

Abstract
Fabry disease represents an X-linked inherited disorder resulting in the accumulation of globotriaosylceramide (Gb3). This review explains the clinical manifestations and the possible therapies for this condition. Fabry disease is considered the second most frequent lysosomal storage disease. More than 1000 mutations of the galactosidase alpha (GLA) gene associated with this disorder have been identified. Pain, either episodic crises or chronic pain, is one of the earliest symptoms in Fabry disease. Gastrointestinal, ocular, ear or skeletal manifestations may complete the clinical picture. Cardiac and renal involvements are the most severe complications leading to organ failure and death. The cerebrovascular lesions may result in severe symptoms including stroke at younger ages. The diagnosis of Fabry disease may be put by enzymatic assays of the α-galactosidase A (AGAL-A) activity in plasma or leukocytes but genetic analysis remains the “gold standard” in identifying the precise mutation and even guiding the treatment. Enzyme replacement therapy (ERT) was the first step in treating subjects with Fabry disease. It proved important decrease of the number of sever clinical events and reduction of symptoms. Chemical chaperone therapy has many advantages including oral administration and was already approved in Europe and US, but it is suitable only for subjects with amenable mutations. Gene therapies (either ex vivo or in vivo) promise to represent a new era for many disorders including Fabry disease, the preliminary data being encouraging. Although many steps were taken in understanding the pathogeny of Fabry disease, future research is needed especially in the field of therapeutic approaches.
Keywords: Fabry disease, lysosome, globotriaosylceramide, kidney failure, heart failure, gene therapy
⧉ Introduction
Fabry disease represents an X-linked inherited disorder affecting the glycosphingolipid metabolism [1]. The disease results from mutations in the galactosidase alpha (GLA) gene, located on the X chromosome [2]. The GLA gene is responsible for encoding a lysosomal enzyme called α-galactosidase (AGAL). The function of AGAL is to metabolize a sphingolipid called globotriaosylceramide or Gb3 or GL3. Therefore, in Fabry disease, Gb3 is not metabolized, and it accumulates in many tissues including within lysosomes. Lysosomes are present in many cell types in the heart (cardiomyocytes and fibroblasts), the kidneys (podocytes, tubular cells, glomerular endothelial), capillary endothelial cells, and nerve cells [3].
Many of the subjects with this disease will have swelling and proliferation of endothelial cells that will lead to kidney failure in the third or fourth decade of life, heart disease, stroke, or premature death [4]. It is considered that the main disease process starts in infancy and some authors indicate that the beginning is during the fetal life [5]. But, despite the evolution in many other lysosomal storage diseases, in Fabry disease, many of the subjects remain asymptomatic during the first years of life. The first signs and symptoms that influence the child’s general status and performance appear during three and 10 years old, usually boys a few years earlier than girls [6]. In time, lysosomal storage and cell damage become progressive affecting vital organs and leading to organ failure. The most severe and life-threatening conditions are end-stage kidney disease and cardiovascular complications [7].
For a long time, Fabry disease was considered to affect mainly males; females were regarded only as “carriers of the affected gene”, but several data indicate that women can have a variety of signs and symptoms of different intensity and variability from nearly asymptomatic to the “classical” phenotype [8]. Males usually develop the “classical” phenotype but subjects experiencing more severe renal manifestations or cardiac involvement were described indicating the idea of a subclassification with a “cardiac variant” and a “renal variant” [9].
⧉ Epidemiology of Fabry disease
Fabry disease is one of more than 50 lysosomal storage diseases that have been identified and characterized biochemically and genetically [10]. By the prevalence of these lysosomal storage diseases, Fabry disease is considered the second most frequent disease after Gaucher disease. The exact prevalence of this disease is unknown and current data may underestimate the prevalence of this disease due to the subjects that are not diagnosed. Because of the limited access to genetic tests, many subjects with Fabry disease do not get a correct diagnose and their symptoms are attributed to other conditions.
A known report indicated the incidence of Fabry hemizygotes as 1:117 000 [11]. It did not indicate the incidence of heterozygotes but by extrapolating the result in hemizygotes, one can determine a combined incidence of 1:58 000 [10].
Other studies indicated other figures, but the method used to determine them also varied. When trying to determine the incidence using newborn screening, a significant high prevalence of the disease was found such as in Italy where one in 3100 had the disease [12]. Other study found that one in 1500 newborn males had Fabry disease, most of them have the IVS4+919G>A mutation considered to determine cardiac phenotype with later-onset [13].
In Netherlands, the prevalence of Fabry disease was considered 1:476 000, lower than the one in other regions [14]. A study conducted by Ozkara & Topçu indicated a much lower prevalence for Fabry disease of one to 6 700 000 but this study included only children under five years of age, with neurological symptoms, therefore many of the subjects with this condition were excluded [15]. In the UK, a study from the beginning of this century estimated a prevalence of one to 366 000 but it is based upon data recorded in the UK clinics from patients who had low AGAL activity. The real prevalence may be underestimated because many of the subjects with Fabry disease may have been omitted [16].
Because renal failure and cardiovascular diseases are the most common manifestations, it was considered to check for subjects with Fabry disease between those with end-stage renal disease (ESRD) on hemodialysis. In a retrospective study that included 105 males on hemodialysis, the prevalence of Fabry disease in patients on hemodialysis was calculated to 0.22% [17]. Having the same premises, a study in Japan indicated that 1.2% of males that received dialysis treatment had Fabry disease [9].
In Romania, by calculating the number of subjects of Fabry disease from a prevalence of approximately 1:100 000 in a country with around 19 million people, the result would be approximately 200 subjects. However, just over 40 subjects with Fabry disease are included in the records of the Expert Center for Rare Cardiovascular Genetic Diseases [2], therefore many persons with this condition remain undiagnosed.
⧉ The role of the lysosome in the cell
Initially described by Christian de Duve, in 1955, lysosomes are now found in all the mammalian cells except red blood cells [10]. They are organelles rich in hydrolases capable of degrading molecules either resulted from biosynthesis either from endocytosis. These organelles can be very heterogeneous in their morphology due to different content, and they are part of a dynamic endocytotic system [18].
After acidification, the proteins follow the process of proteolysis in the lysosomes. The enzymes from the lysosomes are important in other processes, such as antigen processing, degradation of matrix compounds, initiation of apoptosis within the cytosol [19].
Any alteration in the activity of the enzymes or in their activators or transporters may appear due to mutations in the genes that encode these structures. The result will be the accumulation of specific substrates in the lysosomes, and this explains why they are called lysosomal storage diseases. Most of the therapies for these diseases try to replace the deficient enzyme. Other strategies try to reduce the accumulation of storage material by other non-enzymatic means [20].
⧉ The etiology of Fabry disease
Fabry disease represents an inherited X-linked disease. The defective enzyme [the lysosomal α-galactosidase A (AGAL-A)] is coded by a single gene called GLA located on the X chromosome, in Xq22.1 position (Figure 1) [21].
Figure 1.

Graphic representation of X chromosome (the red line represents the location of the GLA gene; the mutations in this gene lead to the development of Fabry disease) (adapted from [21]). GLA: Galactosidase alpha
Because the defective gene is located on the X chromosome, it was considered for many years that women are very little affected by this condition. More evidence suggests that women may present early symptoms and even vital organ failure [22].
Many authors consider that the term X-linked recessive should be avoided, and the use of X-linked inheritance should be recommended. The GLA gene has seven exons and 12 436 base pairs. Fabry disease can be caused by any point mutations, deletion, or insertions [22]. Most of these mutations will lead to the creation of a non-functional enzyme [23]. More than 1000 mutations of the GLA gene have been recorded until now, explaining partly the variability of the clinical manifestations [22,23].
The main metabolic defect is represented by lysosomal AGAL-A deficiency. This enzyme is required for the cleavage of terminal galactose from Gb3; the result is the accumulation of Gb3 in many cells and tissues, such as kidney, heart, nerves, skin, and eyes [4]. Increased endothelial proliferation is frequently followed by vascular accumulation and it may manifest as vascular occlusion, ischemia, and infarction. The most affected vessels are the vertebrobasilar arteries and small cerebral vessels. Usually, subjects that present stroke at younger ages have low levels of thrombomodulin and increased levels of plasminogen activator inhibitor (PAI) indicating that this disease favors a prothrombotic state [4].
The accumulation of Gb3 in the kidneys is located in glomerulus but deposits can be seen also in the distal tubules. The deposits of Gb3 in these locations explain the onset of early proteinuria and polyuria. It is not perfectly understood the mechanism of renal sinus cysts formation [24].
⧉ The clinical presentations of Fabry disease
The first symptoms that appear in early childhood are caused by the accumulation of Gb3 in small nerve fibers of the nervous systems. Pain is one of the earliest symptoms in Fabry disease being described by 60–80% of classically affected boys and girls, usually boys present symptoms at earlier ages [6].
There are two types of pain that characterize the Fabry disease: (i) episodic crises, known as “Fabry crises” described as burning pain that originates in the extreme parts of the body and radiates upwards to the limbs and then to other parts; (ii) chronic pain characterized by paresthesias and burning sensation in the extremities [25]. Some factors like fatigue, stress, exercise, fever, and rapid changes in temperature may precipitate Fabry crises [25]. Pain is considered one of the factors that play an important role in reducing the quality of life (QoL) in subjects with Fabry disease.
After pain, gastrointestinal symptoms are common, starting from childhood and continuing in the adulthood. They consist of nausea, vomiting, diarrhea, abdominal pain (especially after eating). These symptoms may lead to anorexia, and they may be explained by the accumulation of Gb3 in the mesenteric blood vessels [26].
Other frequent sign met in persons with Fabry disease is represented by the absence (anhidrosis) or decreased ability to sweat (hypohidrosis). This is associated with heat, decreased tolerance to warm temperatures and reduced tolerance to physical exercise [22].
Another relatively specific feature and visible from childhood is represented by the presence of skin lesions, such as angiokeratoma and clusters of red purple maculopapular vascular lesions [22]. They can be noticed on the upper thighs, inguinal area, umbilical zone, buttocks and even on mucosal area, such as the mouth. They are small superficial angiomas and their cause is the vessel dilatation in the skin because of the vascular endothelial cells damage [27]. Their size varies from pinpoint to several millimeters, and they increase in size and number with age.
Ear abnormalities are frequently noticed in Fabry subjects. They include hearing loss, tinnitus, and vertigo. It was found a correlation between hearing loss and vascular damage and neuropathy [28]. Corneal opacity (‘cornea verticillata’ or ‘Fabry cataract’) is often noticed during slit lamp examination. Usually, it does not affect the vision and it may be accompanied by tortuous retinal vessels [22]. Other signs of Fabry disease include acute visual loss determined by unilateral central retinal artery occlusion, respiratory complications (chronic cough, wheezing, dyspnea during exercise), anemia, skeletal involvement, arterial remodeling, and intima media thickening, facial dysmorphism, azoospermia, lymphoedema, depression and reduce the QoL [22].
Although they do not develop any major organ failure, these manifestations may significantly reduce the QoL of the children; they may limit the young persons’ physical activity and influence school performance.
Renal involvement in Fabry disease
Signs of renal damage appear from childhood in Fabry disease. The first biochemical manifestations are microalbuminuria and proteinuria, developing from the second decade of life [29]. Kidney biopsy in children may indicate irreversible modifications of glomeruli, interstitial tubules, and renal vessels even before the appearance of microalbuminuria [29]. Other lesions such as podocyte foot processes effacement were cited, and they may indicate focal segmental glomerulosclerosis [20]. Decrease of the glomerular filtration rate (GFR) has rarely been described in childhood.
Similar to other manifestations of Fabry disease, renal damage progresses with age. The accumulation of Gb3 continues in the glomerular endothelium, podocytes, mesangial and interstitial cells. The course of the disease continues to alter the epithelium of Henle’s loop and distal tubules but also the endothelial and smooth muscle cells in the kidney arterioles [30].
Just as like as in diabetic nephropathy, the first signs of renal damage are represented by microalbuminuria and proteinuria, and they appear early in life. Together with continuous Gb3 accumulation, they contribute to the progression of kidney damage. In time, proteinuria increases, and it is associated with tubular damage that affects reabsorption, secretion, and excretion [22]. Just like in diabetic nephrons, at first, hyperfiltration accompanies proteinuria and it may cover the kidney damage. With increasing number of nephrons being deteriorated, renal function measured by GFR begins to decline. Commonly severe chronic kidney disease (CKD) develops in the third to fifth decade of life [31]. Progression to ESRD is common in subjects with renal involvement and it contributes to increasing morbidity and mortality. Uremia represents a frequent cause of death among males with Fabry disease [32].
Because of the common renal damage in Fabry disease, assessment of kidney function should be done early after diagnosing and repeated frequently. Determination of serum creatinine and estimation of GFR using validated formula like Chronic Kidney Disease Epidemiology Collaboration (CKD-EPI) associated with urinary albuminuria or with proteinuria/24 hours or albumin/creatinine ratio in spot urine are useful for the staging of CKD according to The National Kidney Foundation Kidney Disease Outcomes Quality Initiative (NKF K/DOQI) Guidelines. Although the diagnosis of Fabry disease may be put by genetic testing, kidney biopsy is often recommended for baseline evaluation of the kidney and when the renal damage declines despite treatment.
Cardiac involvement
There were described two forms of cardiac involvement in Fabry disease: a variant when the heart is affected together with other organs and another atypical variant where cardiac manifestations are the only signs of disease [33].
Endomyocardial biopsy may help establishing the diagnosis although determining the activity of plasma AGAL-A and genetic testing are easier to perform. The classical description in light microscopy and Hematoxylin–Eosin (HE)-stained tissue is sarcoplasmic vacuolization. Electron microscopy identifies concentric lamellar bodies in the sarcoplasm of myocardial cells [33].
The signs on the electrocardiography include conduction disorders of the atrioventricular (AV) node and shortening of the PR interval (<0.12 ms) [34] because of a decreased P wave, enlarged QRS complex and long QTc interval [35], bundle branch blocks, AV blocks and arrhythmias [36].
Cardiac ultrasonography frequently reveals diastolic dysfunction and concentric left ventricular hypertrophy. Usually, the left ventricular hypertrophy does not become obstructive, and it is more frequently noticed in males than in females [22]. Coronary arteries are soon affected, and myocardial ischemia and infarction may follow [37]. In time, fibrosis affects the myocardium starting from posterior-lateral wall and reduces the cardiac function leading to congestive heart failure [22]. Fibrosis of the sinoatrial node and AV node contribute to the development of arrhythmias and may cause a significant number of deaths in subjects with Fabry disease [37]. Other changes identified by echocardiography are right ventricular hypertrophy, atrial enlargement, valvular thickening, mitral valve prolapse, hypertrophic cardiomyopathy [38].
Studies in subjects with left ventricular hypertrophy identified Fabry disease in an important percent of them. All the subjects with Fabry, presented reduced levels of AGAL-A activity. Although the disease is more severe in males, there were noticed low levels of the enzyme in females that presented left ventricular hypertrophy. Patients with severe symptoms and outflow tract obstruction are referred to cardiac surgery and usually they undergo myectomy [38].
Frequently autonomic dysfunction is observed in subjects with Fabry disease, but it rarely leads to overt signs of orthostatic dysregulation [39]. Because of these conditions involving the heart, the exercise capacity is reduced in subjects with Fabry. This fact contributes to the reduced QoL.
Cerebrovascular lesions
The peripheral neuropathy represents the early neurological sign in Fabry disease. It is rapidly followed by autonomic dysfunction cerebrovascular complications. The cerebrovascular lesions are the result of small arteries affected by deposition of Gb3 [22]. The clinical manifestations of these lesions may vary from mild to severe symptoms such as dizziness, headache, transitory ischemic attacks, and strokes [40]. The presence of stroke is associated with the existence of other complications of Fabry disease like cardiac disease, hypertension, and renal disease. According to Fabry Registry, the prevalence of stroke is 6.9% in males and 4.3% in females with Fabry disease being much higher than in general population [40].
The main cause for stroke is represented by the accumulation of Gb3 in small vessels but a dilatative arteriopathy in the vertebrobasilar arteries may be noticed [22]. The hyperperfusion in the cerebral vessels associated with the adhesion of monocytes and neutrophils to endothelial cell walls may increase thrombus formation [22].
Magnetic resonance imaging (MRI), proton (1H) MR spectroscopy (MRS), positron emission tomography, transcranial Doppler may help exploring cerebrovascular lesions in Fabry disease. A complete neurological examination should be done in all subjects before starting enzyme replacement therapies (ERTs) to evaluate the extent and severity of the disease. Specific tools to assess the pain should be used to evaluate the severity and the frequency of pain [41].
⧉ Diagnosis of Fabry disease
It is important to establish a correct diagnosis from the onset of the signs and symptoms, because of the availability of ERT and other therapies that may reduce the progression of the disease. Unfortunately, an early diagnosis in generally possible only in subjects with relatives with known Fabry disease and it may be late with nearly 15 years in most cases [22]. Frequently, the patients visit many specialists before the suspicion of Fabry to be raised. The early presentation of this disease is very heterogeneous and heart and renal failure rarely develop in children. This may explain the late diagnosis and the reason for immediate biochemical and/or genetic testing when Fabry disease is suspected.
The enzymatic assay assesses the activity of AGAL-A in the blood. It is the main laboratory method, and it must be done in all the subjects with clinical suspicion of Fabry disease. This assay is not very sensitive nor very specific and it must be followed by genetic testing.
Plasma Gb3 was another marker proposed for biochemical diagnosis, but it requires more time and may have false negative results in women where Gb3 levels may be normal [42]. Urinary Gb3 represents a more useful marker, but it may be not elevated in some subjects and with some specific mutations [43].
Because in some persons, especially heterozygotes women, the activity of AGAL-A may be normal the “gold standard” diagnosis must be made by genetic analysis. Direct molecular analysis of the GLA gene may be easily done because this gene is small in size, and this method may help to identify the precise mutation of the gene [22]. This may be a guide for the treatment of the patient.
⧉ Therapeutic approaches in Fabry disease
The first treatment used for Fabry disease was meant to replace the AGAL deficient enzyme (Figure 2). Two substances are known in this class. One is Agalsidase α (Replagal) and the other is Agalsidase β (Fabrazyme). Agalsidase α is produced in a lineage of human fibroblasts. It is administered intravenously at a dose of 0.2 mg/kg every two weeks. Agalsidase β is made in Chinese hamster ovary cells; it is given at a dose of 1 mg/kg as an i.v. infusion at two weeks [44]. Studies have indicated improvement after ERT with clearance of Gb3 and a decrease in the number of sever clinical events and reduction of pain [44].
Figure 2.
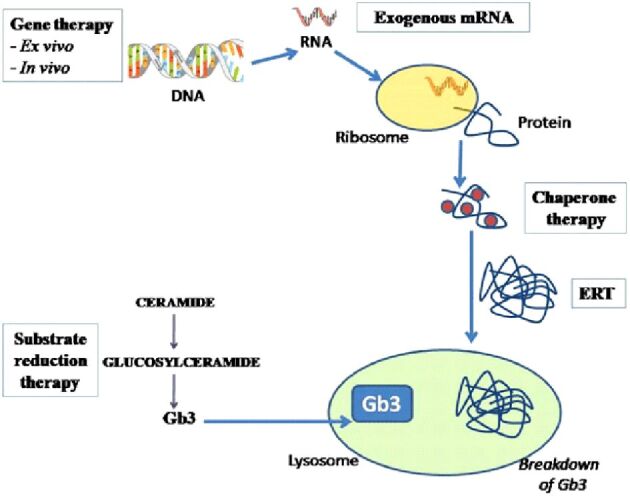
Therapeutic approaches in Fabry disease (adapted from [44]). DNA: Deoxyribonucleic acid; ERT: Enzyme replacement therapy; Gb3: Globotriaosyl-ceramide; mRNA: Messenger ribonucleic acid
Although ERT may be considered the physiological method in treating this condition, it is associated with some risks and side effects. Sometimes, after infusion, reactions like hyperpyrexia, dyspnea and rash may appear. They can be minimized with premedications like steroids or Diphenhydramine. Faster infusion rate of the drug may be associated with intolerance in some subjects [44]. Cardiac MR indicates that cardiac fibrosis progresses despite ERT [45]. ERT may be associated with the development of antibodies, reducing the efficacy of this method.
A new form of AGAL called Pegunigalsidase α was recently produced in a PlantCell Ex system. Preclinical data using this compound have shown good results. Its circulatory half-life is much longer than the one of any existing ERT and increased uptake of the kidney and heart makes it a challenging candidate for a new era in ERT [46]. The idea of plant-based drugs is also challenging and may offer a more efficient production of proteins, antibodies, vaccines at lower cost [46].
Another therapeutic approach is represented by chemical chaperone therapy (Figure 2). The role of chaperones is to bind to defective enzymes and to help with folding, maturation, and movement of the enzyme to the functional site. Therapies with chemical chaperones were used before in other lysosomal storage diseases, such as Gaucher and Pompe disease. In Fabry disease, a new chemical chaperone, Migalastat (Galafold) was approved in Europe in May 2016 and in the US in August 2018.
Migalastat is orally available and is largely distributed in all the body, in the central nervous system because it crosses the blood–brain barrier. It binds to the defective AGAL in the endoplasmic reticulum and helps its folding and its transport to the lysosome [44]. The AT1001 Therapy Compared to Enzyme Replacement in Fabry Patients with AT1001-Responsive Mutations (ATTRACT) study noticed, after 18 months of treatment, significantly decreased ventricular mass index compared to ERT group and low Gb3 plasma levels as in ERT group. The side effects were represented by headache, nausea, pyrexia, and urinary tract infection [47]. Migalastat may be used only for amenable mutations of GLA gene, their number being estimate be between 35% and 50% of the discovered mutations [48].
The amenability is validated in the specific human embryonic kidney (HEK) 293 cells. The amenable mutations are those mutations that render to modified forms of AGAL-A and present a ≥ 1.2-fold increase in AGAL-A activity over baseline and an absolute increase of ≥ 3% over wild-type AGAL-A activity, with 10 μmol/L Migalastat [49]. Administration of Migalastat in subjects with renal impairment, demonstrates an increasing in t1/2 with increasing degrees of renal impairment. Studies with Migalastat did not included patients with Fabry disease and with severe renal impairment or hepatic impairment [50].
Another method that tries to reduce the accumulation of Gb3 is represented by the substrate reduction therapies (Figure 2). Lucerastat, or N-(n-butyl)-deoxygalactonojirimycin, is an iminosugar that acts as a glucosylceramide synthase inhibitor and prevents the accumulation of Gb3 by reducing the amount of ceramide that is transformed to glycosphingolipid. Recent studies indicate that twice daily 1000 mg Lucerastat reduces the circulating levels of Gb3 and other sphingolipids. This new drug is under investigation in a Phase 3 study [the Efficacy and Safety of Lucerastat Oral Monotherapy in Adult Subjects with Fabry Disease (MODIFY) study] having neuropathic pain as the primary end point [51]. Although, Lucerastat is tested in monotherapy, it is thought that it will be used mainly in addition to ERT.
Gene therapies promise to represent a solution for many rare genetic diseases (Figure 2). There are two approaches for these relative new therapies: ex vivo or in vivo. The ex vivo approach presumes that the hematopoietic cells are collected from the patient, they undergo gene editing and transfused back into the patient. In the in vivo way, a vector that edits the genes is infused into the subject and some specialized cell, like hepatocytes, undergo gene editing to express that protein [44].
One of the ex vivo therapies that uses cluster of differentiation 34 (CD34)-positive hematopoietic stem cells is currently tested in a Phase II study. The preliminary data are encouraging because the first subject using this therapy discontinued ERT, being just monitored afterwards [52].
The in vivo approach uses the adeno-associated virus (AAV)-mediated gene transfer in order to maximize the enzyme activity. The preclinical studies in knockout mice indicated increased AGAL-A levels and reduced levels of Gb3 [44]. Besides these gene therapies, the use of AGAL mRNA encapsulated with lipid nanoparticles to enhance the production of AGAL-A in heart, liver and kidney tissues indicated good Gb3 clearance [44].
All these therapies may have some limitations due to side effects and antibodies generation. ERT is frequently associated with formation of antibodies. It is unknown if gene therapies will have long-term effects or immunogenicity; the current vectors usually generate antibodies to the viral capsid [44].
⧉ Conclusions
Although it is considered a rare disease, Fabry disease, like other lysosomal storage diseases, have a serious impact on the QoL, morbidity, and mortality of many subjects. It may affect the heart, the kidneys and the nervous system leading to organ failure. There are several drugs accepted for the treatment of this disease and many others are in trials. The future should focus on (i) identifying most of the persons with this condition by increasing the availability of testing, (ii) finding new therapies that may help these persons to have a normal life, (iii) investigating whether combination therapy represent a better way for achieving the targets, and (iv) what is the optimal time to start the specific therapy in Fabry disease.
Conflict of interest
The authors declare that they have no conflict of interests.
References
- 1.Wanner C, Arad M, Baron R, Burlina A, Elliott PM, Feldt-Rasmussen U, Fomin VV, Germain DP, Hughes DA, Jovanovic A, Kantola I, Linhart A, Mignani R, Monserrat L, Namdar M, Nowak A, Oliveira JP, Ortiz A, Pieroni M, Spada M, Tylki-Szymańska A, Tøndel C, Viana-Baptista M, Weidemann F, Hilz MJ. European Expert Consensus Statement on therapeutic goals in Fabry disease. Mol Genet Metab. 2018;124(3):189–203. doi: 10.1016/j.ymgme.2018.06.004. [DOI] [PubMed] [Google Scholar]
- 2.Militaru S, Adam R, Ismail G, Rusu E, Dulămea A, Jurcut R. Demographic and clinical characteristics of the full 2015-2018 cohort of Romanian Fabry disease patients. Curr Health Sci J. 2019;45(3):272–277. doi: 10.12865/CHSJ.45.03.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Desnick RJ, Ioannou YA, Eng CM. In: The metabolic and molecular bases of inherited disease. 8. Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editors. New York NY USA: McGraw-Hill; 2001. Galactosidase A deficiency Fabry disease; pp. 3733–3774. [Google Scholar]
- 4.Bokhari SRA, Zulfiqar H, Hariz A. In: StatPearls, editor. Treasure Island FL USA: StatPearls Publishing; 2021. Fabry disease. [PubMed] [Google Scholar]
- 5.Vedder AC, Strijland A, vd Bergh Weerman MA, Florquin S, Aerts JMFG, Hollak CEM. Manifestations of Fabry disease in placental tissue. J Inherit Metab Dis. 2006;29(1):106–111. doi: 10.1007/s10545-006-0196-0. [DOI] [PubMed] [Google Scholar]
- 6.Hopkin RJ, Bissler J, Banikazemi M, Clarke L, Eng CM, Germain DP, Lemay R, Tylki-Szymanska A, Wilcox WR. Characterization of Fabry disease in 352 pediatric patients in the Fabry Registry. Pediatr Res. 2008;64(5):550–555. doi: 10.1203/PDR.0b013e318183f132. [DOI] [PubMed] [Google Scholar]
- 7.Mehta A, Beck M, Eyskens F, Feliciani C, Kantola I, Ramaswami U, Rolfs A, Rivera A, Waldek S, Germain DP. Fabry disease: a review of current management strategies. QJM. 2010;103(9):641–659. doi: 10.1093/qjmed/hcq117. [DOI] [PubMed] [Google Scholar]
- 8.Wilcox WR, Oliveira JP, Hopkin RJ, Ortiz A, Banikazemi M, Feldt-Rasmussen U, Sims K, Waldek S, Pastores GM, Lee P, Eng CM, Marodi L, Stanford KE, Breunig F, Wanner C, Warnock DG, Lemay RM, Germain DP. Fabry Registry. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry Registry. Mol Genet Metab. 2008;93(2):112–128. doi: 10.1016/j.ymgme.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 9.Nakao S, Kodama C, Takenaka T, Tanaka A, Yasumoto Y, Yoshida A, Kanzaki T, Enriquez AL, Eng CM, Tanaka H, Tei C, Desnick RJ. Fabry disease: detection of undiagnosed hemodialysis patients and identification of a "renal variant" phenotype. Kidney Int. 2003;64(3):801–807. doi: 10.1046/j.1523-1755.2003.00160.x. [DOI] [PubMed] [Google Scholar]
- 10.Mehta A, Beck M, Sunder-Plassmann G, editors. Fabry disease: perspectives from 5 years of FOS. Oxford: Oxford PharmaGenesis; 2006. [PubMed] [Google Scholar]
- 11.Meikle PJ, Hopwood JJ, Clague AE, Carey WF. Prevalence of lysosomal storage disorders. JAMA. 1999;281(3):249–254. doi: 10.1001/jama.281.3.249. [DOI] [PubMed] [Google Scholar]
- 12.Spada M, Pagliardini S, Yasuda M, Tukel T, Thiagarajan G, Sakuraba H, Ponzone A, Desnick RJ. High incidence of later-onset Fabry disease revealed by newborn screening. Am J Hum Genet. 2006;79(1):31–40. doi: 10.1086/504601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hwu WL, Chien YH, Lee NC, Chiang SC, Dobrovolny R, Huang AC, Yeh HY, Chao MC, Lin SJ, Kitagawa T, Desnick RJ, Hsu LW. Newborn screening for Fabry disease in Taiwan reveals a high incidence of the later-onset GLA mutation c.936+919G>A (IVS4+919G>A) Hum Mutat. 2009;30(10):1397–1405. doi: 10.1002/humu.21074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Poorthuis BJ, Wevers RA, Kleijer WJ, Groener JE, de Jong JG, van Weely S, Niezen-Koning KE, van Diggelen OP. The frequency of lysosomal storage diseases in The Netherlands. Hum Genet. 1999;105(1–2):151–156. doi: 10.1007/s004399900075. [DOI] [PubMed] [Google Scholar]
- 15.Ozkara HA, Topçu M. Sphingolipidoses in Turkey. Brain Dev. 2004;26(6):363–366. doi: 10.1016/j.braindev.2003.09.006. [DOI] [PubMed] [Google Scholar]
- 16.MacDermot KD, Holmes A, Miners AH. Anderson–Fabry disease: clinical manifestations and impact of disease in a cohort of 60 obligate carrier females. J Med Genet. 2001;38(11):769–775. doi: 10.1136/jmg.38.11.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Linthorst GE, Hollak CEM, Korevaar JC, Van Manen JG, Aerts JMFG, Boeschoten EW. alpha-Galactosidase A deficiency in Dutch patients on dialysis: a critical appraisal of screening for Fabry disease. Nephrol Dial Transplant. 2003;18(8):1581–1584. doi: 10.1093/ndt/gfg194. [DOI] [PubMed] [Google Scholar]
- 18.Dell’Angelica EC, Mullins C, Caplan S, Bonifacino JS. Lysosome-related organelles. FASEB J. 2000;14(10):1265–1278. doi: 10.1096/fj.14.10.1265. [DOI] [PubMed] [Google Scholar]
- 19.Brix K. In: Lysosomes Medical Intelligence Unit Book Series. Saftig P, editor. New York NY USA: Springer Science Landes Bioscience; 2005. Lysosomal proteases: revival of a Sleeping Beauty; pp. 50–59. [Google Scholar]
- 20.Matzner U. In: Lysosomes Medical Intelligence Unit Book Series. Saftig P, editor. New York NY USA: Springer Science Landes Bioscience; 2005. Therapy of lysosomal storage diseases; pp. 112–129. [Google Scholar]
- 21.National Fabry Disease Foundation (NFDF). What is Fabry disease? NFDF, Washington, DC, USA. Available from: https://www.fabrydisease.org/index.php/about-fabry-disease/what-is-fabry-disease[ Accessed 27.12.2020]
- 22.Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30–30. doi: 10.1186/1750-1172-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tuttolomondo A, Simonetta I, Duro G, Pecoraro R, Miceli S, Colomba P, Zizzo C, Nucera A, Daidone M, Di Chiara T, Scaglione R, Della Corte V, Corpora F, Vogiatzis D, Pinto A. Inter-familial and intra-familial phenotypic variability in three Sicilian families with Anderson–Fabry disease. Oncotarget. 2017;8(37):61415–61424. doi: 10.18632/oncotarget.18250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Schuller Y, Arends M, Körver S, Langeveld M, Hollak CEM. Adaptive pathway development for Fabry disease: a clinical approach. Drug Discov Today. 2018;23(6):1251–1257. doi: 10.1016/j.drudis.2018.02.004. [DOI] [PubMed] [Google Scholar]
- 25.Hoffmann B, Beck M, Sunder-Plassmann G, Borsini W, Ricci R, Mehta A, FOS European Investigators Nature and prevalence of pain in Fabry disease and its response to enzyme replacement therapy – a retrospective analysis from the Fabry Outcome Survey. Clin J Pain. 2007;23(6):535–542. doi: 10.1097/AJP.0b013e318074c986. [DOI] [PubMed] [Google Scholar]
- 26.Hoffmann B, Schwarz M, Mehta A, Keshav S, Fabry Outcome Survey European Investigators Gastrointestinal symptoms in 342 patients with Fabry disease: prevalence and response to enzyme replacement therapy. Clin Gastroenterol Hepatol. 2007;5(12):1447–1453. doi: 10.1016/j.cgh.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 27.Orteu CH, Jansen T, Lidove O, Jaussaud R, Hughes DA, Pintos-Morell G, Ramaswami U, Parini R, Sunder-Plassman G, Beck M, Mehta AB, FOS Investigators Fabry disease and the skin: data from FOS, the Fabry outcome survey. Br J Dermatol. 2007;157(2):331–337. doi: 10.1111/j.1365-2133.2007.08002.x. [DOI] [PubMed] [Google Scholar]
- 28.Keilmann A, Hajioff D, Ramaswami U, FOS Investigators Ear symptoms in children with Fabry disease: data from the Fabry Outcome Survey. J Inherit Metab Dis. 2009;32(6):739–739. doi: 10.1007/s10545-009-1290-x. [DOI] [PubMed] [Google Scholar]
- 29.Tøndel C, Bostad L, Hirth A, Svarstad E. Renal biopsy findings in children and adolescents with Fabry disease and minimal albuminuria. Am J Kidney Dis. 2008;2009;5153(5)(3):767–776. 567–567. doi: 10.1053/j.ajkd.2007.12.032. [DOI] [PubMed] [Google Scholar]
- 30.Fogo AB, Bostad L, Svarstad E, Cook WJ, Moll S, Barbey F, Geldenhuys L, West M, Ferluga D, Vujkovac B, Howie AJ, Burns A, Reeve R, Waldek S, Noël LH, Grünfeld JP, Valbuena C, Oliveira JP, Müller J, Breunig F, Zhang X, Warnock DG, all members of the International Study Group of Fabry Nephropathy (ISGFN) Scoring system for renal pathology in Fabry disease: report of the International Study Group of Fabry Nephropathy (ISGFN) Nephrol Dial Transplant. 2010;25(7):2168–2177. doi: 10.1093/ndt/gfp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ortiz A, Oliveira JP, Waldek S, Warnock DG, Cianciaruso B, Wanner C. Fabry Registry. Nephropathy in males and females with Fabry disease: cross-sectional description of patients before treatment with enzyme replacement therapy. Nephrol Dial Transplant. 2008;23(5):1600–1607. doi: 10.1093/ndt/gfm848. [DOI] [PubMed] [Google Scholar]
- 32.Schiffmann R, Warnock DG, Banikazemi M, Bultas J, Linthorst GE, Packman S, Sorensen SA, Wilcox WR, Desnick RJ. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol Dial Transplant. 2009;24(7):2102–2111. doi: 10.1093/ndt/gfp031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nagueh SF. Fabry disease. Heart. 2003;89(8):819–820. doi: 10.1136/heart.89.8.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mehta J, Tuna N, Moller JH, Desnick RJ. Electrocardiographic and vector cardiographic abnormalities in Fabry’s disease. Am Heart J. 1977;93(6):699–705. doi: 10.1016/s0002-8703(77)80064-1. [DOI] [PubMed] [Google Scholar]
- 35.Sestito S, Roppa K, Parisi F, Moricca MT, Pensabene L, Chimenz R, Ceravolo MD, Cucinotta U, Ceravolo G, Calabrò MP, Cuppari C, Mondello P, Musolino D, Tallarico V, Concolino D. The heart in Anderson–Fabry disease. J Biol Regul Homeost Agents. 2020;34(4 Suppl 2):63–69. [PubMed] [Google Scholar]
- 36.Shah JS, Hughes DA, Sachdev B, Tome M, Ward D, Lee P, Mehta AB, Elliott PM. Prevalence and clinical significance of cardiac arrhythmia in Anderson–Fabry disease. Am J Cardiol. 2005;96(6):842–846. doi: 10.1016/j.amjcard.2005.05.033. [DOI] [PubMed] [Google Scholar]
- 37.Elliott PM, Kindler H, Shah JS, Sachdev B, Rimoldi OE, Thaman R, Tome MT, McKenna WJ, Lee P, Camici PG. Coronary microvascular dysfunction in male patients with Anderson–Fabry disease and the effect of treatment with alpha galactosidase A. Heart. 2006;92(3):357–360. doi: 10.1136/hrt.2004.054015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sachdev B, Takenaka T, Teraguchi H, Tei C, Lee P, McKenna WJ, Elliott PM. Prevalence of Anderson–Fabry disease in male patients with late onset hypertrophic cardiomyopathy. Circulation. 2002;105(12):1407–1411. doi: 10.1161/01.cir.0000012626.81324.38. [DOI] [PubMed] [Google Scholar]
- 39.Hilz MJ, Marthol H, Schwab S, Kolodny EH, Brys M, Stemper B. Enzyme replacement therapy improves cardiovascular responses to orthostatic challenge in Fabry patients. J Hypertens. 2010;28(7):1438–1448. doi: 10.1097/HJH.0b013e328336a077. [DOI] [PubMed] [Google Scholar]
- 40.Sims K, Politei J, Banikazemi M, Lee P. Stroke in Fabry disease frequently occurs before diagnosis and in the absence of other clinical events: natural history data from the Fabry Registry. Stroke. 2009;40(3):788–794. doi: 10.1161/STROKEAHA.108.526293. [DOI] [PubMed] [Google Scholar]
- 41.Ries M, Kim HJ, Zalewski CK, Mastroianni MA, Moore DF, Brady RO, Dambrosia JM, Schiffmann R, Brewer CC. Neuropathic and cerebrovascular correlates of hearing loss in Fabry disease. Brain. 2007;130(Pt 1):143–150. doi: 10.1093/brain/awl310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vedder AC, Linthorst GE, van Breemen MJ, Groener JEM, Bemelman FJ, Strijland A, Mannens MMAM, Aerts JMFG, Hollak CEM. The Dutch Fabry cohort: diversity of clinical manifestations and Gb3 levels. J Inherit Metab Dis. 2007;30(1):68–78. doi: 10.1007/s10545-006-0484-8. [DOI] [PubMed] [Google Scholar]
- 43.Young E, Mills K, Morris P, Vellodi A, Lee P, Waldek S, Winchester B. Is globotriaosylceramide a useful biomarker in Fabry disease. Acta Paediatr Suppl. 2005;94(447):51–54; discussion 37–38. doi: 10.1111/j.1651-2227.2005.tb02112.x. [DOI] [PubMed] [Google Scholar]
- 44.Felis A, Whitlow M, Kraus A, Warnock DG, Wallace E. Current and investigational therapeutics for Fabry disease. Kidney Int Rep. 2019;5(4):407–413. doi: 10.1016/j.ekir.2019.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hanneman K, Karur GR, Wasim S, Morel CF, Iwanochko RM. Prognostic significance of cardiac magnetic resonance imaging late gadolinium enhancement in Fabry disease. Circulation. 2018;138(22):2579–2581. doi: 10.1161/CIRCULATIONAHA.118.037103. [DOI] [PubMed] [Google Scholar]
- 46.Chen Q, Davis KR. The potential of plants as a system for the development and production of human biologics. F1000Res. 2016;5:F1000 Faculty Rev–912. doi: 10.12688/f1000research.8010.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hughes DA, Nicholls K, Shankar SP, Sunder-Plassmann G, Koeller D, Nedd K, Vockley G, Hamazaki T, Lachmann R, Ohashi T, Olivotto I, Sakai N, Deegan P, Dimmock D, Eyskens F, Germain DP, Goker-Alpan O, Hachulla E, Jovanovic A, Lourenco CM, Narita I, Thomas M, Wilcox WR, Bichet DG, Schiffmann R, Ludington E, Viereck C, Kirk J, Yu J, Johnson F, Boudes P, Benjamin ER, Lockhart DJ, Barlow C, Skuban N, Castelli JP, Barth J, Feldt-Rasmussen U. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. Erratum in: J Med Genet. 2017;2018;5455(4)(6):288–296. 422–429. doi: 10.1136/jmedgenet-2016-104178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lenders M, Stappers F, Niemietz C, Schmitz B, Boutin M, Ballmaier PJ, Zibert A, Schmidt H, Brand SM, Auray-Blais C, Brand E. Mutation-specific Fabry disease patient-derived cell model to evaluate the amenability to chaperone therapy. J Med Genet. 2019;56(8):548–556. doi: 10.1136/jmedgenet-2019-106005. [DOI] [PubMed] [Google Scholar]
- 49.Benjamin ER, Della Valle MC, Wu X, Katz E, Pruthi F, Bond S, Bronfin B, Williams H, Yu J, Bichet DG, Germain DP, Giugliani R, Hughes D, Schiffmann R, Wilcox WR, Desnick RJ, Kirk J, Barth J, Barlow C, Valenzano KJ, Castelli J, Lockhart DJ. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet Med. 2017;19(4):430–438. doi: 10.1038/gim.2016.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.European Medicines Agency (EMA). Galafold (Migalastat): EU summary of product characteristics. EMA, Amsterdam, The Nederlands. Available from: https://www.ema.europa.eu/en/medicines/human/EPAR/galafold[ Accessed 28.12.2020]
- 51.Guérard N, Oder D, Nordbeck P, Zwingelstein C, Morand O, Welford RWD, Dingemanse J, Wanner C. Lucerastat, an iminosugar for substrate reduction therapy: tolerability, pharmacodynamics, and pharmacokinetics in patients with Fabry disease on enzyme replacement. Clin Pharmacol Ther. 2018;103(4):703–711. doi: 10.1002/cpt.790. [DOI] [PubMed] [Google Scholar]
- 52.AVROBIO, Inc. AVROBIO announces updated clinical data for AVR-RD-01 gene therapy in Fabry disease. GlobeNewswire, Cambridge, Massachusetts, USA, October 01, 2018. Available from: https://www.globenewswire.com/news-release/2018/10/01/1587610/0/en/AVROBIO-Announces-Updated-Clinical-Data-for-AVR-RD-01-Gene-Therapy-in-Fabry-Disease.html[ Accessed 28.12.2020]