.2) | 4.4 (5.9) |
EDSS: Expanded Disability Status Scale; IV: intravenous; Q4W: every 4 weeks; Q12W: every 12 weeks; SC: subcutaneous.
MRI data were available for 46 patients in the 150 mg IV Q12W arm and 37 patients in the 150 mg SC Q12W arm.
During the study, the DSMC recommended that the four Q12W treatment arms (n = 191) be closed due to increased disease activity compared with the 300 mg IV Q4W arm. Fifteen patients (7.9%) in the Q12W arms were exposed to all 15 doses of study treatment during the randomized treatment period (Figure 2).
Efficacy
At week 60, the mean (SD) number of combined unique active lesions since baseline was 0.23 (1.262) in the natalizumab 300 mg IV Q4W arm and 0.02 (0.151) in the 300 mg SC Q4W arm (Figure 3(a)). Results of post hoc analyses correcting for T2 lesion volume at baseline indicated that during the 60-week period, the estimated mean number of combined unique active lesions was 0.035 (95% CI: 0.002–0.567) in the 300 mg SC Q4W arm and 0.209 (95% CI: 0.032–1.372) in the 300 mg IV Q4W arm, with an odds ratio of 0.167 (95% CI: 0.004–6.324; p = 0.3348). After 60 weeks of treatment, one patient in the 300 mg SC Q4W arm and four patients in the 300 mg IV Q4W arm exhibited ⩾1 combined unique active lesion since baseline (Figure 3(b)). The estimated proportion of patients with any combined unique active lesion since baseline was 0.023 (95% CI: 0.003–0.170) in the 300 mg SC Q4W arm versus 0.073 (95% CI: 0.024–0.220) in the 300 mg IV Q4W arm, with an odds ratio of 0.320 (95% CI: 0.033–3.056; p = 0.3223).
Figure 3.

MRI results during the study. (a) Combined unique active MRI lesions at week 60. (b) Number of combined unique active lesions since baseline in each patient. (c) Cumulative probability of MRI activity meeting rescue criteria.
IV: intravenous; Q4W: every 4 weeks; Q12W: every 12 weeks; SC: subcutaneous.
Combined unique active lesions were detected in the Q12W treatment arms by week 12. MRI activity increased throughout the randomized period in these arms and was notably greater than in the Q4W arms (Figure 3(a)). The cumulative probability of MRI activity was higher (43.5%–70.3%) in the Q12W arms than the Q4W arms, though patient numbers were small (Figure 3(c)).
In the Q4W arms, the estimated cumulative probability of a protocol-defined relapse was 7.8% for IV treatment and 9.1% for SC treatment at week 60. The cumulative probability of protocol-defined relapse was higher in the Q12W arms (21.2%–58.8%), though patient numbers at later time points were small. At week 60, unadjusted ARRs were comparable in the Q4W treatment arms and were higher in the Q12W treatment arms (Figure 4).
Figure 4.
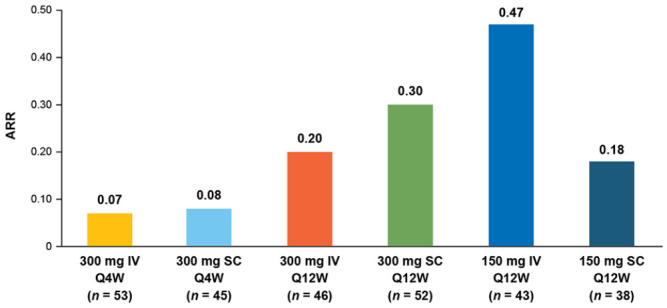
Unadjusted ARRs at week 60.
ARR: annualized relapse rate; IV: intravenous; Q4W: every 4 weeks; Q12W: every 12 weeks; SC: subcutaneous.
At week 60, the estimated proportion of patients with confirmed disability worsening was 5.9% in the 300 mg IV Q4W treatment arm and 4.8% in the 300 mg SC Q4W arm, and mean changes from baseline in EDSS scores were −0.16 and −0.04, respectively.
Ninety of 277 patients in the mITT population (32.5%) met rescue criteria for any reason during the randomized period. The proportions of patients meeting any of the rescue criteria were higher in the Q12W arms than the Q4W arms. Reasons for meeting rescue criteria are detailed in Table 4.
Table 4.
Patients who experienced an event meeting rescue criteria during the randomized treatment period.
| Rescue criteria, n (%) | Natalizumab 300 mg IV Q4W (n = 53) | Natalizumab 300 mg SC Q4W (n = 45) | Natalizumab 300 mg IV Q12W (n = 46) | Natalizumab 300 mg SC Q12W (n = 52) | Natalizumab 150 mg IV Q12W (n = 43) | Natalizumab 150 mg SC Q12W (n = 38) | Total (N = 277) |
|---|---|---|---|---|---|---|---|
| Patients who met rescue criteria for any reason | 9 (17.0) | 7 (15.6) | 21 (45.7) | 21 (40.4) | 17 (39.5) | 15 (39.5) | 90 (32.5) |
| Reason for meeting rescue criteria | |||||||
| Relapse | 4 (7.5) | 4 (8.9) | 8 (17.4) | 10 (19.2) | 10 (23.3) | 3 (7.9) | 39 (14.1) |
| New Gd+ lesions | 1 (1.9) | 0 | 10 (21.7) | 11 (21.2) | 7 (16.3) | 12 (31.6) | 41 (14.8) |
| New or newly enlarging T2 hyperintense lesions | 0 | 0 | 6 (13.0) | 5 (9.6) | 7 (16.3) | 5 (13.2) | 23 (8.3) |
| Confirmed disability worsening | 5 (9.4) | 3 (6.7) | 0 | 1 (1.9) | 0 | 0 | 9 (3.2) |
Gd+: gadolinium-enhancing; IV: intravenous; Q4W: every 4 weeks; Q12W: every 12 weeks; SC: subcutaneous.
PK and PD
Baseline serum natalizumab concentrations were comparable across treatment arms and were considered to reflect steady-state values. 14 Patients in the Q4W treatment arms maintained trough serum concentrations comparable to baseline throughout the randomized period, with mean values ranging from 31.0 to 43.7 μg/mL (Figure 5(a)), and no difference in trough serum concentration was observed between the IV and SC Q4W arms. In the Q12W arms, by contrast, mean trough serum concentrations decreased from baseline values to levels of 2.02–13.36 μg/mL by week 12 and remained low or below the limit of quantitation throughout the remainder of the study.
Figure 5.

Pharmacokinetics and pharmacodynamics. (a) Trough serum natalizumab concentration. (b) Trough α4-integrin saturation. (c) Trough CD49d expression. The mean was not calculated if there were data for ⩽2 patients at any given time point. Missing data are due to values being below the limit of quantitation.
IV: intravenous; Q4W: every 4 weeks; Q12W: every 12 weeks; SC: subcutaneous; SD: standard deviation.
At baseline, mean predose α4-integrin saturation levels were 78.2%–81.8% across all treatment groups. Mean trough α4-integrin saturation levels remained near baseline values throughout the randomized period in the Q4W treatment arms, with values ranging from 76.8% to 83.1% (Figure 5(b)). In the Q12W arms, by contrast, mean trough α4-integrin saturation levels decreased to 16.0%–24.3% by week 12. No difference in α4-integrin saturation was observed between the IV and SC Q4W arms.
Median CD49d expression remained comparable to baseline values through week 60 in the Q4W treatment arms. In the Q12W treatment arms, mean fluorescence intensity (MFI) increased from baseline to week 12 by a mean of 85.6–153.4 and remained elevated throughout the randomization period (Figure 5(c)).
Safety, tolerability, and immunogenicity
AEs were reported for 223 of 289 patients (77.2%), most of which were mild in severity and considered unrelated to study treatment (Table 5). In the overall study population, 27.3% of patients experienced AEs considered related to study treatment during the randomized treatment period. Rates of the most common treatment-related AEs were consistently low in both the IV and SC Q4W treatment arms (Table 6).
Table 5.
Incidence of AEs.
| AE, n (%) | Natalizumab 300 mg IV Q4W (n = 54) | Natalizumab 300 mg SC Q4W (n = 45) | Natalizumab 300 mg IV Q12W (n = 52) | Natalizumab 300 mg SC Q12W (n = 53) | Natalizumab 150 mg IV Q12W (n = 47) | Natalizumab 150 mg SC Q12W (n = 38) | Total (N = 289) |
|---|---|---|---|---|---|---|---|
| Any AE | 48 (88.9) | 37 (82.2) | 41 (78.8) | 41 (77.4) | 34 (72.3) | 22 (57.9) | 223 (77.2) |
| AEs of interest, by preferred term | |||||||
| Drug hypersensitivity | 1 (1.9) | 0 | 0 | 0 | 0 | 0 | 1 (< 1) |
| Hypersensitivity | 0 | 1 (2.2) | 0 | 0 | 0 | 0 | 1 (< 1) |
| Erythema | 2 (3.7) | 1 (2.2) | 0 | 0 | 0 | 0 | 3 (1.0) |
| Pruritus | 0 | 1 (2.2) | 1 (1.9) | 0 | 0 | 0 | 2 (< 1) |
| Dermatitis | 1 (1.9) | 0 | 0 | 0 | 0 | 0 | 1 (< 1) |
| Dermatitis allergic | 0 | 1 (2.2) | 0 | 0 | 0 | 0 | 1 (< 1) |
| Pruritus generalized | 0 | 0 | 0 | 1 (1.9) | 0 | 0 | 1 (< 1) |
| Rash | 0 | 0 | 0 | 0 | 1 (2.1) | 0 | 1 (< 1) |
| Rash generalized | 0 | 0 | 1 (1.9) | 0 | 0 | 0 | 1 (< 1) |
| Rash maculopapular | 0 | 0 | 0 | 1 (1.9) | 0 | 0 | 1 (< 1) |
| Urticaria | 0 | 1 (2.2) | 0 | 0 | 0 | 0 | 1 (< 1) |
| Injection site pain | 0 | 1 (2.2) | 0 | 3 (5.7) | 0 | 3 (7.9) | 7 (2.4) |
| Administration site pain | 0 | 1 (2.2) | 0 | 0 | 0 | 0 | 1 (< 1) |
| Injection site erythema | 0 | 0 | 0 | 1 (1.9) | 0 | 0 | 1 (< 1) |
| Injection site hematoma | 0 | 0 | 0 | 1 (1.9) | 0 | 0 | 1 (< 1) |
| Other injection site reaction | 0 | 1 (2.2) | 0 | 0 | 0 | 0 | 1 (< 1) |
| Patients with severe AEs | 4 (7.4) | 3 (6.7) | 1 (1.9) | 3 (5.7) | 1 (2.1) | 1 (2.6) | 13 (4.5) |
| Patients with serious AEs a | 7 (13.0) | 4 (8.9) | 4 (7.7) | 3 (5.7) | 4 (8.5) | 1 (2.6) | 23 (8.0) |
| Discontinuations from randomized treatment due to AE | 3 (5.6) | 5 (11.1) | 9 (17.3) | 4 (7.5) | 6 (12.8) | 5 (13.2) | 32 (11.1) |
| Study withdrawal due to AE | 2 (3.7) | 3 (6.7) | 3 (5.8) | 1 (1.9) | 1 (2.1) | 1 (2.6) | 11 (3.8) |
| PML b | 1 (1.9) | 0 | 0 | 0 | 0 | 0 | 1 (< 1) |
| Death c | 0 | 0 | 0 | 1 (1.9) | 0 | 0 | 1 (< 1) |
AE: adverse event; IV: intravenous; JCV: JC virus; PML: progressive multifocal leukoencephalopathy; Q4W: every 4 weeks; Q12W: every 12 weeks; SC: subcutaneous.
Serious AEs were defined as any occurrence that required hospitalization or prolongation of existing hospitalization, resulted in significant disability, resulted in death or placed a patient in immediate risk of death (in the view of the investigator) or resulted in a congenital anomaly.
One case of PML was reported and characterized as severe in a patient in the 300 mg IV Q4W treatment arm who had received a total natalizumab exposure of 34 infusions. The patient tested positive for anti-JCV antibodies and had received immunosuppressant therapy before starting natalizumab. The PML was considered related to study treatment and, following immune reconstitution, resolved 145 days after onset.
One patient who received 300 mg SC Q12W died due to metastatic pulmonary adenocarcinoma. This patient had a family history of lung cancer, and the death was considered unrelated to study treatment.
Table 6.
Incidence of treatment-related AEs.
| AE, n (%) | Natalizumab 300 mg IV Q4W (n = 54) | Natalizumab 300 mg SC Q4W (n = 45) | Natalizumab 300 mg IV Q12W (n = 52) | Natalizumab 300 mg SC Q12W (n = 53) | Natalizumab 150 mg IV Q12W (n = 47) | Natalizumab 150 mg SC Q12W (n = 38) | Total (N = 289) |
|---|---|---|---|---|---|---|---|
| Any treatment-related AE | 16 (29.6) | 20 (44.4) | 9 (17.3) | 16 (30.2) | 8 (17.0) | 10 (26.3) | 79 (27.3) |
| Treatment-related AEs that occurred in >2 patients across all treatment arms | |||||||
| Arthralgia | 2 (3.7) | 1 (2.2) | 0 (0) | 0 (0) | 2 (4.3) | 0 (0) | 5 (1.7) |
| Fatigue | 3 (5.6) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 0 (0) | 3 (1.0) |
| Headache | 2 (3.7) | 3 (6.7) | 1 (1.9) | 0 (0) | 0 (0) | 2 (5.3) | 8 (2.8) |
| Injection site pain | 0 (0) | 1 (2.2) | 0 (0) | 3 (5.7) | 0 (0) | 3 (7.9) | 7 (2.4) |
| MS relapse | 0 (0) | 4 (8.9) | 2 (3.8) | 7 (13.2) | 5 (10.6) | 1 (2.6) | 19 (6.6) |
| Nasopharyngitis | 2 (3.7) | 3 (6.7) | 4 (7.7) | 2 (3.8) | 0 (0) | 1 (2.6) | 12 (4.2) |
| Urinary tract infection | 3 (5.6) | 1 (2.2) | 1 (1.9) | 0 (0) | 0 (0) | 0 (0) | 5 (1.7) |
AE: adverse event; IV: intravenous; MS: multiple sclerosis; Q4W: every 4 weeks; Q12W: every 12 weeks; SC: subcutaneous.
Rates of AEs related to IV and SC administration, such as injection site reactions and reports of pain, were low; 10 of 136 patients (7.3%), all in the SC treatment arms, reported mild or moderate injection site reactions considered related to study treatment.
Twenty-three of 277 patients (8.3%) experienced a total of 26 SAEs during the randomized treatment period, with seven patients (2.5%) withdrawing from the study due to SAEs. One case of PML was reported in the 300 mg IV Q4W treatment arm, and one patient in the 300 mg SC Q12W arm died due to metastatic pulmonary adenocarcinoma (Table 5).
No evidence for immunogenicity was observed in patients who received natalizumab 300 mg IV or SC Q4W, as all patients in those treatment arms tested negative for anti-natalizumab antibodies throughout the randomized period. In the Q12W arms, two patients tested persistently positive for anti-natalizumab antibodies.
Discussion
The efficacy and PK/PD results from REFINE indicate that the SC and IV routes of administration for natalizumab appear to be comparable. The incidence of AEs was consistently low in the Q4W arms. Clinical assessments and MRI disease activity were comparable between the IV and SC arms of natalizumab 300 mg Q4W; the essentially “disease-free” efficacy findings were indistinguishable between the IV and SC treatment arms and consistent with the established efficacy of IV natalizumab 300 mg Q4W.2,3 With respect to PK and PD parameters, trough natalizumab concentration and trough α4-integrin saturation were comparable and overlapping in the 300 mg IV and SC Q4W arms.
In contrast, natalizumab administration Q12W (IV and SC) was associated with increased MRI disease activity and a greater number of clinical relapses, which led to premature closure of the four Q12W study arms. Although previous observational studies reported sustained suppression of clinical relapses and MRI disease activity for approximately 12 weeks after the last dose of natalizumab,12,15–17 the loss of efficacy observed in the Q12W arms of this blinded, prospective randomized trial is consistent with other data,14,18 which demonstrated that natalizumab’s effects are reversible and disease activity returns to baseline after approximately 8–12 weeks. The treatment arms with the lowest natalizumab exposure (150 mg Q12W) were closed first based on disease activity, followed by the 300 mg Q12W arms. This study lacked a 150 mg Q4W treatment arm, which would have evaluated an alternative approach to reducing natalizumab exposure.
The design and execution of REFINE predates other efforts that have explored alternative dosing of natalizumab.9,19 Although natalizumab Q12W did not maintain efficacy in REFINE, real-world studies have suggested that efficacy may be maintained with natalizumab administration every 6 weeks (Q6W) or every 8 weeks,18,20–22 which would allow small reductions in drug exposure relative to the current Q4W dosing regimen. In a retrospective analysis of US patients treated with natalizumab within a large longitudinal registry (the TOUCH Prescribing Program), 9 Q6W dosing was associated with significant reduction in PML risk compared with Q4W dosing in anti-JCV antibody positive patients with MS. 9 However, the TOUCH Prescribing Program database did not collect information regarding therapeutic efficacy; thus, no conclusions could be made regarding the benefit-risk profile of Q6W versus Q4W dosing. At the time of publication, a randomized controlled trial is ongoing to evaluate the efficacy of switching to natalizumab 300 mg IV Q6W in patients who are stable on Q4W dosing and to explore Q6W dosing in the context of SC administration (clinicaltrials.gov identifier NCT03689972).
Patients in REFINE were at a low risk of developing anti-natalizumab antibodies, as they had received natalizumab for ⩾1 year before starting the study, though two patients who received IV natalizumab in the Q12W arm became persistently positive for anti-natalizumab antibodies. In DELIVER, the incidence of anti-natalizumab seropositivity was low over 32 weeks, with only one patient in the SC group testing persistently positive. 8
Most AEs in the study were mild and considered unrelated to treatment, with 8% of patients experiencing SAEs. The incidence of injection site reactions in the SC treatment arms (7.3%) fell within or below the range observed for other MS treatments administered SC.23–25 For example, in studies of SC interferon MS treatments, injection site erythema, injection site pain, and injection site pruritus were reported in 32%–56%, 12%–14%, and 2%–11% of patients, respectively.23–25 In REFINE, there was one case of PML in the context of long-term administration of natalizumab prior to study entry.
The findings from REFINE are limited in their ability to inform clinical practice, and interpretation should be guided by the following considerations. Due to the exploratory nature of the study, no formal statistical comparisons were prespecified. In addition, REFINE was not powered to test the equivalence or noninferiority of the SC administration route and patients were not natalizumab naive prior to study enrollment. Notwithstanding these limitations, treatment arms were well balanced at baseline among the treatment groups.
In conclusion, natalizumab 300 mg SC Q4W was comparable to standard 300 mg IV Q4W with respect to clinical and MRI efficacy, PK/PD, safety, and immunogenicity. Administration of natalizumab Q12W was associated with increased MRI and relapse activity, leading to early closure of the Q12W study arms. These results suggest that SC administration of 300 mg natalizumab Q4W could be an alternative option for patients with RRMS.5–7
Acknowledgments
The authors would like to acknowledge the REFINE co-investigators for their efforts and contributions. The authors also thank Veronica Englishby for her contributions. Zheng Ren (Biogen at the time of these analyses) completed the statistical analyses for this study. Autumn Kelly and Alexandra D’Agostino, PhD, of Ashfield Healthcare Communications wrote the first draft of the manuscript based on input from authors, and Joshua Safran of Ashfield Healthcare Communications copyedited and styled the manuscript per journal requirements.
Footnotes
Authors’ note: Authors Stephanie Licata and Zheng Ren were affiliated to Biogen, Cambridge, MA, USA, at the time of these analyses.
Data sharing: The datasets generated and/or analyzed during the current study are not publicly available. The authors fully support sharing whenever possible. Requests for de-identified data should be made to Biogen via established company data-sharing policies and processes detailed on the website http://clinicalresearch.biogen.com/.
Declaration of Conflicting Interest: The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article: M.T. has received compensation for consulting from Biogen, Genzyme, Merck Serono, Novartis, and Roche and speaker honoraria from Biogen, Genzyme, Merck Serono, Novartis, Sanofi, and Teva, and her institution has received research grants from Biogen, Merck Serono, and Novartis. L.R.-T. has received fees for consulting and speaking and his unit has received research grants from Bayer Schering, Biogen, Merck Serono, Novartis, and Teva. L.M.E.G. has received travel funding and/or speaker honoraria from Biogen, Merck Serono, Novartis, Sanofi, and Teva and research support from Biogen and Merck Serono. C.L. has been a local investigator in therapeutic trials for Biogen, Genzyme, Novartis, and Roche; has received fees for speaking or attending advisory board meetings for Biogen, Genzyme, Ipsen, Novartis, Roche, and Vertex; has been involved in scientific collaboration with Vertex; and has received grants for research from Biogen. S.S. received fees for speaking or consulting from Bayer Healthcare, Biogen, Genzyme/Sanofi, Merck, Novartis, and Teva and research grants from Genzyme and Novartis at the time of the study, and he is now an employee of Roche, which was not involved in the study. K.C.E., Z.R., K.K.M., and A.R.G. are employees of and may hold stock and/or stock options in Biogen. S.L. was an employee of Biogen at the time of these analyses and may hold stock and/or stock options in Biogen.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by Biogen. Biogen also provided funding for medical writing support in development of this manuscript. Biogen reviewed the manuscript and provided feedback to the authors. The authors provided final approval of all content.
ORCID iD: Maria Trojano  https://orcid.org/0000-0002-6329-8946
https://orcid.org/0000-0002-6329-8946
Contributor Information
Maria Trojano, Department of Basic Medical Sciences, Neuroscience and Sense Organs, University of Bari “Aldo Moro,” Bari, Italy.
Lluís Ramió-Torrentà, Neurology Department, Dr. Josep Trueta University Hospital, Girona Biomedical Research Institute (IDIBGI), Medical Sciences Department, University of Girona, Girona, Spain.
Luigi ME Grimaldi, Unità Operativa Neurologia, Fondazione Istituto San Raffaele G. Giglio di Cefalù, Cefalù, Italy.
Catherine Lubetzki, Sorbonne University and Paris Brain Institute (ICM), Pitié-Salpêtrière Hospital, Department of Neurology, Assistance Publique–Hôpitaux de Paris, Paris, France.
Sven Schippling, Neuroimmunology and Multiple Sclerosis Research Section, Department of Neurology, University Hospital Zurich and University of Zurich, Zurich, Switzerland.
Karleyton C Evans, Biogen, Cambridge, MA, USA.
Zheng Ren, Biogen, Cambridge, MA, USA.
Kumar Kandadi Muralidharan, Biogen, Cambridge, MA, USA.
Stephanie Licata, Biogen, Cambridge, MA, USA.
Arie R Gafson, Biogen, Cambridge, MA, USA.
References
- 1. TYSABRI (natalizumab) [product insert]. Cambridge, MA: Biogen, 2020. [Google Scholar]
- 2. Polman CH, O’Connor PW, Havrdova E, et al. A randomized, placebo-controlled trial of natalizumab for relapsing multiple sclerosis. N Engl J Med 2006; 354: 899–910. [DOI] [PubMed] [Google Scholar]
- 3. Butzkueven H, Kappos L, Pellegrini F, et al. Efficacy and safety of natalizumab in multiple sclerosis: Interim observational programme results. J Neurol Neurosurg Psychiatry 2014; 85(11): 1190–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Foley J, Carrillo-Infante C, Smith J, et al. The 5-year Tysabri global observational program in safety (TYGRIS) study confirms the long-term safety profile of natalizumab treatment in multiple sclerosis. Mult Scler Relat Disord 2019; 39: 101863. [DOI] [PubMed] [Google Scholar]
- 5. Falanga M, Canzona A, Mazzoni D. Preference for subcutaneous injection or intravenous infusion of biological therapy among Italian patients with SLE. J Patient Exp 2019; 6: 41–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Pivot X, Gligorov J, Müller V, et al. Preference for subcutaneous or intravenous administration of trastuzumab in patients with HER2-positive early breast cancer (PrefHer): An open-label randomised study. Lancet Oncol 2013; 14(10): 962–970. [DOI] [PubMed] [Google Scholar]
- 7. Barbee MS, Harvey RD, Lonial S, et al. Subcutaneous versus intravenous bortezomib: Efficiency practice variables and patient preferences. Ann Pharmacother 2013; 47(9): 1136–1142. [DOI] [PubMed] [Google Scholar]
- 8. Plavina T, Fox EJ, Lucas N, et al. A randomized trial evaluating various administration routes of natalizumab in multiple sclerosis. J Clin Pharmacol 2016; 56(10): 1254–1262. [DOI] [PubMed] [Google Scholar]
- 9. Ryerson LZ, Foley J, Chang I, et al. Risk of natalizumab-associated PML in patients with MS is reduced with extended interval dosing. Neurology 2019; 93: e1452–e1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kerbrat A, Le Page E, Leray E, et al. Natalizumab and drug holiday in clinical practice: An observational study in very active relapsing remitting multiple sclerosis patients. J Neurol Sci 2011; 308: 98–102. [DOI] [PubMed] [Google Scholar]
- 11. Borriello G, Prosperini L, Marinelli F, et al. Observations during an elective interruption of natalizumab treatment: A post-marketing study. Mult Scler 2011; 17(3): 372–375. [DOI] [PubMed] [Google Scholar]
- 12. O’Connor PW, Goodman A, Kappos L, et al. Disease activity return during natalizumab treatment interruption in patients with multiple sclerosis. J Neurol 2011; 76: 1858–1865. [DOI] [PubMed] [Google Scholar]
- 13. West TW, Cree BA. Natalizumab dosage suspension: Are we helping or hurting? Ann Neurol 2010; 68: 395–399. [DOI] [PubMed] [Google Scholar]
- 14. Plavina T, Muralidharan KK, Kuesters G, et al. Reversibility of the effects of natalizumab on peripheral immune cell dynamics in MS patients. J Neurol 2017; 89: 1584–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. O’Connor PW, Goodman AD, Kappos L, et al. Return of disease activity after cessation of natalizumab therapy in patients with multiple sclerosis [abstract]. Mult Scler 2009; 15: S240–S241. [Google Scholar]
- 16. Berger JR, Centonze D, Comi G, et al. Considerations on discontinuing natalizumab for the treatment of multiple sclerosis. Ann Neurol 2010; 68(3): 409–411. [DOI] [PubMed] [Google Scholar]
- 17. Killestein J, Vennegoor A, Strijbis EM, et al. Natalizumab drug holiday in multiple sclerosis: Poorly tolerated. Ann Neurol 2010; 68(3): 392–395. [DOI] [PubMed] [Google Scholar]
- 18. Grimaldi LM, Prosperini L, Vitello G, et al. MRI-based analysis of the natalizumab therapeutic window in multiple sclerosis. Mult Scler 2012; 18(9): 1337–1339. [DOI] [PubMed] [Google Scholar]
- 19. Zhovtis Ryerson L, Li X, Goldberg JD, et al. Pharmacodynamics of natalizumab extended interval dosing in MS. Neurol Neuroimmunol Neuroinflamm 2020; 7(2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Berkovich R, Togasaki DM, Cen SY, et al. CD4 cell response to interval therapy with natalizumab. Ann Clin Transl Neurol 2015; 2(5): 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fox RJ, Cree BA, De Seze J, et al. MS disease activity in RESTORE: A randomized 24-week natalizumab treatment interruption study. Neurology 2014; 82: 1491–1498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Butzkueven H, Kappos L, Spelman T, et al. No significant difference in relapse outcomes in patients switching to natalizumab extended interval dosing or remaining on standard interval dosing: Propensity score comparative effectiveness analysis of patients in the TYSABRI Observational Program. Mult Scler 2019; 25: P1033. [Google Scholar]
- 23. Calabresi PA, Kieseier BC, Arnold DL, et al. Pegylated interferon beta-1a for relapsing-remitting multiple sclerosis (ADVANCE): A randomised, phase 3, double-blind study. Lancet Neurol 2014; 13: 657–665. [DOI] [PubMed] [Google Scholar]
- 24. De Stefano N, Sormani MP, Stubinski B, et al. Efficacy and safety of subcutaneous interferon β-1a in relapsing-remitting multiple sclerosis: Further outcomes from the IMPROVE study. J Neurol Sci 2012; 312: 97–101. [DOI] [PubMed] [Google Scholar]
- 25. Mikol DD, Barkhof F, Chang P, et al. Comparison of subcutaneous interferon beta-1a with glatiramer acetate in patients with relapsing multiple sclerosis (the REbif vs Glatiramer Acetate in Relapsing MS Disease [REGARD] study): A multicentre, randomised, parallel, open-label trial. Lancet Neurol 2008; 7(10): 903–914. [DOI] [PubMed] [Google Scholar]