

Abstract
Paediatric glioblastomas are rapidly growing, devastating brain neoplasms with an invasive phenotype. Radiotherapy and chemotherapy, which are the current therapeutic adjuvant to surgical resection, are still associated with various toxicity profiles and only marginally improve the course of the disease and life expectancy. A considerable body of evidence supports the antitumour and apoptotic effects of certain cannabinoids, such as WIN55,212-2, against a wide spectrum of cancer cells, including gliomas. In fact, we previously highlighted the potent cytotoxic activity of the cannabinoid ligand 5 against glioblastoma KNS42 cells. Taken together, in this study, we designed, synthesised, and evaluated several indoles and indole bioisosteres for their antitumour activities. Compounds 8a, 8c, 8f, 12c, and 24d demonstrated significant inhibitory activities against the viability (IC50 = 2.34–9.06 μM) and proliferation (IC50 = 2.88–9.85 μM) of paediatric glioblastoma KNS42 cells. All five compounds further retained their antitumour activities against two atypical teratoid/rhabdoid tumour (AT/RT) cell lines. When tested against a medulloblastoma DAOY cell line, only 8c, 8f, 12c, and 24d maintained their viability inhibitory activities. The viability assay against non-neoplastic human fibroblast HFF1 cells suggested that compounds 8a, 8c, 8f, and 12c act selectively towards the panel of paediatric brain tumour cells. In contrast, compound 24d and WIN55,212-2 were highly toxic toward HFF1 cells. Due to their structural resemblance to known cannabimimetics, the most potent compounds were tested in cannabinoid 1 and 2 receptor (CB1R and CB2R) functional assays. Compounds 8a, 8c, and 12c failed to activate or antagonise both CB1R and CB2R, whereas compounds 8f and 24d antagonised CB1R and CB2R, respectively. We also performed a transcriptional analysis on KNS42 cells treated with our prototype compound 8a and highlighted a set of seven genes that were significantly downregulated. The expression levels of these genes were previously shown to be positively correlated with tumour growth and progression, indicating their implication in the antitumour activity of 8a. Overall, the drug-like and selective antitumour profiles of indole-2-carboxamides 8a, 8c, 8f, and 12c substantiate the versatility of the indole scaffold in cancer drug discovery.
Indole-2-carboxamides: antitumour potential and selectivity against paediatric glioma.
1. Introduction
Gliomas are brain tumours that originate from non-neuronal supportive cells, called glial cells, which are the most abundant cell types in the central nervous system (CNS).1,2 Paediatric gliomas represent the most common CNS tumours and the leading cause of cancer-related death in children. The average annual incidence rate of CNS tumours in children (0–14 years old) is 5.26 per 100 000 with gliomas accounting for approximately 53% of tumours in this age cohort.3 Gliomas are classified based on the histological criteria of neoplasms into a grading system of malignancy by the World Health Organisation (WHO). The three main categories of gliomas are low grade gliomas (LGGs, WHO grade I and II), high grade gliomas (HGGs, WHO grade III and IV) and diffuse midline gliomas (DMGs).4 These three tumour entities require different treatment and/or management strategies. Paediatric HGGs are nearly universally fatal brain tumours associated with dismal prognosis, with a median survival of 1–2 years.5 A key element hampering the development of new targeted therapies is the relative shortage of paediatric glioma cell lines.6 Despite the clear histopathological similarities between HGGs of all ages, the well-established commonly used adult cell lines are not truly representative of the distinct molecular signatures of childhood HGGs.6–8 Hence, there is a pressing need to develop novel therapeutic agents that effectively target paediatric brain tumours.
We are particularly interested in paediatric glioblastoma multiforme (GBM or grade IV astrocytoma) which is among the most malignant, aggressive and invasive CNS tumours in children.3 The mainstays of treatment of GBM are only palliative, including surgical resection when applicable followed by focal radiotherapy combined with chemotherapy. Many chemotherapeutic agents have been tested including temozolomide, but no remarkable improvement on survival has been achieved in paediatric GBM.5,9 The druggable molecular targets in paediatric HGGs and their corresponding drug candidates that are currently in clinical trials have been recently reviewed.10 In the adult GBM counterpart, Guzman et al. first reported their pilot phase I clinical trial findings on the efficacy of the phytocannabinoid derivative Δ9-tetrahydrocannabinol (Δ9-THC, 1, Fig. 1) delivered intracranially to nine patients.11 This cannabinoid was found to be fairly safe (no overt psychoactive effects) and inhibited tumour cell proliferation in a subset of patients.11
Fig. 1. CB ligands 1–5 as well as the antitumour indoleamide analogue 6 which was identified in our previous report. Δ9-Tetrahydrocannabinol (1) and WIN55,212-2 (2) are mixed CB1R/CB2R agonists, while compounds 3–5 are selective CB2R agonists.

In fact, since the in vitro antitumour potential of Δ9-THC was first reported in 1975,12 an accumulating body of research has demonstrated the antineoplastic effects of many endogenous, naturally occurring, and synthetic cannabinoids against various tumour cells, including gliomas.13–20 These compounds produce most of their biological effects by targeting cannabinoid 1 and 2 receptors (CB1R and CB2R) which are present in the brain and the periphery.21 In particular, two mixed CB1R and CB2R agonists, Δ9-THC (1) and WIN55,212-2 (2), in addition to the selective CB2R agonist JWH-133 (3), Fig. 1, were shown to inhibit the growth of malignant gliomas and impair tumour angiogenesis in vitro and in animal models. They also induced apoptosis in glioma cells in vitro and tumour regression in vivo.22–28 A number of reports ascribed the antitumour activities of cannabinoids to the CB receptors, whilst others demonstrated that their antitumour activities are independent of CB receptors.24–26,29–36 Hence, the mechanism of action of cannabinoids as antitumour agents is still debatable.
In light of the widely reported antitumour activities of cannabinoids, we were interested in exploring the structure–antitumour activity relationship of the indole-containing cannabimimetic agents, exemplified by WIN55,212-2. Indeed, we previously reported several indolecarboxamide derivatives as potent CB1R and/or CB2R ligands.37 The potent CB2R agonist N-(1-adamantyl)-1-(4-hydroxybutyl)-indole-2-carboxamide (4) was the highlight of our previous work (CB2R EC50 = 0.12 μM) which showed no agonist or antagonist activity at CB1R. We also found in the same report that the N-unsubstituted indole 5 exhibited a similar selective CB2R functional activity (EC50 = 0.98 μM) to the N-alkylated indole 4.37 Importantly, we demonstrated the potent inhibitory activity of compound 5 against the viability of paediatric GBM KNS42 cells (IC50 = 0.33 μM).38 In this report, we brought to light the cytotoxic and antiproliferative activities of the indole-2-carboxamide 6 against different malignant brain tumour cells.
Accordingly, in the current study, we describe the design, synthesis, and biological evaluation of several N-unsubstituted indoles and indole bioisosteres as antitumour agents. All final analogues as well as reference ligands WIN55,212-2, JWH-133, and compound 4 were screened in vitro for their viability and proliferation inhibitory activities against paediatric GBM KNS42 cells. KNS42 was the cell line of choice in our primary antitumour screening as it is reported to overexpress CB1R (encoded by the CNR1 gene).39 The antitumour and safety profiles of the most potent compounds were then scrutinised in non-GBM high-grade paediatric brain tumour cells [teratoid/rhabdoid AT/RT (BT12 and BT16) and medulloblastoma (DAOY)] as well as non-neoplastic HFF1 cells. In addition, the functional activities of the top active compounds at CB1R and CB2R were assessed and a transcriptional analysis of KNS42 cells treated with the N-benzylindoleamide analogue 8a was conducted. The drug-likeness of the most active compounds was also predicted using ACD/Labs Percepta.
2. Design
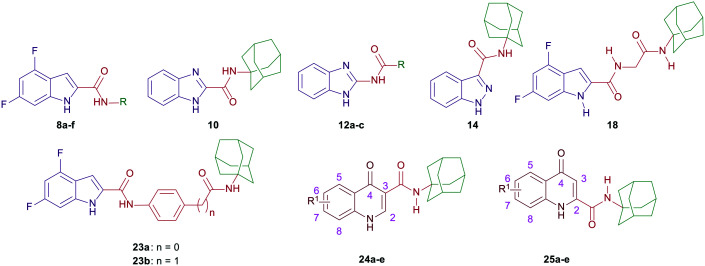
In compounds 8a–f, a 4,6-difluoro-1H-indole nucleus was used instead of the unsubstituted-1H-indole in compound 5, while the adamantane ring was replaced with benzyl, piperonyl, and homopiperonyl motifs. The 4,6-difuoroindole was the scaffold of choice in our study as it has higher lipophilicity (Clog P = 2.6) compared to the unsubstituted indole ring (Clog P = 2.1). In this respect, the slight increase in lipophilicity of the 4,6-difluoroindole-containing compounds may enhance their uptake by tumour cells40 which can lead to an improved antitumour activity profile without significantly impacting the drug-like properties. We also probed the activity of the benzimidazole and indazole cores as bioisosteric replacements to the indole moiety while retaining the adamantane appendage in compounds 10, 12a–c, and 14. In a different approach, we also investigated the activity of some adamantane-derived indoles, wherein extra spacers were introduced to the amide linker between the 4,6-difluoroindole core and the adamantane ring, forming the diamides 18 and 23a and b. It is noteworthy that compound 23a was previously evaluated in our recent report38 and is reincorporated herein to be compared to compound 23b.
On the other hand, N-(1-adamantyl)-1-alkylquinolone-3-carboxamides were widely reported in the literature as cannabinoid receptor modulators.41–44 In addition, some quinolone-3-carboxamides with selective CB2R affinity previously showed potent reduction of the viability of the LNCaP prostatic cancer cell line, displaying IC50 values significantly lower than that of JWH-133 (3).45 Accordingly, in line with the aforementioned strategies, we tested several N-unsubstitutedquinolone-3-carboxamides 24a–e and their analogous quinolone-2-carboxamide counterparts 25a–e for their antitumour activities. Of note, all of the tested quinolonecarboxamides 24a–e and 25a–e were previously evaluated for their antitubercular activity, whereupon most of them were found to be inactive.46
3. Chemistry
The synthetic strategies of all the final compounds are depicted in Schemes 1 and 2. Reference cannabimimetic indoles 4 and 5 were synthesised according to the reported procedure.37 Indoles 8a–f, benzimidazoles 10 and 12a–c, and indazole derivative 14 were synthesised in a one-step amide coupling reaction (method A or B). The indole-2-carboxamides 8a–f and benzimidazole-2-carboxamide 10 were obtained via reacting the carboxylic acid derivatives 7 and 9, respectively, with the corresponding amines in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl), hydroxybenzotriazole hydrate (HOBt) and N,N-diisopropylethylamine (DIPEA) (method A). Method B [EDC·HCl and 4-dimethylaminopyridine (DMAP)] was conducted between compound 11 and the corresponding carboxylic acids to generate the benzimidazole derivatives 12a–c. The indazole-3-carboxamide derivative 14 was formed following the amide coupling protocol A between compound 13 and 1-adamantylamine (Scheme 1).
Scheme 1. General synthetic approach for compounds 8a–f, 10, 12a–c, and 14.

Scheme 2. General synthetic approach for compounds 18 and 23a and b.

On the other hand, the synthesis of the adamantane-derived indole dicarboxamide derivatives 18 and 23a and b is illustrated in Scheme 2. Compound 18 was prepared in three steps starting from coupling 1-adamantylamine (15) with N-(tert-butoxycarbonyl)glycine (Boc-Gly-OH) following a reported procedure47 to give the N-(Boc)adamantane derivative 16. Cleavage of the Boc group thereof using trifluoroacetic acid (TFA) yielded the aminoacetamide intermediate 17. Employing method A of amide coupling, compound 17 was reacted with 4,6-difluoroindole-2-carboxylic acid (7) to afford the final compound 18. Similar to our previously reported compound 23a,38 the dicarboxamide analogue 23b was obtained in a four-step pathway starting from the initial protection of the amino group in the aniline 19b using di-tert-butyl dicarbonate (Boc)2O. Thereafter, the N-Boc derivative 20b was subjected to amide coupling (method A) with 1-adamantylamine to provide compound 21b. Subsequent N-Boc deprotection of the crude product 21b under acidic conditions furnished the key intermediate 22b. Finally, amide coupling of 22b with 7 (method A) delivered the desired dicarboxamide analogue 23b (Scheme 2). Compounds 24a–e and 25a–e were synthesised as reported in our previous work.46
4. Results and discussion
4.1. Biological evaluation and SAR analysis
4.1.1. Cytotoxicity and antiproliferative activity against paediatric KNS42 GBM cells
All the synthesised final compounds 8a–f, 10, 12a–c, 14, 18, 23a and b, 24a–e, and 25a–e were evaluated for their viability inhibition and antiproliferative activities against paediatric GBM KNS42 cells (Table 1). CB receptor ligands WIN55,212-2 (2), JWH-133 (3), 4, and 5 were used as positive controls. Of note, WIN55,212-2 is a non-selective CB1R/CB2R agonist (CB1R EC50 = 0.284 μM, CB2R EC50 = 0.062 μM), whereas JWH-133 (CB2R Ki = 0.0034 μM) and compounds 4 and 5 are selective CB2R agonists (CB2R EC50 = 0.12 and 0.98 μM, respectively).37,48,49 Reference compounds 2, 4, and 5 showed moderate to high cytotoxic activities with low micro- and sub-micromolar inhibitory activities on cell viability (IC50 = 8.02, 4.75, and 0.33 μM, respectively). The selective CB2R agonist JWH-133 (3) showed no inhibitory activity at 10 μM concentration against KNS42 cells in both viability and proliferation studies. WIN55,212-2 was the only active control that inhibited the proliferation of KNS42 cells (IC50 = 8.07 μM). The cytotoxic effects of selective CB2R agonists 4 and 5versus the inactivity of JWH-133 towards KNS42 cells suggest that these compounds may be impacting the cell viability of KNS42 cells via a CB receptor-independent mechanism. This notion is reinforced with many previous reports excluding the involvement of CB receptors in the antiproliferative and apoptotic effects of numerous cannabinoids.22,26,31–36,50
In vitro viability and proliferation inhibitory activities of compounds 8a–f, 10, 12a–c, 14, 18, 23a and b, 24a–e, and 25a–e as well as reference compounds 2–6 against KNS42 cells.
| ||||
|---|---|---|---|---|
| Cpd | R | R1 | Viability IC50a (μM) | Proliferation IC50b (μM) |
| 8a | Benzyl | — | 8.25 ± 1.25 | 9.85 ± 2.76 |
| 8b | 3-Methylbenzyl | — | >10 | >10 |
| 8c | 4-Fluorobenzyl | — | 3.41 ± 0.49 | 4.34 ± 0.46 |
| 8d | 2,3-Dimethoxybenzyl | — | >10 | >10 |
| 8e | Piperonyl | — | >10 | >10 |
| 8f | Homopiperonyl | — | 2.34 ± 0.19 | 2.88 ± 0.25 |
| 10 | — | — | >10 | >10 |
| 12a | Adamantane-1-yl | — | >10 | >10 |
| 12b | 1-Methyladamantane | — | >10 | 6.46 |
| 12c | 4,6-Difluoroindole-2-yl | — | 4.49 ± 0.63 | 4.03 ± 0.01 |
| 14 | >10 | >10 | ||
| 18 | — | — | >10 | >10 |
| 23a | — | — | >10 | >10 |
| 23b | — | — | >10 | 4.90 |
| 24a | — | 6-Chloro | >10 | >10 |
| 24b | — | 7-Bromo | >10 | >10 |
| 24c | — | 5,7-Dichloro | >10 | 9.17 |
| 24d | — | 5,8-Dichloro | 9.06 ± 0.79 | 2.92 ± 0.43 |
| 24e | — | 7,8-Dichloro | >10 | 8.05 ± 4.15 |
| 25a | — | 6-Chloro | >10 | 6.11 ± 0.91 |
| 25b | — | 7-Bromo | >10 | >10 |
| 25c | — | 5,7-Dichloro | >10 | >10 |
| 25d | — | 5,8-Dichloro | >10 | >10 |
| 25e | — | 7,8-Dichloro | >10 | >10 |
| WIN55,212-2 (2) | — | — | 8.02 ± 0.25 | 8.07 ± 0.29 |
| JWH-133 (3) | — | — | >10 | >10 |
| 4 | — | — | 4.75 ± 0.93 | >10 |
| 5 | — | — | 0.33 ± 0.14 | >10 |
| 6 | — | — | 5.04 ± 0.65 | 6.62 ± 2.01 |
Compound dose required to achieve 50% inhibition of KNS42 cell viability, reflecting cytotoxicity.
Compound dose required to achieve 50% inhibition of KNS42 cell proliferation.
The first round of our investigations was focused on exploring the antitumour activity of the 4,6-difluoroindoleamides 8a–f, benzimidazoleamides 10 and 12a–c, and indazoleamide 14. The N-benzyl-indoleamide analogue 8a was previously evaluated in our recently published report.38 It displayed an activity profile similar to WIN55,212-2 against KNS42 cells [IC50 (viability and proliferation) = 8.25 and 9.85 μM, respectively]. We then probed the effect of incorporating different substituents on the benzyl group in 8a on the antitumour activity. Introducing a methyl group at position 3 to the N-linked benzyl group in compound 8b diminished the antitumour activity (IC50 > 10 μM). The N-(4-fluorobenzyl)indoleamide 8c exhibited approximately 2-fold higher cytotoxicity and antiproliferative activities (IC50 = 3.41 and 4.34 μM, respectively) compared to the unsubstituted amidobenzyl counterpart 8a. Similar to 8b, disubstituting the benzyl moiety at positions 2 and 3 with methoxy groups 8d led to a drop in both cytotoxic and antiproliferative activities (IC50 > 10 μM). The N-piperonyl-indoleamide analogue 8e, containing a 1,3-benzodioxole group in lieu of the phenyl ring in 8a, was devoid of activity in the viability and proliferation assays (IC50 > 10 μM). Surprisingly, the N-homopiperonyl analogue 8f, entailing an extra methylene group between the amide linkage and piperonyl motif, showed nearly 3.5-fold higher inhibitory activities (IC50 = 2.34 and 2.88 μM, respectively) compared to 8a against KNS42 cells' viability and proliferation, respectively.
Bioisosteric replacement of the indole ring in reference compound 5 [IC50 (viability) = 0.33 μM] with a benzimidazole scaffold 10 resulted in a dramatic attenuation in the cytotoxic activity against KNS42 cells (IC50 > 10 μM). Of note, compound 10 previously showed high selective agonistic activity towards CB2R (EC50 = 0.52 μM), without observable activity at CB1R.51 Nevertheless, unlike compounds 4 and 5, compound 10 was inactive against KNS42 cells. Next, we evaluated the antitumour activity of benzimidazole derivatives 12a–c, featuring a reversed amide linker. Akin to compound 10, the benzimidazoleamide analogue 12a failed to inhibit the viability and proliferation of KNS42 cells at 10 μM concentration. Introducing a methylene group spacer between the amide linker and the adamantane moiety in 12b led to an improvement in the antiproliferative activity, while the cell viability remained unaffected (IC50 = 6.46 and > 10 μM, respectively). On the other hand, the benzimidazole-indole (BZ-IND) 12c hybrid, linked through an amide group, equally inhibited the viability and proliferation of KNS42 cells (IC50 = 4.49 and 4.03 μM, respectively). In this hybrid, the indole-2-carboxamide framework integrated into compounds 8a–f was preserved, while the benzimidazole nucleus served as the N-linked moiety which may explain the higher activity of this derivative compared to the other benzimidazole analogues 10, 12a, and 12b.
Similar to the N-(adamantyl)benzimidazole derivative 10, integrating an indazole scaffold, in place of the indole nucleus, into compound 14 led to a drastic drop in the cytotoxic activity against KNS42 cells (IC50 > 10 μM). It is worth noting that the same report which documented the CB2R selective activity of compound 10 showed that compound 14 has higher agonistic activity towards CB2R over CB1R (CB2R EC50 = 0.086 μM, CB1R EC50 = 17.1 μM).51 Thus far, the difference in the antitumour activity of 5versus10, 12a, and 14 suggests that the cytotoxic activity of the N-(adamantyl)indoleamide framework against KNS42 cells is superior to the benzimidazole and indazole counterparts. In addition, using the 4,6-difluoroindole scaffold in lieu of the unsubstituted indole ring featured in compound 5, while concomitantly replacing the adamantane group therein with a 4-fluorobenzyl or homopiperonyl motif, was beneficial for the cytotoxic and antiproliferative activities against KNS42 cells.
Based on these findings and the potentiation of the antitumour activity seen in 8f (upon extending the linker connecting the indole ring and the piperonyl motif), in our subsequent round of evaluations, the 4,6-difluoroindole was maintained, while extra spacers were added in between the indole and adamantane moieties (Table 1). First, the adamantane-based indoledicarboxamide 18 showed no inhibitory activity against KNS42 cells in the viability and proliferation studies (IC50 > 10 μM). Similarly, in our previous report, compound 23a, containing a phenyl group as a middle linker between the indole and adamantane rings, showed no activity at 10 μM concentration in both assays.38 However, when we introduced an extra methylene spacer between the phenyl group and the amidoadamantyl motif in compound 23b, a reduction in KNS42 cell proliferation was manifested (IC50 = 4.90 μM), while the cell viability remained unaffected. It is noteworthy that the antiproliferative activity of 23b is comparable to that of our recently reported indoleamide 6 (IC50 = 6.62 μM), entailing a piperazine ring connecting the indole and the adamantane rings. These results suggest that stretching the middle linker connecting the indole and the adamantane moieties could be tolerated in certain structural settings.
The final modification in our study entailed replacing the indole ring in the N-(1-adamantyl)indoleamide framework with a quinolone moiety. Although the majority of the N-(1-adamantyl)quinolone-3-carboxamides that were reported to modulate the activity of CB receptors are N-alkylated at position 1 of the quinolone ring, few N-unsubstituted quinolone analogues were endowed with high affinity and selectivity for CB2R.41–44 Therefore, in accordance with our foregoing strategies, we assessed the cytotoxicity and antiproliferative activities of the adamantane-derived 1H-quinolone-3-carboxamides 24a–e. Monosubstituted quinolones 24a and b failed to inhibit the viability and proliferation of KNS42 cells at 10 μM concentration. Disubstituted quinolones 24c–e, on the other hand, demonstrated good inhibitory activities against proliferation and/or viability of KNS42 cells. The 5,8-dichloroquinolone 24d was the most active compound in this series with cytotoxicity (IC50 = 9.06 μM) and antiproliferative effects (IC50 = 2.92 μM) against KNS42 cells. The 5,7- and 7,8-disubstituted quinolones 24c and 24e exhibited IC50 values of 9.17 and 8.05 μM, respectively, in the proliferation assay, while they manifested lower inhibitory activities in the viability study (IC50 > 10 μM).
Shifting the amidoadamantyl motif from the 3-position of the quinolone scaffold to the 2-position in 25a–e diminished the cytotoxic and antiproliferative activities (IC50 > 10 μM), except for compound 25a. Unlike compound 24a, the 6-chloroquinolone-2-carboxamide analogue 25a showed only moderate antiproliferative activity against KNS42 cells (IC50 = 6.11 μM). Overall, the indole-2-carboxamide architecture appears to be preferable to the benzimidazole, indazole, and quinolone counterparts as antitumour agents.
4.1.2. Cytotoxicity and antiproliferative activities of the most potent compounds against different paediatric brain tumour cells and non-neoplastic fibroblasts
The top potent compounds in the viability and proliferation assays against KNS42 cells, 8a, 8c, 8f, 12c, and 24d (IC50 ≤ 10 μM) as well as WIN55,212-2 (2), were selected for further cytotoxicity and antiproliferative evaluations against a panel of grade IV paediatric brain tumour cells and non-neoplastic human fibroblasts (Table 2). All five compounds and WIN55,212-2 retained their viability and proliferation inhibitory activities (IC50 ≤ 10 μM) against the two AT/RT tumour cell lines (BT12 and BT16). When tested against the medulloblastoma cells DAOY, the N-benzylindoleamide 8a was devoid of antitumour activity (IC50 > 10 μM), as per our previous report.38 However, the N-(4-fluorobenzyl)indole analogue 8c demonstrated moderate cytotoxic activity (IC50 = 4.10 μM) and weak antiproliferative activity (IC50 > 10 μM) against DAOY cells. Similarly, the N-(homopiperonyl)-indoleamide analogue 8f showed appreciable reduction of cell viability and moderate antiproliferative activity against DAOY cells (IC50 = 3.65 and 9.91 μM, respectively). Interestingly, the BZ-IND hybrid 12c exhibited remarkable cytotoxic and antiproliferative activities against all tested paediatric tumour cells (KNS24, BT12, BT16, and DAOY). Compound 12c also showed the most potent viability and proliferation inhibitory activities against DAOY cells (IC50 = 1.02 and 2.31 μM, respectively).
In vitro cytotoxicity and antiproliferative effects of compounds 8a, 8c, 8f, 12c, and 24d as well as WIN55,212-2 (2) against different paediatric brain cancer cell lines (BT12, BT16 and DAOY) and healthy human fibroblasts (HFF1).
| Cpd | BT12 viability IC50a (μM) | BT12 Prolif. IC50b (μM) | BT16 viability IC50a (μM) | BT16 Prolif. IC50b (μM) | DAOY viability IC50a (μM) | DAOY Prolif. IC50b (μM) | HFF1 viability IC5a (μM) | HFF1 Prolif. IC50b (μM) |
|---|---|---|---|---|---|---|---|---|
| 8a | 0.89 ± 0.12 | 7.44 ± 0.59 | 1.81 ± 0.21 | 6.06 ± 0.69 | >10 | >10 | 119 ± 19.78 | 65 ± 8.73 |
| 8c | 9.23 ± 0.35 | 8.87 ± 0.63 | 5.50 ± 0.76 | 9.79 ± 0.89 | 4.10 ± 0.50 | >10 | 281 ± 28.42 | 40 ± 9.81 |
| 8f | 6.08 ± 0.32 | 4.10 ± 0.40 | 2.72 ± 0.33 | 7.53 ± 0.46 | 3.65 ± 0.22 | 9.91 ± 0.97 | 81 ± 20.26 | 6.98 ± 1.23 |
| 12c | 7.65 ± 0.38 | 1.79 ± 0.21 | 1.37 ± 0.13 | 5.41 ± 0.55 | 1.02 ± 0.07 | 2.31 ± 0.38 | 115 ± 11.85 | 3.03 ± 0.55 |
| 24d | 5.15 ± 0.17 | 3.59 ± 0.27 | 1.96 ± 0.39 | 4.88 ± 5.89 | 1.54 ± 0.17 | 4.71 ± 0.24 | 7.04 ± 0.87 | 3.85 ± 0.47 |
| 2 | 8.60 ± 0.78 | 9.32 ± 0.47 | 4.74 ± 3.77 | 4.57 ± 2.71 | >10 ± 3.4 | 6.09 ± 0.22 | 0.023 ± 0.04 | 1.82 ± 0.42 |
Compound dose required to achieve 50% inhibition of tumour/healthy cell viability, reflecting cytotoxicity.
Compound dose required to achieve 50% inhibition of tumour/healthy cell proliferation.
A similar pan-tumour cell viability and/or proliferation inhibition was manifested in the 5,8-dichloroquinolone-3-carboxamide derivative 24d and WIN55,212-2. As we previously reported,38 compound 8a, which was cytotoxic against KNS42, BT12, and BT16 cells, exhibited limited cytotoxicity against healthy human fibroblasts, HFF1 cells (IC50 = 119 μM). Remarkably, compounds 8c, 8f, and 12c, which demonstrated potent cytotoxicity against all paediatric brain tumour cell lines in our study, showed negligible cytotoxicity against HFF1 cells (IC50 = 281, 81, and 115 μM, respectively). However, compounds 8f and 12c inhibited the proliferation of HFF1 cells at concentrations < 10 μM. On the other hand, compound 24d and WIN55,212-2 demonstrated potent viability and proliferation inhibitory activities against non-neoplastic HFF1 cells, indicating the non-selectivity and significant toxicity of these two compounds. Consistent with this observation, Ellert-Miklaszewska et al. also demonstrated that the increase of proapoptotic Bad protein activity is linked to WIN55,212-2's cytotoxic activity.25
4.2. Functional activity at the cannabinoid receptors
Given the structural resemblance of the synthesised molecules to the known cannabinoids, the most active compounds 8a, 8c, 8f, 12c, and 24d in our study in addition to our recently reported indoleamide 6 were further evaluated in vitro for their ability to activate or antagonise human CB1R and CB2R expressed in mouse AtT-20 neuroblastoma cells (Table 3). All six compounds showed no agonistic activity at both CB receptors. These findings were in fact counterintuitive owing to the previously reported potent agonistic profile of the analogous two indoleamide reference compounds 4 and 5 as well as WIN55,212-2 (2). Nonetheless, upon evaluating the antagonistic activity of 8a, 8c, 8f, 12c, and 24d at CB1R and CB2R, we observed some mixed results. The indole-based dicarboxamide analogue 6 and the two N-(benzyl)indoleamide derivatives 8a and 8c failed to antagonise the response of CP55940, a non-selective CB1R/CB2R agonist. On the other hand, the N-(homopiperonyl)indoleamide 8f potently inhibited the CP55940-mediated CB1R activation with an IC50 value of 0.373 μM, whilst the CP55940-mediated CB2R response remained unaffected at 10 μM concentration. The BZ-IND hybrid 12c was inactive as an antagonist at both receptors. Contrary to compound 8f, the quinolone-3-carboxamide 24d failed to inhibit the CP55940-mediated response at CB1R at 10 μM concentration while displaying potent antagonism at CB2R (IC50 = 1.28 μM). These results suggest that the antitumour activities of our most potent compounds are unlikely to be a direct result of activity at CB1R or CB2R.
Functional profile of compounds 6, 8a, 8c, 8f, 12c, and 24d as well as cannabimimetic controls 2, 4, and 5.
| Cpd | Agonism EC50 (μM) | Antagonism IC50 (μM) | ||
|---|---|---|---|---|
| CB1R | CB2R | CB1R | CB2R | |
| 6 | NAa | NAa | NAa | NAa |
| 8a | NAa | NAa | NAa | NAa |
| 8c | NAa | NAa | NAa | NAa |
| 8f | NAa | NAa | 0.373 | NAa |
| 12c | NAa | NAa | NAa | NAa |
| 24d | NAa | NAa | NAa | 1.28 |
| 2 | 0.284 (ref. 48) | 0.062 (ref. 48) | NDb | NDb |
| 4 | NAa | 0.12 (ref. 37) | NAa | NAa |
| 5 | NAa | 0.98 (ref. 37) | NAa | NAa |
NA: not active, defined as <50% activation or inhibition at 10 μM.
ND: not determined.
This was further corroborated by our previous highlights regarding the different cytotoxicity profiles of selective CB2R agonists JWH-133 (3), 10 and 14versus compounds 4 and 5 against KNS42 cells. In fact, the two N-benzylindoleamides 8a and 8c as well as the BZ-IND hybrid 12c which exhibited potent antitumour profiles in our study showed no agonistic or antagonistic activity at both receptors. However, compounds 8f and 24d were shown in the functional assay to be CB ligands. Taken together, it is plausible that the antitumour activities observed in our compounds as well as WIN55,212-2 are not mediated by their action on CB receptors. Indeed, several studies have substantiated the CB-receptor independent induction of tumour cell death and antiproliferative effects exerted by many cannabinoids.
In this respect, in 1998, Sanchez et al. revealed that Δ9-THC-induced apoptosis and sphingomyelin breakdown in C6 glioma cells (expressing CB1R) were not prevented by the CB1R antagonist SR141716. Their results suggested that the observed antitumour effects of Δ9-THC are mediated through a CB1R-independent mechanism.22 This was further supported by Ruiz et al.'s findings in 1999 who also inferred that Δ9-THC-induced apoptosis in human prostate tumour cells is independent of cannabinoid receptors.50 However, Sanchez et al. showed in their subsequent reports that the three cannabinoids [Δ9-THC (1), WIN55,212-2 (2), and JWH-133 (3)] induced apoptosis in C6 glioma cells via a CB1R or CB2R-dependent pathway, as well as sustained accumulation of pro-apoptotic ceramide.24,29,30 Ensuing reports demonstrated that the antitumour activities of cannabinoids are independent of cannabinoid receptors.26,31–36 These conflicting results highlight the elusive mechanism of action of cannabinoids as antitumour agents. Overall, the preceding findings suggest that the antitumour activity observed in our most potent analogues is likely independent of CB receptors.
4.3. Differential gene expression analysis of KNS42 cells treated with compound 8a
In order to unravel the mechanism of action of the indole-2-carboxamides, we previously performed a transcriptional analysis on KNS42 cells treated with compound 6versus the untreated control cell counterparts.38 Upon examining the differential expression of the genes therein, we found that compound 6 downregulated the expression of two key genes, denominated carbonic anhydrase 9 (CA9) and spleen tyrosine kinase (SYK), with statistical significance (p < 0.05).38 Indeed, since knocking down the activity of each of these two genes has been previously shown to inhibit the cell proliferation, invasion, and/or migration of GBM tumours, we concluded in our previous report that the antitumour activities of this indoleamide molecule could be ascribed to repressing the expression of these two genes.38 In the current study, we investigated the gene transcriptional response of KNS42 cells before and after they were treated with the N-benzylindoleamide 8a, employing the DNBSEQ Eukaryotic Stranded Transcriptome Resequencing technique.
Inspecting the differential expression of the genes in the treated cells in comparison to the control cells revealed that compound 8a downregulated the expression of 33 genes (fold change ≥5) with statistical significance (p < 0.05) (Table S1, ESI†). Seven genes were previously shown to promote the progression, proliferation, migration, and/or invasion of various tumours.52–69 Accordingly, compound 8a-induced downregulation of these genes could be the reason behind the antitumour attributes of this indoleamide against GBM KNS42 cells. Interestingly, neither the CA9 nor SYK expression was significantly affected in 8a-treated KNS42 cells.
These seven downregulated genes are: 1) placenta specific protein 1 (PLAC1), 2) Rho GTPase-activating protein 9 (ARHGAP9), 3) apelin early ligand A (APELA) gene, 4) NADH dehydrogenase [ubiquinone] 1 alpha subcomplex subunit 4-like 2 (NDUFA4L2), 5) mitogen-activated protein kinase 4 (MAPK4), 6) l-amino acid transporter 1 (LAT1 or SLC7A5), and 7) angiopoietin-related protein 4 (ANGPTL4). It is noteworthy that the expression levels of CNR1 and CNR2 genes were not significantly altered in KNS42 cells treated with compound 6 or 8a, further corroborating the premise that the CB receptors are presumably uninvolved in our observed antitumour activities. Importantly, upon retrospectively inspecting the differential gene expression data of KNS42 cells treated with our previously reported indoleamide analogue 6, we did not observe any significant alteration in the expression levels of the preceding seven genes. Taken together, these findings suggest that despite both compounds 6 and 8a bearing the same 4,6-difluoroindole-2-carboxamide structure core, the mechanisms through which they exert their antitumour effects are seemingly different.
First, PLAC1, the expression of which is restricted to placental tissues, wherein it plays a key role in the development and function of placenta, was previously found to be highly expressed and aberrantly activated in a wide variety of human cancers.52–54,70 In addition, PLAC1 serves as a biomarker signifying the presence and prognosis of certain tumours.71–73 Indeed, apart from the placenta, no detectable expression of the PLAC1 gene was found in any normal human tissues; therefore it is considered a cancer/placenta-specific gene and was designated as cancer-placenta antigen 1 (CP1).54 A growing body of evidence has demonstrated the oncogenic potential of PLAC1 in various human malignancies, where its expression was shown to be attributed to tumour progression.52–54 Equally important, these studies revealed that silencing PLAC1 in different cancer cells resulted in an inhibition in the proliferation and viability thereof, in addition to induction of apoptosis and cell cycle arrest. Knocking down PLAC1 also impaired the migration and invasion of tumour cells which are metastasis-related phenomena that represent the hallmark of malignancy.52–54 When the KNS42 cells were treated with 8a, PLAC1 was the most downregulated gene with high statistical significance (fold change = 30, p < 0.005). This profound suppression of PLAC1 expression could have contributed to the observed cytotoxic and antiproliferative activities of compound 8a against GBM KNS42 cells.
Two other genes that are potentially involved in the antitumour effects of 8a are ARHGAP9 and MAPK4. The protein encoded by ARHGAP9 belongs to the Rho family of GTPases which principally modulate cytoskeletal dynamics.55 Interestingly, ARHGAP9 was previously shown to serve as a docking protein for mitogen-activated protein kinases (MAPKs).74 Indeed, the interaction observed between ARHGAP9 and MAPKs represents a key crosstalk mechanism between the Rho GTPase and MAPK signalling pathways that is potentially implicated in regulating actin remodelling.74 It was, therefore, intriguing to find that ARHGAP9 and MAPK4 were downregulated in 8a-treated KNS42 cells with high statistical significance (fold change = 12 and 6, respectively, p < 0.0005). However, whether the observed suppression of ARHGAP9 and MAPK4 expression is intertwined, or a mere coincidence requires further investigation. Of note, similar to PLAC1, the abnormal expression of ARHGAP9 and MAPK4 was found to be correlated with poor patient survival as well as the genesis and development/progression of various tumours.55–58 Therefore, it has been proposed that both genes can be used as prognostic biomarkers for these tumours.55,57 Within this context, Wang et al. demonstrated that silencing ARHGAP9 in different human breast cancer cells resulted in a marked decrease in cell proliferation, migration, and invasion, as well as inducing cell cycle arrest and apoptosis thereof.56 Similarly, knocking down the expression of MAPK4 inhibited the proliferation and growth of different human tumour cells and xenografts.57,58 Thus far, although the exact effects of PLAC1, ARHGAP9, and MAPK4 expression in HGGs are yet to be determined, their highly significant downregulation in 8a-treated KNS42 cells suggests, for the first time, their involvement in the tumorigenicity of the GBM tumours.
On the other hand, the oncogenic roles of APELA, NDUFA4L2, SLC7A5, and ANGPTL4 in gliomas, including GBM, were previously demonstrated in the literature.59,60,62,66,75 Accordingly, these four genes were highlighted by researchers as prospective therapeutic targets in malignant gliomas. The protein encoded by the APELA gene binds to the Apelin receptor and promotes the formation of human embryonic vasculature and the growth of embryonic stem cells.59 Since many signalling networks function in both embryogenesis and cancers, Yi et al. investigated the expression of the APELA gene in normal tissues vs. cancer tissues.61 They found that this gene is highly expressed in GBM tissues compared to LGGs. In the same study, the APELA overexpression in ovarian cancers was found to promote cell growth and migration as well as cell cycle progression. Indeed, in the APELA knockout ovarian cancer cells, the loss of APELA led to an inhibition in cell proliferation and migration in vitro. Similar results were discerned in a model of ovarian cancer xenograft bearing mice, in which the size of APELA knockout tumours was significantly decreased compared to the wild-type tumours, supporting the role of APELA in ovarian cancer growth and progression and suggesting its pro-tumorigenic effects in vivo.61
In a subsequent report by Ganguly et al., APELA was found to be expressed at high levels in glioma patients and its upregulation was negatively correlated with patient survival.59 In this respect, there was a significant difference between high and low APELA expressing glioma patients, with high APELA expression being associated with poor patient survival. In fact, the authors showed a direct correlation between APELA expression and glioma grade, wherein the highest APELA expression was found in GBM tumours (grade IV), in resonance with Yi et al.'s findings.59,61 Interestingly, unlike the APELA gene, the expression of the Apelin receptor gene in these patients was not associated with glioma grade or survival rates.59 The significant downregulation of APELA in 8a-treated KNS42 cells (fold change = 11, p < 0.05) accorded with the preceding findings, supporting the role of APELA in the growth of GBM tumours and suggesting that its suppression in KNS42 cells likely contributed to the observed antitumour effects of 8a.
Similar to APELA, NDUFA4L2 was shown to act as an oncogene in different tumours, including GBM.60 NDUFA4L2 is a subunit of the mitochondrial respiratory chain complex I, which is implicated in oxidative stress and metabolic reprograming in various cancers. Indeed, many interesting findings were documented in a very recent study, published in 2021, supporting the role of NDUFA4L2 in promoting GBM progression.60 Chen et al. reported therein that the NDUFA4L2 mRNA and protein were markedly upregulated in human GBM tissues and these elevated levels were associated with shorter survival times in GBM patients. Therefore, they suggested that the high expression of NDUFA4L2 can be regarded as an independent prognostic biological marker for the overall survival of GBM patients.60 Like APELA, the expression levels of NDUFA4L2 in GBM tissues were found to be highly increased, compared to LGGs and normal brain tissues. More importantly, they found that knocking down the NDUFA4L2 gene, both in vitro and in vivo, suppressed tumour cell proliferation and increased apoptosis, whilst protective mitophagy was initiated. They also discovered that apatinib, a multikinase inhibitor that displayed promising antitumour effects in various clinical trials, can effectively target NDUFA4L2, recapitulating the effects brought forth by NDUFA4L2 gene knockdown.60 Indeed, apatinib efficiently reduced the expression of NDUFA4L2, causing cell cycle arrest, enhanced apoptosis, and initiation of protective mitophagy, both in vitro and in vivo. This study was the first to demonstrate the tumorigenic role of NDUFA4L2 in GBM tumours.60 Interestingly, compound 8a inhibited the expression of NDUFA4L2 by 7-fold with high statistical significance (p < 0.0005). Resonating with the preceding antitumour characteristics of apatinib, 8a-induced downregulation of NDUFA4L2 likely contributed to the observed antiproliferative and cytotoxic effects of 8a against GBM cells.
The next gene that we determined to be potentially implicated in 8a-induced antitumour effects is SLC7A5, also referred to as LAT1. This gene belongs to system L transporters, which is accountable for the cellular uptake of most essential amino acids.76LAT1 was found to be upregulated in various types of cancer, with predominant expression in metastatic lesions and primary tumours; therefore it is considered a cancer-type amino acid transporter.76 Indeed, its high expression was previously shown to be closely related to growth, progression, and aggressiveness of different tumours.62–65 Therefore, it was also suggested that LAT1 can be used as an independent marker for poor prognosis in different types of cancer.63 Its tumour-promoting activity was proven in vivo when LAT1 knockdown metastatic prostatic cancer xenografts showed suppressed cell cycle progression, tumour growth, and spontaneous metastasis.64 Importantly, accumulating literature reports have highlighted several LAT1 inhibitors that displayed reduction of cellular uptake of leucine, tumour cell proliferation, and cell cycle progression in various cancer cell lines in vitro, as well as inhibiting tumour growth in vivo.63,76,77 Within this context, Kobayashi et al. demonstrated that 2-aminobicyclo-(2, 2, 1)-heptane-2-carboxylic acid (BCH), a classical LAT1 inhibitor, displayed remarkable cytostatic (reduced proliferation) and cytocidal (increased apoptosis) effects in glioma cells overexpressing the LAT1 gene.62 They also found that increasing the expression of LAT1 significantly enhanced the rate of tumour growth in glioma cells with low endogenous expression of LAT1 in mice. Additionally, LAT1 was found to be expressed at higher levels in human HGGs, including GBM, compared to LGGs, while being undetected in non-neoplastic brain tissues, correlating the expression levels of LAT1 with the malignant status of glioma.62 This was further substantiated by Haining et al.'s study which associated LAT1 upregulation with the histopathological grade, proliferation, and angiogenesis of gliomas, in addition to the poor prognosis of glioma patients.66 The 5-fold downregulation of LAT1 with a very high statistical significance (p < 0.0001) manifested in 8a-treated KNS42 cells, together with the aforementioned compelling findings, supports the possible involvement of LAT1 in the proliferation and viability inhibition of GBM cells induced by 8a.
Finally, we found that ANGPTL4 was 5-fold downregulated in 8a-treated KNS42 cells with statistical significance (p < 0.05). This gene is involved in numerous physiological and pathological functions, such as lipid metabolism, angiogenesis, cell differentiation, and tumorigenesis.78 Several studies demonstrated that high levels of ANGPTL4 are correlated with poorer prognosis in patients with solid tumours, including GBM, suggesting its role in cancer onset, angiogenesis, progression, and metastasis.67–69 Importantly, Katanasaka et al. showed that upregulating the expression of ANGPTL4 promotes tumour angiogenesis in GBM.75 In addition, constitutive knockdown of the ANGPTL4 gene in GBM cells resulted in a significant reduction in the angiogenesis and growth of the corresponding tumour xenografts. This in turn suggests the likely involvement of this gene in the antitumour effects detected in compound 8a.
Overall, we identified seven genes whose expression levels were previously correlated with growth, progression, and poor prognosis of various tumours. Targeting the activity of each of these seven genes genetically and/or chemically was also shown to abrogate tumour growth in several reports. Accordingly, the antitumour activity of 8a could be accredited to modulating the expression levels of these oncogenes.
4.4. ADME profiling
The top potent derivatives 8a, 8c, 8f, 12c, and 24d in addition to WIN55,212-2 (2) were evaluated for their drug-likeness (Table 4) through assessing their conformity to Lipinski's rule of five (RO5) using ACD/Labs Percepta 2016 Build 2911 (13 Jul 2016). The log BB values of these compounds were also predicted in silico to examine their BBB permeability potential. The indole-2-carboxamide derivatives 8a, 8c, 8f, and 12c as well as WIN55,212-2 showed no violation to the RO5, indicating the drug-like attributes of these compounds. The small size and optimum lipophilicity (Log P = 2.69–3.23) of the preceding four indole-2-carboxamides suggest the prospective bioavailability of these analogues. On the contrary, the high lipophilicity of the quinolone derivative 24d (log P = 5.07) accounted for the one minor violation observed therein. All five compounds and WIN55,212-2 (2) are also expected to traverse the BBB, whereupon they may exert their antitumor activities.
Calculated drug-like properties of the top potent compounds 8a, 8c, 8f, 12c, and 24d in addition to reference compound 2 using ACD/Labs Percepta 2016 build 2911 (13 Jul 2016).
| Cpd | MW | HBD | HBA | log P | NRB | TPSA | Log BB |
|---|---|---|---|---|---|---|---|
| 8a | 286.28 | 2 | 3 | 3.17 | 3 | 44.89 | 0.08 |
| 8c | 304.27 | 2 | 3 | 3.23 | 3 | 44.89 | −0.20 |
| 8f | 344.31 | 2 | 5 | 3.02 | 4 | 63.35 | −0.15 |
| 12c | 312.27 | 3 | 5 | 2.69 | 2 | 73.57 | −0.64 |
| 24d | 391.29 | 2 | 4 | 5.07 | 2 | 58.20 | 0.21 |
| 2 | 426.51 | 0 | 5 | 4.13 | 4 | 43.70 | −0.23 |
5. Conclusions
Motivated by the previously reported antitumour activity of the indole-based CB ligands WIN55,212-2 and compound 5, several analogous arylcarboxamide derivatives were designed, synthesised and evaluated for their cytotoxicity and antiproliferative activities against paediatric GBM KNS42 cells. The structure–antitumour activity relationship of our analogues led to the following highlights: (a) the indole-2-carboxamide framework is superior to the benzimidazole, indazole and quinolone counterparts; (b) replacing the adamantane moiety in 5 with benzyl or homopiperonyl groups was favourable; (c) mixed results were manifested when the linker connecting the indole moiety and adamantane group was overextended. Derivatives 8a, 8c, 8f, 12c, and 24d displayed the most potent inhibitory activities against the viability and proliferation of KNS42 cells. WIN55,212-2 and selective CB2R agonists 4 and 5 showed potent cytotoxic activities against KNS42 cells, whilst JWH-133 was devoid of activity. Compounds 8a, 8c, 8f, 12c, and 24d maintained their potent antitumour activities against the other tested grade IV non-GBM paediatric brain tumour cells BT12 and BT16 (AT/RT). When tested against the DAOY (medulloblastoma) cells, these compounds were mostly active, with the exception of 8a. All indole-2-carboxamides 8a, 8c, 8f, and 12c showed no cytotoxicity against non-neoplastic human fibroblasts HFF1, suggesting their selective activity towards tumour cells. On the contrary, the quinolone-3-carboxamide 24d and reference compound WIN55,212-2 were toxic towards HFF-1 cells.
Based on the structural similarities between the newly synthesised compounds and known indole-based cannabinoids, CB functional assays were performed. None of our five most active compounds showed agonistic activity at CB1R or CB2R. In the antagonist mode, compounds 8a, 8c, and 12c failed to inhibit the CP55940-mediated response at both receptors. On the contrary, compounds 8f and 24d behaved as antagonists at CB1R and CB2R, respectively. The discrepancies observed between the antitumour activities of our compounds and their CB functional profiles suggest a complex interplay between CB receptor modulation and CB-independent antitumour mechanisms which remain to be determined. This was further supported by previous reports indicating the non-involvement of CB receptors in the antitumour activity of several cannabinoids.22,26,31–36,50,79 We further substantiated this premise when we performed a transcriptional analysis on KNS42 cells, wherein neither compound 6 nor 8a altered the expression of CNR1 and CNR2 genes. Contrary to our recent findings on compound 6,38 the N-benzylindoleamide 8a did not show a significant modification in the expression of CA9 and SYK genes. Alternatively, we found a set of seven oncogenes that were significantly downregulated in 8a-treated KNS42 cells.
The expression levels of each of these seven genes was previously shown to be positively correlated with the growth, progression, migration, invasion, and/or prognosis of various tumours.52–64,66–69 Since genetically and/or chemically inhibiting the expression of these genes has been previously shown to suppress tumour growth, the antitumour activity of 8a could be attributed to downregulating the expression of these genes. Upon performing a retrospective examination on the differential expression of the genes in compound 6-treated KNS42 cells, we found that the expression levels of the seven genes, potentially implicated in 8a antitumour activity, are not significantly changed. This in turn suggests that despite the structural homology between compounds 6 and 8a, both compounds seemingly inhibit the proliferation and viability of GBM cells via different mechanisms of action. Overall, the drug-like profile, in vitro antitumour activities, and preliminary safety towards non-tumour cells of 8a, 8c, 8f, and 12c establish the indole-2-carboxamides as potential therapeutic agents that can efficiently target malignant brain tumours.
6. Experimental section
6.1. Chemistry
General information
The following starting materials: 4,6-difluoroindole-2-carboxylic acid (7) and 1-adamantylamine (15) were purchased from Fluorochem, while 2-aminobenzimidazole (11) was purchased from AlfaAesar. Benzimidazole-2-carboxylic acid (9), indazole-3-carboxylic acid (13), and 4-aminophenylacetic acid (19b) were purchased from AK Scientific. WIN55,212-2 and JWH-133 were purchased from Cayman Chemical. 1H NMR and 13C NMR spectra were recorded on a Bruker Avance III spectrometer at 400 and 100 MHz, respectively, with TMS as an internal standard. Standard abbreviations indicating multiplicity were as follows: s = singlet, d = doublet, dd = doublet of doublets, t = triplet, q = quadruplet, m = multiplet and br = broad. HRMS experiments were carried out on a Thermo Scientific Q-Exactive Orbitrap mass spectrometer. TLC was performed on Analtech silica gel TLC plates (200 microns, 20 × 20 cm). Flash chromatography was conducted using a Teledyne Isco CombiFlash Rf system with RediSep columns or manually using SiliCycle SiliaFlash® P60 silica gels [40–63 μm (230–400 mesh)]. The final compounds were purified by preparative HPLC unless otherwise stated. The preparative HPLC employed an Omega 5 μm Polar C18 (21.2 × 150 mm) column, with detection at 254 and 280 nm on a Shimadzu SPD-20A detector, flow rate = 25.0 mL min−1. Method 1: 40–100% acetonitrile/H2O in 10 min; 100% acetonitrile in 15 min; 100–40% acetonitrile/H2O in 10 min. Method 2: 50–100% acetonitrile/H2O in 10 min; 100% acetonitrile in 15 min; 100–50% acetonitrile/H2O in 10 min. Method 3: 60–100% acetonitrile/H2O in 10 min; 100% acetonitrile in 15 min; 100–60% acetonitrile/H2O in 10 min. Both solvents contained 0.05 vol% of trifluoroacetic acid (TFA). The purities of the final compounds were established by analytical HPLC, which was carried out using a Waters 1525 HPLC system with a Phenomenex 5 μm C18 (2) (150 × 4.6 mm), on a Waters 2487 dual wavelength detector. Analytical HPLC method: flow rate = 1 mL min−1; gradient elution over 35 min. Gradient: 20–100% acetonitrile/H2O in 15 min; 100% acetonitrile in 10 min; 100–20% acetonitrile/H2O in 5 min. Both solvents incorporated 0.05 vol% of TFA. The purity of all tested compounds was at least 95% as determined by the method described above.
6.1.1. General procedure for amide coupling (method A)
To a solution of the appropriate carboxylic acid (1 equiv.) in anhydrous dimethylformamide (DMF, 10 mL mmol−1), HOBt (2 equiv.) and EDC·HCl (2 equiv.) were added at room temperature (rt). After stirring for 10 min, the corresponding amine (1.2 equiv.) and DIPEA (6 equiv.) were added, and the reaction mixture was stirred at room temperature (rt) until the disappearance of the starting material (usually 60–72 h). After this time, water (50 mL) was added, and the mixture was extracted with EtOAc (3 × 50 mL). The combined organic layers were washed with water (5 × 25 mL) and brine (1 × 25 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using a dichloromethane/methanol (DCM/MeOH) gradient prior to further preparative HPLC purification unless otherwise stated.
6.1.2. General procedure for amide coupling (method B)
A mixture of 2-aminobenzimidazole (1 mmol), EDC·HCl (1.2 mmol), DMAP (1.2 mmol) and the appropriate carboxylic acid (1.2 mmol) in a (1 : 1) 20 mL mixture of anhydrous DCM and anhydrous tetrahydrofuran (THF) was stirred at room temperature (rt) for 72 h. In compounds 12a and 12c, the reaction mixture was quenched with saturated NH4Cl solution (50 mL) and extracted with DCM (3 × 25 mL) and ethyl acetate (3 × 25 mL). The combined organic layers were washed with brine (1 × 50 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using a DCM/MeOH gradient. The two compounds were then further purified via preparative HPLC to attain >95% purity. In compound 12b, after 72 h of stirring at rt, the solvent was evaporated under vacuum and the residue was purified by manual column chromatography. The obtained product was already >95% pure.
6.1.3. General procedure for N-Boc protection (method C)
To a solution of the appropriate amine (3 mmol) in 30 mL of water : dioxane (1 : 2), di-tert-butyl dicarbonate (Boc2O, 6 mmol) and triethylamine (Et3N, 6 mmol) were added and the reaction mixture was stirred at rt for 72 h. Three quarters of the solvent was then evaporated in vacuo and the residue was acidified with 3 M aqueous HCl. The formed precipitate was filtered off, washed with water, and dried.
6.1.4. General procedure for N-Boc deprotection (method D)
To a solution of the N-Boc protected amine (1 mmol) in 5 mL DCM, 2 mL TFA was added. The reaction mixture was stirred for 12 h and concentrated in vacuo then Na2HCO3 solution was added for neutralisation, followed by extraction with DCM (3 × 50 mL). The combined organic phases were washed with brine (1 × 25 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure. The residue was purified by flash chromatography using a DCM/MeOH gradient.
6.1.5. Preparation of synthetic intermediates 20a, 21a, 22a, and final compound 23a
The synthesis of the title compounds and their chemical characterisation are delineated in our recent article.38
6.1.6. Preparation of the tested quinoloneamides
The tested quinolones 24a–e and 25a–e were synthesised and characterised as we previously reported.46
N-Benzyl-4,6-difluoro-1H-indole-2-carboxamide (8a)
This compound was obtained from 4,6-difluoroindole-2-carboxylic acid (7) and benzylamine employing method A. White solid, yield: 98%. Its chemical characterisation is detailed in our recently published report.381H NMR (DMSO-d6) δ 12.05 (s, 1H), 9.13 (t, J = 5.9 Hz, 1H), 7.38–7.28 (m, 5H), 7.28–7.20 (m, 1H), 7.04 (dd, J = 9.4, 1.3 Hz, 1H), 6.87 (td, J = 10.4, 1.9 Hz, 1H), 4.52 (d, J = 6.0 Hz, 2H).
4,6-Difluoro-N-(3-methylbenzyl)-1H-indole-2-carboxamide (8b)
4,6-Difluoroindole-2-carboxylic acid (7) and 3-methylbenzyl amine were used to deliver the title compound following method A. It was >95% pure after flash chromatography. White solid, yield: 90%.1H NMR (DMSO-d6) δ 12.04 (s, 1H), 9.09 (t, J = 6.0 Hz, 1H), 7.30 (s, 1H), 7.22 (t, J = 7.5 Hz, 1H), 7.17–7.00 (m, 4H), 6.87 (td, J = 10.4, 1.8 Hz, 1H), 4.49 (d, J = 6.0 Hz, 2H); 13C NMR (DMSO-d6) δ 160.8, 159.7 (dd, J = 237.0, 12.0 Hz), 156.2 (dd, J = 248.7, 15.5 Hz), 139.7138.1 (dd, J = 15.2, 13.2 Hz), 137.9, 133.1 (d, J = 3.2 Hz), 128.7, 128.3, 128.0, 124.8, 113.6 (d, J = 21.9 Hz), 98.8, 95.7 (dd, J = 29.7, 23.2 Hz), 95.1 (dd, J = 25.9, 4.4 Hz), 42.7, 21.5; HRMS (ESI) m/z calcd for C17H14F2N2O ([M + H]+) m/z 301.1147; found 301.1141.
4,6-Difluoro-N-(4-fluorobenzyl)-1H-indole-2-carboxamide (8c)
The title compound was obtained from 4,6-difluoroindole-2-carboxylic acid (7) and 4-fluorobenzyl amine employing method A. This compound was purified by crystallisation from DMF and was obtained in >95% purity. Off white solid, yield: 76%.1H NMR (DMSO-d6) δ 12.04 (s, 1H), 9.13 (t, J = 6.0 Hz, 1H), 7.46–7.33 (m, 2H), 7.27 (s, 1H), 7.21–7.11 (m, 2H), 7.03 (dd, J = 9.4, 1.5 Hz, 1H), 6.88 (td, J = 10.4, 2.0 Hz, 1H), 4.49 (d, J = 5.9 Hz, 2H); 13C NMR (DMSO-d6) δ 161.7 (d, J = 242.3 Hz), 160.8, 159.7 (dd, J = 237.0, 12.0 Hz), 156.2 (dd, J = 248.8, 15.6 Hz), 138.1 (dd, J = 15.0, 13.3 Hz), 136.0 (d, J = 3.0 Hz), 133.0 (d, J = 3.1 Hz), 129.7 (d, J = 8.1 Hz), 115.5 (d, J = 21.3 Hz), 113.6 (d, J = 21.8 Hz), 98.9, 95.7 (dd, J = 29.7, 23.3 Hz), 95.1 (dd, J = 25.9, 4.3 Hz), 42.0; HRMS (ESI) m/z calcd for C16H11F3N2O ([M + H]+) m/z 305.0896; found 305.0894.
N-(2,3-Dimethoxybenzyl)-4,6-difluoro-1H-indole-2-carboxamide (8d)
This compound was synthesised from 4,6-difluoroindole-2-carboxylic acid (7) and 2,3-dimethoxybenzyl amine following method A. White solid, yield: 78%.1H NMR (DMSO-d6) δ 12.05 (s, 1H), 9.07 (t, J = 5.9 Hz, 1H), 7.29 (s, 1H), 7.03 (dd, J = 9.4, 1.3 Hz, 1H), 6.97 (d, J = 1.5 Hz, 1H), 6.85–6.92 (m, 3H), 4.44 (d, J = 5.9 Hz, 2H), 3.74 (s, 3H), 3.72 (s, 3H); 13C NMR (DMSO-d6) δ 160.7, 159.6 (dd, J = 238.3, 12.1 Hz), 156.2 (dd, J = 248.6, 15.6 Hz), 149.1, 148.3, 138.1 (dd, J = 15.2, 13.3 Hz), 133.2 (d, J = 3.2 Hz), 132.2, 120.0, 113.6 (d, J = 22.0 Hz), 112.3, 112.0, 98.9, 95.6 (dd, J = 29.7, 23.2 Hz), 95.1 (dd, J = 25.9, 4.4 Hz), 56.0, 55.9, 42.5; HRMS (ESI) m/z calcd for C18H16F2N2O3 ([M + H]+) m/z 347.1202; found 347.1202.
N-(Piperonyl)-4,6-difluoro-1H-indole-2-carboxamide (8e)
This compound was obtained from 4,6-difluoroindole-2-carboxylic acid (7) and piperonylamine following method A. It was 95% pure after flash chromatography. Buff solid, yield: 93%.1H NMR (DMSO-d6) δ 12.03 (s, 1H), 9.05 (t, J = 6.0 Hz, 1H), 7.26 (s, 1H), 7.02 (dd, J = 9.4, 1.5 Hz, 1H), 6.93–6.84 (m, 3H), 6.81 (dd, J = 8.0, 1.5 Hz, 1H), 5.98 (s, 2H), 4.41 (d, J = 6.0 Hz, 2H); 13C NMR (DMSO-d6) δ 160.7, 159.7 (dd, J = 238.4, 12.2 Hz), 156.2 (dd, J = 248.7, 15.5 Hz), 147.7, 146.6, 138.1 (dd, J = 15.3, 13.2 Hz), 133.7, 133.1 (d, J = 3.3 Hz), 121.0, 113.6 (d, J = 22.5 Hz), 108.5, 108.4, 101.3, 98.8, 95.7 (dd, J = 29.7, 23.3 Hz), 95.1 (dd, J = 25.8, 4.4 Hz), 42.5; HRMS (ESI) m/z calcd for C17H12F2N2O3 ([M + H]+) m/z 331.0889; found 331.0881.
N-(Homopiperonyl)-4,6-difluoro-1H-indole-2-carboxamide (8f)
The title compound was synthesised from 4,6-difluoroindole-2-carboxylic acid (7) and homopiperonylamine following method A. It was >95% pure after flash chromatography. White solid, yield: 73%.1H NMR (DMSO-d6) δ 11.97 (s, 1H), 8.60 (t, J = 5.6 Hz, 1H), 7.19 (d, J = 1.1 Hz, 1H), 7.02 (dd, J = 9.4, 1.4 Hz, 1H), 6.88 (overlapping td, J = 10.4, 2.0 Hz, 1H), 6.84 (d, J = 1.6 Hz, 1H), 6.81 (d, J = 7.9 Hz, 1H), 6.69 (dd, J = 7.9, 1.6 Hz, 1H), 5.95 (s, 2H), 3.47 (q, J = 6.7 Hz, 2H), 2.77 (t, J = 7.2 Hz, 2H); 13C NMR (DMSO-d6) δ 160.7, 159.6 (dd, J = 238.3, 12.1 Hz), 156.1 (dd, J = 248.5, 15.6 Hz), 147.7, 146.0, 138.0 (dd, J = 15.2, 13.2 Hz), 133.6, 133.3 (d, J = 3.3 Hz), 122.0, 113.6 (d, J = 21.1 Hz), 109.5, 108.6, 101.1, 98.5, 95.6 (dd, J = 29.6, 23.3 Hz), 95.1 (dd, J = 25.9, 4.4 Hz), 41.1, 35.2; HRMS (ESI) m/z calcd for C19H20F2N2O2 ([M + H]+) m/z 345.1045; found 345.1038.
N-(1-Adamantyl)-1H-benzimidazol-2-carboxamide (10)
The title compound was synthesised from benzimidazole-2-carboxylic acid (9) and 1-adamantylamine following method A. The 1H NMR data matched the one reported in the literature.51 It was >95% pure after flash chromatography. White solid, yield: 66%. 1H NMR (DMSO-d6) δ 13.11 (s, 1H), 7.74 (s, 1H), 7.61 (s, 2H), 7.31–7.24 (m, 2H), 2.10 (s, 6H), 2.08 (s, 3H), 1.67 (s, 6H).
N-(1H-Benzimidazol-2-yl)adamantane-1-carboxamide (12a)
The title compound was synthesised from 2-aminobenzimidazole (11) and 1-adamantanecarboxylic acid according to method B and its 1H NMR data matched the one we reported before.80 White solid, yield: 41%. The 1H NMR (DMSO-d6) δ 7.61 (s, 2H), 7.32 (s, 2H), 2.05 (s, 3H), 1.97 (s, 6H), 1.72 (s, 6H).
2-(1-Adamantyl)-N-(1H-benzimidazol-2-yl)acetamide (12b)
This compound was obtained from 2-aminobenzimidazole (11) and 1-adamantaneacetic acid employing method B and it was >95% pure after flash chromatography. Its 1H NMR data matched the one we reported before.80 White solid, yield: 72%.1H NMR (DMSO-d6) δ 12.05 (s, 1H), 11.41 (s, 1H), 7.43 (s, 2H), 7.15–6.95 (m, 2H), 2.19 (s, 2H), 1.91 (s, 3H), 1.74–1.49 (m, 12H).
N-(1H-Benzimidazol-2-yl)-4,6-difluoro-1H-indole-2-carboxamide (12c)
This compound was obtained via amide coupling 2-aminobenzimidazole (11) and 4,6-difluoroindole-2-carboxylic acid (6) following method B. White solid, yield: 35%. 1H NMR (DMSO-d6) δ 12.34 (s, 1H), 7.67 (s, 1H), 7.62 (s, 2H), 7.32 (dd, J = 5.3, 2.2 Hz, 2H), 7.14 (dd, J = 9.3, 1.8 Hz, 1H), 6.96 (td, J = 10.3, 2.0 Hz, 1H); 13C NMR (DMSO-d6) δ 160.8, 160.6 (dd, J = 240.5, 12.0 Hz), 156.6 (dd, J = 250.3, 15.6 Hz), 146.3, 139.1 (overlapping dd, J = 14.0 Hz, 1H), 131.4, 124.0, 113.9, 113.8, 113.7, 102.8, 96.4 (dd, J = 30.0, 23.0 Hz), 95.4 (dd, J = 26.0, 4.1 Hz); HRMS (ESI) m/z calcd for C17H14F2N2O ([M + H]+) m/z 301.1147; found 301.1141.
N-(1-Adamantyl)-1H-indazole-3-carboxamide (14)
The title compound was synthesised from indazole-3-carboxylic acid (13) and 1-adamantylamine following method A. It was >95% pure after flash chromatography. The 1H NMR data matched the one reported in the literature.51 White solid, yield: 62%. 1H NMR (DMSO-d6) δ 13.47 (s, 1H), 8.14 (d, J = 8.2 Hz, 1H), 7.59 (d, J = 8.4 Hz, 1H), 7.40 (t, J = 7.6 Hz, 1H), 7.27–7.17 (m, 2H), 2.11 (s, 6H), 2.07 (s, 3H), 1.68 (s, 6H).
tert-Butyl (2-(adamantan-1-ylamino)-2-oxoethyl)carbamate (16)
The title compound was prepared from 1-adamantylamine (15) according to the reported procedure.47 The obtained crude residue was used without further purification in the next step; white solid, yield: 80%.
N-(1-Adamantyl)-2-aminoacetamide (17)
This compound was synthesised using the crude product 16 employing method D. The 1HNMR data matched the one reported in the literature.47 White solid, yield: 92%. 1H NMR (DMSO-d6) δ 7.48 (s, 1H), 3.12 (s, 2H), 2.01 (s, 3H), 1.92 (d, J = 2.6 Hz, 6H), 1.62 (s, 6H).
N-(2-((Adamantan-1-yl)amino)-2-oxoethyl)-4,6-difluoro-1H-indole-2-carboxamide (18)
This compound was prepared from compound 17 and 4,6-difluoroindole-2-carboxylic acid (7) following method A. White solid, yield: 82%.1H NMR (DMSO-d6) δ 12.02 (s, 1H), 8.71 (t, J = 5.9 Hz, 1H), 7.38 (s, 1H), 7.25 (d, J = 1.7 Hz, 1H), 7.04 (dd, J = 9.3, 1.5 Hz, 1H), 6.88 (td, J = 10.4, 1.8 Hz, 1H), 3.85 (d, J = 5.9 Hz, 2H), 2.01 (s, 3H), 1.94 (s, 6H), 1.62 (s, 6H); 13C NMR (DMSO-d6) δ 168.0, 161.0, 159.7 (dd, J = 238.5, 12.1 Hz), 156.2 (dd, J = 248.7, 15.5 Hz), 138.1 (dd, J = 15.2, 13.2 Hz), 133.0 (d, J = 3.3 Hz), 113.6 (d, J = 21.7 Hz), 99.0, 95.7 (dd, J = 29.6, 23.3 Hz), 95.0 (dd, J = 25.9, 4.4 Hz), 51.3, 42.8, 41.5, 36.5, 29.3; HRMS (ESI) m/z calcd for C21H23F2N3O2 ([M + H]+) m/z 388.1831; found 388.1830.
2-(4-((tert-Butoxycarbonyl)amino)phenyl)acetic acid (20b)
This compound was obtained via N-Boc protection of 4-aminophenylacetic acid 19b following method C. The 1HNMR data matched the one reported in the literature.81 White solid, yield: 84%.1H NMR (DMSO-d6) δ 7.89 (d, J = 8.2 Hz, 2H), 7.45 (t, J = 6.0 Hz, 1H), 7.34 (d, J = 8.2 Hz, 2H), 4.18 (d, J = 6.1 Hz, 2H), 1.39 (s, 9H).
tert-Butyl (4-(2-((-adamantan-1-yl)amino)-2-oxoethyl)phenyl)carbamate (21b)
The title compound was prepared via amide coupling 20b and 1-adamantylamine employing method A. The collected crude product after evaporating the EtOAc extract was used without further purification in the next step; buff solid, yield: 70%.
N-(1-Adamantyl)-2-(4-aminophenyl)acetamide (22b)
This compound was obtained via N-Boc deprotection (method D) of the crude product 21b. Buff solid, yield: 90%.1H NMR (DMSO-d6) δ 10.06 (t, J = 5.7 Hz, 1H), 7.74 (d, J = 8.2 Hz, 2H), 7.54 (s, 1H), 7.31 (d, J = 8.2 Hz, 2H), 4.42 (d, J = 5.8 Hz, 2H), 2.06 (s, 9H), 1.65 (s, 6H); 13C NMR (DMSO-d6) δ 170.8, 147.3, 129.7, 124.4, 114.3, 51.1, 42.9, 41.5, 36.5, 29.3.
N-(4-(2-((-Adamantan-1-yl)amino)-2-oxoethyl)phenyl)-4,6-difluoro-1H-indole-2-carboxamide (23b)
This compound was obtained via amide coupling 4,6-difluoroindole-2-carboxylic acid (7) and 22b employing method A. The product was further crystallised from a 70% DCM/30% MeOH mixture and was obtained in >95% purity. White solid, yield: 96%.1H NMR (DMSO-d6) δ 12.15 (s, 1H), 10.24 (s, 1H), 7.68 (d, J = 8.6 Hz, 2H), 7.52 (s, 2H), 7.23 (d, J = 8.6 Hz, 2H), 7.07 (dd, J = 9.4, 1.4 Hz, 1H), 6.91 (td, J = 10.4, 2.1 Hz, 1H), 3.32 (s, 2H), 1.98 (s, 3H), 1.92 (s, 6H), 1.59 (s, 6H); 13C NMR (DMSO-d6) δ 170.0, 159.9 (dd, J = 239.0, 12.1 Hz), 159.3, 156.3 (dd, J = 249.0, 15.5 Hz), 138.4 (dd, J = 15.3, 13.0 Hz), 137.3, 133.0 (d, J = 3.2 Hz), 132.9, 129.6, 120.6, 113.6 (d, J = 21.9 Hz), 99.9, 95.9 (dd, J = 29.8, 23.2 Hz), 95.1 (dd, J = 25.9, 4.4 Hz), 51.2, 43.0, 41.4, 36.5, 29.3; HRMS (ESI) m/z calcd for C27H27F2N3O2 ([M + H]+) m/z 464.2144; found 464.2138.
6.2. Biological evaluation
6.2.1. Antitumour activity
The four well-established paediatric brain tumour cell lines were all derived from humans and were used to assess the effects on proliferation and viability when treated with the indole and indole bioisostere carboxamide derivatives. KNS42 (glioblastoma multiforme – GBM), BT-12 and BT-16 (atypical teratoid rhabdoid tumour – AT/RT) cell lines were gifts from Dr. Hashizume, Northwestern University, whereas DAOY cells (medulloblastoma – MB) were obtained from ATCC. The human fibroblasts HFF1 (obtained from ATCC) were used as non-neoplastic controls. The cells were cultured and the compounds were screened for their proliferation and viability inhibitory activities following the protocol described in our previous report.38 Each experiment was executed in triplicate.
6.2.2. Transcriptional analysis of KNS42 cells
The KNS42 cells were maintained in Roswell Park Memorial Institute (RPMI) medium supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin and incubated at 37 °C in 5% CO2. The cells were treated with 10 μM compound 8a. The cells were washed with 1× PBS, scraped with a cell scraper and centrifuged to collect cell pellets after 72 hours of treatment. The RNA samples were then prepared according to our previous report38 and were submitted to BGI Americas for DNBSEQ Eukaryotic Stranded Transcriptome Resequencing.
6.2.3. In vitro functional activity assay at CB1R and CB2R
Mouse AtT-20 neuroblastoma cells stably transfected with human CB1R or human CB2R were used for evaluation of membrane potential responses as previously reported.82,83 In antagonist mode, cells were pre-incubated with the vehicle or compounds for 60 minutes, before addition of 500 nM CP 55940. Data were analysed with PRISM (GraphPad Software Inc., SanDiego, CA), using four-parameter non-linear regression to fit agonist (EC50) and antagonist (IC50) concentration response curves.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
SSRA acknowledges Curtin University for the support through a Curtin International Postgraduate Research Scholarship (CIPRS). HG is thankful for the ARC Discovery Early Career Researcher Award DE160100482.
Electronic supplementary information (ESI) available. See DOI: 10.1039/d1md00065a
References
- He F. Sun Y. E. Int. J. Biochem. Cell Biol. 2007;39:661–665. doi: 10.1016/j.biocel.2006.10.022. [DOI] [PubMed] [Google Scholar]
- Jessen K. R. Mirsky R. Nature. 1980;286:736–737. doi: 10.1038/286736a0. [DOI] [PubMed] [Google Scholar]
- Ostrom Q. T. Gittleman H. Liao P. Rouse C. Chen Y. Dowling J. Wolinsky Y. Kruchko C. Barnholtz-Sloan J. Neuro-Oncology. 2014;16(Suppl 4):iv1–iv63. doi: 10.1093/neuonc/nou223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis D. N. Perry A. Reifenberger G. von Deimling A. Figarella-Branger D. Cavenee W. K. Ohgaki H. Wiestler O. D. Kleihues P. Ellison D. W. Acta Neuropathol. 2016;131:803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- Miklja Z. Pasternak A. Stallard S. Nicolaides T. Kline-Nunnally C. Cole B. Beroukhim R. Bandopadhayay P. Chi S. Ramkissoon S. H. Mullan B. Bruzek A. K. Gauthier A. Garcia T. Atchison C. Marini B. Fouladi M. Parsons D. W. Leary S. Mueller S. Ligon K. L. Koschmann C. Neuro-Oncology. 2019;21(8):968–980. doi: 10.1093/neuonc/noz022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bax D. A. Little S. E. Gaspar N. Perryman L. Marshall L. Viana-Pereira M. Jones T. A. Williams R. D. Grigoriadis A. Vassal G. Workman P. Sheer D. Reis R. M. Pearson A. D. Hargrave D. Jones C. PLoS One. 2009;4:e5209. doi: 10.1371/journal.pone.0005209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura K. Nishikawa R. Matsutani M. Neuro-Oncology. 2012;14(Suppl 4):iv90–iv99. doi: 10.1093/neuonc/nos204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones C. Perryman L. Hargrave D. Nat. Rev. Clin. Oncol. 2012;9:400–413. doi: 10.1038/nrclinonc.2012.87. [DOI] [PubMed] [Google Scholar]
- Gaspar N. Marshall L. Perryman L. Bax D. A. Little S. E. Viana-Pereira M. Sharp S. Y. Vassal G. Pearson A. D. Reis R. M. Hargrave D. Workman P. Jones C. Cancer Res. 2010;70:9243–9252. doi: 10.1158/0008-5472.CAN-10-1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller T. Stucklin A. S. G. Postlmayr A. Metzger S. Gerber N. Kline C. Grotzer M. Nazarian J. Mueller S. Curr. Treat. Options Neurol. 2020;22:43. doi: 10.1007/s11940-020-00651-3. [DOI] [Google Scholar]
- Guzman M. Duarte M. J. Blazquez C. Ravina J. Rosa M. C. Galve-Roperh I. Sanchez C. Velasco G. Gonzalez-Feria L. Br. J. Cancer. 2006;95:197–203. doi: 10.1038/sj.bjc.6603236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munson A. E. Harris L. S. Friedman M. A. Dewey W. L. Carchman R. A. J. Natl. Cancer Inst. 1975;55:597–602. doi: 10.1093/jnci/55.3.597. [DOI] [PubMed] [Google Scholar]
- Chakravarti B. Ravi J. Ganju R. K. Oncotarget. 2014;5:5852–5872. doi: 10.18632/oncotarget.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarfaraz S. Adhami V. M. Syed D. N. Afaq F. Mukhtar H. Cancer Res. 2008;68:339–342. doi: 10.1158/0008-5472.CAN-07-2785. [DOI] [PubMed] [Google Scholar]
- Massi P. Valenti M. Solinas M. Parolaro D. Cancers. 2010;2:1013–1026. doi: 10.3390/cancers2021013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parolaro D. Massi P. Expert Rev. Neurother. 2008;8:37–49. doi: 10.1586/14737175.8.1.37. [DOI] [PubMed] [Google Scholar]
- Rocha F. C. Dos Santos Junior J. G. Stefano S. C. da Silveira D. X. J. Neuro-Oncol. 2014;116:11–24. doi: 10.1007/s11060-013-1277-1. [DOI] [PubMed] [Google Scholar]
- Velasco G. Carracedo A. Blazquez C. Lorente M. Aguado T. Haro A. Sanchez C. Galve-Roperh I. Guzman M. Mol. Neurobiol. 2007;36:60–67. doi: 10.1007/s12035-007-0002-5. [DOI] [PubMed] [Google Scholar]
- Velasco G. Sanchez C. Guzman M. Nat. Rev. Cancer. 2012;12:436–444. doi: 10.1038/nrc3247. [DOI] [PubMed] [Google Scholar]
- Velasco G. Sanchez C. Guzman M. Curr. Oncol. 2016;23:S23–S32. doi: 10.3747/co.23.3080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman M. Nat. Rev. Cancer. 2003;3:745–755. doi: 10.1038/nrc1188. [DOI] [PubMed] [Google Scholar]
- Sanchez C. Galve-Roperh I. Canova C. Brachet P. Guzman M. FEBS Lett. 1998;436:6–10. doi: 10.1016/S0014-5793(98)01085-0. [DOI] [PubMed] [Google Scholar]
- Blazquez C. Gonzalez-Feria L. Alvarez L. Haro A. Casanova M. L. Guzman M. Cancer Res. 2004;64:5617–5623. doi: 10.1158/0008-5472.CAN-03-3927. [DOI] [PubMed] [Google Scholar]
- Galve-Roperh I. Sanchez C. Cortes M. L. Gomez del Pulgar T. Izquierdo M. Guzman M. Nat. Med. 2000;6:313–319. doi: 10.1038/73171. [DOI] [PubMed] [Google Scholar]
- Ellert-Miklaszewska A. Kaminska B. Konarska L. Cell. Signalling. 2005;17:25–37. doi: 10.1016/j.cellsig.2004.05.011. [DOI] [PubMed] [Google Scholar]
- Blazquez C. Salazar M. Carracedo A. Lorente M. Egia A. Gonzalez-Feria L. Haro A. Velasco G. Guzman M. Cancer Res. 2008;68:1945–1952. doi: 10.1158/0008-5472.CAN-07-5176. [DOI] [PubMed] [Google Scholar]
- Ramer R. Hinz B. Int. Rev. Cell Mol. Biol. 2015;314:43–116. doi: 10.1016/bs.ircmb.2014.10.005. [DOI] [PubMed] [Google Scholar]
- Blazquez C. Casanova M. L. Planas A. Gomez Del Pulgar T. Villanueva C. Fernandez-Acenero M. J. Aragones J. Huffman J. W. Jorcano J. L. Guzman M. FASEB J. 2003;17:529–531. doi: 10.1096/fj.02-0795fje. [DOI] [PubMed] [Google Scholar]
- Gomez del Pulgar T. Velasco G. Sanchez C. Haro A. Guzman M. Biochem. J. 2002;363:183–188. doi: 10.1042/bj3630183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez C. de Ceballos M. L. Gomez del Pulgar T. Rueda D. Corbacho C. Velasco G. Galve-Roperh I. Huffman J. W. Ramon y Cajal S. Guzman M. Cancer Res. 2001;61:5784–5789. [PubMed] [Google Scholar]
- Fogli S. Nieri P. Chicca A. Adinolfi B. Mariotti V. Iacopetti P. Breschi M. C. Pellegrini S. FEBS Lett. 2006;580:1733–1739. doi: 10.1016/j.febslet.2006.02.024. [DOI] [PubMed] [Google Scholar]
- Gustafsson S. B. Lindgren T. Jonsson M. Jacobsson S. O. Cancer Chemother. Pharmacol. 2009;63:691–701. doi: 10.1007/s00280-008-0788-5. [DOI] [PubMed] [Google Scholar]
- Massi P. Vaccani A. Ceruti S. Colombo A. Abbracchio M. P. Parolaro D. J. Pharmacol. Exp. Ther. 2004;308:838–845. doi: 10.1124/jpet.103.061002. [DOI] [PubMed] [Google Scholar]
- Massi P. Vaccani A. Bianchessi S. Costa B. Macchi P. Parolaro D. Cell. Mol. Life Sci. 2006;63:2057–2066. doi: 10.1007/s00018-006-6156-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarker K. P. Maruyama I. Cell. Mol. Life Sci. 2003;60:1200–1208. doi: 10.1007/s00018-003-3055-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaccani A. Massi P. Colombo A. Rubino T. Parolaro D. Br. J. Pharmacol. 2005;144:1032–1036. doi: 10.1038/sj.bjp.0706134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. Duan Y. H. Ji Y. Y. Wang Z. L. Wu Y. R. Gunosewoyo H. Xie X. Y. Chen J. Z. Yang F. Li J. Tang J. Xie X. Yu L. F. J. Med. Chem. 2017;60:7067–7083. doi: 10.1021/acs.jmedchem.7b00724. [DOI] [PubMed] [Google Scholar]
- Alsayed S. S. R. Lun S. Bailey A. W. Suri A. Huang C.-C. Mocerino M. Payne A. Sredni S. T. Bishai W. R. Gunosewoyo H. RSC Adv. 2021;11:15497–15511. doi: 10.1039/D0RA10728J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouillard A. D. Gundersen G. W. Fernandez N. F. Wang Z. Monteiro C. D. McDermott M. G. Ma’ayan A. Database. 2016;2016:baw100. doi: 10.1093/database/baw100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldfield S. P. Hall M. D. Platts J. A. J. Med. Chem. 2007;50:5227–5237. doi: 10.1021/jm0708275. [DOI] [PubMed] [Google Scholar]
- Mugnaini C. Brizzi A. Ligresti A. Allara M. Lamponi S. Vacondio F. Silva C. Mor M. Di Marzo V. Corelli F. J. Med. Chem. 2016;59:1052–1067. doi: 10.1021/acs.jmedchem.5b01559. [DOI] [PubMed] [Google Scholar]
- Pasquini S. De Rosa M. Pedani V. Mugnaini C. Guida F. Luongo L. De Chiaro M. Maione S. Dragoni S. Frosini M. Ligresti A. Di Marzo V. Corelli F. J. Med. Chem. 2011;54:5444–5453. doi: 10.1021/jm200476p. [DOI] [PubMed] [Google Scholar]
- Pasquini S. Ligresti A. Mugnaini C. Semeraro T. Cicione L. De Rosa M. Guida F. Luongo L. De Chiaro M. Cascio M. G. Bolognini D. Marini P. Pertwee R. Maione S. Di Marzo V. Corelli F. J. Med. Chem. 2010;53:5915–5928. doi: 10.1021/jm100123x. [DOI] [PubMed] [Google Scholar]
- Stern E. Muccioli G. G. Millet R. Goossens J. F. Farce A. Chavatte P. Poupaert J. H. Lambert D. M. Depreux P. Henichart J. P. J. Med. Chem. 2006;49:70–79. doi: 10.1021/jm050467q. [DOI] [PubMed] [Google Scholar]
- Manera C. Malfitano A. M. Parkkari T. Lucchesi V. Carpi S. Fogli S. Bertini S. Laezza C. Ligresti A. Saccomanni G. Savinainen J. R. Ciaglia E. Pisanti S. Gazzerro P. Di Marzo V. Nieri P. Macchia M. Bifulco M. Eur. J. Med. Chem. 2015;97:10–18. doi: 10.1016/j.ejmech.2015.04.034. [DOI] [PubMed] [Google Scholar]
- Alsayed S. S. R. Lun S. Luna G. Beh C. C. Payne A. D. Foster N. Bishai W. R. Gunosewoyo H. RSC Adv. 2020;10:7523–7540. doi: 10.1039/C9RA10663D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kancharla P. Kelly J. X. Reynolds K. A. J. Med. Chem. 2015;58:7286–7309. doi: 10.1021/acs.jmedchem.5b00560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banister S. D. Stuart J. Kevin R. C. Edington A. Longworth M. Wilkinson S. M. Beinat C. Buchanan A. S. Hibbs D. E. Glass M. Connor M. McGregor I. S. Kassiou M. ACS Chem. Neurosci. 2015;6:1445–1458. doi: 10.1021/acschemneuro.5b00107. [DOI] [PubMed] [Google Scholar]
- Pertwee R. G. Expert Opin. Invest. Drugs. 2000;9:1553–1571. doi: 10.1517/13543784.9.7.1553. [DOI] [PubMed] [Google Scholar]
- Ruiz L. Miguel A. Diaz-Laviada I. FEBS Lett. 1999;458:400–404. doi: 10.1016/S0014-5793(99)01073-X. [DOI] [PubMed] [Google Scholar]
- Ji Y. Y. Wang Z. L. Pei F. N. Shi J. J. Li J. J. Gunosewoyo H. Yang F. Tang J. Xie X. Yu L. F. MedChemComm. 2019;10:2131–2139. doi: 10.1039/C9MD00411D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koslowski M. Sahin U. Mitnacht-Kraus R. Seitz G. Huber C. Tureci O. Cancer Res. 2007;67:9528–9534. doi: 10.1158/0008-5472.CAN-07-1350. [DOI] [PubMed] [Google Scholar]
- Lin C. Qian P. Zhang Y. Liu Z. Dai K. Sun D. Tissue Cell. 2021;69:101480. doi: 10.1016/j.tice.2020.101480. [DOI] [PubMed] [Google Scholar]
- Wu Y. Lin X. Di X. Chen Y. Zhao H. Wang X. Oncol. Rep. 2017;37:465–473. doi: 10.3892/or.2016.5272. [DOI] [PubMed] [Google Scholar]
- Han C. He S. Wang R. Gao X. Wang H. Qiao J. Meng X. Li Y. Yu L. J. Transl. Med. 2021;19:65. doi: 10.1186/s12967-021-02733-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T. Ha M. J. Cell. Biochem. 2018;119:7747–7756. doi: 10.1002/jcb.27127. [DOI] [PubMed] [Google Scholar]
- Wang W. Shen T. Dong B. Creighton C. J. Meng Y. Zhou W. Shi Q. Zhou H. Zhang Y. Moore D. D. Yang F. J. Clin. Invest. 2019;129:1015–1029. doi: 10.1172/JCI97712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen T. Wang W. Zhou W. Coleman I. Cai Q. Dong B. Ittmann M. M. Creighton C. J. Bian Y. Meng Y. Rowley D. R. Nelson P. S. Moore D. D. Yang F. J. Clin. Invest. 2021;131(4):e135465. doi: 10.1172/JCI135465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganguly D. Cai C. Sims M. M. Yang C. H. Thomas M. Cheng J. Saad A. G. Pfeffer L. M. Pharmaceuticals. 2019;12(1):45. doi: 10.3390/ph12010045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z. Wei X. Wang X. Zheng X. Chang B. Shen L. Zhu H. Yang M. Li S. Zheng X. Cell Death Dis. 2021;12:377. doi: 10.1038/s41419-021-03646-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi Y. Tsai S. H. Cheng J. C. Wang E. Y. Anglesio M. S. Cochrane D. R. Fuller M. Gibb E. A. Wei W. Huntsman D. G. Karsan A. Hoodless P. A. Gynecol. Oncol. 2017;147:663–671. doi: 10.1016/j.ygyno.2017.10.016. [DOI] [PubMed] [Google Scholar]
- Kobayashi K. Ohnishi A. Promsuk J. Shimizu S. Kanai Y. Shiokawa Y. Nagane M. Neurosurgery. 2008;62(2):493–504. doi: 10.1227/01.neu.0000316018.51292.19. [DOI] [PubMed] [Google Scholar]
- Ohshima Y. Kaira K. Yamaguchi A. Oriuchi N. Tominaga H. Nagamori S. Kanai Y. Yokobori T. Miyazaki T. Asao T. Tsushima Y. Kuwano H. Ishioka N. S. Cancer Sci. 2016;107:1499–1505. doi: 10.1111/cas.13021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q. Tiffen J. Bailey C. G. Lehman M. L. Ritchie W. Fazli L. Metierre C. Feng Y. J. Li E. Gleave M. Buchanan G. Nelson C. C. Rasko J. E. Holst J. J. Natl. Cancer Inst. 2013;105:1463–1473. doi: 10.1093/jnci/djt241. [DOI] [PubMed] [Google Scholar]
- Wang J. Chen X. Su L. Li P. Liu B. Zhu Z. Biomed. Pharmacother. 2013;67:693–699. doi: 10.1016/j.biopha.2013.05.003. [DOI] [PubMed] [Google Scholar]
- Haining Z. Kawai N. Miyake K. Okada M. Okubo S. Zhang X. Fei Z. Tamiya T. BMC Clin. Pathol. 2012;12:4. doi: 10.1186/1472-6890-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama T. Hirakawa H. Shibata K. Nazneen A. Abe K. Nagayasu T. Taguchi T. Oncol. Rep. 2011;25:929–935. doi: 10.3892/or.2011.1176. [DOI] [PubMed] [Google Scholar]
- Tsai Y. T. Wu A. C. Yang W. B. Kao T. J. Chuang J. Y. Chang W. C. Hsu T. I. Int. J. Mol. Sci. 2019;20(22):5625. doi: 10.3390/ijms20225625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. Han B. Zhang Z. Pan J. Xia H. Biomarkers. 2010;15:39–46. doi: 10.3109/13547500903261362. [DOI] [PubMed] [Google Scholar]
- Cocchia M. Huber R. Pantano S. Chen E. Y. Ma P. Forabosco A. Ko M. S. Schlessinger D. Genomics. 2000;68:305–312. doi: 10.1006/geno.2000.6302. [DOI] [PubMed] [Google Scholar]
- Yin Y. Zhu X. Huang S. Zheng J. Zhang M. Kong W. Chen Q. Zhang Y. Chen X. Lin K. Ouyang X. Tumor Biol. 2017;39:1010428317699131. doi: 10.1177/1010428317699131. [DOI] [PubMed] [Google Scholar]
- Yuan H. Chen V. Boisvert M. Isaacs C. Glazer R. I. PLoS One. 2018;13:e0192106. doi: 10.1371/journal.pone.0192106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahmoudian J. Ghods R. Nazari M. Jeddi-Tehrani M. Ghahremani M. H. Ostad S. N. Zarnani A. H. Exp. Oncol. 2019;41:7–13. [PubMed] [Google Scholar]
- Ang B. K. Lim C. Y. Koh S. S. Sivakumar N. Taib S. Lim K. B. Ahmed S. Rajagopal G. Ong S. H. J. Mol. Signaling. 2007;2:1. doi: 10.1186/1750-2187-2-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katanasaka Y. Kodera Y. Kitamura Y. Morimoto T. Tamura T. Koizumi F. Mol. Cancer. 2013;12:31. doi: 10.1186/1476-4598-12-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kongpracha P. Nagamori S. Wiriyasermkul P. Tanaka Y. Kaneda K. Okuda S. Ohgaki R. Kanai Y. J. Pharmacol. Sci. 2017;133:96–102. doi: 10.1016/j.jphs.2017.01.006. [DOI] [PubMed] [Google Scholar]
- Oda K. Hosoda N. Endo H. Saito K. Tsujihara K. Yamamura M. Sakata T. Anzai N. Wempe M. F. Kanai Y. Endou H. Cancer Sci. 2010;101:173–179. doi: 10.1111/j.1349-7006.2009.01386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Paglia L. Listi A. Caruso S. Amodeo V. Passiglia F. Bazan V. Fanale D. PPAR Res. 2017;2017:8187235. doi: 10.1155/2017/8187235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo A. Lorente M. Egia A. Blazquez C. Garcia S. Giroux V. Malicet C. Villuendas R. Gironella M. Gonzalez-Feria L. Piris M. A. Iovanna J. L. Guzman M. Velasco G. Cancer Cell. 2006;9:301–312. doi: 10.1016/j.ccr.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Alsayed S. S. R. Lun S. Payne A. Bishai W. R. Gunosewoyo H. Bioorg. Chem. 2021;106:104486. doi: 10.1016/j.bioorg.2020.104486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urgin K. Jida M. Ehrhardt K. Muller T. Lanzer M. Maes L. Elhabiri M. Davioud-Charvet E. Molecules. 2017;22:161. doi: 10.3390/molecules22010161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knapman A. Santiago M. Du Y. P. Bennallack P. R. Christie M. J. Connor M. J. Biomol. Screening. 2013;18:269–276. doi: 10.1177/1087057112461376. [DOI] [PubMed] [Google Scholar]
- Moir M. Lane S. Lai F. Connor M. Hibbs D. E. Kassiou M. Eur. J. Med. Chem. 2019;180:291–309. doi: 10.1016/j.ejmech.2019.07.036. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.