

Abstract
Histone proteins undergo various modifications that alter chromatin structure, including addition of methyl groups. Enhancer of zeste homolog 2 (EZH2) is a histone methyltransferase that methylates lysine residue 27, and thereby suppresses gene expression. EZH2 plays integral roles in the uterus and other reproductive organs. We have previously shown that conditional deletion of uterine EZH2 results in increased proliferation of luminal and glandular epithelial cells, and RNA-seq analyses reveal several uterine transcriptomic changes in Ezh2 conditional (c) knockout (KO) mice that can affect estrogen signaling pathways. To pinpoint the origin of such gene expression changes, we used the recently developed spatial transcriptomics (ST) method with the hypotheses that Ezh2cKO mice would predominantly demonstrate changes in epithelial cells and/or ablation of this gene would disrupt normal epithelial/stromal gene expression patterns. Uteri were collected from ovariectomized adult WT and Ezh2cKO mice and analyzed by ST. Asb4, Cxcl14, Dio2, and Igfbp5 were increased, Sult1d1, Mt3, and Lcn2 were reduced in Ezh2cKO uterine epithelium vs. WT epithelium. For Ezh2cKO uterine stroma, differentially expressed key hub genes included Cald1, Fbln1, Myh11, Acta2, and Tagln. Conditional loss of uterine Ezh2 also appears to shift the balance of gene expression profiles in epithelial vs. stromal tissue toward uterine epithelial cell and gland development and proliferation, consistent with uterine gland hyperplasia in these mice. Current findings provide further insight into how EZH2 may selectively affect uterine epithelial and stromal compartments. Additionally, these transcriptome data might provide mechanistic understanding and valuable biomarkers for human endometrial disorders with epigenetic underpinnings.
Keywords: Female Reproduction, endometrium, RNA-seq, estrogen, endometrial cancer, epigenetics, uterus, Histone Proteins
Spatial transcriptomics studies reveal how EZH2 selectively affects uterine epithelial and stromal compartments, and findings may provide a mechanistic understanding and biomarkers for human endometrial disorders originating from epigenetic changes.
Graphical Abstract
Graphical Abstract.
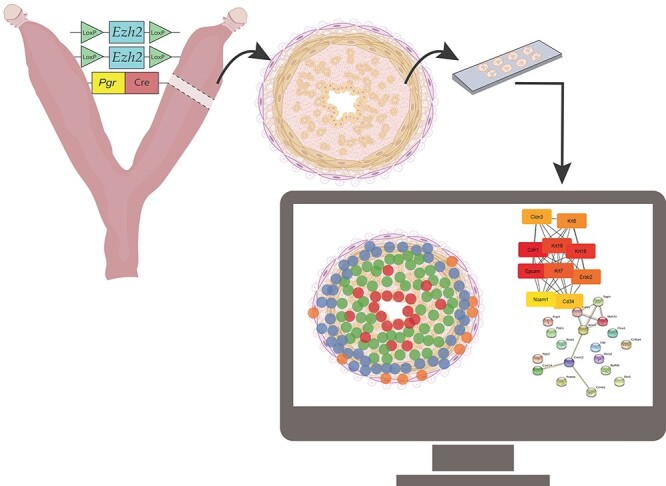
Introduction
Epigenetic modifications regulate gene or protein expression in normal and pathological cells without changing the DNA sequence. Such changes include post-translational modification of histones (e.g. methylation or acetylation), DNA methylation, and small and long non-coding RNAs that target mRNAs before they are translated [1]. Enhancer of zeste homolog 2 (EZH2) is a subunit of the polycomb repressive complex 2 (PRC2), which is a histone methyltransferase critical for histone modifications. This complex regulates gene function by tri-methylating histone 3 on lysine 27 (H2K27me3) [2], resulting in chromatin condensation and inhibition of transcription. Upregulation of EZH2 has been linked to pathologies including endometrial, colorectal, breast, and pancreatic cancer, and its expression has prognostic significance [3–6]. However, loss of function mutations in Ezh2 also occur in conditions such as acute myeloid leukemia, as PRC2 can inhibit the expression of tumor suppressor genes as well as oncogenes [7]. Furthermore, altered EZH2 expression is also associated with endometriosis and uterine fibroids [4, 8, 9], providing further evidence that EZH2 is critical for normal uterine function.
Global deletion of Ezh2 in mice causes embryonic lethality, with mutant embryos exhibiting hyperproliferation of mesoderm cells [10]. Therefore, to understand the role of EZH2 in health and disease, conditional knockout (cKO) mouse models have been created. We and others have generated cKO mice lacking uterine EZH2 (Ezh2cKO) to understand the normal uterine function of this epigenetic modifier [11, 12]. Pathological changes following uterine EZH2 deletion include epithelial hyperproliferation, uterine hypertrophy, cystic endometrial hyperplasia, and E2 hyper-responsiveness [11, 12]. Fang et al. [12] have also reported stratification of uterine epithelium and increased expression of tumor markers in this mutant mouse model. Uterine EZH2 deletion decreased H3K27me3 marks, which in turn could allow for promiscuous gene expression.
We recently showed with RNA-seq analyses that Ezh2cKO mice show global changes in uterine gene expression relative to WT mice that collectively suggested disruption in estrogen receptor signaling pathways [6]. While this study was informative, it did not allow us to pinpoint the specific cellular origins of the gene expression changes in this transgenic model. The recently developed Visium Spatial Transcriptomics (ST) technology [13, 14] permits quantitative mRNA profiles of cells in intact tissue and can determine gene expression changes in various uterine cell lineages without destroying the tissue architecture. Thus, we utilized this cutting-edge technology to define how Ezh2 deletion affects individual uterine cell populations, presumably having important clinical translation to women with uterine diseases originating from different cell lineages. We hypothesized that this innovative method would reveal that Ezh2cKO mice demonstrate transcriptomic changes, especially in epithelial cells and/or disruption in normal distribution of epithelial to stromal transcript patterns. To test this hypothesis, we used ST to analyze uteri of adult ovariectomized WT and Ezh2cKO mice.
Materials and methods
Animals, animal care, and treatments
Transgenic mice expressing Cre recombinase under the control of the progesterone receptor (Pgr) promoter on a mixed C57BL/6 and 129SvEv background were produced at the University of Florida, as described previously [11]. They were housed in standard mouse cages at 25 °C, with 12 h:12 h light/dark cycles and provided ad libitum access to water and rodent commercial diet (Teklad Global 18% Protein Rodent Diet). All procedures and experiments were approved by the University of Florida IACUC and conducted under the NIH Guidelines for the Care and Use of Laboratory Animals. Thus, investigations using experimental animals were conducted in accordance with the relevant institutional and national guidelines and standards.
Transgenic Pgr-Cre mice were bred to mice in which the Ezh2 gene was homozygously floxed (Jackson Laboratories, Bar Harbor, ME). Male and female offspring expressing Pgr-Cre and heterozygous for floxed Ezh2 (Pgrwt/Cre, Ezh2+/flox) were then crossed to obtain Ezh2cKO females expressing Pgr-Cre and homozygous for floxed Ezh2 (Pgrwt/Cre, Ezh2flox/flox), whereas WT controls expressed floxed Ezh2 but no Cre (Pgrwt/wt, Ezh2flox/flox). Four different litters were used for the current studies, with one Ezh2cKO and one WT female randomly selected from each litter. Females were ovariectomized at postnatal day (PND) 71 ± 4. Uteri were collected on PND 112 ± 11. Uteri were embedded and frozen in Tissue-Tek optimal cutting temperature (O.C.T.) compound (Sakura Finetek USA, #25608-930) and then shipped on dry ice to the University of Missouri and stored at –80 °C until processed for spatial transcriptomics followed by RNAScope.
Genotyping
At weaning, genotyping for Pgr-Cre and floxed Ezh2 was performed by multiplex PCR with genomic DNA. Specific primers used; the PCR procedure and the visualization of the Pgr-Cre and floxed Ezh2 PCR products have been described previously [11].
Tissue sectioning, staining, and imaging
Tissue samples were cut using a Thermo Scientific HM525 NX cryostat, and 10 μm thick sections were placed on capture areas of Visium spatial gene expression slides (10X Genomics). Immediately after sectioning, tissues were fixed and stained according to manufacturer’s recommendations. In brief, tissue sections were fixed in prechilled methanol for 30 min at −20 °C, stained with Mayer hematoxylin for 7 min followed by washing in three changes of nuclease-free water and a 2 min incubation in bluing buffer (Dako, Agilent, Santa Clara, CA, USA). Subsequently, sections were counterstained with eosin for 1 min, then briefly washed and dried for 5 min on a slide warmer at 37 °C. Images of individual capture areas with stained sections were acquired using a Zeiss Axiovert 200 M motorized microscope equipped with a 5x Plan Apochromat objective and a Leica (Buffalo Grove, IL USA) DFC290 camera. Image tiles were stitched with Metamorph 7.8.12 software. Pixel size of final composite images was 1.275 μm.
Visium spatial gene expression
Permeabilization, reverse transcription, second strand synthesis, and cDNA PCR amplification
Optimal enzymatic permeabilization time for the tissue type was pre-determined using the 10X Genomics Tissue Optimization kit (Pleasanton, CA, USA). Stained and imaged slides were incubated in permeabilization enzyme solution for 20 min at 37 °C. Following the permeabilization wash step, reverse transcription master mixture was added and incubated for 45 min at 53 °C. After degradation of the mRNA with KOH, the second strand synthesis was carried out for 15 min at 65 °C by adding a second strand mixture containing the second strand enzyme and primer. The second strand cDNA was released by denaturation of the double-stranded cDNA with KOH, neutralized with 1 M Tris-HCl and transferred from the slide section to 8-tube strip. After determining the optimal PCR cycle number by KAPA SYBR FAST qPCR Master Kit, cDNA was amplified for 15 cycles using the PCR cycling conditions according to the 10X Genomics Visium Spatial Gene Expression instructions. The cDNA was purified using 0.6X AxyPrep Mag magnetic beads (Axygen, Tewksbury, MA, USA). The quality and concentration were confirmed by Fragment Analyzer (Agilent, Santa Clara, CA, USA) using the High Sensitivity NGS kit.
Spatial gene expression library construction
The cDNA library for the Illumina platform was prepared by following the 10X Genomics spatial gene expression library construction protocol. Briefly, double stranded cDNA was fragmented, end repaired, and A-tail added in a single step. The A-tailed fragments were size selected using double-sided AxyPrep Mag magnetic beads with a bead to reaction solution ratio of 0.6X and 0.8X for the for the first and second steps, respectively. Next, the adaptor was ligated and purified with 0.8X AxyPrep Mag magnetic beads. The final Illumina libraries were amplified by PCR, which added unique dual indexes to each sample. The PCR-amplified libraries were purified and size selected by using two reactions with double-sided AxyPrep Mag magnetic beads at the bead to reaction solution ratios described above. The individual library was analyzed by Fragment Analyzer (Agilent, Santa Clara, CA, USA) to determine the quality and average fragment size and quantified by Qubit (Invitrogen, Waltham, MA, USA) DNA assay. The libraries were pooled in an equimolar ratio with a total concentration of 5 nM and sequenced on the Illumina Novaseq 6000.
Bioinformatics analyses of RNA-seq data
FASTQ files generated from the sequencing procedure were processed by using the 10X Genomics SpaceRanger 1.2.0 pipeline to demultiplex the spots to their spatial location. One Ezh2cKO section did not contain a full thickness uterine cross section, and thus was excluded from the final analyses. Thus, final ST analyses included 4 WT and 3 Ezh2cKO uterine sections. The remaining seven tissue samples were aggregated using the 10X Genomic SpaceRanger aggregate function grouped by genotype (WT or Ezh2cKO). We detected a minimum of 6600 unique molecular identifiers (UMIs) and over 3000 unique genes per spot for all tissue samples (Supplementary Table S1). In clustering the spots of each ST array based on the principal component scores, we found that the resulting clusters were consistent with the independent histological annotations, supporting the ability to identify distinct spatial regions within a section based on ST gene expression. The aggregated file was then imported into the 10X Genomics Loupe program for further analysis.
Differential gene expression
The 10X Genomics Loupe Browser was used to identify differential gene expression based on sub-regions within the uterus and genotype. These analyses included four comparisons: WT vs. Ezh2cKO epithelium, WT vs. Ezh2cKO stroma, WT epithelium vs. WT stroma, and Ezh2cKO epithelium vs. Ezh2cKO stroma. The latter comparisons were made to determine if conditional Ezh2 deletion alters the normal distribution of epithelium vs. stromal gene expression patterns. For all comparisons, an absolute fold change >1.5 and FDR ≤ 0.05 was considered significant.
To perform these analyses, spots in each tissue region were manually selected based on histology/structure (epithelial cells vs. stromal cells) using the Loupe program for accuracy and confirmation of the different cell types in the seven uterine sections. These data were generated with the uniform manifold approximation and projection feature in the Loupe program, and were determined based on epithelial or stromal categories. Differences between groups were then tested using the sSeq method [15], which employs a negative binomial exact test. The Loupe program computed relative library size based on the total UMI counts for each cluster divided by the median UMI counts per cluster. Normalization was implicit in that the per-cluster library-size parameter is incorporated as a factor in the exact-test probability calculations. The Loupe program was also used to generate t-distributed stochastic neighbor embedding (t-SNE) graphs to visualize the relationship between clusters for the WT and Ezh2cKO groups and examine potential outliers.
Potential protein–protein interactions (PPI) based on differentially expressed genes (DEG) identified for the four above comparisons were determined with STRING [16] (https://string-db.org/). To identify hub genes, the PPI files generated with STRING were imported into the cytohubba application [17] in Cytoscape [18], and the top 10 hub genes, if there were more than 10, were identified. Within this program, hub genes were determined according to the maximal clique centrality (MCC) analysis, as recommended [17]. Pathway enrichment analyses based on DEG for these four comparisons were done by using the over-representation analysis (ORA) and GO biological pathways through WebGestalt (WEB-based GEne SeT AnaLysis Toolkit) [19]. In absence of a mouse database that considers gene expression patterns in uterine tissues, we screened genes that were identified to be DE in our mouse samples with the GTEx Portal (https://www.gtexportal.org/home/). This human database includes a global list of gene expression patterns in the uterus that also allowed us to explore the potential translational of our findings in mice to humans, where EZH2 is abundantly expressed in the uterus and associated with a variety of uterine diseases [3, 6, 20, 21].
Results
Spatial transcriptomics
Supplementary Table S1 summarizes the reads and genes per spot for each tissue section. The average number of mean reads per spot for all seven sections was 167 785. The average genes per spot were 3906. The average total genes detected were 16 056. The median UMI counts per spot were 10 030. These numbers are comparable to other studies that used this approach [22].
To examine how conditional deletion of Ezh2 affected uterine ST patterns, we first compared genes that were differentially expressed (DE) in uterine epithelium of Ezh2cKO vs. WT mice, where 5812 distinct genes were identified. The Loupe program used the gene signature patterns to designate the WT epithelial cells as blue (Supplementary Figure S1) and Ezh2cKO epithelial cells as red (Supplementary Figure S2). To ensure that only epithelial cells were included in the actual comparison, we used the hand select tool feature in the Loupe program to select only epithelial cells in these two groups for comparison based on histology/structure. The t-SNE plots for WT and Ezh2cKO groups are shown in Supplementary Figures S3 and S4, respectively. Based on the cluster arrangement, there was one potential outlier (represented by the last column in Supplementary Figure S2) for the Ezh2cKO group, and this one was excluded from further analyses. This one may be an outlier as we were not able to obtain a representative uterine cross section for it. All the other sections within each of the groups showed tight clustering. These findings suggest that the variation within age for some replicates did not affect the overall gene expression profiles for the WT and Ezh2cKO groups.
The remaining samples were analyzed based on Ezh2cKO epithelium vs. WT epithelium and Ezh2cKO stroma vs. WT stroma. The results of the first comparison are included in Supplementary File 1. For this comparison, seven genes were DE with Asb4, Cxcl14, Dio2, and Igfbp5 increased and Sult1d1, Mt3, and Lcn2 reduced in Ezh2cKO vs. WT uterine epithelium. Of these, the GTEx Portal reveals that IGFBP5 is the most abundant in the human uterus (Figure 1).
Figure 1 .
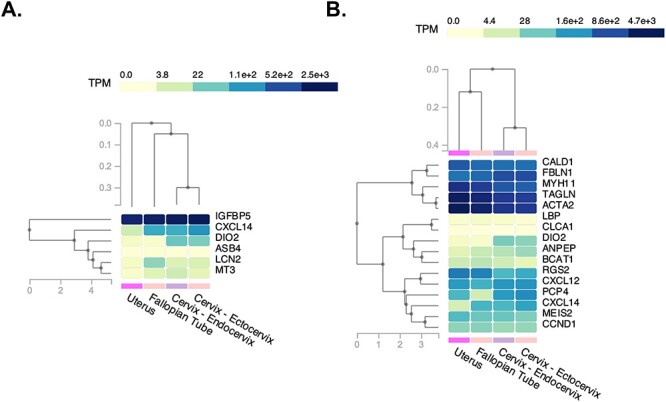
Heatmaps of genes DE for our mouse uterine samples relative to their orthologs that are enriched in human oviduct, uterine, and cervix samples. (A) Ezh2cKO epithelium vs. WT epithelium. (B) Ezh2cKO stroma vs. WT stroma. The GTEx Portal (https://www.gtexportal.org/home/) was used to determine which of the DE genes for the Ezh2cKO epithelium vs. WT epithelium and Ezh2cKO stroma vs. WT stroma are enriched in the human oviduct, uterus, and cervix.
We next compared Ezh2cKO vs. WT stroma and 1192 distinct genes were identified. In Supplementary Figures S1 and S2, this area is depicted in green. However, to ensure that the comparison only included underlying uterine stroma and not myometrial cells, the stromal region in each section was hand selected. Genes that are DE based on this comparison are listed in Supplementary File 2. For this comparison, 18 genes differed in expression between the two groups. Cxcl14, Bcat1, Dio2, Clca1, Bpifb5, Lbp, Ccnd1, Anpep, and Col6a4 were upregulated in Ezh2cKO, but Rgs2, Myh11, Pcp4, Acta2, Meis2, Tagln, Cald1, Cxcl12, and Fbln1 showed reduced expression in this group. CALD1, FBLN1, MYH11, ACTA2, and TAGLN show the greatest expression in the human uterus based on a GTEx Portal search (Figure 1).
Another possibility is that conditional deletion of Ezh2 alters the epithelial to stromal gene expression patterns. Thus, we compared epithelial vs. stromal transcripts in WT and then Ezh2cKO uteri. WT uteri epithelial to stromal gene expression differences are detailed in Supplementary File 3. About 509 genes were DE. The top 10 genes DE based on FDR between these two groups were Igfp3, Cxcl12, Malat1, Dcn, Gpx3, Igfbp5, Tcf4, Vim, Apoe, Tacstd2. All of these were downregulated in WT epithelium relative to WT stromal cells except for Tacstd2, which was significantly elevated in WT epithelium.
Supplementary File 4 lists the genes that were DE in Ezh2cKO epithelium vs. Ezh2cKO stroma. About 271 genes were DE for this comparison. The top 10 DE genes for this comparison were Igfbp3, S100, Lratd1, Cxcl12, Calb1, Sfrrp4, Inmt, Gsto1, Cldn3, and Wfdc2. Igfbp3, Cxcl12, Sfrp4, and Inmt which were reduced in Ezh2cKO epithelium vs. Ezh2cKO stroma. S100g, Lratd1, Calb1, Gsto1, Cldn3, and Wfdc2 were elevated in Ezh2cKO epithelium compared to Ezh2cKO stroma.
We also used these comparisons to validate the two clusters that we defined as uterine epithelium and stroma. We did this by considering the expression pattern for genes characterized as marker genes for uterine epithelium vs. those for uterine stroma in the WT and Ezh2cKO samples. These included Bcat1, Alcam, Cdh1, Krt18, Krt19, Krt8 for uterine epithelium and Vim, Vegfa, Tgfbr2, Cd34 for uterine stroma [23]. We focused just on these two tissues and clusters based on the previously characterized role of EZH2 in uterine epithelium and stromal tissues [11, 21, 24]. As shown in Supplementary Table S2, the expression patterns for these marker genes matched previous reports with epithelial markers enriched in the cluster identified as epithelium for WT and Ezh2cKO, and stromal markers elevated in the cluster identified as uterine stroma for both genotypes.
A comparison of genes DE in Ezh2cKO epithelium vs. WT epithelium and Ezh2cKO stroma vs. WT stroma revealed Dio2 and Cxcl14 to be the only genes that are DE in both comparisons (Figure 2). A Venn diagram shows that 208 genes overlap in WT epithelium vs. WT stroma and Ezh2cKO epithelium vs. Ezh2cKO stromal comparisons (Figure 3). However, 301 and 63 genes, respectively, are unique for these two comparisons, suggesting the conditional loss of Ezh2cKO alters the gene expression patterns between these two cell populations.
Figure 2 .
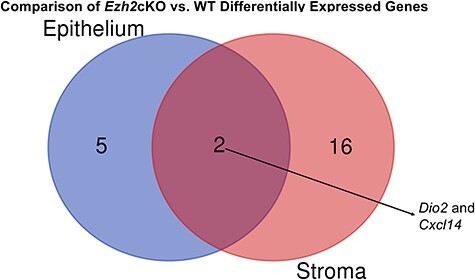
Venn diagram comparison of genes DE in Ezh2cKO epithelium vs. WT epithelium and Ezh2cKO stroma vs. WT stroma.
Figure 3 .
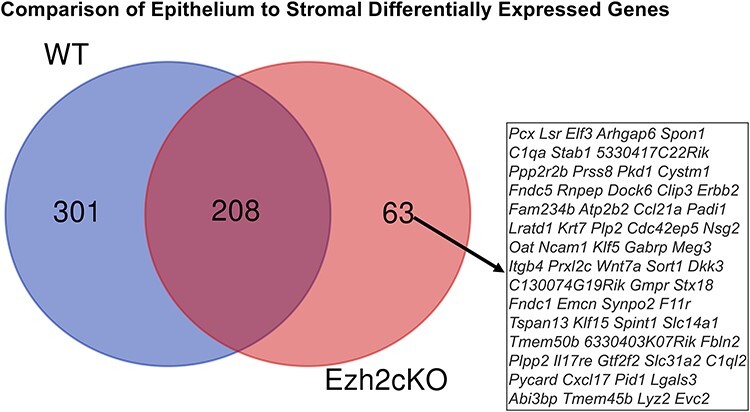
Venn diagram comparison of genes DE in WT epithelium vs. WT stroma and Ezh2cKO epithelium vs. Ezh2cKO stromal comparisons.
Potential PPIs were examined based on proteins encoded for genes DE based on the four comparisons. For the seven DE genes that were DE for Ezh2cKO epithelium vs. WT epithelium and Ezh2cKO stroma vs. WT stroma, no PPI interactions were identified (Figure 4).
Figure 4 .
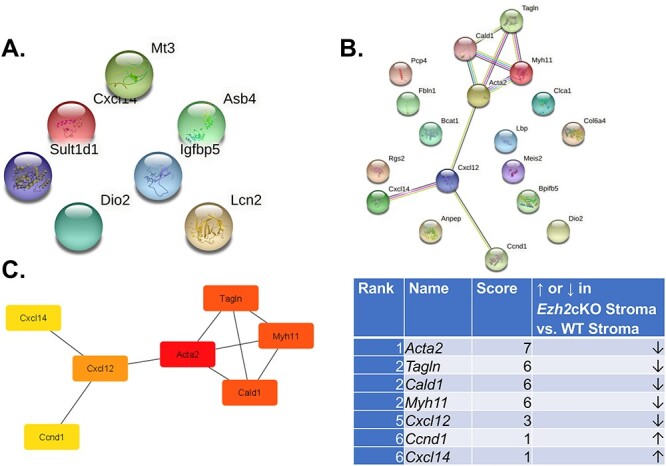
PPI interactions for Ezh2cKO epithelium vs. WT epithelium and Ezh2cKO stroma vs. WT stroma. (A) PPI interactions for Ezh2cKO epithelium vs. WT epithelium. As shown, there are no significant interactions between the seven DE genes for this comparison. (B) PPI interactions for Ezh2cKO stroma vs. WT stroma. Seven of the 18 DE genes showed some form of interactions. (C) Hub genes for the Ezh2cKO stroma vs. WT stroma comparison include Acta2, Tagln, Cald1, Myh11, Cxcl12, Ccnd1, and Cxcl1.
For Ezh2cKO stroma vs. WT stroma, seven proteins encoded by the DE genes were interconnected. These hub genes included Acta2, Tagln, Cald1, Myh11, Cxcl12, Ccnd1, and Cxcl14 (Figure 4). Several PPIs were evident for WT epithelium vs. WT stroma and Ezh2cKO epithelium vs. Ezh2cKO stroma comparisons (Supplementary Figure S5). Based on these PPI, the top 10 hub genes for the WT epithelium to WT stromal comparison are Fst1, Igfb3, Igfbp5, App, Apoe, Igfbp7, C3, Penk, Lgals1, and Mfg8 (Figure 5). The top 10 hub genes for Ezh2cKO epithelium vs. Ezh2cKO stroma are Cdh1, Epcam, Krt18, Krt19, Krt8, Krt19, Krt7, Erbb2, Krt8, Cldn3, Cd34, and Ncam1 (Figure 6).
Figure 5 .
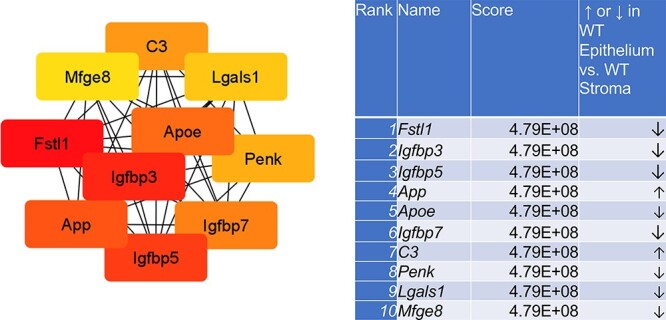
Hub gene analyses for WT epithelium vs. WT stroma comparison. The PPIs were determined by STRING analysis. The PPI files generated with STRING were imported into the cytohubba app [17] in Cytoscape [18] to examine for the top 10 hub genes. Within this program, hub genes were determined with MCC analysis, as recommended [17].
Figure 6 .
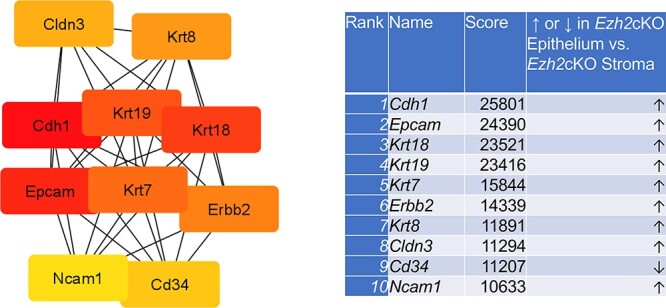
Hub gene analyses for Ezh2cKO epithelium vs. Ezh2cKO stroma comparison. The PPIs were determined by STRING analysis. The PPI files generated with STRING were imported into the cytohubba app [17] in Cytoscape [18] to examine for the top 10 hub genes. Within this program, hub genes were determined with MCC analysis, as recommended [17].
Lastly, we considered those pathways that are likely enriched based on the genes DE for these comparisons. For Ezh2cKO epithelium vs. WT epithelium, some of the pathways that are potentially enriched include adaptive thermogenesis, organic hydroxy compound metabolic process, cellular modified amino acid catabolic process, negative regulation of growth, protein kinase B signaling, cellular response to toxic substance/drug, response to leptin, and transition metal ion homeostasis (Figure 7). For Ezh2cKO stroma vs. WT stroma, select pathways that are predicted to be enriched included positive regulation of response to external stimuli and drugs, regulation of chemotaxis, drug transport, cell chemotaxis, amine transport, monoamine transport, and ammonium transport (Figure 8).
Figure 7 .
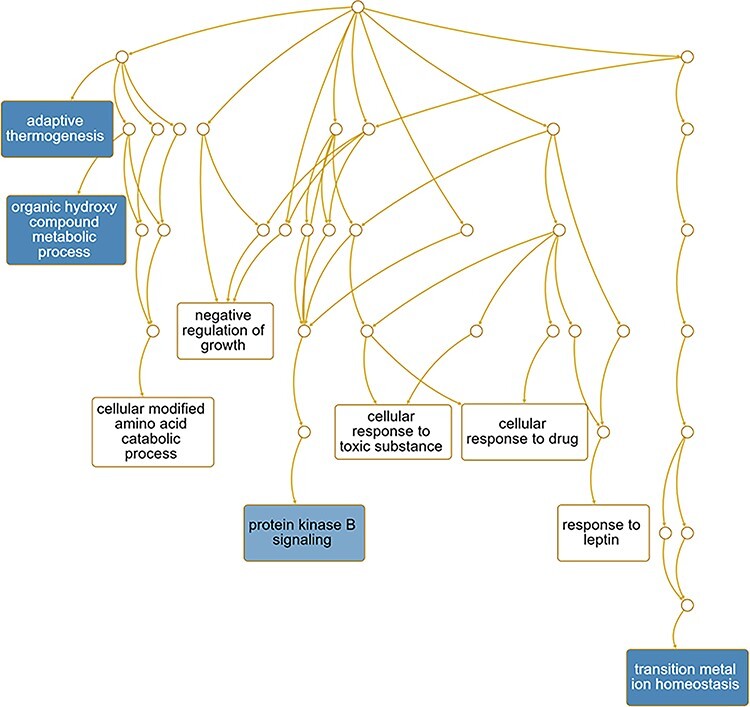
Pathways predicted to be affected based on DE genes for the for Ezh2cKO epithelium vs. WT epithelium comparison. Functional enrichment analyses with the WEB-based Gene SeT AnaLysis Toolkit (WebGestalt) [63] and GO molecular function terms were performed based on DE genes to determine which pathways might be affected for this comparison.
Figure 8 .
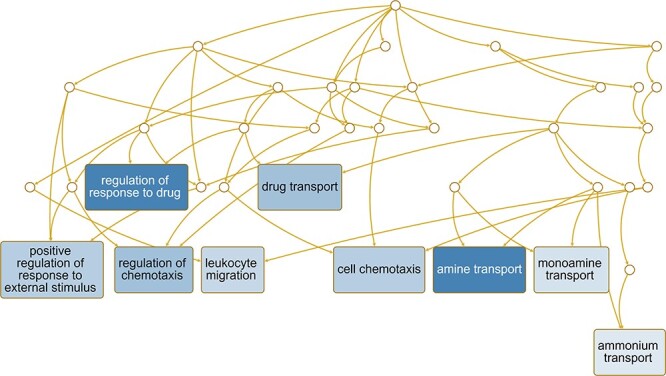
Pathways predicted to be affected based on DE genes for the Ezh2cKO stroma vs. WT stroma comparison. Functional enrichment analyses with the WEB-based Gene SeT AnaLysis Toolkit (WebGestalt) [63] and GO molecular function terms were performed based on DE genes to determine which pathways might be affected for this comparison.
For WT epithelium vs. WT stroma, pathways presumed to be enriched included response to wounding, response to antibiotic, response to alcohol, reproductive system development, urogenital system development, negative regulation of cellular component movement, response to vasculature development and angiogenesis (Figure 9). Pathways enriched for Ezh2cKO epithelium vs. Ezh2cKO stroma included epithelial cell proliferation, morphogenesis of a branching structure, urogenital system development, response to wounding, gland morphogenesis, epithelial tube morphogenesis, epithelial cell development, ameboidal type cell migration, and angiogenesis (Figure 10).
Figure 9 .
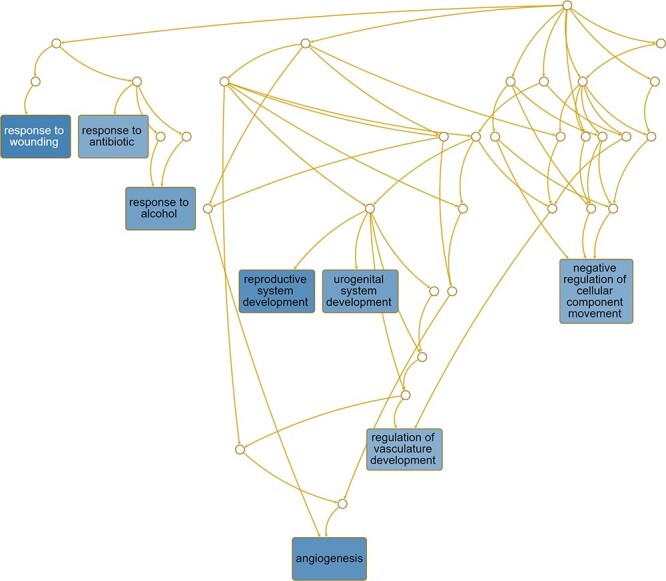
Pathways predicted to be affected based on DE genes for the WT epithelium vs. WT stroma comparison. Functional enrichment analyses with the WEB-based Gene SeT AnaLysis Toolkit (WebGestalt) [63] and GO molecular function terms were performed based on DE genes to determine which pathways might be affected for this comparison.
Figure 10 .
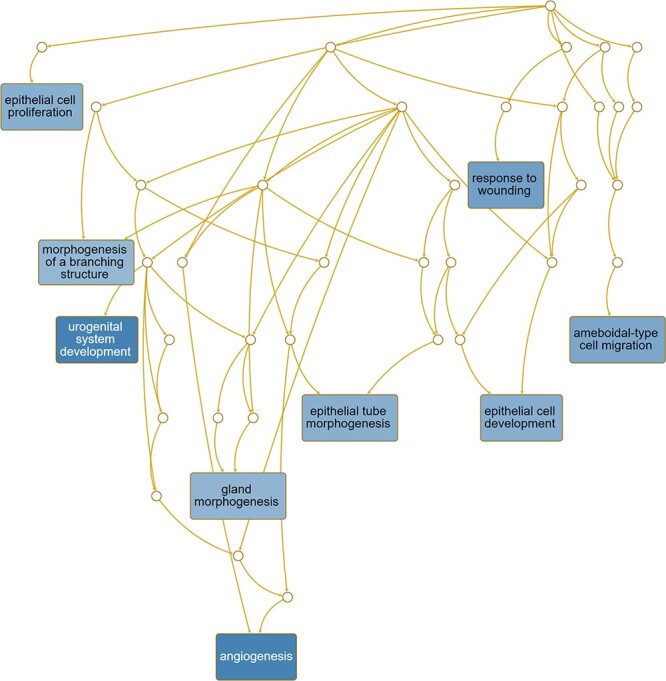
Pathways predicted to be affected based on DE genes for the Ezh2cKO epithelium vs. Ezh2cKO stroma comparison. Functional enrichment analyses with the WEB-based Gene SeT AnaLysis Toolkit (WebGestalt) [63] and GO molecular function terms were performed based on DE genes to determine which pathways might be affected for this comparison.
Discussion
The primary goal of this study was to use the relatively recently developed ST technology [13, 14] to examine how conditional deletion of Ezh2 affected uterine epithelial and stromal gene expression patterns. By using RNA-seq, we found that conditional deletion of Ezh2 results in several gene expression changes in mutant vs. WT mice and may overall impair estrogen receptor signaling pathways [24]. As with any whole organ transcriptome approach though, we could not localize the cellular origin of such gene expression changes. Additionally, some subtle gene expression changes in individual tissue compartments may have been masked or diluted out with this approach. While single cell RNA-seq has been successfully and widely used in the uterus, we opted to use ST for the current studies to maintain the integrity and normal architecture of the uterus such that potential relations between epithelium and stromal compartments could be examined.
We first considered genes that were DE between Ezh2cKO epithelium WT epithelium. Several uterine enriched genes (Asb4, Cxc14, Dio2, and Igfbp5) were upregulated in Ezh2cKO uterine epithelium. Upregulation of these proteins, as detailed below, has been linked to altered cell proliferation and metabolism in other cell types. For example, proteins such as ankyrin repeat (AR) and SOCS box proteins (ASBs) mediate the ubiquitination of numerous proteins [25], specifically an upregulation of ASB4 is associated with migration and invasion capacity in hepatocellular carcinoma cells [26]. Cxc14 encodes a recently characterized chemokine C-X-C Motif Chemokine Ligand 14; its functions have been described as a regulator of immune cell migration and an inflammation modulator [27]. In the uterus, CXCL14 is necessary to recruit natural killer cells, as evidenced by the fact that Cxcl14 knockout show reduced decidual NK cell populations [28]. Further, this protein promotes epithelial–mesenchymal transition by serving as a key regulator of migration and proliferation of oral squamous cell carcinoma cells [29]. Dio2 encodes the deiodinase DIO2 that converts thyroxine (T4) to its active form triiodothyronine (T3). Notably, this gene was also increased in uterine stroma of Ezh2cKO mice. The endometrium appears to synthesize extrathyroidal T3 and T4 [30], T3 and T4 synthesis are upregulated by E2 [31], and altered thyroid hormone metabolism is evident in endometriosis lesions [32]. Igfbp5 mRNA is overexpressed during the proliferative phase in the endometrium [33], which is consistent with the endometrial gland hyperplasia observed in our Ezh2cKO model [11]. IGFBP5 is also important in maintaining epithelial–mesenchymal boundaries [34], a process in which EZH2 is another key player [20].
Sult1d1, Mt3, and Lcn2 were downregulated in Ezh2cKO epithelium relative to WT. When transcriptomes of the whole uterus were previously analyzed, Sult1d1 was consistently downregulated in Ezh2cKO animals treated with vehicle as well as stimulated with E2 [24]. As a sulfotransferase, SULT1D1 is a detoxifying enzyme and has been shown to catalyze the sulfation of several prostaglandins; its expression is an implantation marker in glandular epithelium [35]. Additionally, Sult1d is downregulated in surgically induced endometriosis in mice [36]. Metallothioneins 3 (MT3) is a protein of interest in multiple cancers, as it stimulates tumor cell proliferation, angiogenesis, drug resistance, and metastasis [37]. Specifically, MT3 is overexpressed in prostate cancer cells [38], but downregulated in breast cancer cell lines [39]. Acute-phase protein lipocalin 2 is a secretory protein encoded by the Lcn2 gene; this protein is produced by epithelial cells in response to physiological changes or pathogens. LCN2 is estrogen-responsive, participates in epithelial–mesenchymal transition, and is upregulated in endometrial carcinoma cells and other endometrial lesions [40]. Selective pathways that are likely increased based on the DE genes for the epithelial comparisons include the protein kinase B signaling pathway. This pathway is increased in various uterine diseases, such as uterine fibroids [41] and leiomyomas [42].
The branched-chain amino acid aminotransferase 1 gene (BCAT1) was upregulated in Ezh2cKO stroma relative to epithelial tissue in this genotype. This enzyme supports synthesis of branched-chain amino acids that in turn act to induce the mechanistic target of rapamycin 1 (mTORC1) pathway, which is linked to endometrial cancer progression [43]. Another upregulated stromal gene was Ccnd1, which encodes for cyclin D1. In estrogen receptor-positive, breast cancer cells, EZH2 acts as a gene promoter by binding to ESR1 and β-catenin, activating CCND1 transcription [44], Ccnd1 is a cell cycle regulator enriched in endometrial adenocarcinomas [45]. Stromal Rgs2 was reduced; this protein (regulator of G protein signaling 2) promotes arterial structural and remodeling that has been studied primarily in pregnant mice [46]. Myosin heavy chain 11 encoded by Myh11 was downregulated in our model, but high expression of this protein is correlated with uterine leiomyoma development [47].
Alpha smooth muscle actin encoded by Acta2 was downregulated in stromal cells. Although it is considered a biomarker for smooth muscle cells, it is expressed in uterine stroma [48]. Exposure to activated platelets upregulates its expression, causing smooth muscle metaplasia; alterations in this estrogen-ACTA2 system are associated with endometriosis [49]. Acta2 is highly enriched in human uterus and is considered a key hub gene with the highest interaction score for this comparison. Another hub protein and smooth muscle differentiation marker downregulated in our system was Tagln, encoding transgelin, an actin-binding protein. Upregulation of Tagln occurs in uterine smooth muscle tumors [50]. Some of the pathways enriched in the Ezh2cKO stroma consisted of regulation chemotaxis and leukocyte migration pathways; both pathways are essential for normal endometrial physiology [51, 52].
To gain further understanding into how conditional deletion of Ezh2 affects uterine tissues, we compared our current results where we are able to separate out and resolve gene expression patterns in select tissues to our previous study using RNA-seq on whole uteri from Ezh2cKO and WT mice [24]. Table 1 compares the DE genes identified based on the Ezh2cKO vs. WT epithelium and Ezh2cKO vs. WT stroma. ST technology permitted us to pinpoint which tissues gave rise to the original whole organ changes. For instance, Asb4 was elevated in Ezh2cKO vs. WT uteri. The current findings reveal that this increase presumably originated from the Ezh2cKO epithelium. In contrast, Sult1d1 was reduced in Ezh2cKO uteri, and the ST technology suggests that this is due to decreased expression in Ezh2cKO epithelium vs. WT epithelium. Cxcl12 was reduced in whole uteri of Ezh2cKO mice, and present results reveal that this change in expression originates from Ezh2cKO stroma. While Dio2 and Cxcl14 did not show DE with whole uteri, ST analysis revealed both genes to be upregulated in Ezh2cKO, suggesting that analysis of whole uteri that included the myometrial layer may have masked these key gene expression changes. Likewise, Igfbp5, which is prevalent in human uteri, did not show altered expression in whole uteri, but this gene was upregulated in Ezh2cKO epithelium vs. WT epithelium. Taken together, the ST approach allows more refined resolution of gene expression changes at the tissue and cellular level while preserving the original organ structure. Importantly, this innovative technology has permitted us to identify gene expression changes in tissue specific regions that may have been masked or diluted out with the whole organ approach.
Table 1.
Comparison for current results to previously published results with whole uteri from Ezh2cKO and WT mice
| Epithelial cells | Stromal cells | Whole uterus from [13] | ||||
|---|---|---|---|---|---|---|
| Ezh2cKO to WT | Ezh2cKO to WT | Ezh2cKO to WT | ||||
| Gene | Fold change | Adjusted P-value (FDR) | Fold change | Adjusted P-value (FDR) | Fold change | Adjusted P-value (FDR) |
| Asb4 | 6.506 | 0.000 | 9.718 | 0.000 | ||
| Sult1d1 | 0.247 | 0.001 | 0.490 | 0.000 | ||
| Mt3 | 0.235 | 0.010 | 0.584 | 0.030 | ||
| Igfbp5 | 3.124 | 0.037 | ||||
| Lcn2 | 0.306 | 0.037 | ||||
| Dio2 | 2.869 | 0.016 | 2.937 | 0.004 | ||
| Cxcl14 | 1.446 | 0.010 | 5.932 | 0.000 | ||
| Rgs2 | 0.314 | 0.000 | ||||
| Myh11 | 0.283 | 0.000 | NI | NI | ||
| Bcat1 | 6.250 | 0.000 | 1.442 | 0.014 | ||
| Acta2 | 0.362 | 0.005 | ||||
| Cald1 | 0.424 | 0.005 | ||||
| Clca1 | 7.527 | 0.005 | ||||
| Bpifb5 | 5.916 | 0.005 | ||||
| Lbp | 3.500 | 0.006 | ||||
| Cxcl12 | 0.447 | 0.012 | 0.625 | 0.001 | ||
| Fbln1 | 0.481 | 0.017 | ||||
| Col6a4 | 2.410 | 0.021 | ||||
| Meis2 | 0.374 | 0.031 | ||||
| Tagln | 0.402 | 0.031 | ||||
| Anpep | 2.636 | 0.038 | ||||
| Pcp4 | 0.345 | 0.039 | 0.615 | 0.000 | ||
| Ccnd1 | 2.958 | 0.039 | 2.300 | 0.001 | ||
Blank cells are not significant. NI = not identified.
Sixty-three genes showed differential expression in epithelium relative to stroma only in Ezh2cKO, and these include Wnt7a, C1ql2, Atp2b2, Ccl21a, Lratd1, Cxcl17, Krt7, Fndc1, Aox3, Tagln, Slc14a1, and Fndc5. Of these, key uterine functions have been ascribed to Wnt7a and various Krt forms. Wnt7a is essential in uterine cell specification and epithelial–mesenchymal interactions in mice and humans [53, 54]. Transgenic mice lacking this gene do not develop uterine glands and are infertile [53]. In our previous studies with whole uterine samples, we found that conditional loss of uterine EZH2 generally upregulates various genes associated with uterine disease, including keratin 5 (Krt5) and keratin 15 (Krt15) [24]. Fang et al. [12] also showed that KRT5, KRT6A, and KRT14 protein and mRNA expression were elevated in uteri of Ezh2cKO mice relative to their WT counterparts [12]. Uterine loss of Ezh2 thus also affects Krt7 expression in the epithelium vs. stromal compartment. Notably, this gene, along with Krt18, Krt19, and Krt8, is considered a hub gene for this comparison.
Pathways that were unique to the Ezh2cKO epithelium vs. Ezh2cKO stromal comparison consisted of response to epithelial cell proliferation, epithelial cell development, epithelial tube morphogenesis, morphogenesis of a branching structure, and gland morphogenesis. In contrast, pathways that were enriched in WT epithelium vs. WT stromal comparison include reproductive system development, regulation of vascular development, and negative regulation of cellular component movement. The overall profiles suggest that Ezh2cKO mice exhibit a greater number of gene-sets involved in epithelial cell proliferation and gland development. Such findings are consistent with the endometrial gland hyperplasia that we, and others, have previously reported in these Ezh2cKO mice [11, 12].
This is the first report that utilizes ST, a recently developed high-throughput method, on whole uteri. While other studies have used single cell RNA-seq and laser capture microdissection (LCM) technique in combination with RNA-seq approaches to examine gene expression in select uterine tissues [55–58], these procedures destroy the tissue architecture. These approaches also require pre-existing knowledge about distinct genes to assign gene identities to the different clusters. Moreover, it is not possible to examine in situ relationships between gene expression patterns in neighboring tissues. RNA-seq with whole uteri has been a useful approach in understanding broadly how gene deletion and other factors affect uterine physiology. This global approach though may dilute out and even mask genes that are DE in discrete uterine tissues, especially ones where the gene may be upregulated in one tissue but decreased in another tissue from the same group. This is especially important when considering that the uterus consists of only 5–10% epithelial cells [59]. ST analyses combine histological and transcriptome analyses to permit gene expression patterns in select tissues but in the context of the whole tissue. In so doing, it addresses the limitations of other high-throughput approaches.
As with any transcriptomics approach, one major concern is interpreting gene expression changes and their potential contribution to pathophysiological changes. While we cannot ascertain which, if any, of these changes lead to the uterine gland hyperplasia evident in the Ezh2cKO mice, it is important to note that Igfbp5 was increased in Ezh2cKO epithelium relative to WT epithelium. This gene is abundantly expressed in human uteri, and becomes elevated during the proliferative phase [33], IGFBP5 is upregulated in uterine leiomyoma and is associated with tumorigenesis [60]. All these findings suggest that further studies, such as ChIP-seq, should investigate whether potential interactions exist in the uterus between EZH2 and Igbp5. In mutant breast cancer cells, a relationship exists between EZH2 and IGFBP5 [61]. EZH2 represses IGFBP5, this inhibition is restored by lysine demethylase 6B (KDM6B), a demethylase of H3K27. Analogous findings are present in human umbilical vein endothelial cells [62].
The fact that comparison of epithelial vs. stromal genes in Ezh2cKO reveals overexpression of genes that promote epithelial cell development and proliferation and gland development is consistent with the observation that these conditional mutant mice have increased epithelial proliferation and gland hyperplasia. Lastly, these findings might provide useful biomarkers, namely IGFBP5, for uterine endometrial pathologies.
In conclusion, the current studies used the recently developed ST technology to refine our understanding of how conditional loss of Ezh2 affects expression profiles in uterine epithelium and stroma while preserving the uterine morphology. A handful of genes were DE in uterine epithelium and stroma of Ezh2cKO compared to their WT counterpart tissues. Findings herein might provide additional insight into how EZH2 may selectively affect uterine epithelial and stromal compartments. Moreover, these transcriptome results likely provide mechanistic understanding and useful biomarkers for endometrial disorders in women arising due to EZH2 expression changes.
Supplementary Material
Acknowledgments
We thank Dr Christopher Bottoms of the University of Missouri Informatics Research Core Facility and Dr Michael Campbell at 10X Genomics for biocomputing assistance.
Grant Support: This work was supported by National Institutes of Health (grants HD088006 and HD087528 to PSC, R01 ES025547 CSR, and R01 HD 096083A to GT).
Contributor Information
Ana M Mesa, Department of Physiological Sciences, University of Florida, Gainesville, Florida, USA; Grupo de Investigación en Agrociencias, Biodiversidad y Territorio - GAMMA, Facultad de Ciencias Agrarias, Universidad de Antioquia UdeA, Medellín, Colombia.
Jiude Mao, Christopher S. Bond Life Sciences Center, University of Missouri, Columbia, Missouri, USA; Biomedical Sciences, University of Missouri, Columbia, Missouri, USA.
Theresa I Medrano, Department of Physiological Sciences, University of Florida, Gainesville, Florida, USA.
Nathan J Bivens, Genomics Technology Core, University of Missouri, Columbia, Missouri, USA.
Alexander Jurkevich, Advanced Light Microscopy Core Facility, University of Missouri, Columbia, Missouri, USA.
Geetu Tuteja, Genetics, Development and Cell Biology, Iowa State University, Ames, Iowa, USA.
Paul S Cooke, Department of Physiological Sciences, University of Florida, Gainesville, Florida, USA.
Cheryl S Rosenfeld, Biomedical Sciences, University of Missouri, Columbia, Missouri, USA; Data Science and Informatics Institute, University of Missouri, Columbia, Missouri, USA; Thompson Center for Autism and Neurobehavioral Disorders, University of Missouri, Columbia, Missouri, USA.
Author contributions
A.M.M., J.M., G.T., and P.S.C., and C.S.R. conceived and designed research; A.M.M., J.M., T.I.M., N.G.B, A.J., and C.S.R. performed experiments; A.M.M., J.M., N.G.B., A.J., G.T., and C.S.R. analyzed data; A.M.M., J.M., N.G.B., A.J., P.S.C, and C.S.R. interpreted results of experiments; A.M.M., J.M., and C.S.R. prepared figures; A.M.M., G.T., P.S.C., and C.S.R. drafted manuscript; A.M.M., J.M., T.I.M., N.G.B., A.J., G.T., P.S.C., and C.S.R. edited and revised manuscript; A.M.M., J.M., T.I.M., N.G.B., A.J., G.T., P.S.C., and C.S.R. approved final version of manuscript.
Data availability statement
The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus [64] and are accessible through GEO Series accession number GSE181885 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE181885).
Conflict of interest
The authors have declared that no conflict of interest exists.
References
- 1. Gibney ER, Nolan CM. Epigenetics and gene expression. Heredity 2010; 105:4. [DOI] [PubMed] [Google Scholar]
- 2. Treviño LS, Wang Q, Walker CL. Phosphorylation of epigenetic “readers, writers and erasers”: implications for developmental reprogramming and the epigenetic basis for health and disease. Prog Biophys Mol Biol 2015; 118:8–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Oki S, Sone K, Oda K, Hamamoto R, Ikemura M, Maeda D, Takeuchi M, Tanikawa M, Mori-Uchino M, Nagasaka K, Miyasaka A, Kashiyama T et al. Oncogenic histone methyltransferase EZH2: a novel prognostic marker with therapeutic potential in endometrial cancer. Oncotarget 2017; 8:40402–40411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eskander RN, Ji T, Huynh B, Wardeh R, Randall LM, Hoang B. Inhibition of enhancer of zeste homolog 2 (EZH2) expression is associated with decreased tumor cell proliferation, migration, and invasion in endometrial cancer cell lines. Int J Gynecol Cancer 2013; 23:997–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Eskander RN, Tewari KS. Exploiting the therapeutic potential of the PI3K-AKT-mTOR pathway in enriched populations of gynecologic malignancies. Expert Rev Clin Pharmacol 2014; 7:847–858. [DOI] [PubMed] [Google Scholar]
- 6. Mesa AM, Rosenfeld CS, Tuteja G, Medrano TI, Cooke PS. The roles of the histone protein modifier EZH2 in the uterus and placenta. Epigenomes 2020; 4:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Skoda RC, Schwaller J. Dual roles of EZH2 in acute myeloid leukemia. J Exp Med 2019; 216:725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang Q, Diamond MP, Al-Hendy A. Early life adverse environmental exposures increase the risk of uterine fibroid development: role of epigenetic regulation. Front Pharmacol 2016; 7:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Colon-Caraballo M, Monteiro JB, Flores I. H3K27me3 is an epigenetic mark of relevance in endometriosis. Reprod Sci 2015; 22:1134–1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. O'Carroll D, Erhardt S, Pagani M, Barton SC, Surani MA, Jenuwein T. The polycomb-group gene Ezh2 is required for early mouse development. Mol Cell Biol 2001; 21:4330–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Nanjappa MK, Mesa AM, Medrano TI, Jefferson WN, DeMayo FJ, Williams CJ, Lydon JP, Levin ER, Cooke PS. The histone methyltransferase EZH2 is required for normal uterine development and function in mice. Biol Reprod 2019; 101:306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fang X, Ni N, Lydon JP, Ivanov I, Bayless KJ, Rijnkels M, Li Q. Enhancer of zeste 2 polycomb repressive complex 2 subunit is required for uterine epithelial integrity. Am J Pathol 2019; 189:1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Berglund E, Maaskola J, Schultz N, Friedrich S, Marklund M, Bergenstrahle J, Tarish F, Tanoglidi A, Vickovic S, Larsson L, Salmen F, Ogris C et al. Spatial maps of prostate cancer transcriptomes reveal an unexplored landscape of heterogeneity. Nat Commun 2018; 9:2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Maniatis S, Aijo T, Vickovic S, Braine C, Kang K, Mollbrink A, Fagegaltier D, Andrusivova Z, Saarenpaa S, Saiz-Castro G, Cuevas M, Watters A et al. Spatiotemporal dynamics of molecular pathology in amyotrophic lateral sclerosis. Science 2019; 364:89–93. [DOI] [PubMed] [Google Scholar]
- 15. Yu D, Huber W, Vitek O. Shrinkage estimation of dispersion in negative binomial models for RNA-seq experiments with small sample size. Bioinformatics 2013; 29:1275–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2014; 43:D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chin C-H, Chen S-H, Wu H-H, Ho C-W, Ko M-T, Lin C-Y. Cytohubba: identifying hub objects and sub-networks from complex interactome. BMC Syst Biol 2014; 8:S11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003; 13:2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang J, Vasaikar S, Shi Z, Greer M, Zhang B. WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res 2017; 45:W130–W137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Q, Dong P, Liu X, Sakuragi N, Guo SW. Enhancer of Zeste homolog 2 (EZH2) induces epithelial-mesenchymal transition in endometriosis. Sci Rep 2017; 7:6804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Colón-Caraballo M, Torres-Reverón A, Soto-Vargas JL, Young SL, Lessey B, Mendoza A, Urrutia R, Flores I. Effects of histone methyltransferase inhibition in endometriosis. Biol Reprod 2018; 99:293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Moncada R, Wagner F, Chiodin M, Devlin JC, Baron M, Hajdu CH, Simeone DM, Yanai I. Integrating single-cell RNA-Seq with spatial transcriptomics in pancreatic ductal adenocarcinoma using multimodal intersection analysis . bioRxiv. 2019; 254375. 26 January 2018. 10.1101/254375 preprint: not peer reviewed. [DOI]
- 23. Valdez-Morales FJ, Gamboa-Domínguez A, Vital-Reyes VS, Cruz JCH, Chimal-Monroy J, Franco-Murillo Y, Cerbón M. Changes in receptivity epithelial cell markers of endometrium after ovarian stimulation treatments: its role during implantation window. Reprod Health 2015; 12:45–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Mesa AM, Mao J, Nanjappa MK, Medrano TI, Tevosian S, Yu F, Kinkade J, Lyu Z, Liu Y, Joshi T, Wang D, Rosenfeld CS et al. Mice lacking uterine enhancer of zeste homolog 2 have transcriptomic changes associated with uterine epithelial proliferation. Physiol Genomics 2020; 52:81–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ferguson JE 3rd, Wu Y, Smith K, Charles P, Powers K, Wang H, Patterson C. ASB4 is a hydroxylation substrate of FIH and promotes vascular differentiation via an oxygen-dependent mechanism. Mol Cell Biol 2007; 27:6407–6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Au V, Tsang FH, Man K, Fan ST, Poon RT, Lee NP. Expression of ankyrin repeat and SOCS box containing 4 (ASB4) confers migration and invasion properties of hepatocellular carcinoma cells. Biosci Trends 2014; 8:101–110. [DOI] [PubMed] [Google Scholar]
- 27. Lu J, Chatterjee M, Schmid H, Beck S, Gawaz M. CXCL14 as an emerging immune and inflammatory modulator. J Inflamm (Lond) 2016; 13:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cao Q, Chen H, Deng Z, Yue J, Chen Q, Cao Y, Ning L, Lei X, Duan E. Genetic deletion of Cxcl14 in mice alters uterine NK cells. Biochem Biophys Res Commun 2013; 435:664–670. [DOI] [PubMed] [Google Scholar]
- 29. Rong L, Wang L, Shuai Y, Guo H, Liu K. CXCL14 regulates cell proliferation, invasion, migration and epithelial-mesenchymal transition of oral squamous cell carcinoma. Biotechnol Biotechnol Equip 2019; 33:1335–1342. [Google Scholar]
- 30. Detti L, Uhlmann RA, Fletcher NM, Diamond MP, Saed GM. Endometrial signaling pathways during ovarian stimulation for assisted reproduction technology. Fertil Steril 2013; 100:889–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Santin AP, Furlanetto TW. Role of estrogen in thyroid function and growth regulation. J Thyroid Res 2011; 2011:875125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Peyneau M, Kavian N, Chouzenoux S, Nicco C, Jeljeli M, Toullec L, Reboul-Marty J, Chenevier-Gobeaux C, Reis FM, Santulli P, Doridot L, Chapron C et al. Role of thyroid dysimmunity and thyroid hormones in endometriosis. Proc Natl Acad Sci U S A 2019; 116:11894–11899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhou J, Dsupin BA, Giudice LC, Bondy CA. Insulin-like growth factor system gene expression in human endometrium during the menstrual cycle. J Clin Endocrinol Metab 1994; 79:1723–1734. [DOI] [PubMed] [Google Scholar]
- 34. Vijayan A, Guha D, Ameer F, Kaziri I, Mooney CC, Bennett L, Sureshbabu A, Tonner E, Beattie J, Allan GJ, Edwards J, Flint DJ. IGFBP-5 enhances epithelial cell adhesion and protects epithelial cells from TGFβ1-induced mesenchymal invasion. Int J Biochem Cell Biol 2013; 45:2774–2785. [DOI] [PubMed] [Google Scholar]
- 35. Niklaus AL, Pollard JW. Mining the mouse transcriptome of receptive endometrium reveals distinct molecular signatures for the luminal and glandular epithelium. Endocrinology 2006; 147:3375–3390. [DOI] [PubMed] [Google Scholar]
- 36. Pelch KE, Schroder AL, Kimball PA, Sharpe-Timms KL, Davis JW, Nagel SC. Aberrant gene expression profile in a mouse model of endometriosis mirrors that observed in women. Fertil Steril 2010; 93:1615, e1618–1627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Sun S, Liu F, Xian S, Cai D. miR-325-3p overexpression inhibits proliferation and metastasis of bladder cancer cells by regulating MT3. Med Sci Monit 2020; 26:e920331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Garrett SH, Sens MA, Shukla D, Nestor S, Somji S, Todd JH, Sens DA. Metallothionein isoform 3 expression in the human prostate and cancer-derived cell lines. The Prostate 1999; 41:196–202. [DOI] [PubMed] [Google Scholar]
- 39. Gomulkiewicz A, Jablonska K, Pula B, Grzegrzolka J, Borska S, Podhorska-Okolow M, Wojnar A, Rys J, Ambicka A, Ugorski M, Zabel M, Dziegiel P. Expression of metallothionein 3 in ductal breast cancer. Int J Oncol 2016; 49:2487–2497. [DOI] [PubMed] [Google Scholar]
- 40. Liao C-J, Li P-T, Lee Y-C, Li S-H, Chu ST. Lipocalin 2 induces the epithelial–mesenchymal transition in stressed endometrial epithelial cells: possible correlation with endometriosis development in a mouse model. Reproduction 2014; 147:179–187. [DOI] [PubMed] [Google Scholar]
- 41. Varghese BV, Koohestani F, McWilliams M, Colvin A, Gunewardena S, Kinsey WH, Nowak RA, Nothnick WB, Chennathukuzhi VM. Loss of the repressor REST in uterine fibroids promotes aberrant G protein-coupled receptor 10 expression and activates mammalian target of rapamycin pathway. Proc Natl Acad Sci U S A 2013; 110:2187–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liu S, Yin P, Dotts AJ, Kujawa SA, Coon VJ, Wei JJ, Chakravarti D, Bulun SE. Activation of protein kinase B by WNT4 as a regulator of uterine leiomyoma stem cell function. Fertil Steril 2020; 114:1339–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wang P, Wu S, Zeng X, Zhang Y, Zhou Y, Su L, Lin W. BCAT1 promotes proliferation of endometrial cancer cells through reprogrammed BCAA metabolism. Int J Clin Exp Pathol 2018; 11:5536–5546. [PMC free article] [PubMed] [Google Scholar]
- 44. Chan K-M, Fang D, Gan H, Hashizume R, Yu C, Schroeder M, Gupta N, Mueller S, James CD, Jenkins R. The histone H3. 3K27M mutation in pediatric glioma reprograms H3K27 methylation and gene expression. Genes Devt 2013; 27:985–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Xu J, Lin DI. Oncogenic c-terminal cyclin D1 (CCND1) mutations are enriched in endometrioid endometrial adenocarcinomas. PLoS One 2018; 13:e0199688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Koch JN, Dahlen SA, Owens EA, Osei-Owusu P. Regulator of g protein signaling 2 facilitates uterine artery adaptation during pregnancy in mice. J Am Heart Assoc 2019; 8:e010917–e010917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhang W, Cheng Z, Qu X, Dai H, Ke X, Chen Z. Overexpression of myosin is associated with the development of uterine myoma. J Obstet Gynaecol Res 2014; 40:2051–2057. [DOI] [PubMed] [Google Scholar]
- 48. Musolf K, Meindl S, Larsen AL, Kalcounis-Rueppell MC, Penn DJ. Ultrasonic vocalizations of male mice differ among species and females show assortative preferences for male calls. PLoS One 2015; 10:e0134123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Xu Z, Zhang L, Yu Q, Zhang Y, Yan L, Chen Z-J. The estrogen-regulated lncRNA H19/miR-216a-5p axis alters stromal cell invasion and migration via ACTA2 in endometriosis. Mol Hum Reprod 2019; 25:550–561. [DOI] [PubMed] [Google Scholar]
- 50. Tawfik O, Rao D, Nothnick WB, Graham A, Mau B, Fan F. Transgelin, a novel marker of smooth muscle differentiation, effectively distinguishes endometrial stromal tumors from uterine smooth muscle tumors. Int J Gynecol Obstet Reprod Med Res 2014; 1:26–31. [PMC free article] [PubMed] [Google Scholar]
- 51. Santamaria X, Massasa EE, Taylor HS. Migration of cells from experimental endometriosis to the uterine endometrium. Endocrinology 2012; 153:5566–5574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Weil SJ, Wang S, Perez MC, Lyttle CR. Chemotaxis of macrophages by a peritoneal fluid protein in women with endometriosis. Fertil Steril 1997; 67:865–869. [DOI] [PubMed] [Google Scholar]
- 53. Miller C, Sassoon DA. Wnt-7a maintains appropriate uterine patterning during the development of the mouse female reproductive tract. Development 1998; 125:3201–3211. [DOI] [PubMed] [Google Scholar]
- 54. Tulac S, Nayak NR, Kao LC, Van Waes M, Huang J, Lobo S, Germeyer A, Lessey BA, Taylor RN, Suchanek E, Giudice LC. Identification, characterization, and regulation of the canonical Wnt signaling pathway in human endometrium. J Clin Endocrinol Metab 2003; 88:3860–3866. [DOI] [PubMed] [Google Scholar]
- 55. Huang J, Yang Y, Tian M, Deng D, Yu M. Spatial transcriptomic and miRNA analyses revealed genes involved in the mesometrial-biased implantation in pigs. Genes 2019; 10:808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang F, Zhao S, Deng D, Wang W, Xu X, Liu X, Zhao S, Yu M. Integrating lcm-based spatio-temporal transcriptomics uncovers conceptus and endometrial luminal epithelium communication that coordinates the conceptus attachment in pigs. Int J Mol Sci 2021; 22:1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mucenski ML, Mahoney R, Adam M, Potter AS, Potter SS. Single cell RNA-seq study of wild type and Hox9,10,11 mutant developing uterus. Sci Rep 2019; 9:4557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wu B, An C, Li Y, Yin Z, Gong L, Li Z, Liu Y, Heng BC, Zhang D, Ouyang H. Reconstructing lineage hierarchies of mouse uterus epithelial development using single-cell analysis. Stem Cell Reports 2017; 9:381–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. McCormack SA, Glasser SR. Differential response of individual uterine cell types from immature rats treated with estradiol. Endocrinology 1980; 106:1634–1649. [DOI] [PubMed] [Google Scholar]
- 60. Cirilo PD, Marchi FA, Barros Filho Mde C, Rocha RM, Domingues MA, Jurisica I, Pontes A, Rogatto SR. An integrative genomic and transcriptomic analysis reveals potential targets associated with cell proliferation in uterine leiomyomas. PLoS One 2013; 8:57901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wang W, Lim KG, Feng M, Bao Y, Lee PL, Cai Y, Chen Y, Zhang H, Marzese D, Hoon DSB, Yu Q. KDM6B counteracts EZH2-mediated suppression of IGFBP5 to confer resistance to PI3K/AKT inhibitor treatment in breast cancer. Mol Cancer Ther 2018; 17:1973–1983. [DOI] [PubMed] [Google Scholar]
- 62. Xu S, Xu Y, Yin M, Zhang S, Liu P, Koroleva M, Si S, Little PJ, Pelisek J, Jin ZG. Flow-dependent epigenetic regulation of IGFBP5 expression by H3K27me3 contributes to endothelial anti-inflammatory effects. Theranostics 2018; 8:3007–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Wang J, Duncan D, Shi Z, Zhang B. WEB-based gene set analysis toolkit (WebGestalt): update 2013. Nucleic Acids Res 2013; 41:W77–W83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002; 30:207–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data discussed in this publication have been deposited in NCBI's Gene Expression Omnibus [64] and are accessible through GEO Series accession number GSE181885 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE181885).