

Abstract
Pulmonary arterial hypertension (PAH) is characterized by impaired regulation of pulmonary hemodynamics and vascular growth. Alterations of metabolism and bioenergetics are increasingly recognized as universal hallmarks of PAH, as metabolic abnormalities are identified in lungs and hearts of patients, animal models of the disease, and in cells derived from lungs of patients. Mitochondria are the primary organelle critically mediating the complex and integrative metabolic pathways in bioenergetics, biosynthetic pathways, and cell signaling. Here, we review the alterations in metabolic pathways that are linked to the pathologic vascular phenotype of PAH, including abnormalities in glycolysis and glucose oxidation, fatty acid oxidation, glutaminolysis, arginine metabolism, one carbon metabolism, reducing and oxidizing (REDOX) cell environment, and tricarboxylic acid (TCA) cycle, as well as the effects of PAH-associated nuclear and mitochondrial mutations on metabolism. Understanding of the metabolic mechanisms underlying PAH provides important knowledge for design of new therapeutics for treatment of patients.
Keywords: Pulmonary hypertension, right ventricle, metabolism, glycolysis, fatty acid oxidation, glutaminolysis
I. INTRODUCTION
Pulmonary arterial hypertension (PAH) is a progressive lethal disorder disproportionately afflicting women, characterized by impaired regulation of pulmonary hemodynamics and vascular growth, in which right ventricular failure leads to death. PAH is defined clinically by pulmonary artery pressures (PAP) >20 mm Hg at rest (1, 2) and classified into five categories by the World Symposium on Pulmonary Hypertension (WSPH). This review focuses on category 1 disease, which includes idiopathic, heritable, and drug- and toxin-induced PAH, as well as PAH associated with connective tissue diseases and congenital heart diseases (Table 1) (2, 3). Pathologically, PAH is defined as panvasculopathy of the pulmonary arteries because abnormalities are found in all three types of pulmonary arteries: elastic, muscular, and nonmuscular, and all cell types of the artery, including endothelial and smooth muscle cells (4, 5). Plexiform lesions, which are seen in PAH lungs, are composed of proliferative endothelial cells and arise only in muscular arteries, or supernumerary muscular arteries, ~200 um in diameter (Figure 1) (4, 6). Increased pulmonary vascular resistance (PVR) due to obstructive lung panvasculopathy leads to progressive right ventricular failure.
Table 1.
World Symposium on Pulmonary Hypertension (WSPH) grouping of pulmonary hypertension patients
| WSPH 1 / Pulmonary Arterial Hypertension (PAH) |
| 1.1 Idiopathic |
| 1.2 Heritable |
| 1.2.1 BMPR2 |
| 1.2.2 ALK-1, ENG, SMAD9, CAV1, KCNK3 |
| 1.2.3 Unknown |
| 1.3 Drug and toxin-induced |
| 1.4 Associated with |
| 1.4.1 Connective tissue disease |
| 1.4.2 HIV infection |
| 1.4.3 Portal hypertension |
| 1.4.4 Congenital heart diseases |
| 1.4.5 Schistosomiasis |
| 1.5 PAH long-term responders to calcium channel blockers |
| 1.6 PAH with overt features of venous/capillaries (PVOD/PCH) involvement |
| 1.7 Persistent PH of the newborn syndrome |
|
WSPH 2 / Pulmonary Hypertension (PH) due to left heart disease |
| 2.1 PH due to heart failure with preserved LVEF |
| 2.2 PH due to heart failure with reduced LVEF |
| 2.3 Valvular heart disease |
| 2.4 Congenital/acquired cardiovascular conditions leading to post-capillary PH |
|
WSPH 3 / PH due to Lung Disease and/or Hypoxia |
| 3.1 Obstructive lung disease |
| 3.2 Restrictive lung disease |
| 3.3 Other lung disease with mixed restrictive/obstructive pattern |
| 3.4 Hypoxia without lung disease |
| 3.5 Developmental lung disorders |
|
WSPH 4 / PH due to pulmonary artery obstructions |
| 4.1 Chronic thromboembolic PH |
| 4.2 Other pulmonary artery obstructions |
|
WSPH 5 / PH with unclear and/or multifactorial mechanisms: |
| 5.1 Hematological disorders |
| 5.2 Systemic and metabolic disorders |
| 5.3 Others |
| 5.4 Complex congenital heart disease |
Abbreviation: BMPR2, bone morphogenetic protein receptor type-II, ALK-1, activin A receptor like type 1; ENG, endoglin; SMAD9, SMAD family member 9; CAV1, caveolin-1; KCNK3, potassium channel subfamily K member 3
Figure 1:
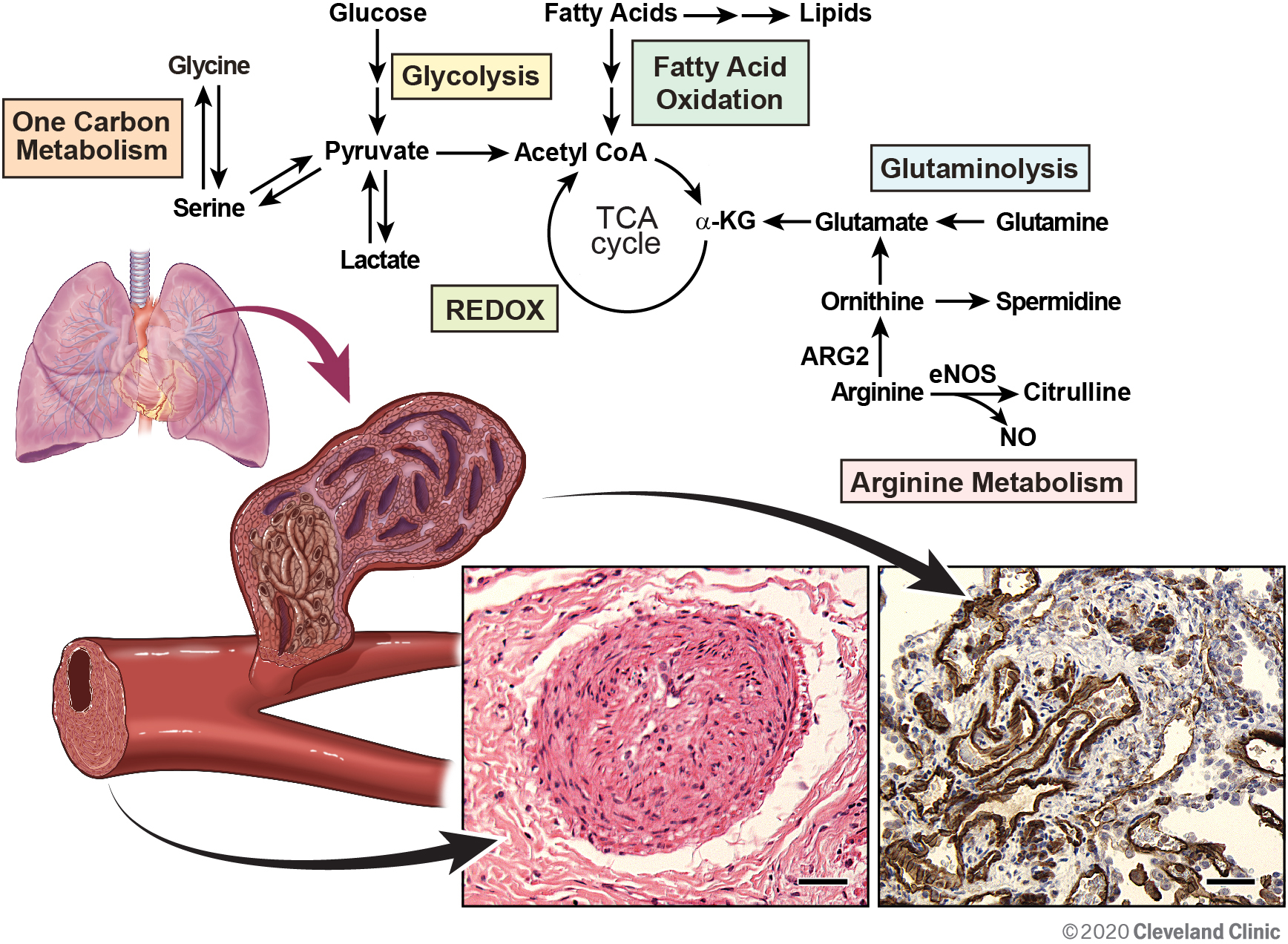
[Top panel] An overview of metabolism in the lung and heart
Glucose is converted to pyruvate and lactate via glycolysis. Pyruvate can be further converted to acetyl coenzyme A (acetyl-CoA) for entry into tricarboxylic acid (TCA) cycle. Fatty acids supply acetyl-CoA via β-oxidation. Serine and glycine required for one carbon metabolism are biosynthetically linked and can be used to supply pyruvate. Glutamine is converted to glutamate and αKG to fuel TCA cycle via glutaminolysis. Nitric oxide (NO) is produced in the lung by endothelial nitric oxide synthase (eNOS). Arginine is the substrate for both endothelial NOS (eNOS) and arginase (ARG). Mitochondrial arginase 2 (ARG2) catabolizes arginine to ornithine, which can provide glutamate and α-ketoglutarate (αKG). The reducing and oxidizing (REDOX) environment affects cell function and metabolism.
[Bottom panel] Model of abnormal pulmonary vasculature, including the plexiform lesion, and the hypertrophic right ventricle in PAH and histopathology of endothelial cell lesions in the PAH lung. [left histopathology] A concentric laminar intimal lesion obliterating the pulmonary blood vessel. [right histopathology] CD31 positive staining in proliferative endothelial cells lining the vascular lumen of plexiform lesion. Scale bars: 40 μm. Histopathology figures are modified from Figure 4G–4H in Reference 9.
Endothelial and smooth muscle cell dysfunction in pulmonary arteries is mechanistically linked to the pathobiology of PAH (7–16). Previous studies identify down-regulation of endothelium-derived vasodilators, e.g. prostacyclin (PGI2) and nitric oxide (NO), and up-regulation of endothelium-derived vasoconstrictors, e.g. endothelin-1, in models of PH and in human PAH (7, 10, 17–20). Existing therapies target vasodilatory pathways and have improved survival, i.e. 5-year survival for newly diagnosed patients is 70% (21). It is increasingly recognized that alterations of metabolism and bioenergetics are universal hallmarks of PAH in patients and animal models of the disease, but the metabolic pathways have not yet been targeted by therapies (7–16, 22–31). Here, we review the metabolic pathways and mechanisms that are linked to the pathophysiology of PAH (Figure 1).
II. METABOLIC PATHWAYS
Mitochondria are remarkable organelles that integrate energy production, biosynthetic pathways, and signal transduction. The pathways localized in mitochondria are dependent on cytosolic reactions and/or molecules. Some mitochondrial pathways are also found in the cytosol. In this review, seven major metabolic pathways that are implicated in PAH are considered, including glucose and fatty acid oxidation, glutaminolysis, arginine metabolism, one carbon metabolism, reducing and oxidizing (REDOX) reactions, and the tricarboxylic acid (TCA) cycle and electron transport chain (ETC). The TCA cycle is (1) the central hub for all the metabolic pathways in the mitochondria, (2) necessary for life-sustaining energy production, and (3) essential for cell biosynthetic functions in which TCA intermediates mediate signal transduction and regulate cell functions (32). Fueling the TCA cycle is acetyl-CoA derived from glucose via glycolysis and fatty acids via β-oxidation, and α-ketoglutarate (αKG) derived from the amino acids glutamate and arginine. Reactive oxygen species (ROS) are produced as a byproduct of oxidative phosphorylation and are imperative in the REDOX homeostasis of the cell. One-carbon metabolism in the mitochondria delivers methyl groups for synthesis of essential components of cells and tissues to maintain integrity of human health and body repair (Figure 2). The following details of the pathways identify the magnitude of effects of the mitochondrion for human health and in the pathobiology of PAH.
Figure 2:

Alterations of metabolic pathways in PAH described in this review. [a] Glucose uptake and glycolysis, [b] Fatty acid uptake and oxidation, [c] Glutamine uptake and glutaminolysis, [d] Arginine metabolism, [e] One carbon metabolism, and [f] REDOX. The line delineates those molecules and reactions found in the mitochondria. Some proteins, e.g., SOD1, eNOS, are located in both mitochondria and cytosol. Abbreviations: αKG, α-ketoglutarate; ACACA, acetyl-CoA carboxylase 1; ACAT, acetyl-CoA Acetyl transferase; Acyl-CoA, acyl-coenzyme A; Acetyl-CoA, acetyl coenzyme A; ACSL1, fatty acetyl CoA L1; ALDH18A1, aldehyde dehydrogenase 18 family, member A1; ARG2, arginase 2; CPT1, carnitine palmitoyltransferase 1; eNOS, endothelial nitric oxide synthase; ENO, enolase; ETC, electron transport chain; FBP, fructose 1,6-bisphosphate; FDG-PET, fluorodeoxyglucose positron emission tomography; fMet, N-formylmethionine; G6PC3, glucose-6-phosphatase catalytic subunit 3; GLS1, glutaminase 1; GLUT, glucose transporter; GPx, glutathione peroxidases; GSH, glutathione; H2O2, hydrogen peroxide; HK, hexokinase; LDHB, lactate dehydrogenase B, ME2, malic enzyme 2; MCD, malonyl-CoA decarboxylase; MTHFD1L, monofunctional C1-tetrahydrofolate (THF) synthase; NADPH, reduced nicotinamide adenine dinucleotide phosphate; NO, nitric oxide; P5C, Δ1-pyrroline-5-carboxylate; PFKFB, 6-phosphofructo-2-kinase/fructose-2, 6-biphosphatase; PFKM, phosphofructokinase; •OH, hydroxyl radicals; O2•−, superoxide; REDOX, reducing and oxidizing; SHMT2, mitochondrial serine hydroxymethyltransferase; SLC1A5, solute carrier family 1 member 5; SLC25A1, solute carrier family 25A1; SOD, superoxide dismutase; TCA, tricarboxylic acid; THF, tetrahydrofolate. Red ↑ indicates increase, and blue ↓ indicates reduction.
Glycolysis and Glucose Oxidation
Glucose is converted to pyruvate via the glycolytic pathway (Figures 1, 2a). Pyruvate can subsequently be converted to lactate or further oxidized in the mitochondria. Glycolysis is an important source for energy in vascular endothelial cells but much less so in cardiomyocytes (33). In PAH, glucose metabolism is shifted away from complete mitochondrial oxidative phosphorylation and towards greater cytoplasmic glycolysis to pyruvate and ultimately lactate in primary pulmonary artery endothelial cells (PAEC) and pulmonary artery smooth muscle cells (PASMC) derived from PAH lungs in culture, in hearts and lungs of patients with PAH in vivo, and animal models of PAH (Table 2) (Figures 1, 2a)(7, 8, 10, 11, 15, 16, 26, 29, 31, 34–45).
Table 2.
Increased glycolysis and hypoxia-inducible factor (HIF) expression in pulmonary hypertension
| Sources | Parameters | References |
|---|---|---|
| Human data | ||
| Pulmonary arterial endothelial cells (PAEC) in vitro | Hypoxia-inducible factor (HIF) presence in nuclear extract by Western blot analyses, ↑5-3H-glucose to 3H2O, ↑13C-fructose bisphosphate from 13C-glucose [↑proximate glycolysis pathway intermediates], ↑Lactate by spectrophotometry, ↑Hepatocyte growth factor (HGF) and stromal-derived factor (SDF)-1 by ELISA |
(10, 11, 16, 26) |
| Pulmonary artery smooth muscle cells (PASMC) in vitro |
↑13C-pyruvate and 13C-lactate from 13C-glucose [↑ glucose uptake and utilization], ↑Enolase (ENO)1 by Western blot analyses |
(16, 35) |
| Lung | HIF presence by immunohistochemistry, ↑18Fluorodeoxyglucose (FDG) uptake, ↑6-phosphofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB)3 and lactate dehydrogenase (LDH)B by Western blot analyses, ↑HGF and SDF-1 by immunohistochemistry, ↑PFKFB2 and LDHB by gene microarray analyses, ↑Glucose to sorbitol, fructose, and fructose 6-phosphate, ↓Fructose 1,6-bisphosphate (FBP), 3-phosphoglycerate (3-PGA), and phosphoenolpyruvate (PEP) by metabolomics, ↓Glucose-6-phosphatase catalytic subunit 3 (G6PC3) by Western blot analyses, immunohistochemistry, and gene microarray analyses |
(10, 11, 15, 26, 31, 34, 36, 37) |
| Small pulmonary arteries | HIF presence by immunofluorescence | (14, 15) |
| Right ventricle | HIF presence in nuclei of cardiomyocytes by immunohistochemistry, ↑18FDG uptake, ↑Glycolytic gene, e.g. Hexokinase (HK)2 and solute carrier family 2 member 3 (SLC2A3), expression by microarray analyses |
(29, 31, 38, 39, 47, 48) |
| Animal models | ||
| Shunt lambsa | ||
| Lung | ↑Lactate/pyruvate ratio by spectrophotometry, | (40, 45) |
| Fawn hooded ratsb | ||
| PASMC | HIF presence by immunofluorescence | (14, 15) |
| Small rat pulmonary arteries | HIF presence by Western blot analyses | (14, 15) |
| Hypoxia/Sugen ratsc | ||
| Lung |
↑ENO, ↑PFKFB3 mRNA by qRT-PCR, protein by Western blot analyses, and activity by F-2,6-P2 assay |
(35, 37, 41) |
| Right ventricle | ↑HK1 and phosphofructokinase (PFKM) by gene microarray analyses | (41) |
| Monocrotaline (MCT) ratsd | ||
| Lung | ↑ Glucose transporter (GLUT)1 and LDHA mRNA by qRT-PCR and protein by Western blot | (44) |
| Right ventricle |
↑18FDG uptake, ↑GLUT1 protein expression by Western blot analyses, ↑GLUT1, HK2 and LDHA mRNA by qRT-PCR |
(42–44) |
| Liver | ↑Metabolic fluxes including glucose, lactate, and pyruvate in liver perfusion | (55) |
Shunt lambs, with pulmonary hypertension secondary to increased pulmonary blood flow with surgically created heart defect;
Fawn hooded rats, a spontaneously pulmonary hypertensive strain with a hereditary bleeding tendency due to a genetic defect in platelet aggregation;
Hypoxia/Sugen rats, with hypoxia or sugen-induced pulmonary hypertension;
MCT rats, a maladaptive model (heart failure model) combined with PAH and right ventricular hypertrophy induced by MCT
Many studies have identified increased glycolysis in PAH PAEC and PASMC (Table 2), including definitive studies using radioisotopes and stable isotopes (10, 16). Despite greater glucose metabolism to pyruvate, PAH PAEC have less mitochondrial respiration than PAEC from control lungs (Table 3) (10, 12). Oxygen consumption is decreased in PAH PAEC compared to control PAEC when measured using tricarboxylic acid (TCA) cycle intermediates glutamate-malate or succinate as substrates (10). Furthermore, PAH PAEC have lower oxygen consumption than control cells for any glucose concentration provided, supporting the idea that the shift away from oxidative metabolism of glucose in PAH endothelial cells is independent of oxygen availability, i.e. the Warburg effect (12). Overall, there is abundant evidence that PAH PAEC have less oxidative metabolism of glucose (12).
Table 3.
Decreased glucose oxidation and abnormal mitochondria in pulmonary hypertension
| Sources | Parameters | References |
|---|---|---|
| Human data | ||
| PAEC in vitro |
↓Oxygen consumption with a Seahorse XF24 analyzer, ↓Oxygen consumption under state 3 and state 4 respiration using the classical method of circulator chambers and a 5300A biological oxygen monitor and Clark-type polarographic oxygen electrode, ↓Complex IV activity by spectrophotometry and mitochondrial dehydrogenase activity by 3-(4,5-dimethylthiazolyl-2)-2,5-diphenyltetrazolium bromide (MTT) assay, ↓mtDNA by Southern blot, ↓Mitochondrial numbers by quantitation of mitochondria in electron microscopy images of cells |
(10, 12) |
| PASMC in vitro | ↓ Mitochondria by immunofluorescence | (14, 15) |
| Small pulmonary arteries | ↓Complex I by immunofluorescence | (14, 15) |
| Animal models | ||
| Shunt lambs | ||
| Lung | ↑Uncoupling protein (UCP)2-expression by Western blot analyses | (40) |
| Chickens with pulmonary hypertension syndromea | ||
| Lung | ↓Oxygen consumption under state 3 and state 4 respiration using the classical method | (50, 51) |
| Breast and heart muscle |
↓Oxygen consumption under state 3 respiration using the classical method, ↓Complex I activity by spectrophotometry, ↑Succinate metabolism |
(50, 51) |
| Fawn hooded rats | ||
| PASMC |
↓Complexes I by immunofluorescence, Small, dense, dysmorphic mitochondria (swollen cristae) by transmission electron microscopy |
(14, 15) |
| Small rat pulmonary arteries | ↓Complexes I, III, and IV by Western blot analyses | (14, 15) |
| Monocrotaline (MCT) rats | ||
| Right ventricle |
↓Oxygen consumption by high-resolution respirometry, ↓14C-glucose oxidation |
(42, 43) |
| Endothelial nitric oxide synthase (eNOS)−/− miceb | ||
| Lung | ↓Oxygen consumption with Oxymax metabolic chamber | (52) |
| Brain, liver, heart tissue and brown adipocytes or brown adipose tissue |
↓Complex IV and cytochrome complex (Cyt c) by Western blot analyses, ↓UCP1 and peroxisome proliferator-activated receptor gamma coactivator (PGC)-1α, nuclear factor erythroid 2–related factor (NRF)-1, and mitochondrial transcription factor A (mtTFA) mRNA by qRT-PCR, ↓mtDNA by Southern blot |
(52) |
| Soleus muscles | ↓Oxygen consumption by a Clark electrode in an oxygraphic cell | (53) |
| Brown adipocytes or brown adipose tissue | ↓Number of mitochondria by MitoTracker fluorescence | (52) |
Chickens with pulmonary hypertension syndrome and increased RV/total ventricle weight ratio; chickens developed pulmonary hypertension syndrome under cool temperatures combined with feed to support rapid growth rate;
eNOS−/− mice, with eNOS knockout
Unlike in the vascular endothelium, in healthy adult hearts mitochondrial fatty acid β-oxidation is the major source of energy production and oxygen consumption (46). In right ventricular hypertrophy, as occurs in PAH, energy production is increasingly dependent on glycolysis. Glucose uptake, as a measure of cardiac glucose use, can be quantitatively measured by 18F-fluorodeoxyglucose uptake in the heart using positron emission tomography (FDG-PET) in vivo. FDG-PET studies show much greater FDG uptake in the hearts and lungs of patients with PAH compared to healthy individuals (10, 29, 31, 36, 38). High levels of right ventricular FDG uptake are associated with a poor heart rate recovery after a 6-minute walk, higher right ventricular systolic pressure (RVSP), higher pulmonary vascular resistance (PVR), and lower cardiac index, all of which are clinical indicators of more severe disease (47, 48). The relationship of FDG uptake to clinical phenotype of severe disease suggests that functional metabolic imaging may be useful in monitoring patients over time for response to therapy and/or progression of disease in anticipation of transplantation (29). In accord with the greater FDG uptake in lungs and hearts of PAH patients, increased levels of glycolysis-related enzymes have been reported in PAH lungs, i.e., 6-phosphofructo-2-kinase/fructose-2, 6-biphosphatase (PFKFB)2 and lactate dehydrogenase (LDH)B (34); and in biopsies of right ventricles from PAH patients, i.e., hexokinase (HK)2 and solute carrier family 2 member 3 (SLC2A3), which encodes glucose transporter (GLUT)3 (Table 2) (Figure 2a) (39, 49). Similar to findings in patients, there is reduced mitochondrial oxygen consumption in the lungs and hearts of PH animal models (Table 3) (42, 43, 50–53).
Among molecular mechanisms that likely promote the shift to glycolysis and away from oxidative glucose metabolism in PAH is the pathologic accumulation of hypoxia-inducible factor (HIF)-1α. HIF-1α is a transcription factor that plays a role in a variety of cellular functions, including proliferation, angiogenesis, survival, and metabolism (11, 14, 15, 29, 48, 49, 54). Upregulation of HIF-1α is found in plexiform lesions, pulmonary arteries, and cardiomyocytes from PAH patients (11, 13–15, 29). Secretion of the HIF-regulated factors hepatocyte growth factor (HGF) and stromal-derived factor (SDF)-1 is significantly increased in PAH PAEC as compared to healthy control cells (26). Increased glycolysis, greater FDG uptake, and increased levels of glycolysis-related enzymes and activity in lungs and right ventricles in PH animal models have been mechanistically associated with HIF accumulation (Table 2) (14, 15, 35, 37, 40–45, 55).
Fatty Acid Oxidation
Fatty acid metabolism involves pathways such as cellular fatty acid uptake and storage, fatty acid transport into mitochondria, mitochondrial fatty acid β-oxidation, and fatty acid synthesis. Imbalanced fatty acid metabolism is reported in hearts and lungs of PAH patients as shown (Table 4) (Figure 2b) (12, 16, 34, 39, 40, 44, 52, 56–64). The healthy adult heart relies on fatty acids as the main energy source to power contractile function with approximately 60–90% of ATP generated from fatty acid oxidation and the remaining 10–40% from oxidation of glucose, lactate, amino acids, or ketone bodies (65, 66). Disruption in fatty acid metabolism contributes to cardiac contractile dysfunction and hypertrophy (39, 56, 57, 59, 60). Although glycolysis and glutaminolysis can help maintain normal function in hypertrophic cardiomyocytes during the initial stages of heart failure, these pathways are inadequate in progressive right ventricular hypertrophy, as occurs in PAH (39, 56). PAH patients show higher right ventricular lipid accumulation in the form of triglycerides, diacylglycerols, and ceramides that associate with right ventricular dysfunction and failure (39, 64). Excess cellular lipid uptake mediated by CD36, the main transporter responsible for fatty acid uptake into contracting cardiomyocytes, results in lipid accumulation in right ventricular tissue from individuals with heritable PAH with a Bone Morphogenetic Protein Receptor type-II (BMPR2) mutation as well as mice genetically deficient in BMPR2 (39, 56, 61). CD36 mRNA and protein expression is also increased in the lungs and hearts of the monocrotaline (MCT)-induced PH rat model (44). On the other hand, single photon emission computed tomography (SPECT) using the fatty acid analog 123I-β-iodophenyl pentadecanoic acid suggests impaired fatty acid uptake in the right ventricles of patients with very severe PAH (60, 62), perhaps suggesting that the failing heart has limited ability to uptake fatty acid. Once taken up, fatty acids are converted into acyl-carnitine and transported into the mitochondria by carnitine palmitoyltransferase (CPT), the rate limiting enzyme in the fatty acid oxidation pathway. Expression of CPT1 in the right heart is upregulated in the MCT-induced PH rat model (44, 63), but the transport of fatty acids into mitochondria is not accompanied by enhanced mitochondrial fatty acid oxidation. Several reports suggest disruption in mitochondrial fatty acid oxidation in BMPR2-mutant cardiomyocytes (39, 56, 64). Abnormalities of cardiac mitochondrial fatty acid oxidation in patients are supported by findings of increased levels of carnitine and acylcarnitine in plasma from PAH patients (44), which suggest incomplete mitochondrial fatty acid oxidation. Abnormalities of carnitine metabolism are also linked to mechanisms of PH in a lamb model (40, 45). In the MCT-induced PH rat model, right heart hypertrophy, physiological signs of heart failure, and myocardial lipid peroxidation are found in the late stages, 6 weeks after MCT administration (58). As in cell studies of glucose metabolic abnormalities, increased HIF-1α accumulation in the right ventricle is associated with decreased fatty acid oxidation and cardiomyocyte lipid accumulation in PAH (67, 68).
Table 4.
Abnormalities of fatty acid synthesis/oxidation in pulmonary hypertension
| Sources | Parameters | References |
|---|---|---|
| Human data | ||
| PAEC in vitro |
↓13C-α-ketoglutarate (αKG) from 13C-long chain fatty acids (LCFAs), ↓Acetyl-CoA acetyltransferase (ACAT)2 by global proteomics, ↑Enoyl-CoA delta isomerase (ECI)1 by global proteomics |
(12, 16) |
| Lung |
↑Carnitine, dicarboxylic acid, and long- and medium-chain free fatty acid products including caproate, caprylate, myristate, and palmitoleate by metabolomics, ↑Adrenate [a pro-thrombotic lipid that can cause obstructions] by metabolomics, ↑Expression of genes coding the enzymes fatty acetyl CoA L1 (ACSL1) and Acetyl CoA Carboxylase 1 (ACACA) by gene microarray |
(34) |
| Right ventricle |
↓Fatty acid uptake by 123I-labeled l5-(p-iodophenyl)-3-(R,S)-methylpentadecanoic acid (BMIPP) single-photon emission computerized tomography (SPECT), ↓Fatty acid oxidation by gene microarray analyses, ↓Long-chain acylcarnitine by liquid chromatography/mass spectrometric analysis, ↑Lipid content by oil red O staining, ↑Ceramide by liquid chromatography/mass spectrometric analysis, ↑Triglycerides by cardiac imaging with proton spectroscopy |
(39, 60, 62, 64) |
| Plasma |
↑Free fatty acids by enzymatic assay, ↑Long-chain acylcarnitines by liquid chromatography/mass spectrometric analysis |
(64) |
| Animal models | ||
| Shunt lambs | ||
| Lung | ↑Acylcarnitines by FPLC | (40) |
| MCT rats | ||
| PASMC in vitro | ↑Carnitine palmitoyltransferase (CPT)1 by Western blot and immunofluorescence | (63) |
| Lung | ↑CPT1 and CD36 mRNA by qRT-PCR and protein by Western blot | (44, 63) |
| Right ventricle |
↑CPT1 and CD36 mRNA by qRT-PCR and protein by Western blot, ↑Myocardial lipid peroxidation in right ventricle in late stage PH by thiobarbituric acid reactive substances (TBARS) assay |
(44, 58, 63) |
| Severe pulmonary hypertension (SPH) ratsa | ||
| Right ventricle | ↓14-(R,S)-[18F]fluoro-6-thia-heptadecanoic acid (18F-FTHA) [fatty acid analog] uptake | (59) |
| BMPR2−/− miceb | ||
| Right ventricle |
↓Fatty acid β-oxidation of palmitate by oxygen consumption measurement using Oroboros O2k Oxygraph, ↑Fatty acid uptake by 14C-palmitate, ↑CD36 fatty acid transporter molecule by immunofluorescence and Western blot analyses, ↑Fatty acid metabolites by metabolomics, ↑Lipid accumulation in the form of triglycerides, diacylglycerol, and ceramides by metabolomics, ↑Lipid by oil red O staining |
(39, 56, 61, 64) |
| CAV1−/− micec | ||
| Heart |
↓Triglycerides, fatty acids, and cholesterol measured enzymatically, ↑Lipid uptake by measure the uptake of 14C-palmitate, ↑14C-palmitate oxidation, ↑cAMP by cAMP [3H] assay |
(57) |
| eNOS−/− mice | ||
| Brown adipocytes or brown adipose tissue |
↑Lipid droplets in adipocytes by oil red O staining |
(52) |
SPH rats, SPH induced by treatment with the VEGF receptor inhibitor SU5416 and under hypoxia;
BMPR2−/− mice, with BMPR2 knockout, do not develop pulmonary hypertension spontaneously but are more susceptible to hypoxic or chemically induced PH
CAV1−/− mice, with caveolin-1 knockout with pulmonary hypertension and left ventricular hypertrophy
Fatty acid abnormalities in the lung have also been described. Accumulation of carnitine and dicarboxylic acid in PAH lung, smooth muscle, and endothelial cells are associated with enhanced expression of genes involved in fatty acid oxidation such as acetyl-CoA carboxylase 1 (ACACA), which is associated with proliferation in cancer cells (12, 34, 69–71). Using in vitro stable isotope metabolic flux analysis, PAH PAEC have less mitochondrial fatty acid oxidation, which results in less contribution of fatty acid-derived carbons to support TCA cycle in PAH cells (16). Similar to findings in the PAH heart, CPT1 is also upregulated in lungs and pulmonary arteries of the MCT-rat model (63). Upregulation of fatty acid oxidation by overexpressing CPT1 in PASMC increases cellular ATP and promotes cellular proliferation, while CPT1 inhibition abrogates PASMC proliferation (63). In murine studies, abolishing fatty acid oxidation by genetic deletion of malonyl-CoA decarboxylase (MCD) prevents development of PH in mice exposed to MCT or hypoxia, as compared to wild-type mice (72). Taken together, there are fatty acid metabolic disturbances in PAH that are not fully understood. Additional studies in cells, tissues, and animal models will help to define the relationship to PAH pathophysiology.
Glutaminolysis
Glutamine is an important source for a variety of biochemical functions including as a carbon donor in TCA cycle, protein and lipid synthesis, cell energy, nitrogen donor for purine synthesis, and biosynthesis of purine nucleosides. With a shift away from glucose oxidation in PAH, an alternative source of carbons for macromolecule synthesis may be provided via glutamine metabolism. Glutaminolysis starts with a two-step process of (1) deamination of glutamine to glutamate by glutaminase (GLS; 1 and 2), followed by (2) conversion of glutamate to α-ketoglutarate (αKG) by glutamate dehydrogenase (Figure 2c). This process is critical for rapid growth of cancer and other proliferating cells (73–75). HIF-1α promotes glutamine metabolism towards biosynthetic pathways (76).
Glutamine metabolism is increased in the PAH right ventricle. Glutamine uptake is mediated by the alanine-serine-cysteine-transporter ASCT2 (solute carrier family 1 member 5, SLC1A5) (77). SLC1A5 is upregulated in hypertrophic right ventricle of PAH patients and in the right ventricle of the MCT-rat model (Table 5) (43). The glutamine antagonist 6-Diazo-5-oxo-L-norleucine decreases glutaminolysis, reduces right ventricular hypertrophy, and increases cardiac output in MCT-induced right ventricular hypertrophy. Cardiac glutaminolysis is associated with microvascular rarefaction/ischemia in the right ventricle of the MCT rat; capillary rarefaction is similarly found in the right ventricle of PAH individuals, suggesting increased cardiac glutaminolysis (43). Overall, glutamine metabolism and glutamine-derived metabolites are linked to maladaptive remodeling in PAH right heart.
Table 5.
Increased glutamate metabolism
| Sources | Parameters | References |
|---|---|---|
| Human data | ||
| PAEC in vitro |
↑13C-succinate from 13C-glutamine [↑glutamine-derived anaplerosis], ↑Aldehyde dehydrogenase 18 family, member A1 (ALDH18A1) by global proteomics, ↑Spermidine by metabolomics |
(12, 16) |
| Lung |
↑Glutamine uptake measured at right heart catheterization, ↑Glutaminase (GLS)1 by immunofluorescence, ↑ALDH18A1 by metabolomics and Western blot analyses |
(34, 76, 78) |
| Right ventricle | ↑Glutamine transporter solute carrier family (SLC)1A5 by immunofluorescence | (43) |
| Plasma |
↑Glutamine, ↑Glutamate by metabolomics |
(12, 76) |
| Animal models | ||
| MCT rats | ||
| Right ventricle |
↑14C-glutamine metabolism, ↑Solute carrier family 1 member 5 (SLC1A5) by Western blot analyses, ↑SLC1A5 and mitochondrial malic enzyme (ME)2 by qRT-PCR |
(43) |
| Plasma | ↑Glutamine and malate | (43) |
| BMPR2−/− mice |
↑13C-glutamine metabolism, ↑Glutamine-supported ATP-linked mitochondrial respiration with a Seahorse XF96 analyzer |
(76) |
Increased GLS1 and glutamine metabolism are reported in pulmonary arteries of PAH lungs (78). Increased glutamine levels in the circulation are related to severity of PAH. PAH PAEC but not PASMC have increased glutamine consumption (Table 5) (Figure 2c) (12, 16, 76, 78). Genetic inhibition of GLS1 (the main isoform of GLS) in endothelial cells or the genetic deletion in mice, or inhibition of GLS1 in MCT-rats, suppress endothelial cell proliferation and angiogenesis and abrogates the severity of PH in these models (78, 79). These findings support the importance of glutamine metabolism in the proliferative endothelial phenotype of PAH.
Glutamine also has other metabolic fates, including conversion to Δ1-pyrroline-5-carboxylate (P5C) by delta-1-pyrroline-5-carboxylate synthetase (P5CS) encoded by aldehyde dehydrogenase 18 family, member A1 (ALDH18A1) gene, a major step in the biosynthesis of proline, ornithine, and arginine (80, 81). Proteomics study found higher levels of ALDH18A1 in PAH PAEC compared to control PAEC (12). Consistent with these results, there is increased expression of ALDH18A1 in PAH lungs (34). High levels of plasma glutamate, a substrate of ALDH18A1, and spermidine, downstream of ornithine, are found in PAH (12). Upregulated ALDH18A1 and consequent elevated spermidine in PAH are similar to findings in rapidly growing malignant cells. Altogether, these studies support a role for glutamine metabolism in cell survival and proliferation in PAH.
Arginine Metabolism
Arginine, a semi-essential amino acid, is the substrate for both nitric oxide synthase (NOS) and arginase (ARG) (Figure 2d). Although arginine may be taken into cells by cationic amino acid transporter (CAT) proteins, de novo arginine synthesis is the major intracellular source for arginine (82, 83). Endothelial nitric oxide synthase (eNOS), the predominant NOS isoform in the pulmonary vasculature, converts arginine to nitric oxide (NO) and citrulline. NO is a potent vasodilator that is deficient in PAH (10, 17–20). Under certain in vivo conditions, arginine bioavailability may limit the production of NO, e.g., arginine utilized by other enzymes, such as ARG (30, 84, 85). ARG catabolizes arginine to ornithine and urea (30). ARG1 is present exclusively in the cytosol of hepatic cells as part of the urea cycle, but ARG2 is found in mitochondria of many tissues without a functioning complete urea cycle, including the lungs and heart.
PAH patients, animal models, and pulmonary arterial endothelial cells (PAEC) derived from human PAH lungs are deficient in NO production by eNOS (Figure 2d) (17, 30). Loss of NO production in PAH and the underlying mechanisms have been identified: (i) phosphorylation inactivation of eNOS and (ii) decreased eNOS substrate arginine bioavailability due to increased mitochondrial ARG2 (8, 10, 17, 19, 25, 26, 28, 30, 86). A recent study found differential expression of proteins in the eNOS pathway in PAH PAEC, confirming impaired eNOS in PAH (12).
While eNOS activity is low, serum arginase activity is higher in PAH patients as compared to controls (8, 30). ARG2 is expressed at higher levels in PAEC from PAH than controls and is present at high levels in the endothelium of PAH lungs in vivo, particularly in plexiform lesions (8, 30, 87). Addition of excess arginine to PAH cells in culture does not recover NO synthesis unless ARG is inhibited by (S)-(2-boronoethyl)-l-cysteine-HCl (BEC) (8). ARG inhibition results in arginine bioavailability and increased pulmonary NO production in neonatal rat lungs of a bleomycin-induced pulmonary hypertension model (88). ARG inhibition in mouse and rat models of PH leads to lower right ventricular systolic pressure (RVSP), reduced lung tissue remodeling, and improved NO bioavailability (89, 90), suggesting a potential treatment strategy in PAH. Arginine and citrulline levels and arginine-to-ornithine ratio are significantly lower in plasma of PAH patients, confirming substrate limitation for NOS in PAH in vivo (8, 12, 30). However, arginine metabolic fate impacts PAH beyond loss of vasodilatory NO. Arginine metabolism to ornithine via ARG2 can provide glutamate and αKG to fuel TCA cycle (Figure 2d). Increased mitochondrial ARG2 production of ornithine provides a steady supply of αKG to TCA (10, 30), which affects cell metabolism and mitochondrial bioenergetics (electron transport system and oxygen consumption) in PAH and accumulation of HIF (11, 49, 91). Mitochondrial arginine metabolism is a biochemical link between the increased vasoconstriction, i.e., loss of NO production, defining the hemodynamic component of PAH with the abnormalities of TCA cycle and bioenergetics that drive the proliferative phenotype of pulmonary vascular cells.
One Carbon Metabolism and Glycine Metabolism
Rapidly proliferating cells upregulate other metabolic pathways including one carbon metabolism, which is essential for biosynthetic processes including purine and thymidine synthesis and homocysteine remethylation and pivotal for reducing and oxidizing (REDOX) balance during hypoxia (92, 93). While mitochondrial generation of one-carbon units from both serine and glycine is not a major metabolic process in non-proliferative adult tissues, one carbon metabolism is required in proliferating cells. Increased one-carbon metabolism has been observed in the mouse model of PH (94, 95) and in human PAH (12)(Figure 2e).
Serine catabolism is initiated by serine hydroxymethyltransferase (SHMT) activity, catalyzed in the cytosol by SHMT1 and in the mitochondrion by SHMT2. SHMT catalyzes a reversible reaction converting serine to glycine with concurrent 5,10-methylene–tetrahydrofolate (5,10-methylene-THF) generation (Figure 2e). THF is an essential precursor for purine and N-formylmethionine (fMet) synthesis. One-carbon transformations producing and consuming REDOX equivalents are also important in mitochondrial REDOX homeostasis (96, 97). In cancer cells, the transcription factors HIF-1 and MYC cooperate to upregulate SHMT2 during hypoxia to prevent uncontrolled levels of hydrogen peroxide (H2O2) in the mitochondrial matrix (93, 98). Proteomic studies reveal that two key enzymes in the one carbon pathway [SHMT2 and monofunctional C1-tetrahydrofolate (THF) synthase (MTHFD1L)], are elevated in PAH PAEC (12). Global metabolomics of plasma from PAH patients reveal lower glycine levels, supporting the notion of higher enzyme activity of SHMT2. Global plasma metabolomics also identify an increase in purine metabolites in PAH, including fMet, guanosine, adenosine, inosine, xanthosine, and hypoxanthine. Further studies are needed, but these early studies point to one carbon metabolism as a mechanism to support the proliferative angiopathy of PAH.
Reducing and Oxidizing (REDOX) Cell Environment
Reactive oxygen species (ROS), including hydroxyl radicals (•OH), superoxide (O2•−), and H2O2, are byproducts of normal cellular metabolism. Mitochondria are the largest producers of ROS through the process of cellular respiration in which mitochondria catalyze one-electron reduction of oxygen to superoxide radical followed by formation of H2O2 in addition to the four-electron reduction of oxygen to water. Antioxidant enzymes include superoxide dismutases (SOD), catalase, glutathione peroxidases (GPx), glutathione S-transferase, and thioredoxin. The primary antioxidants for removal of superoxide are the superoxide dismutases [MnSOD (SOD2) in the mitochondria and CuZnSOD (SOD1) in both cytosol and mitochondria] (99).
While the transition to aerobic glycolysis minimizes creation of ROS through diminished oxidative metabolism, increased oxidative stress and decreased SOD activity in are present in PAH lungs and PAEC, and animal models of pulmonary hypertension, and contribute to the pathophysiology of PAH (Table 6) (Figure 2f) (7, 11, 12, 14, 15, 40, 50, 51, 100–105). Excessive ROS generation and loss of SOD activity have been linked to HIF activation (11, 12, 14, 15, 49, 100, 103, 106).
Table 6.
Reduced antioxidants and increased reactive oxygen species (ROS)
| Sources | Parameters | References |
|---|---|---|
| Human data | ||
| PAEC in vitro |
↓Superoxide dismutase (SOD) activity by the rate of reduction of cytochrome c, ↓SOD2 by Western blot analyses, ↓SOD1 by global proteomics and Western blot analyses, ↑ROS using CellROX staining |
(11, 12) |
| Lung |
↓SOD activity by the rate of reduction of cytochrome c, ↓SOD2 in lung by Western blot analyses, ↓Glutathione peroxidase (GPx) activity by spectrophotometry, ↑8-hydroxy guanosine by immunohistochemistry |
(100, 103) |
| Small pulmonary arteries |
↓SOD2 by immunofluorescence, ↓ROS by MitoSOX fluorescence staining, ↓Hydrogen peroxide (H2O2) by Amplex Red |
(14, 15) |
| Plasma | ↑Oxidative stress by measure of levels of cysteine and its oxidized form, cystine, glutathione (GSH) and glutathione disulfide (GSSG) using HPLC | (105) |
| Animal models | ||
| Shunt lambs | ||
| Lung |
↓SOD2 by Western blot analyses, ↑Superoxide by MitoSOX fluorescence staining, ↑ROS by dihydroethidium (DHE) or dichlorodihydrofluorescein diacetate (H2DCF-DA) fluorescence staining |
(40, 102) |
| Chickens with pulmonary hypertension syndrome | ||
| Lung |
↓Mitochondrial GSH by HPLC, ↑GSSG/GSH ratio by HPLC |
(51) |
| Breast and heart muscle | ↑H2O2 using 2’, 7’-dichlorofluorescin diacetate (DCFHDA) | (50, 104) |
| Fawn hooded rats | ||
| PASMC |
↓SOD activity with a colorimetric assay, ↓SOD2 by immunofluorescence and Western blot analyses, ↓ROS by MitoSOX fluorescence staining, ↓ H2O2 by Amplex Red |
(14, 15) |
| Small pulmonary arteries |
↓SOD2 by immunofluorescence, ↓ROS by MitoSOX fluorescence staining, ↓ H2O2 by Amplex Red |
(14, 15) |
| MCT rats | ||
| Right ventricle |
↓SOD activity by determination of percent inhibition of pyrogallol autooxidation, ↓GPx and catalase activity by spectrophotometry |
(58) |
In addition to reduced expression/activity of SOD, the antioxidants glutathione (GSH), GPx, and catalase are downregulated in PAH and animal models of pulmonary hypertension (Table 6) (12, 50, 51, 58, 100, 105). A significant increase in myocardial lipid peroxidation along with a decrease in antioxidant reserve are found in the right ventricle in the late stage of MCT-induced pulmonary hypertension (58).
Taken together, loss of antioxidant response contributes to greater capacity for ROS generation and increased oxidative stress in PAH. Targeted therapies to decrease ROS generation may be beneficial in PAH.
TCA Cycle and Electron Transport Chain (ETC)
Within the mitochondria, when metabolic processes are used for energy production, the final step is oxidative phosphorylation. The electron transport chain (ETC), located in the mitochondrial inner membrane, consists of a series of complexes that receive NADH and FADH2 from TCA to generate ATP. Complexes I and II, along with the flavoprotein-ubiquinone oxidoreductase, transfer electrons from different sources to ubiquinone (coenzyme Q). Next, electrons are transferred sequentially to Complex III, cytochrome c, Complex IV, and finally to molecular oxygen, the terminal electron acceptor. Protons are pumped by Complexes I, III, and IV into the mitochondrial intermembrane space, generating an electrochemical gradient. These protons are then used by mitochondrial F0F1 ATP synthase to synthesize ATP from ADP.
There is some evidence for deficiencies in the ETC in PAH. Expression and/or activity of mitochondrial complexes are reduced in PAEC (10) and small pulmonary arteries (14, 15) from patients with PAH, as well as in PH animal models (14, 15, 50–52) (Table 3)(Figure 3). While CoQ levels are similar between PAH patients and controls, CoQ levels rise to significantly higher levels in PAH patients with supplementation as compared to healthy controls. Supplementation with CoQ improves echocardiographic markers of right ventricular function (107), suggesting therapies targeting ETC may benefit patients.
Figure 3:
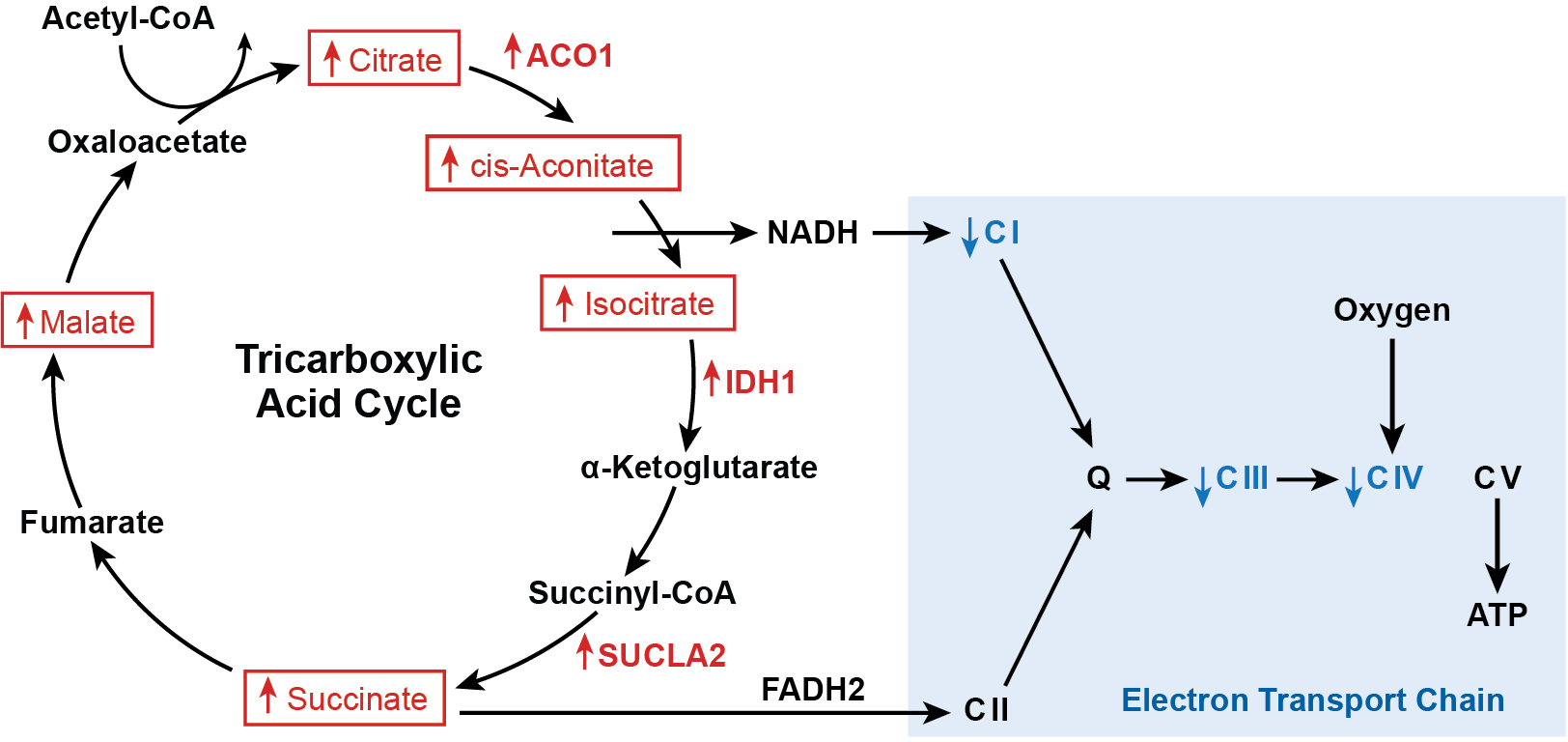
TCA cycle and ETC in PAH. Increased TCA intermediates and downregulated ETC Complexes I, III, and IV. Abbreviations: ACO1, aconitase 1; IDH1, isocitrate dehydrogenase 1; SUCLA2, succinate coA ligase beta subunit; CI, Complex I; CII, Complex II; Q, coenzyme Q; CIII, Complex III; CIV, Complex IV; CV, Complex V.
While the TCA cycle is important for the oxidation of substrates for energy production (ATP), it is also the biochemical hub that facilitates the traffic flow of carbon in the form of acetyl coenzyme A (acetyl-CoA), receiving inputs from all metabolic pathways and providing the biomolecules necessary for cell function, proliferation, and signal transduction. Nontargeted metabolomics, proteomics and transcriptomics analyses indicate that PAH PAEC have abnormal TCA cycle mitochondrial metabolic pathways (12). Metabolomics study reveals higher TCA cycle metabolites such as citrate, isocitrate, cis-aconitate, succinate, and malate in PAH plasma and lung when compared to control (Figure 3) (12, 34, 44). The higher levels are accompanied by increased expression of genes of TCA cycle enzymes such as aconitase (ACO)1, isocitrate dehydrogenase (IDH)1, and succinate-CoA ligase beta subunit (SUCLA2) (Figure 3) in PAH lung (34) and biopsies of right ventricles from individuals with PAH (39, 56). Proteomic study of PAEC shows lower levels of the mitochondrial citrate carrier solute carrier family 25A1 (SLC25A1) (12, 108). SLC25A1 exchanges mitochondrial citrate to cytosolic malate. Transporting citrate from the mitochondrial to the cytoplasmic compartment is essential to form acetyl-CoA to be used for fatty acid and sterol synthesis in the cell (109) (Figure 2c). Finally, HIF accumulation is adjusted in response to nonhypoxic stimuli of TCA intermediates (49). Prolyl hydroxylases target HIF for proteasomal degradation using both oxygen and αKG as substrates, while succinate and fumarate are able to compete with αKG for binding to the catalytic center to decrease degradation of HIF. Thus, TCA metabolism and HIF activity are closely intertwined.
III. GENETICS AND METABOLISM IN PAH
Heritable PAH (HPAH, WSPH 1.2) includes patients with an identifiable genetic mutation and/or a family history of the disease. Genetic mutations are found in 25 − 30% of Idiopathic PAH patients (IPAH, WSPH 1.1) (Table 1) (2, 3). Heterozygous germline mutations of the BMPR2 gene, a serine-threonine kinase receptor of transforming growth factor (TGF)-β superfamily that signals to inhibit cell growth, are identifiable in approximately 75% of HPAH cases and in approximately 20% of IPAH cases (22, 110, 111). Human PAEC with genetic knockdown of BMPR2 using siRNA have higher levels of glycolysis (112) (Table 7). Similarly, human pulmonary microvascular endothelial cells stably transfected with mutant BMPR2 have greater glycolysis, pentose phosphate, nucleotide salvage, and polyamine biosynthesis pathways. In addition, the stably transfected cells have less carnitine metabolism, fatty acid β-oxidation, and TCA cycle intermediates, i.e., malate, fumarate, and succinate (108). Activity of the TCA cycle enzyme IDH is increased in cells stably expressing mutant BMPR2 and importantly in the serum of PAH patients with heritable or idiopathic PAH (108). Increased glutamine metabolism is also present in BMPR2-mutant pulmonary microvascular endothelial cells from genetically modified mice, suggesting that the BMPR2 mutation leads to abnormal metabolic pathways (76). Lung endothelial cells of BMPR2-deletion mice have elevation of mitochondrial ROS (112). The right ventricle of the BMPR2-mutant mouse has increased lipid accumulation, decreased acylcarnitines, and decreased fatty acid oxidation (39, 56). HIF-1α accumulation is increased in lungs of the BMPR2-mutant mice exposed to bleomycin (113), and in endothelial cells of BMPR2-deleted mice with hypoxia-induced PH (114). All these studies link BMPR2, HIF-1α, and metabolism in the pathophysiology of PH.
Table 7.
Pathogenic mutations in PAH
| Gene | Located | Parameters | References |
|---|---|---|---|
| BMPR2 | Chromosome 2 |
↑Citrate, ↑Glutamine metabolism, ↑Glycolysis, ↑HIF-1α expression, ↑Isocitrate dehydrogenase (IDH) activity, ↑Lipid accumulation, ↑Mitochondrial ROS, ↓Acylcarnitines, ↓Fatty acid β-oxidation, ↓Malate, fumarate, and succinate |
(39, 56, 76, 108, 112–114) |
| ALK-1 | Chromosome 12 | glucose and lipid metabolism | (119) |
| ENG | Chromosome 9 |
↓Hepatic triglyceride content, ↓Insulin levels, ↓NO production [↓eNOS] |
(120, 121) |
| SMAD9 | Chromosome 13 |
||
| CAV1 | Chromosome 7 |
↑Lipid uptake, ↑14C-palmitate oxidation, ↑cAMP, ↑eNOS-derived NO synthesis and vasodilation, ↓Triglycerides, fatty acids, and cholesterol |
(57, 125) |
| KCNK3 | Chromosome 2 |
↑cAMP, ↑Lipolysis rate, ↓Lipid accumulation |
(124) |
Other mutations are also associated with PAH (Tables 1, 7), but there are few studies of associated metabolic abnormalities. Mutations in genes encoding other components of the TGF-β/bone morphogenetic protein signalling pathways include activin A receptor like type 1 (ALK-1), endoglin (ENG), and SMAD Family Member 9 (SMAD9 also known as SMAD8) (115–119). Compared with wild-type mice, mice deficient in ENG show decreased hepatic triglyceride content, lower insulin levels (120), and decreased NO production due to decreased levels of eNOS (121). Mutations in potassium channel subfamily K member 3 (KCNK3) (122) and caveolin-1 (CAV1) (123) also are associated with PAH. KCNK3-deficient cells show increased membrane potential, lipolysis rates, and cAMP production (124). Mutation in CAV1, which encodes the predominant protein of caveolae plasma membranes, increases lipid uptake but decreases myocardial triglyceride, fatty acid, and cholesterol levels (57), and enhances eNOS-derived NO synthesis and vasodilation (125).
Recent work also supports a role for mitochondrial genetics in PAH, mitochondria contain ~1200 proteins, most encoded by nuclear DNA and imported into the mitochondria (99). Mitochondria have their own circular DNA (mtDNA), which contains 13 genes that encode oxidative phosphorylation proteins crucial for bioenergetic function and that are transmitted to subsequent generations exclusively through the maternal lineage. Due to limited repair, mutations occur with greater frequency in mtDNA than in the nuclear genome (126, 127). Adding to the complexity of studying mtDNA mutations or variants, cells have many mitochondria, and each mitochondrion has ~10 copies of mtDNA. Thus, mtDNA mutations can be heteroplasmic, when different mtDNA variants co-exist within a cell, or homoplasmic, when mutations occur in all the mtDNA within the cell. Mitochondrial haplogroups are defined collections of homoplasmic mutations that are maternally inherited. While haplogroups do not cause disease, they are associated with increased or decreased risk of metabolic, degenerative, infectious, and autoimmune diseases and have been identified as at risk or protective from cardiovascular disease, type 2 diabetes mellitus, neurodegenerative diseases and several types of cancer (27, 126, 128). Several mitochondrial haplogroups M and HV, JT and UK of the N macro-haplogroup are associated with the risk of PAH (27). mtDNA copy number is associated with cardiovascular disease and human cancers (129, 130). In a meta-analysis comparing individuals with cardiovascular disease to controls, mtDNA copy number relative to nuclear DNA is a biomarker of mitochondrial function and could predict cardiovascular events and mortality in longitudinal studies, i.e., lower mtDNA copy number in blood predicts more cardiovascular events (131). mtDNA copy numbers are decreased in PAH PAEC (10) but have not been evaluated in PAH patients.
The impact of nuclear and mitochondrial mutations on metabolism in PAH is relatively underexplored. The roles of mitochondrial haplogroups, hetero- and homoplasmy, mtDNA copy numbers, and the crosstalk between the nuclear and mitochondrial genomes needs further investigation. Discovering the connection between genetics and metabolism could be beneficial for further understanding the genetic mechanisms that underlie the disease.
IV. METABOLISM AND POTENTIAL THERAPEUTICS FOR PAH
Metabolic changes in PAH suggest new metabolic avenues for therapeutics. Preclinical and early clinical data support targeting many of the mitochondrial pathways to improve disease severity and outcomes. Specifically targeting glucose metabolism has been proposed in the treatment of PAH and shows some success. Dichloroacetate, an inhibitor of pyruvate dehydrogenase kinase, improves RV function in animal PH models and increases mitochondrial respiration, lowers the mean PA pressure and PVR, and improves functional capacity (6-min walk) in patients with PAH (36, 132, 133). Targeting the glycolytic enzymes ENO and PFKFB3 decreases glucose metabolism and improves RV function in rat PH models (35, 37). mTOR inhibitors, which target HIF-1α and block glycolysis, may also be viable treatments for PAH (134–136). Carvedilol, an adrenergic receptor blocker, decreases expression of glycolytic enzymes, and reverses RV remodeling in a rat PH model (41, 137). Carvedilol also decreases RV glycolysis measured by FDG uptake and improves cardiac function in patients with PAH (47, 48).
Decreasing long-chain fatty acid oxidation with trimetazidine and ranolazine leads to improved cardiac output and treadmill distance in a RV hypertrophy rat model (138). Metformin, an antihyperglycemic agent, enhances fatty acid oxidation and reduces lipid deposition in the RV of BMPR2-mutant transgenic mouse models (39). The glutamine antagonist 6-Diazo-5-oxo-L-norleucine (DON) inhibits glutaminolysis, abrogates RV hypertrophy, and improves cardiac function and treadmill distance in a RV hypertrophy rat model (43). Inhibitors of GLS1 decrease pulmonary vascular cell proliferation in the monocrotaline rat model of PH (78). Likewise, 2-hydroxyben-zylamine (2HOBA), which decreases glutamine metabolism, improves cardiac output, and decreases RVSP in a BMPR2-mutant mouse PH model (76). Arginase inhibitors reverse lung tissue remodeling, reduce RVSP, and improve NO bioavailability in animal PH models (89, 90). Oral arginine therapy lowers pulmonary artery systolic pressures in PH patients with sickle cell disease (139).
Improving REDOX balance is another highly interesting avenue for treatment. High dietary vitamin E decreases mitochondrial H2O2 production and mitochondrial oxidative stress in avian species with pulmonary hypertension syndrome (104). Treatment with MnTBAP, a synthetic SOD-mimetic, lowers mean PA pressure and PVR, and improves treadmill distance in a PH rat model (132). In humans, supplementation with Coenzyme Q, critical in mitochondria functions and REDOX, promotes greater mitochondrial respiration and improves echocardiographic markers of RV function in PAH (107).
These reports of metabolic interventions successes are encouraging. However, further work is needed to define whether metabolic interventions will improve survival and/or reverse the cardiopulmonary remodeling of the disease. Almost certainly a precision approach for care will be needed for metabolic interventions as well as close observation over time to ensure that there are no, or limited, adverse effects. There is also a gap in our knowledge of how to best combine metabolic therapies in the context of current PH therapeutic approaches. Given the continued morbidity and mortality of PAH despite available therapies, these new metabolic concepts are likely to provide further advances in the care of patient.
V. SUMMARY AND FUTURE DIRECTIONS
Summary Points
Metabolic abnormalities in PAH vary by cell type (PAEC vs PASMC) and organ (heart vs lung).
It is uncertain as to whether metabolic derangements are causative or compensatory for the disease.
HIF stabilization is mechanistically linked to altered metabolism.
Arginine metabolic fate connects metabolism to the vasoconstrictive phenotype of PAH.
Nuclear and mitochondrial genetics influence metabolism and risks for PAH.
Metabolic abnormalities are found in human PAH and animal models of PH. Key hallmarks of PAH metabolism include a shift to glycolysis, increased glutamine utilization and one carbon metabolism, and decreased fatty acid oxidation. Increased ROS generation and changes in TCA cycle intermediates may contribute to HIF stabilization, driving the metabolic changes seen. Arginine metabolism, which links NO production and TCA cycle, has a key role linking the vasoconstrictive phenotype of the disease with the metabolic abnormalities. The metabolic phenotype of PAH is characteristic of proliferative cells such as cancer. Thus, therapies being used to target metabolism in cancer might have relevance in addressing the hyperproliferation seen in vascular cells in PAH.
The interconnectedness of metabolic pathways, genetics, and substrate availability and utilization are difficult to study in vivo and in vitro. Novel bioinformatics methods, including genomics, transcriptomics, proteomics, metabolomics, network analysis, and systems biology, are powerful tools for a deeper understanding of metabolism in silico. Big data and computational modeling will provide tools by which we can address the questions of how the metabolic abnormalities in PAH develop and determine which pathways are ideal therapeutic targets.
While traditional therapies targeting the vasodilator–vasoconstrictor imbalance in PAH have resulted in improved survival, they are insufficient to cure the disease. Fully understanding mitochondrial dysfunction and metabolic abnormalities of PAH over the course of the disease are the essential next step for developing therapies.
Acknowledgments:
We are thankful to A. Mulya, S. Comhair, K. Asosingh and S. Farha for critical input to the manuscript and D. Schumick for artwork. WX, AJJ, and SCE are supported by HL060917.
ABBREVIATIONS:
- 3-PGA
3-phosphoglycerate
- αKG
α-ketoglutarate
- ACACA
acetyl-CoA carboxylase 1
- ACAT
acetyl-CoA Acetyl transferase
- acetyl-CoA
acetyl coenzyme A
- ACO
aconitase
- ACSL1
fatty acetyl CoA L1
- acyl-CoA
acyl-coenzyme A
- ALDH18A1
aldehyde dehydrogenase 18 family, member A1
- ALK-1
activin A receptor like type 1
- ARG
arginase
- BEC
(S)-(2-boronoethyl)-l-cysteine-HCl
- BMPR2
bone morphogenetic protein receptor type-II
- CAT
cationic amino acid transporter
- CAV1
caveolin-1
- CPT
carnitine palmitoyltransferase
- ECI
enoyl-CoA delta isomerase
- ENG
endoglin
- ENO
enolase
- eNOS
endothelial nitric oxide synthase
- ETC
electron transport chain
- FBP
fructose 1,6-bisphosphate
- FDG-PET
fluorodeoxyglucose positron emission tomography
- fMet
N-formylmethionine
- G6PC3
glucose-6-phosphatase catalytic subunit 3
- GLS
glutaminase
- GLUT
glucose transporter
- GPx
glutathione peroxidases
- GSH
glutathione
- GSSG
glutathione disulfide
- H2O2
hydrogen peroxide
- HIF
hypoxia-inducible factor
- HK
hexokinase
- HPAH
heritable pulmonary arterial hypertension
- IDH
isocitrate dehydrogenase
- KCNK3
potassium channel subfamily K member 3
- LDH
lactate dehydrogenase
- MCT
monocrotaline
- ME
malic enzyme
- mtDNA
mitochondrial DNA
- MTHFD1L
monofunctional C1-tetrahydrofolate (THF) synthase
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- NO
nitric oxide
- NRF
nuclear factor erythroid 2–related factor
- P5C
Δ1-pyrroline-5-carboxylate
- PAEC
pulmonary arterial endothelial cells
- PAH
pulmonary arterial hypertension
- PAP
pulmonary artery pressure
- PASMC
pulmonary artery smooth muscle cells
- PEP
phosphoenolpyruvate
- PFKFB
6-phosphofructo-2-kinase/fructose-2, 6-biphosphatase
- PFKM
phosphofructokinase
- PGC
peroxisome proliferator-activated receptor gamma coactivator
- PGI2
prostacyclin
- PVR
pulmonary vascular resistance
- •OH
hydroxyl radicals
- O2•−
superoxide
- REDOX
reducing and oxidizing
- ROS
reactive oxygen species
- RVSP
right ventricular systolic pressure
- SHMT
serine hydroxymethyltransferase
- SLC1A5
solute carrier family 1 member 5
- SLC25A1
solute carrier family 25A1
- SMAD9
SMAD family member 9
- SOD
superoxide dismutase
- SUCLA2
succinate-CoA ligase beta subunit
- TCA
tricarboxylic acid
- TFAM
mitochondrial transcription factor A
- THF
tetrahydrofolate
- UCP
uncoupling protein
- WSPH
World Symposium on Pulmonary Hypertension
Footnotes
Conflicts of interest: The authors have declared that no conflict of interest exists.
REFERENCES
- 1.Rabinovitch M 2008. Molecular pathogenesis of pulmonary arterial hypertension. J Clin Invest 118: 2372–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simonneau G, Montani D, Celermajer DS, Denton CP, Gatzoulis MA, et al. 2019. Haemodynamic definitions and updated clinical classification of pulmonary hypertension. Eur Respir J 53: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Morrell NW, Aldred MA, Chung WK, Elliott CG, Nichols WC, et al. 2019. Genetics and genomics of pulmonary arterial hypertension. Eur Respir J 53: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tuder RM, Marecki JC, Richter A, Fijalkowska I, Flores S. 2007. Pathology of pulmonary hypertension. Clin Chest Med 28: 23–42, vii [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Humbert M, Guignabert C, Bonnet S, Dorfmuller P, Klinger JR, et al. 2019. Pathology and pathobiology of pulmonary hypertension: state of the art and research perspectives. Eur Respir J 53: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yoder MC, Mead LE, Prater D, Krier TR, Mroueh KN, et al. 2007. Redefining endothelial progenitor cells via clonal analysis and hematopoietic stem/progenitor cell principals. Blood 109: 1801–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Xu W, Erzurum SC. 2011. Endothelial cell energy metabolism, proliferation, and apoptosis in pulmonary hypertension. In American Physiological Society. Compr Physiol 1: 357–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu W, Kaneko FT, Zheng S, Comhair SA, Janocha AJ, et al. 2004. Increased arginase II and decreased NO synthesis in endothelial cells of patients with pulmonary arterial hypertension. Faseb J 18: 1746–8 [DOI] [PubMed] [Google Scholar]
- 9.Masri FA, Xu W, Comhair SA, Asosingh K, Koo M, et al. 2007. Hyperproliferative apoptosis-resistant endothelial cells in idiopathic pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 293: L548–54 [DOI] [PubMed] [Google Scholar]
- 10.Xu W, Koeck T, Lara AR, Neumann D, DiFilippo FP, et al. 2007. Alterations of cellular bioenergetics in pulmonary artery endothelial cells. Proc Natl Acad Sci U S A 104: 1342–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fijalkowska I, Xu W, Comhair SA, Janocha AJ, Mavrakis LA, et al. 2010. Hypoxia inducible-factor1alpha regulates the metabolic shift of pulmonary hypertensive endothelial cells. Am J Pathol 176: 1130–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xu W, Comhair SAA, Chen R, Hu B, Hou Y, et al. 2019. Integrative proteomics and phosphoproteomics in pulmonary arterial hypertension. Sci Rep 9: 18623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Archer S, Rich S. 2000. Primary pulmonary hypertension: a vascular biology and translational research “Work in progress”. Circulation 102: 2781–91 [DOI] [PubMed] [Google Scholar]
- 14.Archer SL, Gomberg-Maitland M, Maitland ML, Rich S, Garcia JG, Weir EK. 2008. Mitochondrial metabolism, redox signaling, and fusion: a mitochondria-ROS-HIF-1alpha-Kv1.5 O2-sensing pathway at the intersection of pulmonary hypertension and cancer. Am J Physiol Heart Circ Physiol 294: H570–8 [DOI] [PubMed] [Google Scholar]
- 15.Bonnet S, Michelakis ED, Porter CJ, Andrade-Navarro MA, Thebaud B, et al. 2006. An abnormal mitochondrial-hypoxia inducible factor-1alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats: similarities to human pulmonary arterial hypertension. Circulation 113: 2630–41 [DOI] [PubMed] [Google Scholar]
- 16.Hernandez-Saavedra D, Sanders L, Freeman S, Reisz JA, Lee MH, et al. 2020. Stable isotope metabolomics of pulmonary artery smooth muscle and endothelial cells in pulmonary hypertension and with TGF-beta treatment. Sci Rep 10: 413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaneko FT, Arroliga AC, Dweik RA, Comhair SA, Laskowski D, et al. 1998. Biochemical reaction products of nitric oxide as quantitative markers of primary pulmonary hypertension. Am J Respir Crit Care Med 158: 917–23 [DOI] [PubMed] [Google Scholar]
- 18.Fagan KA, McMurtry I, Rodman DM. 2000. Nitric oxide synthase in pulmonary hypertension: lessons from knockout mice. Physiol Res 49: 539–48 [PubMed] [Google Scholar]
- 19.Machado RF, Londhe Nerkar MV, Dweik RA, Hammel J, Janocha A, et al. 2004. Nitric oxide and pulmonary arterial pressures in pulmonary hypertension. Free Radic Biol Med 37: 1010–7 [DOI] [PubMed] [Google Scholar]
- 20.Girgis RE, Champion HC, Diette GB, Johns RA, Permutt S, Sylvester JT. 2005. Decreased exhaled nitric oxide in pulmonary arterial hypertension: response to bosentan therapy. Am J Respir Crit Care Med 172: 352–7 [DOI] [PubMed] [Google Scholar]
- 21.Farber HW, Miller DP, Poms AD, Badesch DB, Frost AE, et al. 2015. Five-Year outcomes of patients enrolled in the REVEAL Registry. Chest 148: 1043–54 [DOI] [PubMed] [Google Scholar]
- 22.Aldred MA, Comhair SA, Varella-Garcia M, Asosingh K, Xu W, et al. 2010. Somatic chromosome abnormalities in the lungs of patients with pulmonary arterial hypertension. Am J Respir Crit Care Med 182: 1153–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Asosingh K, Aldred MA, Vasanji A, Drazba J, Sharp J, et al. 2008. Circulating angiogenic precursors in idiopathic pulmonary arterial hypertension. Am J Pathol 172: 615–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Asosingh K, Erzurum S. 2018. Mechanisms of right heart disease in pulmonary hypertension (2017 Grover Conference Series). Pulm Circ 8: 2045893217753121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cheong HI, Asosingh K, Stephens OR, Queisser KA, Xu W, et al. 2016. Hypoxia sensing through beta-adrenergic receptors. JCI Insight 1: e90240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farha S, Asosingh K, Xu W, Sharp J, George D, et al. 2011. Hypoxia-inducible factors in human pulmonary arterial hypertension: a link to the intrinsic myeloid abnormalities. Blood 117: 3485–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Farha S, Hu B, Comhair S, Zein J, Dweik R, et al. 2016. Mitochondrial Haplogroups and Risk of Pulmonary Arterial Hypertension. PLoS One 11: e0156042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ghosh S, Gupta M, Xu W, Mavrakis DA, Janocha AJ, et al. 2016. Phosphorylation inactivation of endothelial nitric oxide synthesis in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 310: L1199–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lundgrin EL, Park MM, Sharp J, Tang WH, Thomas JD, et al. 2013. Fasting 2-deoxy-2-[18F]fluoro-D-glucose positron emission tomography to detect metabolic changes in pulmonary arterial hypertension hearts over 1 year. Ann Am Thorac Soc 10: 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kao CC, Wedes SH, Hsu JW, Bohren KM, Comhair SA, et al. 2015. Arginine metabolic endotypes in pulmonary arterial hypertension. Pulm Circ 5: 124–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saygin D, Highland KB, Farha S, Park M, Sharp J, et al. 2017. Metabolic and Functional Evaluation of the Heart and Lungs in Pulmonary Hypertension by Gated 2-[18F]-Fluoro-2-deoxy-D-glucose Positron Emission Tomography. Pulm Circ 7: 428–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Owen OE, Kalhan SC, Hanson RW. 2002. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem 277: 30409–12 [DOI] [PubMed] [Google Scholar]
- 33.Potente M, Carmeliet P. 2017. The Link Between Angiogenesis and Endothelial Metabolism. Annu Rev Physiol 79: 43–66 [DOI] [PubMed] [Google Scholar]
- 34.Zhao Y, Peng J, Lu C, Hsin M, Mura M, et al. 2014. Metabolomic heterogeneity of pulmonary arterial hypertension. PLoS One 9: e88727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dai J, Zhou Q, Chen J, Rexius-Hall ML, Rehman J, Zhou G. 2018. Alpha-enolase regulates the malignant phenotype of pulmonary artery smooth muscle cells via the AMPK-Akt pathway. Nat Commun 9: 3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao L, Ashek A, Wang L, Fang W, Dabral S, et al. 2013. Heterogeneity in lung (18)FDG uptake in pulmonary arterial hypertension: potential of dynamic (18)FDG positron emission tomography with kinetic analysis as a bridging biomarker for pulmonary vascular remodeling targeted treatments. Circulation 128: 1214–24 [DOI] [PubMed] [Google Scholar]
- 37.Cao Y, Zhang X, Wang L, Yang Q, Ma Q, et al. 2019. PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc Natl Acad Sci U S A 116: 13394–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Can MM, Kaymaz C, Tanboga IH, Tokgoz HC, Canpolat N, et al. 2011. Increased right ventricular glucose metabolism in patients with pulmonary arterial hypertension. Clin Nucl Med 36: 743–8 [DOI] [PubMed] [Google Scholar]
- 39.Hemnes AR, Brittain EL, Trammell AW, Fessel JP, Austin ED, et al. 2014. Evidence for right ventricular lipotoxicity in heritable pulmonary arterial hypertension. Am J Respir Crit Care Med 189: 325–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sharma S, Sud N, Wiseman DA, Carter AL, Kumar S, et al. 2008. Altered carnitine homeostasis is associated with decreased mitochondrial function and altered nitric oxide signaling in lambs with pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 294: L46–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Drake JI, Bogaard HJ, Mizuno S, Clifton B, Xie B, et al. 2011. Molecular signature of a right heart failure program in chronic severe pulmonary hypertension. Am J Respir Cell Mol Biol 45: 1239–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Piao L, Fang YH, Cadete VJ, Wietholt C, Urboniene D, et al. 2010. The inhibition of pyruvate dehydrogenase kinase improves impaired cardiac function and electrical remodeling in two models of right ventricular hypertrophy: resuscitating the hibernating right ventricle. J Mol Med (Berl) 88: 47–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Piao L, Fang YH, Parikh K, Ryan JJ, Toth PT, Archer SL. 2013. Cardiac glutaminolysis: a maladaptive cancer metabolism pathway in the right ventricle in pulmonary hypertension. J Mol Med (Berl) 91: 1185–97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen C, Luo F, Wu P, Huang Y, Das A, et al. 2020. Metabolomics reveals metabolite changes of patients with pulmonary arterial hypertension in China. J Cell Mol Med 24: 2484–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sharma S, Aramburo A, Rafikov R, Sun X, Kumar S, et al. 2013. L-carnitine preserves endothelial function in a lamb model of increased pulmonary blood flow. Pediatr Res 74: 39–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Stanley WC, Recchia FA, Lopaschuk GD. 2005. Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85: 1093–129 [DOI] [PubMed] [Google Scholar]
- 47.Farha S, Saygin D, Park MM, Cheong HI, Asosingh K, et al. 2017. Pulmonary arterial hypertension treatment with carvedilol for heart failure: a randomized controlled trial. JCI Insight 2: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stephens OR, Weiss K, Frimel M, Rose JA, Sun Y, et al. 2019. Interdependence of hypoxia and beta-adrenergic receptor signaling in pulmonary arterial hypertension. Am J Physiol Lung Cell Mol Physiol 317: L369–L80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Semenza GL. 2010. HIF-1: upstream and downstream of cancer metabolism. Curr Opin Genet Dev 20: 51–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tang Z, Iqbal M, Cawthon D, Bottje WG. 2002. Heart and breast muscle mitochondrial dysfunction in pulmonary hypertension syndrome in broilers (Gallus domesticus). Comp Biochem Physiol A Mol Integr Physiol 132: 527–40 [DOI] [PubMed] [Google Scholar]
- 51.Iqbal M, Cawthon D, Wideman RF Jr., Bottje WG. 2001. Lung mitochondrial dysfunction in pulmonary hypertension syndrome. II. Oxidative stress and inability to improve function with repeated additions of adenosine diphosphate. Poult Sci 80: 656–65 [DOI] [PubMed] [Google Scholar]
- 52.Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, et al. 2003. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science 299: 896–9 [DOI] [PubMed] [Google Scholar]
- 53.Momken I, Fortin D, Serrurier B, Bigard X, Ventura-Clapier R, Veksler V. 2002. Endothelial nitric oxide synthase (NOS) deficiency affects energy metabolism pattern in murine oxidative skeletal muscle. Biochem J 368: 341–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Semenza GL. 2003. Targeting HIF-1 for cancer therapy. Nat Rev Cancer 3: 721–32 [DOI] [PubMed] [Google Scholar]
- 55.Mingatto FE, Maioli MA, Bracht A, Ishii-Iwamoto EL. 2008. Effects of monocrotaline on energy metabolism in the rat liver. Toxicol Lett 182: 115–20 [DOI] [PubMed] [Google Scholar]
- 56.Talati MH, Brittain EL, Fessel JP, Penner N, Atkinson J, et al. 2016. Mechanisms of Lipid Accumulation in the Bone Morphogenetic Protein Receptor Type 2 Mutant Right Ventricle. Am J Respir Crit Care Med 194: 719–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Augustus AS, Buchanan J, Gutman E, Rengo G, Pestell RG, et al. 2008. Hearts lacking caveolin-1 develop hypertrophy with normal cardiac substrate metabolism. Cell Cycle 7: 2509–18 [DOI] [PubMed] [Google Scholar]
- 58.Farahmand F, Hill MF, Singal PK. 2004. Antioxidant and oxidative stress changes in experimental cor pulmonale. Mol Cell Biochem 260: 21–9 [DOI] [PubMed] [Google Scholar]
- 59.Graham BB, Kumar R, Mickael C, Sanders L, Gebreab L, et al. 2015. Severe pulmonary hypertension is associated with altered right ventricle metabolic substrate uptake. Am J Physiol Lung Cell Mol Physiol 309: L435–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Nagaya N, Goto Y, Satoh T, Uematsu M, Hamada S, et al. 1998. Impaired regional fatty acid uptake and systolic dysfunction in hypertrophied right ventricle. J Nucl Med 39: 1676–80 [PubMed] [Google Scholar]
- 61.Habets DD, Coumans WA, Voshol PJ, den Boer MA, Febbraio M, et al. 2007. AMPK-mediated increase in myocardial long-chain fatty acid uptake critically depends on sarcolemmal CD36. Biochem Biophys Res Commun 355: 204–10 [DOI] [PubMed] [Google Scholar]
- 62.Kim Y, Goto H, Kobayashi K, Sawada Y, Miyake Y, et al. 1997. Detection of impaired fatty acid metabolism in right ventricular hypertrophy: assessment by I-123 beta-methyl iodophenyl pentadecanoic acid (BMIPP) myocardial single-photon emission computed tomography. Ann Nucl Med 11: 207–12 [DOI] [PubMed] [Google Scholar]
- 63.Zhuang W, Lian G, Huang B, Du A, Gong J, et al. 2019. CPT1 regulates the proliferation of pulmonary artery smooth muscle cells through the AMPK-p53-p21 pathway in pulmonary arterial hypertension. Mol Cell Biochem 455: 169–83 [DOI] [PubMed] [Google Scholar]
- 64.Brittain EL, Talati M, Fessel JP, Zhu H, Penner N, et al. 2016. Fatty Acid Metabolic Defects and Right Ventricular Lipotoxicity in Human Pulmonary Arterial Hypertension. Circulation 133: 1936–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stanley WC, Lopaschuk GD, Hall JL, McCormack JG. 1997. Regulation of myocardial carbohydrate metabolism under normal and ischaemic conditions. Potential for pharmacological interventions. Cardiovasc Res 33: 243–57 [DOI] [PubMed] [Google Scholar]
- 66.van der Vusse GJ, van Bilsen M, Glatz JF. 2000. Cardiac fatty acid uptake and transport in health and disease. Cardiovasc Res 45: 279–93 [DOI] [PubMed] [Google Scholar]
- 67.Krishnan J, Suter M, Windak R, Krebs T, Felley A, et al. 2009. Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab 9: 512–24 [DOI] [PubMed] [Google Scholar]
- 68.Mylonis I, Simos G, Paraskeva E. 2019. Hypoxia-Inducible Factors and the Regulation of Lipid Metabolism. Cells 8: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Brusselmans K, De Schrijver E, Verhoeven G, Swinnen JV. 2005. RNA interference-mediated silencing of the acetyl-CoA-carboxylase-alpha gene induces growth inhibition and apoptosis of prostate cancer cells. Cancer Res 65: 6719–25 [DOI] [PubMed] [Google Scholar]
- 70.Chajes V, Cambot M, Moreau K, Lenoir GM, Joulin V. 2006. Acetyl-CoA carboxylase alpha is essential to breast cancer cell survival. Cancer Res 66: 5287–94 [DOI] [PubMed] [Google Scholar]
- 71.Vazquez-Martin A, Corominas-Faja B, Oliveras-Ferraros C, Cufi S, Dalla Venezia N, Menendez JA. 2013. Serine79-phosphorylated acetyl-CoA carboxylase, a downstream target of AMPK, localizes to the mitotic spindle poles and the cytokinesis furrow. Cell Cycle 12: 1639–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sutendra G, Bonnet S, Rochefort G, Haromy A, Folmes KD, et al. 2010. Fatty acid oxidation and malonyl-CoA decarboxylase in the vascular remodeling of pulmonary hypertension. Sci Transl Med 2: 44ra58. [DOI] [PubMed] [Google Scholar]
- 73.Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, et al. 2011. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 481: 385–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.DeBerardinis RJ, Mancuso A, Daikhin E, Nissim I, Yudkoff M, et al. 2007. Beyond aerobic glycolysis: transformed cells can engage in glutamine metabolism that exceeds the requirement for protein and nucleotide synthesis. Proc Natl Acad Sci U S A 104: 19345–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Perez-Escuredo J, Dadhich RK, Dhup S, Cacace A, Van Hee VF, et al. 2016. Lactate promotes glutamine uptake and metabolism in oxidative cancer cells. Cell Cycle 15: 72–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Egnatchik RA, Brittain EL, Shah AT, Fares WH, Ford HJ, et al. 2017. Dysfunctional BMPR2 signaling drives an abnormal endothelial requirement for glutamine in pulmonary arterial hypertension. Pulm Circ 7: 186–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wise DR, Thompson CB. 2010. Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 35: 427–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bertero T, Oldham WM, Cottrill KA, Pisano S, Vanderpool RR, et al. 2016. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J Clin Invest 126: 3313–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kim B, Li J, Jang C, Arany Z. 2017. Glutamine fuels proliferation but not migration of endothelial cells. EMBO J 36: 2321–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fischer-Zirnsak B, Escande-Beillard N, Ganesh J, Tan YX, Al Bughaili M, et al. 2015. Recurrent De Novo Mutations Affecting Residue Arg138 of Pyrroline-5-Carboxylate Synthase Cause a Progeroid Form of Autosomal-Dominant Cutis Laxa. Am J Hum Genet 97: 483–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Skidmore DL, Chitayat D, Morgan T, Hinek A, Fischer B, et al. 2011. Further expansion of the phenotypic spectrum associated with mutations in ALDH18A1, encoding Delta(1)-pyrroline-5-carboxylate synthase (P5CS). Am J Med Genet A 155A: 1848–56 [DOI] [PubMed] [Google Scholar]
- 82.Morris SM Jr. 2006. Arginine: beyond protein. Am J Clin Nutr 83: 508S–12S [DOI] [PubMed] [Google Scholar]
- 83.Hecker M, Sessa WC, Harris HJ, Anggard EE, Vane JR. 1990. The metabolism of L-arginine and its significance for the biosynthesis of endothelium-derived relaxing factor: cultured endothelial cells recycle L-citrulline to L-arginine. Proc Natl Acad Sci U S A 87: 8612–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Leiper J, Vallance P. 1999. Biological significance of endogenous methylarginines that inhibit nitric oxide synthases. Cardiovasc Res 43: 542–8 [DOI] [PubMed] [Google Scholar]
- 85.Leiper JM, Santa Maria J, Chubb A, MacAllister RJ, Charles IG, et al. 1999. Identification of two human dimethylarginine dimethylaminohydrolases with distinct tissue distributions and homology with microbial arginine deiminases. Biochem J 343 Pt 1: 209–14 [PMC free article] [PubMed] [Google Scholar]
- 86.Sandqvist A, Schneede J, Kylhammar D, Henrohn D, Lundgren J, et al. 2018. Plasma L-arginine levels distinguish pulmonary arterial hypertension from left ventricular systolic dysfunction. Heart Vessels 33: 255–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, et al. 2005. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. JAMA 294: 81–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Grasemann H, Dhaliwal R, Ivanovska J, Kantores C, McNamara PJ, et al. 2015. Arginase inhibition prevents bleomycin-induced pulmonary hypertension, vascular remodeling, and collagen deposition in neonatal rat lungs. Am J Physiol Lung Cell Mol Physiol 308: L503–10 [DOI] [PubMed] [Google Scholar]
- 89.Jung C, Grun K, Betge S, Pernow J, Kelm M, et al. 2017. Arginase Inhibition Reverses Monocrotaline-Induced Pulmonary Hypertension. Int J Mol Sci 18: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Steppan J, Tran HT, Bead VR, Oh YJ, Sikka G, et al. 2016. Arginase Inhibition Reverses Endothelial Dysfunction, Pulmonary Hypertension, and Vascular Stiffness in Transgenic Sickle Cell Mice. Anesth Analg 123: 652–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xu W, Ghosh S, Comhair SA, Asosingh K, Janocha AJ, et al. 2016. Increased mitochondrial arginine metabolism supports bioenergetics in asthma. J Clin Invest 126: 2465–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jain M, Nilsson R, Sharma S, Madhusudhan N, Kitami T, et al. 2012. Metabolite profiling identifies a key role for glycine in rapid cancer cell proliferation. Science 336: 1040–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Martinez-Reyes I, Chandel NS. 2014. Mitochondrial one-carbon metabolism maintains redox balance during hypoxia. Cancer Discov 4: 1371–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Izquierdo-Garcia JL, Arias T, Rojas Y, Garcia-Ruiz V, Santos A, et al. 2018. Metabolic Reprogramming in the Heart and Lung in a Murine Model of Pulmonary Arterial Hypertension. Front Cardiovasc Med 5: 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Padron-Barthe L, Villalba-Orero M, Gomez-Salinero JM, Acin-Perez R, Cogliati S, et al. 2018. Activation of Serine One-Carbon Metabolism by Calcineurin Abeta1 Reduces Myocardial Hypertrophy and Improves Ventricular Function. J Am Coll Cardiol 71: 654–67 [DOI] [PubMed] [Google Scholar]
- 96.Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, Rabinowitz JD. 2014. Quantitative flux analysis reveals folate-dependent NADPH production. Nature 510: 298–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, et al. 2015. Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527: 186–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ye J, Fan J, Venneti S, Wan YW, Pawel BR, et al. 2014. Serine catabolism regulates mitochondrial redox control during hypoxia. Cancer Discov 4: 1406–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kraja AT, Liu C, Fetterman JL, Graff M, Have CT, et al. 2019. Associations of Mitochondrial and Nuclear Mitochondrial Variants and Genes with Seven Metabolic Traits. Am J Hum Genet 104: 112–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Masri FA, Comhair SAA, Dostanic-Larson I, Kaneko FT, Dweik RA, et al. 2008. Deficiency of Lung Antioxidants in Idiopathic Pulmonary Arterial Hypertension. Clinical and Translational Science 1: 99–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xu W, Erzurum SC. 2007. Airways inflammation and reactive oxygen/nitrogen species in pulmonary hypertension. In Oxidative Stress: Clinical and Biomedical Implications. Nova Science Publishers, Inc. : 259–76 [Google Scholar]
- 102.Grobe AC, Wells SM, Benavidez E, Oishi P, Azakie A, et al. 2006. Increased oxidative stress in lambs with increased pulmonary blood flow and pulmonary hypertension: role of NADPH oxidase and endothelial NO synthase. Am J Physiol Lung Cell Mol Physiol 290: L1069–77 [DOI] [PubMed] [Google Scholar]
- 103.Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, et al. 2004. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med 169: 764–9 [DOI] [PubMed] [Google Scholar]
- 104.Iqbal M, Cawthon D, Wideman RF Jr., Bottje WG. 2001. Lung mitochondrial dysfunction in pulmonary hypertension syndrome. I. Site-specific defects in the electron transport chain. Poult Sci 80: 485–95 [DOI] [PubMed] [Google Scholar]
- 105.Ghasemzadeh N, Patel RS, Eapen DJ, Veledar E, Al Kassem H, et al. 2014. Oxidative stress is associated with increased pulmonary artery systolic pressure in humans. Hypertension 63: 1270–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Guzy RD, Hoyos B, Robin E, Chen H, Liu L, et al. 2005. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab 1: 401–8 [DOI] [PubMed] [Google Scholar]
- 107.Sharp J, Farha S, Park MM, Comhair SA, Lundgrin EL, et al. 2014. Coenzyme Q supplementation in pulmonary arterial hypertension. Redox Biol 2: 884–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fessel JP, Hamid R, Wittmann BM, Robinson LJ, Blackwell T, et al. 2012. Metabolomic analysis of bone morphogenetic protein receptor type 2 mutations in human pulmonary endothelium reveals widespread metabolic reprogramming. Pulm Circ 2: 201–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Nota B, Struys EA, Pop A, Jansen EE, Fernandez Ojeda MR, et al. 2013. Deficiency in SLC25A1, encoding the mitochondrial citrate carrier, causes combined D-2- and L-2-hydroxyglutaric aciduria. Am J Hum Genet 92: 627–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, et al. 2000. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet 67: 737–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lane KB, Machado RD, Pauciulo MW, Thomson JR, Phillips JA 3rd, et al. 2000. Heterozygous germline mutations in BMPR2, encoding a TGF-beta receptor, cause familial primary pulmonary hypertension. The International PPH Consortium. Nat Genet 26: 81–4 [DOI] [PubMed] [Google Scholar]
- 112.Diebold I, Hennigs JK, Miyagawa K, Li CG, Nickel NP, et al. 2015. BMPR2 preserves mitochondrial function and DNA during reoxygenation to promote endothelial cell survival and reverse pulmonary hypertension. Cell Metab 21: 596–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bryant AJ, Robinson LJ, Moore CS, Blackwell TR, Gladson S, et al. 2015. Expression of mutant bone morphogenetic protein receptor II worsens pulmonary hypertension secondary to pulmonary fibrosis. Pulm Circ 5: 681–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Spiekerkoetter E, Tian X, Cai J, Hopper RK, Sudheendra D, et al. 2013. FK506 activates BMPR2, rescues endothelial dysfunction, and reverses pulmonary hypertension. J Clin Invest 123: 3600–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, et al. 2001. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 345: 325–34 [DOI] [PubMed] [Google Scholar]
- 116.Mache CJ, Gamillscheg A, Popper HH, Haworth SG. 2008. Early-life pulmonary arterial hypertension with subsequent development of diffuse pulmonary arteriovenous malformations in hereditary haemorrhagic telangiectasia type 1. Thorax 63: 85–6 [DOI] [PubMed] [Google Scholar]
- 117.Nasim MT, Ogo T, Ahmed M, Randall R, Chowdhury HM, et al. 2011. Molecular genetic characterization of SMAD signaling molecules in pulmonary arterial hypertension. Hum Mutat 32: 1385–9 [DOI] [PubMed] [Google Scholar]
- 118.Shintani M, Yagi H, Nakayama T, Saji T, Matsuoka R. 2009. A new nonsense mutation of SMAD8 associated with pulmonary arterial hypertension. J Med Genet 46: 331–7 [DOI] [PubMed] [Google Scholar]
- 119.Chen C, Grzegorzewski KJ, Barash S, Zhao Q, Schneider H, et al. 2003. An integrated functional genomics screening program reveals a role for BMP-9 in glucose homeostasis. Nat Biotechnol 21: 294–301 [DOI] [PubMed] [Google Scholar]
- 120.Beiroa D, Romero-Pico A, Langa C, Bernabeu C, Lopez M, et al. 2013. Heterozygous deficiency of endoglin decreases insulin and hepatic triglyceride levels during high fat diet. PLoS One 8: e54591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Jerkic M, Rivas-Elena JV, Prieto M, Carron R, Sanz-Rodriguez F, et al. 2004. Endoglin regulates nitric oxide-dependent vasodilatation. FASEB J 18: 609–11 [DOI] [PubMed] [Google Scholar]
- 122.Ma L, Roman-Campos D, Austin ED, Eyries M, Sampson KS, et al. 2013. A novel channelopathy in pulmonary arterial hypertension. N Engl J Med 369: 351–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Austin ED, Ma L, LeDuc C, Berman Rosenzweig E, Borczuk A, et al. 2012. Whole exome sequencing to identify a novel gene (caveolin-1) associated with human pulmonary arterial hypertension. Circ Cardiovasc Genet 5: 336–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chen Y, Zeng X, Huang X, Serag S, Woolf CJ, Spiegelman BM. 2017. Crosstalk between KCNK3-Mediated Ion Current and Adrenergic Signaling Regulates Adipose Thermogenesis and Obesity. Cell 171: 836–48 e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Razani B, Engelman JA, Wang XB, Schubert W, Zhang XL, et al. 2001. Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J Biol Chem 276: 38121–38 [DOI] [PubMed] [Google Scholar]
- 126.Wallace DC. 2015. Mitochondrial DNA variation in human radiation and disease. Cell 163: 33–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liu C, Fetterman JL, Liu P, Luo Y, Larson MG, et al. 2018. Deep sequencing of the mitochondrial genome reveals common heteroplasmic sites in NADH dehydrogenase genes. Hum Genet 137: 203–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Wallace DC. 2013. A mitochondrial bioenergetic etiology of disease. J Clin Invest 123: 1405–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Huang J, Tan L, Shen R, Zhang L, Zuo H, Wang DW. 2016. Decreased Peripheral Mitochondrial DNA Copy Number is Associated with the Risk of Heart Failure and Long-term Outcomes. Medicine (Baltimore) 95: e3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Reznik E, Miller ML, Senbabaoglu Y, Riaz N, Sarungbam J, et al. 2016. Mitochondrial DNA copy number variation across human cancers. Elife 5: [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Yue P, Jing S, Liu L, Ma F, Zhang Y, et al. 2018. Association between mitochondrial DNA copy number and cardiovascular disease: Current evidence based on a systematic review and meta-analysis. PLoS One 13: e0206003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Archer SL, Marsboom G, Kim GH, Zhang HJ, Toth PT, et al. 2010. Epigenetic attenuation of mitochondrial superoxide dismutase 2 in pulmonary arterial hypertension: a basis for excessive cell proliferation and a new therapeutic target. Circulation 121: 2661–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Michelakis ED, Gurtu V, Webster L, Barnes G, Watson G, et al. 2017. Inhibition of pyruvate dehydrogenase kinase improves pulmonary arterial hypertension in genetically susceptible patients. Sci Transl Med 9: [DOI] [PubMed] [Google Scholar]
- 134.Houssaini A, Abid S, Mouraret N, Wan F, Rideau D, et al. 2013. Rapamycin reverses pulmonary artery smooth muscle cell proliferation in pulmonary hypertension. Am J Respir Cell Mol Biol 48: 568–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Paddenberg R, Stieger P, von Lilien AL, Faulhammer P, Goldenberg A, et al. 2007. Rapamycin attenuates hypoxia-induced pulmonary vascular remodeling and right ventricular hypertrophy in mice. Respir Res 8: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Seyfarth HJ, Hammerschmidt S, Halank M, Neuhaus P, Wirtz HR. 2013. Everolimus in patients with severe pulmonary hypertension: a safety and efficacy pilot trial. Pulm Circ 3: 632–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bogaard HJ, Natarajan R, Mizuno S, Abbate A, Chang PJ, et al. 2010. Adrenergic receptor blockade reverses right heart remodeling and dysfunction in pulmonary hypertensive rats. Am J Respir Crit Care Med 182: 652–60 [DOI] [PubMed] [Google Scholar]
- 138.Fang YH, Piao L, Hong Z, Toth PT, Marsboom G, et al. 2012. Therapeutic inhibition of fatty acid oxidation in right ventricular hypertrophy: exploiting Randle’s cycle. J Mol Med (Berl) 90: 31–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Morris CR, Morris SM Jr., Hagar W, Van Warmerdam J, Claster S, et al. 2003. Arginine therapy: a new treatment for pulmonary hypertension in sickle cell disease? Am J Respir Crit Care Med 168: 63–9 [DOI] [PubMed] [Google Scholar]