

Abstract
Hand, foot, and mouth disease (HFMD) is a common global epidemic. From 2008 onwards, many HFMD outbreaks caused by coxsackievirus A6 (CV-A6) have been reported worldwide. Since 2013, with a dramatically increasing number of CV-A6-related HFMD cases, CV-A6 has become the predominant HFMD pathogen in mainland China. Phylogenetic analysis based on the VP1 capsid gene revealed that subtype D3 dominated the CV-A6 outbreaks. Here, we performed a large-scale (near) full-length genetic analysis of global and Chinese CV-A6 variants, including 158 newly sequenced samples collected extensively in mainland China between 2010 and 2018. During the global transmission of subtype D3 of CV-A6, the noncapsid gene continued recombining, giving rise to a series of viable recombinant hybrids designated evolutionary lineages, and each lineage displayed internal consistency in both genetic and epidemiological features. The emergence of lineage –A since 2005 has triggered CV-A6 outbreaks worldwide, with a rate of evolution estimated at 4.17 × 10−3 substitutions site-1 year−1 based on a large number of monophyletic open reading frame sequences, and created a series of lineages chronologically through varied noncapsid recombination events. In mainland China, lineage –A has generated another two novel widespread lineages (–J and –L) through recombination within the enterovirus A gene pool, with robust estimates of occurrence time. Lineage –A, –J, and –L infections presented dissimilar clinical manifestations, indicating that the conservation of the CV-A6 capsid gene resulted in high transmissibility, but the lineage-specific noncapsid gene might influence pathogenicity. Potentially important amino acid substitutions were further predicted among CV-A6 variants. The evolutionary phenomenon of noncapsid polymorphism within the same subtype observed in CV-A6 was uncommon in other leading HFMD pathogens; such frequent recombination happened in fast-spreading CV-A6, indicating that the recovery of deleterious genomes may still be ongoing within CV-A6 quasispecies. CV-A6-related HFMD outbreaks have caused a significant public health burden and pose a great threat to children’s health; therefore, further surveillance is greatly needed to understand the full genetic diversity of CV-A6 in mainland China.
Keywords: Coxsackievirus A6, hand, foot and mouth disease, whole-genome analysis, evolutionary dynamics, genetic recombination, phylogeny, pathogenicity
1. Introduction
Hand, foot, and mouth disease (HFMD) is a common global epidemic that is mainly caused by enterovirus A (EV-A) pathogens (Oberste et al. 2004). As mainland China incorporated HFMD into the National Notifiable Disease Surveillance System in 2008, HFMD has had the highest yearly incidence among all national notifiable diseases since 2010, with over 1.5 million cases annually reported (Yang et al. 2017; Liu et al. 2018). Even though enterovirus A71 (EV-A71) and coxsackievirus A16 (CV-A16) have been commonly regarded as the leading pathogens causing HFMD worldwide (Zhang et al. 2009, 2010; Xing et al. 2014; Ji et al. 2019), frequently reported CV-A6-associated HFMD outbreaks in recent years have changed the view on the major HFMD pathogens (Hayman et al. 2014; Osterback et al. 2014; Gaunt et al. 2015; Ceylan et al. 2019; Cisterna et al. 2019; Ji et al. 2019). Similar to all EVs, CV-A6 is a small, nonenveloped, single-stranded, positive-sense RNA virus. The genome, which has approximately 7,534 nucleotides (nt), contains a long open reading frame (ORF) flanked by a 5' untranslated region (UTR) and a 3' UTR. The ORF can be translated into a 2,201-amino acid (aa)-long polyprotein and then cleaved into the three polyprotein precursors P1, P2, and P3, which encode the capsid proteins VP4, VP2, VP3, and VP1 and the noncapsid proteins 2A, 2B, 2C, 3A, 3B, 3C, and 3Dpol (Bruu 2002).
The first reported CV-A6-related HFMD outbreak was in Finland in 2008 (Osterback et al. 2009, 2014). Within the past decade, CV-A6-related HFMD has become a common global epidemic, especially in many Asian countries (Fujimoto et al. 2012; Puenpa et al. 2013; Ang et al. 2015; Song et al. 2017; Anh et al. 2018). Since 2013, numerous cases have revealed repeated large-scale HFMD outbreaks caused by CV-A6 in mainland China, where CV-A6 has even surpassed EV-A71 and CV-A16 to become the leading HFMD pathogen (Han et al. 2014; Li et al. 2014a; Yang et al. 2014; Tan et al. 2015; Zeng et al. 2015; Song et al. 2017; Wang et al. 2019). In addition, atypical clinical manifestations are always reported in CV-A6-related HFMD cases, which are characterized by the extension of lesions beyond the typical sites of HFMD, onychomadesis that causes nail loss, occasional infection in adults, and other features (Yasui et al. 2013; Chong and Aan 2014; Sinclair et al. 2014; Buttery et al. 2015; Laga, Shroba, and Hanna 2016; Magnelli et al. 2017; Broccolo et al. 2019). Many severe cases caused by CV-A6 have also been reported (defined as neurological or cardiopulmonary complications according to the National Guideline for Diagnosis and Management of HFMD cases) (Centers for Disease, Control and Prevention 2012; Yang et al. 2014; Broccolo et al. 2019); therefore, CV-A6-related HFMD outbreaks have caused a significant public health burden in mainland China, posing a great threat to children's health.
Phylogenetic analysis based on the VP1 capsid region of EV has been widely adopted as a methodology, as VP1 contains many important neutralization epitopes and is serotype-specific (Oberste et al. 1999). Based on 807 entire CV-A6 VP1 sequences, we segregated CV-A6 into four genotypes in a previous study, namely, A, B, C, and D, and genotype D was further divided into the three subtypes D1, D2, and D3 based on an intergroup nucleotide pairwise distance greater than 8 per cent (Song et al. 2017). Genotype A and genotypes B and C are composed of the prototype strain and a few sporadic isolates, respectively. The transmission of subtype D3 has been responsible for CV-A6-related HFMD outbreaks worldwide. Only subtypes D2 and D3 have been found to circulate in mainland China since 2010 and D2 became almost undetectable in approximately 2013. Nevertheless, studies using VP1 capsid sequences may have some research limitations because recombination events in EVs always occur in noncapsid genes. Recombination is a frequently observed phenomenon among EVs and, more importantly, a driving force of EV evolution. The 3Dpol error-prone RNA-dependent RNA polymerases (RdRps) of EV always lead to misincorporations during genome replication; thus, ongoing recombination may be the main process preventing EV genomes from deleterious mutation accumulation (Lukashev et al. 2004, 2005; Bouslama et al. 2007; Chen et al. 2010; Schibler et al. 2012; Kok and Au 2013; Zhang et al. 2013, 2015; Lin et al. 2015). In addition, particular recombination events may have medically important consequences, such as the existence of virulence determinants in noncapsid proteins of some EVs (Macadam et al. 1994; Pfeiffer and Kirkegaard 2005; Li et al. 2017, 2018).
In some recent EV studies, different bootstrap-supported clusters in the 3Dpol phylogeny were designated recombination forms (RFs) by scholars because 3Dpol represents the noncapsid coding region farthest from VP1 (McWilliam Leitch et al. 2010, 2012; Gaunt et al. 2015; Puenpa et al. 2016). Previous studies categorized global CV-A6 variants into 13 RFs, alphabetically termed RF –A to –M (Gaunt et al. 2015; Puenpa et al. 2016; Lau et al. 2018), but included only a few strains from mainland China. However, investigations of the genetic characteristics of CV-A6 using VP1, 3Dpol or other regions would be incomplete. Since fast-spreading CV-A6 has become an important public health problem worldwide, studies limited to partial sequence data are insufficient. In this study, we characterized the (near) whole genomes of emerging CV-A6 in depth, including 158 new sequences collected from 2010 to 2018 that covered 22 provinces of mainland China. In addition, the genetic diversity of global CV-A6 recombination variants designated evolutionary lineages in this study was also described. Based on a large number of full-length CV-A6 sequences, we unveiled the potential influences of genetic recombination on the evolutionary dynamics and pathogenic features of fast-spreading CV-A6 variants.
2. Materials and methods
2.1 Ethical considerations and virus isolation
This study did not involve human participants or human experimentation; the only human materials used were stool samples, throat swab samples, and vesicles collected from HFMD patients from 2010 to 2018 for public health purposes at the urging of the Ministry of Health, P.R. of China. The clinical HFMD samples were collected from the HFMD Surveillance Network established in our laboratory. The samples were processed based on standard protocols (Xu and Zhang 2016) and were first confirmed as positive for CV-A6 by a commercial real-time PCR assay (Shuoshi Biotech, Jiangsu, China). All CV-A6-positive samples were then inoculated into human rhabdomyosarcoma (RD) and human laryngeal epidermoid carcinoma (HEp-2) cell lines, which were obtained from the WHO Global Poliovirus Specialized Laboratory (USA) and originally purchased from the American Type Culture Collection, for virus propagation and purification. Infected cell cultures were harvested after a complete cytopathic effect was observed.
2.2 CV-A6 representative samples selection and whole-genome sequencing
Viral RNA was extracted using a QIAamp Viral RNA Mini Kit (Qiagen, Valencia, CA, USA). First, we performed reverse transcription polymerase chain reaction (RT-PCR) to amplify the entire VP1 capsid region (915 nt) using a PrimeScript One Step RT-PCR Kit Ver. 2 (TaKaRa, Dalian, China) with previously designed primers (Song et al. 2017). The PCR products were purified using a QIAquick PCR Purification Kit (Qiagen, Germany), and then amplicons were bidirectionally sequenced using an ABI 3130 Genetic Analyzer (Applied Biosystems, USA). Second, a neighbor-joining tree of VP1 sequences was constructed using MEGA (v7.0) for genotyping and representative isolate selection (Kumar, Stecher, and Tamura 2016; Song et al. 2017).
We randomly selected the representative strains of CV-A6 from each branch of the VP1 tree and evaluated the representation of geographic locations and dates for each cluster. A total of 158 representative CV-A6 samples were selected between 2010 and 2018 from 22 provinces (municipalities and autonomous regions) of mainland China (Supplementary Fig. S1). The 5′ end of the genome sequence was amplified using a 5′-Full RACE Kit (Takara Biomedicals, China), and the 3′-end sequence was obtained using an oligo-dT primer (primer 7500A) (Yang et al. 2003) as the downstream primer for amplification. The primers used for PCR amplification and sequencing of the remaining genome in this study were designed based on the primer walking method (Supplementary Table S1). PCR products were purified using a QIAquick PCR Purification Kit (Qiagen, Germany), and amplicons were bidirectionally sequenced using an ABI 3130 Genetic Analyzer (Applied Biosystems, USA).
2.3 Datasets construction of worldwide and Chinese CV-A6
In addition to the 158 samples sequenced in this study, all the CV-A6 (near) whole-genome sequences (sequence length of 6,600–7,500 nt, dated to 30 September 2019) in the GenBank database were retrieved. Questionable and low-quality sequences were eliminated, including laboratory-adapted strains, clones, strains with high passage numbers, sequences that contained many undetermined bases, and misnamed sequences that belonged to other serotypes. A total of 431 near full-length sequences were finally recruited from GenBank. Combined 158 sequences from this study and 431 sequences from GenBank together, we selected 142 out of a total of 589 worldwide sequences based on the ORF, VP1 and 3Dpol phylogenies and the years and geographic regions of isolation to describe the CV-A6 genetic diversity on a global scale (Supplementary Figs S2 and S3). For the dataset of Chinese CV-A6, all the sequences from mainland China were used, including 178 GenBank sequences isolated between 2010 and 2016 from 8 provinces (municipalities and autonomous regions) and 158 sequences selected from this study. A total of 336 (near) full-length sequences composed the dataset of Chinese CV-A6 (Supplementary Figs S2 and S3).
2.4 Phylogenetic and evolutionary analyses of global CV-A6
Sequence alignment was conducted using the Muscle tool in MEGA (v7.0), and ORF sequences were acquired for the analysis, as many sequences lacked a 3′ or 5' UTR. RAxML (v8.2.12) was used to construct maximum-likelihood trees of each region, and the best nucleotide substitution models were selected for different datasets using jModelTest (Darriba et al. 2012; Stamatakis 2014). Support was estimated with 1,000 bootstrap replicates, and the results were visualized using FigTree (v1.4.4) (http://beast.community/figtree). The global evolutionary dynamics of CV-A6 over time were inferred based on the whole P1 capsid region. The correlation coefficient and regression value of each dataset were calculated using TempEst (v1.5.1) to estimate the correlation between sequence divergence and the date of isolation in each dataset (Rambaut et al. 2016). The Markov chain Monte Carlo (MCMC) method implemented in BEAST (v1.7.5) was used to estimate the temporal phylogenies and rates of evolution (Drummond and Rambaut 2007). All 142 P1-region sequences were analyzed using the uncorrected lognormal clock (UCLD) and constant site tree prior with the GTR +G +I nucleotide substitution model. A Bayesian MCMC run consisted of 2 × 108 generations to ensure that each parameter could converge. The sampling frequency was set to 2 × 104 generations. The output from BEAST was analyzed using TRACER (v1.7.1) (http://beast.community/tracer) (with estimated sample size (ESS) values higher than 200). A maximum clade credibility (MCC) tree was constructed using TreeAnnotator, with the burn-in option used to remove the first 10 per cent of sampled trees, and the resulting tree was visualized by FigTree (v1.4.4). A total of 400 ORF sequences of lineage –A of CV-A6 were further analyzed using a strict clock and a constant site tree prior with the GTR +G nucleotide substitution model. The Bayesian MCMC run consisted of 5 × 108 generations, and the sampling frequency was set to 5 × 104 generations.
SimPlot (v3.5.1) was used to produce similarity plots with a 200-nt window moving in 20-nt steps in order to evaluate genetic diversity and detect recombination breakpoints (Salminen et al. 1995).
2.5 Bioinformatic analyses of Chinese CV-A6
The maximum-likelihood trees of sequences for each region were generated using RAxML with 1,000 bootstrap replicates (v8.2.12), and the CV-A6 prototype Gdula strain was used as an outgroup. All 336 P1 region sequences were analyzed using the UCLD and a constant site tree prior with the GTR +G nucleotide substitution model. We performed the analysis using 3 × 108 generations, and the sampling frequency was set to 3 × 104 generations. The burn-in option was used to remove the first 10 per cent of sampled trees. A Gaussian Markov random field (GMRF) skyride plot was inferred (Katsuki, Torii, and Inoue 2012) using the same clock and nucleotide substitution model to reconstruct the evolutionary history of Chinese CV-A6 from 2010 to 2018. Individual datasets of each evolutionary lineage (corresponding to P1, 3Dpol, and ORF) were analyzed using a strict clock and a constant site tree prior with the GTR +G model to estimate the evolutionary rate. A geographic map of China was taken from Highcharts (grant number: 0321912045738052), which was used to display the geographic distribution of CV-A6 variants.
The average pairwise genetic diversity along all the CV-A6 genomes was calculated using DnaSP 6 (Rozas et al. 2017) software with a sliding window of 200 nt and a step size of 20 nt. Recombination was detected in whole CV-A6 genomes using seven algorithms (RDP, Geneconv, BootScan, MaxChi, Chimaera, SiScan, and 3Seq) implemented in RDP4 (Martin et al. 2017). SimPlot was used to produce similarity plots and bootscan analyses with a 200-nt window moving in 20-nt steps. Bootscan analyses were run using the neighbor-joining method. Statistical analysis (chi-square test) was conducted using SPSS. Mega (v7.0) was used to obtain nucleotide and aa sequence similarities, the web-based application WebLogo was used for generating aa sequences logos (Crooks et al. 2004) (http://weblogo.threeplusone.com/). The mixed effects model of evolution (MEME) was used to estimate the role of natural selection pressure (Murrell et al. 2012).
2.6 Nucleotide sequence accession numbers
All 158 sequences obtained for this study were deposited in the GenBank database under the accession numbers MK106189–MK106191, MK106193–MK106216, and MN845761–MN845891.
3. Results
3.1 Summary of the CV-A6 datasets
A total of 142 representative global (near) whole-genome sequences were analyzed to evaluate the evolutionary dynamics of CV-A6 worldwide over time. The strains were isolated between 1949 (the prototype strains) and 2018 from fifteen countries and regions, including mainland China (n = 55), Japan (n = 22), Thailand (n = 11), the United Kingdom (n = 11), Australia (n = 8), Vietnam (n = 7), Taiwan of China (n = 5), Denmark (n = 4), Hong Kong of China (n = 4), Germany (n = 3), Spain (n = 3), Finland (n = 3), India (n = 2), Madagascar (n = 2) and the United States (n = 2), representing wide temporal and regional distributions.
All 178 (near) full-length sequences from GenBank and 158 from this study composed the Chinese CV-A6 dataset. These 336 strains were isolated from twenty-three provinces (municipalities and autonomous regions), which were represented by seven geographical regions (North, Northwest, Northeast, South, Southwest, East, and Central China) of mainland China from 2010 to 2018. Among the 158 new samples from this study, 2 were reported as fatal cases, 17 were severe cases, and 139 were mild cases. Dataset information can be found in Supplementary Tables S2–S4 and Figs S1–S4.
3.2 Evolutionary lineage diversification of global CV-A6
The overall mean nucleotide distance among these 142 ORF sequences was 10.9 per cent; specifically, the distances for VP4, VP1, VP3, VP2, 2A, 2B, 2C, 3AB, 3C, and 3Dpol sequences were 7.7 per cent, 7.4 per cent, 7.5 per cent, 7.6 per cent, 8.6 per cent, 11.0 per cent, 13.2 per cent, 16.4 per cent, 14.6 per cent, and 14.7 per cent, respectively, which indicated high nucleotide dissimilarity in the noncapsid region. Phylogenetic trees of the capsid coding region (P1) and noncapsid coding region (P2 and P3) were generated (Fig. 1). Referring to the phylogeny of VP1 and 3Dpol (Supplementary Fig. S5), the phylogeny of P1 was consistent with that of VP1 (Fig. 1A) with a new genotype (designated genotype E) composed of two Malagasy strains; the noncapsid region sequences formed a series of bootstrap-supported phylogenetic lineages (Fig. 1B), which were designated evolutionary lineages in this study. The strains that consisted of each evolutionary lineage based on the noncapsid phylogeny were almost the same as those that formed RFs based on the 3Dpol phylogeny, with one exception: RF-K was composed two lineages. Therefore, we categorized global CV-A6 into seventeen evolutionary lineages: –A, –B, –C, –D, –E, –F, –G, –H, –I, –J, –K1, –K2, –L, –M, –N, –O, and –P. Except for lineages –I (formed solely by prototype strains), –M, –O, and –P, all the lineages contained more than one sequence each. The two phylogenetic trees showed quite different topologies, and some of the lineages that clustered together in the P1 phylogeny displayed high divergence in the noncapsid region. The predominant subtype D3 consisted of nine lineages, –A, –F, –G, –H, –J, –K2, –L, –M, and –N. Among these nine lineages, in the noncapsid phylogeny, hardly any mapped to the P1 capsid region, which revealed obvious phylogenetic inconsistencies. This finding suggested that during the transmission of fast-spreading D3-subtype CV-A6, the noncapsid gene continued recombining in the EV gene pool, giving rise to a series of viable interserotypic recombinant hybrids. Detailed topological transformation for each lineage based on all the coding-region phylogenetic trees can be observed in Supplementary Fig. S6.
Figure 1.

Maximum-likelihood phylogenetic trees of 142 global CV-A6 variants constructed based on the (A) P1 capsid region and (B) noncapsid region; the lineages are differentiated by distinct colors, which are indicated in the bottom left, and each subtype is indicated on the right side of the P1 tree. The branches in each phylogeny are colored according to lineage. (C) Intralineage pairwise similarity comparison based on ORF sequences computed between each sequence and the group mean using sliding window nucleotide similarity analysis with a 200-nt window moving in 20-nt steps; similarity was not calculated for lineages containing only one sequence.
We further employed a sliding window nucleotide similarity analysis of intralineage ORF sequences. Pairwise nucleotide similarity was assessed between each sequence and the group mean. As the similarity plot indicated (Fig. 1C), all the strains displayed high intralineage homology throughout the ORF sequences. Lineage –A comprised most of the CV-A6 strains, while lineages –D, –E, –K1, –K2, –M, –N, –O, and –P contained very few sequences and were detected in the same region within a limited period of time (Table 1), indicating sporadic recombinants that were minor and short-lived components of the circulating CV-A6 population. Seven lineages have been detected in mainland China during the past decade, including –A, –C, –D, –K1, –K2, –J, and –L, with most of the Chinese CV-A6 strains belonging to lineages –A, –J, and –L (Table 1).
Table 1.
Information on global CV-A6 evolutionary lineages. Mainland China is indicated as bold characters.
| Genotype/subtype | Lineage | n | Nucleotide mean distances (%) | Isolated countries/regions | Isolated years | Occurrence time | Recombination breakpoints position (within subtype) |
|---|---|---|---|---|---|---|---|
| Subtype D1 | B | 8 | 4.2 | Australia; Japan | 1999–2006 | 1996–1999 | Original lineage of subtype D1 (query) |
| E | 4 | 1.2 | Taiwan of China | 2007 | 1997–2007 | End of 2C | |
| Subtype D2 | C | 10 | 4.4 | Mainland China; Japan | 2005–2012 | 1996–2005 | Original lineage of Subtype D2 (query) |
| D | 4 | 2.8 | Mainland China | 2011–2013 | 2008–2011 | Middle of 3Dpol | |
| K1 | 2 | 1.7 | Mainland China | 2013 | 2008–2013 | Beginning of 2C | |
| Subtype D3 | A | 59 | 4.8 | Australia; Mainland China; Germany; Denmark; Finland; Hong Kong of China; India; Japan; Thailand; Taiwan of China; United Kingdom; United State; Vietnam | 2008–2018 | 2003–2008 | Original lineage of subtype D3 (query) |
| F | 8 | 1.6 | Germany; Spain; Thailand; United Kingdom | 2012–2014 | 2007–2012 | Beginning of 2B | |
| G | 5 | 1.9 | Denmark; United Kingdom | 2011–2014 | 2003–2011 | Original lineage of subtype D3 | |
| H | 6 | 0.9 | Denmark; Spain; United Kingdom | 2013–2014 | 2011–2013 | Beginning of 2C | |
| J | 13 | 2.3 | Mainland China | 2012–2017 | 2010–2012 | End of 2A | |
| K2 | 2 | 0.5 | Mainland China | 2013 | 2010–2013 | End of 2C | |
| L | 12 | 2.6 | Mainland China; Hong Kong of China | 2014–2018 | 2013–2014 | Middle of 3Dpol | |
| M | 1 | NA | Hong Kong of China | 2015 | 2012–2015 | End of 3C | |
| N | 4 | 2 | Australia | 2016–2017 | 2011–2016 | Middle of 2C | |
| Genotype E | O | 1 | NA | Madagascar | 2011 | 2004–2011 | NA |
| P | 1 | NA | Madagascar | 2011 | 2004–2011 | NA | |
| Genotype A | I(prototype) | 1 | NA | United State | 1949 | 1930–1949 | NA |
3.3 Evolutionary dynamics and recombination breakpoints of global CV-A6
With an uncorrelated relaxed molecular clock model and constant site tree prior, an MCC tree based on entire P1 capsid region sequences of 142 globally distributed CV-A6 strains was obtained using BEAST (Fig. 2A). The MCMC method was used to estimate the time to the most recent common ancestor (tMRCA) and substitution rate; more importantly, the chronology of each evolutionary lineage was calculated. The average nucleotide substitution rate for the P1 region in all CV-A6 strains worldwide was 4.73 × 10−3 [95% highest posterior density (HPD): 4.14–5.30] ×10−3 substitutions site−1 year−1, with a predicted date of tMRCA of 1931 (95% HPD: 1916–1945). The two Malagasy strains had been evolving for a very long time, with a distant evolutionary route from the remaining CV-A6 strains. In contrast to the prototype strain (lineage –I) and the two Malagasy strains (lineages –O and –P), the other 139 CV-A6 strains formed genotype D with a tMRCA estimated at 1995. Genotype D evolved into subtypes D1, D2, and D3 in 1996, 2000, and 2003, respectively, and subtypes D1 and D2 formed a clade with sporadic strains, suggestive of their closer evolutionary relationship and lower transmissibility. The date of recombination events was estimated as between the tMRCA shared by the original lineage and newly emerged lineage and the isolation date of the first clinical sample with the new lineage (Table 1). In subtype D1, lineage –B emerged in approximately late 1996 in Japan, underwent recombination events during transmission to Taiwan of China, and generated lineage –E; likewise, in subtype D2, original lineage –C first circulated in Japan in 2000, was transmitted to mainland China while recombining, and then gave rise to lineage –D and lineage –K1 (Table 1).
Figure 2.

The MCC phylogenetic tree generated using the MCMC method based on the entire P1 sequences of 142 global CV-A6 variants and colored according to different lineages. The scale bar represents time in years. The tree was node-labeled with inferred dates of lineage splits. Each subtype is shaded in light gray, except for the independent lineage –G. Recombination breakpoints based on the ORFs of different lineages were detected within (B) subtype D1, (C) subtype D2, and (D) subtype D3 using the original lineage of each subtype as the query group with a 200-nt window moving in 20-nt steps.
Recombination among fast-spreading subtype D3 variants created two clusters. The small cluster corresponded to lineage –G, suggesting its independent evolution in northwestern Europe; the other, massive cluster contained most of the evolutionary lineages detected within the decade. The predominant lineage –A, which emerged in 2005, has been responsible for CV-A6 outbreaks worldwide. During global transmission, lineage –A underwent a succession of recombination events, generating the seven lineages –F, –J, –K2, –H, –N, –M, and –L in different periods of time (Table 1). Recombination breakpoints of different lineages were further detected in the same subtype using the first appearing lineage as the query group. The similarity plot reflected obvious intrasubtype recombination breakpoints at different positions in the noncapsid region, with the P1 capsid region showing high nucleotide homology (Fig. 2B–D and Table 1).
As lineage –A of subtype D3 CV-A6 turned out to be the predominant type worldwide, MCMC analysis based on all the ORF sequences of lineage –A was further conducted (Table 2). A total of 400 out of 589 (67.9%) sequences have been identified as belonging to lineage –A to date, with an overall mean distance of 4.4 per cent and a pairwise distance varying from 0 per cent to 9.6 per cent. Regression (R = 0.922) and correlation coefficient (cc = 0.960) analyses reflected a strong correlation between sequence divergence and the date of isolation. The rate of evolution of global lineage –A of CV-A6 was estimated to be 4.17 × 10−3 [95% HPD: 3.97–4.37] ×10−3 substitutions site−1 year−1, with a tMRCA of 13.30 (95% HPD: 12.84–13.77). This result reflected the evolutionary information for the real predominant CV-A6 strains, since it was obtained from a large-scale dataset of monophyletic ORF sequences of lineage –A but not whole VP1 sequences of subtype D3, offering more complete genetic information and ruling out polyphyletism being influenced by recombination in the noncapsid gene.
Table 2.
Markov chain Monte Carlo (MCMC) results and other parameters for different CV-A6 datasets.
| Geographic set and datasets | Number of strains |
Regression (R) |
dN/dS |
MCMC (BEAST)
a
|
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All | From this study |
Substitution rate
(10-−3)(95%HPD) |
TMRCA(95%HPD) |
||||||||||||
| Mild cases | Severe/fatal cases | P1 | 3D | ORF | P1 | 3D | ORF | P1 | 3D | ORF | P1 | 3D | ORF | ||
| Worldwide | |||||||||||||||
| Selected | 142 | NA | NA | 0.69 | 0.05 | 0.22 | 0.017 | 0.020 | 0.019 | 4.73 (4.14, 5.30) | NA | NA | 87.63(69.06, 146.15) | NA | NA |
| Lineage –A | 400 | NA | NA | 0.91 | 0.82 | 0.92 | 0.022 | 0.031 | 0.022 | 5.48(4.86, 6.10) | 4.83(4.41, 5.24) | 4.16(3.97, 4.37) | 13.01(12.18, 13.93) | 12.91(12.06, 13.88) | 13.30(12.84, 13.77) |
| Mainland China | |||||||||||||||
| All | 336 | 137 | 19 | 0.81 | 0.44 | 0.51 | 0.023 | 0.022 | 0.020 | 5.20(4.74, 5.68) | NA | NA | 22.08(16.56, 28.53) | NA | NA |
| Lineage –A | 222 | 116 | 12 | 0.88 | 0.82 | 0.90 | 0.025 | 0.024 | 0.020 | 4.40(4.01, 4.81) | 3.83(3.41, 4.26) | 3.89(3.66, 4.11) | 13.38(12.36, 14.48) | 13.36(11.96, 14.91) | 14.21(13.51, 15.05) |
| Lineage –J | 62 | 8 | 2 | 0.74 | 0.73 | 0.87 | 0.027 | 0.062 | 0.042 | 5.21(4.52, 5.94) | 5.17(4.18, 6.21) | 4.68(4.25, 5.11) | 6.28(5.48 7.31) | 5.86(5.24, 6.66) | 5.95(5.62, 6.30) |
| Lineage –L | 32 | 13 | 5 | 0.93 | 0.85 | 0.93 | 0.033 | 0.030 | 0.031 | 4.85(4.07, 5.59) | 5.09(3.92, 6.20) | 4.72(4.26 5.24) | 5.76(5.27, 6.30) | 5.68(5.04, 6.38) | 5.49(5.20 5.78) |
NA, MCMC values were not calculated for the 3Dpol and ORF datasets, which were not monophyletic.
3.4 Genetic diversity, timescale, and evolutionary dynamics of Chinese CV-A6
Among all 336 Chinese CV-A6 strains isolated from 2010 to 2018, lineage –A comprised the largest number of the strains (n = 222), and lineages –J and –L comprised the second (n = 62) and third (n = 32) largest numbers, respectively (Supplementary Fig. S7). Only a few strains were detected in lineages –D (n = 5), –C (n = 10), –K1 (n = 2) and –K2 (n = 3). The intralineage nucleotide similarity of lineages –A, –C, –D, –K1, –K2, –J, and –L in mainland China was 96.1 per cent, 95.9 per cent, 97.5 per cent, 98.3 per cent, 99.3 per cent, 98.1 per cent, and 97.4 per cent, respectively, indicating high nucleotide homology. The sliding window analysis performed across intralineage sequences revealed low pairwise genetic diversity; however, when all the lineages were considered together, the diversity was obviously higher in the noncapsid region (Supplementary Fig. S8A). We then further compared pairwise genetic diversity between lineage –A and the other six lineages, revealing apparent distinctions (Supplementary Fig. S8B).
All 336 entire P1 regions of Chinese CV-A6 were analyzed using the MCMC method. The MCC tree contained subtypes D2 and D3 (Fig. 3A). The substitution rate of Chinese CV-A6 P1 was estimated at 5.20 × 10−3 [95% HPD: 4.74–5.68] ×10−3 substitutions site−1 year−1, comparable to but slightly higher than that estimated worldwide, and the tMRCA estimated for Chinese CV-A6 based on the P1 region was 1996 (95% HPD: 1989–2006) (Fig. 3A).
Figure 3.

(A) Time-scaled phylogenetic tree generated using the MCMC method for 336 complete CV-A6 P1 sequences from mainland China. The seven lineages from mainland China are indicated by different branch colors. The scale bar represents time in years. The tree was node-labeled with inferred dates of lineage splits. (B) The geographical distribution of seven lineages in mainland China. The proportion and number of cases caused by each lineage are indicated by the pie chart located in each geographic region on the map. The geographic map of China was taken from Highcharts (grant number: 0321912045738052), with regions denoted by different colors. Similarity plots were constructed and bootscan analyses were performed for parental recombination detection among screened EV-A strains and (C) lineage –K2, (D) lineage –J, and (E) lineage –L. Using lineage –A as the comparative group, prototype strains of CV-A6 and CV-A2 were used as outgroups. (F) A Gaussian Markov random field (GMRF) skyride plot of the P1 region of Chinese CV-A6, reflecting the relative genetic diversity from 2010 to 2018. The x-axis is the time scale (years), and the y-axis is the effective population size. The solid line indicates the median estimates, and blue shading indicates the 95 per cent HPD.
In terms of the geographical distribution of Chinese CV-A6 lineages (Fig. 3B), subtype D2 first emerged as lineage –C in northern parts of China in approximately 2005, which was imported from Japan according to the global phylogeny. Lineages –K1 and –D then successively originated as sporadic viruses from lineage –C within a limited geographical region. In subtype D3, lineage –A was also the main reason for the CV-A6 outbreaks in mainland China. During its large-scale and long-term circulation since 2005, lineage –A has undergone three recombination events, generating one sporadic lineage (lineage –K2) and two epidemic lineages (lineages –J and –L). Lineage –J comprised strains isolated mostly from East China because of an outbreak that reportedly happened there (Feng et al. 2015a,b) but still spread to other regions; lineage –L probably originated in Central and Southwest China, was transmitted north and eventually spread across the country. This finding suggested that novel recombinants could be highly transmissible.
Lineages –K2, –J, and –L, which were shown to have arisen from recombination with lineage –A, were detected only in China. It could be speculated that the parental strains were acquired from the national EV-A gene pool. The other EV-A-serotype strains with high similarity in the noncapsid region with lineage –K2, –J, and –L strains were screened from GenBank for parental recombination detection. As a result, several recombination events were detected using RDP4 (at least four methods supported the recombination results). For further visualization of the recombination donor strains, similarity plots and bootscan analyses were performed. Lineage –A was used as the comparative group, and the CV-A6 Gdula and CV-A2 Fleetwood prototype strains were used as outgroups (Fig. 3C–E). In comparison to lineage –A, lineage –J had three obvious recombination events, corresponding to the Guangdong EV-A71 strain (JF799986/2009), the Shenzhen CV-A4 strain (HQ728260/2009), and the Shenzhen CV-A8 strain (KM609478/2012). Lineage –K2 was recombined from the Jiangsu CV-A14 strain (KP036482/2012) in the whole P3 region. Furthermore, lineage –L was found to have two recombination breakpoints with two different EV-A strains, the Shenzhen CV-A4 (HQ728260/2009), and Shenzhen CV-A8 (KM609478/2012) strains.
Additionally, a GMRF skyride plot analysis was performed to reconstruct the demographic history of Chinese CV-A6 based on whole P1 region sequences (Fig. 3F). The plot reflected detailed changes in genetic diversity from 2010 to 2018. Before 2011, the effective population size had been gradually increasing, as this was a period with cocirculation of the seven lineages. There was a small decline in approximately 2012; at that time, strains from subtype D2 had decreased, and the outbreaks of the lineage –A CV-A6 strain had not yet taken place. Since the large-scale outbreaks in 2013, the population size obviously increased and fluctuated annually until 2018, which corresponded to the yearly incidences of CV-A6-related HFMD cases reported in China over this time period.
3.5 Evolutionary correlates in recombinant lineages
According to recombination breakpoint detection, the 3Dpol region was specific among all the lineages. The pairwise distances of the seven lineages among the sequences of the P1 and 3Dpol regions were further compared (Fig. 4). Similar dynamics of sequence drift between the P1 and 3Dpol regions within the lineages and parted distances of recombination among the lineages were apparent in Chinese CV-A6. Within the same lineage, comparisons reflected a positive linear correlation between P1 and 3Dpol divergence. However, distributions of pairwise distances between lineage –A and the other six lineages revealed obviously higher divergence in 3Dpol than in P1, indicating an interlineage discrepancy on account of different recombination events. In addition, lineages –J, –K2, and –L, which originated from lineage –A, were similarly divergent from lineage –A in P1; in contrast, lineages from subtype D2 had higher divergence.
Figure 4.
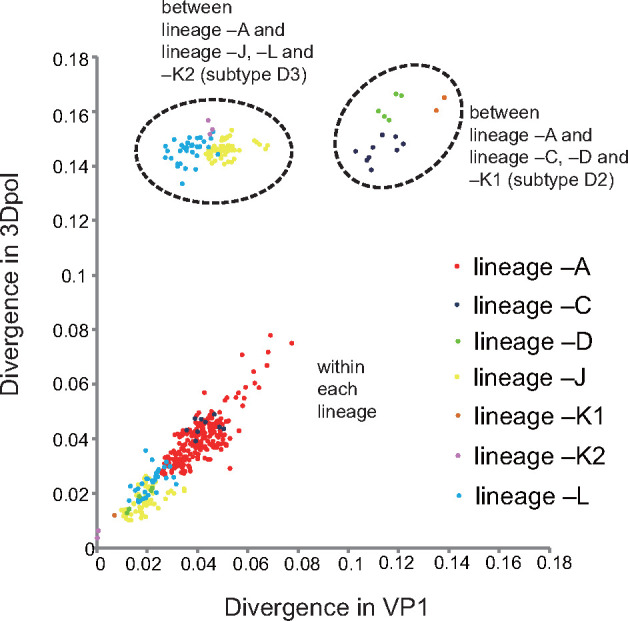
Comparisons of pairwise distance in P1- and 3Dpol-region sequences within seven lineages; between lineage –A and lineages –J, L, and –K2 (from subtype D3) and between lineage –A and lineages –C,–D, and –K1 (from subtype D2). Axes depict nucleotide divergence between the two genomic regions. Each lineage is indicated by a differently colored dot in the scatterplot.
To evaluate the relationship between recombination diversification and evolutionary differences among CV-A6 lineages, separate MCMC analyses were further conducted for the three most widely distributed CV-A6 lineages (–A, –J, and –L). Among the three lineages, the tMRCA and substitution rate were further calculated for the 3Dpol and ORF of each lineage group, in addition to P1. As the results indicated, markedly similar tMRCAs and substitution rates were determined for the ORF, 3Dpol, and P1 sequences in each lineage (Table 2). This was consistent with the similar sequence drift patterns observed between the P1 and 3Dpol regions in the pairwise distance comparison. These data reflected robust estimates for the dates of the recombination events that created the widespread lineages in mainland China. The MCC trees were further visualized based on the ORF sequences of lineages –A, –J, and –L to see the geographical transmission of each lineage in mainland China (Fig. 5).
Figure 5.

MCC phylogenetic trees based on the ORF sequences of (A) lineage –A, (b) lineage –J, and (C) lineage –L from mainland China. The branches are colored according to the seven geographic regions of mainland China, which are indicated in the bottom left. The scale bar represents time in years. The tree was node-labeled with inferred dates of lineage splits.
3.6 Association of recombinant lineages with pathogenic severity
In addition to lineage –A, novel lineages –J and –L were also widespread in mainland China, indicative of their potential high transmissibility. Cases of lineage –J infection presented more generalized rashes than those of lineage –A infection according to a previous statistical study in Shanghai (Feng et al. 2015a). Apart from that, comparing the numbers and proportions of severe/fatal cases among the three lineages detected in this study, we found that lineage –L may be more likely to cause severe HFMD than lineage –A of CV-A6 (chi-square test, P value = 0.03) (Table 2). This finding suggested that the noncapsid region might affect the pathogenicity of CV-A6.
3.7 Important amino acid sites prediction in Chinese CV-A6
The natural selection pressure analysis suggested that strong negative selection occurred over most of the codons in the P1 capsid region during D3-subtype CV-A6 transmission in the Chinese population. Nevertheless, elevated dN to dS ratios were detected for some codons, and statistically significant evidence (P value < 0.1) for positive selection was uncovered for three codons located at aa VP3180, VP130, and VP1268. These positive sites could play an important role in CV-A6 adaptation to new hosts.
In comparison to the CV-A6 prototype Gdula strain, a total of 101, 97, 97, 98, and 101 aa substitutions were detected in consensus sequences (substitutional rate greater than 70% at the same position) of lineages –A, –C, –D, –J, and –L, respectively (lineages –K1 and –K2 included too few sequences for analysis, Supplementary Table S4). We further compared the consensus sequences of subtypes D3 to D2 based on the whole P1 capsid region and identified ten aa substitutions, including four in VP4, four in VP2, one in VP3, and two in VP1 (Fig. 6A). Notably, the four substitutions in the VP2 region were all embedded in the assumed epitope EF loop, and one substitution in VP1 was in the assumed epitope HI loop (Xu et al. 2017; Chen et al. 2018). These five changes might represent adaptations to the host and play an important role in the high pathogenicity and transmissibility of fast-spreading subtype D3 CV-A6 in mainland China. In addition, even if lineages –A, –J, and –L shared monophyletic capsids, six polymorphic residues of capsids displayed obviously dissimilar compositions among these lineages, including VP2244 and VP15, 27, 30, 137, 174, 242 (Fig. 6B). These differences, especially those that occurred at the potential receptor-binding sites, indicated that lineages –J and –L might have undergone some imperceptible changes in capsid structure through the recombination process with lineage –A. Furthermore, 3Dpol was lineage-specific, and aa mutations in RdRp proteins might have influenced the outcomes of CV-A6 infections. Seventeen and eleven obvious aa changes were detected in RdRp proteins of lineage –J and –L strains in comparison to lineage –A strains, respectively. The above aa differences may functionally affect the virulence of each lineage (Supplementary Table S4).
Figure 6.

The aa polymorphisms in the P1 capsid region (A) between subtypes D2 and D3 and (B) among lineages –A, –J, and –L within subtype D3. The positions of aa polymorphisms detected when comparing subtype D3 with subtype D2 are marked in blue, those detected when comparing lineages –A, –J, and –L within subtype D3 are marked in crimson, and position 242 is marked in both colors. The positions of aa polymorphisms that are embedded in the assumed epitope loops are marked with a superscript asterisk. The web-based application WebLogo was used for generating aa sequences logos (Crooks et al. 2004) (http://weblogo.threeplusone.com/).
4. Discussion
CV-A6-associated HFMD epidemics in mainland China have become a serious public health problem in recent years. Although our previous study of CV-A6 provided useful molecular epidemiological information based on 807 entire VP1 sequences isolated between 2008 and 2015 from mainland China, recombination-related factors were not considered. Likewise, most of the phylogenetic and evolutionary studies of EVs were based on the VP1 capsid coding region. Recombination has long been recognized to act as a driving force of EV evolution by eradicating deleterious mutations; it creates chimeric molecules from parental genomes with different phylogenetic origins and may also help EVs attain combined advantageous features from various genomes during the process of evolution. This may generate new recombinants with higher virulence and transmissibility, such as vaccine-derived polioviruses and the C4a evolutionary branch of EV-A71 (Minor 2009; Zhang et al. 2009, 2013; Plotkin 2010). The epidemic pattern of CV-A6 was different from that of EV-A71. The documented outbreaks of EV-A71 date back to the 1970s, which suggests long-term transmission and ongoing outbreaks within several decades (Blomberg et al. 1974; Schmidt, Lennette, and Ho 1974; Ishimaru et al. 1980; Melnick et al. 1980). Moreover, different outbreaks were always associated with different subtypes; for example, EV-A71 outbreaks occurring from 1997 to 2001 worldwide belonged to the B3 and B4 subtypes; outbreaks reported since 2000 in Malaysia, Thailand, Vietnam, and Taiwan of China were mainly caused by subtype B5 strains, but C4 strains were predominant in mainland China (Wang et al. 2002; van der Sanden et al. 2010; Zhang et al. 2013; Yee et al. 2017; Vakulenko, Deviatkin, and Lukashev 2019). The EV-A71 strains from the same subtypes (monophyletic VP1 capsid gene) were also clustered together in the phylogeny of 2C and 3Dpol noncapsid regions (with only a few exceptions), presenting a degree of genetic conservation. EV-A71 subtypes almost ceased recombining with the rest of the EV-A gene pool upon their emergence; therefore, it would be reasonable to analyze the phylodynamics of EV-A71 based on capsid genes (McWilliam Leitch et al. 2012; Lukashev et al. 2014). The low apparent recombination in EV-A71 is the opposite of that in CV-A6. The first reported CV-A6 outbreak occurred in Finland in 2008; since then, CV-A6 has rapidly triggered a series of ongoing outbreaks worldwide within the last decade and has been the predominant HFMD pathogen in mainland China since 2013. Reported CV-A6 outbreaks that have occurred worldwide since 2008 were dominated only by subtype D3; notwithstanding homogeneous capsids, frequent recombination events occurring in noncapsid regions suggest active evolution. The evolutionary phenomenon of noncapsid polymorphism in the same subtype of CV-A6 compared to EV-A71 might explain the rapid spread of CV-A6 within a short period of time.
We allocated the worldwide CV-A6 variants to seventeen different evolutionary recombinant lineages based on noncapsid sequences, nine of which belonged to the dominant subtype D3. Research limitations existed, as genotypes B (n = 8 to date) and C (n = 2 to date) were published in GenBank only as VP1 sequences, lacking whole-genome information; therefore, the actual number of CV-A6 evolutionary lineages should be more than seventeen. According to the MCMC analysis, each subtype contained more than one lineage, and the emergence of subsequent lineage(s) resulted from the recombination events of the primary lineage. Such frequent recombination happened in fast-spreading CV-A6, indicating that the recovery of deleterious genomes may still be ongoing within CV-A6 quasispecies. However, the predominant lineage has not changed within the past decade, indicating that lineage –A of CV-A6 is currently the most adapted to the host environment. In mainland China, the widespread CV-A6 recombinants detected from lineages –J and –L revealed that in recent years, novel CV-A6 variants that originated from lineage –A, with relatively high transmissibility and unique to China, have emerged. To determine whether the predominant lineage will change in the future, greater surveillance will be required.
Previous studies of CV-A6 with clinical descriptions indicated a potential correlation between clinical phenotypes and recombination. It was reported that some cases associated with atypical HFMD in the Finnish outbreak belonged to lineage –A, and cases of eczema herpeticum reported from Edinburgh in 2014 belonged to lineage –H (Osterback et al. 2009; Sinclair et al. 2014). A previous study revealed that lineage –J strains detected in Shanghai between 2012 and 2013 may result in more severe clinical features of widespread skin lesions (Feng et al. 2015a). Most importantly, we observed in this study that lineage –L was more likely to cause severe HFMD. This new finding further suggested that lineage-specific recombination events may play a potential role in the pathogenicity of CV-A6. In summary, the rapid and wide spread of subtype D3 strains suggested that host adaptation in CV-A6 may be involved mainly by capsids, but noncapsid differences may influence pathogenicity. The infection outcomes of CV-A6 are influenced by host factors, such as herd immunity and the age of infected people, but the evidence for its rapid emergence worldwide argues strongly against population-wide changes in host disease susceptibility to CV-A6. Therefore, virus-specific factors may be considered the main reason for the outcome of CV-A6 infection. Although we lack experimental models with which to precisely identify determinants of pathogenicity in CV-A6 infections and it is difficult to acquire specific clinical data from patients to conduct robust statistical analysis, we still provide some insights into the potential correlation between pathogenicity and genetic recombination for future studies of CV-A6. Whether the correlation is influenced indirectly through the functional proteins generated from noncapsids or directly through the interaction of specific molecular structures between noncapsids and capsids, the mechanism still needs to be identified.
Specific receptors have been identified for many EVs, such as EV-A71 and CV-A16. Many molecular structure and cellular receptor studies of EV-A71 and CV-A16 have been conducted; however, apparent structural differences were found in the surface-exposed loops of CV-A6 compared with EV-A71 and CV-A16, which resulted in some obstacles to identifying the specific antigenic receptors for CV-A6 (Xu et al. 2017; Chen et al. 2018; Anasir and Poh 2019). Nevertheless, two major molecular structural studies on CV-A6 still predicted important surface-exposed loop structures in the P1 capsid that serve as potential viral-neutralizing epitopes for CV-A6 (Xu et al. 2017; Chen et al. 2018; Anasir and Poh 2019). Hence, aa substitutions of subtype D3 from D2 occurred at VP2132, VP2172, VP2179, and VP2180 embedded in the EF loop, and VP1242 in the HI loop may enhance CV-A6 adaptation to the host. In addition, the substitutions from lineage –A to –J of VP1N137S and VP1V242I in the assumed receptor-binding sites of the DE loop and HI loop, respectively, may influence the pathogenicity of lineage –J strains. Recent studies on recombination mechanisms of PV and EV-A71 revealed that recombination is primarily a replicative process mediated by RdRp and that the triggers for template switching may be sequence independent (Kempf, Peersen, and Barton 2016; Woodman et al. 2019). These findings suggested that the most widespread EV recombinants may be significantly influenced by RdRp 3Dpol gene function. The RdRp of the most widespread lineage –A CV-A6 strains may affect their recombination capacity. The significant 3Dpol aa sequence variation among lineages –A, –J, and –L may explain their dissimilar pathogenicities.
For the prevention and control of HFMD, vaccination may be the most effective strategy. Recently, three inactive EV-A71 vaccines went through clinical trials and were approved for marketing in mainland China (Meng et al. 2012; Li et al. 2014b; Zhu et al. 2014; Hu et al. 2018). These vaccines may help decrease morbidity caused by EV-A71 but induce outbreaks caused by other EVs in recent years. As CV-A6 is emerging as the predominant causative pathogen of HFMD, with a very large number of severe cases in mainland China, there is an urgent need to develop an effective vaccine against CV-A6 infection. Unlike conserved EV-A71, CV-A6 was characterized by highly frequent recombination events correlated with pathogenicity. This finding may provide new insight into the selection of virus seeds for vaccine development.
In conclusion, we revealed the genetic diversity of global CV-A6 strains based on their ORF sequences, characterized the evolutionary dynamics of emerging CV-A6 in mainland China based on 336 (near) whole-genome sequences and initially discussed the correlation between recombination and pathogenicity in CV-A6. The influences of noncapsid genes might have been underestimated in studies on the evolution and pathogenicity of CV-A6. Identifying the underlying interactions between the recombinant variations and pathogenic characteristics of the repeated outbreaks of Chinese CV-A6 is essential. Studies based on whole genomes of CV-A6 could provide more comprehensive and detailed information than those based only on VP1; therefore, further surveillance is greatly needed to understand the full genetic diversity of CV-A6 in mainland China.
Supplementary data
Supplementary data are available at Virus Evolution online.
Supplementary Material
Funding
The study was supported by the National Science and Technology Major Project (project no. 2017ZX10104001, 2018ZX10713002, 2018ZX10305409-004-002, and 2018ZX10711001), the Key Technologies R&D Program of the National Ministry of Science (project no. 2018ZX10713002 and 2018ZX10713001-003) and Beijing Natural Science Foundation (Project No. L192014).
Data availability
All sequences used in this article are publicly accessible through the NCBI database. The GenBank Accession numbers of all sequences used in this article are available in Materials and methods section
Conflict of interest: None declared.
References
- Anasir M., Poh C. (2019) ‘Advances in Antigenic Peptide-Based Vaccine and Neutralizing Antibodies against Viruses Causing Hand, Foot, and Mouth Disease’, International Journal of Molecular Sciences, 20: 1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ang L. W. et al. (2015) ‘Seroepidemiology of Coxsackievirus A6, Coxsackievirus A16, and Enterovirus 71 Infections among Children and Adolescents in Singapore, 2008–2010’, PLoS One, 10: e0127999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anh N. T. et al. (2018) ‘Emerging Coxsackievirus A6 Causing Hand, Foot and Mouth Disease, Vietnam’, Emerging Infectious Diseases, 24: 654–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg J. et al. (1974) ‘Letter: New Enterovirus Type Associated with Epidemic of Aseptic Meningitis and-or Hand, Foot, and Mouth Disease’, The Lancet, 304: 112. [DOI] [PubMed] [Google Scholar]
- Bouslama L. et al. (2007) ‘Natural Recombination Event within the Capsid Genomic Region Leading to a Chimeric Strain of Human Enterovirus B’, Journal of Virology, 81: 8944–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broccolo F. et al. (2019) ‘Severe Atypical Hand-Foot-and-Mouth Disease in Adults due to Coxsackievirus A6: Clinical Presentation and Phylogenesis of CV-A6 Strains’, Journal of Clinical Virology, 110: 1–6. [DOI] [PubMed] [Google Scholar]
- Bruu A. L. (2002) ‘Enteroviruses: Polioviruses, Coxsackieviruses, Echoviruses and Newer Enteroviruses', A Practical Guide to Clinical Virology, 44. [Google Scholar]
- Buttery V. W. et al. (2015) ‘Atypical Presentations of Hand, Foot, and Mouth Disease Caused by Coxsackievirus A6–Minnesota, 2014’, Morbidity and Mortality Weekly Report, 64: 805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease, Control and Prevention (2012) ‘Notes from the Field: Severe Hand, Foot, and Mouth Disease Associated with Coxsackievirus A6 - Alabama’, Morbidity and Mortality Weekly Report, 61: 213–4. Connecticut, California, and Nevada, ', (12), [PubMed] [Google Scholar]
- Ceylan A. N. et al. (2019) ‘Hand, Foot, and Mouth Disease Caused by Coxsackievirus A6: A Preliminary Report from Istanbul’, Polish Journal of Microbiology, 68: 165–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J. et al. (2018) ‘A 3.0-Angstrom Resolution Cryo-Electron Microscopy Structure and Antigenic Sites of Coxsackievirus A6-like Particles’, Journal of Virology, 92: e01257–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. et al. (2010) ‘Analysis of Recombination and Natural Selection in Human Enterovirus 71’, Virology, 398: 251–61. [DOI] [PubMed] [Google Scholar]
- Chong J. H., Aan M. K. (2014) ‘An Atypical Dermatologic Presentation of a Child with Hand, Foot and Mouth Disease Caused by Coxsackievirus A6’, The Pediatric Infectious Disease Journal, 33: 889. [DOI] [PubMed] [Google Scholar]
- Cisterna D. M. et al. (2019) ‘Atypical Hand, Foot, and Mouth Disease Caused by Coxsackievirus A6 in Argentina in 2015’, Revista Argentina de Microbiología, 51: 140–43. [DOI] [PubMed] [Google Scholar]
- Crooks G. E. et al. (2004) ‘WebLogo: A Sequence Logo Generator’, Genome Research, 14: 1188–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darriba D. et al. (2012) ‘jModelTest 2: More Models, New Heuristics and Parallel Computing’, Nature Methods, 9: 772–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond A. J., Rambaut A. (2007) ‘BEAST: Bayesian Evolutionary Analysis by Sampling Trees’, BMC Evolutionary Biology, 7: 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X. et al. (2015. a) ‘A Novel Recombinant Lineage’s Contribution to the Outbreak of Coxsackievirus A6-Associated Hand, Foot and Mouth Disease in Shanghai, China, 2012–2013’, Scientific Reports, 5: 11700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng X. et al. (2015. b) ‘Genome Sequence of a Novel Recombinant Coxsackievirus a6 Strain from Shanghai, China, 2013’, Genome Announc, 3: e01347–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto T. et al. (2012) ‘Hand, Foot, and Mouth Disease Caused by Coxsackievirus A6, Japan, 2011’, Emerging Infectious Diseases, 18: 337–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaunt E. et al. (2015) ‘Genetic Characterization of Human Coxsackievirus A6 Variants Associated with Atypical Hand, Foot and Mouth Disease: A Potential Role of Recombination in Emergence and Pathogenicity’, Journal of General Virology, 96: 1067–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J. F. et al. (2014) ‘Hand, Foot, and Mouth Disease Outbreak Caused by Coxsackievirus A6, China, 2013’, Journal of Infection, 69: 303–5. [DOI] [PubMed] [Google Scholar]
- Hayman R. et al. (2014) ‘Outbreak of Variant Hand-Foot-and-Mouth Disease Caused by Coxsackievirus A6 in Auckland, New Zealand’, Journal of Paediatrics and Child Health, 50: 751–5. [DOI] [PubMed] [Google Scholar]
- Hu Y. et al. (2018) ‘Five-Year Immunity Persistence following Immunization with Inactivated Enterovirus 71 Type (EV71) Vaccine in Healthy Children: A Further Observation’, Human Vaccines & Immunotherapeutics, 14: 1517–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimaru Y. et al. (1980) ‘Outbreaks of Hand, Foot, and Mouth Disease by Enterovirus 71. High Incidence of Complication Disorders of Central Nervous System’, Archives of Disease in Childhood, 55: 583–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji T. et al. (2019) ‘Surveillance, Epidemiology, and Pathogen Spectrum of Hand, Foot, and Mouth Disease in Mainland of China from 2008 to 2017’, Biosafety and Health, 1: 32–40. [Google Scholar]
- Katsuki T., Torii A., Inoue M. (2012) ‘Posterior-Mean Super-Resolution with a Causal Gaussian Markov Random Field Prior’, IEEE Transactions on Image Processing, 21: 3182–93. [DOI] [PubMed] [Google Scholar]
- Kempf B. J., Peersen O. B., Barton D. J. (2016) ‘Poliovirus Polymerase Leu420 Facilitates RNA Recombination and Ribavirin Resistance’, Journal of Virology, 90: 8410–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok C. C., Au G. G. (2013) ‘Novel Marker for Recombination in the 3'-Untranslated Region of Members of the Species Human Enterovirus A’, Archives of Virology, 158: 765–73. [DOI] [PubMed] [Google Scholar]
- Kumar S., Stecher G., Tamura K. (2016) ‘MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets’, Molecular Biology and Evolution, 33: 1870–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laga A. C., Shroba S. M., Hanna J. (2016) ‘Atypical Hand, Foot and Mouth Disease in Adults Associated with Coxsackievirus A6: A Clinico-Pathologic Study’, Journal of Cutaneous Pathology, 43: 940–45. [DOI] [PubMed] [Google Scholar]
- Lau S. K. P. et al. (2018) ‘Molecular Epidemiology of Coxsackievirus A6 Circulating in Hong Kong Reveals Common Neurological Manifestations and Emergence of Novel Recombinant Groups’, Journal of Clinical Virology, 108: 43–49. [DOI] [PubMed] [Google Scholar]
- Li B. et al. (2017) ‘A Novel Enterovirus 71 (EV71) Virulence Determinant: The 69th Residue of 3C Protease Modulates Pathogenicity’, Frontiers in Cellular and Infection Microbiology, 7: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C. et al. (2018) ‘Nonstructural Protein 2A Modulates Replication and Virulence of Enterovirus’, Virus Research, 244: 262–69. [DOI] [PubMed] [Google Scholar]
- Li J. L. et al. (2014. a) ‘Epidemic Characteristics of Hand, Foot, and Mouth Disease in Southern China, 2013: Coxsackievirus A6 Has Emerged as the Predominant Causative Agent’, Journal of Infection, 69: 299–303. [DOI] [PubMed] [Google Scholar]
- Li R. et al. (2014. b) ‘An Inactivated Enterovirus 71 Vaccine in Healthy Children’, New England Journal of Medicine, 370: 829–37. [DOI] [PubMed] [Google Scholar]
- Lin C. H. et al. (2015) ‘Precise Genotyping and Recombination Detection of Enterovirus’, BMC Genomics, 16: S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q. et al. (2018) ‘Landscape of Emerging and Re-Emerging Infectious Diseases in China: Impact of Ecology, Climate, and Behavior’, Frontiers of Medicine, 12: 3–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukashev A. N. et al. (2004) ‘Recombination in Uveitis-Causing Enterovirus Strains’, Journal of General Virology, 85: 463–70. [DOI] [PubMed] [Google Scholar]
- Lukashev A. N. et al. (2005) ‘Recombination in Circulating Human Enterovirus B: Independent Evolution of Structural and Non-Structural Genome Regions’, Journal of General Virology, 86: 3281–90. [DOI] [PubMed] [Google Scholar]
- Lukashev A. N. et al. (2014) ‘Recombination Strategies and Evolutionary Dynamics of the Human Enterovirus a Global Gene Pool’, Journal of General Virology, 95: 868–73. [DOI] [PubMed] [Google Scholar]
- Macadam A. J. et al. (1994) ‘The 5' Noncoding Region and Virulence of Poliovirus Vaccine Strains’, Trends in Microbiology, 2: 449–54. [DOI] [PubMed] [Google Scholar]
- Magnelli D. et al. (2017) ‘Atypical Hand, Foot and Mouth Disease Due to Coxsackievirus A6 in a Traveler Returning from Indonesia to Italy’, Journal of Travel Medicine, 24: tax029. [DOI] [PubMed] [Google Scholar]
- Martin D. P. et al. (2017) ‘Detecting and Analyzing Genetic Recombination Using RDP4’, Methods in Molecular Biology, 1525: 433–60. [DOI] [PubMed] [Google Scholar]
- McWilliam Leitch E. C. et al. (2010) ‘Evolutionary Dynamics and Temporal/Geographical Correlates of Recombination in the Human Enterovirus Echovirus Types 9, 11, and 30’, Journal of Virology, 84: 9292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliam Leitch E. C. et al. (2012) ‘The Association of Recombination Events in the Founding and Emergence of Subgenogroup Evolutionary Lineages of Human Enterovirus 71’, Journal of Virology, 86: 2676–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melnick J. L. et al. (1980) ‘Identification of Bulgarian Strain 258 of Enterovirus 71’, Intervirology, 12: 297–302. [DOI] [PubMed] [Google Scholar]
- Meng F. Y. et al. (2012) ‘Tolerability and Immunogenicity of an Inactivated Enterovirus 71 Vaccine in Chinese Healthy Adults and Children: An Open Label, Phase 1 Clinical Trial’, Human Vaccines & Immunotherapeutics, 8: 668–74. [DOI] [PubMed] [Google Scholar]
- Minor P. (2009) ‘Vaccine-Derived Poliovirus (VDPV): Impact on Poliomyelitis Eradication’, Vaccine, 27: 2649–52. [DOI] [PubMed] [Google Scholar]
- Murrell B. et al. (2012) ‘Detecting Individual Sites Subject to Episodic Diversifying Selection’, PLoS Genetics, 8: e1002764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberste M. S. et al. (1999) ‘Molecular Evolution of the Human Enteroviruses: Correlation of Serotype with VP1 Sequence and Application to Picornavirus Classification’, Journal of Virology, 73: 1941–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberste M. S. et al. (2004) ‘Complete Genome Sequences of All Members of the Species Human Enterovirus A’, Journal of General Virology, 85: 1597–607. [DOI] [PubMed] [Google Scholar]
- Osterback R. et al. (2009) ‘Coxsackievirus A6 and Hand, Foot, and Mouth Disease’, Finland', Emerging Infectious Diseases, 15: (9), 1485–1488.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterback R. et al. (2014) ‘Genome Sequence of Coxsackievirus A6, Isolated during a Hand-Foot-and-Mouth Disease Outbreak in Finland in 2008’, Genome Announc, 2: e01004–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer J. K., Kirkegaard K. (2005) ‘Increased Fidelity Reduces Poliovirus Fitness and Virulence under Selective Pressure in Mice’, PLoS Pathogens, 1: e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotkin S. A. (2010) ‘Poliovirus Vaccine and Vaccine-Derived Polioviruses’, The New England Journal of Medicine, 363: 1870.Author reply 70–1. [DOI] [PubMed] [Google Scholar]
- Puenpa J. et al. (2013) ‘Hand, Foot, and Mouth Disease Caused by Coxsackievirus A6, Thailand, 2012’, Emerging Infectious Diseases, 19: 641–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puenpa J. et al. (2016) ‘Molecular Epidemiology and the Evolution of Human Coxsackievirus A6’, Journal of General Virology, 97: 3225–31. [DOI] [PubMed] [Google Scholar]
- Rambaut A. et al. (2016) ‘Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen)’, Virus Evolution, 2: vew007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas J. et al. (2017) ‘DnaSP 6: DNA Sequence Polymorphism Analysis of Large Data Sets’, Molecular Biology and Evolution, 34: 3299–302. [DOI] [PubMed] [Google Scholar]
- Salminen M. O. et al. (1995) ‘Identification of Breakpoints in Intergenotypic Recombinants of HIV Type 1 by Bootscanning’, AIDS Research and Human Retroviruses, 11: 1423–5. [DOI] [PubMed] [Google Scholar]
- Schibler M. et al. (2012) ‘Experimental Human Rhinovirus and Enterovirus Interspecies Recombination’, Journal of General Virology, 93: 93–101. [DOI] [PubMed] [Google Scholar]
- Schmidt N. J., Lennette E. H., Ho H. H. (1974) ‘An Apparently New Enterovirus Isolated from Patients with Disease of the Central Nervous System’, Journal of Infectious Diseases, 129: 304–9. [DOI] [PubMed] [Google Scholar]
- Sinclair C. et al. (2014) ‘Atypical Hand, Foot, and Mouth Disease Associated with Coxsackievirus A6 Infection, Edinburgh, United Kingdom, January to February 2014’, Eurosurveillance, 19: 20745. [DOI] [PubMed] [Google Scholar]
- Song Y. et al. (2017) ‘Persistent Circulation of Coxsackievirus A6 of Genotype D3 in Mainland of China between 2008 and 2015’, Scientific Reports, 7: 5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. (2014) ‘RAxML Version 8: A Tool for Phylogenetic Analysis and Post-Analysis of Large Phylogenies’, Bioinformatics, 30: 1312–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan X. et al. (2015) ‘Molecular Epidemiology of Coxsackievirus A6 Associated with Outbreaks of Hand, Foot, and Mouth Disease in Tianjin, China, in 2013’, Archives of Virology, 160: 1097–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakulenko Y., Deviatkin A., Lukashev A. (2019) ‘Using Statistical Phylogenetics for Investigation of Enterovirus 71 Genotype a Reintroduction into Circulation’, Viruses, 11: 895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Sanden S. et al. (2010) ‘Evolutionary Trajectory of the VP1 Gene of Human Enterovirus 71 Genogroup B and C Viruses’, Journal of General Virology, 91: 1949–58. [DOI] [PubMed] [Google Scholar]
- Wang H. et al. (2019) ‘Molecular Characteristic Analysis for the VP1 Region of Coxsackievirus A6 Strains Isolated in Jiujiang Area, China, from 2012 to 2013’, Medicine, 98: e15077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J. R. et al. (2002) ‘Change of Major Genotype of Enterovirus 71 in Outbreaks of Hand-Foot-and-Mouth Disease in Taiwan between 1998 and 2000’, Journal of Clinical Microbiology, 40: 10–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodman A. et al. (2019) ‘Predicting Intraserotypic Recombination in Enterovirus 71’, Journal of Virology, 93: e02057–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing W. et al. (2014) ‘Hand, Foot, and Mouth Disease in China, 2008-12: An Epidemiological Study’, The Lancet Infectious Diseases, 14: 308–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L. et al. (2017) ‘Atomic Structures of Coxsackievirus A6 and Its Complex with a Neutralizing Antibody’, Nature Communications, 8: 505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W., Zhang Y. (2016) ‘Isolation and Characterization of Vaccine-Derived Polioviruses, Relevance for the Global Polio Eradication Initiative’, Methods in Molecular Biology, 1387: 213–26. [DOI] [PubMed] [Google Scholar]
- Yang C. F. et al. (2003) ‘Circulation of Endemic Type 2 Vaccine-Derived Poliovirus in Egypt from 1983 to 1993’, Journal of Virology, 77: 8366–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang F. et al. (2014) ‘Severe Hand, Foot, and Mouth Disease and Coxsackievirus A6-Shenzhen, China’, Clinical Infectious Diseases, 59: 1504–5. [DOI] [PubMed] [Google Scholar]
- Yang S. et al. (2017) ‘Epidemiological Features of and Changes in Incidence of Infectious Diseases in China in the First Decade after the SARS Outbreak: An Observational Trend Study’, The Lancet Infectious Diseases, 17: 716–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui Y. et al. (2013) ‘A Case of Atypical Hand-Foot-and-Mouth Disease Caused by Coxsackievirus A6: Differential Diagnosis from Varicella in a Pediatric Intensive Care Unit’, Japanese Journal of Infectious Diseases, 66: 564–6. [DOI] [PubMed] [Google Scholar]
- Yee P. T. I. et al. (2017) ‘Characterization of Significant Molecular Determinants of Virulence of Enterovirus 71 Sub-Genotype B4 in Rhabdomyosarcoma Cells’, Virus Research, 238: 243–52. [DOI] [PubMed] [Google Scholar]
- Zeng H. et al. (2015) ‘The Epidemiological Study of Coxsackievirus A6 Revealing Hand, Foot and Mouth Disease Epidemic Patterns in Guangdong’, Scientific Reports, 5: 10550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. et al. (2009) ‘An Outbreak of Hand, Foot, and Mouth Disease Associated with Subgenotype C4 of Human Enterovirus 71 in Shandong, China’, Journal of Clinical Virology, 44: 262–7. [DOI] [PubMed] [Google Scholar]
- Zhang Y. et al. (2010) ‘Molecular Evidence of Persistent Epidemic and Evolution of Subgenotype B1 Coxsackievirus A16-Associated Hand, Foot, and Mouth Disease in China’, Journal of Clinical Microbiology, 48: 619–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. et al. (2013) ‘Complete Genome Analysis of the C4 Subgenotype Strains of Enterovirus 71: Predominant Recombination C4 Viruses Persistently Circulating in China for 14 Years’, PLoS One, 8: e56341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. et al. (2015) ‘An Insight into Recombination with Enterovirus Species C and Nucleotide G-480 Reversion from the Viewpoint of Neurovirulence of Vaccine-Derived Polioviruses’, Scientific Reports, 5: 17291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F. et al. (2014) ‘Efficacy, Safety, and Immunogenicity of an Enterovirus 71 Vaccine in China’, New England Journal of Medicine, 370: 818–28. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequences used in this article are publicly accessible through the NCBI database. The GenBank Accession numbers of all sequences used in this article are available in Materials and methods section
Conflict of interest: None declared.