

Abstract
Covering: 2016 to 2021
With genetic information available for hundreds of thousands of organisms in publicly accessible databases, scientists have an unprecedented opportunity to meticulously survey the diversity and inner workings of life. The natural product research community has harnessed this breadth of sequence information to mine microbes, plants, and animals for biosynthetic enzymes capable of producing bioactive compounds. Several orthogonal genome mining strategies have been developed in recent years to target specific chemical features or biological properties of bioactive molecules using biosynthetic, resistance, or transporter proteins. These “biosynthetic hooks” allow researchers to query for biosynthetic gene clusters with a high probability of encoding previously undiscovered, bioactive compounds. This review highlights recent case studies that feature orthogonal approaches that exploit genomic information to specifically discover bioactive natural products and their gene clusters.
The continual growth of publicly available genomic databases offers researchers unprecedented bioinformatic opportunities. This review examines different approaches to mining genomic data for the targeted discovery of bioactive natural products.
1. Introduction
In the natural world, communication is largely chemical. Organisms produce small chemical compounds, which we refer to as natural products, to interact with the biological and physical world around them.1 While much remains to be discovered about the ecological roles of these compounds, humankind has harnessed the incredible chemical intricacies produced by nature for a variety of purposes.2 These molecules have evolved over millennia to interact with a specialized biological target, resulting in complex chemical structures and potent bioactivities that are difficult to replicate in the laboratory. Most famously, natural products play a critical role in modern medicine, serving as the source of and inspiration for many lifesaving drugs.3
For decades, the discovery of natural products was largely accomplished by a bioactivity-guided isolation approach. Since bioactivity was used as the guiding force during isolation, this method naturally had a very high success rate of identifying bioactive natural products, but this approach also came with its own set of challenges. Dereplication of an active compound from a complex mixture can be incredibly time consuming, and this untargeted approach often resulted in the re-isolation of the same compounds.4 However, the field of natural products discovery underwent a fundamental shift in the early 2000s, when the first Streptomyces bacterial genomes were sequenced.5,6 These genomes revealed that the vast majority of small molecules produced by microbes had yet to be discovered,7 thus opening the door for future discovery efforts by complementary genome-focused approaches.
Researchers have increasingly relied on genome mining strategies to discover new natural products, which classically has referred to using genomic sequence data to identify and predict genes that encode the production of novel compounds.8 While chemical structures can be remarkably diverse, nature often converges on a few mechanisms to generate the same chemical building blocks, thereby allowing researchers to exploit genetic signatures of enzymes to identify new biosynthetic pathways. Once the genetic basis behind production of a molecule is identified, it opens the door to further discovery. Scientists are now incredibly competent at surveying genomic sequence, predicting whether or not it encodes a biosynthetic gene cluster (BGC), and targeting that BGC for isolation of the encoded product.9,10 However, unlike the bioactivity-guided isolation approach, genome-based methods to discovery do not obviously reveal whether the encoded natural product is likely to be bioactive. Bioactivity is in fact often the last feature known about a molecule; only once it is isolated and characterized does the compound get tested in a bioassay.
While many recent reviews have focused on genome mining strategies,11–15 the purpose of this review is to highlight recent examples in the literature that utilize genome mining strategies that target molecules suspected to have bioactivity. We have organized this review into four sections, each encompassing a distinct strategy commonly used for genome mining with the goal of bioactive natural product discovery. Moreover, while there are many genome mining reports that successfully mine a single bacterial genome or a handful of genomes for the presence of a BGC encoding a likely bioactive compound, this review instead focuses on larger-scale genome mining efforts. We highlight examples where researchers used targeted, hypothesis-driven approaches to mine large datasets with the express purpose of identifying new bioactive natural products. While not exhaustive, the examples described here aim to cover a variety of bioactive chemical features, compound families, and pharmacological properties.
2. Bioactive feature targeting
While natural products can be large and complex, they often contain smaller chemical features that directly lead to bioactivity, which we refer to as the “bioactive feature” of a molecule. For the purposes of this article, we have categorized these bioactive features into two distinct groups. The first group encompasses reactive features, which includes functional groups with electrophilic, radical, or nucleophilic reactivity that often result in covalent binding of the ligand to the protein target (Section 2.1). The second group includes structural features important for the natural product's ability to bind non-covalently to a biological or chemical target, which can vary from a macromolecular protein to small metal ions (Section 2.2). For both types of bioactive features, reactive and ligand binding, detailed studies have shown how a diversity of these chemical features are biosynthesized and installed into natural products. These insights can be utilized for genome mining efforts to identify orphan BGCs predicted to produce natural products with the queried bioactivity-associated chemical moiety (Fig. 1). The examples described here all successfully utilize an enzyme responsible for the installation of a bioactive chemical feature as a genome mining hook to discover new natural products with that target moiety. Importantly, the resulting molecule may exist in an entirely different compound family (peptide versus polyketide, for example) but still contain the cognate ligand feature (β-lactone, for example). We refer to this strategy as “bioactive feature targeting”.
Fig. 1. Overall scheme for genome mining new natural products with different target chemical features including (A) reactive chemical features, (B) ligand binding features, and (C) compound family defining features. In each case, the diagnostic biosynthetic genes are used to bioinformatically identify candidate gene clusters that putatively encode the production of a target natural product. Subsequent production, isolation, characterization, and bioactivity assay tests validate the bioinformatic prediction.

Because the diversity of bioactive functional groups is particularly broad, we have tabulated a selection of reactive chemical features commonly found in natural products and their associated biosynthetic routes (Table 1). Some of these chemical features (i.e., enediynes, β-lactones, epoxyketones, and disulfides) have been the target of large-scale genome mining efforts, resulting in the discovery of new bioactive molecules. Those are elaborated on in Section 2.1. However, for some well-known reactive chemical features, such as β-lactams and α,β-unsaturated carbonyl Michael acceptors, dedicated large-scale genome mining efforts have yet to be reported.
Reactive chemical features found in natural products.
| Reactive chemical feature | Structure | Enzymatic route(s) to installation | Genome mining efforts using reactive chemical feature biosynthetic enzymes |
|---|---|---|---|
| Enediyne |
|
PKS16,17 | Large scale genome mining18,19 |
| β-Lactam |
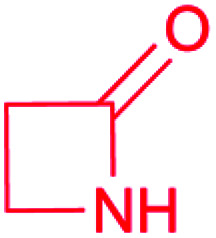
|
(1) β-Lactam synthetase (β-LS)20 | Single genome mining24 |
| (2) Carbapenam synthetase (CPS)21 | |||
| (3) IPNS22 | |||
| (4) Condensation domain23 | |||
| β-Lactone |
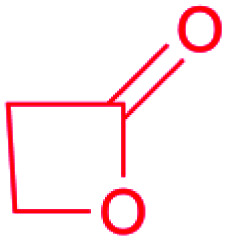
|
(1) β-Lactone synthetase25,26 | Large scale genome mining26 |
| (2) Thioesterase (TE)27 | |||
| (3) Hydrolase28 | Single genome mining29 | ||
| Michael acceptor: α,β-unsaturated carbonyl |
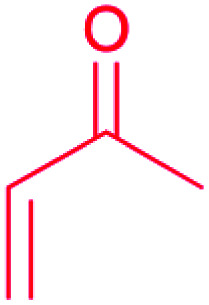
|
(1) Terpenes synthase30 | |
| (2) PKSs31 | |||
| (3) Hybrid NRPS-PKS32 | |||
| Epoxyketone |
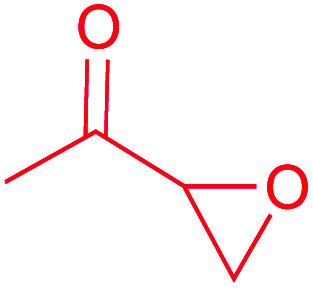
|
(1) Flavin-dependent decarboxylase–dehydrogenase–monooxygenase33,34 | Large scale genome mining35 |
| Epoxide |

|
(1) P450 (epothilone)36 | Single genome mining41 |
| (2) Flavin-dependent epoxidases37 | Single genome mining42 | ||
| (3) Dioxygenases (epoxyquinones)38 | |||
| (4) Non-heme iron-dependent epoxidases (fosfomycin)39,40 | |||
| Aziridine |

|
Unknown | |
| Maleimide |
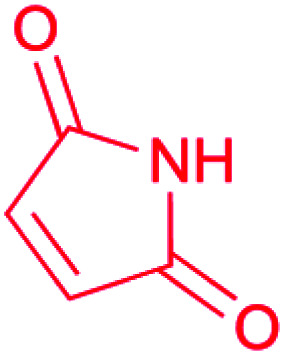
|
(1) Flavin-dependent oxidase43 | |
| (2) PKS-NRPS44 | |||
| Sulfonamide/sulfone |
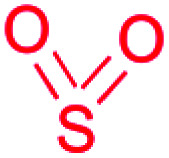
|
(1) Radical-forming, SO2 incorporating flavoprotein45 | |
| Furan |
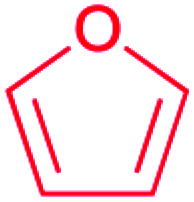
|
Terpene oxidation46 | Large scale genome mining47 |
| Disulfide |

|
(1) FAD-dependent dithiol oxidase (holomycin, gliotoxin, FK228)48–50 | Single genome mining52 |
| (2) DUF-SH didomain51 | Large scale genome mining53 | ||
| Isothiocyanate |

|
Putative isonitrile synthase54 | Large scale genome mining54 |
| Carbamate |
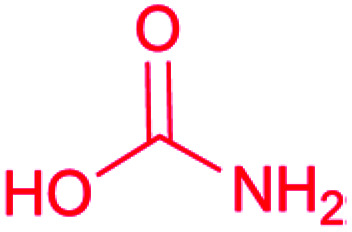
|
Carbamoyltransferase55 |
2.1. Reactive chemical features
2.1.1. Enediynes
Enediynes are a highly reactive class of polyketide synthase (PKS)-derived natural products characterized by an alkene flanked by a pair of alkynes within a 9- or 10-membered ring.56 This reactive chemical feature undergoes a Bergman cycloaromatization reaction to form an aromatic diradical species that can interact with the minor groove of DNA thereby facilitating interstrand cross links or double strand breaks. Both actions are mutagenic and lead to cytotoxicity. As of 2016, 11 enediyne-contain natural products, such as calicheamicin γ1 (1), had been characterized with four either approved as drugs or as drug candidates in clinical trials as antibody–drug conjugates.19 This high translational success rate motivated the Shen lab to search for new enediyne natural products using a genome mining approach.19 Previous sequence alignments of enediyne biosynthetic gene clusters revealed a conserved set of PKS genes responsible for installation of the enediyne warhead.17,57,58 An initial survey of the NCBI and JGI genomic databases using these biosynthetic genes suggested that enediyne containing natural products are not especially rare as they identified 87 putative enediyne BGCs across 78 bacterial strains.59 Using these insights, the Shen lab surveyed the 3400 strain actinomycete collection housed at the Scripps Research Institute (TSRI) to prioritize strains for isolation of enediynes.19 Because genomic information was not available for the entire collection, a real-time PCR method was employed to target two different enediyne biosynthetic genes. This analysis revealed 81 producing strains, and phylogenetic analysis of a 1 kb gene fragment suggested that many of the clusters were distinct from known ones. To confirm this observation, whole genome sequencing was completed for 31 representative strains, and a Genome Neighborhood Network (GNN) composed of the newly sequenced clusters revealed the potential for new chemistry. Of particular interest was a gene cluster from Streptomyces sp. strain CB03234 that appeared to encode a 10-membered enediyene related to uncialamycin.60 Subsequent isolation work identified tiancimycin A (2) as a new uncialamycin analog. As with other characterized enediynes, tiancimycin A was highly cytotoxic with IC50s in the subnanomolar range across different cancer lines and with potential to be developed as an antibody–drug conjugate. The potent activity of enediynes and presence of many promising uncharacterized BGCs make this reactive feature a promising target for future discovery efforts.
2.1.2. β-Lactones
Electrophilic moieties in natural products are often responsible for the bioactivity of a molecule due to their ability to form covalent interactions with the nucleophilic centers present in a wide variety of biological targets.61 Well known examples include Michael acceptor systems such as α,β-unsaturated ketones, β-lactams, epoxyketones, and β-lactones (Table 1). β-Lactones are often referred to as “privileged” structures due to their highly strained and reactive nature.62 These electrophilic warheads are particularly intriguing as they react with nucleophiles via ring opening reactions and covalently bind their biological target. Therefore, the presence of a β-lactone often endows a molecule with bioactivity. However, this inherent reactivity has made their isolation and characterization difficult, which has hampered efforts towards understanding their biosynthesis. Three examples of β-lactone biosynthesis have been biochemically validated, and all proceed via a different enzymatic mechanism (Table 1). The first enzymatic route to β-lactone biosynthesis was discovered only recently in the pathway for olefinic hydrocarbons.25 The enzyme OleC is a member of the Acyl-CoA ligases, Nonribosomal peptide synthetases (NRPSs), and Luciferases (ANL) superfamily. In the olefin pathway OleC instead acts as an ATP-dependent β-lactone synthetase that converts syn- and anti-β-hydroxy acids into cis- and trans β-lactones. Following this discovery, the Wackett group generated a web-based predictive tool to take a genome mining approach to discover novel β-lactone natural products that are biosynthesized via similar β-lactone synthetase chemistry.26 Their tool, AdenylPred, utilizes machine learning to predict ANL superfamily enzyme function and substrate specificity. Using AdenylPred, they mined their collection of over 50 000 BGCs for β-lactone synthetases and identified over 90 candidates. They proceeded to experimentally validate one of these predicted enzymes found in an uncharacterized BGC in pathogenic Nocardia bacteria. In vitro reconstitution of the four gene pathway led to the production of nocardiolactone (3), a previously isolated natural product63 but one for which no BGC had yet been characterized. By focusing on the enzyme responsible for the synthesis of the β-lactone warhead, the Wackett group was able to use genome mining to not only link a previously isolated natural product to its orphan gene cluster, but also identify a treasure trove of uncharacterized pathways predicted to encode novel β-lactone natural products that await discovery.
2.1.3. Epoxyketones
Epoxyketones are well known reactive chemical features that perform covalent modifications of their protein target. Natural products bearing epoxyketones may be rare, but their potent bioactivity has been well recognized.64 Eponemycin and epoxomicin (4) were the first epoxyketone-bearing proteasome inhibitors to be linked to their biosynthetic genes,65,66 and while neither of these compounds are used clinically, derivatives such as carfilzomib are FDA-approved chemotherapeutics.67 With the discovery of a biosynthetic route to epoxyketone construction established in 2014,34 genome mining for bioactive natural products containing epoxyketones became possible.
To find new epoxyketone-bearing molecules, the Brady lab utilized their previously developed informatics platform eSNaPD (environmental Surveyor of Natural Product Diversity).35,68 This platform compares PCR generated sequences from metagenomes that target conserved biosynthetic motifs, called natural product sequence tags (NPSTs), to a reference set of gene clusters. This comparison reveals the biosynthetic potential of the original metagenomic sample. To apply this approach specifically to mine for epoxyketones, they utilized the lone ketosynthase (KS) domain essential for the biosynthesis of the conserved epoxyketone in eponemycin and epoxomicin as their NPST target.34 By using that sequence as a genome mining hook, they used the eSNaPD platform to mine 185 soil metagenomes, which likely include tens of thousands of unique bacterial genomes. This search returned 99 hits indicative of a sequence encoding an enzyme capable of installing an epoxyketone moiety. Four of the metagenomes with epoxyketone hits had cosmid libraries that allowed the authors to identify 11 putative BGCs that contained the hit sequence. Out of these 11 BGCs, 9 were suggested by in silico analysis to produce an epoxyketone-containing natural product, which was further verified by the proteasome inhibitory activity of culture extracts of these BGCs. The Brady lab was ultimately able to identify seven novel compounds from these strains, the clarepoxcins (clarepoxcin A, 5) and the landepoxcins (landepoxcin A, 6), all nanomolar inhibitors of the human 20S proteasome. Both series of compounds contain an epoxyketone moiety attached to a hydrophobic peptidic backbone, but the structures differ from any previously described molecule. This example demonstrates that enzymes responsible for installation of a specific bioactive chemical feature can prioritize BGCs of interest from even massive metagenomic datasets, ultimately leading to the discovery of new bioactive natural products.
2.1.4. Disulfide bonds
Disulfide bonds are well recognized structural components in proteins, but they can also serve as highly reactive features in small molecules leading to pronounced bioactivity.69 Disulfide bonds act as prodrugs – the inactive disulfide bond can be readily transformed to an active dithiol via cleavage in the cellular environment.70 Leinamycin (7) is one such disulfide-containing small molecule with potent bioactivity.71 Isolated from Streptomyces atroolivaceus, leinamycin contains a unique 1,3-dioxo-1,2-dithiolane moiety spirofused to a macrolactam ring and displays potent anticancer bioactivity via an unusual mechanism of action.71,72 Because leinamycin's bioactivity is directly linked to the reactive 1,3-dioxo-1,2-dithiolane moiety, we will refer to this as the reactive chemical feature. Cellular thiols reduce the 1,3-dioxo-1,2-dithiolane moiety thereby forming an episulfonium ion capable of alkylating DNA, ultimately resulting in DNA cleavage and death.73,74
Despite its unusual chemical features and potent bioactivity, leinamycin remained the only member of its family until 2017, when the Shen lab aimed to add to the leinamycin family of natural products via a genome mining approach.53 Because they knew that leinamycin's bioactivity was linked to the 1,3-dioxo-1,2-dithiolane moiety, they used a bioactive feature mining-based approach to identify new compounds containing this reactive chemical feature. Previous biochemical work on the leinamycin pathway (lnm) had revealed a domain of unknown function (DUF) and a cysteine lyase domain (SH), referred to as the DUF-SH didomain, was responsible for sulfur incorporation.51,75 Therefore the authors mined all public databases using the DUF-SH didomain and identified 19 potential lnm BGCs. An additional 30 BGCs were selected by mining TSRI's strain collection. Phylogenetic analysis of this DUF-SH protein revealed 18 distinct clades containing 28 unique BGCs. A series of bioinformatic analyses were used to make structural prediction and aid in strain prioritization. Ultimately, two strains were chosen for future analysis. Using gene knockouts, Shen and coworkers identified two series of leinamycin analogs, the guangnanmycins from the gnm BGC and the weishanmycins from the wsm BGC. Guangnanmycin A (8) had the same reactivity towards oxidants as leinamycin, suggesting it could possess cytotoxic activity using a similar episulfonium ion mediated DNA alkylating mechanism. Weishanmycin A lacks the disulfide bond found in leinamycin. Instead, the free thiol may be activated by reactive oxygen species similar to leinamycin E1.73 After 30 years without the discovery of any leinamycin analog, a bioactive feature-based genome mining approach ultimately resulted in the expansion of the leinamycin family.
2.2. Ligand binding features
2.2.1. Diazeniumdiolates
Metallophores are small molecules with electron-rich ligand features that bind metal ions, such as iron and cobalt, that are critical for an organism's survival. While this ecological role is well recognized, these compounds are also important from a biomedical perspective. Their metal mobilization abilities have been used clinically to treat metal toxicity,76 and are now utilized to create siderophore–antibiotic conjugates, capable of stealthily entering bacterial cells via the endogenous metal transport system where they can then deploy an antibiotic payload.77,78 Despite the fact that over 500 different siderophores have been isolated, showcasing tremendous structural diversity, these small molecule metallophores employ just a small set of functional groups for binding trace metals,79 which endow the molecule with its bioactivity.
In 2018, Hertweck and coworkers discovered a new bacterial siderophore with a novel iron-binding functional group produced by plant-associated symbiotic bacteria.80 This metallophone, named gramibactin A (9), is unusual among siderophores because it harbors two diazeniumdiolate (N-nitrosohydroxylamine) moieties. The diazeniumdiolate moiety, while never previously recognized as an iron-binding feature, was shown to be directly involved in iron complex formation, and thus represents the bioactive feature critical for the ligand's metal-binding ability. Because of this important biological activity and unusual chemical structure, the Hertweck group took a genome mining approach to identify novel diazeniumdiolate-containing siderophores.81
Using a targeted knockout approach, two enzymes, GrbD and GrbE, were identified as essential for graminine biosynthesis. In the hopes of identifying novel natural products that contain the diazeniumdiolate ligand, they used BLAST to query GrbD against the non-redundant UniProt protein sequence database. Genome neighborhood analysis identified 37 probable gene clusters. Notably, all clusters were found in organisms from the Burkholderiaceae family, but the strains harboring these BGCs were divided into two distinct groups. One group of BGCs was found in plant symbiotic bacterial strains, while the other group was found in plant-pathogenic strains. The authors cultured a selection of these strains and screened via MS/MS for the characteristic fragment resulting from cleavage of the diazeniumdiolate moieties. From the symbiotic bacteria, they ultimately identified a gramibactin congener, gramibactin B, and a new family of six peptide compounds bearing the diazeniumdiolate moiety that they named the megapolibactins A–F. From the culture extract of a strain from the group of plant-pathogenic strains they isolated a new compound they named plantaribactin. Bioactivity testing of megapolibactin B (10) and plantaribactin (11) revealed that beyond just iron chelation, these diazeniumdiolate-containing compounds are also capable of serving as nitric oxide (NO) donors. The discovery of new diazeniumdiolate siderophores from bacteria further suggests that ligand binding-based genome mining is an attractive approach for the targeted discovery of natural products with specific bioactivities, and that this approach transcends the nature of the ligand's target. As this example showed, bioactive feature targeting is not limited to features important for macromolecular binding but can be utilized to target bioactive features important for metal ion binding as well.
2.2.2. FKBP12-binding compounds
Protein–protein interactions have been seen as a promising target for new drugs. Unfortunately, it has proven difficult to design or discover small molecules that can alter how proteins interact with each other.82 This issue is so problematic that some promising targets are considered “undruggable”.83 A particularly interesting strategy to overcome this challenge is observed in the FK506/rapamycin family of natural products.84 These NRPS/PKS hybrid natural products possess immunosuppressive bioactivity by modulating the kinase mTOR or phosphatase calcineurin. The bioactivity of rapamycin (12)/FK506 (13) can be attributed to two structural elements. The first is a conserved region that contains a key pipecolate moiety. This portion of the molecule tightly binds to the common cellular protein peptidyl prolyl isomerase FK506-binding protein (FKBP12).83 The rapamycin-FKBP12 binary complex then displays the second structural feature of rapamycin, a PKS derived variable region that directly interacts with mTOR and modulates its activity. Changes to this variable region allow rapamycin-like molecules to target different proteins, making this class of molecules a promising source of new bioactive compounds. Critically, rapamycin-like natural products cannot bind to their protein targets, such as mTOR, alone. Instead, the initial binding to FKBP12 via the pipecolate-containing conserved region is necessary. Thus the conserved ligand binding motif is critically important for the class defining bioactivity.
By exploiting the two structural features of rapamycin, a recent study conducted by scientists at Warp Drive Bio sought to use a genome mining approach to discover new FK506/rapamycin family members with new bioactivity.85 To find new FK506/rapamycin analogs, the authors focused on the pipecolate moiety found in the ligand binding feature that binds in the FKBP12 active site. The pipecolate residue is biosynthesized by lysine cyclodeaminase and is subsequently incorporated as the final step of the NRPS assembly line.86 The authors therefore reasoned that the lysine cyclodeaminase would be an ideal search query for a genome mining based discovery effort. With an inhouse strain database of ∼135 000 Actinomycetes, it was not feasible to assemble and search each genome for lysine cyclodeaminases in an efficient way. Instead, the authors pooled between 40 and 480 strains at a time and conducted initial high throughput sequencing and assembly. The resulting contigs were then searched for the presence of a lysine cyclodeaminase analog. Strains in the positive pools were subsequently screened by PCR and followed up with complete genome assemblies. This process rediscovered the four known rapamycin family members along with seven additional clusters that appeared to produce new analogs with similar conserved regions and diverse variable regions. Five of the clusters were over expressed and their products were structurally characterized. A cluster that was designated as X1 from Streptomyces malaysiensis DSM 41697 proved promising. X1 produced three new analogs (WDB001-003) with the predicted conserved and variable regions (14–16). Detailed studies demonstrated that this series of natural products behaved similar to FK506/rapamycin in that they tightly bind to FKBP12 via the conserved region. The variable region is then exposed to interact with the protein target. Rigorous studies revealed the target to be a coiled-coil region of a centrosome-associated protein called CEP250. Cell-based assays demonstrated that WDB002 recruits FKBP12 to CEP250 in the centromere and decreases centromere separation. A recent screening assay indicated that CEP250 may interact with Nef13 from SARS-CoV-2,87 the virus responsible for COVID-19. This suggests that WDB002 may warrant further study as antiviral.
2.2.3. Nucleotidyl phosphoramidates
Enzymatic reactions often make use of chemically activated, high energy intermediates to facilitate catalysis. This is most often observed with ATP-dependent enzymes that utilize phosphorylated or adenylated intermediates. Bioactive natural products that mimic these activated intermediates with non-hydrolyzable analogs of the labile phosphoester bond can therefore tightly bind to and inhibit a target enzyme.88 For example, the ribosomally synthesized and post-translationally modified peptide (RiPP) natural product microcin C7 (McC, 17) utilizes this approach to inhibit aspartyl-tRNA synthetase. McC is a seven amino acid long peptide that contains a C-terminal aspartyl-adenylate formed with a stable phosphoramidate bond, the critical ligand binding feature.89 Upon import of McC into the cell90 and its subsequent hydrolysis into constitutive amino acids, the modified aspartyl residue is released and specifically targets aspartyl-tRNA synthetase because it mimics the aspartyl adenylate mechanistic intermediate.91 Mining for amino acid nucleotidyl phosphoramidates should therefore reveal new natural products that similarly target and inhibit tRNA-synthetases.
Detailed study of the McC biosynthetic gene cluster revealed that MccB, a ThiF-like adenyltransferase, is responsible for installation of critical phosphoramidate bond from an essential C-terminal asparagine residue of the precursor.92–94 Genome mining for MccB, along with a peptide exporter, identified homologs from diverse bacterial species.95,96 Sequence analysis suggested that many of these clusters contained precursor peptides with the obligate C-terminal asparagine, indicating that these strains could be producing McC-like antibiotics that still target aspartyl-tRNA synthetase.95 Some of these new clusters appeared to contain additional enzymes that were not present in the original Escherichia coli mcC operon, indicating that further modifications to the precursor peptide could be found in the resulting natural products. In particular, the Severinov and Dubiley groups focused on a cluster found in Bacillus amyloliquefaciens.97 Because metabolite analysis of B. amyloliquefaciens DSM 7 did not indicate the presence of McC analogs, the gene cluster was heterologously expressed in Bacillus subtilis 168. Mass-spectrometric analysis revealed the presence of a modified precursor peptide with an unknown nucleobase. In vitro reconstitution of the biosynthetic pathway revealed that B. amyloliquefaciens MccB favors CTP instead of ATP to form an aspartyl-cytidylate mimic that retains the non-hydrolyzable phosphoramidate bond (18). Moreover, this nucleobase is further modified with a carboxymethyl group. Activity assays demonstrated that the new McC analog was still a potent aspartyl-tRNA synthetase inhibitor despite the change in nucleobase due to the conserved ligand binding feature. Notably, B. amyloliquefaciens McC was active in the presence of the acylating McC resistance gene mccE, allowing it to overcome one of the two modes of McC resistance found in E. coli.97 This combination of genome mining and biochemical study could lead to the discovery of other novel McC antibiotics that use modified peptidyl-nucleobases to inhibit tRNA synthetases.
2.2.4. Indolizidines
The examples discussed in this section all showcase genome mining studies that targeted specific features important for a ligand's ability to bind a known target, whether that target be a cellular protein or extracellular iron. However, in some cases a specific feature is identified as important for ligand binding to a biological target, but that target remains unknown. Such is the case for the indolizidine alkaloids, a diverse class of compounds characterized by an indolizidine ring system: fused 5- and 6-membered rings sharing a nitrogen atom at the bridgehead position.98 This ring structure is a privileged feature associated with bioactive molecules,99 and structure–activity relationship studies have shown the bioactivity of indolizidine alkaloids is associated with the indolizidine moiety.100 This potent bioactivity has created tremendous interest in this family of natural products and inspired the search for new members of this class of compounds, resulting in the discovery of curvulamine (19) and curindolizine, two compounds isolated from a fish-associated fungi with antibacterial and anti-inflammatory activities, respectively.101,102
Dai et al., identified the BGC responsible for curvulamine production using a combination of antiSMASH, RT-PCR experiments, and targeted gene deletions.103 Ultimately, heterologous expression of a PLP-dependent aminotransferase, CuaB, revealed its role as a bifunctional aminotransferase capable of both a Claisen condensation to form new C–C bonds and the α-hydroxylation of an amino acid. The unusual biosynthetic ability of this enzyme to install a potent pharmacophore prompted the authors to use it as a genome mining hook to discover new antibacterial indolizidine alkaloids. By searching the NCBI and JGI databases, they uncovered five distinct BGCs that encode CuaB homologs as well as other putative biosynthetic enzymes. These included a BGC from Bipolaris maydis ATCC 48331 that they referred to as the bip cluster. Upregulation of the putative transcription factor bipF using a strong promoter resulted in the production of nine new polyketide indolizidine alkaloids they named bipolamines A–I. Antibacterial bioactivity assays revealed that many of the bipolamines were bioactive, with bipolamine G (20) exhibiting even more potency than curvulamine, the small molecule that initially originated this search. This work thus demonstrates that even when a ligand's binding partner is unknown, genome mining using a bioactive feature targeting approach can still be effective for the discovery of new bioactive natural products.
3. Compound family mining
The bioactivity-guided isolation approach that dominated natural product discovery for decades, while untargeted and time consuming, resulted in the discovery of a tremendous number of bioactive small molecules, many of which are still used clinically today.104 This discovery work revealed that specific compound families have proven time and time again to likely possess bioactivity. We define ‘compound families’ as a structurally related group of compounds that share a biosynthetic origin. With the advent of genome mining, researchers can now utilize a targeted, hypothesis-driven approach to search for a new member of a bioactive compound family, with the assumption that related members of this compound family will also be bioactive and likely have the same molecular target.
In a compound family genome mining approach, biosynthetic genes responsible for the overall chemical structure of a bioactive molecule are used as genome mining hooks to identify related analogs (Fig. 1C). This differs from the bioactive feature mining approach (Section 2) because the goal is to extend a structural series of compounds to better understand and optimize structure–activity relationships around a lead molecule rather than to mine for the presence of a specific functional group. To search for new analogs of a known bioactive compound family, the genome mining hook might be a single gene, a small biosynthetic cassette, or even an entire BGC. In the case of single gene hooks, sequence similarity networks such as a those generated by the Enzyme Function Initiative Enzyme-Similarity Tool (EFI-EST) can rapidly identify protein homologs for consideration.105,106 While not inclusive, the diversity of examples included in this section is intended to highlight the utility of this genome mining approach to discover new bioactive molecules across compound classes.
3.1. Glycopeptide antibiotics
Glycopeptide antibiotics are an important family of natural products and include the clinically valuable drugs vancomycin, teicoplanin, and their semi-synthetic derivatives.107 These NRPS derived products are composed of seven amino acids that are modified into typically tri- or tetra-cyclic structures. Additional tailoring reactions, such as glycosylations and halogenations, are also common.108 Glycopeptide antibiotics often have a conserved mode of action wherein they bind to lipid II of the bacterial cell wall, inhibiting the transpeptidation or transglycosylation reactions required for bacterial cell wall synthesis. Because of this potent bioactivity, the Wright lab sought to identify new glycopeptide antibiotics that function via a novel mode of action instead of the well-known lipid II binding activity.109
To accomplish this, Wright and coworkers focused on 71 glycopeptide antibiotic BGCs. Hypothesizing that phylogenetic diversity leads to molecular diversity, they generated phylogenetic trees that were based on condensation domains of the C-2 module rather than the phylogeny of the organism itself.109,110 The presence of known antibiotic resistance genes in the cluster was then mapped onto the phylogenetic tree, revealing a large conserved clade of traditional glycopeptide antibiotics with the anticipated lipid II binding mode of action. Further examination of the tree revealed two divergent clades that lacked an identifiable resistance gene. One of these produced the known molecule complestatin (21),111 while the other produced a novel compound that was named corbomycin (22). Even though these compounds belonged to a known family of glycopeptide alkaloids, the lack of a recognizable resistance gene suggested a novel mode of action. Indeed, corbomycin and complestatin do not bind to lipid II and inhibit peptidoglycan biosynthesis. Instead, they bind to peptidoglycan itself and prevent cell wall remodeling by peptidoglycan hydrolases called autolysins.112 These enzymes are essential for cell growth and division, and their inhibition represents a novel mode of action. Overall, this study demonstrates that a combination of genome mining and knowledge of the resistance mechanism can facilitate discovery of new molecules from a known compound family with unprecedented modes of action.
3.2. Cationic nonribosomal peptides
Recently, a major public health crisis has revolved around Gram-negative bacteria and their growing resistance to many antibiotics. Cationic non-ribosomal peptides (CNRPs) have become a prominent contender for treatment of these pathogens.113 Members of this natural product family are rich in positively charged amino acids, allowing them to cross the outer membrane and interact with anionic targets. To discover new NRPS products rich in cationic residues, the Qian group utilized an antiSMASH9 based genome mining approach.114 They analyzed roughly 7400 complete or draft bacterial genomes from public databases using antiSMASH to search for putative NRPS biosynthetic gene clusters. Based on the predicted substrate specificity of the adenylation domains found in the gene clusters, they anticipated that 807 of the non-ribosomal peptides could be classified as CNRPs. They were all at least 6 amino acids in size and contained at least two positively charged amino acids, such as arginine, lysine, and histidine.
Using the results from the antiSMASH data, the Qian group generated a protein sequence similarity network (SSN) of the predicted peptide sequences and concluded that 91% of these cationic peptides were not closely related to a known CNRP.114 Amongst all CNRPs, some N-acylated CNRPs (cationic lipopeptides), such as the tridecaptins, hold particular promise as a treatment for Gram-negative pathogens.115 A specific search for cationic lipopeptides revealed that they represent 32% of all cationic non-ribosomal peptides and 85% of these did not cluster with a known CNRP. Following this genome mining, the Qian group focused their isolation efforts on clusters from two bacilli, Brevibacillus laterosporus and Paenibacillus alvei. They successfully isolated and characterized two cationic lipopeptides, later named brevicidine (23) and laterocidine (24).114 Both consist of a linear, positively charged section with three ornithine residues and a small hydrophobic peptide ring, representing a novel class of cyclic cationic peptides.
To fully characterize these newly isolated compounds, bioactivity assays were completed against ESKAPE pathogens, which are often the cause of nosocomial infections.116 Both brevicidine and laterocidine showed significant in vitro efficacy against these Gram-negative pathogens with no resistance development. Further in vivo studies in a mouse model demonstrated these compounds were effective in treating an E. coli infection. While the exact mechanism by which they operate is still being researched, brevicidine and laterocidine appear to be promising leads due to their in vivo efficacy, selectivity, and a low rate of bacterial resistance.
3.3. Cinnamoyl-containing nonribosomal peptides
In some cases, an unusual chemical moiety is not the pharmacophore itself, but is often associated with bioactive natural products, and therefore becomes the defining feature of a compound family. An interesting example of this correlation is the 2-[1-(Z)-propenyl]-cinnamoyl moiety. This substructure is found in only a small number of PKS/NRPS natural products, but each compound in this family has a different bioactivity. For instance, skyllamycin (25) is a platelet-derived growth factor inhibitor,117 WS9326 is a tachykinin receptor agonist,118 mohangamides inhibit isocitrate lyase,119 and coprisamides enhance quinone reductase activity.119 The Ge Lab capitalized on the bioactivity of this compound family and conducted a study to search for new cinnamoyl-containing nonribosomal peptides.120 The cinnamoyl moiety itself is constructed from a type II PKS as a linear polyene that is isomerized and then cyclized.117,121,122 To mine for new examples of this family of natural products, a series of SSNs were created that included different PKS subunits from polyene producing type II PKSs, type I PKSs, aromatic type II PKSs, and type II fatty acid synthases (FASs). The Ge Lab found that not all the polyene PKS subunits that generate the cinnamoyl structure are consistently clustered together. Instead, only the KSα subunit was a reliable predictor. In addition to the PKS architecture, they found that both the isomerase responsible for introducing the cis bond into the polyene chain and ketoreductase domain was diagnostic of the cinnamoyl motif compared to simple linear polyenes. Using these three domains, the Ge Lab then conducted a bioinformatic search for bacteria in the NCBI and JGI databases that contained both the KSα subunit and isomerase. These three genes were present in 192 bacteria and further filtering with antiSMASH identified 51 gene clusters that contained an intact type II PKS adjacent to an NRPS. A GNN was then generated and revealed that only two of the 51 gene clusters appeared similar to known natural products and suggested there were opportunities for discovery of novel bioactive molecules. The Ge Lab chose to focus on a glycosyltransferase containing cluster from Kitasatospora sp. CGMCC 16924. Gene knockout and isolation studies lead to the discovery of six compounds named kitacinnamycin A–F. The isolated kitacinnamycins A–F along with intermediates obtained from the knockout and biosynthetic studies were tested for stimulator of interferon genes (STING) activation. One of the intermediates, kitacinnamycin H (26) was an effective activator of the STING signaling pathway. These results further solidify the value of targeting the cinnamoyl containing NRPS family for the discovery of diverse bioactive molecules.
3.4. Calcium dependent antibiotics
Calcium dependent antibiotics are NRPS derived natural products that require calcium for activity.123 They typically contain an aspartate rich motif that is responsible for calcium binding, which is required for penetration of the bacterial cell membrane.124 Because calcium binding defines this class of peptidic natural products, members have diverse modes of action including targeting of cell wall biosynthesis or cell membrane integrity.123 They also hold great promise as new antibiotics. The most well-known calcium dependent antibiotic, daptomycin, has been clinically proven to be an effective treatment of Gram-positive bacterial infections125 and has been classified as a critically important antimicrobial for human medicine by the World Health.
A recent study by the Brady Lab sought to discover new calcium dependent antibiotics by mining soil metagenomes for BGCs that should produce non-ribosomal peptides with the characteristic Asp–X–Asp–Gly calcium binding motif.126 However, simply shotgun genome sequencing each soil sample would not provide enough sequencing depth to reliably detect low abundance BGCs. Instead, the Brady lab used a PCR based approach to enhance the sensitivity of detection of target biosynthetic gene clusters within a particular soil sample.127,128 Specifically, they targeted conserved regions in the adenylation domains of non-ribosomal peptide synthetases to generate next-generation sequencing reads.126 Using eSNaPD,68 the Brady lab analyzed the sequencing data for a conserved adenylation domain responsible for the first Asp in the Asp–X–Asp–Gly calcium binding sequence. Phylogenetic analysis indicated numerous sequencing tags fell into new clades, suggesting novel products. For detailed study, they decided to pursue a particularly abundant clade found in 19% of the metagenomes and named it the malacidins (metagenomic acidic lipopeptide antibiotic-cidins). Traditional cloning followed by cosmid screening led to the recovery of the malacidin gene cluster. Heterologous expression permitted isolation and structure elucidation of malacidin A and B (27 and 28), cyclic lipopeptides that typically differ by a methylene on the lipid tail terminus and four non-coded amino acids at the core. Although the malacidins were found to be calcium dependent, they do not contain this conserved Asp–X–Asp–Gly motif, but rather have a different motif, 3-hydroxylAsp–Asp–Gly. Further experiments demonstrated that the malacidins target lipid II, have a low rate of developed resistance, and could be effective against multidrug resistant Gram-positive pathogens.126
3.5. Thioamide RiPPs
Typically used by synthetic chemists to form stable isosteres, thioamides are a rare functional group in natural products, but one with recognized bioactivity.129,130 Famously thioamide-containing drugs have been used to treat thyroid disorders.131 Because of this, the biological activity tied to thioamide moieties found in natural products is currently under investigation. Thioviridamide (29) is one such thioamide-containing RiPP natural product with potent anticancer activity.132 Due to the unusual chemical structure and intriguing bioactivity of thioviridamide, two research groups in 2017 almost simultaneously undertook very similar genome mining approaches to expand the thioviridamide family of natural products.133,134
The Truman group chose to mine bacterial pathways using a YcaO domain protein, TvaH, found in the recently identified thioviridamide BGC (tva) as a genome mining hook.133,135 While TvaH was not fully functionally characterized at the time, YcaO proteins are typically involved in the biosynthesis of thiazoline and (methyl)oxazoline moieties via an ATP-dependent cyclodehydration reaction, and so TvaH was predicted to play a role in thioamide biosynthesis.136 A BLAST search for TvaH homologs followed by MultiGeneBlast ultimately identified 14 thioviridamide-like BGCs from bacterial genomes. Five publicly available strains were selected for fermentation, and three thioviridamide-like molecules were isolated and structurally characterized. These compounds were designated thioalbamide (30), isolated from Amycolatopsis alba DSM 44262, and thiostreptamides S4 and S87, isolated from Streptomyces sp. NRRL S-4 and NRRL S-87, respectively. All three compounds contained characteristic thioamide moieties present in the peptide backbone. Importantly, thioalbamide was found to be highly bioactive, with an IC50 in the nanomolar range, but with high levels of selectivity for tumor cells as opposed to healthy human cells.
Concurrent with this study, the Müller group utilized a similar approach to identify novel members of the thioviridamide compound class.134 Rather than genome mining using the TvaH enzyme putatively involved in thioamide synthesis, they utilized the thioviridamide precursor peptide TvaA to probe sequence databases. This mining effort yielded 13 hits that resembled the thioviridamide BGC, and they obtained four of these strains for further investigation. Notably, three of these four strains were also identified by the Truman group. However, fermentation of the fourth strain, Streptomyces malaysiense MUSC 13657, resulted in the production, isolation, and structural characterization of two novel compounds, thioholgamide A and B. Thioholgamide A (31) showed anticancer activity more pronounced than that of thioviridamide A. Both groups were thus independently able to use a genome mining-based approach, utilizing different biosynthetic enzymes, to expand a family of bioactive compounds.
3.6. Proteusins
Some bioactive natural products do not have a single important chemical feature, but instead the entire structure acts in concert to produce the observed bioactivity. An example of natural products that fall into this category are pore forming molecules, such as the polytheonamides. These highly modified peptides were first isolated from sponges and shown to be highly cytotoxic.137–139 While initially hypothesized to be biosynthesized by NRPS machinery, they were later demonstrated to actually be members of the proteusin class of RiPP natural products.140,141 These long peptides are nearly 50 amino acids in size with extensive epimerizations and methylations that promote formation of a β-helix.142 This lipophilic structure can act as a cation-channel that inserts into membranes.143 However, efforts to study and engineer the entire biosynthetic pathway were hindered by the fact that the producing organism, Candidatus Entotheonella factor, is a bacterial sponge symbiont and unculturable. To solve this problem, the Piel lab sought to find alternative sources of proteusins from culturable organisms that still retained the potent cytotoxicity observed in polytheonamide.144 Previous NMR142 and molecular dynamics145 studies indicated that the bioactive β-helix structure is stabilized by evenly spaced N-methylated Asn residues defined by an N–X5–N motif (Fig. 2). Therefore, the Piel lab conducted a bioinformatic search for clusters that contained an N–X5–N–proteusin precursor along with an epimerase and methyltransferase.144 Three clusters from diverse bacteria were identified, one of which came from the culturable bacteria Microvirgula aerodenitrificans. After establishing a conjugation-based plasmid system, production of the expected aeronamide A (32) product was confirmed using a hybrid in vivo/in vitro production system. Because aeronamide A contained the critical repeating N–X5–N motif that promotes β-helix formation, it possessed high cytotoxic activity (IC50 of 1.48 nM in HeLa cells). Moreover, the ability to create a genetic system for M. aerodenitrificans enabled processing of alternative precursor peptides to make non-native products.
Fig. 2. NRM solution structure of polytheonamide B (PDB: 2RQO) reveals N-methyl Asns are found on one face of the β-helix. This repeating N–X5–N motif was targeted for genome mining efforts.

3.7. Meroterpenoids
Meroterpenoids are hybrid natural products that arise from a combination of terpenoid biosynthesis coupled with structural features originating from alternative biosynthetic origin.146–148 These compounds are ubiquitous in nature, found across all domains of life, and comprise a tremendous variety of structural diversity.149 Importantly, many of these compounds are well recognized for their bioactivity. Fungal meroterpenoids, such as the clinically used immunosuppressant mycophenolic acid (33), demonstrate the potential utility of this compound class.148 Because of the tremendous structural diversity and high rate of bioactivity in this compound class, Zhang et al., decided to capitalize on the known biosynthetic origins of this family to discover new, likely bioactive, fungal meroterpenoids.150
The fungal biosynthesis of meroterpenoids includes the co-clustering of genes encoding polyketide synthases, prenyltransferases (PTs), and terpenoid cyclases (TCs).151 By exploiting this knowledge, the authors were able to mine fungal genomes from their extensive strain collection for the co-occurrence of these genes, which led to the discovery of two homologous BGCs containing all three required biosynthetic elements.150 Fermentation of these strains resulted in the production of ten related meroterpenoids, eight of which represent novel compounds. These molecules were then definitively linked back to their gene clusters via gene inactivation experiments. Six of these meroterpenoids, isolated from Arthrinium sp. NF2194, were named arthripenoid A–F and were used as a model system to explore the biosynthesis of this compound family. A series of gene inactivation experiments coupled with in vitro protein expression and enzymatic activity assays were used to parse out the biosynthetic route to these compounds. Finally, assays demonstrated that arthripenoid C (34) is capable of immunosuppressive activity on CD4+ T cells. By targeting proximal genes for meroterpenoid biosynthesis, the authors were able to identify two similar BGCs in phylogenetically distinct fungi and isolate bioactive natural products. Thus, genome mining using the key enzymes that biosynthesize a compound family previously recognized for its bioactivity as a hook has proven successful in prioritizing BGCs of interest in the hunt for novel therapeutics.
3.8. Phosphonates
The phosphonate class of natural products are characterized by the presence of a stable C–P bond that mimics the labile phosphoester bond found in numerous phosphorylated primary metabolites.152 These natural products have diverse bioactivities including antifungal, antimalarial, antibacterial, and herbicidal. Several commercial products, such as the clinical antibiotic fosfomycin (35), are also members of this class. While the presence of a phosphonate bond defines this class, this molecular feature is not necessarily a pharmacophore unlike the microcin C7 compounds where the stable phosphoramidate serves as the critical ligand binding feature for bioactivity (Section 2.2.3). For example, the epoxide ring in fosfomycin serves as an electrophile to form a covalent adduct with UDP-GlcNAc enolpyruvyl transferase (MurA),153 thereby granting the molecule its potent bioactivity. In contrast, FR900098 and fosfomidimycin are tight binding competitive inhibitors of d-1-deoxyxylulose-5-phosphate reductoisomerase, a key enzyme in the non-mevalonate pathway for isoprenoid biosynthesis.154
Because phosphonates have been a prolific source of potent natural products that utilize diverse modes of action, there have been concerted efforts to identify novel members of this natural product family. Genome mining for phosphonates relies on the first committed step of the biosynthesis which is nearly always catalyzed by phosphoenolpyruvate mutase (PepM).155 A detailed investigation and discovery by the Metcalf group effort revealed that the phylogeny and sequence identity of this single gene can be diagnostic for identifying new classes of phosphonates. Specifically, pepM genes that share at least 80% sequence identity are likely to be found in gene clusters that produce related compounds.156,157 This observation led to strain prioritization and discovery of several new bioactive phosphonates.157 A recent report from the Ju lab applied this observation using a genome first approach. They first mined pepM sequences from all public databases to identify 448 putative phosphonate producing gene clusters.158 A SSN was then generated using the 80% sequence identity threshold to generate 113 gene cluster families predicted to produce novel compounds. The PepM reaction is thermodynamically unfavorable and requires a coupled secondary reaction, such as decarboxylation, to push the reaction forward.152,155 Therefore, to identify a novel phosphonate scaffold, the Ju lab examined each of the 113 gene cluster families for enzymes that catalyze the thermodynamically favored biosynthetic step. In 6% of gene cluster families, no candidate enzyme was identified. Examination of the largest of these, an 11-membered gene cluster family, lead to the discovery of the phosphonoalamides (36), a new class of phosphonates that contains a phosphoalanine moiety and demonstrates antibacterial activity.
4. Target directed genome mining
Both the bioactive feature targeting approach (Section 2) and the compound family strategy (Section 3) take a “chemistry first” approach to genome mining, where a bioactive chemical feature or family is known prior to any genome mining attempts. However, researchers can also take a “gene first” approach to genome mining to prioritize specific BGCs over others for further investigation. In this strategy, there is no known compound guiding the genome mining. Instead BGCs are prioritized from sequence data alone that portend function or bioactivity. Recent prioritization methods have relied on the fact that bioactive molecule producing organisms must be resistant to their own products to avoid inadvertent suicide.159,160 Microorganisms typically utilize three main strategies to avoid self-toxicity: (1) product detoxification, (2) binding and removal of the product via transports and (3) target duplication or modification, where an organism encodes a duplicated but resistant copy of the target gene. In each case, genes encoding these resistance strategies are often colocalized in the cognate BGC. The third mechanism described above has led to a new strategy for genome mining that enables prioritization of BGCs with potential bioactivity. By searching for duplicated housekeeping genes located near BGCs, it is possible to identify BGCs with predicted bioactivity towards that target (Fig. 3). This approach is therefore chemistry independent, as the BGC is not identified for any specific chemical feature, but rather by the presence of a colocalized duplicated housekeeping gene. This method to prioritize BGCs with potential bioactivity has been termed “target-directed genome mining” as well as “resistance-gene genome mining”. In this section we will discuss four examples of target-directed genome mining that led to the directed discovery of natural products with anticipated bioactivity.
Fig. 3. By mining for the presence of duplicate housekeeping genes, it is possible to discover natural products and their biological target simultaneously.

4.1. Thiotetronic acid
In 2015, the Moore lab successfully used a target-directed genome mining approach with the express purpose of natural product discovery for the first time.161 They proposed that by identifying gene clusters that also contained a duplicated copy of a housekeeping gene that serves as a mechanism of self-resistance, they could prioritize bioactive BGCs and discover natural products with bioactivity towards that particular target.162,163 By querying the genomes of 86 Salinispora strains, the Moore group was able to identify 912 orthologous groups (OGs) that were both duplicated copies of core housekeeping genes and were associated with previously identified BGCs. They were particularly interested in BGCs that contained a duplicated copy of proteins in the fatty acid synthase (FASII) complex, as this is an attractive drug target due to its lack of similarity to fatty acid synthesis in mammals, which would result in ideal selectivity for a potential antibiotic.164–166 One gene (tlmE) encoding a FabB/F homolog was promising due to its high sequence similarity to the previously characterized self-resistance proteins PtmP3 and PtnP3 found in other BGCs that produce FASII inhibitors.167 That suggested that this gene was indeed a resistance gene, and not involved in biosynthesis.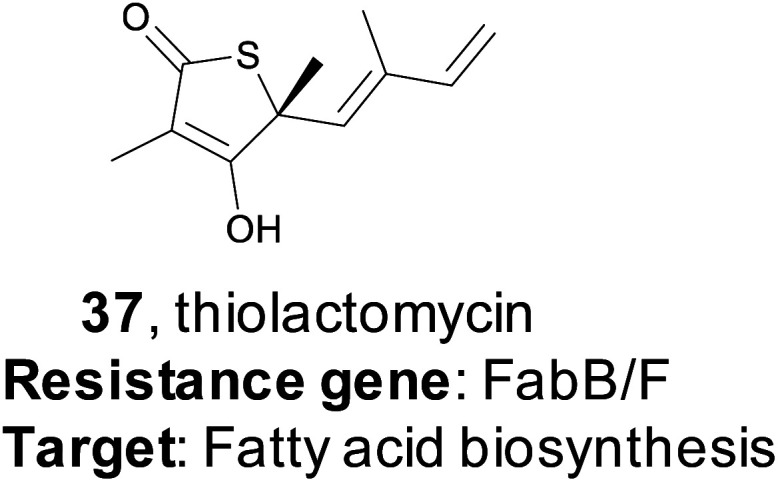
The putative FASII resistance gene was found co-localized within a hybrid ∼22 kb PKS/NRPS BGC that was captured by transformation-associated recombination (TAR) cloning in yeast and expressed in a Streptomyces heterologous host. This BGC produced four natural products that share a rare thiotetronic acid moiety, including the previously characterized antibiotic thiolactomycin (TLM, 37).168,169 This discovery ultimately allowed the authors to use this resistance gene (tlmE) as a genome mining hook to discover other, related, natural products from Streptomyces. A second gene cluster (ttm) contained two copies of a tlmE homolog, and subsequent TAR cloning and heterologous expression resulted in the production of a new series of TLM analogs. This work utilized a systematic approach to identify BGCs that might produce bioactive natural products based on the presence of a duplicated housekeeping gene found within the cluster that confers resistance to the BGC product. This target-directed genome mining strategy allows for the discovery of BGCs without prior knowledge of the molecules produced, and it has since been used extensively to prioritize BGCs that potentially produce bioactive compounds. Finally, this method is now the basis of the ARTs (Antibiotic Resistant Target Seeker) program, which allows for efficient in silico genome mining based on putative bioactivity.170
4.2. Pyxidicyclines
Myxobacteria are ubiquitous but incredibly interesting soil dwelling bacteria with large genomes and complex life cycles, including the tendency to form multicellular fruiting bodies.171 These organisms have long been overlooked as sources of natural products, but genomic analysis suggests they are a prolific source for new molecules.172 The Müller group utilized a target-directed approach to mine the genomes of 93 myxobacterial strains to find novel topoisomerase inhibitors.173 Topoisomerase inhibitors are used as antibiotics as well as chemotherapeutics, depending on the specificity of the inhibitor. The Müller group utilized CysO, a topoisomerase-targeting pentapeptide repeat protein responsible for self-resistance against the gyrase inhibitor cystobactamid, as their genetic search hook.174 This pentapeptide repeat protein mimics the structure of DNA, thereby protecting DNA gyrase from inhibitors like cystobactamid. Their search revealed 31 candidate sequences, eight of which were located near uncharacterized BGCs. They became particularly interested in a candidate gene, which they named pcyT, located next to a BGC that putatively encoded a type II PKS. Type II PKS BGCs are rare and uncharacterized in myxobacteria, making this cluster an interesting target for further investigation. Because the gene cluster was silent in the wild type organism, the authors pursued an activation via promoter exchange strategy. By replacing two native promoters in the BGC, the authors were ultimately able to isolate two novel type II PKS-derived nitrogen-containing tetracene quinones which they named pyxidicycline A and B (38 and 39). The pyxidicyclines exhibited strong cytotoxicity with activity in the nanomolar range, which prompted the authors to pursue a heterologous expression strategy to facilitate investigation of this BGC. The Müller lab was thus able to use a target directed approach to identify and prioritize a BGC predicted to encode a topoisomerase inhibitor, isolate two chemically novel products, and confirm they had the expected topoisomerase inhibitory activity.
4.3. Fellutamide B
While the resistance gene genome mining strategy was initially utilized in bacteria, it has also been successfully applied to fungi as well. A 2016 study from the Wang and Oakley groups applied a target-directed genome mining approach to fungi by examining uncharacterized BGCs in Aspergillus nidulans.175 They identified one BGC of interest containing a gene (inpE) that encodes a putative proteasome subunit. This suggested that the gene cluster might encode a proteasome inhibitor, which is of particular interest due to the clinical success of proteasome inhibitors such as the marine natural product salinosporamide A.176 However, this cluster was silent in the native organism, and no small molecule products had been identified. The authors utilized a serial promoter replacement strategy to activate this BGC in the native producing organism, replacing every native promoter in the cluster with the regulatable alcA promoter. Ultimately, this led to the production of the tripeptide fellutamide B (40), a potent proteasome inhibitor that had been previously isolated from fungi,177 although no gene cluster had been described. Interestingly, the authors also found that the resistance gene, inpE, was constitutively expressed and not coregulated with the other genes of the inp cluster, which indicates that the fellutamide-resistant proteasome subunit is always expressed. This work shows that target-directed genome mining is useful for the discovery, or rediscovery, of natural products from eukaryotic organisms as well.
4.4. Aspterric acid
Resistance gene-based genome mining is valuable not only because it can efficiently link a natural product to a target, but it also provides a mechanism of action. The utility of this approach was recently demonstrated by the Tang group.178 Their study specifically targeted dihydroxyacid dehydratase (DHAD), an enzyme in the branched chain amino acid (BCAA) biosynthetic pathway. No natural product inhibitors of DHAD were known at that time, but DHAD is a particularly compelling potential target for an herbicide as the BCAA pathway is essential for plants but not found in animals. DHAD, despite being highly conserved, has never been targeted by an herbicide.179 The authors proposed that a fungal gene cluster which encodes a DHAD inhibitor may contain an additional copy of DHAD that provides a mechanism of self-resistance, as fungi do synthesize BCAAs de novo. Via genome mining, the authors identified three genes responsible for the biosynthesis of the tricyclic terpenoid aspterric acid (41) found co-clustered with a duplicated copy of DHAD, named astD. Although aspterric acid was a previously isolated molecule, until this work the molecular target of aspterric acid remained unknown, as did its biosynthesis.180
Additional structural biology work of the wild type fungal DHAD as well as the duplicated copy astD allowed the researchers to understand the mechanism of astD resistance to aspterric acid on a molecular level.178 This understanding enabled their development of a trans gene system, where astD was deployed in Arabidopsis thaliana to provide resistance to aspterric acid, which could then be used as an effective and selective herbicide, in the vein of the RoundUp/Roundup Ready glyphosate system.181 Although aspterric acid is a previously known compound, this paper is an excellent example of the ability to repurpose known compounds by uncovering their mechanism of action through a target-directed genome mining approach.
5. (Bio)synthetic production of genome mined natural products
With the abundance of available bioinformatic tools, it has become relatively straightforward to identify a new putative natural product gene cluster in silico. The challenge then becomes obtaining the producing organism, growing it under conditions that permit production of the compound, and isolating enough of the natural product for characterization and bioactivity testing. If interesting bioactivity is observed, the compound must then be produced on scale using chemical synthesis, fermentation, or a mixture of the two approaches. Recent studies that we discuss here have demonstrated that this workflow can be avoided all together. Instead of growing the producing organism to isolate the target compound, the natural product is bioinformatically predicted and then directly produced, either by chemical synthesis or with promiscuous biosynthetic enzymes (Fig. 4). This approach avoids the issues of production, isolation, and scalability. The caveat to this methodology is that there needs to be sufficient understanding of the biosynthetic enzymes to predict a product. Moreover, the new compound may not directly match the authentic natural product but should at least closely resemble it. Here we describe examples that demonstrate the utility of this (bio)synthetic production approach to discover potent antibacterial agents from mined gene clusters.
Fig. 4. Bioinformatically identified natural products can be produced either synthetically or biosynthetically to generate new molecules that mimic the authentic compound.

5.1. Antibacterial syn-BNPs from the human microbiome
The human gut microbiome is home to a vast array of diverse bacterial species that produce an extensive collection of small molecules. Some of these compounds appear to directly interact with our bodies and have important consequences for our health. Others may be antibiotics that help provide a growth advantage against the competitive human gut environment. Studying these small molecules can be challenging because many of the bacteria are obligate anaerobes or difficult to cultivate. However, the extensive analysis of sequencing data from the human microbiome demonstrates that numerous biosynthetic gene clusters do exist and could provide a wealth of useful compounds.182 In 2016, the Brady lab described their development of the synthetic-bioinformatic natural products (syn-BNPs) approach to address this problem.183 They decided to focus on the NRPS class of natural products for several reasons. First, the bioinformatic prediction of NRPS clusters and adenylation module specificity is relatively mature and accurate.184,185 This allows for successful in silico prediction of at least a close approximation of the authentic natural product. Second, the predicted molecules can be rapidly produced by solid phase peptide synthesis or purchased at a low cost. Therefore, the Brady lab mined genomes from both the Human Microbiome Project and Human Oral Microbiome Database for the presence of NRPSs.183 They culled the mined NRPSs to only include those that produced products at least five amino acids in size. These larger products were thought to be less heavily modified and therefore more amenable to solid phase synthesis. In total, 25 of the 30 targeted peptides were successfully synthesized. After screening against a panel of bacterial strains, two syn-BNPs possessed promising antibacterial activity and were named humimycin A and B (42 and 43). Critically, these two related peptides were not detected in the host organisms, demonstrating the utility of this approach is accessing natural products without relying on the host organism.
5.2. Design of cyclic syn-BNPs
Initial studies on syn-BNPs focused on synthesizing and testing only linear NRPS derived peptides. However, the majority of NRPS natural products are cyclized in some way. Therefore, the Brady lab expanded upon their initial work with two studies that examine cyclic syn-BNPs. In the first study, the genomes of human-associated bacteria were again mined for the presence of large NRPS gene clusters to generate a list of 25 candidate peptides.186 Bioinformatic analysis of NRPS domains can predict the peptide sequence of the resulting natural product with a relatively high degree of certainty. However, current software programs are unable to accurately predict the mode of cyclization. Multiple different moieties can serve as the nucleophile in the offloading process, such as the N-terminal amine or a Ser/Thr sidechain.187 To account for this ambiguity, each peptide was synthesized with up to three cyclic scaffolds to generate 72 syn-BNPs. These compounds were used in assays targeting both Gram-positive and Gram-negative bacteria, fungi, and HeLa cells. While no compounds were found to be active in the antibacterial or antifungal assays, five of the cyclic syn-BNPs elicited a response in the HeLa cell MTT metabolic activity assay.
In a second study, the Brady lab completed a larger scale analysis of cyclic syn-BNPs.188 Analysis of 3000 bacterial genomes led to 96 NRPS natural product predictions. Instead of synthesizing every possible cyclic scaffold for the 96 peptide targets, a series of rules was created based on examining authentic NRPS natural products. Of 171 designed peptides, 157 were successfully synthesized and tested in a semi-pure form for antibiotic activity against a panel of bacteria that included the ESKAPE pathogens. After initial testing and subsequent validation, nine of the cyclic syn-BNPs demonstrated promising activity. Further analysis indicated that only two of the nine compounds possessed cytotoxic activity, indicating that the majority are specifically bacterial antibiotics. Detailed mode of action studies revealed diverse mechanisms of bioactivity including cell lysis, inhibition of cell wall biosynthesis, cell membrane depolarization, and dysregulation of the ClpP protease. Amongst the compounds, mucilasyn (44) was particularly promising due to an unidentified mode of action, low rate of resistance, and efficacy against Acinetobacter baumannii.
A recent study by the Parkinson lab used a related strategy to synthesize a library of predicted natural products.189 By focusing on NRPs that are cyclized by penicillin binding protein-like cyclases, the authors could better hypothesize the authentic cyclic scaffold and were able to ultimately identify 14 cyclic peptides with antibiotic activity out of a library of 51.
5.3. Lanthipeptides
RiPP natural products are particularly intriguing from a genome mining perspective because the substrates for the biosynthetic pathways (precursors peptides) are directly encoded for in the gene cluster. Several published software programs have taken advantage of this and can rapidly discover thousands of putative precursor peptides in silico.190 However, unlike the NRPS derived syn-BNPs, total synthesis of most RiPPs is incredibly challenging and cannot be completed in an automated, high-throughput manner. Instead, researchers have focused on using the inherent promiscuity of many RiPP biosynthetic enzymes to produce new RiPP compounds. Several studies have demonstrated this approach can work both in a random high throughput manner191,192 or targeted approach using genome mined precursor peptides193,194 to generate new bioactive lanthipeptide analogs.
Lanthipeptide natural products are defined by the presence of a (methyl)lanthionine bridge between a cysteine residue and a dehydrated serine or threonine.195 They also typically contain additional dehydroalanine or dehydrobutyrine residues derived from serine or threonine respectively. To explore the activity of bioinformatically identified lanthipeptides, the Kuipers lab used a heterologous production method to evaluate 54 putative precursor peptides.193 They employed an E. coli strain that was first transformed with a plasmid containing the core class I lanthipeptide biosynthetic genes (dehydratase, cyclase, and transporter). They added a second plasmid that contained a gene for one of the mined precursor peptides. The 54 target precursor peptides were chosen based on several features, such as the ability to predict the location of the leader cleavage site, and represented diverse examples. As with the syn-BNPs, these heterologously produced lanthipeptides will not necessarily be structurally identical to the authentic natural product but may be similar enough to still be bioactive. Of the 54 precursor peptides, 31 could be detected analytically. Of those, 18 appeared to be good substrates for the biosynthetic enzymes and were heavily modified. Bioactivity testing revealed that five were active against a panel of Gram-positive pathogens. This represents a nearly 10% success rate of the tested precursor peptide to bioactive molecule pipeline. MS/MS based structural analysis of one of the newly produced lanthipeptides, flavucin (45), demonstrated that (methyl)lanthionine bridges were formed and matched the predicted pattern observed in known lanthipeptides. Efforts to isolate the authentic flavucin from its native host, Corynebacterium lipophiloflavum DSM 44219, were unsuccessful as no production was observed. This again highlights the value of a (bio)synthetic production approach to study genome mined natural products because the host organism often does not produce the desired compound under laboratory conditions.
6. Conclusions and outlook
The 2020s have ushered in an era of biological revolutions, largely heralded by advancements in so-called ‘omic’ technologies. The convergence of these biological and technological advances has resulted in a sharp drop in the cost of sequencing DNA; in less than 20 years the cost of sequencing the human genome has plummeted, from millions of dollars in 2003 to just around $1000 today.196 Sequencing a single bacterial genome costs just a fraction of that. Because of this unprecedented access to genomic data, genome mining has become integral to the field of natural products12 and birthed a renaissance in natural product discovery.197
This review focused on four approaches taken by researchers for genome mining large genomic datasets with a particular focus on the discovery of bioactive compounds. Two of these strategies, bioactive feature mining and compound family mining, we categorize as “chemistry first” approaches. Both strategies require previously isolated chemistry to have exhibited interesting bioactivity, which allows known enzymatic reactions to be exploited for mining large databases for similar BGCs and their products, under the assumption that related compounds are likely to be bioactive as well. The other two strategies, target-directed genome mining and the (bio)synthetic production approach to genome mining natural products, we classify as “gene first” methods. These strategies rely on sequence data alone to either help prioritize a BGC based on the presence of a putative biological target, or to directly predict a chemical structure ultimately made via chemical or enzymatic synthesis.
By prioritizing bioactivity, these four strategies serve as the 21st century genome-oriented answer to traditional bioactivity-guided isolation strategies. These examples also make clear the continued importance of collaboration between the field of natural product discovery and other research disciplines. Target-directed genome mining will only improve as new protein targets for drug discovery are identified by cell biologists. Bioactive feature targeting relies on the identification of these specific chemical moieties/pharmacophores, which speaks to the continued importance of mechanism of action and structure–activity relationship studies of natural products to explicitly determine what chemical feature endows a molecule with a specific bioactivity. As Table 1 indicates, there is room for bioinformaticians to introduce pipelines to mine for specific pharmacophores, much like AdenylPred did for β-lactone mining.26 Similar programs could be developed for other bioactive features, such as epoxyketones or β-lactams. While programs like antiSMASH can indicate gene clusters that encode enzymes responsible for the installation of these features, the program is not oriented towards this purpose like AdenylPred is for β-lactones. Table 1 is designed to help the reader understand where these genome mining gaps may exist, and where feature-specific pipelines could be developed. To complement this, dedicated genome mining algorithms could be specifically designed to rapidly search through large genomic data sets and expand known bioactive natural product (sub)families. This approach has gained significant attention in the RiPP community with several family specific genome mining programs available.189 Together, these types of specific tools could aid discovery pipelines and help identify new bioactive natural products.
Additional approaches could further exploit the fact that genes responsible for the installation of specific chemical features can sometimes be found in so-called “sub-clusters” within a larger BGC (Fig. 5).198 These smaller biosynthetic units are interesting from an evolutionary perspective as they are thought to consist of co-evolving genes required for construction of a specific biosynthetic feature and they function almost like an independent unit. These sub-clusters may be found in otherwise unrelated BGCs, resulting in the same chemical feature appearing across compound classes. There is a functional aspect to this sub-clustering phenomenon as well; from a discovery perspective sub-clusters allow MultiGeneBlast199 or GNNs106 to identify co-localization of genes and guide identification in new genomic contexts. Beyond discovery, sub-clustering of genes often results in coordinated expression which can provide gene cassettes for experimentation and isolation of new molecules in the lab.
Fig. 5. Sub-clustering of genes is responsible for the installation of bioactive chemical features and class-defining chemical features. Gene clusters not shown to scale. (A) Sub-clustering of the DUF and cysteine lyase domains (red) is responsible for the installation of reactive 1,3-dioxo-1,2-dithiolane moiety and is seen in leinamycin (lnm) BGC, guangnanmycin (gnm) BGC, and weishanmycin (wsm) BGC (Section 2.1.4). (B) Sub-clustering of two genes grbD and grbE (blue) that are responsible for the installation of the diazeniumdiolate moiety is evident in the gramibactin BGC (grb) as well as the BGCs responsible for production of megapolibactin from a symbiotic bacterial strain and gladiobactin/plantaribactin from a pathogenic bacterial strain (Section 2.2.1). C. Genes required for installation of the cinnamoyl-moiety (green) are found sub-clustered in the BGCs for WS9326 (cal), skyllamycin (sky), and kitacinnamycin (kcn) (Section 3.3).

Finally, genome mining rests on detailed mechanistic enzymology that identifies and characterizes the enzymes responsible for biosynthetic installation of specific chemical features. The leinamycin story perfectly exemplifies this continued need for collaboration; it took 30 years of cell biology experiments, mechanism of action studies, bioinformatics advances, and mechanistic enzymology to develop the knowledge and tools for researchers to identify related leinamycin analogs via a genome mining approach.53 The interdisciplinary nature of natural products research is a real strength of the field, and it is precisely this position at the interface of chemistry, biology, informatics, and medicine that will help expand the role of genome mining in natural product discovery in the years to come.
7. Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We thank Ajalaye Sykes for her work on the table of contents image. This work was partially supported by the US National Institutes of Health (F31-HD101307 to K. D. B and R01-GM085770 to B. S. M.), University of North Carolina at Greensboro (research start-up funds to J. R. C.), and the National Center for Complementary and Integrative Health, a component of the US National Institutes of Health (T32AT008938 to K. S. B.).
Biographies
Biography
Katherine D. Bauman.

Katherine D. Bauman is an NIH NRSA F31 predoctoral fellow in the lab of Bradley S. Moore at the Scripps Institution of Oceanography, UC San Diego. She is pursuing a PhD in Marine Chemical Biology where her research focuses on the biosynthesis of natural products. Prior to her graduate studies, Katherine studied chemistry at Middlebury College where she studied natural products with Professor Lesley-Ann Giddings.
Biography
Keelie S. Butler.

Keelie S. Butler is a NIH NCCIH T32 trainee in the lab of Jonathan R. Chekan at the University of North Carolina at Greensboro. She is pursuing a PhD in Chemistry and Biochemistry, with a research focus on the biosynthesis of natural products. Prior to her graduate studies, Keelie studied chemistry and biochemistry at Campbell University, where she studied biosurfactant production in marine microbes with Dr Michelle S. Thomas.
Biography
Bradley S. Moore.

Bradley Moore is a Distinguished Professor of Marine Chemical Biology and Pharmaceutical Chemistry at UC San Diego. His research focuses on the molecular and genomic basis of natural product biosynthesis and the application of new genetic tools and biocatalysts to produce bioactive small molecules. He is the former chair of the editorial board of Natural Product Reports and the recipient of the Natural Product Chemistry Award from the Royal Society of Chemistry in 2018 and the Ernest Guenther Award in the Chemistry of Natural Products from the American Chemical Society in 2021.
Biography
Jonathan R. Chekan.

Jonathan R. Chekan is an Assistant Professor of Chemistry and Biochemistry at the University of North Carolina at Greensboro. He received his PhD in 2016 from the University of Illinois Urbana-Champaign under the supervision of Satish K. Nair. As the Simons Foundation Post-Doctoral Fellow of the Life Science Research Foundation in the laboratory of Bradley S. Moore, he investigated the biosynthesis of neurotoxic marine natural products. In 2020, he joined University of North Carolina at Greensboro where his research group focuses on the bioinformatic guided discovery of new natural products and characterization of their biosynthetic pathways.
9. References
- Demain A. L. Fang A. Adv. Biochem. Eng./Biotechnol. 2000;132:1–39. doi: 10.1007/3-540-44964-7_1. [DOI] [PubMed] [Google Scholar]
- Berdi J. J. Antibiot. 2005;58:1–26. doi: 10.1038/ja.2005.1. [DOI] [PubMed] [Google Scholar]
- Newman D. J. Cragg G. M. J. Nat. Prod. 2020;83:770–803. doi: 10.1021/acs.jnatprod.9b01285. [DOI] [PubMed] [Google Scholar]
- Atanasov A. G. Zotchev S. B. Dirsch V. M. Orhan I. E. Banach M. Rollinger J. M. Barreca D. Weckwerth W. Bauer R. Bayer E. A. Majeed M. Bishayee A. Bochkov V. Bonn G. K. Braidy N. Bucar F. Cifuentes A. D'Onofrio G. Bodkin M. Diederich M. Dinkova-Kostova A. T. Efferth T. El Bairi K. Arkells N. Fan T. P. Fiebich B. L. Freissmuth M. Georgiev M. I. Gibbons S. Godfrey K. M. Gruber C. W. Heer J. Huber L. A. Ibanez E. Kijjoa A. Kiss A. K. Lu A. Macias F. A. Miller M. J. S. Mocan A. Müller R. Nicoletti F. Perry G. Pittalà V. Rastrelli L. Ristow M. Russo G. L. Silva A. S. Schuster D. Sheridan H. Skalicka-Woźniak K. Skaltsounis L. Sobarzo-Sánchez E. Bredt D. S. Stuppner H. Sureda A. Tzvetkov N. T. Vacca R. A. Aggarwal B. B. Battino M. Giampieri F. Wink M. Wolfender J. L. Xiao J. Yeung A. W. K. Lizard G. Popp M. A. Heinrich M. Berindan-Neagoe I. Stadler M. Daglia M. Verpoorte R. Supuran C. T. Nat. Rev. Drug Discovery. 2021;20:200–216. doi: 10.1038/s41573-020-00114-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda H. Ishikawa J. Hanamoto A. Shinose M. Kikuchi H. Shiba T. Sakaki Y. Hattori M. Omura S. Nat. Biotechnol. 2003;21:526–531. doi: 10.1038/nbt820. [DOI] [PubMed] [Google Scholar]
- Bentley S. D. Chater K. F. Cerdeño-Tárraga A. M. Challis G. L. Thomson N. R. James K. D. Harris D. E. Quail M. A. Kieser H. Harper D. Bateman A. Brown S. Chandra G. Chen C. W. Collins M. Cronin A. Fraser A. Goble A. Hidalgo J. Hornsby T. Howarth S. Huang C. H. Kieser T. Larke L. Murphy L. Oliver K. O'Neil S. Rabbinowitsch E. Rajandream M. A. Rutherford K. Rutter S. Seeger K. Saunders D. Sharp S. Squares R. Squares S. Taylor K. Warren T. Wietzorrek A. Woodward J. Barrell B. G. Parkhill J. Hopwood D. A. Nature. 2002;417:141–147. doi: 10.1038/417141a. [DOI] [PubMed] [Google Scholar]
- Nett M. Ikeda H. Moore B. S. Nat. Prod. Rep. 2009;26:1362–1384. doi: 10.1039/B817069J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemert N. Alanjary M. Weber T. Nat. Prod. Rep. 2016;33:988–1005. doi: 10.1039/C6NP00025H. [DOI] [PubMed] [Google Scholar]
- Blin K. Shaw S. Kloosterman A. M. Charlop-Powers Z. van Wezel G. P. Medema M. H. Weber T. Nucleic Acids Res. 2021;49:W29–W35. doi: 10.1093/nar/gkab335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skinnider M. A. Johnston C. W. Gunabalasingam M. Merwin N. J. Kieliszek A. M. MacLellan R. J. Li H. Ranieri M. R. M. Webster A. L. H. Cao M. P. T. Pfeifle A. Spencer N. To Q. H. Wallace D. P. Dejong C. A. Magarvey N. A. Nat. Commun. 2020;11:6058. doi: 10.1038/s41467-020-19986-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherlach K. Hertweck C. Nat. Commun. 2021;12:3864. doi: 10.1038/s41467-021-24133-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema M. H. de Rond T. Moore B. S. Nat. Rev. Genet. 2021;22:553–571. doi: 10.1038/s41576-021-00363-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albarano L. Esposito R. Ruocco N. Costantini M. Mar. Drugs. 2020;18:199. doi: 10.3390/md18040199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenshole E. Herisse M. Michael M. Pidot S. J. Curr. Opin. Chem. Biol. 2021;60:47–54. doi: 10.1016/j.cbpa.2020.07.010. [DOI] [PubMed] [Google Scholar]
- Baltz R. H. J. Ind. Microbiol. Biotechnol. 2021:kuab044. doi: 10.1093/jimb/kuab044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlert J. Shepard E. Lomovskaya N. Zazopoulos E. Staffa A. Bachmann B. O. Huang K. Fonstein L. Czisny A. Whitwam R. E. Farnet C. M. Thorson J. S. Science. 2002;297:1173–1176. doi: 10.1126/science.1072105. [DOI] [PubMed] [Google Scholar]
- Liu W. Christenson S. D. Standage S. Shen B. Science. 2002;297:1170–1173. doi: 10.1126/science.1072110. [DOI] [PubMed] [Google Scholar]
- Yan X. Chen J. J. Adhikari A. Yang D. Crnovcic I. Wang N. Chang C. Y. Rader C. Shen B. Org. Lett. 2017;19:6192–6195. doi: 10.1021/acs.orglett.7b03120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X. Ge H. Huang T. Hindra Yang D. Teng Q. Crnovčić I. Li X. Rudolf J. D. Lohman J. R. Gansemans Y. Zhu X. Huang Y. Zhao L. X. Jiang Y. van Nieuwerburgh F. Rader C. Duan Y. Shen B. mBio. 2016;7:e02104–e02116. doi: 10.1128/mBio.02104-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann B. O. Li R. Townsend C. A. Proc. Natl. Acad. Sci. U. S. A. 1998;95:9082–9086. doi: 10.1073/pnas.95.16.9082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerratana B. Stapon A. Townsend C. A. Biochemistry. 2003;42:7836–7847. doi: 10.1021/bi034361d. [DOI] [PubMed] [Google Scholar]
- Weigel B. J. Burgett S. G. Chen V. J. Skatrud P. L. Frolik C. A. Queener S. W. Ingolia T. D. J. Bacteriol. 1988;170:3817–3826. doi: 10.1128/jb.170.9.3817-3826.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelli N. M. Long D. H. Townsend C. A. Nature. 2015;520:383–387. doi: 10.1038/nature14100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco G. Arch. Microbiol. 2012;194:549–555. doi: 10.1007/s00203-011-0781-y. [DOI] [PubMed] [Google Scholar]
- Christenson J. K. Richman J. E. Jensen M. R. Neufeld J. Y. Wilmot C. M. Wackett L. P. Biochemistry. 2017;56:348–351. doi: 10.1021/acs.biochem.6b01199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson S. L. Terlouw B. R. Smith M. D. Pidot S. J. Stinear T. P. Medema M. H. Wackett L. P. J. Biol. Chem. 2020;295:14826–14839. doi: 10.1074/jbc.RA120.013528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer J. E. Reck M. R. Prasad N. K. Wencewicz T. A. Nat. Chem. Biol. 2017;13:737–744. doi: 10.1038/nchembio.2374. [DOI] [PubMed] [Google Scholar]
- Feng K. N. Yang Y. L. Xu Y. X. Zhang Y. Feng T. Huang S. X. Liu J. K. Zeng Y. Angew. Chem., Int. Ed. 2020;59:7209–7213. doi: 10.1002/anie.202000710. [DOI] [PubMed] [Google Scholar]
- Eustáquio A. S. Nam S. J. Penn K. Lechner A. Wilson M. C. Fenical W. Jensen P. R. Moore B. S. ChemBioChem. 2011;12:61–64. doi: 10.1002/cbic.201000564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaurasiya N. D. Sangwan N. S. Sabir F. Misra L. Sangwan R. S. Plant Cell Rep. 2012;31:1889–1897. doi: 10.1007/s00299-012-1302-4. [DOI] [PubMed] [Google Scholar]
- Wu J. Zaleski T. J. Valenzano C. Khosla C. Cane D. E. J. Am. Chem. Soc. 2005;127:17393–17404. doi: 10.1021/ja055672+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J. J. Tang X. Huan T. Ross A. C. Moore B. S. Nat. Chem. Biol. 2020;16:42–49. doi: 10.1038/s41589-019-0385-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zabala D. Cartwright J. W. Roberts D. M. Law B. J. C. Song L. Samborskyy M. Leadlay P. F. Micklefield J. Challis G. L. J. Am. Chem. Soc. 2016;138:4342–4345. doi: 10.1021/jacs.6b01619. [DOI] [PubMed] [Google Scholar]
- Schorn M. Zettler J. Noel J. P. Dorrestein P. C. Moore B. S. Kaysser L. ACS Chem. Biol. 2014;9:301–309. doi: 10.1021/cb400699p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen J. G. Charlop-Powers Z. Smith A. G. Ternei M. A. Calle P. Y. Reddy B. V. B. Montiel D. Brady S. F. Proc. Natl. Acad. Sci. U. S. A. 2015;112:4221–4226. doi: 10.1073/pnas.1501124112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L. Shah S. Chung L. Carney J. Katz L. Khosla C. Julien B. Science. 2000;287:640–642. doi: 10.1126/science.287.5453.640. [DOI] [PubMed] [Google Scholar]
- Thibodeaux C. J. Chang W. C. Liu H. W. Chem. Rev. 2012;112:1681–1709. doi: 10.1021/cr200073d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gould S. J. Kirchmeier M. J. LaFever R. E. J. Am. Chem. Soc. 1996;118:7663–7666. doi: 10.1021/ja960696p. [DOI] [Google Scholar]
- Liu P. Murakami K. Seki T. He X. Yeung S. M. Kuzuyama T. Seto H. Liu H. J. Am. Chem. Soc. 2001;123:4619–4620. doi: 10.1021/ja004153y. [DOI] [PubMed] [Google Scholar]
- Kuzuyama T. Seki T. Kobayashi S. Hidaka T. Seto H. Biosci., Biotechnol., Biochem. 1999;63:2222–2224. doi: 10.1271/bbb.63.2222. [DOI] [PubMed] [Google Scholar]
- Ueoka R. Bortfeld-Miller M. Morinaka B. I. Vorholt J. A. Piel J. Angew. Chem., Int. Ed. 2018;57:977–981. doi: 10.1002/anie.201709056. [DOI] [PubMed] [Google Scholar]
- Liu X. Biswas S. Berg M. G. Antapli C. M. Xie F. Wang Q. Tang M. C. Tang G. L. Zhang L. Dreyfuss G. Cheng Y. Q. J. Nat. Prod. 2013;76:685–693. doi: 10.1021/np300913h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren D. Kim M. Wang S. A. Liu H. Angew. Chem., Int. Ed. 2021;60:17148–17154. doi: 10.1002/anie.202105667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M. Dander J. E. Sato C. Hung Y. S. Gao S. S. Tang M. C. Hang L. Winter J. M. Garg N. K. Watanabe K. Tang Y. J. Am. Chem. Soc. 2017;139:5317–5320. doi: 10.1021/jacs.7b02432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baunach M. Ding L. Willing K. Hertweck C. Angew. Chem., Int. Ed. 2015;54:13279–13283. doi: 10.1002/anie.201506541. [DOI] [PubMed] [Google Scholar]
- Wang G.-Q. Chen G.-D. Qin S.-Y. Hu D. Awakawa T. Li S.-Y. Lv J.-M. Wang C.-X. Yao X.-S. Abe I. Gao H. Nat. Commun. 2018;9:1838. doi: 10.1038/s41467-018-04298-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belknap K. C. Park C. J. Barth B. M. Andam C. P. Sci. Rep. 2020;10:2003. doi: 10.1038/s41598-020-58904-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B. Walsh C. T. Biochemistry. 2011;50:4615–4622. doi: 10.1021/bi200321c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharf D. H. Remme N. Heinekamp T. Hortschansky P. Brakhage A. A. Hertweck C. J. Am. Chem. Soc. 2010;132:10136–10141. doi: 10.1021/ja103262m. [DOI] [PubMed] [Google Scholar]
- Wang C. Wesener S. R. Zhang H. Cheng Y. Q. Chem. Biol. 2009;16:585–593. doi: 10.1016/j.chembiol.2009.05.005. [DOI] [PubMed] [Google Scholar]
- Ma M. Lohman J. R. Liu T. Shen B. Proc. Natl. Acad. Sci. U. S. A. 2015;112:10359–10364. doi: 10.1073/pnas.1508437112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H. Fan J. Zhang P. Hu Y. Liu X. Li S. M. Yin W. B. Chem. Sci. 2021;12:4132–4138. doi: 10.1039/D0SC06647H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan G. Xu Z. Guo Z. Hindra Ma M. Yang D. Zhou H. Gansemans Y. Zhu X. Huang Y. Zhao L.-X. X. Jiang Y. Cheng J. Van Nieuwerburgh F. Suh J.-W. W. Duan Y. Shen B. Proc. Natl. Acad. Sci. U. S. A. 2017;114:E11131–E11140. doi: 10.1073/pnas.1716245115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dose B. Niehs S. P. Scherlach K. Shahda S. Flórez L. V. Kaltenpoth M. Hertweck C. ChemBioChem. 2021;22:1920–1924. doi: 10.1002/cbic.202100089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. Ng T. Rui Z. Ad O. Zhang W. Angew. Chem., Int. Ed. 2014;53:136–139. doi: 10.1002/anie.201308069. [DOI] [PubMed] [Google Scholar]
- Liang Z. Nat. Prod. Rep. 2010;27:499. doi: 10.1039/B908165H. [DOI] [PubMed] [Google Scholar]
- Zazopoulos E. Huang K. Staffa A. Liu W. Bachmann B. O. Nonaka K. Ahlert J. Thorson J. S. Shen B. Farnet C. M. Nat. Biotechnol. 2003;21:187–190. doi: 10.1038/nbt784. [DOI] [PubMed] [Google Scholar]
- Shen B. Yan X. Huang T. Ge H. Yang D. Teng Q. Rudolf J. D. Lohman J. R. Bioorg. Med. Chem. Lett. 2015;25:9–15. doi: 10.1016/j.bmcl.2014.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudolf J. D. Yan X. Shen B. J. Ind. Microbiol. Biotechnol. 2016;43:261–276. doi: 10.1007/s10295-015-1671-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies J. Wang H. Taylor T. Warabi K. Huang X. H. Andersen R. J. Org. Lett. 2005;7:5233–5236. doi: 10.1021/ol052081f. [DOI] [PubMed] [Google Scholar]
- Gersch M. Kreuzer J. Sieber S. A. Nat. Prod. Rep. 2012;29:659–682. doi: 10.1039/C2NP20012K. [DOI] [PubMed] [Google Scholar]
- Robinson S. L. Christenson J. K. Wackett L. P. Nat. Prod. Rep. 2019;36:458–475. doi: 10.1039/C8NP00052B. [DOI] [PubMed] [Google Scholar]
- Mikami Y. Yazawa Y. Tanaka Y. Ritzau M. Gräfe U. Nat. Prod. Lett. 1999;13:277–284. doi: 10.1080/10575639908048798. [DOI] [Google Scholar]
- Zeeck A. Schröder K. Frobel K. Grote R. Thiericke R. J. Antibiot. 1987;40:1530–1540. doi: 10.7164/antibiotics.40.1530. [DOI] [PubMed] [Google Scholar]
- Hanada M. Sugawara K. Kaneta K. Toda S. Nishiyama Y. Tomita K. Yamamoto H. Konishi M. Oki T. J. Antibiot. 1992;45:1746–1752. doi: 10.7164/antibiotics.45.1746. [DOI] [PubMed] [Google Scholar]
- Sugawara K. Hatori M. Nishiyama Y. Tomita K. Kamei H. Konishi M. Oki T. J. Antibiot. 1990;43:8–18. doi: 10.7164/antibiotics.43.8. [DOI] [PubMed] [Google Scholar]
- Kuhn D. J. Chen Q. Voorhees P. M. Strader J. S. Shenk K. D. Sun C. M. Demo S. D. Bennett M. K. Van Leeuwen F. W. B. Chanan-Khan A. A. Orlowski R. Z. Blood. 2007;110:3281–3290. doi: 10.1182/blood-2007-01-065888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy B. V. B. Milshteyn A. Charlop-Powers Z. Brady S. F. Chem. Biol. 2014;21:1023–1033. doi: 10.1016/j.chembiol.2014.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. H. Arch. Pharmacal Res. 2009;32:299–315. doi: 10.1007/s12272-009-1300-4. [DOI] [PubMed] [Google Scholar]
- Yi M. C. Khosla C. Annu. Rev. Chem. Biomol. Eng. 2016;7:197–222. doi: 10.1146/annurev-chembioeng-080615-033553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kara M. Asano K. Kawamoto I. Nakano H. Takiouchi T. Katsumata S. Takahashi K. J. Antibiot. 1989;42:1768–1774. doi: 10.7164/antibiotics.42.1768. [DOI] [PubMed] [Google Scholar]
- Hara M. Takahashi I. Yoshida M. Asano K. Kawamoto I. Nakano H. Morimoto M. J. Antibiot. 1989;42:333–335. doi: 10.7164/antibiotics.42.333. [DOI] [PubMed] [Google Scholar]
- Huang S. X. Yun B. S. Ma M. Basu H. S. Church D. R. Ingenhorst G. Huang Y. Yang D. Lohman J. R. Tang G. L. Ju J. Liu T. Wilding G. Shen B. Proc. Natl. Acad. Sci. U. S. A. 2015;112:8278–8283. doi: 10.1073/pnas.1506761112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viswesh V. Gates K. Sun D. Chem. Res. Toxicol. 2010;23:99–107. doi: 10.1021/tx900301r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G.-L. Cheng Y.-Q. Shen B. Chem. Biol. 2004;11:33–45. doi: 10.1016/j.chembiol.2003.12.014. [DOI] [PubMed] [Google Scholar]
- Yang L. P. H. Keam S. J. Keating G. M. Drugs. 2007;67:2211–2230. doi: 10.2165/00003495-200767150-00007. [DOI] [PubMed] [Google Scholar]
- Zhanel G. G. Golden A. R. Zelenitsky S. Wiebe K. Lawrence C. K. Adam H. J. Idowu T. Domalaon R. Schweizer F. Zhanel M. A. Lagacé-Wiens P. R. S. Walkty A. J. Noreddin A. Lynch J. P. Karlowsky J. A. Drugs. 2019;79:271–289. doi: 10.1007/s40265-019-1055-2. [DOI] [PubMed] [Google Scholar]
- Negash K. H. Norris J. K. S. Hodgkinson J. T. Molecules. 2019;24:3314. doi: 10.3390/molecules24183314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hider R. C. Kong X. Nat. Prod. Rep. 2010;27:637–657. doi: 10.1039/B906679A. [DOI] [PubMed] [Google Scholar]
- Hermenau R. Ishida K. Gama S. Hoffmann B. Pfeifer-Leeg M. Plass W. Mohr J. F. Wichard T. Saluz H. P. Hertweck C. Nat. Chem. Biol. 2018;14:841–843. doi: 10.1038/s41589-018-0101-9. [DOI] [PubMed] [Google Scholar]
- Hermenau R. Mehl J. L. Ishida K. Dose B. Pidot S. J. Stinear T. P. Hertweck C. Angew. Chem., Int. Ed. 2019;58:13024–13029. doi: 10.1002/anie.201906326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott D. E. Bayly A. R. Abell C. Skidmore J. Nat. Rev. Drug Discovery. 2016;15:533–550. doi: 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- Mabonga L. Kappo A. P. Biophys. Rev. 2019;11:559–581. doi: 10.1007/s12551-019-00570-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo Y. J. Kim H. Park S. R. Yoon Y. J. J. Ind. Microbiol. Biotechnol. 2017;44:537–553. doi: 10.1007/s10295-016-1834-7. [DOI] [PubMed] [Google Scholar]
- Shigdel U. K. Lee S. J. Sowa M. E. Bowman B. R. Robison K. Zhou M. Pua K. H. Stiles D. T. Blodgett J. A. V. Udwary D. W. Rajczewski A. T. Mann A. S. Mostafavi S. Hardy T. Arya S. Weng Z. Stewart M. Kenyon K. Morgenstern J. P. Pan E. Gray D. C. Pollock R. M. Fry A. M. Klausner R. D. Townson S. A. Verdine G. L. Proc. Natl. Acad. Sci. U. S. A. 2020;117:17195–17203. doi: 10.1073/pnas.2006560117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gatto G. J. Boyne M. T. Kelleher N. L. Walsh C. T. J. Am. Chem. Soc. 2006;128:3838–3847. doi: 10.1021/ja0587603. [DOI] [PubMed] [Google Scholar]
- Gordon D. E. Jang G. M. Bouhaddou M. Xu J. Obernier K. White K. M. O'Meara M. J. Rezelj V. V. Guo J. Z. Swaney D. L. Tummino T. A. Hüttenhain R. Kaake R. M. Richards A. L. Tutuncuoglu B. Foussard H. Batra J. Haas K. Modak M. Kim M. Haas P. Polacco B. J. Braberg H. Fabius J. M. Eckhardt M. Soucheray M. Bennett M. J. Cakir M. McGregor M. J. Li Q. Meyer B. Roesch F. Vallet T. Mac Kain A. Miorin L. Moreno E. Naing Z. Z. C. Zhou Y. Peng S. Shi Y. Zhang Z. Shen W. Kirby I. T. Melnyk J. E. Chorba J. S. Lou K. Dai S. A. Barrio-Hernandez I. Memon D. Hernandez-Armenta C. Lyu J. Mathy C. J. P. Perica T. Pilla K. B. Ganesan S. J. Saltzberg D. J. Rakesh R. Liu X. Rosenthal S. B. Calviello L. Venkataramanan S. Liboy-Lugo J. Lin Y. Huang X. P. Liu Y. F. Wankowicz S. A. Bohn M. Safari M. Ugur F. S. Koh C. Savar N. S. Tran Q. D. Shengjuler D. Fletcher S. J. O'Neal M. C. Cai Y. Chang J. C. J. Broadhurst D. J. Klippsten S. Sharp P. P. Wenzell N. A. Kuzuoglu-Ozturk D. Wang H. Y. Trenker R. Young J. M. Cavero D. A. Hiatt J. Roth T. L. Rathore U. Subramanian A. Noack J. Hubert M. Stroud R. M. Frankel A. D. Rosenberg O. S. Verba K. A. Agard D. A. Ott M. Emerman M. Jura N. von Zastrow M. Verdin E. Ashworth A. Schwartz O. d'Enfert C. Mukherjee S. Jacobson M. Malik H. S. Fujimori D. G. Ideker T. Craik C. S. Floor S. N. Fraser J. S. Gross J. D. Sali A. Roth B. L. Ruggero D. Taunton J. Kortemme T. Beltrao P. Vignuzzi M. García-Sastre A. Shokat K. M. Shoichet B. K. Krogan N. J. Nature. 2020;583:459–468. doi: 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travin D. Y. Severinov K. Dubiley S. RSC Chem. Biol. 2021;2:468–485. doi: 10.1039/D0CB00208A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guijarro J. I. Gonzalez-Pastor J. E. Baleux F. San Millan J. L. Castilla M. A. Rico M. Moreno F. Delepierre M. J. Biol. Chem. 1995;270:23520–23532. doi: 10.1074/jbc.270.40.23520. [DOI] [PubMed] [Google Scholar]
- Novikova M. Metlitskaya A. Datsenko K. Kazakov T. Kazakov A. Wanner B. Severinov K. J. Bacteriol. 2007;189:8361–8365. doi: 10.1128/JB.01028-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metlitskaya A. Kazakov T. Kommer A. Pavlova O. Praetorius-Ibba M. Ibba M. Krasheninnikov I. Kolb V. Khmel I. Severinov K. J. Biol. Chem. 2006;281:18033–18042. doi: 10.1074/jbc.M513174200. [DOI] [PubMed] [Google Scholar]
- Roush R. F. Nolan E. M. Löhr F. Walsh C. T. J. Am. Chem. Soc. 2008;130:3603–3609. doi: 10.1021/ja7101949. [DOI] [PubMed] [Google Scholar]
- Dong S. H. Kulikovsky A. Zukher I. Estrada P. Dubiley S. Severinov K. Nair S. K. Chem. Sci. 2019;10:2391–2395. doi: 10.1039/C8SC03173H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regni C. A. Roush R. F. Miller D. J. Nourse A. Walsh C. T. Schulman B. A. EMBO J. 2009;28:1953–1964. doi: 10.1038/emboj.2009.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severinov K. Semenova E. Kazakov A. Kazakov T. Gelfand M. S. Mol. Microbiol. 2007;65:1380–1394. doi: 10.1111/j.1365-2958.2007.05874.x. [DOI] [PubMed] [Google Scholar]
- Bantysh O. Serebryakova M. Makarova K. S. Dubiley S. Datsenko K. A. Severinov K. mBio. 2014;5:e01059. doi: 10.1128/mBio.01059-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serebryakova M. Tsibulskaya D. Mokina O. Kulikovsky A. Nautiyal M. Van Aerschot A. Severinov K. Dubiley S. J. Am. Chem. Soc. 2016;138:15690–15698. doi: 10.1021/jacs.6b09853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michael J. P. Nat. Prod. Rep. 2003;20:476–493. doi: 10.1039/B208140G. [DOI] [PubMed] [Google Scholar]
- Zhang J. Morris-Natschke S. L. Ma D. Shang X. F. Yang C. J. Liu Y. Q. Lee K. H. Med. Res. Rev. 2021;41:928–960. doi: 10.1002/med.21747. [DOI] [PubMed] [Google Scholar]
- Chemler S. Curr. Bioact. Compd. 2009;5:2–19. doi: 10.2174/157340709787580928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han W. B. Zhang A. H. Deng X. Z. Lei X. Tan R. X. Org. Lett. 2016;18:1816–1819. doi: 10.1021/acs.orglett.6b00549. [DOI] [PubMed] [Google Scholar]
- Han W. B. Lu Y. H. Zhang A. H. Zhang G. F. Mei Y. N. Jiang N. Lei X. Song Y. C. Ng S. W. Tan R. X. Org. Lett. 2014;16:5366–5369. doi: 10.1021/ol502572g. [DOI] [PubMed] [Google Scholar]
- Dai G. Z. Han W. B. Mei Y. N. Xu K. Jiao R. H. Ge H. M. Tan R. X. Proc. Natl. Acad. Sci. U. S. A. 2020;117:1174–1180. doi: 10.1073/pnas.1914777117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dias D. A. Urban S. Roessner U. Metabolites. 2012;2:303–336. doi: 10.3390/metabo2020303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlt J. A. Bouvier J. T. Davidson D. B. Imker H. J. Sadkhin B. Slater D. R. Whalen K. L. Biochim. Biophys. Acta. 2015;1854:1019–1037. doi: 10.1016/j.bbapap.2015.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zallot R. Oberg N. O. Gerlt J. A. Curr. Opin. Chem. Biol. 2018;47:77–85. doi: 10.1016/j.cbpa.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler M. S. Hansford K. A. Blaskovich M. A. T. Halai R. Cooper M. A. J. Antibiot. 2014;67:631–644. doi: 10.1038/ja.2014.111. [DOI] [PubMed] [Google Scholar]
- Yim G. Thaker M. N. Koteva K. Wright G. J. Antibiot. 2014;67:31–41. doi: 10.1038/ja.2013.117. [DOI] [PubMed] [Google Scholar]
- Culp E. J. Waglechner N. Wang W. Fiebig-Comyn A. A. Hsu Y.-P. Koteva K. Sychantha D. Coombes B. K. Van Nieuwenhze M. S. Brun Y. V. Wright G. D. Nature. 2020;578:582–587. doi: 10.1038/s41586-020-1990-9. [DOI] [PubMed] [Google Scholar]
- Waglechner N. McArthur A. G. Wright G. D. Nat. Microbiol. 2019;4:1862–1871. doi: 10.1038/s41564-019-0531-5. [DOI] [PubMed] [Google Scholar]
- Chiu H. T. Hubbard B. K. Shah A. N. Eide J. Fredenburg R. A. Walsh C. T. Khosla C. Proc. Natl. Acad. Sci. U. S. A. 2001;98:8548–8553. doi: 10.1073/pnas.151246498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T. J. Blackman S. A. Foster S. J. Microbiology. 2000;146:249–262. doi: 10.1099/00221287-146-2-249. [DOI] [PubMed] [Google Scholar]
- Hancock R. E. W. Lehrer R. Trends Biotechnol. 1998;16:82–88. doi: 10.1016/S0167-7799(97)01156-6. [DOI] [PubMed] [Google Scholar]
- Li Y. X. Zhong Z. Zhang W. P. Qian P. Y. Nat. Commun. 2018;9:2–10. doi: 10.1038/s41467-017-02294-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cochrane S. A. Vederas J. C. Med. Res. Rev. 2016;36:4–31. doi: 10.1002/med.21321. [DOI] [PubMed] [Google Scholar]
- Navidinia M. J. Paramed. Sci. 2016;7:43–57. [Google Scholar]
- Pohle S. Appelt C. Roux M. Fiedler H. P. Süssmuth R. D. J. Am. Chem. Soc. 2011;133:6194–6205. doi: 10.1021/ja108971p. [DOI] [PubMed] [Google Scholar]
- Hayashi K. Hashimoto M. Shigematsu N. Nishikawa M. Ezaki M. Yamashita M. Kiyoto S. Okuhara M. Kohsaka M. Imanaka H. J. Antibiot. 1992;45:1055–1063. doi: 10.7164/antibiotics.45.1055. [DOI] [PubMed] [Google Scholar]
- Bae M. Kim H. Moon K. Nam S. J. Shin J. Oh K. B. Oh D. C. Org. Lett. 2015;17:712–715. doi: 10.1021/ol5037248. [DOI] [PubMed] [Google Scholar]
- Shi J. Liu C. L. Zhang B. Guo W. J. Zhu J. Chang C. Y. Zhao E. J. Jiao R. H. Tan R. X. Ge H. M. Chem. Sci. 2019;10:4839–4846. doi: 10.1039/C9SC00815B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rickards R. W. Skropeta D. Tetrahedron. 2002;58:3793–3800. doi: 10.1016/S0040-4020(02)00320-4. [DOI] [Google Scholar]
- Deng Z. Liu J. Li T. Li H. Liu Z. Dong Y. Li W. Angew. Chem., Int. Ed. 2021;60:153–158. doi: 10.1002/anie.202007777. [DOI] [PubMed] [Google Scholar]
- Wood T. M. Martin N. I. MedChemComm. 2019;10:634–646. doi: 10.1039/C9MD00126C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung D. Rozek A. Okon M. Hancock R. E. Chem. Biol. 2004;11:949–957. doi: 10.1016/j.chembiol.2004.04.020. [DOI] [PubMed] [Google Scholar]
- Sauermann R. Rothenburger M. Graninger W. Joukhadar C. Pharmacology. 2008;81:79–91. doi: 10.1159/000109868. [DOI] [PubMed] [Google Scholar]
- Hover B. M. Kim S.-H. Katz M. Charlop-Powers Z. Owen J. G. Ternei M. A. Maniko J. Estrela A. B. Molina H. Park S. Perlin D. S. Brady S. F. Nat. Microbiol. 2018;3:415–422. doi: 10.1038/s41564-018-0110-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen J. G. Reddy B. V. B. Ternei M. A. Charlop-Powers Z. Calle P. Y. Kim J. H. Brady S. F. Proc. Natl. Acad. Sci. U. S. A. 2013;110:11797–11802. doi: 10.1073/pnas.1222159110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz M. Hover B. M. Brady S. F. J. Ind. Microbiol. Biotechnol. 2016;43:129–141. doi: 10.1007/s10295-015-1706-6. [DOI] [PubMed] [Google Scholar]
- Banala S. Süssmuth R. D. ChemBioChem. 2010;11:1335–1337. doi: 10.1002/cbic.201000266. [DOI] [PubMed] [Google Scholar]
- Mahanta N. Szantai-Kis D. M. Petersson E. J. Mitchell D. A. ACS Chem. Biol. 2019;14:142–163. doi: 10.1021/acschembio.8b01022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipsky J. J. Gallego M. O. Drug Metab. Drug Interact. 1988;6:317–326. doi: 10.1515/dmdi.1988.6.3-4.317. [DOI] [PubMed] [Google Scholar]
- Hayakawa Y. Sasaki K. Nagai K. Shin-ya K. Furihata K. J. Antibiot. 2006;59:6–10. doi: 10.1038/ja.2006.2. [DOI] [PubMed] [Google Scholar]
- Frattaruolo L. Lacret R. Cappello A. R. Truman A. W. ACS Chem. Biol. 2017;12:2815–2822. doi: 10.1021/acschembio.7b00677. [DOI] [PubMed] [Google Scholar]
- Kjaerulff L. Sikandar A. Zaburannyi N. Adam S. Herrmann J. Koehnke J. Müller R. ACS Chem. Biol. 2017;12:2837–2841. doi: 10.1021/acschembio.7b00676. [DOI] [PubMed] [Google Scholar]
- Izawa M. Kawasaki T. Hayakawa Y. Appl. Environ. Microbiol. 2013;79:7110–7113. doi: 10.1128/AEM.01978-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhart B. J. Schwalen C. J. Mann G. Naismith J. H. Mitchell D. A. Chem. Rev. 2017;117:5389–5456. doi: 10.1021/acs.chemrev.6b00623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamada T. Sugawara T. Matsunaga S. Fusetani N. Tetrahedron Lett. 1994;35:609–612. doi: 10.1016/S0040-4039(00)75851-5. [DOI] [Google Scholar]
- Hamada T. Sugawara T. Matsunaga S. Fusetani N. Tetrahedron Lett. 1994;35:719–720. doi: 10.1016/S0040-4039(00)75799-6. [DOI] [Google Scholar]
- Hamada T. Matsunaga S. Yano G. Fusetani N. J. Am. Chem. Soc. 2005;127:110–118. doi: 10.1021/ja045749e. [DOI] [PubMed] [Google Scholar]
- Freeman M. F. Gurgui C. Helf M. J. Morinaka B. I. Uria A. R. Oldham N. J. Sahl H.-G. Matsunaga S. Piel J. Science. 2012;338:387–390. doi: 10.1126/science.1226121. [DOI] [PubMed] [Google Scholar]
- Freeman M. F. Helf M. J. Bhushan A. Morinaka B. I. Piel J. Nat. Chem. 2017;9:387–395. doi: 10.1038/nchem.2666. [DOI] [PubMed] [Google Scholar]
- Hamada T. Matsunaga S. Fujiwara M. Fujita K. Hirota H. Schmucki R. Güntert P. Fusetani N. J. Am. Chem. Soc. 2010;132:12941–12945. doi: 10.1021/ja104616z. [DOI] [PubMed] [Google Scholar]
- Oiki S. Muramatsu I. Matsunaga S. Fusetani N. Folia Pharmacol. Jpn. 1997;110:195–198. doi: 10.1254/fpj.110.supplement_195. [DOI] [PubMed] [Google Scholar]
- Bhushan A. Egli P. J. Peters E. E. Freeman M. F. Piel J. Nat. Chem. 2019;11:931–939. doi: 10.1038/s41557-019-0323-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renevey A. Riniker S. Eur. Biophys. J. 2017;46:363–374. doi: 10.1007/s00249-016-1179-1. [DOI] [PubMed] [Google Scholar]
- Gozari M. Alborz M. El-Seedi H. R. Jassbi A. R. Eur. J. Med. Chem. 2021;210:112957. doi: 10.1016/j.ejmech.2020.112957. [DOI] [PubMed] [Google Scholar]
- Murray L. A. M. McKinnie S. M. K. Moore B. S. George J. H. Nat. Prod. Rep. 2020;37:1334–1366. doi: 10.1039/D0NP00018C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geris R. Simpson T. J. Nat. Prod. Rep. 2009;26:1063–1094. doi: 10.1039/B820413F. [DOI] [PubMed] [Google Scholar]
- Teufel R., in Methods in Enzymology, Elsevier Inc., 1st edn, 2018, vol. 604, pp. 425–439 [DOI] [PubMed] [Google Scholar]
- Zhang X. Wang T. T. Xu Q. L. Xiong Y. Zhang L. Han H. Xu K. Guo W. J. Xu Q. Tan R. X. Ge H. M. Angew. Chem., Int. Ed. 2018;57:8184–8188. doi: 10.1002/anie.201804317. [DOI] [PubMed] [Google Scholar]
- Matsuda Y. Abe I. Nat. Prod. Rep. 2016;33:26–53. doi: 10.1039/C5NP00090D. [DOI] [PubMed] [Google Scholar]
- Horsman G. P. Zechel D. L. Chem. Rev. 2017;117:5704–5783. doi: 10.1021/acs.chemrev.6b00536. [DOI] [PubMed] [Google Scholar]
- Marquardt J. L. Brown E. D. Lane W. S. Haley T. M. Ichikawa Y. Wong C. H. Walsh C. T. Biochemistry. 1994;33:10646–10651. doi: 10.1021/bi00201a011. [DOI] [PubMed] [Google Scholar]
- Kuzuyama T. Shimizu T. Takahashi S. Seto H. Tetrahedron Lett. 1998;39:7913–7916. doi: 10.1016/S0040-4039(98)01755-9. [DOI] [Google Scholar]
- Bowman E. McQueney M. Barry R. J. Dunaway-Mariano D. J. Am. Chem. Soc. 1988;110:5575–5576. doi: 10.1021/ja00224a054. [DOI] [Google Scholar]
- Yu X. Doroghazi J. R. Janga S. C. Zhang J. K. Circello B. Griffin B. M. Labeda D. P. Metcalf W. W. Proc. Natl. Acad. Sci. U. S. A. 2013;110:20759–20764. doi: 10.1073/pnas.1315107110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju K. S. Gao J. Doroghazi J. R. Wang K. K. A. Thibodeaux C. J. Li S. Metzger E. Fudala J. Su J. Zhang J. K. Lee J. Cioni J. P. Evans B. S. Hirota R. Labeda D. P. van der Donk W. A. Metcalf W. W. Proc. Natl. Acad. Sci. U. S. A. 2015;112:12175–12180. doi: 10.1073/pnas.1500873112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayrouz C. M. Zhang Y. Pham T. M. Ju K. S. ACS Chem. Biol. 2020;15:1921–1929. doi: 10.1021/acschembio.0c00256. [DOI] [PubMed] [Google Scholar]
- O'Neill E. C. Schorn M. Larson C. B. Millán-Aguiñaga N. Crit. Rev. Microbiol. 2019;45:255–277. doi: 10.1080/1040841X.2019.1590307. [DOI] [PubMed] [Google Scholar]
- Davies J. Davies D. Microbiol. Mol. Biol. Rev. 2010;74:417–433. doi: 10.1128/MMBR.00016-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X. Li J. Millán-Aguiñaga N. Zhang J. J. O'Neill E. C. Ugalde J. A. Jensen P. R. Mantovani S. M. Moore B. S. ACS Chem. Biol. 2015;10:2841–2849. doi: 10.1021/acschembio.5b00658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Costa V. M. McGrann K. M. Hughes D. W. Wright G. D. Science. 2006;311:374–377. doi: 10.1126/science.1120800. [DOI] [PubMed] [Google Scholar]
- Thaker M. N. Wang W. Spanogiannopoulos P. Waglechner N. King A. M. Medina R. Wright G. D. Nat. Biotechnol. 2013;31:922–927. doi: 10.1038/nbt.2685. [DOI] [PubMed] [Google Scholar]
- Parsons J. B. Rock C. O. Curr. Opin. Microbiol. 2011;14:544–549. doi: 10.1016/j.mib.2011.07.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. M. White S. W. Rock C. O. J. Biol. Chem. 2006;281:17541–17544. doi: 10.1074/jbc.R600004200. [DOI] [PubMed] [Google Scholar]
- Young K. Jayasuriya H. Ondeyka J. G. Herath K. Zhang C. Kodali S. Galgoci A. Painter R. Brown-Driver V. Yamamoto R. Silver L. L. Zheng Y. Ventura J. I. Sigmund J. Ha S. Basilio A. Vicente F. Tormo J. R. Pelaez F. Youngman P. Cully D. Barrett J. F. Schmatz D. Singh S. B. Wang J. Antimicrob. Agents Chemother. 2006;50:519–526. doi: 10.1128/AAC.50.2.519-526.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson R. M. Huang T. Rudolf J. D. Smanski M. J. Shen B. Chem. Biol. 2014;21:389–397. doi: 10.1016/j.chembiol.2014.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oishi H. Noto T. Sasaki H. Suzuki K. Hayashi T. Okazaki H. Ando K. Sawada M. J. Antibiot. 1982;35:391–395. doi: 10.7164/antibiotics.35.391. [DOI] [PubMed] [Google Scholar]
- Sasaki H. Oishi H. Hayashi T. Matsuura I. Ando K. Sawada M. J. Antibiot. 1982;35:396–400. doi: 10.7164/antibiotics.35.396. [DOI] [PubMed] [Google Scholar]
- Mungan M. D. Alanjary M. Blin K. Weber T. Medema M. H. Ziemert N. Nucleic Acids Res. 2020;48:W546–W552. doi: 10.1093/nar/gkaa374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sozinova O. Jiang Y. Kaiser D. Alber M. Proc. Natl. Acad. Sci. U. S. A. 2006;103:17255–17259. doi: 10.1073/pnas.0605555103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann J. Fayad A. A. Müller R. Nat. Prod. Rep. 2017;34:135–160. doi: 10.1039/C6NP00106H. [DOI] [PubMed] [Google Scholar]
- Panter F. Krug D. Baumann S. Müller R. Chem. Sci. 2018;9:4898–4908. doi: 10.1039/C8SC01325J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumann S. Herrmann J. Raju R. Steinmetz H. Mohr K. I. Hüttel S. Harmrolfs K. Stadler M. Müller R. Angew. Chem., Int. Ed. 2014;53:14605–14609. doi: 10.1002/anie.201409964. [DOI] [PubMed] [Google Scholar]
- Yeh H. H. Ahuja M. Chiang Y. M. Oakley C. E. Moore S. Yoon O. Hajovsky H. Bok J. W. Keller N. P. Wang C. C. C. Oakley B. R. ACS Chem. Biol. 2016;11:2275–2284. doi: 10.1021/acschembio.6b00213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenical W. Jensen P. R. Palladino M. A. Lam K. S. Lloyd G. K. Potts B. C. Bioorg. Med. Chem. 2009;17:2175–2180. doi: 10.1016/j.bmc.2008.10.075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin G. Li D. Chidawanyika T. Nathan C. Li H. Arch. Biochem. Biophys. 2010;501:214–220. doi: 10.1016/j.abb.2010.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan Y. Liu Q. Zang X. Yuan S. Bat-Erdene U. Nguyen C. Gan J. Zhou J. Jacobsen S. E. Tang Y. Nature. 2018;559:415–418. doi: 10.1038/s41586-018-0319-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amorim Franco T. M. Blanchard J. S. Biochemistry. 2017;56:5849–5865. doi: 10.1021/acs.biochem.7b00849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuda Y. Kaneda M. Tada A. Nitta K. Yamamoto Y. Yoichi I. J. Chem. Soc., Chem. Commun. 1978:160–161. doi: 10.1039/C39780000160. [DOI] [Google Scholar]
- Dill G. M. Pest Manage. Sci. 2005;61:219–224. doi: 10.1002/ps.1008. [DOI] [PubMed] [Google Scholar]
- Milshteyn A. Colosimo D. A. Brady S. F. Cell Host Microbe. 2018;23:725–736. doi: 10.1016/j.chom.2018.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J. Vila-Farres X. Inoyama D. Ternei M. Cohen L. J. Gordon E. A. Reddy B. V. B. Charlop-Powers Z. Zebroski H. A. Gallardo-Macias R. Jaskowski M. Satish S. Park S. Perlin D. S. Freundlich J. S. Brady S. F. Nat. Chem. Biol. 2016;12:1004–1006. doi: 10.1038/nchembio.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch C. Weber T. Kohlbacher O. Wohlleben W. Huson D. H. Nucleic Acids Res. 2005;33:5799–5808. doi: 10.1093/nar/gki885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röttig M. Medema M. H. Blin K. Weber T. Rausch C. Kohlbacher O. Nucleic Acids Res. 2011;39:362–367. doi: 10.1093/nar/gkr323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J. Vila-Farres X. Brady S. F. J. Am. Chem. Soc. 2019;141:15737–15741. doi: 10.1021/jacs.9b07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du L. Lou L. Nat. Prod. Rep. 2010;27:255–278. doi: 10.1039/B912037H. [DOI] [PubMed] [Google Scholar]
- Chu J. Koirala B. Forelli N. Vila-Farres X. Ternei M. A. Ali T. Colosimo D. A. Brady S. F. J. Am. Chem. Soc. 2020;142:14158–14168. doi: 10.1021/jacs.0c04376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hostetler M. A. Smith C. Nelson S. Budimir Z. Modi R. Woolsey I. Frerk A. Baker B. Gantt J. Parkinson E. I. ACS Chem. Biol. 2021 doi: 10.1021/acschembio.1c00641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell A. H. Truman A. W. Comput. Struct. Biotechnol. J. 2020;18:1838–1851. doi: 10.1016/j.csbj.2020.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X. Lennard K. R. He C. Walker M. C. Ball A. T. Doigneaux C. Tavassoli A. van der Donk W. A. Nat. Chem. Biol. 2018;14:375–380. doi: 10.1038/s41589-018-0008-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt S. Montalbán-López M. Peterhoff D. Deng J. Wagner R. Held M. Kuipers O. P. Panke S. Nat. Chem. Biol. 2019;15:437–443. doi: 10.1038/s41589-019-0250-5. [DOI] [PubMed] [Google Scholar]
- van Heel A. J. Kloosterman T. G. Montalban-Lopez M. Deng J. Plat A. Baudu B. Hendriks D. Moll G. N. Kuipers O. P. ACS Synth. Biol. 2016;5:1146–1154. doi: 10.1021/acssynbio.6b00033. [DOI] [PubMed] [Google Scholar]
- Liu R. Zhang Y. Zhai G. Fu S. Xia Y. Hu B. Cai X. Zhang Y. Li Y. Deng Z. Liu T. Adv. Sci. 2020;7:2001616. doi: 10.1002/advs.202001616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Repka L. M. Chekan J. R. Nair S. K. van der Donk W. A. Chem. Rev. 2017;117:5457–5520. doi: 10.1021/acs.chemrev.6b00591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetterstrand K. A., The Cost of Sequencing a Human Genome, https://www.genome.gov/about-genomics/fact-sheets/Sequencing-Human-Genome-cost, accessed 1 September 2021 [Google Scholar]
- Paterson I. Anderson E. A. Science. 2005;310:451–453. doi: 10.1126/science.1116364. [DOI] [PubMed] [Google Scholar]
- Medema M. H. Cimermancic P. Sali A. Takano E. Fischbach M. A. PLoS Comput. Biol. 2014;10:e1004016. doi: 10.1371/journal.pcbi.1004016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medema M. H. Takano E. Breitling R. Mol. Biol. Evol. 2013;30:1218–1223. doi: 10.1093/molbev/mst025. [DOI] [PMC free article] [PubMed] [Google Scholar]