

ABSTRACT
Although prokaryotic DNA methylation investigations have long focused on immunity against exogenous DNA, it has been recently recognized that DNA methylation impacts gene expression and phase variation in Streptococcus pneumoniae and Streptococcus suis. A comprehensive analysis of DNA methylation is lacking for beta-hemolytic streptococci, and thus we sought to examine DNA methylation in the major human pathogen group A Streptococcus (GAS). Using a database of 224 GAS genomes encompassing 80 emm types, we found that nearly all GAS strains encode a type I restriction modification (RM) system that lacks the hsdS′ alleles responsible for impacting gene expression in S. pneumoniae and S. suis. The GAS type I system is located on the core chromosome, while sporadically present type II orphan methyltransferases were identified on prophages. By combining single-molecule real-time (SMRT) analyses of 10 distinct emm types along with phylogenomics of 224 strains, we were able to assign 13 methylation patterns to the GAS population. Inactivation of the type I RM system, occurring either naturally through phage insertion or through laboratory-induced gene deletion, abrogated DNA methylation detectable via either SMRT or MinION sequencing. Contrary to a previous report, inactivation of the type I system did not impact transcript levels of the gene (mga) encoding the key multigene activator protein (Mga) or Mga-regulated genes. Inactivation of the type I system significantly increased plasmid transformation rates. These data delineate the breadth of the core chromosomal type I RM system in the GAS population and clarify its role in immunity rather than impacting Mga regulon expression.
IMPORTANCE The advent of whole-genome approaches capable of detecting DNA methylation has markedly expanded appreciation of the diverse roles of epigenetic modification in prokaryotic physiology. For example, recent studies have suggested that DNA methylation impacts gene expression in some streptococci. The data described herein are from the first systematic analysis of DNA methylation in a beta-hemolytic streptococcus and one of the few analyses to comprehensively characterize DNA methylation across hundreds of strains of the same bacterial species. We clarify that DNA methylation in group A Streptococcus (GAS) is primarily due to a type I restriction modification (RM) system present in the core genome and does not impact mga-regulated virulence gene expression, but does impact immunity against exogenous DNA. The identification of the DNA motifs recognized by each type I RM system may assist with optimizing methods for GAS genetic manipulation and help us understand how bacterial pathogens acquire exogenous DNA elements.
KEYWORDS: Streptococcus pyogenes, type I RM system, immunity
INTRODUCTION
Methylation of DNA impacts a broad variety of physiological functions in both prokaryotic and eukaryotic organisms. In prokaryotes, DNA methylation has mainly been investigated for its role in innate immunity although there is increasing recognition that DNA methylation can also modulate gene expression. Eukaryotes typically methylate cytosines on C5 (m5C). While prokaryotes also contain m5C, N6-methyladenine (m6A) is the predominant form of prokaryotic methylation, and N4-methylcytosine (m4C) is found exclusively in bacterial genomes (1, 2). In prokaryotes, base methylation is catalyzed by methyltransferases (MTases) that are typically components of restriction modification (RM) systems (3, 4). RM systems are widespread, being found in >90% of all sequenced prokaryotes (5, 6).
Bacterial RM systems are classified into four types based on components, sequence specificity, cofactors, and cleavage position (7, 8). The type I RM systems are hetero-oligomeric and contain an MTase (HsdM), a restriction endonuclease (REase [HsdR]), and a DNA specificity subunit (HsdS). HsdS combines with HsdM and HsdR to methylate and cleave unmethylated DNA targets, respectively. The DNA motifs targeted by type I RM systems are bipartite, and cleavage occurs at large distances from their binding sites (9). Type II RM systems bind short motifs, cleave within or close to the binding site, and are the most-studied systems, commercialized and used for genetic manipulation of DNA in laboratory protocols (10). While some type II systems consist of a single protein that functions as an MTase and REase (type IIG, IIB, and IIC), most have separate MTases and REases (11). Additionally, a large number of type II systems consist of only an orphan MTase (11, 12). Of these, the Dam and CcrM methylases are well-studied examples (13, 14). In type III systems, res and mod encode the REase and MTase, respectively. These systems recognize short palindromic motifs and cleave outside the binding site (15). Some type I and type III RM systems are phase variable and can alter their methylation patterns reversibly by swapping specificity components (16). In contrast to the groups described above, type IV systems have only an REase. DNA targets containing methylated, hydroxymethylated, or glucosyl-hydroxymethylated bases are cleaved (7).
In addition to distinguishing between self and nonself, prokaryotic methylation affects a variety of cellular functions, such as DNA mismatch repair, cell cycle control, transcriptional regulation, and virulence (13, 17). Phase-variable RM systems impact the virulence of numerous clinically important organisms, including Helicobacter pylori (18), Neisseria meningitidis (19), Haemophilus influenzae (20), and Streptococcus pneumoniae (21, 22). Phase variation in Streptococcus pneumoniae and Streptococcus suis is achieved by recombination between tandemly arranged HsdS subunits in the type I RM system (21, 23). In contrast, there has been less study of DNA methylation in beta-hemolytic streptococci. A phage-borne type II m5C MTase was the first characterized MTase in Streptococcus pyogenes, also known as group A Streptococcus (GAS). It was found to protect genomic DNA from digestion by SmaI and was speculated to function in maintaining erythromycin-resistant GAS populations (24). Nye et al. (25) recently reported that a type I RM system in emm28 GAS was responsible for the majority of DNA methylation and that inactivation of the system decreased transcription of the gene (mga) encoding the key multigene activator (Mga) transcriptional regulator. Conversely, inactivation of the type I RM system in an emm1 GAS strain did not impact virulence gene transcript levels (26). Currently, a systematic examination of RM systems in GAS is not available.
The large number of available GAS genome sequences and the clustering of GAS strains by emm types potentially make GAS a useful organism for understanding the distribution and function of RM systems among beta-hemolytic streptococci. Herein, we took a combined bioinformatic and biological approach to study GAS RM systems. We found that the core GAS genome contains a single type I RM system and that variable type II RM systems are located on prophages. Through PacBio single-molecule real-time (SMRT) sequencing, we compiled the various DNA motifs methylated by the type I RM systems in different GAS strains and found that GAS emm types that cluster based on their core gene alignment do not necessarily harbor similar methylation target specificities. Finally, we inactivated the type I MTase in two distinct emm types and found that the type I MTases did not contribute to mga or Mga-regulated virulence gene transcript levels but did impact efficiency of DNA uptake. Taken together, these data are the first comprehensive analysis of RM systems in GAS and suggest that the type I RM systems ubiquitously present in GAS are likely primarily involved in innate immunity rather than modulation of genes that are Mga regulated.
RESULTS
Overview of RM systems in GAS and closely related streptococci.
Only a handful of studies of RM systems using a large number of strains from the same bacterial species have been reported, and none in beta-hemolytic streptococci (27, 28). We analyzed 224 GAS genomes from 80 different emm types to understand the diversity of RM systems (see Table S1 in the supplemental material). We found that GAS strains generally contain a single, core chromosomal type I RM system, along with variable type II systems located in prophages (Fig. 1A). We found no evidence of type IIG, type III, or type IV systems. The type I RM system was consistently present at the same location in the GAS genome (Fig. 1A) and exhibited an hsdRSM arrangement. There were isolated GAS strains in which the type I system was either interrupted by a prophage or completely deleted, as has been previously reported (29, 30).
FIG 1.
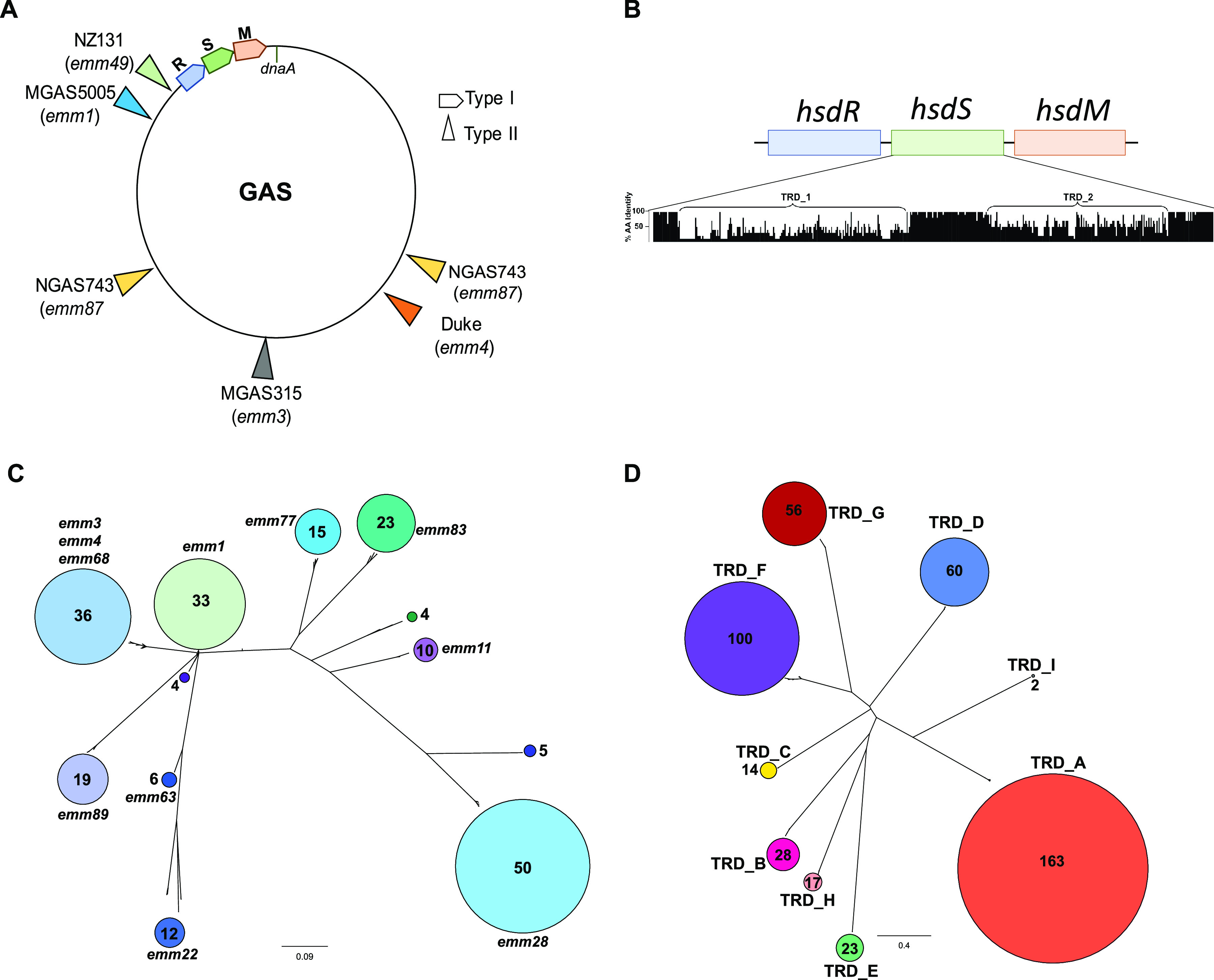
Overview of GAS type I RM systems. (A) A schematic figure shows the location of the type I RM system on the GAS chromosome. Locations of selected type II RM systems relative to dnaA are also shown, and the respective GAS strain and emm type are indicated in parentheses. (B) Alignment of HsdS sequences from the 10 most common target recognition domain (TRD) patterns identified in our GAS population. The percentage of amino acid identity across each position was calculated using Jalview (72) and is displayed (0 to 100%) on the y axis of the histogram. The x axis indicates amino acid positions. (C and D) Unrooted, neighborhood-joining trees based on (C) full-length HsdS sequences and (D) TRD sequence alignment from the 224 GAS strains in this study. GAS emm types dominating each HsdS cluster are indicated. The number of strains (C) and TRDs (D) in each cluster is indicated within the colored circles and reflected by circle size.
List of GAS strains used for TRD and phylogenomic analysis. Download Table S1, XLSX file, 0.03 MB (35.6KB, xlsx) .
Copyright © 2021 DebRoy et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
HsdR and HsdM proteins were highly conserved among GAS strains (>95% identity at the amino acid level for both proteins across the 224 genomes), while GAS HsdS was highly variable, as will be discussed later. GAS HsdR is nearly identical (93% identical and 96% similar at the amino acid level) to HsdR present in 10 Streptococcus agalactiae (also known as group B Streptococcus [GBS]) strains, although the vast majority of sequenced GBS strains did not contain a GAS HsdR homolog (Table 1). GAS HsdR was ∼70% similar to HsdR homologs present in various viridans group streptococci (VGS) (i.e., S. pneumoniae) and 64/78% identical/similar to a single strain of Streptococcus dysgalactiae (Streptococcus sp. strain 714). Analogous to HsdR, GAS HsdM was 97/99% identical/similar to HsdM homologs present in ∼30 GBS strains, although like HsdR, most GBS strains did not contain a GAS HsdM homolog (Table 1). Many GBS strains that contained a GAS HsdM homolog lacked an HsdR homolog and evidenced loss of genetic material at the location of the type I RM system site (see Fig. S1 in the supplemental material). GAS HsdM had ∼74/86% identity/similarity to three S. dygalactiae strains as well as many other VGS (Table 1). Taken together, these data showed that the GAS HsdR and HsdM proteins are not widely shared among closely related beta-hemolytic streptococci such as Streptococcus equi or S. dysgalactiae subsp. equisimilis but do occasionally have close homologs in sporadic GBS isolates.
TABLE 1.
Comparison of HsdR and HsdM between GAS and other streptococci
| S. pyogenes/GAS | % of identity or similarity to GAS HsdR or HsdMa |
|||||
|---|---|---|---|---|---|---|
|
S. agalactiae/GBSb |
VGS |
S. dysgalactiae
b
|
||||
| Identity | Similarity | Identity | Similarity | Identity | Similarity | |
| HsdR | 93 | 96 | 70 | 70 | 64 | 78 |
| HsdM | 97 | 99 | 74 | 86 | 47 | 86 |
The percentages of identity and similarity indicated are at the amino acid level. VGS, viridans group streptococci.
Homologs are present in only a few strains.
Schematic representation comparing the architecture of the GAS type I RM system to that of Streptococcus agalactiae (also known as group B Streptococcus). Download FIG S1, EPS file, 1.2 MB (1.2MB, eps) .
Copyright © 2021 DebRoy et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Analysis of the GAS HsdS protein and its tandem recognition domains.
The GAS type I RM system contains a single hsdS gene. In contrast, the type I RM system from many S. pneumoniae and some S. suis strains previously shown to affect gene expression contains two pseudogenes, hsdS′ and hsdS′′, which provide a scaffold for recombination and thus alteration of HdsS targeting (see Fig. S2 in the supplemental material) (21, 23). Alignment of the HsdS protein from 224 GAS genomes revealed several main findings. First, there was significantly less HsdS protein homology shared across the GAS genomes relative to HsdR and HsdM, with HsdS from different strains having as little as 37% identity over the full length of the protein (Fig. 1B). Second, comparison of the HsdS protein revealed clear clustering, with particular clusters often harboring strains of more than one emm type (Fig. 1C). Finally, the diversity of the HsdS protein was located in two distinct regions, which we will refer to as the TRD1 and TRD2 positions, consistent with these areas being target recognition domains (TRDs), which function to detect a specific combination of a bipartite DNA target sequence (Fig. 1B) (9). The presumed TRDs are flanked by and separated by relatively conserved regions with the length of the inter-TRD conserved region dictating the distance between the two halves of the target sequence (31).
Comparison of the type I RM systems of GAS, Streptococcus pneumoniae, and Streptococcus suis. Two examples of S. pneumoniae are included to display different arrangements of the hsdS′ and hsdS′′ genes as observed in sequenced strains. Download FIG S2, EPS file, 1.2 MB (1.2MB, eps) .
Copyright © 2021 DebRoy et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Given that the presumed TRDs account for HsdS diversity, we next focused on analyzing these individually. Based on TRD sequence alignment and subsequent clustering analysis, we identified nine distinct TRDs that varied widely in their prevalence (Fig. 1D). Within clusters, the TRD amino acid compositions were highly similar—generally 99 to 100% identity but always >92%. Conversely, the amino acid identity levels were typically 15% or less between TRDs from distinct clusters. Of the 9 unique TRDs, 7 occurred in either the N-terminus (i.e., TRD1) or C-terminus (i.e., TRD2) position. TRD_B, -C, -D, -E, and -I occurred only at position 1, and the TRD_G and -H alleles were only found at position 2. TRD_A and TRD_F exhibited domain movement, as previously described (32). Specifically, TRD_A, which was typically found in the TRD1 position, was located in the TRD2 position for several strains from different GAS emm types (Fig. 2; see Fig. S3 in the supplemental material). Conversely, TRD_F, which usually occurred at a TRD2 position, was present in the TRD1 position for all five emm5 strains (Fig. 2). Interestingly, TRD_F was the only TRD allele that was found to occur in positions 1 and 2 of the same HsdS protein (TRD1/2 [F/F]) in four different GAS emm types (Fig. 2).
FIG 2.
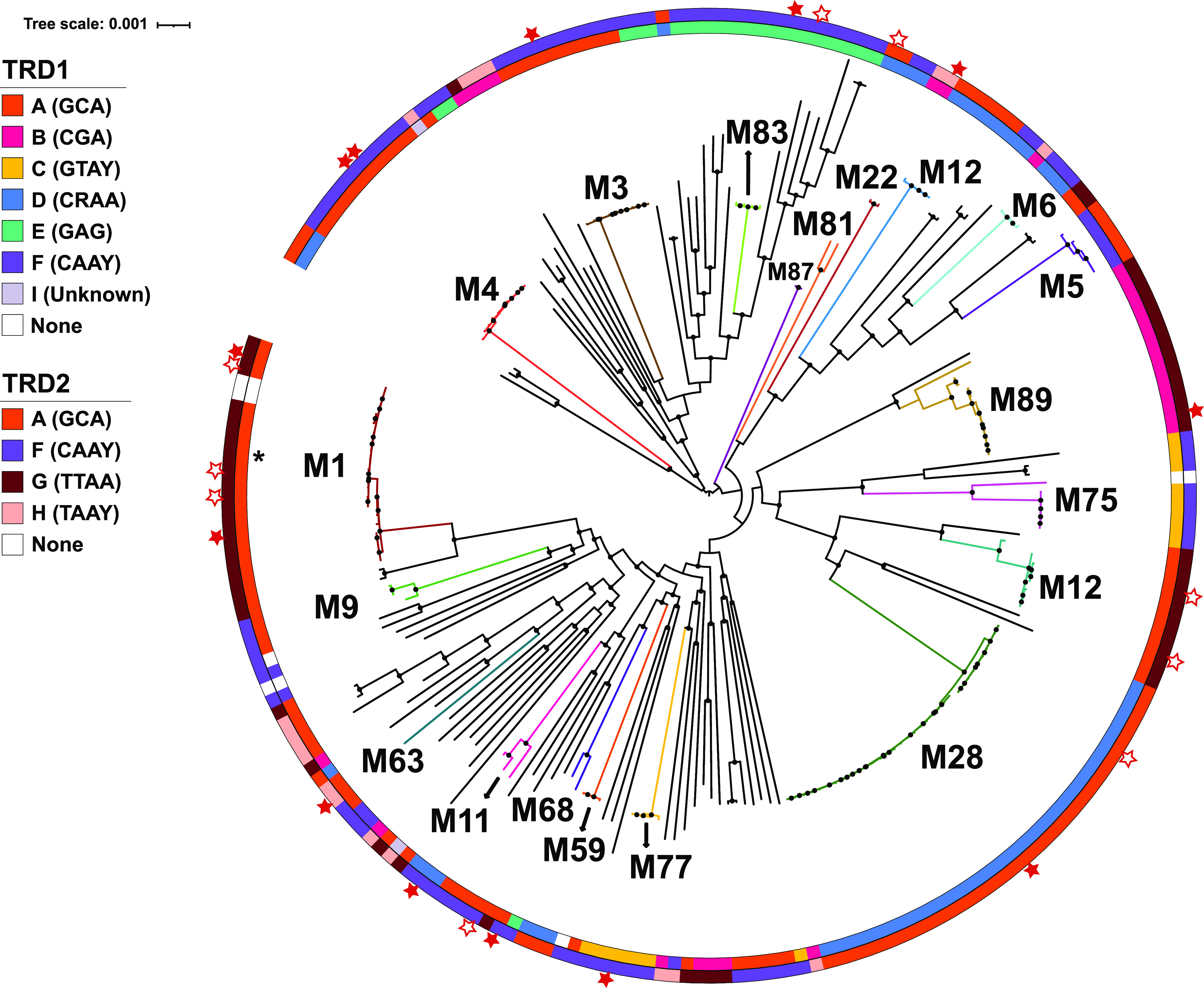
Correlation between GAS TRD alleles and phylogeny inferred from core gene alignment. The maximum likelihood phylogenetic tree was created from a core gene alignment of the 224 GAS genomes. Inner and outer circles are color coordinated to indicate the TRD present in positions 1 and 2 of HsdS, respectively. The half-site of the target recognized by each TRD is listed in parentheses within the legend. Closed stars indicate strains that were sequenced by PacBio in this study. Open stars are GAS strains for which PacBio data are available on REBASE (73). Major GAS emm types are indicated, and isolates of the same emm type tend to cluster, with the exception of emm12. Black dots on internal nodes indicate >95% bootstrap support. The single black star along the inner circle denotes a GAS strain that clusters with the emm1 strains but belongs to emm204.
A schematic representation of the movement of TRD domains within GAS strains (emm1/emm28) and between GAS and GBS strains compared using EasyFig (67). Download FIG S3, EPS file, 0.7 MB (747KB, eps) .
Copyright © 2021 DebRoy et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
There were 13 distinct TRD combinations among the analyzed GAS strains, with occasional strains lacking one or more TRDs or even the entire HsdRSM system (Fig. 2; Table S1). The majority of strains of the same emm type contained the same TRD1/2 combination, with the main exception being emm12 strains, which had two completely different TRD1/2 combinations (AG and DA). Similar variation of TRD1/2 combination between strains of the same emm type was also observed for emm22, emm25, emm44, emm64, emm68, emm70, emm75, emm77, emm78, and emm92 isolates. When analyzed by emm type, the EF combination was the most common, being present in 17 emm types, with AF (16 emm types), AG (13 emm types), and DA (10 emm types) being observed in at least 10 emm types. Conversely, the FA and FH combinations were only observed in a single emm type.
To determine whether the TRD combinations correlated with core genome phylogenies, we created a maximum likelihood phylogenetic tree inferred from core gene alignment and then layered the TRD combinations on top of this tree (Fig. 2). Interestingly, GAS strains that are quite distinct at the whole-genome level (e.g., emm1 and emm12) share identical TRD1/2 alleles. Conversely, emm types that are closely related based on core gene phylogeny (e.g., emm5 and emm6) can have completely different TRD1/2 alleles, consistent with the occurrence of horizontal gene transfer (HGT) among different GAS emm types. The two groups of aforementioned emm12 strains with distinct TRD combinations also were significantly different at the core gene level, suggesting that horizontal transfer of the emm gene may have occurred (33).
Accessory genome elements account for ∼10% of the average GAS genome and encode a variety of critical GAS virulence factors, such as pili, cell surface molecules, and superantigens (34). The exogenous nature of many of these non-core chromosomal elements suggests that their presence could be influenced by the activity of the type I RM system. Therefore, we next asked whether there was a relationship between TRD composition and accessory gene content. As shown in Fig. S4 in the supplemental material, we did observe some clustering of accessory genes by TRD group, which was primarily driven by strains of identical emm types. When strains of distinct emm types were considered, we did not discern clustering of accessory gene content relative to TRD composition (e.g., yellow dots representing the AF TRD combination are present both in the middle and lower left quadrants of Fig. S4). We conclude that TRD composition alone is not determinative of accessory gene content.
GAS TRD alleles do not correlate with accessory genome composition. A t-SNE plot indicates the distribution of TRD1/2 combinations in the accessory genomes of 224 GAS strains. Each dot indicates a single isolate’s accessory genome (gene content shared in <99% genomes), with color indicating TRD pattern assignment per the legend. emm types that clustered by accessory genome content are labeled on the figure. An asterisk indicates one isolate belonging to emm204 that clusters with emm1 isolates. Download FIG S4, EPS file, 0.5 MB (482KB, eps) .
Copyright © 2021 DebRoy et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
GAS TRD composition and recombination.
Recombination events within GAS strains as well as between GAS and other hemolytic streptococci have been reported (35, 36). Given that type I RM systems are thought to limit recombination (28), we next sought to determine whether the compositions of the GAS TRDs were identical between emm types previously identified as having undergone recombination. The best-described example of recombination to date among GAS is between emm1 and emm12 strains and involves a 36-kb segment of DNA, including the nga-slo region (35). As noted above, we identified two distinct emm12 TRD combinations. Examination of the emm12 genomes showed significant variation in the nga-slo locus between the two clusters. The emm12 strains that contain an nga-slo region nearly identical to emm1 strains also have the same TRD combination (AG) as emm1 strains. Another major identified recombination occurred between GBS and emm28 strains (36). Consistent with TRD composition being important for recombination events, emm28 strains contain TRD_D in the HsdS TRD1 position, which is the TRD allele present with high homology in a limited number of GBS isolates. The TRD2 emm28 allele TRD_A was not identified in any GBS isolates (Fig. S3). No highly similar TRDs were identified in strains of streptococci closely related at the whole-genome level to GAS such as S. dysgalactiae.
Use of SMRT sequencing to identify HsdS TRD allele methylation specificity.
Given that there were significant clusters of GAS HsdS proteins, we next sought to determine the target recognition sites (TRSs) for methylation by the various HsdS isoforms. We selected for genome-wide methylation analysis 10 different GAS strains that included representatives of each cluster that had more than five isolates in our HsdS analysis (Fig. 1C). All reads from the SMRT sequencing were aligned to the respective reference genomes (see Table S2a in the supplemental material) to identify the location and type of methylation. Methylation was detected in all GAS genomes sequenced, and the most prevalent type of methylation observed was m6A (see Table 3 below). For several of the sequenced strains, we identified additional methylation events (Table S2b) with low modification quality values (modQV) compared to that observed for m6A. A previous study reported similar observations and suggested that these events likely reflect “noise” in the SMRT data set (25).
(a) List of strains analyzed by PacBio and reference genomes used. (b) List of DNA motifs with non-m6A base modification detected by PacBio. Download Table S2, XLSX file, 0.01 MB (13.5KB, xlsx) .
Copyright © 2021 DebRoy et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We then analyzed sequence motifs that recurred at these m6A sites to derive the target recognition site (TRS) for the MTase in each genome. We identified 9 different TRSs with more than 97% of the identified motifs being methylated in each sequenced genome (Table 3). Given the bipartite nature of the TRS and the correspondence of each half of the motif to TRD1 and -2 in HsdS, we were able to assign the half-motif recognized by all the TRDs identified from our HsdS analysis, with the exception of TRD_I. The distributions of TRDs and TRSs are shown in Fig. 2.
The type I RM system is the major source of methylation.
Given that GAS strains consistently harbor only the single type I RM system, we sought to determine if all of the observed methylation was due to these MTases. To this end, we performed SMRT sequencing of an emm1 GAS strain with a prophage insertion in hsdRSM (37). We found that this strain, M1-SC, completely lacked m6A, consistent with the idea that the type I system is the major methylation system in GAS (Table 3; Fig. 3A). To investigate further, we generated targeted knockouts of hsdM in emm28 (MGAS6180) and emm87 (TSPY1057) strains (Table 2) and confirmed the absence of spurious mutations by whole-genome sequencing. Deletion of the hsdM gene did not impact the growth of these strains in standard laboratory media (see Fig. S5A in the supplemental material). We also confirmed the absence of hsdM transcripts in the mutant strains by TaqMan quantitative real-time PCR (qRT-PCR) (Fig. S5B). We used Oxford Nanopore Technologies (ONT) sequencing and a neural network classifier (38) to detect m6A methylation and determine if hsdM knockouts would reduce methylation detected in GAS genomes. There was m6A methylation in both wild-type strains MGAS6180 and TSPY1057, while deletion of hsdM resulted in statistically significant reduction in methylation for both (Fig. 4A). We next assessed the ability of the emm87 strain TSPY1057 and its hsdM mutant to incorporate DNA by electroporation. We found that the TSPY1057 ΔhsdM mutant yielded ∼500-fold more transformants when electroporated with the pLZ12 plasmid (39) compared to the wild-type strain (Fig. 4B). Similar increases in transformation efficiency of strains lacking the type I RM system have been reported for emm1 and emm28 GAS (25, 26, 30). Taken together, we conclude that the type I RM system is responsible for methylation observed in GAS strains, and disruption of this system improves entry of exogenous DNA under laboratory conditions.
FIG 3.
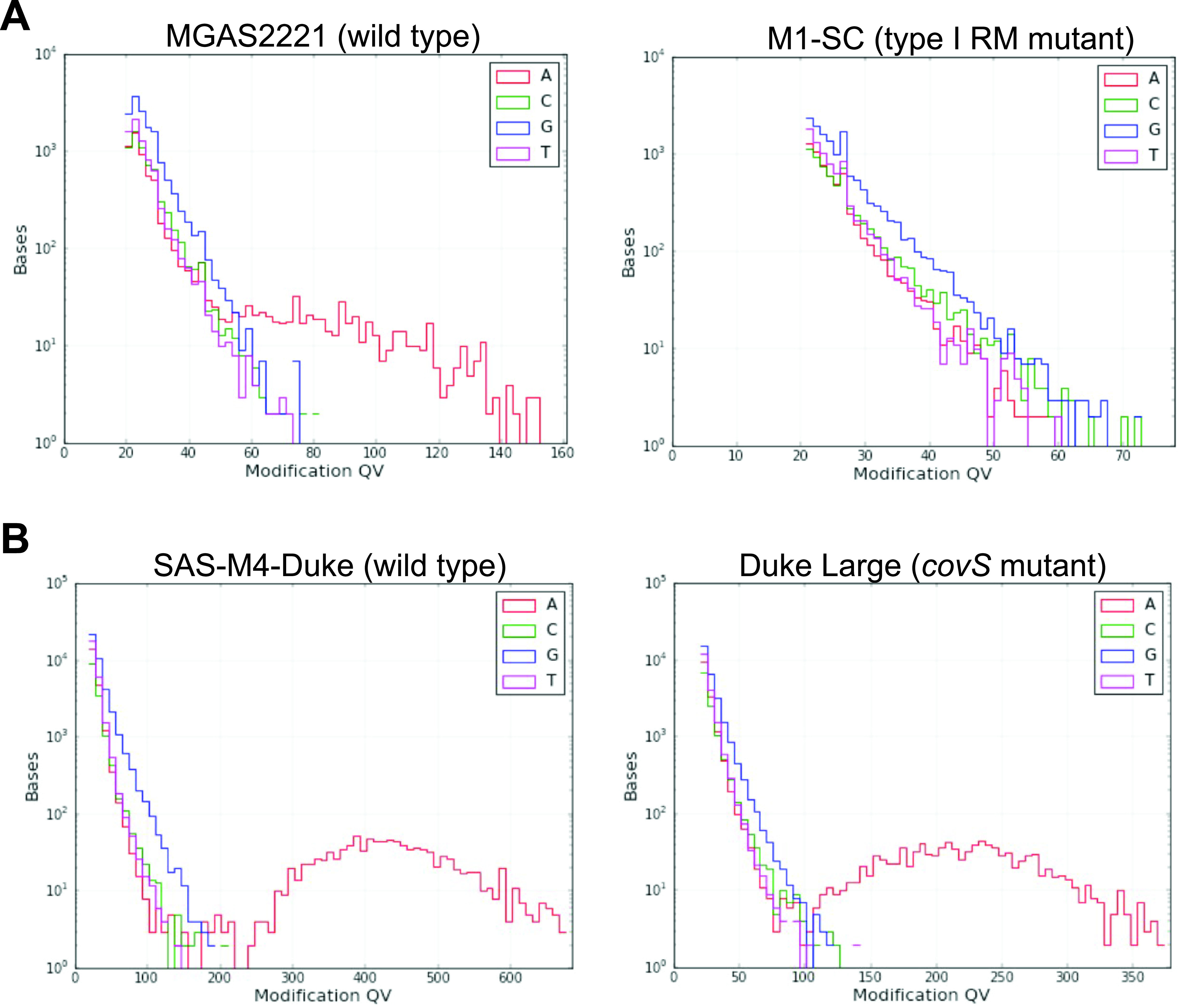
Methylation events detected in various GAS strains. Comparison of modification quality value (modQV) histograms indicating methylation events detected by PacBio sequencing between (A) emm1 strains MGAS2221 and M1-SC and (B) emm4 strains SAS-M4-Duke and Duke Large. modQV values are indicated on the x axis, and the numbers of bases are displayed on the y axis. The lines are color-coded for each nucleotide.
TABLE 2.
Bacterial strains used in this study
| Strain | Description | Reference |
|---|---|---|
| MGAS2221 | Invasive clinical isolate, emm1 | 41 |
| M1-SC-1 | Clinical isolate, emm1 | 37 |
| MSPY1 | Clinical isolate, emm89 | 69 |
| SAS-M4-Duke | Clinical isolate, emm4 | 43 |
| RLGH | Clinical isolate, emm4 | 43 |
| Duke Large | Invasive clinical isolate, coisolated with SAS-M4-Duke, inactive CovS | 44 |
| Duke ΔcovS | Isogenic mutant of SAS-M4-Duke, CovS inactive | 44 |
| MGAS10870 | Clinical isolate, emm3 | 70 |
| TSPY416 | emm68 | This study |
| TSPY125 | Clinical isolate, emm83 | This study |
| TSPY155 | Clinical isolate, emm11 | 62 |
| TSPY453 | Clinical isolate, emm77 | 62 |
| TSPY1309 | Clinical isolate, emm63 | This study |
| TSPY136 | Clinical isolate, emm22 | This study |
| MGAS6180 | Clinical isolate, emm28 | 36 |
| MGAS6180 ΔhsdM | Isogenic hsdM mutant | This study |
| TSPY1057 | Clinical isolate, emm87 | 71 |
| TSPY1057 ΔhsdM | Isogenic hsdM mutant | This study |
FIG 4.
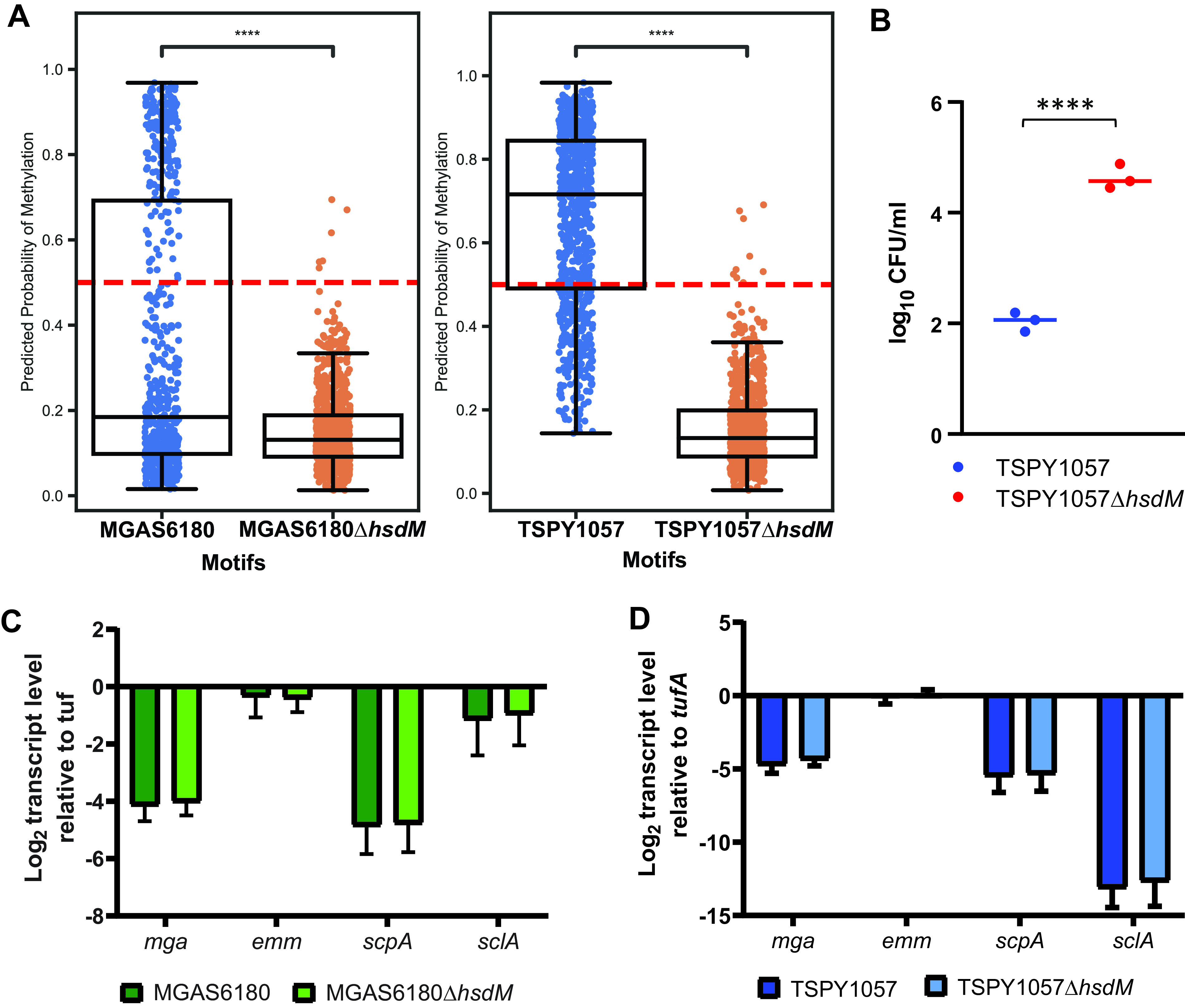
Characterization of impact of hsdM inactivation on GAS methylation, transformation, and gene expression. (A) Comparison of methylation events detected by the Caller neural network model using ONT long-read sequencing data between wild-type (blue dots) and ΔhsdM mutant (orange dots) strains of MGAS6180 (emm28) and TSPY1057 (emm87), respectively. The dotted red line in each subpanel indicates a probability of 0.5. Welch’s t test of independent samples with a multiple-sample Bonferroni correction was performed for each wild-type strain and respective ΔhsdM group, with **** indicating statistically significant difference between the strains at P ≤ 0.0001. (B) Comparison of transformation efficiency between TSPY1057 and its ΔhsdM mutant. GAS cells were transformed on three separate occasions, and the numbers of colonies detected are shown as log10 CFU/ml per μg DNA. ****, statistically significant difference between the strains at P ≤ 0.0001 by unpaired t test (C and D) TaqMan qRT-PCR analysis of the impact of hsdM deletion on transcript levels of the gene encoding the multigene activator (mga) and Mga-regulated genes in (C) MGAS6180 and (D) TSPY1057. TaqMan qRT-PCR data are means ± standard deviations from two biological replicates, with two technical replicates, done on 2 separate days.
Isogenic hsdM mutants of emm28 and emm87 GAS. (A) Growth of MGAS6180 and TSPY1057 and their isogenic hsdM mutants in THY broth. (B) TaqMan qRT-PCR confirms lack of hsdM transcripts in isogenic ΔhsdM strains. Data show the mean ± standard deviation from two biological replicates, with two technical replicates, done on separate days. Download FIG S5, EPS file, 0.9 MB (937.9KB, eps) .
Copyright © 2021 DebRoy et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Inactivation of the type I RM system does not alter transcript levels of mga or Mga-regulated virulence genes.
A previous study of MEW123 (emm28) demonstrated reduced transcript levels of mga and other genes in the Mga regulon upon inactivation of the type I RM system (25). However, a recent report found no differences in the transcript levels of known GAS virulence factor-encoding genes when comparing an isogenic emm1 hsdM mutant to its wild-type parent (26). To address a potential role of methylation in expression of mga and its regulon and the possibility of emm-specific differences, we performed targeted gene transcript-level analysis of both MGAS6180 (emm28) and TSPY1057 (emm87) compared to their respective isogenic ΔhsdM mutants. In accordance with the emm1 study and unlike the emm28 report, we found no difference in the transcript levels of mga or Mga-regulated genes upon inactivation of the type I RM system (Fig. 4C and D).
Analysis of intra-emm type methylation patterns.
It is well known that GAS strains belonging to the same emm type (and likely with identical TRD1/2 alleles) can exhibit differing virulence attributes, either inherently or due to naturally occurring alterations (40). Given the known relationship between epigenetics and virulence in other streptococci (21, 22), it is possible that epigenetics might play a role in GAS virulence. To address this question, we performed SMRT sequencing to compare GAS strains of the same emm type and TRD1/2 combination, but with distinct virulence phenotypes. First, we compared the methylation patterns of the emm1 strains SF370 and MGAS2221, which represent two distinct emm1 clades that are well known to differ markedly in their virulence (40). Another established difference between these two strains is that MGAS2221 readily develops hypervirulent mutants that harbor changes in the control of virulence (CovRS) two-component gene regulatory system, while the same is not observed in SF370 (41, 42). Consistent with the TRDs being the determinative factor of GAS methylation, we found that the methylation patterns of SF370 and MGAS2221 were nearly identical (Table 3; see Fig. S6A in the supplemental material). We also compared the emm4 strains SAS-M4-Duke and RLGH, which like the emm1 strains differ in the spontaneous occurrence of hypervirulent CovRS mutants, and again found identical methylation patterns (Fig. S6B). To address whether a CovS mutant has altered methylation, we studied three emm4 strains, SAS-M4-Duke (wild type), a spontaneous CovS mutant derived from Duke (Duke Large), and an isoallelic CovS mutant of Duke (Duke ΔcovS) (43, 44). SMRT analysis of the three strains showed no difference in the extent of methylation or in the motifs targeted, suggesting that genome-wide methylation is not altered upon inactivation of the CovRS system (Table 3; Fig. 3B). Taken together, we conclude that methylation variation does not account for differences in virulence attributes observed among the tested GAS strains with identical TRD alleles.
TABLE 3.
Methylation identified in the genomes of GAS strains from different emm types
| GAS strain | emm type | Motif string | Position modified | Type | % of motifs detected | No. of motifs detected | No. of motifs in genome | Mean modQV |
|---|---|---|---|---|---|---|---|---|
| MGAS2221 | 1 | GCANNNNNNTTAA | 3 | m6A | 98.0 | 337 | 344 | 79.09 |
| TTAANNNNNNTGC | 3 | m6A | 97.7 | 336 | 344 | 77.17 | ||
| SF370 | 1 | TTAANNNNNNTGC | 3 | m6A | 98.8 | 331 | 335 | 75.26 |
| GCANNNNNNTTAA | 3 | m6A | 97.9 | 328 | 335 | 76.43 | ||
| M1-SC | 1 | ND | ||||||
| MGAS10870 | 3 | CAAYNNNNNNTGC | 3 | m6A | 100.0 | 524 | 524 | 513.45 |
| GCANNNNNNRTTG | 3 | m6A | 99.8 | 523 | 524 | 493.54 | ||
| SAS-M4-Duke | 4 | CAAYNNNNNNTGC | 3 | m6A | 100.0 | 542 | 542 | 143.86 |
| GCANNNNNNRTTG | 3 | m6A | 100.0 | 542 | 542 | 142.56 | ||
| RLGH | 4 | CAAYNNNNNNTGC | 3 | m6A | 100.0 | 542 | 542 | 307.29 |
| GCANNNNNNRTTG | 3 | m6A | 99.8 | 541 | 542 | 299.71 | ||
| Duke Large | 4 | CAAYNNNNNNTGC | 3 | m6A | 100.0 | 542 | 542 | 228.72 |
| GCANNNNNNRTTG | 3 | m6A | 100.0 | 542 | 542 | 225.93 | ||
| Duke ΔcovS | 4 | GCANNNNNNRTTG | 3 | m6A | 98.2 | 532 | 542 | 68.37 |
| CAAYNNNNNNTGC | 3 | m6A | 96.5 | 523 | 542 | 68.08 | ||
| TSPY155 | 11 | CRAANNNNNNRTTG | 4 | m6A | 99.8 | 462 | 463 | 283.43 |
| CAAYNNNNNNTTYG | 3 | m6A | 99.6 | 461 | 463 | 312.70 | ||
| TSPY136 | 22 | TAAYNNNNNNTCG | 3 | m6A | 100.0 | 241 | 241 | 285.17 |
| CGANNNNNNRTTA | 3 | m6A | 100.0 | 241 | 241 | 258.73 | ||
| MGAS6180 | 28 | CRAANNNNNNNTGC | 4 | m6A | 99.5 | 414 | 416 | 203.92 |
| GCANNNNNNNTTYG | 3 | m6A | 99.0 | 412 | 416 | 229.83 | ||
| TSPY1309 | 63 | TAAYNNNNNNTGC | 3 | m6A | 100.0 | 537 | 537 | 525.49 |
| GCANNNNNNRTTA | 3 | m6A | 99.8 | 536 | 537 | 474.90 | ||
| TSPY416 | 68 | CAAYNNNNNNTGC | 3 | m6A | 100.0 | 513 | 513 | 270.36 |
| GCANNNNNNRTTG | 3 | m6A | 99.8 | 512 | 513 | 259.95 | ||
| TSPY453 | 77 | GTAYNNNNNRTTG | 3 | m6A | 100.0 | 190 | 190 | 412.97 |
| CAAYNNNNNRTAC | 3 | m6A | 100.0 | 190 | 190 | 404.43 | ||
| TSPY125 | 83 | CAAYNNNNNNCTC | 3 | m6A | 98.9 | 345 | 349 | 476.50 |
| GAGNNNNNNRTTG | 2 | m6A | 98.0 | 342 | 349 | 458.08 | ||
| MSPY1 | 89 | TTAANNNNNNTCG | 3 | m6A | 100.0 | 179 | 179 | 312.57 |
| CGANNNNNNTTAA | 3 | m6A | 99.4 | 178 | 179 | 280.25 | ||
Modification quality value (QV) histograms indicating methylation events detected by PacBio sequencing between (A) emm1 strains MGAS2221 and SF370 and (B) emm4 strains SAS-M4-Duke and RLGH. Modification quality values (modQV) are indicated on the x axis, and the number of bases is displayed on the y axis. The lines are color-coded for each nucleotide. Download FIG S6, TIF file, 2.5 MB (2.5MB, tif) .
Copyright © 2021 DebRoy et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
GAS type II RM systems.
Type II RM systems typically consist of an MTase and an REase, encoded by a single or separate genes (11). However, it is well recognized that a large number of type II orphan methyltransferases can be present on bacteriophages and other mobile genetic elements (11, 12, 45). We identified only a single type II system in our 224 GAS strains that contained both REase and MTase enzymes. This system was present on a prophage in the emm6 strain MGAS10394, the emm1 strain 10-85, and the emm75 strain TSPY208 and has been previously characterized as important for cytosine methylation (m5C) in strain MGAS10394 (24). The vast majority of sequenced GAS strains contained at least one and up to three orphan type II methyltransferases present on endogenous prophages. The presence of these type II MTase-containing prophages varies both among and within emm types (Fig. 1A), and these prophages also typically contain virulence genes encoding superantigens (e.g., SpeC or Ssa) or DNases (e.g., SdaI or SpdI) (46, 47). In one strain (MGAS10750 [emm4]), a prophage encoding a type II orphan methyltransferase had integrated into and inactivated the type I RM system (29). Despite the widespread presence of the type II orphan MTases, we did not observe any m5C methylation in our SMRT analyses. It is well known that m5C is not as readily detected by SMRT sequencing as m6A, and thus increasing sequencing depth is needed for confident detection (48). We achieved sequencing depths of >250 recommended for m5C and still did not identify any m5C modification (49). We hypothesized that the lack of m5C might be due to very low or absent expression of the type II MTases under the conditions in which samples were collected for SMRT sequencing. Consistent with this hypothesis, we found little to no transcripts of the genes encoding type II MTase genes of MGAS2221 (emm1) and MGAS1870 (emm3), respectively (Fig. 5). Given that genes carried on prophages can be induced (50, 51), it remains formally possible that the type II orphan MTases do contribute to the GAS methylome under different conditions.
FIG 5.

Genes encoding the type I MTase have higher transcript levels than those encoding type II RM MTases. Shown are the results from analysis of transcript levels of genes encoding the MTase of the type I and type II RM systems in representative emm1 (MGAS2221) and emm3 (MGAS10870) strains. The data shown are means ± standard deviations from two biological replicates, with two technical replicates, done on 2 separate days.
DISCUSSION
The advent of whole-genome sequencing (WGS) approaches capable of detecting DNA methylation has facilitated high-throughput analyses of epigenetic modifications, which in turn has greatly facilitated understanding of how DNA methylation impacts different aspects of prokaryotic physiology. Herein, we used a combination of WGS and large-scale phylogenomics to systematically characterize the restriction modification (RM) systems of 224 strains of the major human pathogen group A Streptococcus (GAS). We found that a single type I RM system present in the GAS core genome is responsible for methylation detectable by different WGS approaches and is distinct from the type I systems in S. pneumoniae and S. suis recently shown to impact global gene expression. Our analysis of the type I system defined the DNA methylation motifs of 13 combinations of nine TRDs present and correlated GAS TRD composition with the GAS pangenome.
Our conclusion that the type I RM system present on the GAS chromosome is responsible for most, if not all, methylation detectable via SMRT and MinION sequencing approaches is based on the following. First, only adenines, which are the target of the type I system, were consistently detected as methylated (m6A) in numerous GAS strains via SMRT sequencing. Second, no methylation was detected by SMRT sequencing in a GAS strain naturally lacking an intact type I RM system due to phage insertion. Finally, genetic inactivation of the type I MTase in two emm types dramatically decreased the methylation signal by MinION sequencing. These data are in accord with a previous study of a single emm28 strain using SMRT sequencing in which inactivation of the type I RM system abolished genome-wide m6A methylation (25). Given the nature of predicting methylation sites using long-read sequencing data, there is not a binary answer to whether any methylation is detected, but the non-m6A methylation detected in our SMRT sequencing was inconsistent, likely indicating “noise” rather than true methylation, as has been previously described (52). Inasmuch as SMRT sequencing does not readily detect m5C methylation (53), the presumed target of type II RM systems that showed low transcript levels under the conditions studied, it remains a formal possibility that the phage-encoded, variably present type II systems do contribute to GAS methylation. The recent development of a WGS approach to detect m5C methylation could help to clarify the role of type II RM systems in GAS (53).
By performing SMRT sequencing on numerous GAS emm types, we deduced the target sequences of the type I system for nearly all of the publicly available, fully sequenced GAS strains in our database, with the exception of two strains that carry a rare TRD_I allele. In turn, these TRD assignments in combination with phylogenetic clustering allowed for discernment that GAS hsdS gene composition has likely been shaped by horizontal gene transfer (HGT) of TRD-encoding subunits. This stands in contrast to the HsdS structure observed in Staphylococus aureus, in which HsdS composition tracks with clonal complexes (54). The HsdS conservation among related S. aureus strains is thought to explain the limited exchange of genetic material between various clonal complexes (54). The GAS type I HsdS population structure seems more closely related to those of Staphylococcus epidermidis (52) and S. pneumoniae (55), in which HsdS composition does not align with whole-genome relatedness and is thought to facilitate recombination among genetically diverse strains. It is tempting to hypothesize that the presence of identical TRDs in genetically distinct GAS isolates would permit interstrain transfer of genetic material, and indeed, we identified that emm1 and emm12 isolates, which share nearly identical nga-slo regions, also have the same TRD combination (35). However, when more broadly applied to the GAS population, we did not find a clear signal that TRD composition correlated with accessory genome content, suggesting that additional factors are important for the acquisition of key GAS adaptive genes encoding superantigens and DNases. GAS also harbors CRISPR-Cas systems, another major mechanism used by bacteria to distinguish between self and nonself (56), and this might explain, in part, the incongruency observed between non-core chromosomal content and TRD distribution. Knowledge of the TRD combinations in various emm types could assist with the choice of vectors for genetic manipulation of GAS and even facilitate designing genetic changes to permit vector use when there are incompatibilities with particular TRD target sequences (28, 57).
Another key finding of our work was that the conserved GAS type I RM system is distinct from those of S. pneumoniae (21, 58) and S. suis (23), which contain multiple HsdS genes arranged in tandem at the 3′ end of the hsdRSM operon. Recombination among the hsdS genes results in strains with distinct methylation patterns and distinct transcriptomes, presumably through differential methylation of promoter DNA that in turn influences transcription (21, 23, 58). In contrast, the GAS hsdRSM operon contains a single hsdS gene. To date, the ability of prokaryotic type I RM systems to influence gene expression has been limited to those with the capacity to switch between various hsdS alleles, as seen in S. pneumoniae and S. suis (59, 60). Deletion of the type I RM system in an emm28 GAS strain was reported to strongly reduce the expression of mga and Mga-regulated genes (25). However, a recent study of emm1 GAS reported no impact of the loss of the type I system on gene expression (26). In accordance with Finn et al. (26), we found that inactivation of the type I MTase did not impact mga or Mga-regulated gene transcript levels in two distinct GAS emm types. It is well established that laboratory manipulation of GAS can result in downregulation of mga and Mga-regulated genes through unclear mechanisms, so it is possible that the previous observation resulted from such a phenomenon (42). Alternatively, the difference between the previous study and our findings could have resulted from strain-specific findings. The identification of clinical isolates from multiple GAS emm types that contain inactivation or even absence of the type I system also argues against a significant contribution of the type I RM system to GAS virulence (30). Nevertheless, the fact that vast majority of GAS strains do contain a type I RM system suggests that it is important for the overall fitness of the bacteria from an evolutionary standpoint.
We also sought to discern the origin of the type I GAS system through a comparative sequence approach. Surprisingly, streptococcal strains closely related to GAS, such as S. dysgalactiae or S. equi, did not contain clear homologs of the type I system, indicating that GAS may have acquired the system through HGT or that loss of the system has occurred in closely related streptococci. Interestingly, nearly identical type I systems were found in occasional S. agalactiae (also known as GBS) strains, which despite the nomenclature, are not closely related to GAS. A major recombination event between GBS and GAS has been identified involving emm28 strains (36), and the type I system in GBS does contain a TRD1 allele nearly identical to that present in emm28. This finding raises the possibility that the presence of identical or near-identical TRD combinations in GAS and GBS might have facilitated the recombination event that seems to have been critical in GAS emm28 strains being the major cause of puerperal sepsis, an infection typically caused by GBS (61). The scattered nature of the type I system in GBS suggests it may have been imported from GAS rather than serving as the source. Thus, at present, the origin of the GAS type I system remains obscure.
Finally, our analysis also revealed that all but 3 of the 224 GAS strains analyzed possessed orphan type II methyltransferases present in mobile genetic elements. Methylation (m5C) activity has been reported for a type II system in an emm6 GAS strain that contains both an REase and MTase (24). However, we did not detect any m5C modifications in the strains sequenced by PacBio in this study, all of which harbor orphan type II MTases. The impact of these orphan type II MTases in GAS remains unclear since they have very low expression under the conditions studied herein.
In summary, we have characterized from a bioinformatic and biologic standpoint a type I RM system that is the lone RM system consistently found in group A Streptococcus. Unlike other streptococci, the system seems primarily involved in protection against exogenous DNA. Knowledge of the type I RM system may facilitate future efforts to genetically manipulate this important pathogen.
MATERIALS AND METHODS
Growth and DNA isolation, mutant construction, and GAS electroporation.
Bacterial strains listed in Table 2 were routinely grown in Todd-Hewitt (THY) broth at 37°C with 5% CO2. Isogenic hsdM mutations in MGAS6180 and TSPY1057 were obtained by nonpolar insertional mutagenesis with a spectinomycin cassette as described before (44). To determine electroporation efficiency, competent GAS cells were transformed with 1 μg of pLZ12 plasmid DNA (39) that carries a spectinomycin resistance cassette. Cells were allowed to recover for 2 h, and dilutions were plated on THY agar plates with spectinomycin (150 mg/ml) and enumerated after overnight growth. Primers are listed in Table S3 in the supplemental material.
Primers and probes used in this study. Download Table S3, XLSX file, 0.01 MB (10.7KB, xlsx) .
Copyright © 2021 DebRoy et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Generation of reference genomes.
Complete genome sequences were determined for several GAS strains in this study using a combination of short-read (Illumina) and long-read (Oxford Nanopore) sequence data (Table 2; Table S2a). Genomic DNA extraction, library construction, and sequencing (Illumina MiSeq or ONT GridION) were performed as previously described (62, 63). Average depths of coverage for completed genomes were >100-fold and a minimum of 50-fold for both short- and long-read sequences. Hybrid genome assemblies were determined using Unicycler v0.4.6 (64) and annotated using PGAP at NCBI (65). Accession numbers for completed genomes are provided in Table S2a.
SMRT and MinION sequencing.
GAS strains were harvested at mid-exponential phase, and high-quality genomic DNA was isolated using the Master Pure kit (Lucigen) for sequencing by the PacBio or MinION system. SMRT sequencing was performed at the Johns Hopkins Deep Sequencing and Microarray Core. For PacBio RS II sequencing, 10 to 20 libraries were prepared following the manufacturer’s recommended procedure using the PacBio SMRTBell Template Prep kit v1.0 with BluePippin size selection. Each library was sequenced using polymerase binding kit P6v2 and sequencing kit 4v2 (C4 chemistry) on one SMRTcell. The sequencing data were analyzed using PacBio smrtanalysis software v2.3.1 base modification and motif analysis pipeline.
ONT library preparation was performed using the Rapid barcoding kit (SQK-RBK004) with 400 ng of genomic DNA (gDNA) as input using the manufacturer’s protocol. A MinION (R9.4.1) flow cell was used on the GridION platform (Oxford Nanopore Technologies) with base calling and demultiplexing performed offline with Guppy v4.5.2. Flye v2.8 was used to create long-read assemblies using the ONT long-read data. In order to detect methylation using ONT data, a neural network classifier was trained using the MGAS6180 PacBio data as the “gold standard” with the mCaller software package. A 50% data set split of detected m6A motif sites identified by PacBio in the MGAS6180 genome (1,222 sites) was used for training and testing, respectively. We performed 5-fold cross validation training on the mCaller neural network model using the MGAS6180 train set and obtained a cross validation accuracy of 0.82 ± 0.03. The test set of detected m6A positions for MGAS6180 was used to determine the test data accuracy of the MGAS6180 ONT data (180/607 = 30%). The motif recognition argument of 'CRAANNNNNNNTGC' was used for methylation prediction of the 3 remaining constructs with motifs detected using their respective de novo Flye genome assemblies. The accuracies of MGAS6180 ΔhsdM (motifs predicted = 10/835 = 1.1%), TSPY1057 (604/818 = 73.8%), and TSPY1057 ΔhsdM (9/850 = 1.0%) were determined using a 50% predicted probability of methylation threshold with a minimum read depth of 10 reads. The prediction model is available on the mCaller GitHub page (38; https://github.com/al-mcintyre/mCaller).
Pangenome and phylogenetic analysis.
There were 224 GAS complete and draft genomes that were included from NCBI as well as our study for the purpose of performing a pangenome analysis using Panaroo 1.2.4 (66). Gene content in >99% of the 224 GAS genomes was used as the cutoff threshold for the core genome. The gene cluster output was then aligned using Mafft v7.471 to create a multiple-sequence alignment. A maximum likelihood phylogenetic tree inferred from the core gene alignment was created using IQ-TREE multicore version 2.0.6 using ModelFinder, which selected a generalized time-reversible nucleotide substitution model with a FreeRate model for heterogeneity across sites using 3 categories. Additionally, bootstrap support for the maximum likelihood phylogenetic tree was added using an unbiased estimate with UFBoot2. The web-based iTol v6 software was used for phylogenetic tree visualization. The binary gene presence/absence matrix generated from Panaroo was used as input for PANINI to perform a t-SNE analysis to explore patterns of relatedness within the accessory genomes of GAS genomes and grouped by TRD pattern and emm type.
Characterization of hsdRSM operon.
To identify homologs of GAS HsdM and HsdR in other prokaryotes, we performed a blastp search of the nonredundant protein sequences in the NCBI database using the GAS HsdRSM from MGAS2221 (emm1) while excluding GAS.
Conserved regions from the consensus sequence of a Mafft nucleotide alignment of the hsdS gene from 221 GAS isolates were used to create in silico primers to extract the TRD1 and TRD2 regions, respectively, using Cutadapt. The amino acid sequences of TRD1 and TRD2 were then subsequently aligned with Mafft, and the multiple-sequence alignment was used to create a phylogenetic tree using a neighbor-joining method with Geneious software. The determination of TRD groups (e.g., TRD_A) was accomplished using the RhierBAPS clustering algorithm tool, with each cluster assigned to a sequence and annotated on the neighbor-joining tree. TRD site motifs were identified using REBASE prediction, along with blastp results that have 100% identity and 100% coverage of the hsdS gene against previously characterized hsdS genes that are archived in REBASE, as well as blastp results against hsdS that have been characterized in our study using the PacBio SMRT analysis software. We were able to infer TRD site motifs from PacBio data with hsdS genes that clustered at >92% identity. JalView software was used to visualize the alignment of hsdS for 10 representative GAS genomes that had unique TRD patterns. A MUSCLE nucleotide alignment was generated from the 36-kbp nga-slo region for four isolates (MGAS5005 [emm1], SF370 [emm1], NCTC8332 [emm12], and MGAS9429 [emm12]). SNP-dists was used to convert the fasta alignment file into a single nucleotide polymorphism (SNP) distance matrix, where pairwise SNP distances were calculated. The comparison of hsdRSM operons between GAS and GBS isolates was performed using EasyFig (67).
TaqMan qRT-PCR.
For qRT-PCR, samples were grown in duplicate on 2 separate days as described above. Cells were harvested at mid-exponential phase. RNA was prepared using the Qiagen RNeasy kit and processed as described earlier (68). Primers and probes used are listed in Table S3.
Data availability.
The data that support the findings of this study will be shared upon publication. Whole genome sequences for GAS strains TSPY136 (CP060647), TSPY1309 (CP060644), TSPY416 (CP060643), and TSPY125 (CP007562.1) were obtained during this study and submitted to GenBank.
Contributor Information
Samuel A. Shelburne, Email: sshelburne@mdanderson.org.
Paul D. Fey, University of Nebraska Medical Center
REFERENCES
- 1.Cheng X. 1995. DNA modification by methyltransferases. Curr Opin Struct Biol 5:4–10. doi: 10.1016/0959-440X(95)80003-J. [DOI] [PubMed] [Google Scholar]
- 2.Sanchez-Romero MA, Cota I, Casadesus J. 2015. DNA methylation in bacteria: from the methyl group to the methylome. Curr Opin Microbiol 25:9–16. doi: 10.1016/j.mib.2015.03.004. [DOI] [PubMed] [Google Scholar]
- 3.Cheng X. 1995. Structure and function of DNA methyltransferases. Annu Rev Biophys Biomol Struct 24:293–318. doi: 10.1146/annurev.bb.24.060195.001453. [DOI] [PubMed] [Google Scholar]
- 4.Jeltsch A. 2002. Beyond Watson and Crick: DNA methylation and molecular enzymology of DNA methyltransferases. Chembiochem 3:274–293. doi:. [DOI] [PubMed] [Google Scholar]
- 5.Blow MJ, Clark TA, Daum CG, Deutschbauer AM, Fomenkov A, Fries R, Froula J, Kang DD, Malmstrom RR, Morgan RD, Posfai J, Singh K, Visel A, Wetmore K, Zhao Z, Rubin EM, Korlach J, Pennacchio LA, Roberts RJ. 2016. The epigenomic landscape of prokaryotes. PLoS Genet 12:e1005854. doi: 10.1371/journal.pgen.1005854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts RJ, Vincze T, Posfai J, Macelis D. 2010. REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res 38:D234–D236. doi: 10.1093/nar/gkp874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts RJ, Belfort M, Bestor T, Bhagwat AS, Bickle TA, Bitinaite J, Blumenthal RM, Degtyarev SK, Dryden DTF, Dybvig K, Firman K, Gromova ES, Gumport RI, Halford SE, Hattman S, Heitman J, Hornby DP, Janulaitis A, Jeltsch A, Josephsen J, Kiss A, Klaenhammer TR, Kobayashi I, Kong H, Krüger DH, Lacks S, Marinus MG, Miyahara M, Morgan RD, Murray NE, Nagaraja V, Piekarowicz A, Pingoud A, Raleigh E, Rao DN, Reich N, Repin VE, Selker EU, Shaw P-C, Stein DC, Stoddard BL, Szybalski W, Trautner TA, Van Etten JL, Vitor JMB, Wilson GG, Xu S-y. 2003. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res 31:1805–1812. doi: 10.1093/nar/gkg274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wilson GG, Murray NE. 1991. Restriction and modification systems. Annu Rev Genet 25:585–627. doi: 10.1146/annurev.ge.25.120191.003101. [DOI] [PubMed] [Google Scholar]
- 9.Murray NE. 2000. Type I restriction systems: sophisticated molecular machines (a legacy of Bertani and Weigle). Microbiol Mol Biol Rev 64:412–434. doi: 10.1128/MMBR.64.2.412-434.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Williams RJ. 2003. Restriction endonucleases: classification, properties, and applications. Mol Bitechnol 23:225–243. doi: 10.1385/MB:23:3:225. [DOI] [PubMed] [Google Scholar]
- 11.Anton BP, Roberts RJ. 2021. Beyond restriction modification: epigenomic roles of DNA methylation in prokaryotes. Annu Rev Microbiol 75:129–149. doi: 10.1146/annurev-micro-040521-035040. [DOI] [PubMed] [Google Scholar]
- 12.Seshasayee AS, Singh P, Krishna S. 2012. Context-dependent conservation of DNA methyltransferases in bacteria. Nucleic Acids Res 40:7066–7073. doi: 10.1093/nar/gks390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marinus MG, Casadesus J. 2009. Roles of DNA adenine methylation in host-pathogen interactions: mismatch repair, transcriptional regulation, and more. FEMS Microbiol Rev 33:488–503. doi: 10.1111/j.1574-6976.2008.00159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palmer BR, Marinus MG. 1994. The dam and dcm strains of Escherichia coli—a review. Gene 143:1–12. doi: 10.1016/0378-1119(94)90597-5. [DOI] [PubMed] [Google Scholar]
- 15.Bickle TA, Kruger DH. 1993. Biology of DNA restriction. Microbiol Rev 57:434–450. doi: 10.1128/mr.57.2.434-450.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seib KL, Srikhanta YN, Atack JM, Jennings MP. 2020. Epigenetic regulation of virulence and immunoevasion by phase-variable restriction-modification systems in bacterial pathogens. Annu Rev Microbiol 74:655–671. doi: 10.1146/annurev-micro-090817-062346. [DOI] [PubMed] [Google Scholar]
- 17.Heithoff DM, Sinsheimer RL, Low DA, Mahan MJ. 1999. An essential role for DNA adenine methylation in bacterial virulence. Science 284:967–970. doi: 10.1126/science.284.5416.967. [DOI] [PubMed] [Google Scholar]
- 18.Srikhanta YN, Gorrell RJ, Steen JA, Gawthorne JA, Kwok T, Grimmond SM, Robins-Browne RM, Jennings MP. 2011. Phasevarion mediated epigenetic gene regulation in Helicobacter pylori. PLoS One 6:e27569. doi: 10.1371/journal.pone.0027569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan A, Hill DMC, Harrison OB, Srikhanta YN, Jennings MP, Maiden MCJ, Seib KL. 2016. Distribution of the type III DNA methyltransferases modA, modB and modD among Neisseria meningitidis genotypes: implications for gene regulation and virulence. Sci Rep 6:21015. doi: 10.1038/srep21015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Atack JM, Srikhanta YN, Fox KL, Jurcisek JA, Brockman KL, Clark TA, Boitano M, Power PM, Jen FE-C, McEwan AG, Grimmond SM, Smith AL, Barenkamp SJ, Korlach J, Bakaletz LO, Jennings MP. 2015. A biphasic epigenetic switch controls immunoevasion, virulence and niche adaptation in non-typeable Haemophilus influenzae. Nat Commun 6:7828. doi: 10.1038/ncomms8828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manso AS, Chai MH, Atack JM, Furi L, De Ste Croix M, Haigh R, Trappetti C, Ogunniyi AD, Shewell LK, Boitano M, Clark TA, Korlach J, Blades M, Mirkes E, Gorban AN, Paton JC, Jennings MP, Oggioni MR. 2014. A random six-phase switch regulates pneumococcal virulence via global epigenetic changes. Nat Commun 5:5055. doi: 10.1038/ncomms6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oliver MB, Basu Roy A, Kumar R, Lefkowitz EJ, Swords WE. 2017. Streptococcus pneumoniae TIGR4 phase-locked opacity variants differ in virulence phenotypes. mSphere 2:e00386-17. doi: 10.1128/mSphere.00386-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Atack JM, Weinert LA, Tucker AW, Husna AU, Wileman TM, Hadjirin NF, Hoa NT, Parkhill J, Maskell DJ, Blackall PJ, Jennings MP. 2018. Streptococcus suis contains multiple phase-variable methyltransferases that show a discrete lineage distribution. Nucleic Acids Res 46:11466–11476. doi: 10.1093/nar/gky913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Euler CW, Ryan PA, Martin JM, Fischetti VA. 2007. M.SpyI, a DNA methyltransferase encoded on a mefA chimeric element, modifies the genome of Streptococcus pyogenes. J Bacteriol 189:1044–1054. doi: 10.1128/JB.01411-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nye TM, Jacob KM, Holley EK, Nevarez JM, Dawid S, Simmons LA, Watson ME. 2019. DNA methylation from a type I restriction modification system influences gene expression and virulence in Streptococcus pyogenes. PLoS Pathog 15:e1007841. doi: 10.1371/journal.ppat.1007841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Finn MB, Ramsey KM, Tolliver HJ, Dove SL, Wessels MR. 2021. Improved transformation efficiency of group A Streptococcus by inactivation of a type I restriction modification system. PLoS One 16:e0248201. doi: 10.1371/journal.pone.0248201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen P, den Bakker HC, Korlach J, Kong N, Storey DB, Paxinos EE, Ashby M, Clark T, Luong K, Wiedmann M, Weimer BC. 2017. Comparative genomics reveals the diversity of restriction-modification systems and DNA methylation sites in Listeria monocytogenes. Appl Environ Microbiol 83:e02091-16. doi: 10.1128/AEM.02091-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Monk IR, Tree JJ, Howden BP, Stinear TP, Foster TJ. 2015. Complete bypass of restriction systems for major Staphylococcus aureus lineages. mBio 6:e00308-15. doi: 10.1128/mBio.00308-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beres SB, Musser JM. 2007. Contribution of exogenous genetic elements to the group A Streptococcus metagenome. PLoS One 2:e800. doi: 10.1371/journal.pone.0000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okada R, Matsumoto M, Zhang Y, Isaka M, Tatsuno I, Hasegawa T. 2014. Emergence of type I restriction modification system-negative emm1 type Streptococcus pyogenes clinical isolates in Japan. APMIS 122:914–921. doi: 10.1111/apm.12230. [DOI] [PubMed] [Google Scholar]
- 31.Calisto BM, Pich OQ, Piñol J, Fita I, Querol E, Carpena X. 2005. Crystal structure of a putative type I restriction-modification S subunit from Mycoplasma genitalium. J Mol Biol 351:749–762. doi: 10.1016/j.jmb.2005.06.050. [DOI] [PubMed] [Google Scholar]
- 32.Furuta Y, Kawai M, Uchiyama I, Kobayashi I. 2011. Domain movement within a gene: a novel evolutionary mechanism for protein diversification. PLoS One 6:e18819. doi: 10.1371/journal.pone.0018819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Simpson WJ, Musser JM, Cleary PP. 1992. Evidence consistent with horizontal transfer of the gene (emm12) encoding serotype M12 protein between group A and group G pathogenic streptococci. Infect Immun 60:1890–1893. doi: 10.1128/iai.60.5.1890-1893.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bessen DE, McShan WM, Nguyen SV, Shetty A, Agrawal S, Tettelin H. 2015. Molecular epidemiology and genomics of group A Streptococcus. Infect Genet Evol 33:393–418. doi: 10.1016/j.meegid.2014.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sumby P, Porcella SF, Madrigal AG, Barbian KD, Virtaneva K, Ricklefs SM, Sturdevant DE, Graham MR, Vuopio-Varkila J, Hoe NP, Musser JM. 2005. Evolutionary origin and emergence of a highly successful clone of serotype M1 group A Streptococcus involved multiple horizontal gene transfer events. J Infect Dis 192:771–782. doi: 10.1086/432514. [DOI] [PubMed] [Google Scholar]
- 36.Green NM, Zhang S, Porcella SF, Nagiec MJ, Barbian KD, Beres SB, LeFebvre RB, Musser JM. 2005. Genome sequence of a serotype M28 strain of group A Streptococcus: potential new insights into puerperal sepsis and bacterial disease specificity. J Infect Dis 192:760–770. doi: 10.1086/430618. [DOI] [PubMed] [Google Scholar]
- 37.Flores AR, Sahasrabhojane P, Saldaña M, Galloway-Peña J, Olsen RJ, Musser JM, Shelburne SA. 2014. Molecular characterization of an invasive phenotype of group A Streptococcus arising during human infection using whole genome sequencing of multiple isolates from the same patient. J Infect Dis 209:1520–1523. doi: 10.1093/infdis/jit674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McIntyre ABR, Alexander N, Grigorev K, Bezdan D, Sichtig H, Chiu CY, Mason CE. 2019. Single-molecule sequencing detection of N6-methyladenine in microbial reference materials. Nat Commun 10:579. doi: 10.1038/s41467-019-08289-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hanski E, Horwitz PA, Caparon MG. 1992. Expression of protein F, the fibronectin-binding protein of Streptococcus pyogenes JRS4, in heterologous streptococcal and enterococcal strains promotes their adherence to respiratory epithelial cells. Infect Immun 60:5119–5125. doi: 10.1128/iai.60.12.5119-5125.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu L, Olsen RJ, Nasser W, Beres SB, Vuopio J, Kristinsson KG, Gottfredsson M, Porter AR, DeLeo FR, Musser JM. 2015. A molecular trigger for intercontinental epidemics of group A Streptococcus. J Clin Invest 125:3545–3559. doi: 10.1172/JCI82478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sumby P, Whitney AR, Graviss EA, DeLeo FR, Musser JM. 2006. Genome-wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Pathog 2:e5. doi: 10.1371/journal.ppat.0020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Liu G, Feng W, Li D, Liu M, Nelson DC, Lei B. 2015. The Mga regulon but not deoxyribonuclease Sda1 of invasive M1T1 group A Streptococcus contributes to in vivo selection of CovRS mutations and resistance to innate immune killing mechanisms. Infect Immun 83:4293–4303. doi: 10.1128/IAI.00857-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Galloway-Peña J, Clement ME, Sharma Kuinkel BK, Ruffin F, Flores AR, Levinson H, Shelburne SA, Moore Z, Fowler VG, Jr.. 2016. Application of whole-genome sequencing to an unusual outbreak of invasive group A streptococcal disease. Open Forum Infect Dis 3:ofw042. doi: 10.1093/ofid/ofw042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Galloway-Peña J, DebRoy S, Brumlow C, Li X, Tran TT, Horstmann N, Yao H, Chen K, Wang F, Pan B-F, Hawke DH, Thompson EJ, Arias CA, Fowler VG, Bhatti MM, Kalia A, Flores AR, Shelburne SA. 2018. Hypervirulent group A Streptococcus emergence in an acaspular background is associated with marked remodeling of the bacterial cell surface. PLoS One 13:e0207897. doi: 10.1371/journal.pone.0207897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murphy J, Mahony J, Ainsworth S, Nauta A, van Sinderen D. 2013. Bacteriophage orphan DNA methyltransferases: insights from their bacterial origin, function, and occurrence. Appl Environ Microbiol 79:7547–7555. doi: 10.1128/AEM.02229-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tse H, Bao JYJ, Davies MR, Maamary P, Tsoi H-W, Tong AHY, Ho TCC, Lin C-H, Gillen CM, Barnett TC, Chen JHK, Lee M, Yam W-C, Wong C-K, Ong C-LY, Chan Y-W, Wu C-W, Ng T, Lim WWL, Tsang THF, Tse CWS, Dougan G, Walker MJ, Lok S, Yuen K-Y. 2012. Molecular characterization of the 2011 Hong Kong scarlet fever outbreak. J Infect Dis 206:341–351. doi: 10.1093/infdis/jis362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ben Zakour NL, Davies MR, You Y, Chen JHK, Forde BM, Stanton-Cook M, Yang R, Cui Y, Barnett TC, Venturini C, Ong C-lY, Tse H, Dougan G, Zhang J, Yuen K-Y, Beatson SA, Walker MJ. 2015. Transfer of scarlet fever-associated elements into the group A Streptococcus M1T1 clone. Sci Rep 5:15877. doi: 10.1038/srep15877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Flusberg BA, Webster DR, Lee JH, Travers KJ, Olivares EC, Clark TA, Korlach J, Turner SW. 2010. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods 7:461–465. doi: 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pacific Biosciences. 2015. Detecting DNA base modifications using single molecule, real-time sequencing. https://www.pacb.com/wp-content/uploads/2015/09/WP_Detecting_DNA_Base_Modifications_Using_SMRT_Sequencing.pdf.
- 50.Broudy TB, Pancholi V, Fischetti VA. 2002. The in vitro interaction of Streptococcus pyogenes with human pharyngeal cells induces a phage-encoded extracellular DNase. Infect Immun 70:2805–2811. doi: 10.1128/IAI.70.6.2805-2811.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Banks DJ, Lei B, Musser JM. 2003. Prophage induction and expression of prophage-encoded virulence factors in group A Streptococcus serotype M3 strain MGAS315. Infect Immun 71:7079–7086. doi: 10.1128/IAI.71.12.7079-7086.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee JYH, Carter GP, Pidot SJ, Guérillot R, Seemann T, Gonçalves da Silva A, Foster TJ, Howden BP, Stinear TP, Monk IR. 2019. Mining the methylome reveals extensive diversity in Staphylococcus epidermidis restriction modification. mBio 10:e02451-19. doi: 10.1128/mBio.02451-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anton BP, Fomenkov A, Wu V, Roberts RJ. 2021. Genome-wide identification of 5-methylcytosine sites in bacterial genomes by high-throughput sequencing of MspJI restriction fragments. PLoS One 16:e0247541. doi: 10.1371/journal.pone.0247541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cooper LP, Roberts GA, White JH, Luyten YA, Bower EKM, Morgan RD, Roberts RJ, Lindsay JA, Dryden DTF. 2017. DNA target recognition domains in the type I restriction and modification systems of Staphylococcus aureus. Nucleic Acids Res 45:3395–3406. doi: 10.1093/nar/gkx067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Croucher NJ, Coupland PG, Stevenson AE, Callendrello A, Bentley SD, Hanage WP. 2014. Diversification of bacterial genome content through distinct mechanisms over different timescales. Nat Commun 5:5471. doi: 10.1038/ncomms6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Karginov FV, Hannon GJ. 2010. The CRISPR system: small RNA-guided defense in bacteria and archaea. Mol Cell 37:7–19. doi: 10.1016/j.molcel.2009.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roberts GA, Houston PJ, White JH, Chen K, Stephanou AS, Cooper LP, Dryden DTF, Lindsay JA. 2013. Impact of target site distribution for type I restriction enzymes on the evolution of methicillin-resistant Staphylococcus aureus (MRSA) populations. Nucleic Acids Res 41:7472–7484. doi: 10.1093/nar/gkt535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li J, Li J-W, Feng Z, Wang J, An H, Liu Y, Wang Y, Wang K, Zhang X, Miao Z, Liang W, Sebra R, Wang G, Wang W-C, Zhang J-R. 2016. Epigenetic switch driven by DNA inversions dictates phase variation in Streptococcus pneumoniae. PLoS Pathog 12:e1005762. doi: 10.1371/journal.ppat.1005762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huang X, Wang J, Li J, Liu Y, Liu X, Li Z, Kurniyati K, Deng Y, Wang G, Ralph JD, De Ste Croix M, Escobar-Gonzalez S, Roberts RJ, Veening J-W, Lan X, Oggioni MR, Li C, Zhang J-R. 2020. Prevalence of phase variable epigenetic invertons among host-associated bacteria. Nucleic Acids Res 48:11468–11485. doi: 10.1093/nar/gkaa907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fagerlund A, Langsrud S, Schirmer BCT, Møretrø T, Heir E. 2016. Genome analysis of Listeria monocytogenes sequence type 8 strains persisting in salmon and poultry processing environments and comparison with related strains. PLoS One 11:e0151117. doi: 10.1371/journal.pone.0151117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Eraso JM, Kachroo P, Olsen RJ, Beres SB, Zhu L, Badu T, Shannon S, Cantu CC, Saavedra MO, Kubiak SL, Porter AR, DeLeo FR, Musser JM. 2020. Genetic heterogeneity of the Spy1336/R28-Spy1337 virulence axis in Streptococcus pyogenes and effect on gene transcript levels and pathogenesis. PLoS One 15:e0229064. doi: 10.1371/journal.pone.0229064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sanson MA, Macias OR, Shah BJ, Vega LA, Alaramat Z, Flores AR. 2019. Unexpected relationships between frequency of antimicrobial resistance, disease phenotype and emm type in group A Streptococcus. Microb Genom 5:e000316. doi: 10.1099/mgen.0.000316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.DebRoy S, Sanson M, Shah B, Regmi S, Vega LA, Odo C, Sahasrabhojane P, McGeer A, Tyrrell GJ, Fittipaldi N, Shelburne SA, Flores AR. 2021. Population genomics of emm4 group A Streptococcus reveals progressive replacement with a hypervirulent clone in North America. mSystems 6:e0049521. doi: 10.1128/mSystems.00495-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wick RR, Judd LM, Gorrie CL, Holt KE. 2017. Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput Biol 13:e1005595. doi: 10.1371/journal.pcbi.1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tatusova T, DiCuccio M, Badretdin A, Chetvernin V, Nawrocki EP, Zaslavsky L, Lomsadze A, Pruitt KD, Borodovsky M, Ostell J. 2016. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res 44:6614–6624. doi: 10.1093/nar/gkw569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tonkin-Hill G, MacAlasdair N, Ruis C, Weimann A, Horesh G, Lees JA, Gladstone RA, Lo S, Beaudoin C, Floto RA, Frost SDW, Corander J, Bentley SD, Parkhill J. 2020. Producing polished prokaryotic pangenomes with the Panaroo pipeline. Genome Biol 21:180. doi: 10.1186/s13059-020-02090-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sullivan MJ, Petty NK, Beatson SA. 2011. Easyfig: a genome comparison visualizer. Bioinformatics 27:1009–1010. doi: 10.1093/bioinformatics/btr039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.DebRoy S, Aliaga-Tobar V, Galvez G, Arora S, Liang X, Horstmann N, Maracaja-Coutinho V, Latorre M, Hook M, Flores AR, Shelburne SA. 2021. Genome-wide analysis of in vivo CcpA binding with and without its key co-factor HPr in the major human pathogen group A Streptococcus. Mol Microbiol 115:1207–1228. doi: 10.1111/mmi.14667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Alramadhan M, Heresi GP, Flores AR. 2019. Severe emm89 group A streptococcal disease characterized by toxic shock and endocarditis. Case Rep Infect Dis 2019:6568732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Beres SB, Carroll RK, Shea PR, Sitkiewicz I, Martinez-Gutierrez JC, Low DE, McGeer A, Willey BM, Green K, Tyrrell GJ, Goldman TD, Feldgarden M, Birren BW, Fofanov Y, Boos J, Wheaton WD, Honisch C, Musser JM. 2010. Molecular complexity of successive bacterial epidemics deconvoluted by comparative pathogenomics. Proc Natl Acad Sci USA 107:4371–4376. doi: 10.1073/pnas.0911295107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Flores AR, Luna RA, Runge JK, Shelburne SA, Baker CJ. 2017. Cluster of fatal group A streptococcal emm87 infections in a single family: molecular basis for invasion and transmission. J Infect Dis 215:1648–1652. doi: 10.1093/infdis/jix177. [DOI] [PubMed] [Google Scholar]
- 72.Waterhouse AM, Procter JB, Martin DMA, Clamp M, Barton GJ. 2009. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Roberts RJ, Vincze T, Posfai J, Macelis D. 2015. REBASE—a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res 43:D298–D299. doi: 10.1093/nar/gku1046. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of GAS strains used for TRD and phylogenomic analysis. Download Table S1, XLSX file, 0.03 MB (35.6KB, xlsx) .
Copyright © 2021 DebRoy et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Schematic representation comparing the architecture of the GAS type I RM system to that of Streptococcus agalactiae (also known as group B Streptococcus). Download FIG S1, EPS file, 1.2 MB (1.2MB, eps) .
Copyright © 2021 DebRoy et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Comparison of the type I RM systems of GAS, Streptococcus pneumoniae, and Streptococcus suis. Two examples of S. pneumoniae are included to display different arrangements of the hsdS′ and hsdS′′ genes as observed in sequenced strains. Download FIG S2, EPS file, 1.2 MB (1.2MB, eps) .
Copyright © 2021 DebRoy et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
A schematic representation of the movement of TRD domains within GAS strains (emm1/emm28) and between GAS and GBS strains compared using EasyFig (67). Download FIG S3, EPS file, 0.7 MB (747KB, eps) .
Copyright © 2021 DebRoy et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
GAS TRD alleles do not correlate with accessory genome composition. A t-SNE plot indicates the distribution of TRD1/2 combinations in the accessory genomes of 224 GAS strains. Each dot indicates a single isolate’s accessory genome (gene content shared in <99% genomes), with color indicating TRD pattern assignment per the legend. emm types that clustered by accessory genome content are labeled on the figure. An asterisk indicates one isolate belonging to emm204 that clusters with emm1 isolates. Download FIG S4, EPS file, 0.5 MB (482KB, eps) .
Copyright © 2021 DebRoy et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
(a) List of strains analyzed by PacBio and reference genomes used. (b) List of DNA motifs with non-m6A base modification detected by PacBio. Download Table S2, XLSX file, 0.01 MB (13.5KB, xlsx) .
Copyright © 2021 DebRoy et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Isogenic hsdM mutants of emm28 and emm87 GAS. (A) Growth of MGAS6180 and TSPY1057 and their isogenic hsdM mutants in THY broth. (B) TaqMan qRT-PCR confirms lack of hsdM transcripts in isogenic ΔhsdM strains. Data show the mean ± standard deviation from two biological replicates, with two technical replicates, done on separate days. Download FIG S5, EPS file, 0.9 MB (937.9KB, eps) .
Copyright © 2021 DebRoy et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Modification quality value (QV) histograms indicating methylation events detected by PacBio sequencing between (A) emm1 strains MGAS2221 and SF370 and (B) emm4 strains SAS-M4-Duke and RLGH. Modification quality values (modQV) are indicated on the x axis, and the number of bases is displayed on the y axis. The lines are color-coded for each nucleotide. Download FIG S6, TIF file, 2.5 MB (2.5MB, tif) .
Copyright © 2021 DebRoy et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Primers and probes used in this study. Download Table S3, XLSX file, 0.01 MB (10.7KB, xlsx) .
Copyright © 2021 DebRoy et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Data Availability Statement
The data that support the findings of this study will be shared upon publication. Whole genome sequences for GAS strains TSPY136 (CP060647), TSPY1309 (CP060644), TSPY416 (CP060643), and TSPY125 (CP007562.1) were obtained during this study and submitted to GenBank.