

Abstract
BACKGROUND AND AIMS:
Histological and clinical outcomes in HBV-HIV coinfection in the era of combination antiretroviral therapy (cART) are poorly defined.
APPROACH AND RESULTS:
Adult patients co-infected with HBV-HIV from eight North American sites were enrolled in this National Institutes of Health (NIH)–funded prospective observational study (n = 139). Demographic, clinical, serological, and virological data were collected at entry and every 24 weeks for ≤ 192 weeks. Paired liver biopsies were obtained at study entry and at ≥ 3 years of follow-up. Biopsies were assessed by a central pathology committee using the modified Ishak scoring system. Clinical outcome rate and changes in histology are reported. Among participants with follow-up data (n = 114), median age was 49 years, 91% were male, 51% were non-Hispanic Black, and 13% had at-risk alcohol use, with a median infection of 20 years. At entry, 95% were on anti-HBV cART. Median CD4 count was 562 cells/mm3 and 93% had HIV < 400 copies/mL. HBeAg was positive in 61%, and HBV DNA was below the limit of quantification (< 20 IU/mL) in 61% and < 1,000 IU/mL in 80%. Clinical events were uncommon across follow-up: one hepatic decompensation, two HCC, no liver transplants, and one HBV-related deaths, with a composite endpoint rate of 0.61/100 person-years. Incident cirrhosis (n = 1), alanine aminotransferase flare (n = 2), and HBeAg loss (n = 13) rates were 0.40, 0.65, and 6.86 per 100 person-years, respectively. No participants had HBsAg loss. Paired biopsy (n = 62; median 3.6 years apart) revealed minimal improvement in Histologic Activity Index (median [interquartile range]: 3 [2–4] to 3 [1–3]; P = 0.02) and no significant change in fibrosis score (1 [1–2] to 1 [0–3]; P = 0.58).
CONCLUSIONS:
In a North American cohort of adults with HBV-HIV on cART with virological suppression, clinical outcomes and worsening histological disease were uncommon. (Hepatology 2021;74:1174–1189).
Due to shared routes of transmission, co-infection with HBV and HIV is common.(1–7) Advances in combination antiretroviral therapy (cART) have greatly reduced acquired immunodeficiency syndrome (AIDS)–related mortality. As a result, liver-related morbidity and mortality is now a major concern in HBV-HIV co-infection.(8,9) Despite the widespread use of effective antiviral agents for both HBV and HIV, liver-related mortality remains the second-leading cause of death among co-infected patients.(5,8)
Current HIV treatment guidelines from the Department of Health and Human Services recommend that all persons with HBV-HIV co-infection be treated with cART containing tenofovir plus lamivudine or emtricitabine.(1–3,10,11) Despite the well-established guidelines on antiviral regimens in the management of individuals with HBV-HIV co-infection,(1–3,10,11) there are limited prospective data on the clinical and histological impact of effective HBV suppression over time in those on cART.(12) Furthermore, because liver biopsy is not recommended in patients with HBV-HIV before or following initiation of cART, the changes in HBV-related histological disease in the era of tenofovir-containing cART is unknown. To address this knowledge gap, the HBV-HIV cohort, an NIH-funded ancillary study of the Hepatitis B Research Network (HBRN),(13) was initiated. The primary aim of this multicenter prospective cohort study was to assess histological changes by paired liver biopsy, in a well-characterized cohort of patients with HBV-HIV on cART in North America. Secondary aims were to describe clinical and virological outcomes in these participants to better define the natural history of clinical, biochemical, and serological events in this understudied population.
Materials and Methods
STUDY DESIGN
Adult patients with HBV-HIV co-infection were recruited from eight HBRN sites in the United States and Canada (Virginia Commonwealth University; University of California, San Francisco; University of Texas, Southwestern; Johns Hopkins University; University Health Network, Toronto; Washington University Saint Louis; Massachusetts General Hospital; Liver Diseases Branch, NIDDK; NIH) to participate in this prospective observational study. The study protocol specified that study participants be at least 18 years old, be anti-HIV positive, and HBsAg positive for at least 6 months, on cART including an anti-HBV nucleoside or nucleotide analogue, and agree to undergo a liver biopsy within 1 year of study entry and approximately 3–4 years later, regardless of clinical or laboratory tests. Although prior infection with HCV or delta was allowed, detectable HCV RNA less than 6 months from entry, decompensated cirrhosis, and HCC were exclusion criteria. Full eligibility criteria are reported in the Appendix. During follow-up, antiretroviral treatment could be stopped, initiated, or changed per standard of care at the discretion of a treating physician.
Following the baseline assessment, participants underwent evaluation every 24 weeks thereafter. Additional assessments occurred when participants experienced a flare in alanine aminotransferase (ALT) level, pregnancy, or had a liver biopsy that did not align with a per-protocol assessment. Study flow and measures by time point are provided in Supporting Figs. S1 and S2, respectively. Data were entered by study coordinators or the central lab and transmitted the HBRN Data Coordinating Center (University of Pittsburgh). Follow-up ended with liver transplant or death. Otherwise, participants were followed up to approximately 4 years (192 weeks) or January 31, 2020, whichever came first. The institutional review board at each center approved the protocol, and participants gave written, informed consent. The study is registered at ClinicalTrials.gov (NCT01924455).
ASSESSMENTS
The baseline and serial follow-up evaluations included assessments of demographics, medical history, and current health status, with self-report and interviewer-administered questionnaires, a physical examination, and blood tests, as previously described.(14) Relevant clinical, laboratory, and radiological data were extracted from medical records, including standard-of-care test from local laboratories (i.e., liver panel, clusters of differentiation [CD] 4 count and percentage, CD8 count and percentage, HIV RNA, anti-HCV, anti-HDV, HBV-DNA level, and HBV serologies [HBsAg, antibody against HBsAg (anti-HBs), HBeAg, and antibody against HBeAg (anti-HBe)]). In general, anti-HBe testing was limited to HBeAg-negative participants. HIV-RNA suppression was defined as <20 copies/mL. HIV stage (1–4) was defined by CD4 count at entry (≥500, 350–499, 200–349, and <200 cells/mm3, respectively) according to 2005 World Health Organization Guidelines.(15) HBV DNA was categorized as <20 (unquantifiable), 20 to <1,000 (suppressed), 1,000 to <20,000, and ≥20,000 IU/mL (not suppressed). We further categorized those as HBV/HIV-suppressed (HBV DNA < 1,000 IU/mL, HIV RNA < 20 copies/mL), not suppressed (HBV DNA ≥ 1,000 IU/mL, HIV RNA ≥ 20 copies/mL), and incomplete suppression (HBV DNA was ≥1,000 IU/mL, while HIV RNA was <20 copies/mL). Upper limit of normal (ULN) for ALT was defined as 30 U/L for men and 19 U/L for women.(16) Aspartate aminotransferase (AST) to platelet (PLT) ratio index (APRI)(17) and the fibrosis-4 (FIB-4) score ([age (year) × AST (U/L)]/[PLT (109/L) × ALT (U/L)1/2])(18) were calculated. Baseline age was used in the FIB-4 calculation at all time points, to avoid the impact of increasing age over time independent of disease progress.
Liver stiffness was measured through vibration-controlled transient elastography (VCTE) (FibroScan; EchoSens, Paris, France). All scans were performed by trained study personnel at each site. Participants were instructed to fast for at least 3 hours before assessment. At least 10 successful measurements were required to calculate the data point, median liver stiffness expressed in kilopascals (referred to simply as liver stiffness from here forward), across measurements. VCTE results were excluded due to fasting <3 hours, inadequate data (i.e., <10 measurements), or an interquartile range (IQR)/median liver stiffness ratio >0.3.
Duration of HIV and/or HBV as well as current and past cART use was collected but could not be verified in many subjects due to the fragmented care received from different health providers at various sites between the time of diagnosis and enrollment. Alcohol use was assessed with the alcohol use disorder identification test; increased risk of alcohol use disorder was defined as a score of 8–15, and high risk as a score ≥ 16.(19) Clinical assessment included waist circumference, height and weight, used to calculated body mass index (BMI), presence of diabetes mellitus (DM), and lipodystrophy grade (none, mild, moderate, or severe).(20) DM was defined by self-report, antidiabetic medications, or fasting glucose ≥126 mg/dL, and hyperlipidemia was defined by self-report, current use of lipid-lowering medication, or LDL ≥ 160 mg/dL.
Research blood samples were collected at each assessment, processed and stored at −70°C at each site, and shipped in batches to a central repository for subsequent transfer to central testing laboratories (University of Washington, Seattle, WA). Quantitative HBV DNA and HBeAg (tested every 24 weeks) and quantitative HBsAg (tested every 48 weeks) were performed at a central laboratory. HBV-DNA levels were determined using a real-time PCR assay (COBAS Ampliprep/COBAS TaqMan HBV Test, v2.0; Roche Molecular Diagnostics, Branchburg, NJ) with a lower limit of detection (LLOD) of 10 IU/mL and lower limit of quantification (LLOQ) of 20 IU/mL. Quantitative HBsAg and HBeAg were tested using the Roche Diagnostics Elecsys platform with LLOD of 0.05 IU/mL for HBsAg and LLOD of 0.3 IU/mL for HBeAg. When central laboratory results were missing, the following local laboratory results were used: qualitative HBsAg and HBeAg determined using commercially available enzyme immunoassays and genotype.
The non-histological primary clinical outcomes were hepatic decompensation, HCC, liver transplant, and HBV-related death. Given the low occurrence of individual outcomes, they were grouped and termed “composite clinical outcome.” Secondary clinical outcomes were incident cirrhosis (among participants without baseline cirrhosis), incident ALT flare (defined as ≥10 × ULN),(21) ever becoming HBsAg-negative, ever becoming HBeAg-negative (among participants who were HBeAg-positive at baseline), and non-HBV deaths. All outcomes were predefined, and the occurrence and timing of the clinical outcomes were adjudicated by an independent committee of HBRN clinical investigators to determine whether criteria for these outcomes were met. Cirrhosis was based on histology if available, and in the absence of biopsy, by presence of two of the following criteria: splenomegaly or nodular liver on radiological imaging performed for clinical care, or platelet count < 120,000/mm3. Participants meeting these criteria within 24 weeks after enrollment were considered to have baseline cirrhosis.
HISTOLOGY
Histological findings were scored blindly with respect to clinical data by the HBRN Pathology Committee (D.E.K., chair) using the modified Ishak scoring system.(22) The histologic activity index (HAI) is a global scale of hepatic inflammation ranging from 0 to 18, calculated as the sum of lobular (0–4), peri-portal (0–4) and portal inflammation (0–4), and confluent necrosis scores (0–6). The Ishak fibrosis score stages fibrosis from 0 to 6, with 0 indicating absence of fibrosis and 6 indicating likely or definite cirrhosis; ≥ 2 is considered significant fibrosis or worse; and ≥ 3 is considered advanced fibrosis or worse. Fatty liver disease (steatosis/steatohepatitis) was graded using the Kleiner scoring system.(23) Total length of biopsy was recorded. Although the number of portal tracts was not recorded, each biopsy was assessed for adequacy by the central pathology committee with a minimum of three portal tracts required to be adequate.(14)
ANALYSIS
Analyses were conducted using SAS, version 9.4 (SAS Institute Inc., Cary, NC). Descriptive statistics were used to report characteristics of the full analysis sample (n = 114) and the histology subsample (n = 62). The Pearson chi-square test or Fisher exact test for categorical variables, the Wilcoxon rank-sum test for continuous variables, and the Cochran-Armitage trend test for ordinal variables were used to compare characteristics of the main analysis sample to those excluded due to lack of follow-up data, and those in the histology subsample to those in the main sample excluded due to lack of histology data.
For each outcome measured throughout follow-up (hepatic decompensation, HCC, liver transplant, HBV-related death, composite clinical outcome, cirrhosis, ALT flare, HBsAg loss, and HBeAg loss), event rates per 100 person-years and corresponding CIs, assuming a Poisson distribution, are reported. Event rates for incident cirrhosis and HBeAg loss were calculated among the appropriate subgroups (n = 106 without cirrhosis, and n = 67 who were HBeAg-positive, respectively, at baseline).
Most outcomes were too rare for a time-to-event analysis. However, the Kaplan-Meier method was used to estimate the cumulative probability of HBeAg loss over time, and HBeAg loss rate is reported by select baseline factors (age, sex, race, ALT, AST, platelets, APRI, FIB-4, quantitative HBsAg [qHBsAg], HBV DNA, CD4, CD4%, HIV RNA, HAI, Ishak fibrosis score, cirrhosis, and fatty liver disease). Variables were categorized using standard thresholds when available. Adjacent categories were collapsed (e.g., age < 30, 30 to < 40, and age 40 to < 50 years) due to low frequency and similar estimates among categories.
Linear mixed models were used to estimate the mean of select viral (qHBsAg, quantitative HBeAg [qHBeAg], and HBV DNA) and clinical markers (ALT, AST, AST/ALT, platelets, APRI, FIB-4, and VCTE), with each outcome as a repeated measure, time, and site (which were related to missing follow-up data) entered as discrete fixed effects, and a random intercept. HBV-DNA classification schemes were also evaluated over time with an ordinal mixed model.
Among the paired biopsy subsample (n = 62), descriptive statistics were used to summarize inflammation and fibrosis by time point. Group-level changes were tested with mixed-effects models (generalized linear for continuous, ordinal logistic for ordinal, binomial for binary outcomes), with each outcome as a repeated measure, time (i.e., days since baseline biopsy) as a continuous fixed effect, and random intercept. To display these data graphically, the distribution of change in the HAI and fibrosis score between baseline and follow-up were each standardized per the maximum follow-up of 192 weeks (192 weeks*[follow-up value minus baseline value]/weeks between biopsies).(24) Individual-level changes are also shown with cross tabulations. Additionally, we determined the percentage (with 95% CIs) of participants who had at least a two-point worsening and two-point improvement in fibrosis score standardized per 192 weeks.
Spearman correlation was used to test and quantify associations between (1) change in each inflammation measure with change in fibrosis, (2) change in ALT with change in each inflammation, and (3) change in APRI and FIB-4, respectively, with change in fibrosis. Analysis including histology data and clinical data excludes participants whose clinical data were >24 weeks from the date of biopsy.
Generalized linear-mixed models were used to evaluate baseline predictors of change in HAI and fibrosis score, respectively, between biopsies (i.e., HAI or fibrosis score at second biopsy minus index/score at first biopsy), with adjustment for time between biopsies. Based on our previous analysis of factors cross-sectionally associated with HAI and fibrosis score,(14) the following characteristics were first investigated in basic models: age, sex, coffee consumption, alcohol use, BMI, lipodystrophy grade, history of diabetes, history of hyperlipidemia, ALT, AST, AST/ALT ratio, platelets, APRI, FIB-4, HIV RNA, HIV stage, CD4, CD4%, CD8, CD8%, HBV or HIV duration, HBeAg-positive status, qHBeAg level among HBeAg-positive participants, HBV DNA, qHBsAg level, HAI, portal inflammation, periportal inflammation, lobular inflammation, confluent necrosis score, Ishak fibrosis score, cirrhosis, and fatty liver disease. Among categorical variables, several adjacent categories were collapsed (e.g., high-risk and increased-risk alcohol use) due to low frequency and similar estimates between categories. Because qHBeAg levels and qHBsAg were skewed, they were log-transformed (natural log for qHBeAg, 10-base for qHBsAg) to reduce the impact of skewness. To identify factors with independent associations, factors associated at P < 0.20 were entered into a multivariable model for each outcome, and variables with P > 0.10 were removed using a step-wise variable selection method. Quantitative HBeAg level was not considered for multivariable models, as it was only available for the HBeAg-positive subgroup of participants. APRI and FIB-4 were not considered, as the Ishak fibrosis score was available. Among independent variables with high correlation (e.g., ALT, AST), the variable with the lowest Bayesian information criteria from the basic models was selected.(25) However, if a selected variable was removed (i.e., P > 0.10), a replacement with overlap that had not initially been considered with P < 0.20 from a basic model was considered. Beta coefficients with corresponding 95% CIs and P values are reported for independent variables.
Results
By close of study enrollment, 139 participants attended a baseline research assessment. However, 4 were discovered to be HBsAg-negative at baseline through central laboratory testing. Of the 135 HBsAg-positive participants, 114 had at least 24 weeks of follow-up that was required for the longitudinal analysis in this report. Despite study entry criteria, 55 participants did not undergo planned liver biopsies (n = 3 initial and n = 52 follow-up). Evaluation of change in histology was limited to 62 participants with paired liver biopsies (Supporting Fig. S1).
BASELINE PARTICIPANT CHARACTERISTICS
Demographic and clinical characteristics of the full longitudinal analysis sample and the biopsy subsample at study entry are reported in Table 1. Participants ranged in age from 18 to 70 years (median, 49 years) and had HIV or HBV infection for an estimated 1–52 years (median, 20 years). Most (91.2%; n = 104) participants were male and just over half (51.4%; n = 57) were non-Hispanic Black, followed by non-Hispanic White (32.4%; n = 36). Despite study entry criteria, 6 participants were not on cART, including an anti-HBV nucleoside or nucleotide analogue at the baseline assessment, but remained in the study, given that throughout the study antiretroviral treatment could be stopped, initiated, or changed per standard of care at the discretion of a treating physician. One of these 6 participants had a history of tenofovir use but reported not currently being on antiviral therapy, 2 reported anti-HIV medication only (ritonavir and darunavir, respectively), and 3 were on lamivudine alone as part of cART. Most of the participants on cART including an anti-HBV nucleoside or nucleotide analogue were on tenofovir alone or in combination with lamivudine or emtricitabine (n = 96), while a minority were on entecavir alone or in combination with lamivudine (n = 12).
TABLE 1.
Characteristics of HBV-HIV Co-infected North American Adult Sample and the Biopsy Subsample
| Analysis Sample (n = 114)* | Biopsy Subsample (n = 62)* | |
|---|---|---|
| Variable | n (%)† | n (%)† |
| Age (years) | ||
| Median (IQR) | 49 (45: 55) | 50 (46: 54) |
| Sex | ||
| Male | 104 (91.2%) | 58 (93.5%) |
| Female | 10 (8.8%) | 4 (6.5%) |
| Race | n = 111 | n = 60 |
| Non-Hispanic White | 36 (32.4%) | 17 (28.3%) |
| Non-Hispanic Black | 57 (51.4%) | 32 (53.3%) |
| Non-Hispanic Asian | 5 (4.5%) | 4 (6.7%) |
| Other | 13 (11.7%) | 7 (11.7%) |
| Coffee of cups per day | n = 111 | n = 60 |
| None to <1 per day | 56 (50.5%) | 31 (51.7%) |
| 1 to 2 per day | 37 (33.3%) | 17 (28.3%) |
| 3 or more per day | 18 (16.2%) | 12 (20.0%) |
| Alcohol | n = 113 | n = 61 |
| None | 63 (55.8%) | 31 (50.8%) |
| Moderate | 35 (31.0%) | 24 (39.3%) |
| At risk | 15 (13.3%) | 6 (9.8%) |
| BMI (kg/m2) | n = 109 | n = 60 |
| Median (IQR) | 25.9 (22.5–30.3) | 25.8 (22.7–29.1) |
| Weight status (race-adjusted) | n = 109 | n = 60 |
| Under/normal | 44 (40.4%) | 24 (40.0%) |
| Overweight | 37 (33.9%) | 25 (41.7%) |
| Obese | 28 (25.7%) | 11 (18.3%) |
| Lipodystrophy grade | n = 103 | n = 56 |
| None | 87 (84.5%) | 45 (80.4%) |
| Mild | 10 (9.7%) | 7 (12.5%) |
| Moderate or severe | 6 (5.8%) | 4 (7.1%) |
| DM | 10 (8.8%) | 4 (6.5%) |
| Hyperlipidemia | 34 (29.8%) | 17 (27.4%) |
| Sexually transmitted HBV or HIV | n = 106 | n = 57 |
| 101 (95.3%) | 57 (100.0%) | |
| Estimated duration of HIV or HBV infection (years) | n = 107 | n = 57 |
| Median (IQR) | 20 (13–26) | 22 (16–28) |
| HBV treatment | ||
| None‡ | 3 (2.6%) | 0 (0.0%) |
| Lamivudine alone | 3 (2.6%) | 0 (0.0%) |
| Tenofovir alone or in combination | 96 (84.2%) | 56 (90.3%) |
| Entecavir alone or in combination | 12 (10.5%) | 6 (9.7%) |
| cART including an anti-HBV nucleoside or nucleotide analogue, n (%) | 108 (94.7%) | 62 (100.0%) |
| Nucleoside/nucleote reverse transcriptase inhibitors | 109 (95.6%) | 60 (96.8%) |
| Non-nucleoside reverse transcriptase inhibitors | 38 (33.3%) | 23 (37.1%) |
| Protease inhibitors | 53 (46.5%) | 31 (50.0%) |
Data presented among this sample unless a subset is indicated due to missing data.
Unless otherwise indicated.
One participant with a history of tenofovir use was not on any antiviral therapy at the time of the baseline assessment. One was on ritonavir, darunavir, emtricitabine, and dolutegravir. Another was on ritonavir alone.
Ten (8.8%) participants had cirrhosis at baseline (3 of whom were diagnosed based on their initial study biopsy). Liver-related tests, HIV-related tests, and viral serologies at study entry are reported in Table 2. Most subjects had normal liver enzymes (AST, ALT, and alkaline phosphatase), low noninvasive indices (APRI, FIB-4, and VCTE), and well-controlled HIV, while 62% were HBeAg-positive and 76% had suppressed HBV DNA.
TABLE 2.
Laboratory Characteristics of HIV-HBV Co-infected North American Adult Sample
| Variable | Longitudinal Sample (n = 114) | Biopsy Sample (n = 62) |
|---|---|---|
| Liver-related tests | ||
| ALT (U/L) | n = 110 | n = 61 |
| Median (IQR) | 27 (19–42) | 24 (18–36) |
| AST (U/L) | n = 110 | n = 61 |
| Median (IQR) | 29 (22–41) | 27 (22–41) |
| AST/ALT ratio | n = 110 | n = 61 |
| Median (IQR) | 1.0 (0.8–1.3) | 1.0 (0.9–1.4) |
| Alkaline phosphatase (U/L) | n = 109 | n = 60 |
| Median (IQR) | 85 (68–107) | 82 (63–110) |
| Total bilirubin (mg/dL) | n = 109 | n = 60 |
| Median (IQR) | 0.4 (0.3–0.7) | 0.5 (0.3–0.7) |
| Albumin (g/dL) | n = 110 | n = 61 |
| Median (IQR) | 4.3 (4.1–4.6) | 4.4 (4.2–4.6) |
| Platelets (×103/mm3) | n = 112 | n = 61 |
| Median (IQR) | 200.5 (175–238) | 200 (174–248) |
| Noninvasive indices of fibrosis APRI | n = 110 | n = 61 |
| Median (IQR) | 0.3 (0.3–0.5) | 0.3 (0.3–0.5) |
| FIB-4 | n = 110 | n = 61 |
| Median (IQR) | 1.4 (1.0–1.9) | 1.4 (1.0–1.9) |
| VCTE (kPa) | n = 69 | n = 46 |
| Median (IQR) | 5.4 (4.0–6.9) | 5.4 (4.1–6.4) |
| HIV-related tests | ||
| CD4 (cells/mm3) | n = 101 | n = 58 |
| Median (IQR) | 562 (366–707) | 564.5 (374–678) |
| CD4 % | n = 102 | n = 59 |
| Median (IQR) | 25.7 (18.0–36.0) | 26.0 (21.0–36.3) |
| CD8 (cells/mm3) | n = 66 | n = 39 |
| Median (IQR) | 878.5 (595.0–243.0) | 843.0 (560.0–1,139.0) |
| CD8 % | n = 67 | n = 40 |
| Median (IQR) | 44.0 (36.4–53.0) | 43.0 (35.0–53.0) |
| HIV stage (1–4), n (%) | n = 86 | n = 45 |
| 1 (≥500 cells/mm3) | 59 (68.6%) | 31 (68.9%) |
| 2 (250–499 cells/mm3) | 13 (15.1%) | 7 (15.6%) |
| 3 (200–349 cells/mm3) | 6 (7.0%) | 3 (6.7%) |
| 4 (<200 cells/mm3) | 8 (9.3%) | 4 (8.9%) |
| HIV RNA (copies/mL), n (%) | n = 103 | n = 55 |
| <20 | 80 (77.7%) | 45 (81.8%) |
| 20 to <400 | 16 (15.5%) | 7 (12.7%) |
| 400 to <10,000 | 4 (3.9%) | 1 (1.8%) |
| ≥10,000 | 3 (2.9%) | 2 (3.6%) |
| Hepatitis viral serologies | ||
| Anti-HCV positive/RNA negative, n (%) | n = 105 | n = 58 |
| Positive | 4 (3.8%) | 2 (3.4%) |
| Anti-HDV,n (%) | n = 71 | n = 41 |
| Positive | 1 (1.4%) | 0 (0.0%) |
| Anti-HBe, n (%) | n = 103 | n = 56 |
| Positive | 28 (27.2%) | 13 (23.2%) |
| HBeAg, n (%) | ||
| Positive | 70 (61.4%) | 39 (62.9%) |
| HBeAg (log10 IU/mL) among HBeAg-positive | n = 69 | n = 39 |
| Median (IQR) | 1.3 (0.3–2.5) | 0.8 (0.1–1.8) |
| HBsAg (log10 IU/mL) | n = 111 | n = 60 |
| Median (IQR) | 3.3 (2.6–4.0) | 3.2 (2.6–3.5) |
| HBV DNA (IU/mL), n (%) | ||
| <20 | 70 (61.4%) | 43 (69.4%) |
| 20–999 | 21 (18.4%) | 10 (16.1%) |
| 1,000–20,000 | 8 (7.0%) | 2 (3.2%) |
| 20,000 | 15 (13.2%) | 7 (11.3%) |
| HBV DNA (log10 IU/mL) among ≥ 20 IU/mL | n = 44 | n = 19 |
| Median (IQR) | 3.4 (1.8–5.3) | 2.1 (1.7–6.3) |
| Range | 1.3:10.1 | 1.3:8.5 |
| HBV-DNA and HIV-RNA suppression status, n (%) | n = 94 | n = 52 |
| Suppressed | 72 (76.6%) | 43 (82.7%) |
| Incomplete suppression | 8 (8.5%) | 2 (3.8%) |
| Not suppressed | 14 (14.9%) | 7 (13.5%) |
Note: Data are presented among this sample unless a subset is indicated due to missing data.
The only apparent difference between the paired biopsy subgroup (n = 62) versus those in the full longitudinal analysis sample without paired biopsies (n = 52) was viral transmission mode (100% vs. 89.8% sexual; P = 0.01), duration of HBV or HIV (median 22 vs. 18 years; P = 0.04), and HBeAg level among HBeAg-positive participants (median 0.8 vs. 1.9 log10 IU/mL; P = 0.02) (Supporting Table S1). In comparison to those in the longitudinal analysis, the 21 participants with less than 24 weeks of follow-up were older (median age 49 vs. 44; P = 0.02), had HBV or HIV longer (median 20 vs. 13.5 years; P = 0.03), had higher rates of HIV-RNA suppression (e.g., 77.9% vs. 50.0% undetectable; P < 0.01), risk of acquiring HIV or HBV by sexual transmission (95.3% vs. 76.2%; P = 0.003), and HBeAg positivity (61.4% vs. 38.1%; P = 0.047). Other characteristics were similar among groups (Supporting Table S1).
FOLLOW-UP
Follow-up data were available for 95.6% (n = 109) of the full longitudinal analysis sample at week 24, 87.7% (n = 100) at week 48, 85.1% (n = 97) at week 72, 76.3% (n = 87) at week 96, 71.9% (n = 82) at week 120, 67.5% (n = 77) at week 144, 56.1% (n = 64) at week 168, and 43.9% (n = 50) at week 192. Over the follow-up, use of cART including an anti-HBV nucleoside or nucleotide analogue was common (92.7%−100% across time points; Supporting Table S2). However, 13 participants were not on any anti-HBV medication at one or more assessments, and 15 were not cART, including an anti-HBV nucleoside or nucleotide analogue at one or more assessments. Reasons for breaks in HBV therapy were not recorded and may have been clinician-initiated or patient-initiated.
COMPOSITE CLINICAL OUTCOME
Incidence rates of clinical outcomes are reported in Table 3. Among 114 participants, 10 (8.8%) had baseline cirrhosis, and 2—both with baseline cirrhosis—developed the composite clinical outcome during 326 person-years of follow-up (0.61 [95% CI, 0.15–2.45] per 100 person-years). One developed hepatic decompensation, followed by HCC and an HBV-related death (Supporting Table S3). The other developed HCC and had suppressed HBV DNA and normal ALT at the time of diagnosis. No participants underwent a liver transplantation.
TABLE 3.
Incidence Rates of Non-histological Outcomes Among 114 HBV-HIV Co-infected North American Adults With > 24 Weeks of Follow-up
| No. of Participants With the Outcome (Person-Years¶) | Incidence per 100 Person-Years (95% CI) | |
|---|---|---|
| Primary clinical outcomes (n = 114*) | ||
| Hepatic decompensation | 1 (329.5) | 0.30 (0.04, 2.15) |
| HCC | 2 (326.1) | 0.61 (0.15, 2.45) |
| Liver transplant† | 0 (331.3) | — |
| HBV-related death† | 1 (332.1) | 0.30 (0.04, 2.14) |
| Composite clinical outcome‡ | 2 (326.1) | 0.61 (0.15, 2.45) |
| Secondary outcomes (n indicated below) | ||
| Incident cirrhosis (n = 104 without baseline cirrhosis) | 1 (248.5) | 0.40 (0.06, 2.86) |
| Incident ALT flare (n = 111 with follow-up ALT) | 2 (305.5) | 0.65 (0.16, 2.62) |
| Ever become HBsAg-negative (n = 104 HBsAg-positive with follow-up HBsAg) | 0 (296.2) | NA |
| Ever become HBeAg-negative (n = 67 HBeAg-positive with follow-up HBeAg) | 13 (189.5) | 6.86 (3.98, 11.81) |
| Non-HBV-related death (n = 114)§ | 4 (333.0) | 1.20 (0.45, 3.20) |
Includes those with baseline cirrhosis (developed before or within 24 weeks after enrollment).
No transplants or deaths occurred within 24 weeks of study entry.
Defined as first of decompensation, HCC, liver transplant, or HBV-related death.
The 4 participants with non-HBV deaths did not have any of the other outcomes presented in this table
Time to event or end of follow-up.
Abbreviation: NA, not available.
Among those without baseline cirrhosis (n = 104), 1 participant developed incident cirrhosis during 248.5 person-years of follow-up (a rate of 0.40 [95% CI, 0.06–2.86] per 100 person-years). The cirrhosis diagnosis was adjudicated per HBRN study protocol based on presence of ascites, splenomegaly, and nodular liver documented by CT, MR, or liver ultra-sound report. However, HBV DNA was suppressed, ALT was normal, and platelet count (123,000 mm3) did not meet the cirrhosis criteria at the time of diagnosis. Additionally, the follow-up Ishak fibrosis score was 3. No participants had an ALT flare at study entry, and ALT flares were rare during follow-up (n = 2; 0.65 [95% CI, 0.16, 2.62] per 100 person-years). No one experienced HBsAg loss over 296.2 person-years of follow-up, and among those who were HBeAg-positive at study entry (n = 67), HBeAg loss occurred in 13 participants at a rate of 6.86 (95% CI, 3.98, 11.81) per 100 person-years. Four participants died of causes unrelated to HBV, a rate of 1.20 (95% CI, 0.45, 3.20) per 100 person-years. One participant had a pregnancy during follow-up; she had none of the outcomes reported in Table 3. Because delta infection can affect the natural history of HBV, including those on suppressive therapy, outcomes of the 1 participant with anti-delta antibodies were evaluated. The 1 participant with anti-delta antibodies did not have any HBV-related outcomes, but did experience a death of unknown cause 75 weeks after their baseline assessment. He was not included in the histology analysis.
The cumulative probability of ever becoming HBeAg-negative among participants who were HBeAg-positive at baseline (n = 67) is shown in Fig. 1. The HBeAg loss rate by baseline characteristic is provided in Supporting Table S4. Even for variables in which categories appeared to have clinically meaningful different rates (e.g., age > 50 years, AST > 1 × ULN, platelets ≥ 200 ×103/mm3, qHBsAg < 3 log10 IU/mL, CD4 ≥ 20%, HIV-RNA suppression, Ishak fibrosis score ≥ 3, and cirrhosis), the 95% CIs between categories overlapped, partially reflecting the small sample size (n = 67) and rarity of the outcome (n = 13).
FIG. 1.
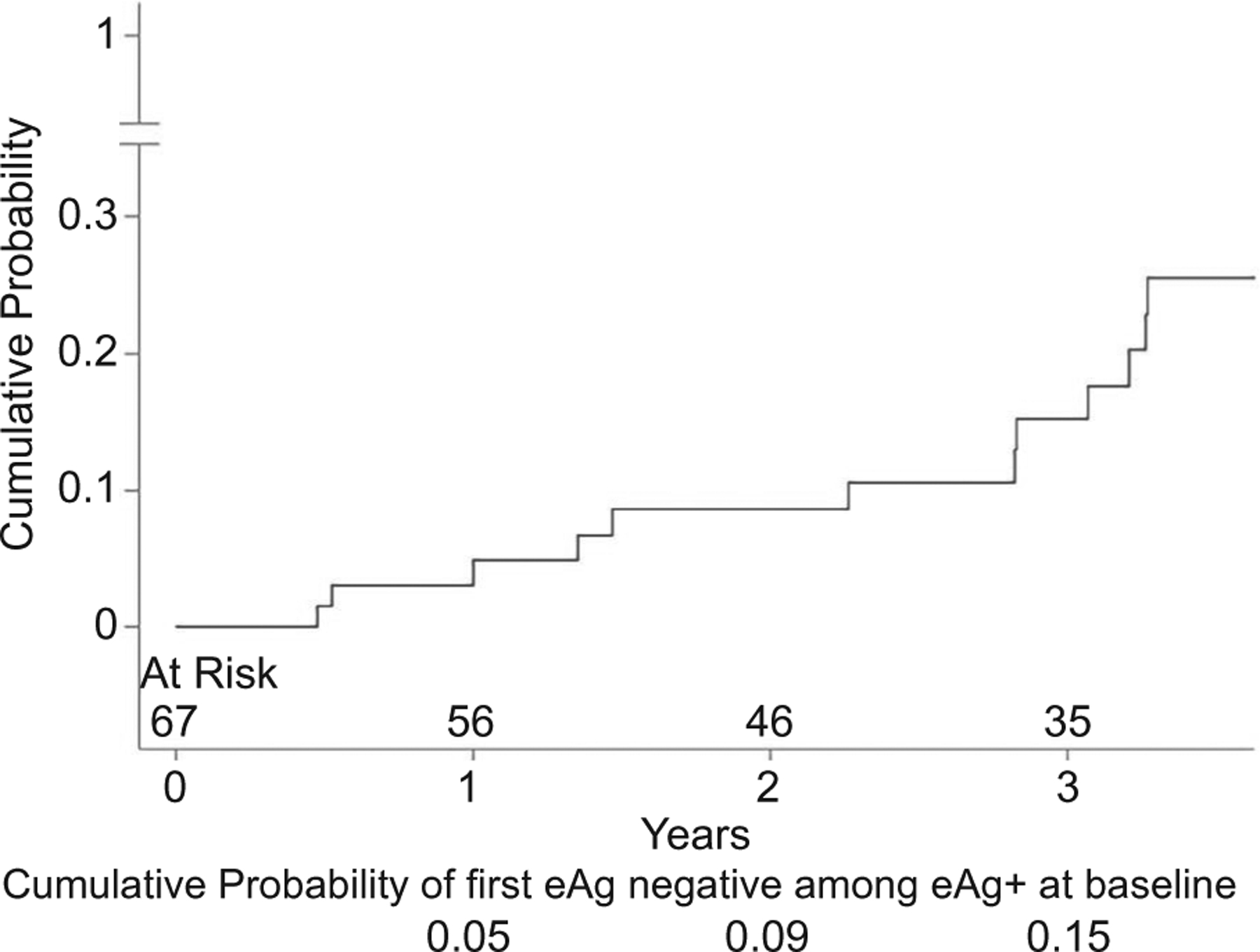
Cumulative probability of ever becoming HBeAg-negative among participants who were HBeAg-positive at baseline (13 events).
Select viral and clinical markers are shown by time point in Fig. 2 and reported in Supporting Table S5. Mean HBeAg (log10 IU/L), HbsAg (log10 IU/L), and HBV DNA (log10 IU/L) decreased over time (P ≤ 0.01) (Fig. 2A). Likewise, the percentage of participants with suppressed HBV DNA increased over time (Fig. 2B; P < 0.001). Although mean AST and platelet values did not significantly differ over time (P = 0.80 and 0.10, respectively), mean ALT (log2 U/L) decreased over time (P = 0.046) and ALT/AST ratio increased over time (P < 0.01). Despite using baseline age in the FIB-4 calculation, there was a significant trend of FIB-4 increasing over time (P = 0.009), and indication of a similar trend with APRI (P = 0.07; Fig. 2C). There was not a significant difference in VCTE over time (P = 0.24; Supporting Table S5).
FIG. 2.
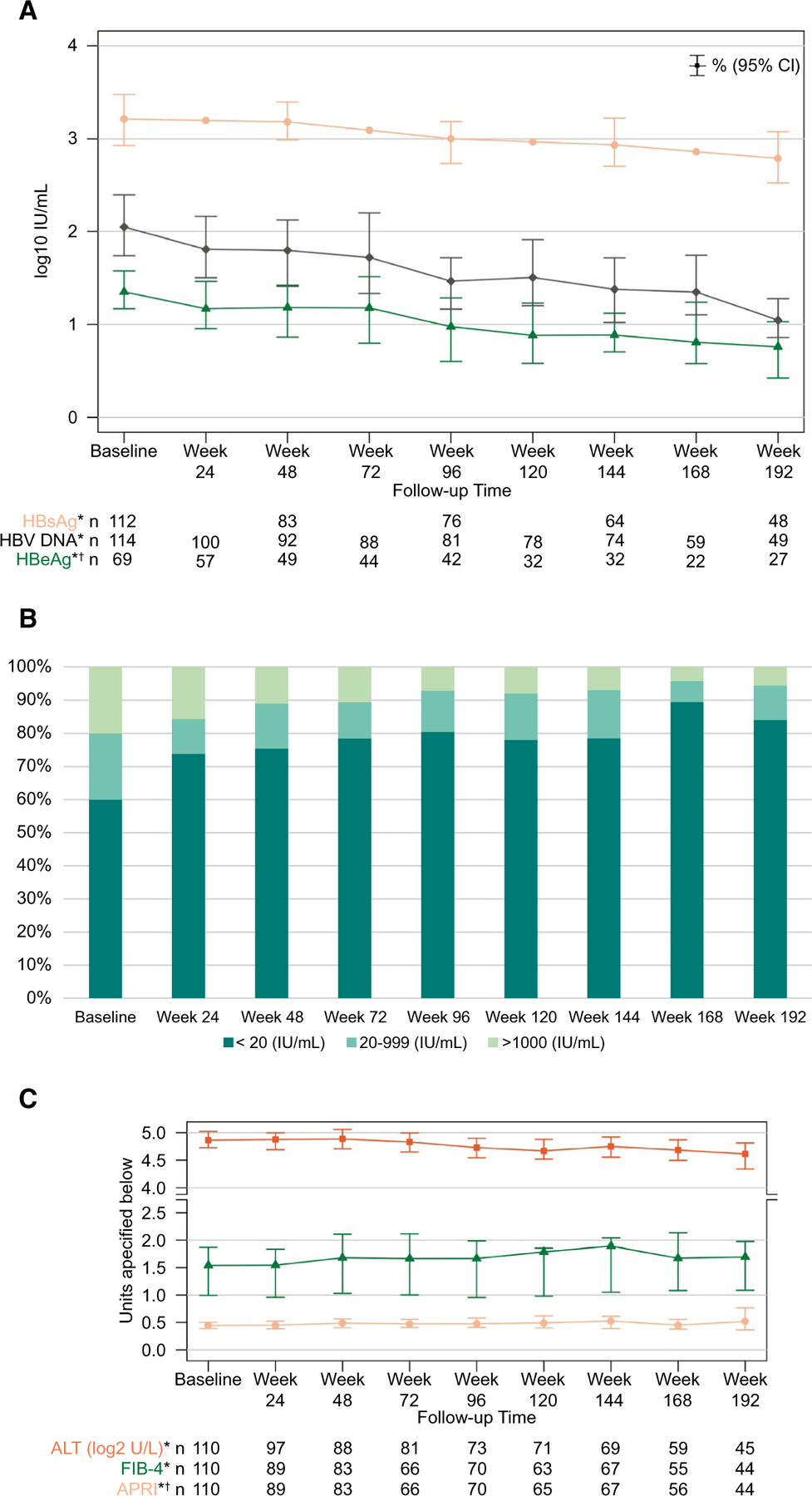
Select viral and clinical markers by assessment. (A) Modeled means (95%) of HBeAg, HbsAg, and HBV DNA (log10 IU/mL). (B) HBV DNA categories. (C) Modeled means (95%) of ALT (log2 U/L), APRI, and FIB-4. *HBsAg log10 IU/mL (P < 0.001), HBV DNA log10 IU/mL (P < 0.001), HBeAg log10 IU/mL (P = 0.01), HBV DNA category (P < 0.001), ALT (P = 0.046), FIB-4 (P = 0.009), APRI (P = 0.07). †HBeAg is only among HBeAg-positive participants.
HISTOLOGIC OUTCOMES
Paired biopsies occurred a median of 3.6 (IQR, 3.1–3.7; range, 2.6–6.1) years apart (n = 62). Inflammation and fibrosis measures at study entry and follow-up are provided in Table 4. Five (8.1%) participants had a confluent necrosis score ≥ 0 at baseline versus none at follow-up, and there was a small but statistically significant decrease in HAI (P = 0.02), but the distributions of other histology measures were not significantly different between time points, including the Ishak fibrosis score (continuous, P = 0.35) or binary measures of significant (33.9% vs. 31.2%; P = 0.90) or advanced fibrosis (22.6% vs. 26.2%; P = 0.49), respectively. The distributions of change in the HAI and Ishak fibrosis score standardized per 192 weeks are shown in Fig. 3.
TABLE 4.
Histology-Determined Liver Inflammation and Fibrosis in HBV-HIV Co-infected North American Adults at Study Entry and Approximately 3–4 Years Later
| Baseline (n = 62) | Follow-up (n = 62) | P Value | |
|---|---|---|---|
| HAI | 0.02 | ||
| Median (25th–75th) | 3 (2–4) | 3 (1–3) | |
| Range | 0–14 | 0–8 | |
| Portal inflammation score, n (%) | 0.47 | ||
| 0 | 7 (11.3%) | 14 (22.6%) | |
| 1 | 45 (72.6%) | 40 (64.5%) | |
| 2 | 9 (14.5%) | 8 (12.9%) | |
| 3 | 1 (1.6%) | 0 (0.0%) | |
| 4 | |||
| Periportal inflammation score, n (%) | 0.45 | ||
| 0 | 20 (32.3%) | 27 (43.5%) | |
| 1 | 36 (58.1%) | 29 (46.8%) | |
| 2 | 3 (4.8%) | 5 (8.1%) | |
| 3 | 2 (3.2%) | 1 (1.6%) | |
| 4 | 1 (1.6%) | 0 (0.0%) | |
| Lobular inflammation score, n (%) | 0.71 | ||
| 0 | 7 (11.3%) | 7 (11.3%) | |
| 1 | 38 (61.3%) | 42 (67.7%) | |
| 2 | 12 (19.4%) | 11 (17.7%) | |
| 3 | 4 (6.5%) | 2 (3.2%) | |
| 4 | 1 (1.6%) | 0 (0.0%) | |
| Confluent necrosis score, n (%) | — | ||
| 0 | 57 (91.9%) | 62 (100.0%) | |
| 1 | 2 (3.2%) | 0 (0.0%) | |
| 2 | 1 (1.6%) | 0 (0.0%) | |
| 3 | 1 (1.6%) | 0 (0.0%) | |
| 6 | 1 (1.6%) | 0 (0.0%) | |
| Fibrosis score | n = 61 | n = 61 | 0.58 |
| Median (25th–75th) | 1 (1–2) | 1 (0–3) | |
| Range | 0–6 | 0–6 |
Note: Changes in histological-determined inflammation and fibrosis were tested with mixed-effects models (linear for continuous, ordinal logistic for ordinal, binomial for binary outcomes) with a repeated outcome, time (i.e., days since first biopsy) as a continuous fixed effect, and random intercept. Median of time between biopsies was: 3.6 (IQR, 3.1–3.7; range, 2.6–6.1) years.
FIG. 3.
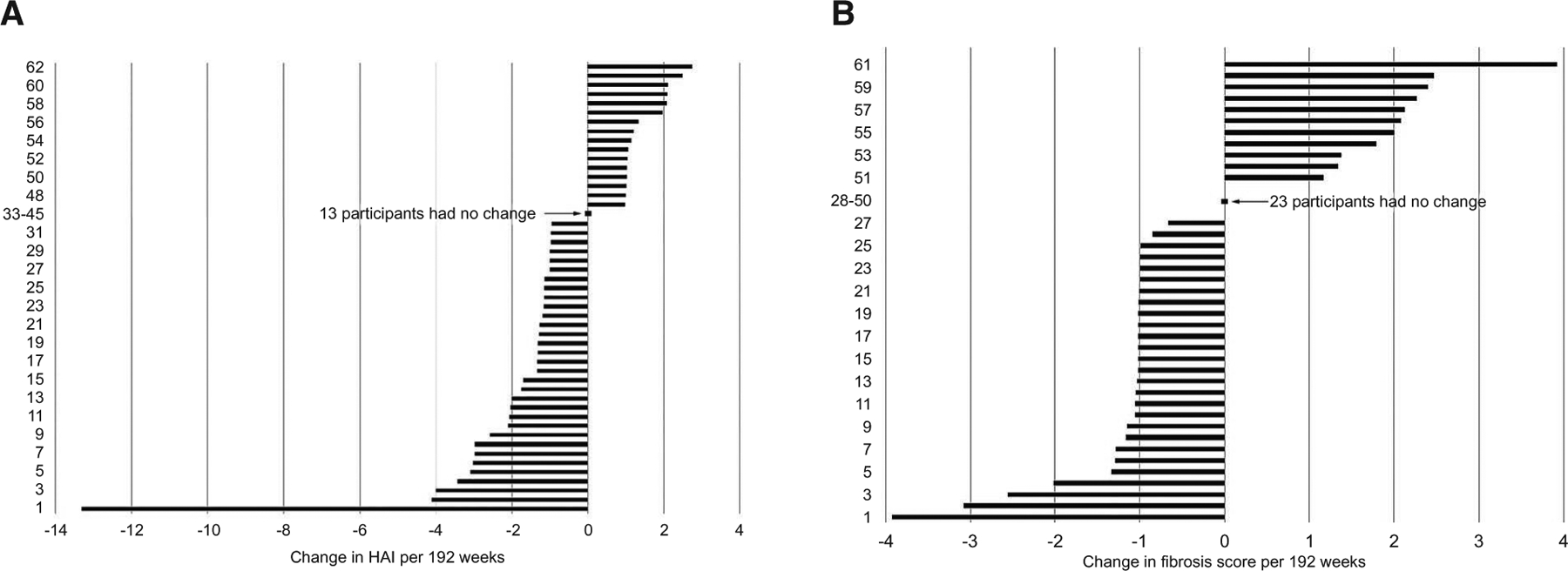
Change in HAI and Fibrosis Score standardized per 192 weeks. (A) HAI (n = 62). (B) Fibrosis score (n = 61). *The distribution of change in the HAI and fibrosis score between baseline and follow-up were each standardized per 192 weeks (192 weeks*[follow-up value minus baseline value]/weeks between biopsies).
Individual-level change between time points (shown with cross tables of inflammation and fibrosis measures by time point) is provided in Supporting Tables S6–S11. The largest increase in HAI across follow-up was 2, from 0–2 (n = 1), 1–3 (n = 2), 3–5 (n = 2), and 4–6 (n = 1), while the largest decrease was 10, from 14–4 (Supporting Table S6). The largest increase in fibrosis score was 4, from 0–4 (n = 1) and 2–6 (n = 1), while the largest decrease was 3, from 3–0 (n = 2) (Supporting Table S11). Of the 61 participants, only 7 (11.5% [95% CI, 5.7–21.8]) had a rate of progression of at least 2 Ishak fibrosis points per 192 weeks, while 4 (6.6% [95%CI, 2.6–15.7]) had an improvement of at least 2 points.
Correlations between change in fibrosis score and change in each inflammation marker (n = 61) were HAI (ρ = 0.24; P = 0.058), portal inflammation score (ρ = 0.34; P = 0.008), periportal inflammation score (ρ = 0.27; P = 0.04), lobular inflammation score (ρ = 0.08; P = 0.53), and confluent necrosis score (ρ = −0.12; P = 0.36). Scatter plots are available in Supporting Fig. S3.
After excluding 2 participants with baseline biopsies at > 24 weeks from their first research assessment, the median time between the clinical evaluation date and first biopsy was 3.9 weeks (n = 60). Correlations between change in ALT and change in inflammation markers were not significant: HAI (ρ = 0.12; P = 0.36), portal inflammation score (ρ = −0.09; P = 0.50), peri-portal inflammation score (ρ = −0.01; P = 0.95), lobular inflammation score (ρ = 0.19; P = 0.16), and confluent necrosis score (ρ = 0.10; P = 0.46). Correlations between change in noninvasive measures of fibrosis and change in fibrosis score were also not significant: APRI (ρ = −0.01; P = 0.97) and FIB-4 (ρ = −0.06; P = 0.63).
Associations between baseline characteristics and change in the HAI (n = 60) and the fibrosis score (n = 59), respectively, adjusted for time between biopsies only, are reported in Table 5. In the full HAI multivariable model, every 1 point higher in the HAI at baseline was associated with a 0.3 (95% CI, 0.1–0.5; P < 0.01) decrease in the HAI of at follow-up. In the full fibrosis score multivariable model, every 1 point higher in the fibrosis score at baseline was associated with a 0.4 (95% CI, 0.1–0.6; P < 0.01) decrease in the fibrosis score at follow-up, whereas lipodystrophy ≥ 2 versus 0–1 (1.5 [95% CI, 0.3–2.7]; P = 0.02) and steatohepatitis versus no fatty liver disease (2.4 [95% CI, 1.3–3.6]; P < 0.001) at baseline were associated with increases in the fibrosis score. No other baseline factors were retained in the multivariable models.
TABLE 5.
Associations Among Baseline Demographic, Clinical and Serologic Characteristics, and Change in the HAI and Ishak Fibrosis Score Across Follow-up, Adjusted for Time
| Baseline Status | Comparison Group | Change in HAI (n = 60) | Change in Fibrosis Score (n = 59) | ||
|---|---|---|---|---|---|
| Beta (95% CI)* | Overall P Value | Beta (95% CI)* | Overall P Value | ||
| Age, per 5 years | −0.11 (−0.44, 0.21) | 0.48 | 0.07 (−0.14, 0.27) | 0.51 | |
| Sex (ref = male) | Female | −0.67 (−3.03, 1.70) | 0.58 | −0.46 (−1.95, 1.02) | 0.54 |
| Coffee, cups per day (ref = none or occasional) | 1 to 2 | 0.38 (−0.88, 1.64) | 0.56 | 0.55 (−0.25, 1.35) | 0.31 |
| 3 or more | 0.72 (−0.66, 2.10) | −0.11 (−1.01, 0.79) | |||
| Alcohol use (ref = none) | Moderate/at risk | −0.33 (−1.37, 0.71) | 0.53 | −0.68 (−1.33, −0.04) | 0.04 |
| BMI, kg/m2 | −0.01 (−0.12, 0.10) | 0.86 | −0.04 (−0.11, 0.03) | 0.25 | |
| Lipodystrophy grade (ref = 0 or 1) | 2 or above | −0.9 (−3.35, 1.55) | 0.47 | 1.05 (−0.42, 2.52) | 0.16 |
| Diabetes (ref = no) | Yes | −0.60 (−2.67, 1.46) | 0.56 | 0.51 (−0.79, 1.80) | 0.44 |
| Hyperlipidemia (ref = no) | Yes | −0.03 (−1.18, 1.11) | 0.95 | 0.30 (−0.43, 1.03) | 0.41 |
| ALT, per 10 IU/L | −0.13 (−0.36, 0.10) | 0.27 | 0.16 (0.01, 0.3) | 0.04 | |
| High ALT (ref = no) | Yes (>1 × ULN) | −0.15 (−1.25, 0.96) | 0.79 | 0.36 (−0.33, 1.06) | 0.30 |
| AST, per 10 IU/L | −0.33 (−0.68, 0.02) | 0.07 | 0.23 (0.02, 0.45) | 0.04 | |
| AST/ALT ratio, per 0.1 | −0.03 (−0.15, 0.08) | 0.58 | −0.01 (−0.09, 0.06) | 0.72 | |
| Platelets, per 200 mm3 | 1.59 (−0.34, 3.52) | 0.10 | −0.43 (−1.65, 0.8) | 0.49 | |
| APRI, per 0.1 unit | −0.31 (−0.49, −0.13) | 0.001 | 0.13 (0.01, 0.25) | 0.03 | |
| FIB-4, per 1 unit | −1.00 (−1.56, −0.45) | 0.001 | 0.20 (−0.18, 0.58) | 0.29 | |
| Detectable HIV RNA (ref = no) | Yes | −0.22 (−1.78, 1.33) | 0.77 | −0.41 (−1.39, 0.58) | 0.41 |
| HIV stage (ref = 1) | ≥ 2 | −0.89 (−1.78, 0.01) | 0.052 | 0.13 (−0.72, 0.98) | 0.76 |
| CD4, per 100 cells/mm3 | 0.14 (−0.05, 0.34) | 0.15 | 0.08 (−0.04, 0.21) | 0.18 | |
| CD4%, per 1% | 0.05 (0.01, 0.10) | 0.03 | 0.01 (−0.02, 0.04) | 0.37 | |
| CD8, per 100 cells/mm3 | −0.01 (−0.12, 0.10) | 0.79 | −0.03 (−0.14, 0.08) | 0.55 | |
| CD8%, per 1% | 0.01 (−0.02, 0.05) | 0.45 | 0.01 (−0.03, 0.05) | 0.63 | |
| HBV or HIV duration, per year | 0.02 (−0.04, 0.08) | 0.46 | −0.002 (−0.04, 0.04) | 0.99 | |
| HBeAg (ref = negative) | Positive | 0.58 (−0.48, 1.63) | 0.28 | −0.22 (−0.9, 0.46) | 0.52 |
| HBeAg, per log10 IU/mL | (HBeAg + n = 39) | −0.05 (−0.65, 0.55) | 0.86 | 0.09 (−0.35, 0.52) | 0.69 |
| HBV DNA < 1,000 IU/mL (ref = no) | Yes | 0.52 (−1.08, 2.12) | 0.52 | 0.72 (−0.27, 1.72) | 0.15 |
| HBV-DNA and HIV-RNA suppression status (ref = suppressed) | Incomplete suppression | −1.42 (−4.41, 1.57) | 0.63 | −0.95 (−2.86, 0.95) | 0.32 |
| Not suppressed | −0.22 (−2.17, 1.74) | −0.75 (−2.00, 0.49) | |||
| HBV DNA, per log10 IU/mL | −0.15 (−0.44, 0.15) | 0.32 | −0.14 (−0.33, 0.04) | 0.13 | |
| HBsAg, per log10 IU/mL | 0.16 (−0.30, 0.62) | 0.49 | −0.10 (−0.39, 0.19) | 0.49 | |
| HAI, per 1 | −0.61 (−0.77, −0.45) | <0.001 | −0.003 (−0.15, 0.15) | 0.99 | |
| Portal inflammation (ref = 0) | 1 | −1.01 (−2.40, 0.38) | <0.001 | −0.55 (−1.74, 0.63) | 0.29 |
| ≥2 | −3.75 (−5.39, −2.10) | −1.06 (−2.43, 0.3) | |||
| Periportal inflammation (ref = 0) | 1 | −0.88 (−1.84, 0.08) | <0.001 | −0.42 (−1.2, 0.36) | 0.55 |
| ≥2 | −4.02 (−5.57, −2.47) | −0.30 (−1.54, 0.94) | |||
| Lobular inflammation (ref = 0) | 1 | −0.89 (−2.43, 0.66) | 0.02 | −0.03 (−1.13, 1.06) | 0.76 |
| ≥2 | −2.21 (−3.90, −0.52) | 0.25 (−0.95, 1.45) | |||
| Confluent necrosis (ref = 0) | ≥1 | −2.98 (−4.71, −1.24) | 0.001 | 0.63 (−0.62, 1.88) | 0.32 |
| Ishak fibrosis score, per 1 | −0.76 (−1.13, −0.40) | <0.001 | −0.24 (−0.51, 0.03) | 0.08 | |
| Cirrhosis (ref = no) | Yes | −2.12 (−3.91, −0.34) | 0.02 | 0.23 (−0.94, 1.41) | 0.69 |
| Fatty liver disease (ref = no) | Steatosis | 0.07 (−1.17, 1.30) | 0.65 | 0.54 (−0.18, 1.27) | 0.001 |
| Steatohepatitis | 0.81 (−0.94, 2.57) | 2.10 (1.07, 3.13) | |||
Associations between baseline factors and change in HAI and fibrosis score (value at follow-up minus value at baseline), respectively, were tested with a linear mixed-effects model, time (i.e., days between biopsy) as a continuous fixed effect, and random intercept. Median of time between biopsies was 3.6 (IQR, 3.1–3.7; range, 2.6–6.1) years. A positive beta represents worsening in inflammation or fibrosis, respectively.
Discussion
In this longitudinal analysis that included protocol-directed paired liver biopsy of North American patients with HBV-HIV, primarily treated with cART, we observed few clinical events (i.e., hepatic decompensation, HCC, liver transplant, ALT flare, incident cirrhosis, or HBV-related death) or histologic changes (HAI and fibrosis) over a median interval of 3.6 years. Additionally, we observed declines in ALT, qHBeAg, and qHBsAg over time, an HBeAg loss rate of 6.9 per 100 person-years, and no HBsAg loss.
When we evaluated components of inflammation, we observed no significant changes in distributions of peri-portal, portal, or lobular inflammation, but confluent necrosis was less common, which resulted in a small group-level improvement in mean HAI during follow-up. We did not see group-level changes in the distribution of the fibrosis score or proportions with significant (≥ F2) or advanced (≥ F3) fibrosis between time points. Furthermore, overlap of the 95% CI of the percentage with progression (11%) and improvement (7%) in fibrosis, when defined by changes of at least 2 Ishak fibrosis stages per 192 weeks (3.7 years), indicated that there was not a significant difference in the percentage with fibrosis improvement versus worsening. Although there are few studies with comparable data, our fibrosis results are similar to observations among 38 co-infected patients with paired biopsy (median time interval of 2.7 years) after initiating cART.(26) The possible reasons for lack of histological improvements include other HIV-related factors contributing to liver disease, or the fact that participants had been treated for years, and benefit conferred by suppression had plateaued.
In line with our histology results, liver stiffness measured by VCTE did not significantly differ over time in our cohort. However, serum-based markers of fibrosis yielded slightly different results. We observed a mild group-level increase in AST/ALT ratio (due to ALT decreasing) and FIB-4 (independent of aging, as baseline age was used to calculate FIB-4 at all time points) and a similar, but not statistically significant, increase in APRI across follow-up, despite no significant change in platelet count over time. Compared with our study, other studies in patients with HBV-HIV using serum-based noninvasive methods to assess changes in liver disease severity observed higher proportions with regression of fibrosis.(27–30) However, because noninvasive measures may not be accurate in the treated patient with HBV-HIV,(31) these improvements in serum-based noninvasive models over time may reflect improvements in ALT, a major component of serum-based fibrosis scores, rather than changes in fibrosis itself.
Although there was little or no group-level change in HAI and fibrosis score, respectively, variability at the individual level allowed us to explore baseline predictors of change. Both baseline HAI and baseline fibrosis were independent predictors of change in the respective outcome across follow-up, such that worse versus better scores at baseline were associated with better changes (i.e., a decrease in inflammation or fibrosis). Additionally, baseline steatohepatitis and lipodystrophy were positively associated with fibrosis progression, indicating the importance of identifying their presence in this population, as they may counteract long-term HBV suppression. Another report focused on fatty liver disease will evaluate its impact on HBV progression in more detail. Interestingly, we also observed that HIV stage ≥ 2 versus 1 at baseline might be associated with a decrease in HAI. However, these results might be spurious and would need to be confirmed in other studies. Other histologic and virologic factors were not related to either outcome.
Of the non-histologic outcomes, we observed two cases of HCC (both with baseline cirrhosis), a rate of 0.61 per 100 person-years. Although infrequent, this finding underscores the need for patients with HBV-HIV with cirrhosis to be in HCC surveillance. We observed one case of incident cirrhosis, a rate of 0.40 per 100 person-years. Although data for a direct comparison with other cohorts with HBV-HIV using histology are not available, our cirrhosis incidence is similar to some,(27,32) but not all studies,(26) and is in line with the higher rate reported, in general, in the HBV-HIV literature.(33) However, it should be noted that due to our small sample size and the rarity of the outcome, our estimate had a wide 95% CI (0.1–2.9). Unlike studies of liver-related outcomes of those initiating cART(26,27,34), most of our cohort was on stable cART at study entry, suggesting that ongoing cART containing anti-HBV agents results in stable liver disease, similar to those treated for HBV alone.(35–37) It is difficult to compare the HBeAg loss rate in our study with other longitudinal studies of patients initiating cART, which generally have reported the percentage of participants with HBeAg loss (range 7%−57%) without taking length of follow-up into account.(27,29,32,33,38–40) We did not observe any patients who lost HBsAg during almost 4 years of follow-up on stable cART. This differs with other studies of patients initiating cART, which observed HBsAg clearance percentages of 3%−22% among participants over 2–6 years.(27,29,32,33,38–40 ) This difference may, in part, be due to our patients being on stable cART at study entry with long-term viral suppression.
STRENGTHS AND LIMITATIONS
Most observational studies of HBV liver disease in patients with HIV co-infection included those who were not yet on effective HBV therapy(28–30,32,40) or did not use liver histology(26,27,29,30,39,41) as an endpoint. The strengths of our study are the prospective study design with careful follow-up, the experience of the investigators, paired liver biopsy analysis, and the central reading of liver biopsies by the HBRN pathology committee.
Both study enrollment and obtaining a second liver biopsy among enrolled participants were more challenging than our research team anticipated, based on prior experiences with patients with HCV-HIV, and in patients with HBV-HIV with elevated liver enzymes. Thus, a potential limitation of our study is the self-selection of patients who were willing to undergo two liver biopsies, which may not reflect all adults with HBV-HIV co-infection. In particular, Caucasians, Asians, and women may be underrepresented in our cohort. Incomplete data collection at each time point, the effect of sampling error, and the small sample size of the histology subsample should also be considered.
Because our cohort was mostly on cART at entry, we were unable to assess the effect of initiating cART on liver histology. As such, medication-related improvements in liver histology may have occurred before the initial biopsy. Likewise, we were unable to evaluate to effect of HBV-DNA suppression over time, and in particular, incomplete suppression, as most of the participants were continuously suppressed. Perhaps due to widespread cART use and HBV-DNA suppression, this study reported few clinical events. However, given our limited sample size, the low event rate should be interpreted with caution. Finally, we had limited power to identify predictors of HBeAg loss and insufficient power to predict other clinical outcomes. Notwithstanding, our study is one of a few studies using paired biopsies in patients with HBV-HIV on stable cART. The lack of willingness of patients with HBV-HIV with suppressed HBV and normal ALT to undergo liver biopsy should be considered in future studies in this population.
In conclusion, we have shown that patients with HBV-HIV, mostly on cART including a nucleoside or nucleotide analogues, experience a low incidence rate of clinical events (HCC, hepatic decompensation, liver transplant, HBV-related death, or incident cirrhosis) and experience minimal histological change over almost 4 years of observation. Given the rarity of outcomes, future studies with larger samples, use of novel therapies, and longer follow-up are needed to better define and compare the natural history of liver disease in this understudied population to those with HBV alone.
Supplementary Material
Potential conflict of interest:
Dr. Chung received grants from Gilead, AbbVie, Merck, Bristol-Myers Squibb, Boehringer-Ingelheim, Roche, and GlaxoSmithKline. Dr. Lisker-Melman is on the speakers’ bureau for Gilead and AbbVie. Dr. Sulkowski consults and received grants from Assembly. He consults for Gilead and Arbutus. Dr. Khalili consults and received grants from Gilead. She received grants from Intercept. Dr. Jain advises and received grants from Gilead. She received grants from Merck and Janssen. Dr. Wong received grants from AbbVie. Dr. Sterling received grants from Abbott, Roche, AbbVie, and Gilead. He is on the data security monitoring board for AskBio and Pfizer. Dr. King received grants from Abbott.
Supported by the National Institute of Diabetes and Digestive and Kidney Diseases at the National Institutes of Health (R01-DK94818) as an ancillary study (NCT01924455) of the Hepatitis B Research Network; National Institutes of Health (NIH; K24AA022523); and the Intramural Research Program of the NIH, National Cancer Institute.
Abbreviations:
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- APRI
aspartate aminotransferase to platelet ratio index
- BMI
body mass index
- cART
combination antiretroviral therapy
- CD
clusters of differentiation
- DM
diabetes mellitus
- FIB-4
fibrosis index based on four factors
- HAI
histologic activity index
- HBRN
Hepatitis B Research Network
- IQR
interquartile range
- LLOD
lower limit of detection
- NIH
National Institutes of Health
- NIDDK
National Institute of Diabetes and Digestive and Kidney Diseases
- qHBeAg
quantitative HBeAg
- qHBsAg
quantitative HBsAg
- TDF
tenofovir disoproxil fumarate
- ULN
upper limit of normal
- VCTE
vibration-controlled transient elastography
Footnotes
In a cohort of 114 adults with HBV-HIV followed for approximately 4 years, the composite clinical endpoint (hepatic decompensation/HCC/liver transplant/HBV-related death) and HBeAg loss were uncommon. In those with paired liver biopsies, there was slight improvement in liver inflammation but no significant change in fibrosis severity.
Supporting Information
Additional Supporting Information may be found at onlinelibrary.wiley.com/doi/10.1002/hep.31823/suppinfo.
REFERENCES
- 1).Benhamou Y Hepatitis B in the HIV-coinfected patient. J Acquir Immune Defic Syndr 2007;45(Suppl. 2):S57–S65; discussion S66-S67. [DOI] [PubMed] [Google Scholar]
- 2).Soriano V, Puoti M, Peters M, Benhamou Y, Sulkowski M, Zoulim F, et al. Care of HIV patients with chronic hepatitis B: updated recommendations from the HIV-hepatitis B virus international panel. AIDS 2008;22:1399–1410. [DOI] [PubMed] [Google Scholar]
- 3).Sulkowski MS. Viral hepatitis and HIV coinfection. J Hepatol 2008;48:353–367. [DOI] [PubMed] [Google Scholar]
- 4).Ockenga J, Tillmann HL, Trautwein C, Stoll M, Manns MP, Schmidt RE. Hepatitis B and C in HIV-infected patients. Prevalence and prognostic value. J Hepatol 1997;27:18–24. [DOI] [PubMed] [Google Scholar]
- 5).Thio CL. Hepatitis B in the human immunodeficiency virus-infected patient: epidemiology, natural history, and treatment. Semin Liver Dis 2003;23:125–136. [DOI] [PubMed] [Google Scholar]
- 6).Gaglio PJ, Sterling R, Daniels E, Tedaldi E. Hepatitis B virus and HIV coinfection: results of a survey on treatment practices and recommendations for therapy. Clin Infect Dis 2007;45:618–623. [DOI] [PubMed] [Google Scholar]
- 7).Ioannou GN. Hepatitis B virus in the United States: infection, exposure, and immunity rates in a nationally representative survey. Ann Intern Med 2011;154:319–328. [DOI] [PubMed] [Google Scholar]
- 8).Smith C Factors associated with specific causes of death amongst HIV-positive individuals in the D:A:D Study. AIDS 2010;24:1537–1548. [DOI] [PubMed] [Google Scholar]
- 9).Hoffmann CJ, Seaberg EC, Young S, Witt MD, D’Acunto K, Phair J, et al. Hepatitis B and long-term HIV outcomes in coinfected HAART recipients. AIDS 2009;23:1881–1889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10).Rockstroh JK, Bhagani S, Benhamou Y, Bruno R, Mauss S, Peters L, et al. European AIDS Clinical Society (EACS) guidelines for the clinical management and treatment of chronic hepatitis B and C coinfection in HIV-infected adults. HIV Med 2008;9:82–88. [DOI] [PubMed] [Google Scholar]
- 11).Iser DM, Sasadeusz JJ. Current treatment of HIV/hepatitis B virus coinfection. J Gastroenterol Hepatol 2008;23:699–706. [DOI] [PubMed] [Google Scholar]
- 12).Martin-Carbonero L, Teixeira T, Poveda E, Plaza Z, Vispo E, Gonzalez-Lahoz J, et al. Clinical and virological outcomes in HIV-infected patients with chronic hepatitis B on long-term nucleos(t)ide analogues. AIDS 2011;25:73–79. [DOI] [PubMed] [Google Scholar]
- 13).Ghany MG, Perrillo R, Li R, Belle SH, Janssen HL, Terrault NA, et al. Characteristics of adults in the hepatitis B research network in North America reflect their country of origin and hepatitis B virus genotype. Clin Gastroenterol Hepatol 2015;13:183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14).Sterling RK, Wahed AS, King WC, Kleiner DE, Khalili M, Sulkowski M, et al. Spectrum of liver disease in hepatitis B virus (HBV) patients co-infected with human immunodeficiency virus (HIV): results of the HBV-HIV cohort study. Am J Gastroenterol 2019;114:746–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15).WHO. Report of the consensus meeting on WHO antiretroviral therapy guidelines for adults and adolescents 2009. http://www.who.int/hiv/topics/treatment/art_consensus_meeting_091016.pdf. Accessed October 14-16, 2009.
- 16).Prati D, Taioli E, Zanella A, Torre ED, Butelli S, Del Vecchio E, et al. Updated definitions of healthy ranges for serum alanine aminotransferase levels. Ann Intern Med 2002;137:1–10. [DOI] [PubMed] [Google Scholar]
- 17).Wai CT, Greenson JK, Fontana RJ, Kalbfleisch JD, Marrero JA, Conjeevaram HS, et al. A simple noninvasive index can predict both significant fibrosis and cirrhosis in patients with chronic hepatitis C. Hepatology 2003;38:518–526. [DOI] [PubMed] [Google Scholar]
- 18).Sterling RK, Lissen E, Clumeck N, Sola R, Correa MC, Montaner J, et al. ; APRICOT Clinical Investigators. Development of a simple noninvasive index to predict significant fibrosis in patients with HIV/HCV coinfection. Hepatology 2006;43:1317–1325. [DOI] [PubMed] [Google Scholar]
- 19).Bohn MJ, Babor TF, Kranzler HR. The Alcohol Use Disorders Identification Test (AUDIT): validation of a screening instrument for use in medical settings. J Stud Alcohol 1995;56:423–432. [DOI] [PubMed] [Google Scholar]
- 20).Carr A, Law M. Objective lipodystrophy severity grading scale derived from the lipodystrophy case definition score. J Acquir Immune Defic Syndr 2003;33:571–576. [DOI] [PubMed] [Google Scholar]
- 21).Brahmania M, Lombardero M, Hansen BE, Terrault NA, Lok AS, Perrillo RP, et al. Association between severe serum alanine aminotransferase flares and hepatitis B e antigen seroconversion and HBV DNA decrease in untreated patients with chronic HBV infection. Clin Gastroenterol Hepatol 2019;17:2541–2551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Ishak K, Baptista A, Bianchi L, Callea F, De Groote J, Gudat F, et al. Histological grading and staging of chronic hepatitis. J Hepatol 1995;22:696–699. [DOI] [PubMed] [Google Scholar]
- 23).Lok AS, Everhart JE, Chung RT, Padmanabhan L, Greenson JK, Shiffman ML, et al. Hepatic steatosis in hepatitis C: comparison of diabetic and nondiabetic patients in the hepatitis C antiviral long-term treatment against cirrhosis trial. Clin Gastroenterol Hepatol 2007;5:245–254. [DOI] [PubMed] [Google Scholar]
- 24).Sterling RK, Wegelin JA, Smith PG, Stravitz RT, Luketic VA, Fuchs M, et al. Similar progression of fibrosis between HIV/HCV- and HCV-infected patients: analysis of paired liver biopsy samples. Clin Gastroenterol Hepatol 2010;8:1070–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25).Kass RE, Raftery AE. Bayes factors. J Am Stat Assoc 1995;90:773–795. [Google Scholar]
- 26).Boyd A, Lasnier E, Molina JM, et al. Liver fibrosis changes in HIV-HBV-coinfected patients: clinical, biochemical, and histologic effect of long-term tenofovir disoproxil fumarate use. Antiviral Ther 2010;15:963–974. [DOI] [PubMed] [Google Scholar]
- 27).Boyd A, Bottero J, Miailhes P, Lascoux-Combe C, Rougier H, Girard PM, et al. Liver fibrosis regression and progression during controlled hepatitis B virus infectioln among HIV-HBV patients treated with tenofovir disoproxil fumarate in France: a prospective cohort study. JIAS 2017;20:21426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Audsley J, Robson C, Aitchison S, Matthews GV, Iser D, Sasadeusz J, et al. Liver fibrosis regression measured by transient elastography in human immunodeficiency virus (HIV)-hepatitis B virus (HBV)-coinfected individuals on long-term HBV-active combination antiretrovial therapy. Open Forum Infect Dis 2016;3:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Vinikoor MJ, Sinkala E, Chilengi R, Mulenga LB, Chi BH, Zyambo Z, et al. Impact of antiretroviral therapy on liver fibrosis among human immunodeficiency virus-infected adults with and without HBV coinfection in Zambia. CID 2017;64:1343–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30).Rana U, Driedger M, Serda P, Pan S, Ding E, Wong A, et al. Characteristics and outcomes of antiretroviral-treated HIV-HBV co-infected patients in Canada? BMC Infect Dis 2019;16:982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Sterling RK, King WC, Wahed AS, Kleiner DE, Khalili M, Sulkowski M, et al. Evaluating non-invasive markers to identify advanced fibrosis by liver biopsy in HBV/HIV coinfected adults. Hepatology 2020;71:411–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32).Piroth L, Pol S, Lacombe K, Miailhes P, Rami A, Rey D, et al. Management and treatment of chronic hepatitis B virus infection in HIV positive and negative patients: the EPIB 2008 study. J Hep 2010;53:1006–1012. [DOI] [PubMed] [Google Scholar]
- 33).Singh KP, Crane M, Audsley J, Lewin SR. HIV-hepatitis virus co-infection: epidemiology, pathogenesis and treatment. AIDS 2017;31:2035–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Boyd A, Gozlan J, Maylin S, Delaugerre C, Peytavin G, Girard PM, et al. Persistent viremia in human immunodeficiency virus/hepatitis B coinfected patients undergoing long-term tenofovir: virologic and clinical implications. Hepatology 2014;60:497–507. [DOI] [PubMed] [Google Scholar]
- 35).Ching-Lung L, Yuen MF. Prevention of hepatitis B virus-related hepatocellular carcinoma with antiviral therapy. Hepatology 2013;57:399–408. [DOI] [PubMed] [Google Scholar]
- 36).Marcellin P, Gane E, Buti M, Afdhal N, Sievert W, Jacobson IM, et al. Regression of cirrhosis during treatment with tenofovir disoproxil fumarate for chronic hepatitis B: a 5-year open-label follow-up study. Lancet 2013;381:468–475. [DOI] [PubMed] [Google Scholar]
- 37).Lok AS, Perrillo R, Lalama CM, Fried MW, Belle SH, Ghany MG, et al. medLow incidence of adverse outcomes in adults with chronic hepatitis B virus infection in the era of antiviral therapy. Hepatology 2020. September 16. 10.1002/hep.31554. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38).Zoutendilk R, Zaaijer HL, de Vries-Sluijs TEMS, Reijnders JG, Mulder JW, Kroon FP, et al. Hepatitis B surface antigen decline and clearance during long-term tenofovir therapy in patients coinfected with HBV and HIV. JID 2012;206:974–980. [DOI] [PubMed] [Google Scholar]
- 39).Chihota BV, Wandeler G, Chilengi R, Mulenga L, Chung RT, Bhattacharya D, et al. High rates of hepatitis B virus (HBV) functional cure among human immunodeficiency virus-HBV coinfected patients on antiretroviral therapy in Zambia. JID 2020;221:218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40).Martin-Carbonero L, Teixeira T, Poveda E, Plaza Z, Vispo E, González-Lahoz J, et al. Clinical outcomes in HIV-infected patients with chronic hepatitis B on long-term nucleos(t)ide analogues. AIDS 2011;25:73–79. [DOI] [PubMed] [Google Scholar]
- 41).Bonacini M. Virologic and clinical outcomes in HIV/HBV coinfected patients on tenofovir-containing HAART. Gastroenterology 2010;1827–1829. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.