

Abstract
Induction of nucleic-acid sensing-mediated type I interferon (IFN) has emerged as a novel approach to activate the immune system against cancer. Here we show that the depletion of DEAD-box RNA helicase 3X (DDX3X) triggers a tumor-intrinsic type I IFN response in breast cancer cells. Depletion or inhibition of DDX3X activity led to aberrant cytoplasmic accumulation of cellular endogenous double-stranded RNAs (dsRNA), which triggered type I IFN production through the melanoma differentiation-associated gene 5 (MDA5)-mediated dsRNA sensing pathway. Furthermore, DDX3X interacted with dsRNA-editing ADAR1 and dual depletion of DDX3X and ADAR1 synergistically activated the cytosolic dsRNA pathway in breast cancer cells. Loss of DDX3X in mouse mammary tumors enhanced anti-tumor activity by increasing the tumor-intrinsic type I IFN response, antigen presentation, and tumor-infiltration of cytotoxic T and dendritic cells. These findings may lead to the development of a novel therapeutic approach for breast cancer by targeting DDX3X in combination with immune checkpoint blockade.
Keywords: DDX3X, Double-stranded RNAs, Type I interferon, Cancer immunity
Introduction
Recent advanced transcriptomics studies have shown that metazoan cells express various types of endogenous “self” dsRNAs, such as endogenous retroviral elements (ERVs), repetitive RNA elements, mitochondrial dsRNAs, mRNAs with inverted Alu-containing 3’ UTRs, and structural dsRNAs with long dsRNA stem (1–6). Malignant cells may have elevated levels of dsRNAs due to loss of suppressive epigenetic modifications in repetitive elements, genomic instability, or oxidative stress-induced mitochondrial damage, which would increase dsRNA burden in cancer cells (7,8). For example, ERVs constitute more than 8% of the human genome, and most ERVs are silenced in normal somatic cells by promoter DNA methylation (9–11). Some cancers lose ERV DNA methylation and aberrantly overexpress ERVs, and their bi-directional transcription has been shown to increase the formation of dsRNAs (12,13).
Accumulating evidence has revealed that induced intracellular dsRNA accumulation in cancer cells stimulates the induction of cancer-derived type I IFN, which enhances anti-tumor immunity (14–17). Abnormal accumulation of the endogenous dsRNAs triggers an antiviral innate immune response through activation of dsRNA sensing pathways, such as melanoma differentiation-associated gene 5 (MDA5) or protein kinase R (PKR), and this response could cause chronic inflammation and related human diseases (8,18,19). Human ADARs (adenosine deaminases) are known to be critical regulators of endogenous cellular dsRNAs (2,20). ADAR1 edits adenosine (A), within the double-stranded regions, to inosine (I) (known as A-to-I RNA editing), which results in the disruption of dsRNA structures or the retention of edited dsRNAs within the nucleus (2). In the mitochondria, RNA degradosome components SUV3 and polynucleotide phosphorylase (PNPase) prevent the accumulation of mitochondrial dsRNAs (21).
DEAD-box RNA helicase 3X (DDX3X) belongs to the large DEAD-box (Asp-Glu-Ala-Asp) family of ATP-dependent RNA helicase superfamily 2 (22). DDX3X has an RNA helicase activity to unwind RNA duplexes, and participates in multiple aspects of RNA metabolism, such as transcription, RNA splicing, RNA transport, and initiation of translation (22,23). In particular, increased DDX3X levels have been reported in primary and metastatic breast cancer, and high expression of DDX3X is correlated with worse survival, suggesting DDX3X as a potential therapeutic target for breast cancer (24–27).
Here, we found that knockdown or inhibition of DDX3X resulted in the aberrant cytoplasmic accumulation of cellular dsRNAs in breast cancer cells, which triggered type I IFN production through MDA5 cytosolic dsRNA sensing pathway. The depletion of DDX3X in a syngeneic mouse breast tumor model suppressed tumor progression and enhanced anti-tumor activity by inducing the dsRNA-derived type I IFN response.
Materials and Methods
Cell culture and Generation of stable cell lines
The human breast cancer cell lines MCF7, MDA-MB-453, MDA-MB-231, as well as HEK293T human embryonic kidney, and 4T1 and E077 mouse mammary cancer cell lines were obtained from the American Type Culture Collection and cultured under standard conditions specified by the manufacturer. B16-OVA cells were kindly provided by Dr. Charles G. Drake (Columbia University Medical Center, NY, USA) and maintained in DMEM medium supplemented with 10% fetal bovine serum, 1% penicillin/streptomycin, and 250 μg/ml G418 (Invitrogen) at 37°C with 5% CO2. All of the cell lines tested negative for mycoplasma using a Mycoplasma Detection kit (Lonza). For generation of the stable or inducible DDX3X knockdown cell lines, lentiviral pGIPZ, inducible TRIPZ, and SMARTvector (Inducible Mouse Ddx3x shRNA) were obtained from Dharmacon.
Kaplan-Meier (KM) plotter analysis
KM survival analysis of DDX3X was performed with an online tool (http://kmplot.com/analysis) using breast cancer microarray, RNA-seq, and protein datasets (28). The Affymetrix ID: 201210_at (JeSet best optimal probe for DDX3X) was selected for microarray analysis. The cutoff value of DDX3X was determined by an auto-select cutoff algorithm embedded in the KM plotter website. Statistically, a p-value < 0.05 (log rank p-value) was considered a significant Hazard ratio (HR), and the corresponding 95% confidence intervals (95% CI) are displayed within the figure.
qRT-PCR
RNA was extracted using TRIzol reagent (15596026, Thermo Scientific) or TRIzol LS Reagent (10296010, Thermo Scientific), according to the manufacturer’s protocol. RNA concentration was measured by NanoDrop analysis. 1 μg of total RNA was used to generate cDNA with the High Capacity cDNA Reverse Transcription Kit (4368814, Thermo Scientific). Quantitative reverse transcription PCR (qRT-PCR) was performed using TaqMan Fast Advanced Master Mix (4444557, Applied Biosystems) or SYBR Green master mix (A25742, Applied Biosystems). All qRT-PCR was performed on a QuantStudio 3 Real-Time PCR system (Applied Biosystems). GAPDH or 18s were used as reference genes. All qRT-PCR assays were carried out in triplicate and then repeated with new cDNA synthesis. Primer information is provided in Supplementary Table 1.
TASA-TD strand-specific PCR
First strand cDNA synthesis and strand specific PCR for detection of sense and antisense ERV transcripts using TASA-TD methodology was performed according to Henke et al. (29).
dsRNA dot blot
The dsRNA dot blot analysis was performed as previously described (30,31). Briefly, total cellular RNA or the RNA of cytoplasmic and nuclear fractions were extracted using TRIzol reagent (15596026, Thermo Scientific) or TRIzol LS Reagent (10296010, Thermo Scientific), respectively, according to the manufacturer’s protocol. Isolated RNA was dotted on PVDF membrane and incubated with dsRNA-specific J2 or K1 antibody overnight at 4°C. Membranes were incubated with secondary antibody for 1 hour at room temperature. ECL was applied to the membrane for visualization (WBKLS0500, Millipore or #1705061, Bio-Rad).
Western blot
Cell lysates were prepared with Cell lysis buffer (#9803, Cell Signaling Technology) containing a protease inhibitor cocktail (87785, Thermo Fisher). Cell lysates were kept on ice for 30 minutes and centrifuged at 13,200 rpm for 10 minutes at 4°C. Protein concentrations were measured by the Bio-Rad protein assay (#5000006, Bio-Rad). 4X Laemmli Sample Buffer (#1610747, Bio-Rad) containing 2-mercaptoethanol (#1610710, Bio-Rad) was added to the cell lysates and boiled at 95°C for 10 minutes. Protein samples were subjected to SDS-PAGE and transferred to polyvinylidene difluoride membranes (#1620177, Bio-Rad). The membranes were blocked with 5% skim milk in TBS-T (0.1% Tween-20) for 1 hour at room temperature and incubated with indicated primary antibodies at 4°C overnight. The next day, membranes were washed with TBS-T (0.1% Tween-20) and incubated with appropriate secondary antibodies for 1 hour at room temperature. ECL (WBKLS0500, Millipore or #1705061, Bio-Rad) was applied for membrane development. Antibody information is provided in Supplementary Table S2.
Immunohistochemistry and immunofluorescence.
For immunohistochemistry, paraffin embedded tumor samples and sections were prepared as previously described (32). Samples were incubated with primary antibodies, including CD8 (#98941, Cell Signaling Technology), Ki-67 (#12202, ell Signaling Technology), perforin (#31647, Cell Signaling Technology), and J2 (10010200, Scicons) at 4°C overnight. Sections were incubated with biotinylated goat anti-rabbit IgG (PK-4000, Vector laboratories) followed by washing three times with TBS-T (0.05% Tween-20). After washing, sections were incubated with VECTASTAIN ABC Reagent (PK-4000, Vector laboratories) for 30 minutes and then developed using a DAB peroxidase substrate kit (ImmPACT DAB, SK-4105, Vector laboratories). Sections were counterstained with hematoxylin (3801570, Leica). For nuclease treatment, cells were treated with RNase I (50 U/ml), RNase A (10 μg/ml), or RNase III (20 U/ml at 37°C for 30 minutes before fixation. For J2 immunofluorescence, cells were plated on glass coverslips. Cells were washed twice with PBS and then fixed with 4% (v/v) formaldehyde in PBS at room temperature for 15 minutes and permeabilized with 0.25% Triton-X100 in PBS for 20 minutes at room temperature. Cells were incubated in the J2 antibody, diluted in PBS (1: 100), at 4°C overnight. The following day, cells were washed three times with PBS and incubated with rat anti-mouse IgG2a conjugated with APC (407109, BioLegend) for 1 hour at room temperature. Imaging was performed with a Leica SP8 confocal microscope.
Dual reporter assay
Cells with single or double knockdown of DDX3X and ADAR1 were transfected with ADAR1 editing reporter plasmids (including positive and negative controls) using ViaFect transfection reagent (E4981, Promega). Reporter activity was measured using the Nano-Glo Dual-Luciferase Reporter Assay System (N1610, Promega), according to manufacturer’s instructions.
ELISA
ELISA assays were performed according to manufacturer’s instructions. The supernatant of cell cultures, or tumor lysates, was collected and the concentration of IFNs were determined using the Human IFN-α (41100-1, PBL assay science), Human IFN-β (DIFNB0, R&D systems), Human IFN-γ (DY285B, R&D systems), Mouse IFN-α (42120-1, PBL assay science), Mouse IFN-β (DY8234–05, R&D systems), and Mouse IFN-γ (DY485–05, R&D systems) ELISA Kit.
Animal experiments
All studies were approved and supervised by the Institutional Animal Care and Use Committee at Georgetown University. Female Balb/c mice (6-8 weeks old) were purchased from Jackson Laboratories. To generate the syngeneic mouse mammary tumor model, 1×105 4T1 mouse mammary cancer cells, stably expressing tet-inducible DDX3X shRNA were implanted into the mammary fat pad of female Balb/c mice after being mixed (1:1 ratio by volume) with matrigel (Corning). Prior to the induction of DDX3X knockdown, mice were divided into control and DDX3X knockdown groups of equal average tumor volume. Doxycycline water (5% sucrose with 2 mg/ml of doxycycline; Sigma) was provided to the DDX3X knockdown group to induce knockdown of DDX3X. The doxycycline water was changed every other day. Tumor size was measured every 3–5 days by caliper. Tumor volumes were calculated using the formula: Volume = (Width2 × Length)/2.
Results
Depletion of DDX3X upregulates genes involved in the antiviral innate immune response pathway.
Previous reports suggest that high levels of DDX3X expression correlated with worse survival in breast cancer patients (25,26,33). In our Kaplan-Meier (KM) plotter analysis using a Pan-cancer RNA-sequencing dataset of breast cancer, high expression of DDX3X mRNA correlated with worse Overall Survival (OS, p=0.019) and worse Post-Progression Survival (PPS, p=0.00063) in breast cancer patients (Fig.1A). High DDX3X protein expression also was correlated with worse survival in patients with breast cancer (p=0.017) (Fig.1A). We further analyzed the prognostic significance of DDX3X expression in “intrinsic” subtypes luminal A, luminal B, HER2-enriched, and basal-like using KM plotter with microarray datasets (Affymetrix ID: 201210_at). The results showed that higher DDX3X expression was associated with a worse Relapse-Free Survival (RFS) in luminal A (p=0.00041), luminal B (p=0.038), and basal (p=0.00049) types of breast cancer (Supplementary Figs. S1A and S1B). Higher DDX3X expression also was associated with a worse PPS in luminal A (p=0.00017) breast cancer patients (Supplementary Fig. S1C).
Figure 1. Depletion of DDX3X upregulates genes in the antiviral innate immune response.
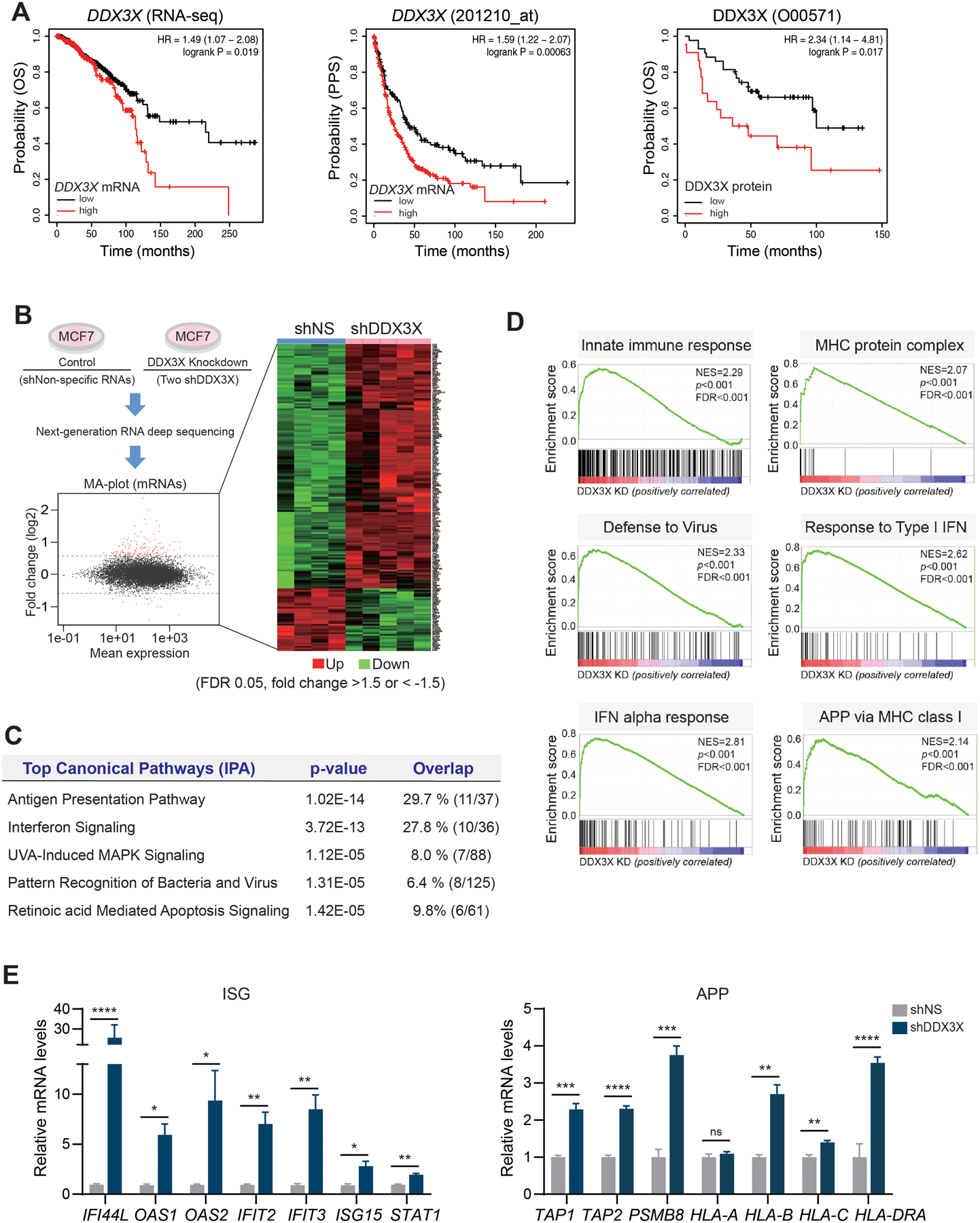
A. KM-plotter analysis shows that mRNA and protein levels of DDX3X are associated with a poor survival outcome in breast cancer. Overall survival (pan-cancer RNA seq), OS; Post- Progression Survival, PPS.
B. Genome-wide transcriptome analysis in DDX3X-control (shNS) or -knockdown (shDDX3X) MCF7 cells using a next-generation RNA deep sequencing. Differentially expressed genes (DEGs) (FDR 0.05, fold change >1.5 or < −1.5) in shNS vs. shDDX3X are shown in an MA-plot (labeled in red) and in a heat map.
C. Top canonical pathways of DEGs between DDX3X-control and -knockdown (KD) MCF7 cells.
D. GSEA analysis of DEGs between DDX3X-control and -KD MCF7 cells.
E. qRT-PCR of ISGs and APP gene expression in DDX3X-control or -KD MCF7 cells.
A non-specific shRNA (shNS); DDX3X targeting shRNAs (shDDX3X). Data are representative of three independent experiments. Statistical significance was calculated using unpaired t-tests. *p < 0.05; **p<0.01; ***p < 0.001; ****p < 0.0001; ns, not significant.
To assess the impact of DDX3X on gene expression in breast cancer, we performed a genome-wide transcriptome analysis in DDX3X-control (using non-specific shRNA) or -knockdown (using two different shRNAs) MCF7 human breast cancer cells (Fig. 1B and Supplementary Fig. S1D). The knockdown (KD) of DDX3X significantly increased the expression of genes involved in the innate antiviral immune response (Fig.1B, Supplementary Table 3, GEO accession number GSE157323). Ingenuity pathway analysis (IPA) of differentially expressed genes (DEGs, cut off >1.5-fold, FDR <0.05) in DDX3X-control versus (vs.) -KD MCF7 identified the top 5 canonical pathways, including type I IFN signaling, antigen presentation pathway (APP), and pattern recognition receptors (PRRs) response to bacteria and virus (Fig.1C). In the gene-set enrichment analysis (GSEA) of differentially expressed genes (DEGs), the gene sets co-enriched in the DDX3X KD cells included type I IFN response, antigen presentation via major histocompatibility complex (MHC) class I, and viral defense response (Fig.1D). Next, using a quantitative RT-PCR (qRT-PCR) analysis, we validated the upregulation of genes in IFN-stimulated genes (ISGs) and APP in DDX3X-KD MCF7 or DDX3X-KD MDA-MB-453 cells (Fig.1E and Supplementary Fig. S1E). As a control, we depleted other DDX proteins, such as DDX1 or DDX5 in MCF7 cells. The depletion of DDX1 or DDX5-KD did not impact the expression of the ISGs (Supplementary Fig. S1F).
To analyze the relationship between DDX3X and the anti-viral innate immune response in various cancers, we selected DDX3X-highly expressing (DDX3Xhi, 253 cell lines) and DDX3X-low expressing cancer cell lines (DDX3Xlow, 253 cell lines) in the Cancer Cell Line Encyclopedia (CCLE) (Supplementary Fig. S2A). The DDX3Xhi or DDX3Xlow groups consisted of various cancer types derived from both male and female patients (Supplementary Fig. S2B). The transcript level of DDX3X was similar between males and females within each group (Supplementary Fig. S2C). 386 DEGs were identified in DDX3Xhi vs. DDX3Xlow cancer cells (cut off at p-value < 0.01 and fold change > 1.5). Forty-nine genes in the DDX3Xhi cell group and 337 genes in the DDX3Xlow cell group were upregulated, respectively. We have analyzed gene set enrichment of DEGs in DDX3Xhi vs. DDX3Xlow cancer cells (Supplementary Fig. S2D). The DDX3Xlow cell group revealed a significant gene set enrichment for innate immune responses, such as the inflammatory response, IFN-alpha response, and allograft rejection pathways (Supplementary Fig. S2E). In contrast, UV response gene set enrichment was found in DDX3Xhi cell group. Elevated levels of MHC class genes, types I IFNs (IFNB1, IFNE), and the proinflammatory cytokine IL6 were found in the DDX3Xlow cells (Supplementary Figs. S2F). Collectively, these data suggest that the expression of DDX3X is inversely correlated with the intrinsic type I IFN and innate immune response activation in various solid cancers, including breast cancer.
Loss of DDX3X induces type I IFN production and STAT1 activation.
Canonical type I IFN signaling leads to the transcription of hundreds of IFN-stimulated genes (ISGs) through the activation of the Janus kinase-signal transducer and activator of transcription (JAK/STAT) pathway (34). Therefore, we examined the level of IFNs in the culture media of DDX3X-control or -KD breast cancer cells, including human MCF7 and MDA-MB-453 cells, as well as E0771 mouse mammary cancer cells. Media from DDX3X-KD cells showed a higher level of type I IFN (interferon-alpha, α; interferon-beta, β), while the level of type II IFN (interferon-gamma, γ) was not changed by DDX3X KD (Fig.2A and Supplementary Fig. S3A). The depletion of DDX3X increased the level of phosphorylated STAT1 (p-STAT1) in MCF7 and MDA-MB-453 cells, while the level of phosphorylated STAT2 was lower than our ability to detect (Fig.2B, and Supplementary Figs.S3B and S3C). Upregulation of non-phosphorylated STAT1 and STAT2 also are known to induce downstream IFN signaling (35,36). In DDX3X KD cells, non-phosphorylated STAT1 and STAT2 levels also were increased (Fig.2B and Supplementary Fig. S3B).
Figure 2. Loss of DDX3X induces type I IFN production, STAT1 activation, and antigen processing and presentation.
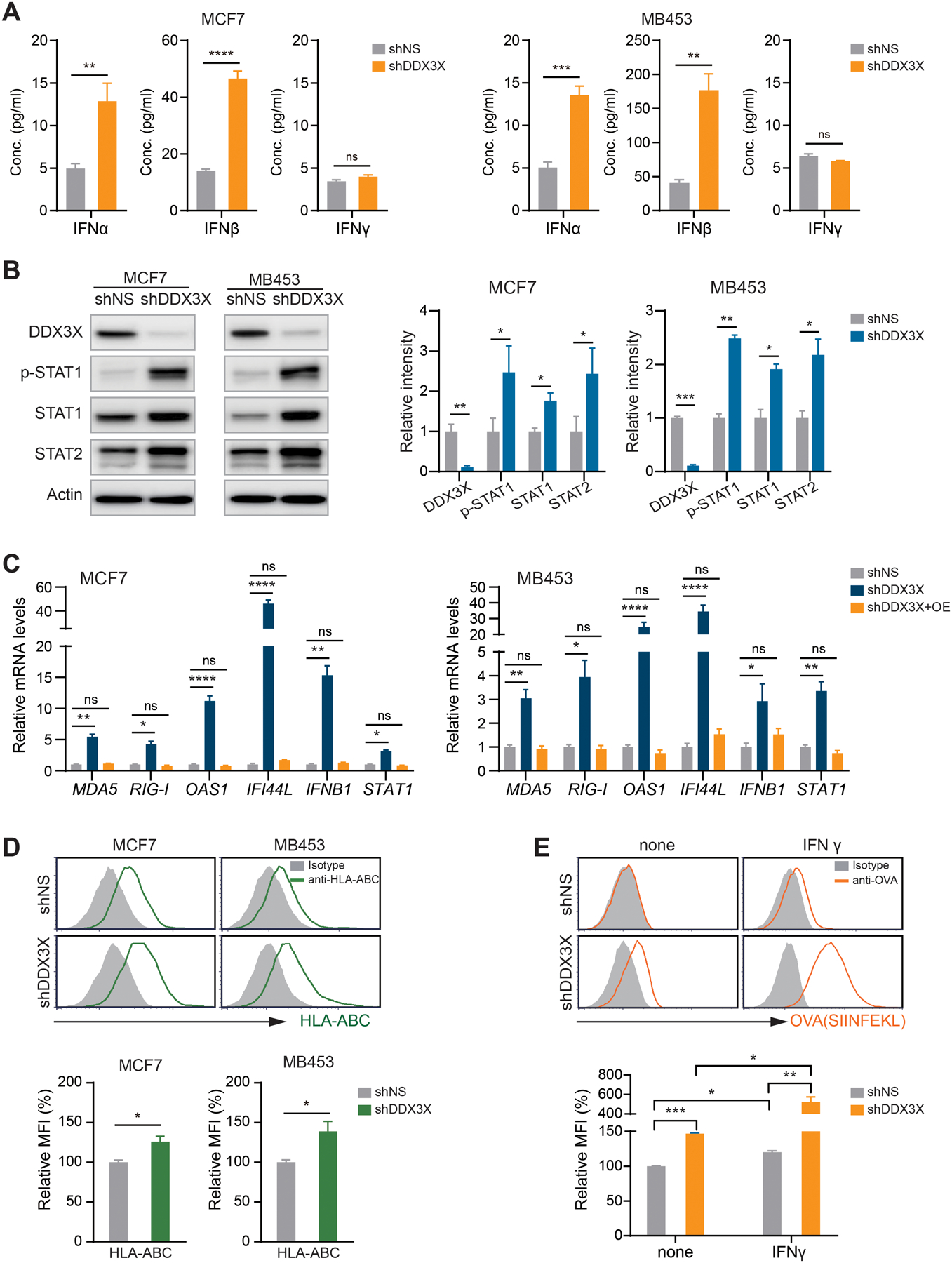
A. ELISA of IFN-α, -β, and -γ in the culture supernatants from DDX3X-control or -KD MCF7 and MDA-MB-453 cells.
B. Western blot analysis in the DDX3X-control or -KD MCF7 or MDA-MB-453 cells.
C. qRT-PCR of ISGs in DDX3X-KD MCF7 or MDA-MB-453 cells with DDX3X overexpression.
D. Representative flow histograms (upper) and relative MFI quantification (bottom) of MHC class I (HLA-ABC) expression in the DDX3X-KD MCF7 or DDX3X-KD MDA-MB-453 cells.
E. Representative histograms and a relative MFI quantification of OVA peptide (SIINFEK) bound to MHC class I (H-2Kb) on DDX3X-KD B16-OVA cells. Cells were treated 100 ng/ml of IFNγ for 48 hours.
Mean fluorescence intensity (MFI); Data are shown as mean ± SEM of three independent experiments. Statistical significance was calculated using unpaired t-tests (Fig. 2A, 2B, 2D, and 2E) or one-way ANOVA with uncorrected Fisher’s LSD test (Fig. 2C). *p < 0.05; **p< 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant.
To validate the specific role of DDX3X in type I IFN activation, we performed a rescue experiment by overexpressing DDX3X in the DDX3X-control or -KD cells (Supplementary Fig. S3D). DDX3X overexpression (OE) successfully diminished the induction of ISGs as well as upregulation of IFNB1 and STAT1 in DDX3X-KD MCF7 and DDX3X-KD MDA-MB-453 cells (Fig.2C). DDX3X-KD MCF-7 cells demonstrated a more robust induction of ISG expression than DDX3X-control MCF7 cells in response to treatment with IFN-α (Supplementary Fig. S3E). DDX3X KD also significantly suppressed colony-formation ability in MCF7 cells (Supplementary Fig. S3F).
We examined cell surface expression of MHC class I proteins of DDDX3X-KD cells, using flow cytometry analysis. DDX3X-KD MCF7 and MDA-MB-453 cells displayed enhanced expression of cell surface MHC class I proteins (HLA-ABC) (Fig.2D). Although qRT-PCR analysis showed an increased expression of HLA-DRA, one of the subunits of the MHC class II molecule, in DDX3X KD cells (Fig.1E), we did not observe the increased surface expression of the MHC II protein complex on the surface of DDX3X-KD MCF7 and MDA-MB-453 cells (Supplementary Fig. S3G). Furthermore, DDX3X KD increased the presentation of the ovalbumin (OVA) epitope (SIINFEKL) on MHC I molecules on the cell surface of OVA-expressing mouse melanoma cancer cells (Fig.2E). OVA peptide presentation was dramatically increased in the DDX3X KD cells by IFN-γ stimulation (Fig.2E). These data indicate that DDX3X KD enhances antigen processing, as well as antigen presentation via MHC I, in cancer cells.
dsRNA sensing MDA5-NFκB signaling axis leads to type I IFN production in DDX3X-depleted breast cancer cells.
To determine which signaling pathway is responsible for inducing type I IFN expression in the DDX3X-KD breast cancer cells, we examined the cytosolic DNA sensing-mediated innate immune response through the stimulator of interferon genes (STING) pathway (37). Control MCF7 and MDA-MB-453 cells showed an intact STING activation in response to synthetic dsDNA (Poly (dA:dT) (Supplementary Fig. S4A). DDX3X KD did not induce the phosphorylation of STING (Ser366), which is a downstream target of cyclic GMP-AMP synthetase (cGAS), a central receptor of cytosolic DNA (Fig.3A). No phosphorylation was detected in TANK-binding kinase 1 (TBK1) or in transcription factors, such as interferon regulatory factor 3 (IRF3) or interferon regulatory factor 7 (IRF7) (Fig.3B). These data indicate that the STING pathway is not involved in DDX3X KD-mediated type I IFN production. However, we observed increased phosphorylation of NF-κB p65 (Ser536) and IκB alpha (Ser32) in DDX3X-KD cells (Fig.3C and Supplementary Fig. S4B), suggesting that NFκB may be a central transcription factor regulating type IFN expression in DDX3X-KD cells. Interestingly, treatment with a synthetic dsRNA mimic analog, polyinosinic–polycytidylic acid (poly I:C), was able to induce the phosphorylation of both IRF3 and IRF7 in MCF7 and MDA-MB-453 (Supplementary Fig. S4C), suggesting that DDX3X KD may activate a unique endogenous dsRNAs-mediated pathway, which is different than the dsRNA-sensing response to exogenous synthetic dsRNAs.
Figure 3. dsRNA sensing MDA5-NFκB signaling axis leads to type I IFN production in DDX3X-depleted breast cancer cells.
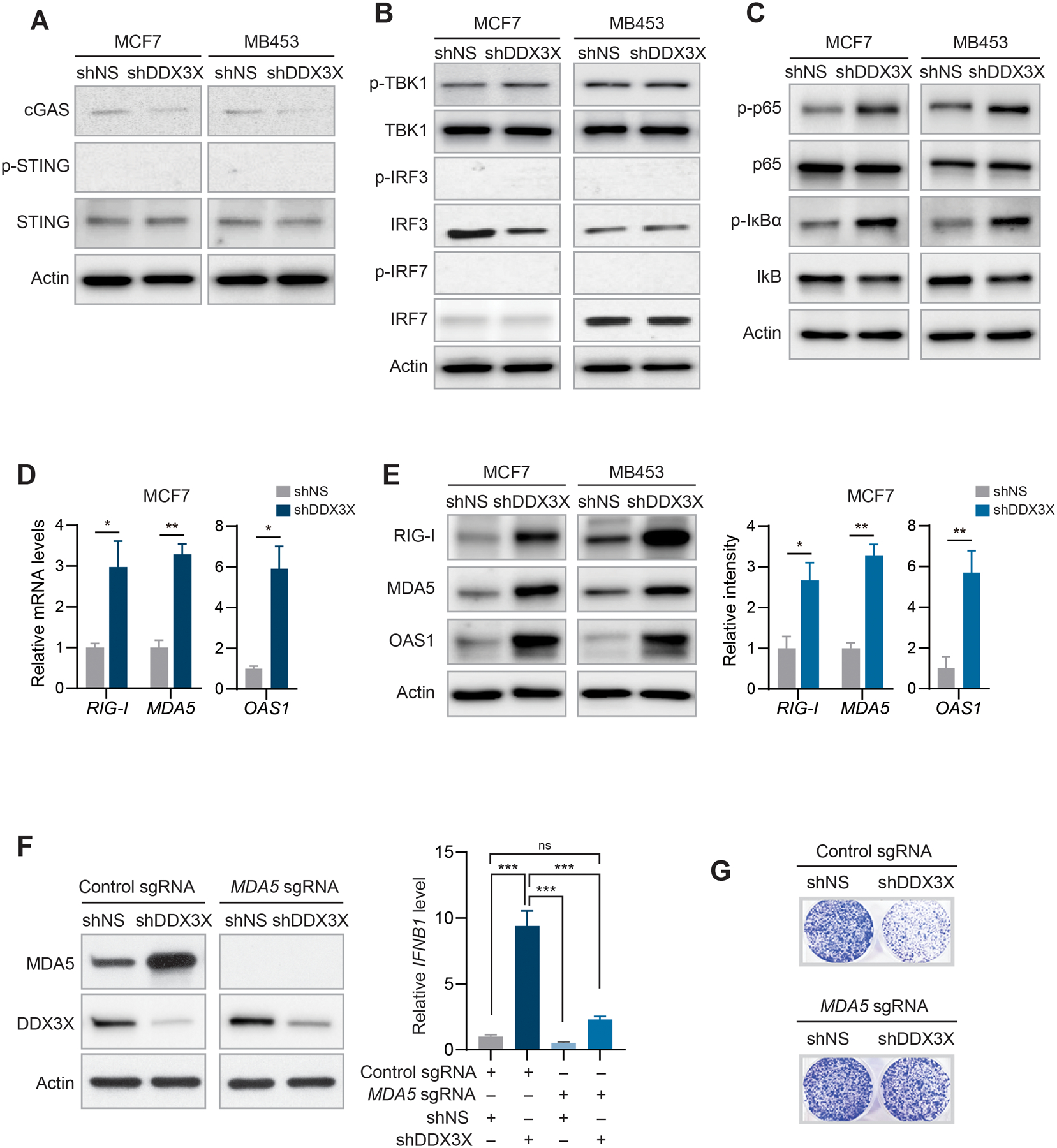
A. Western blot analysis of STING, phosphorylation of STING, and cGAS in DDX3X-control or -KD MCF7 or MDA-MB-453 cells.
B. Western blot analysis of phosphorylation of TBK1, IRF3, and IRF7 in DDX3X-control or -KD MCF7 or MDA-MB-453 cells.
C. Western blot analysis of phosphorylation of NFκB p65 and IκBα in DDX3X-control or -KD MCF7 or MDA-MB-453 cells.
D. qRT-PCR of RIG-I, MDA5, and OAS1 in DDX3X-control or KD-MCF7 cells.
E. Western blot analysis of RIG-I, MDA5, and OAS1 in DDX3X-control or -KD MCF7 or MDA-MB-453 cells (left). Bar graphs of relative protein band intensity (right).
F. Western blot validation of CRISPR/Cas9-mediated MDA5 knockout (KO) in DDX3X-control or -KD MCF7 cells (left). qRT-PCR of IFNB1 in MDA5 wildtype or KO cells followed by DDX3X-control or -KD.
G. Colony formation assay in CRISPR/Cas9-mediated MDA5-KO MCF7 cells followed by DDX3X-control or -KD.
Data are shown as mean ± SEM of three independent experiments. Statistical significance was calculated using unpaired t-tests (Fig. 3D and 3E) or one-way ANOVA with uncorrected Fisher’s LSD test (Fig. 3F). *p < 0.05; **p< 0.01.
Next, we examined the protein kinase R (PKR) pathway, and no activation of PKR signaling was found in DDX3X-KD cells (Supplementary Fig. S4D). DDX3X KD greatly upregulated the mRNA and protein levels of cytosolic dsRNA binding sensors, including MDA5, retinoic acid inducible gene-I (RIG-I), and 2’−5’-oligoadenylate synthetase 1 (OAS1) (Figs.3D and 3E and Supplementary Fig. S4E). Importantly, these cytosolic dsRNA binding sensors themselves belong to the ISG family, constituting a feed-forward loop in pro-inflammatory signaling. Therefore, to determine which dsRNA sensor is critical for type I IFN production in the DDX3X-KD cells, we deleted MDA5, RIG-I, or OAS1 in MCF7 cells by using the CRISPR/Cas9 system followed by knockdown of DDX3X. MDA5 knockout (KO) significantly diminished the induction of IFN-β expression when combined with DDX3X KD (Fig.3F). In contrast, RIG-I KO or OAS1 KO did not impair IFN-β induction by DDX3X KD (Supplementary Fig. S4F). MDA5 KO also rescued the reduced survival of DDX3X KD cells in a colony formation assay (Fig.3G and Supplementary Fig. S4G). Collectively, these data suggest that the depletion of DDX3X activates the cytosolic dsRNA sensing pathway, mainly through the MDA5-NFκB signaling axis, which leads to type I IFN production in breast cancer cells.
DDX3X depletion induces the cytoplasmic accumulation of endogenous dsRNAs.
Failure to degrade excess endogenous dsRNAs is known to activate the cytoplasmic dsRNA sensors and trigger type I IFN response in several types of cancer cells (2,4,8,18). We investigated the level of endogenous dsRNAs in DDX3X-control or -KD cells using a dsRNA-specific antibody. Anti-dsRNA-specific J2 antibody (Scion, Hungary) is widely used to recognize viral or cellular dsRNA of more than 40-bp length with no sequence specificity (38–40). First, we validated that the J2 antibody specifically recognizes authentic cellular dsRNAs by treating MCF7 cells, or isolated total RNAs, with RNases or DNase I, and performing immunostaining or dot blot analysis with the J2 antibody. The J2 signals were sensitive to the dsRNA-specific RNase III or RNase A, but not affected by the treatment of the single-stranded RNA (ssRNA)-specific RNase I or DNase I (Fig.4A and Supplementary Fig. S5A). J2 antibody also recognized the synthetic dsRNA mimic analog, poly (I:C) in vitro as well as in the poly I:C transfected cells (Supplementary Fig. S5B). DNA methyltransferase inhibitors are known to increase the bi-directional transcription of endogenous retroviral elements (ERVs) or repetitive RNA elements, which increase the RNA duplexes (Supplementary Fig. S5C) (13,17,41). J2 antibody was able to distinguish the increased level of endogenous dsRNAs in the several cancer cells in response to the treatment of DNA methyltransferase inhibitor (5-AzaC) using dot blot (Supplementary Fig. S5D) and flow cytometry (Supplementary Fig. S5E). These data confirmed that the J2 antibody specifically recognizes authentic cellular dsRNAs.
Figure 4. Knockdown DDX3X triggers the cytoplasmic accumulation of endogenous dsRNAs including ERVs.
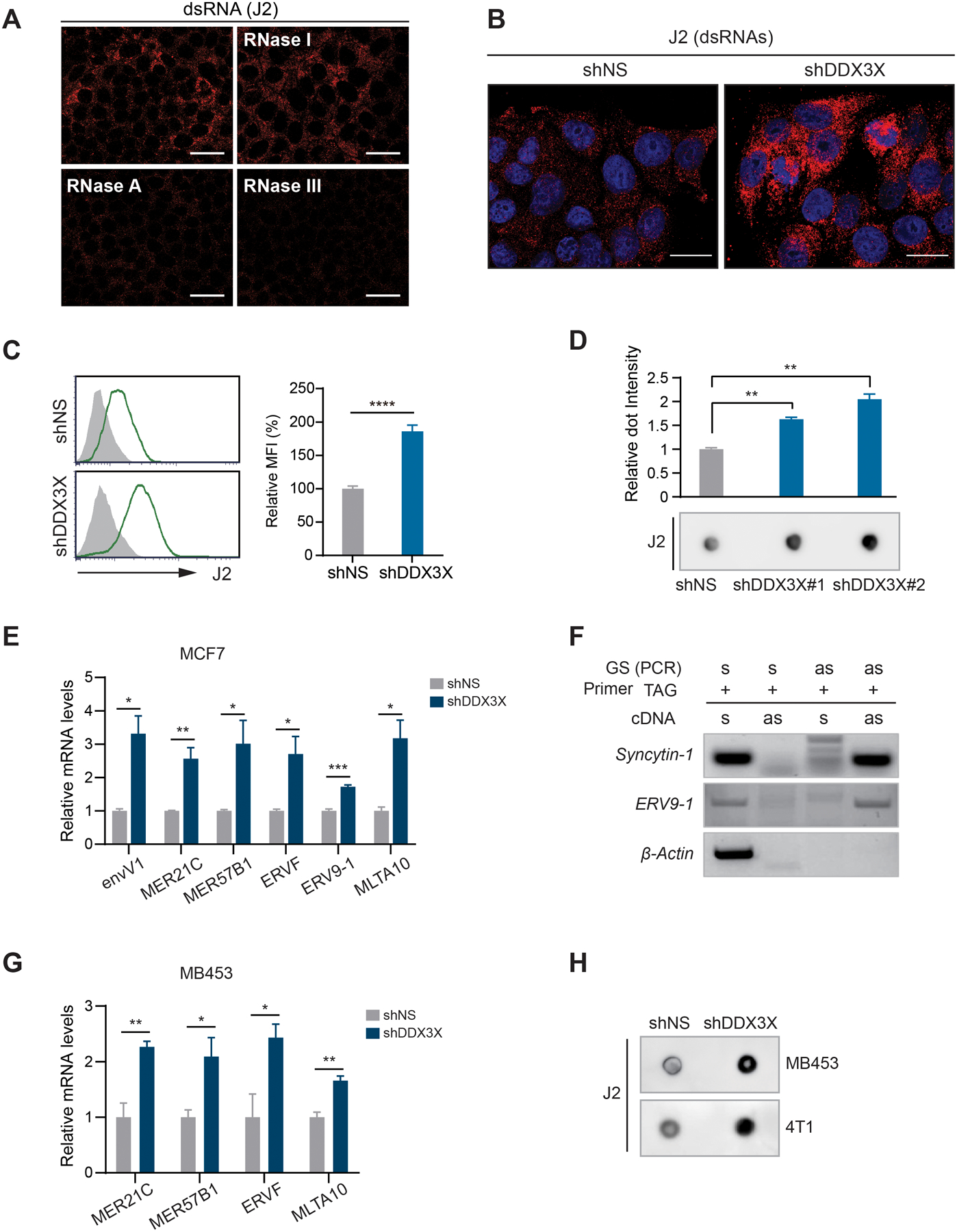
A. Immunofluorescence analysis using J2 antibody in MCF7 cells treated with indicated RNases. Scale bars, 50 μm
B. Endogenous dsRNAs accumulation in DDX3X-control or -KD MCF7 cells was detected by immunofluorescence with an anti-dsRNA specific J2 antibody. Scale bars, 20 μm
C. Endogenous dsRNAs accumulation in DDX3X-control or -KD MCF7 cells was detected by flow cytometry with an anti-dsRNA specific J2 antibody.
D. J2 dot blot analysis with total RNAs extracted from DDX3X-control or -KD MCF7 cells. A graph shows relative dot intensity.
E. qRT-PCR of ERV genes in DDX3X-KD MCF7 cells.
F. TASA-TD PCR of ERV9–1 gene. β-Actin was used for sense transcript amplification. Syncytin-1 was used for a positive ERV control for a bi-directional transcript. s, sense; as, anti-sense direction.
G. qRT-PCR of ERV genes in DDX3X-control or -KD MDA-MB-453 cells.
H. J2 dot blot analysis with total RNAs extracted from DDX3X-control or -KD MDA-MB-453 and 4T1 cells.
Data are shown as mean ± SEM of three independent experiments. Statistical significance was calculated using unpaired t-tests. *p < 0.05; **p< 0.01; ***p < 0.001; ****p < 0.0001.
We found that the depletion of DDX3X substantially increased the level of endogenous dsRNAs in MCF7 cells by immunostaining using the validated dsRNA-specific J2 antibody (Fig.4B). The enhanced dsRNA signals also were detected in DDX3X-KD MCF7 cells using flow cytometry (Fig.4C) and dot blot (Fig.4D) approaches. In addition to the J2 antibody, we also confirmed similar dsRNA induction using another dsRNA-specific K1 antibody (Scion, Hungary) (39) (Supplementary Fig. S5F). Next, we examined the expression level of ERVs that were previously identified to trigger the type I IFN response (13,17,41,42). An upregulated level of ERVs was detected in DDX3X-KD MCF7 or MDA-MB-453 cells (Fig.4E and Supplementary Fig. S5G). We validated a dsRNA form of ERV9–1 using TAG-aided sense/antisense transcript detection PCR (TASA-TD PCR) that detects the bi-directional expression of ERVs (Fig.4F). The expression levels of SINE (Alu) and LINE-1 (L1HS) elements were not affected by DDX3X knockdown (Supplementary Fig. S5H). The induction of dsRNAs also was detected in the DDX3X-KD MDA-MB-453 and mouse breast cancer cells 4T1cells (Figs. 4G and 4H).
DDX3X helicase activity is critical for preventing the accumulation of dsRNAs and induction of dsRNAs-mediated type I IFN activation.
To determine if inhibiting DDX3X activity affects dsRNA regulation, we treated MCF7 cells with the DDX3X inhibitor RK-33 (Selleck Chemicals). RK-33 specifically binds to the ATP-binding cleft of DDX3X and inhibits the helicase activity of DDX3X in unwinding double-stranded RNA to single-stranded RNA in vitro and in a cell-based assay (43–47). We found that effects of RK-33 treatment are similar to shRNA-mediated DDX3X knockdown. RK-33 treatment in MCF7 cells led to the accumulation of dsRNAs (Fig.5A). Type I IFN signaling (phospho-STAT1, STAT1, and STAT2) was activated by the RK-33 treatment in MCF7 cells (Fig.5B and Supplementary Fig. S6A). RK-33 dramatically increased the expression of the IFNB1 gene (Fig.5C). The levels of IGSs also were significantly increased by RK-33 treatment (Fig.5D). Lastly, we treated the MDA5 knockout MCF7 cells with RK-33. MDA5 knockout abrogated the effect of RK-33 on STAT1 activation (Fig.5E).
Figure 5. DDX3X helicase activity is critical for preventing accumulation of dsRNAs and dsRNAs-mediated type I IFN activation.
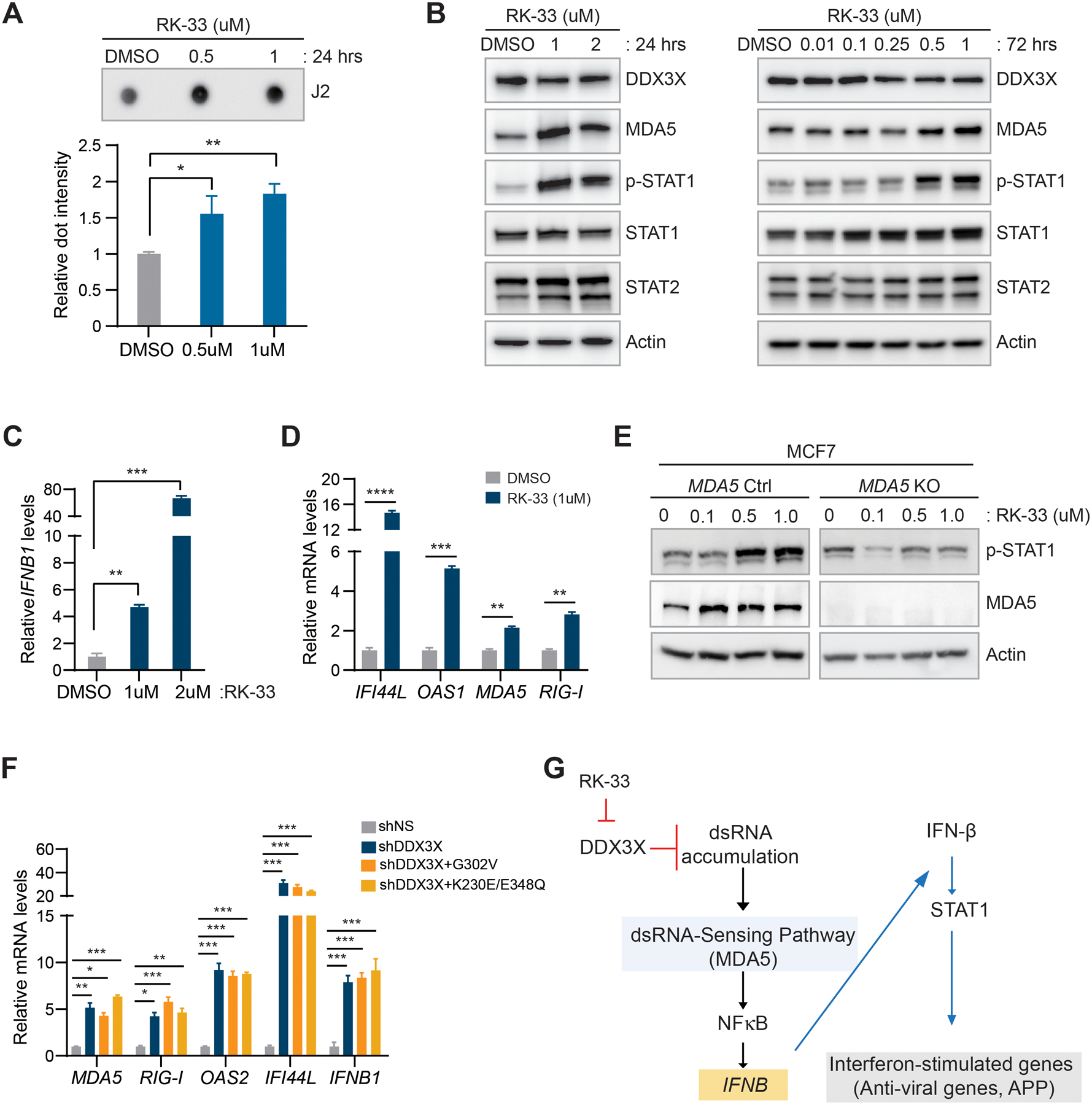
A. J2 dot blot analysis with total RNAs extracted from MCF7 cells treated with RK-33 for 24 hours. A bar graph shows relative dsRNA dot intensity.
B. Western blot analysis in RK-33 treated MCF7 cells (24 hours and 72 hours).
C. qRT-PCR of IFNB1 in RK-33 treated MCF7cells (72 hours).
D. qRT-PCR of ISGs in RK-33 treated MCF7 cells (72 hours).
E. Western blot analysis of MDA5 and phospho-STAT1 in MDA5 KO MCF7 cells followed by RK-33 treatment (24 hours).
F. qRT-PCR of ISGs in DDX3X-KD MCF7 cells followed by overexpression of DDX3X mutants (G302V and K230E/E348Q).
G. Schema of the activation of dsRNA-mediated type I IFN signaling in DDX3X-KD or DDX3X-inhibited (RK-33) cancer cells.
Data are shown as mean ± SEM of three independent experiments. Statistical significance was calculated using unpaired t-tests (Fig. 5A, 5C, and 5D) or one-way ANOVA with uncorrected Fisher’s LSD test (Fig. 5F). *p < 0.05; **p< 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant.
Next, we performed rescue experiments in DDX3X-control or -KD MCF7 cells by expressing two DDX3X mutants, G302V and K230E/E348Q (gift from Dr. Sun), which have mutations in their RNA binding or ATP binding sites (48). These DDX3X mutants are defective in RNA-stimulated ATP hydrolysis activity (49,50). Both mutant forms of DDX3X failed to reduce the expression of ISGs in DDX3X-KD MCF7 cells (Fig. 5F and Supplementary Fig. S6B). Together, these data suggest that DDX3X is critical in regulating endogenous cellular dsRNAs to prevent activation of type I IFN response. Conversely, knockdown or inhibition of DDX3X activates the MDA5-mediated dsRNA sensing pathway (Fig.5G).
DDX3X interacts with ADAR1 and promotes ADAR1-mediated A-to-I dsRNA editing.
To examine whether DDX3X is directly associated with endogenous dsRNAs in the cells, we pulled down dsRNAs with a dsRNA-specific J2 antibody in MCF7 cells. DDX3X co-precipitated with dsRNAs, but not with the control IgG (Fig.6A), indicating DDX3X association with dsRNAs. 5-AzaC treatment, which induces the transcription of endogenous dsRNAs, further enhanced the association of DDX3X with dsRNAs (Fig.6A). DDX3X colocalized with endogenous dsRNAs in 5-AzaC treated-MCF7 cells (Fig.6B). Notably, DDX3X interacted with a dsRNA editing enzyme ADAR1 (Fig.6C and Supplementary Figs. S6C and S6D). RNase A treatment maintained the interaction between DDX3X and ADAR1 (Supplementary Fig. S6E). DDX3X is known to be a shuttling protein between the cytoplasm and nucleus, whose steady-state localization is mostly cytoplasmic (Supplementary Fig. S6F) (51). ADAR1 has two isoforms, ADAR1-p110 (110 kDa) and ADAR-1-p150 (150 kDa). ADAR1-p110 edits dsRNAs from Adenosine (A) to Inosine (I) mostly in the nucleus, while ADAR1-p150 functions in the cytoplasm (2,52). Interestingly, DDX3X primarily interacted with ADAR1-p150 in the cytoplasmic fraction of MCF7 cells (Fig. 6C).
Figure 6. DDX3X interacts with ADAR1 and promotes ADAR1-mediated A-to-I dsRNA editing.
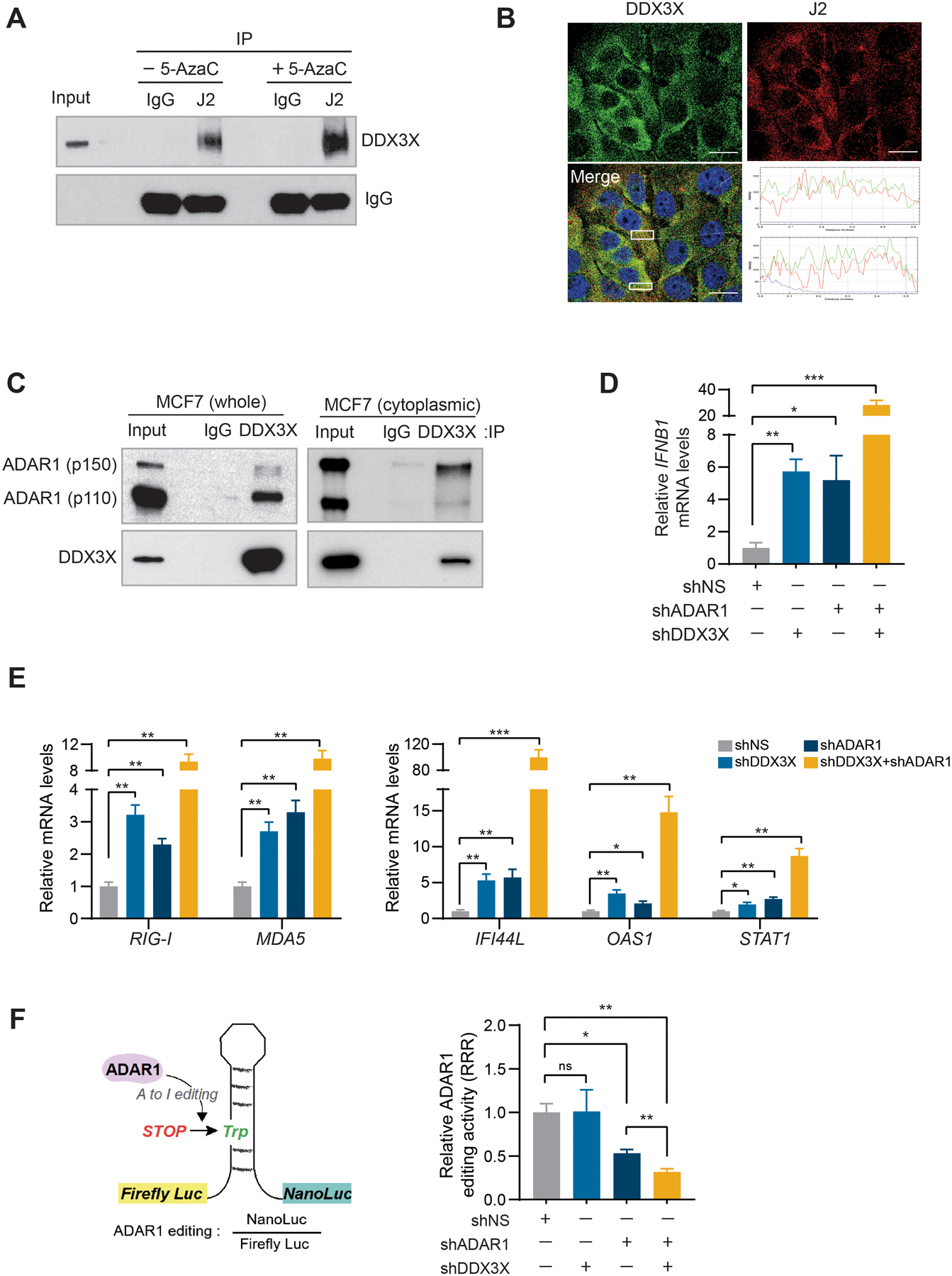
A. J2 Immunoprecipitation (IP) in MCF7 cells treated with or without 5-AzaC. Western blot analyzed using anti-DDX3X or anti-IgG (light chain) antibodies.
B. Immunofluorescence analysis of DDX3X and dsRNA (J2) in MCF7 cells treated with 5-AzaC. RGB profiles for the region of interest selected with white boxes are shown on the lower right. Scale bars, 20 μm.
C. IP in MCF7 whole-cell lysate or cytoplasmic fraction with DDX3X antibody or control IgG. Western blot analyzed using anti-ADAR1 and anti-DDX3X antibodies.
D. qRT-PCR of IFNB1 in DDX3X and ADAR1 single or double KD MCF7 cells.
E. qRT-PCR of ISGs in DDX3X and ADAR1 single or double KD MCF7 cells.
F. ADAR1 editing activity was measured as the ratio between luminescence from Nano luciferase/Firefly luciferase (left). Data was calculated as a relative response ratio (RRR) = (experimental editing assay ratio-negative control ratio) / (positive control ratio-negative control ratio).
Data are representative of three independent experiments. Data represented as mean ± SEM.
Statistical significance was calculated using unpaired t-tests. *p < 0.05; **p< 0.01; ***p < 0.001; ns, not significant.
DDX3X KD alone increased the expression of IFNB1, comparable to the effect of ADAR1 KD (Figs.6D and Supplementary Fig. S6G). Dual KD of DDX3X and ADAR1 produced a greater effect than single knockdown of each gene in upregulating the expression of IFNB1, as well as ISGs (Figs.6D and 6E). To assess the effect of DDX3X effect on ADAR1-mediated A-to-I dsRNA editing, we used a dual-luciferase reporter to monitor the ADAR1-dsRNA editing efficiency (gift from Dr. Ohman) (53). The dsRNA editing activity was determined as the ratio between luminescence from the Nano luciferase/Firefly luciferase (Fig.6F, left). We transiently expressed a negative (0% editing), a positive (100% editing), and an editing reporter (A can be edited to I) vectors, respectively, to measure ADAR1 editing activity (Supplementary Fig. S6H). While DDX3X KD alone in MCF7 cells did not change the activity of ADAR1-mediated A-to-I editing (Fig.6F), the double knockdown of ADAR1 and DDX3X significantly reduced the dsRNA editing activity compared to ADAR1 KD alone (Fig.6F). These data suggest that double-depletion of DDX3X and ADAR1 synergistically facilitates ADAR1-mediated dsRNA editing and increases the dsRNAs-mediated IFN signaling pathway.
Loss of DDX3X augments the dsRNA sensing pathway and IFN signaling in tumor and enhances anti-tumor immunity.
To investigate the impact of DDX3X on the breast tumor immune microenvironment, we inoculated 1×105 4T1 syngeneic cells expressing Tet-inducible shDDX3X into the mammary fat pads of immune-competent BALB/c female mice. When tumors became palpable (20 to 25 mm²), randomized groups were treated with doxycycline (Dox+) water to induce DDX3X KD. Tumor growth was monitored with calipers every three days (Fig.7A). We found that knockdown of DDX3X substantially suppressed 4T1 tumor growth (Fig.7A and Supplementary Fig. S7A), tumor volume and weight (Fig.7B), as well as metastatic spread to the lungs (Supplementary Fig. S7B). DDX3X-inducible KD tumors (Dox+) exhibited increased protein expression of IFN-β (Fig.7C and Supplementary Fig.S7C), MDA5, phospho-STAT1, STAT1, and STAT2 s (Fig.7D and Supplementary Fig. S7D). Upregulated mRNA expression of ISGs as well as STAT1 and STAT2 were found in DDX3X-inducible KD (Dox+) tumors (Fig.7E).
Figure 7. DDX3X-depleted tumor innate immunity is primed by dsRNA-stimulated tumor-intrinsic type I IFN.
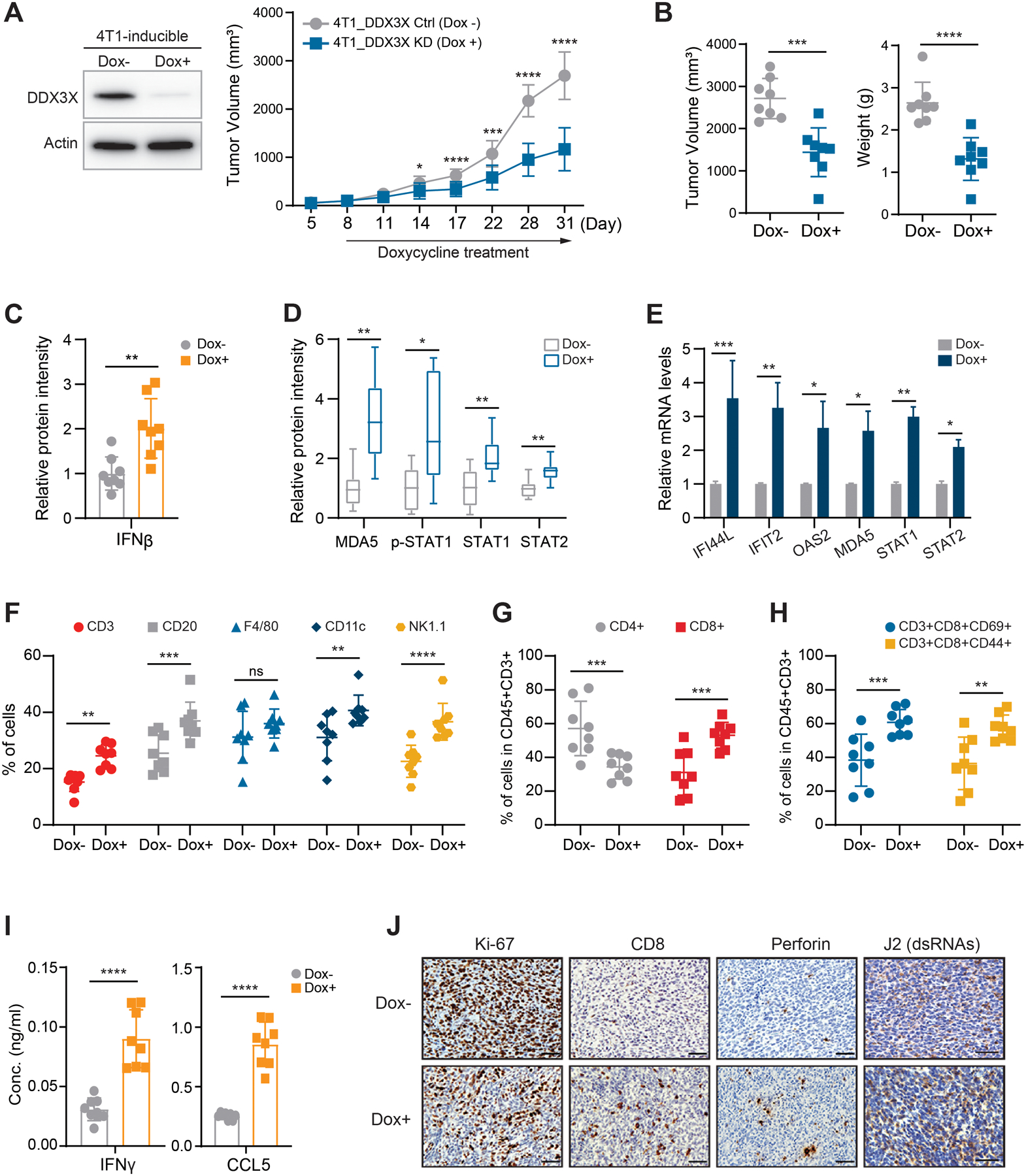
A. 4T1 cells expressing tet-inducible shDDX3X with 2 ug/ml doxycycline (Dox) for 72 hours (left). Tumor growth (mm3) of 4T1 cells expressing tet-inducible shDDX3X in BALC/c mice treated with or without Dox water (right). 8 mice/group
B. Tumor volumes (mm3) and weights (g) of DDX3X-control (Dox-) or -inducible KD (Dox+) 4T1 breast tumors.
C. Western blot protein intensity of IFN-β in DDX3X-control or -inducible KD 4T1 breast tumors.
D. Western blot protein intensity of MDA5, p-STAT1, STAT1, and STAT2 in DDX3X-control or -inducible KD 4T1 breast tumors.
E. qRT-PCR of ISGs in DDX3X-control or -inducible KD 4T1 breast tumors.
F. Flow cytometry analysis of CD3+; T cells, CD20+; B cells, F4/80+; Macrophage, CD11c; dendritic cells, NK1.1; NK cells from tumors.
G. Flow cytometry analysis of CD4+ T cells or CD8+ T cells of CD45+CD3+ cells from tumors.
H. Flow cytometry analysis of T-cell activation markers (CD44 or CD69) of CD45+CD3+ cells from tumors.
I. ELISA of IFN-γ and CCL5 in tumor lysates.
J. Representative images of immunohistochemistry analysis for Ki67, CD8, perforin, and J2. Scale bars, 20 μm
Data are represented as mean ± SEM. n=8 mice per group. Statistic was calculated using unpaired t-tests (Fig. 7A–E and 7I) or two-way ANOVA with uncorrected Fisher’s LSD test (Fig. 7F–H). *p < 0.05; **p< 0.01; ***p < 0.001; ****p< 0.0001; ns, not significant.
Flow cytometry analysis of DDX3X-KD (Dox+) tumors revealed a significant increase in tumor infiltration of immune cells, including T cells and dendritic cells (DCs) (Fig.7F and Supplementary Fig.S8A). Cytotoxic CD8+ T cells were increased in the DDX3X-KD (Dox+) tumors, whereas the proportions of helper CD4+ T cells were present at a similar level between DDX3X -KD and -control tumors (Fig.7G and Supplementary Fig.S8B). Intratumoral CD8+ T cells in the DDX3X inducible-KD (Dox+) 4T1 tumors displayed markedly increased expression of the activated cytotoxic CD8+ T cells markers, CD69 and CD44 (Fig.7H and Supplementary Fig.S8C). ELISA analysis showed an increase in the level of IFN-γ and CCL5 in the DDX3X-inducible KD (Dox+) 4T1 tumor lysates (Fig.7I). IFN-γ is mainly produced by tumor-infiltrating T cells and NK cells in tumors (54). Increased tumor-infiltrating cytotoxic CD8+ T cells in the DDX3X depleted tumors could be the main contributor of IFN-γ. CCL5 is a known T cell chemoattractant, and it also could recruit T cells to tumors (55). Consistent with the flow cytometry results, the increased CD8+ T cell infiltration was observed in the paraffin-embedded DDX3X-inducible KD (Dox+) tumors (Fig.7J and Supplementary Fig. S8D). DDX3X-inducible KD (Dox+) tumors exhibited decreased expression of the proliferation marker Ki67 and increased expression of the cytotoxic lymphocyte-secreted pore-forming protein perforin (Fig.7J and Supplementary Fig.S8D). Furthermore, DDX3X-inducible KD (Dox+) tumors exhibited a higher expression of dsRNAs (Fig.7J and Supplementary Fig.S8D).
We also inoculated 1×105 syngeneic 4T1 cells stably expressing DDX3X shRNA (shDDX3X) or non-specific RNA (shNS) into the mammary fat pad of immune-competent BALB/c female mice. Similar to the inducible DDX3X KD tumors, DDX3X-stable KD 4T1 tumors exhibited suppressed tumor growth and metastasis (Supplementary Figs.S8E and S8F). DDX3X-stable KD 4T1 tumors also showed significantly increased tumor infiltration of immune cells, including cytotoxic T cells (CD3+CD8+CD69+, CD3+CD8+CD44+) (Supplementary Figs.S8G and S8H). Altogether, these data suggest that DDX3X-depleted tumors activated a dsRNA response, thus inducing type I IFN signaling in the tumor microenvironment. Also, in vivo results suggest that innate immunity could be primed by dsRNA-stimulated tumor-intrinsic type I IFN and production of IFN‐γ and CCL5 in DDX3X-depleted tumors.
Discussion
The present study shows that the depletion or inhibition of DDX3X leads to the abnormal accumulation of cellular dsRNAs in the cytoplasm of breast cancer cells, which induces the activation of a type I IFN response via MDA5. Previous studies have reported DDX3X as a positive regulator of type I IFN production through the direct association with anti-viral signaling pathways (56–58). In contrast, Loureiro et al. reported that DDX3X suppressed type I IFN production, suggesting a pro-viral effect of DDX3X (59). Szappanos et al. showed that DDX3X is essential for innate immunity against the infection of pathogenic bacteria, but DDX3X also is important for hematopoiesis and maintenance of immune cells in mice (60). Recently, Samir et al. showed a role for DDX3X in driving NLRP3 inflammasome and stress granule assembly in bone-marrow-derived macrophages stimulated with lipopolysaccharide (61). Our study demonstrates that DDX3X prevents type I IFN production against endogenous dsRNAs in the steady-state cell condition without pathogenic infection, suggesting a role of DDX3X in anti-autoimmunity against cellular dsRNAs. Collectively, these findings, including the present study, propose a more complex role for DDX3X in the dsRNA biology network and in antiviral immunity.
We found that DDX3X helicase activity is important in preventing the accumulation of dsRNAs, which suggests that DDX3X could act directly on dsRNAs by unwinding the dsRNA structures. Or DDX3X could facilitate the efficient dsRNA editing by unwinding of complex three-dimensional RNA structures and exposing dsRNA extension to ADAR1. To better understand the molecular mechanism by which DDX3X is involved in the regulation of cellular dsRNAs, it will be important to identify the cellular dsRNA species that are directly regulated by DDX3X in future studies.
Notably, our DDX3X-KD 4T1 tumor-bearing syngeneic mouse study suggests that inhibiting DDX3X could lead to both direct anti-proliferative effects as well as indirect anti-tumor immune response effects in vivo. DDX3X-KD tumors showed increased tumor-infiltrating CD8+ T cells and DCs. The upregulation of cancer cell-intrinsic type I IFNs and the CCL5 chemokine in DDX3X-KD tumors may activate effector T cells as well as other immune cells that could further prime T cells in the tumor. Increased tumor cell antigen presentation, coupled with anti-tumor T-cell responses, also suggests that immune checkpoint blockade might further enhance the anti-cancer efficacy of DDX3X inhibition.
Several small-molecule inhibitors have been developed to inhibit DDX3X as a therapeutic target in cancer (62). In particular, RK-33, a small molecule inhibitor, is a first-in-class pharmaceutical for targeting DDX3X (24,33,45,46). In in vitro and cell-based assays, RK-33 inhibits the DDX3X helicase activity of unwinding double-stranded RNA to single-stranded RNA (43–45). Previous in vivo studies, including medulloblastoma, breast, and prostate cancer animal models, have demonstrated anti-tumor effects of RK-33 with no significant toxicity (43,44,47). Combination studies using RK-33 and radiation exhibited synergistic anti-tumor effects in a preclinical lung cancer mouse model (47). The present study reveals that RK-33 treatment in MCF7 cells resulted in the accumulation of cellular dsRNAs, STAT1 activation, and IFN-β upregulation. Notably, a 0.5~1 μM range of RK-33 was sufficient for induction of type I IFN and anti-proliferative effects in MCF7 cells. Serum levels of around 1 μM are reasonable to achieve with a small molecule, so in theory, RK-33 could achieve an anti-tumor effect in humans. This requires pharmacokinetic data from clinical trials, and subsequent clinical development should consider the impact of DDX3X inhibition on the tumor immune microenvironment. In addition, combination therapies to target cancer cells with high intrinsic dsRNA stress or with combination therapy may open a therapeutic window and reduce potential toxicity.
Supplementary Material
Statement of Significance.
This study elucidates the novel role of DDX3X in regulating endogenous cellular dsRNA homeostasis and type I IFN signaling in breast cancer.
Acknowledgements
We thank Dr. Xiongbin Lu for a critical reading of the manuscript. We thank Drs. Louis Weiner, Aykut Uren, and Marta Catalfamo for their helpful scientific discussion. We thank Dr. Marie Öhman for the kind gift of the ADAR1 editing vectors. We thank Drs. Charles G. Drake and Mark Smyth for sharing their the B16-OVA cells. We thank Dr. Shuying Sun for sharing DDX3X-mutant vectors. Graphic Abstract was created with BioRender.com. This work was supported, in part, by an NIH/NCI Pathway to Independence Award (K99/R00 CA197487).
Footnotes
Conflict of interest
The authors declare no competing interests.
Data Availability
RNA deep sequencing information (related to Figure 1) is available in GEO repository.
GEO accession number: GSE157323
Reference
- 1.Bhate A, Sun T, Li JB. ADAR1: A New Target for Immuno-oncology Therapy. Mol Cell 2019;73:866–8 [DOI] [PubMed] [Google Scholar]
- 2.Reich DP, Bass BL. Mapping the dsRNA World. Cold Spring Harb Perspect Biol 2019;11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Portal MM, Pavet V, Erb C, Gronemeyer H. Human cells contain natural double-stranded RNAs with potential regulatory functions. Nat Struct Mol Biol 2015;22:89–97 [DOI] [PubMed] [Google Scholar]
- 4.Linder A, Hornung V. Mitochondrial dsRNA: A New DAMP for MDA5. Dev Cell 2018;46:530–2 [DOI] [PubMed] [Google Scholar]
- 5.Riley JS, Tait SW. Mitochondrial DNA in inflammation and immunity. EMBO Rep 2020;21:e49799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Katayama S, Tomaru Y, Kasukawa T, Waki K, Nakanishi M, Nakamura M, et al. Antisense transcription in the mammalian transcriptome. Science 2005;309:1564–6 [DOI] [PubMed] [Google Scholar]
- 7.Lamers MM, van den Hoogen BG, Haagmans BL. ADAR1: “Editor-in-Chief” of Cytoplasmic Innate Immunity. Front Immunol 2019;10:1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hur S Double-Stranded RNA Sensors and Modulators in Innate Immunity. Annu Rev Immunol 2019;37:349–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, et al. Initial sequencing and analysis of the human genome. Nature 2001;409:860–921 [DOI] [PubMed] [Google Scholar]
- 10.Tokuyama M, Kong Y, Song E, Jayewickreme T, Kang I, Iwasaki A. ERVmap analysis reveals genome-wide transcription of human endogenous retroviruses. Proc Natl Acad Sci U S A 2018;115:12565–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bannert N, Kurth R. Retroelements and the human genome: new perspectives on an old relation. Proc Natl Acad Sci U S A 2004;101 Suppl 2:14572–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jones PA, Ohtani H, Chakravarthy A, De Carvalho DD. Epigenetic therapy in immune-oncology. Nat Rev Cancer 2019;19:151–61 [DOI] [PubMed] [Google Scholar]
- 13.Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, et al. Inhibiting DNA Methylation Causes an Interferon Response in Cancer via dsRNA Including Endogenous Retroviruses. Cell 2015;162:974–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goel S, DeCristo MJ, Watt AC, BrinJones H, Sceneay J, Li BB, et al. CDK4/6 inhibition triggers anti-tumour immunity. Nature 2017;548:471–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, et al. LSD1 Ablation Stimulates Anti-tumor Immunity and Enables Checkpoint Blockade. Cell 2018;174:549–63 e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu H, Golji J, Brodeur LK, Chung FS, Chen JT, deBeaumont RS, et al. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat Med 2019;25:95–102 [DOI] [PubMed] [Google Scholar]
- 17.Liu M, Ohtani H, Zhou W, Orskov AD, Charlet J, Zhang YW, et al. Vitamin C increases viral mimicry induced by 5-aza-2’-deoxycytidine. Proc Natl Acad Sci U S A 2016;113:10238–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uggenti C, Lepelley A, Crow YJ. Self-Awareness: Nucleic Acid-Driven Inflammation and the Type I Interferonopathies. Annu Rev Immunol 2019;37:247–67 [DOI] [PubMed] [Google Scholar]
- 19.Ahmad S, Mu X, Yang F, Greenwald E, Park JW, Jacob E, et al. Breaching Self-Tolerance to Alu Duplex RNA Underlies MDA5-Mediated Inflammation. Cell 2018;172:797–810 e13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eisenberg E, Levanon EY. A-to-I RNA editing - immune protector and transcriptome diversifier. Nat Rev Genet 2018;19:473–90 [DOI] [PubMed] [Google Scholar]
- 21.Dhir A, Dhir S, Borowski LS, Jimenez L, Teitell M, Rotig A, et al. Mitochondrial double-stranded RNA triggers antiviral signalling in humans. Nature 2018;560:238–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Linder P, Jankowsky E. From unwinding to clamping - the DEAD box RNA helicase family. Nat Rev Mol Cell Biol 2011;12:505–16 [DOI] [PubMed] [Google Scholar]
- 23.Song H, Ji X. The mechanism of RNA duplex recognition and unwinding by DEAD-box helicase DDX3X. Nat Commun 2019;10:3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xie M, Vesuna F, Botlagunta M, Bol GM, Irving A, Bergman Y, et al. NZ51, a ring-expanded nucleoside analog, inhibits motility and viability of breast cancer cells by targeting the RNA helicase DDX3. Oncotarget 2015;6:29901–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Heerma van Voss MR, Brilliant JD, Vesuna F, Bol GM, van der Wall E, van Diest PJ, et al. Combination treatment using DDX3 and PARP inhibitors induces synthetic lethality in BRCA1-proficient breast cancer. Med Oncol 2017;34:33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Botlagunta M, Vesuna F, Mironchik Y, Raman A, Lisok A, Winnard P Jr., et al. Oncogenic role of DDX3 in breast cancer biogenesis. Oncogene 2008;27:3912–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heerma van Voss MR, Schrijver WA, Ter Hoeve ND, Hoefnagel LD, Manson QF, van der Wall E, et al. The prognostic effect of DDX3 upregulation in distant breast cancer metastases. Clin Exp Metastasis 2017;34:85–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagy A, Lanczky A, Menyhart O, Gyorffy B. Validation of miRNA prognostic power in hepatocellular carcinoma using expression data of independent datasets. Sci Rep 2018;8:9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Henke C, Strissel PL, Schubert MT, Mitchell M, Stolt CC, Faschingbauer F, et al. Selective expression of sense and antisense transcripts of the sushi-ichi-related retrotransposon--derived family during mouse placentogenesis. Retrovirology 2015;12:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qiu W, Zhang Q, Zhang R, Lu Y, Wang X, Tian H, et al. N(6)-methyladenosine RNA modification suppresses antiviral innate sensing pathways via reshaping double-stranded RNA. Nat Commun 2021;12:1582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hartono SR, Malapert A, Legros P, Bernard P, Chedin F, Vanoosthuyse V. The Affinity of the S9.6 Antibody for Double-Stranded RNAs Impacts the Accurate Mapping of R-Loops in Fission Yeast. J Mol Biol 2018;430:272–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Han C, Yang L, Choi HH, Baddour J, Achreja A, Liu Y, et al. Amplification of USP13 drives ovarian cancer metabolism. Nat Commun 2016;7:13525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bol GM, Xie M, Raman V. DDX3, a potential target for cancer treatment. Mol Cancer 2015;14:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ivashkiv LB, Donlin LT. Regulation of type I interferon responses. Nat Rev Immunol 2014;14:36–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheon H, Holvey-Bates EG, Schoggins JW, Forster S, Hertzog P, Imanaka N, et al. IFNbeta-dependent increases in STAT1, STAT2, and IRF9 mediate resistance to viruses and DNA damage. EMBO J 2013;32:2751–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Provance OK, Lewis-Wambi J. Deciphering the role of interferon alpha signaling and microenvironment crosstalk in inflammatory breast cancer. Breast Cancer Res 2019;21:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woo SR, Fuertes MB, Corrales L, Spranger S, Furdyna MJ, Leung MY, et al. STING-dependent cytosolic DNA sensing mediates innate immune recognition of immunogenic tumors. Immunity 2014;41:830–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lukacs N Detection of virus infection in plants and differentiation between coexisting viruses by monoclonal antibodies to double-stranded RNA. J Virol Methods 1994;47:255–72 [DOI] [PubMed] [Google Scholar]
- 39.Schonborn J, Oberstrass J, Breyel E, Tittgen J, Schumacher J, Lukacs N. Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res 1991;19:2993–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J Virol 2006;80:5059–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, et al. DNA-Demethylating Agents Target Colorectal Cancer Cells by Inducing Viral Mimicry by Endogenous Transcripts. Cell 2015;162:961–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Canadas I, Thummalapalli R, Kim JW, Kitajima S, Jenkins RW, Christensen CL, et al. Tumor innate immunity primed by specific interferon-stimulated endogenous retroviruses. Nat Med 2018;24:1143–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tantravedi S, Vesuna F, Winnard PT Jr., Martin A, Lim M, Eberhart CG, et al. Targeting DDX3 in Medulloblastoma Using the Small Molecule Inhibitor RK-33. Transl Oncol 2019;12:96–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie M, Vesuna F, Tantravedi S, Bol GM, Heerma van Voss MR, Nugent K, et al. RK-33 Radiosensitizes Prostate Cancer Cells by Blocking the RNA Helicase DDX3. Cancer Res 2016;76:6340–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang SNY, Atkinson SC, Audsley MD, Heaton SM, Jans DA, Borg NA. RK-33 Is a Broad-Spectrum Antiviral Agent That Targets DEAD-Box RNA Helicase DDX3X. Cells 2020;9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kondaskar A, Kondaskar S, Kumar R, Fishbein JC, Muvarak N, Lapidus RG, et al. Novel, Broad Spectrum Anti-Cancer Agents Containing the Tricyclic 5:7:5-Fused Diimidazodiazepine Ring System. ACS Med Chem Lett 2010;2:252–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bol GM, Vesuna F, Xie M, Zeng J, Aziz K, Gandhi N, et al. Targeting DDX3 with a small molecule inhibitor for lung cancer therapy. EMBO Mol Med 2015;7:648–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheng W, Wang S, Zhang Z, Morgens DW, Hayes LR, Lee S, et al. CRISPR-Cas9 Screens Identify the RNA Helicase DDX3X as a Repressor of C9ORF72 (GGGGCC)n Repeat-Associated Non-AUG Translation. Neuron 2019;104:885–98 e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Russell R Unwinding the mechanisms of a DEAD-box RNA helicase in cancer. J Mol Biol 2015;427:1797–800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yedavalli VS, Neuveut C, Chi YH, Kleiman L, Jeang KT. Requirement of DDX3 DEAD box RNA helicase for HIV-1 Rev-RRE export function. Cell 2004;119:381–92 [DOI] [PubMed] [Google Scholar]
- 51.Frohlich A, Rojas-Araya B, Pereira-Montecinos C, Dellarossa A, Toro-Ascuy D, Prades-Perez Y, et al. DEAD-box RNA helicase DDX3 connects CRM1-dependent nuclear export and translation of the HIV-1 unspliced mRNA through its N-terminal domain. Biochim Biophys Acta 2016;1859:719–30 [DOI] [PubMed] [Google Scholar]
- 52.Bass BL. RNA editing by adenosine deaminases that act on RNA. Annu Rev Biochem 2002;71:817–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fritzell K, Xu LD, Otrocka M, Andreasson C, Ohman M. Sensitive ADAR editing reporter in cancer cells enables high-throughput screening of small molecule libraries. Nucleic Acids Res 2019;47:e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ni L, Lu J. Interferon gamma in cancer immunotherapy. Cancer Med 2018;7:4509–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 2009;69:3077–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Soulat D, Burckstummer T, Westermayer S, Goncalves A, Bauch A, Stefanovic A, et al. The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. EMBO J 2008;27:2135–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Schroder M, Baran M, Bowie AG. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J 2008;27:2147–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gu L, Fullam A, McCormack N, Hohn Y, Schroder M. DDX3 directly regulates TRAF3 ubiquitination and acts as a scaffold to co-ordinate assembly of signalling complexes downstream from MAVS. Biochem J 2017;474:571–87 [DOI] [PubMed] [Google Scholar]
- 59.Loureiro ME, Zorzetto-Fernandes AL, Radoshitzky S, Chi X, Dallari S, Marooki N, et al. DDX3 suppresses type I interferons and favors viral replication during Arenavirus infection. PLoS Pathog 2018;14:e1007125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Szappanos D, Tschismarov R, Perlot T, Westermayer S, Fischer K, Platanitis E, et al. The RNA helicase DDX3X is an essential mediator of innate antimicrobial immunity. PLoS Pathog 2018;14:e1007397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Samir P, Kesavardhana S, Patmore DM, Gingras S, Malireddi RKS, Karki R, et al. DDX3X acts as a live-or-die checkpoint in stressed cells by regulating NLRP3 inflammasome. Nature 2019;573:590–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kukhanova MK, Karpenko IL, Ivanov AV. DEAD-box RNA Helicase DDX3: Functional Properties and Development of DDX3 Inhibitors as Antiviral and Anticancer Drugs. Molecules 2020;25 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA deep sequencing information (related to Figure 1) is available in GEO repository.
GEO accession number: GSE157323