

Abstract
De novo loss‐of‐function mutations in methyl‐CpG‐binding protein 2 (MeCP2) lead to the neurodevelopmental disorder Rett syndrome (RTT). Despite promising results from strategies aimed at increasing MeCP2 levels, additional studies exploring how hypomorphic MeCP2 mutations impact the therapeutic window are needed. Here, we investigated the consequences of genetically introducing a wild‐type MECP2 transgene in the Mecp2 R133C mouse model of RTT. The MECP2 transgene reversed the majority of RTT‐like phenotypes exhibited by male and female Mecp2 R133C mice. However, three core symptom domains were adversely affected in female Mecp2 R133C/+ animals; these phenotypes resemble those observed in disease contexts of excess MeCP2. Parallel control experiments in Mecp2 Null/+ mice linked these adverse effects to the hypomorphic R133C mutation. Collectively, these data provide evidence regarding the safety and efficacy of genetically overexpressing functional MeCP2 in Mecp2 R133C mice and suggest that personalized approaches may warrant consideration for the clinical assessment of MeCP2‐targeted therapies.
Keywords: MeCP2, MECP2 duplication syndrome, mouse model, neurodevelopment, Rett syndrome
Introducing a functional MeCP2 in a Rett syndrome mouse model with a hypomorphic MECP2 mutation is contingent on mutation and symptom domain.
1. INTRODUCTION
Rett syndrome (RTT) is a rare neurodevelopmental disorder that is predominantly seen in female individuals and is characterized by a period of developmental regression in early childhood. Although it has been 20 years since the discovery of the gene causing RTT, 1 therapeutic treatments remain elusive. The majority of RTT cases (90%–95%) are due to loss‐of‐function (LOF) mutations in Methyl‐CpG‐Binding Protein 2 (MECP2), a gene that is expressed from the X‐chromosome and encodes for the methyl reader protein (MeCP2). Seminal findings in preclinical hemizygous Mecp2 null models have described that RTT symptoms are reversible if MeCP2 levels are restored to those of wild‐type mice, even after disease onset. 2 , 3 Several studies have since shown that viral delivery of human MECP2 in hemizygous and heterozygous Mecp2 null mice can achieve similar effects in reducing symptom severity and increasing survival. 4 , 5 , 6 , 7 , 8 , 9 These data support the hypothesis that restoring MeCP2 function by supplying the wild‐type protein using gene therapy could be a viable treatment option for RTT.
Three primary challenges arise with assessing the value of gene therapy as a therapeutic strategy for RTT. First, proper brain function has remarkably precise requirements for MeCP2 dosage, as even 2x MeCP2 overexpression is also detrimental, and is the molecular basis of another neurodevelopmental disorder known as MECP2 Duplication syndrome (MDS). MDS has been modeled in mice and, interestingly, there are clusters of symptoms that appear to overlap with those in RTT models, while additional symptoms oppose those seen in RTT. 10 , 11 For example, seizures are common clinically in both disorders as well as in mouse models of the two diseases. In contrast, anxiety, motor coordination and learning and memory phenotypes are antiparallel, with deficits observed in RTT models that oppose phenotypes seen in MDS model mice.
The second and third challenges arise from the fact that RTT patients are a heterogeneous clinical population, and their diversity has the potential to impact the therapeutic window. In contrast to the Mecp2 null mice that have traditionally been used to assess the efficacy of prospective therapeutics, the RTT patient population is variable in regard to symptom severity, which is impacted by X‐chromosome inactivation (XCI) and the specific pathological MECP2 mutation. XCI, a random process of silencing one X‐chromosome in somatic cells, 12 leaves female RTT patients and Mecp2 heterozygous mice mosaic for the wild‐type (WT) and mutant MeCP2. Although the majority of RTT patients display a random XCI pattern, XCI can be skewed, whereby the expression of the X chromosome with the mutant MECP2 allele can be expressed at substantially higher or lower levels compared to the WT allele. Several studies have reported skewed XCI in RTT patients, including familial (e.g., 13 , 14 ) and sporadic (e.g., 15 ) cases, with the majority suggesting that a relationship exists between skewed XCI (i.e., favoring WT MeCP2) and milder disease severity. This has also been illustrated in Mecp2 heterozygous mice, whereby more WT MeCP2 cells leads to milder phenotypic severity. 16 , 17 , 18
Similarly, correlations exist between each patient's specific MECP2 mutation and symptom severity. 19 , 20 , 21 Pathogenic MECP2 mutations can render the protein completely nonfunctional via truncation or diminished stability, or partially functional through subtle disruption of key domains. The latter situation creates a hypomorphic mutation, in which some critical functions of MeCP2 are retained, and include the most prominent pathological mutations such as R133C, R306C and R294X, as well as other sporadic mutations, for example, P152A. 22 , 23 , 24 , 25 These retained features of MeCP2 include the ability to bind to DNA or recruit a complex of the co‐repressors nuclear receptor co‐repressor 2 (NCoR)/silencing mediator for retinoid or thyroid‐hormone receptors (SMRT) to regulate gene transcription. Thus far, the majority of preclinical studies regarding gene therapy have addressed the efficacy and safety of this treatment in models of complete LOF mutations, including T158M and R255X. 6 , 26 , 27 With the exception of a study that investigated the effects of increased MeCP2 dosage in the context of the hypomorphic mutation R306C, 23 the feasibility of such treatment for patients bearing hypomorphic mutations has been understudied and would provide valuable information to the field.
We focused here on the hypomorphic mutation R133C, which is (1) one of the eight most common mutations found in patients, (2) accounts for 7% of the current patient population, 28 and (3) leads to a mild form of RTT. 20 , 21 , 29 , 30 This mild severity is thought to be attributed to the R133C mutant MeCP2 protein retaining some ability to bind to DNA. 22 , 31 Mecp2 R133C mice have been previously shown to exhibit some RTT‐like phenotypes, in particular increased phenotypic score and decreased survival, but these phenotypes are milder in comparison to Mecp2 null mice. 22
In this current report, we further validated the Mecp2 R133C model of RTT by conducting an extensive behavioral phenotypic characterization of male and female mice. We then crossed MDS mice (MECP2 Tg1/o ) and Mecp2 R133C mice to introduce a human MECP2 transgene (termed MECP2 Tg1/o ; Mecp2 R133C/y or MECP2 Tg1/o ; Mecp2 R133C/+ ) and demonstrate reversibility of several symptoms in male and female mice. Interestingly, however, we observed that female MECP2 Tg1/o ; Mecp2 R133C/+ mice demonstrate some phenotypes that are similar to MDS mice, suggesting a potential safety risk for MeCP2 overexpression in the R133C context. In a parallel experiment, heterozygous Mecp2 null (Mecp2 Null/+ ) mice with the MECP2 transgene were indistinguishable from WT littermates, further supporting that the MDS‐like phenotypes observed in the female Mecp2 R133C/+ mice are attributable to the hypomorphic R133C mutation. In short, we present the first empirical evidence demonstrating that the consequences of introducing a functional MeCP2 in a female mouse model of RTT bearing a hypomorphic MECP2 mutation are contingent on mutation and symptom.
2. MATERIALS AND METHODS
2.1. Animals
All animals used in the present study were group housed with food and water given ad libitum and maintained on a 12 h light/dark cycle. Animals were cared for in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All studies were approved by the Institutional Animal Care and Use Committee for Vanderbilt University School of Medicine and took place during the light phase. Mecp2 R133C (B6.129P2[C]‐Mecp2 tm6.1Bird /J, stock no. 026848) mice were cryorecovered and Mecp2 Null/+ (B6.129P2[C]‐Mecp2 tm1.1Bird /J, stock no. 003890) were obtained from The Jackson Laboratory (Bar Harbor, ME, USA). MECP2 Tg1/o mice (C57Bl6 background) were generously shared by Dr Jeffrey Neul (Vanderbilt University). Experimental mice were obtained by crossing female Mecp2 R133C/+ and male MECP2 Tg1/o mice. Male and female mice were aged until the predicted symptomatic ages (6 and 20 weeks old, respectively) for all experiments. Analogously, female Mecp2 Null/+ were crossed with male MECP2 Tg1/o mice, and female mice were aged to 20 weeks old for experiments.
2.2. Behavioral assays
All behavioral experiments were conducted during predicted symptomatic ages (6–13 weeks male mice; 20–28 weeks female mice) at the Vanderbilt Mouse Neurobehavioral Core. Each mouse was utilized in multiple assays and conducted in the following order: acoustic startle response, accelerated rotarod, open field, elevated zero maze, 3‐chamber social preference, contextual fear conditioning, and whole‐body plethysmography; a minimum of 4 days elapsed between each assay. For each assay, mice were habituated to the testing room for at least 30 min prior to the experiment. Quantification was performed either by a researcher blinded to the genotype or by automated software.
2.2.1. Hindlimb clasping
Mice were suspended by their tail for 1 min, which was video recorded and analyzed by a blinded reviewer. Recording occurred every 2 weeks within a 6‐week span starting from 6 weeks old (male) or 20 weeks old (females). Clasping was defined as the number of seconds spent either clasping one or more paws, or knuckling the digits of the paw.
2.2.2. Acoustic startle response and pre‐pulse inhibition
Mice were placed in individual acoustic startle chambers (Med Associates Inc, St. Albans, VT) and, after a 5 min acclimation period, followed the testing paradigm of 61 trials previously described in Reference 32. Mice were first presented with five 120 dB startle stimulus alone, which was averaged and presented as the acoustic startle response (in arbitrary units). Then, animals were exposed to nine rounds of pseudo‐randomized presentations of the following trials (intertrial interval varied pseudo‐randomly between 9 and 21 s): No stimulus, startle pulse alone (120 dB), highest pre‐pulse noise alone (80 dB), and three varying pre‐pulses (70, 75, or 80 dB; 20 ms) followed by a startle pulse (120 dB, 50 ms interstimulus interval). Background noise of 65 dB was presented continuously. Percent pre‐pulse inhibition (PPI) was calculated as 100 × (mean acoustic startle response [ASR] − mean ASR in pre‐pulse plus pulse trials)/mean ASR in startle pulse trials.
2.2.3. Accelerated rotarod
Mice were placed on a rotarod (Ugo Basile, Med Associates Inc, St. Albans, VT, USA) that accelerated from 4 to 40 rpm over 5 min with a 10 min maximum per test. Each animal was tested three times a day for 3 days with 1 h between trials. The latency to fall was recorded as the time that the mice fall from the rod or time at which the mice turn with the rod twice. Data are presented as an average latency to fall of the three testing days.
2.2.4. Open field
Mice were placed in the activity chamber for 30 min and locomotor activity was quantified as beam breaks in the X, Y, and Z axis using the Activity Monitor software (Med Associates Inc, St. Albans, VT, USA).
2.2.5. Elevated zero maze
Mice were placed on a continuous circular platform with two closed and two open regions for 5 min under full light conditions (~ 700 lux in the open regions, ~ 400 lux in the closed regions). The time spent exploring the open regions, as well as distanced traveled in both open and closed regions, was quantified by ANY‐maze software (Stoelting, Wood Dale, IL, USA).
2.2.6. Three‐chamber social preference assay
Mice were placed in a standard three‐chamber apparatus and allowed to habituate for 5 min. The mice were then exposed to a novel mouse (Stranger 1) and an empty cup for 7 min. Immediately after, the animals were exposed to a novel stranger (“novel mouse”) in addition to Stranger 1 (“familiar mouse”) for 7 min. Stranger mice were of the same sex and strain, as well as ≤5 weeks younger as experimental mice. In between experimental mice, chamber placement of stranger mice and cups were switched. Time spent in the chambers were quantified using the ANY‐maze software (Stoelting, Wood Dale, IL, USA).
2.2.7. Contextual fear conditioning
Mice were habituated to the room for 2 h on the day prior to fear conditioning and for 1 h before conditioning and contextual testing. On conditioning day, mice were placed into an operant chamber with a shock grid (Med Associates Inc, St. Albans, VT, USA) in the presence of a 10% vanilla odor cue. Following a 3 min habituation period, mice were exposed to two mild (0.7 mA) 1 s foot shocks spaced 30 s apart that were preceded by a tone. Mice remained in the context for an additional 30 s after the second foot shock. On test day, 24 h after conditioning, mice were placed back into the same operant chamber with a 10% vanilla odor cue for 5 min, and the percentage of time spent freezing was measured by Video Freeze software (Med Associates Inc, St. Albans, VT, USA). Due to high baseline freezing (pre‐tone shock), the freezing percentage on the contextual test day was subtracted by the baseline freezing on conditioning day.
2.2.8. Whole body plethysmography
Unrestrained mice were placed in a whole body plethysmography (WBP) recording chamber (Buxco, two‐site system, DSI, New Brighton, MN, USA) with a continuous inflow of air (1 L/min). After a habituation period of 1 h, respiratory measurements were made for 30 min. Analysis was performed using the FinePointe ResearchSuite (version 2.3.1.9). Apneas, defined as pauses spanning twice the average expiratory time of the previous 2 min, were quantified using the FinePointe apnea software patch. Only points of motion‐free recording were analyzed. Periods of movement were removed (i) automatically by the FinePointe apnea software, (ii) manually by identifying points where the D‐chamber volume exceeded the normal breath range, and (iii) at points where the researcher was present during the recording noted activity.
2.3. Total protein preparation
The cortex, cerebellum and hippocampus were microdissected from naïve 6‐week male and 20‐week female mice that were of separate cohorts from the mice that underwent behavior to avoid changes in gene expression that may occur after behavioral testing. Total protein was prepared as previously described in Reference 33. Briefly, tissue samples were homogenized using a hand‐held motorized mortar and pestle in radioimmunoprecipitation assay buffer (RIPA) containing 10 mM Tris–HCl, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 0.1% sodium dodecyl sulfate (SDS), 1% Triton X‐100, and 1% deoxycholate (Sigma, St. Louis, MO, USA). After homogenization, samples were centrifuged and the supernatant was collected. Protein concentration was determined using a bicinchoninic acid (BCA) protein assay (Pierce, ThermoFisher, Waltham, MA, USA).
2.4. SDS‐Page and Western blotting
As previously described in Reference 33, 50 μg of total protein was electrophoretically separated using a 4%–20% SDS polyacrylamide gel and transferred onto a nitrocellulose membrane (Bio‐Rad, Hercules, CA, USA). Membranes were blocked in Odyssey blocking buffer (LI‐COR, Lincoln, NE, USA) for 1 h at room temperature. Membranes were probed with primary antibodies overnight at 4°C: rabbit anti‐MeCP2 (1:1000, Millipore cat no. 07‐013, Burlington, MA, USA) and mouse anti‐Gapdh (1:1000, ThermoFisher cat. no. MA5‐15738, Waltham, MA, USA), followed by the fluorescent secondary antibodies: goat anti‐rabbit (800 nm, 1:5000, LI‐COR, Lincoln, NE, USA) and goat anti‐mouse (680 nm, 1:10,000, LI‐COR, Lincoln, NE, USA). Fluorescence was detected using the Odyssey (LI‐COR, Lincoln, NE, USA) imaging system at the Vanderbilt University Medical Center Molecular Cell Biology Resource (MCBR) Core and then quantified using the Image Studio Lite software (LI‐COR, Lincoln, NE, USA). Values were normalized to Gapdh and compared relative to wild‐type controls.
2.5. Statistical analyses
Statistics were carried out using the Prism 8 (GraphPad) and Excel (Microsoft). All data shown represent mean ± SEM. Statistical significance between genotypes was determined using mixed‐effects analysis, or 1‐ or 2‐way analysis of variance (ANOVA) with Sidak's, Tukey's or t‐test post‐hoc, or unpaired t‐test. Sample size and statistical tests are specified in each figure legend with p‐values represented as *p < 0.05, **p < 0.01, ***p < 0.001, and ****p < 0.0001 for within‐genotype comparison.
3. RESULTS
3.1. Mecp2 R133C mice exhibit RTT‐like phenotypes
Male mice harboring the R133C mutation (Mecp2 R133C/y ) have been previously characterized as exhibiting RTT‐like phenotypes such as decreased survival, increased phenotypic score (an aggregate measurement of six parameters including general condition, breathing and hindlimb clasping 34 ) and altered anxiety behavior compared to WT mice. 22 To expand these observations to more quantitative phenotypes, we began by assessing weight deficits in both male and female (Mecp2 R133C/+ ) Mecp2 R133C mice. We found that, regardless of sex, decreased weight was observed as early as 5 weeks old and maintained at older ages (Figure 1A, mixed‐effects analysis, male: F (1, 47) = 93.09, p < 0.0001, female: F (1, 66) = 26.65, p < 0.0001). Similarly, hindlimb clasping was pronounced in male and female Mecp2 R133C mice, as shown in the representative images (Figure 1B). Blinded scoring of video recordings illustrated the increased hindlimb clasping in Mecp2 R133C mice compared to WT littermates (Figure 1B, t‐test, male: t (44) = 6.630, p < 0.0001, female: t (61) = 5.362, p < 0.0001). The clasping phenotype was also maintained from Weeks 6 to 13 in male Mecp2 +/y and 20–28 in female Mecp2 R133C/+ animals (Supplementary Figure 1A‐B, ANOVA, male: F (1, 44) = 43.95, p < 0.0001, female: F (1, 61) = 28.83, p < 0.0001).
FIGURE 1.
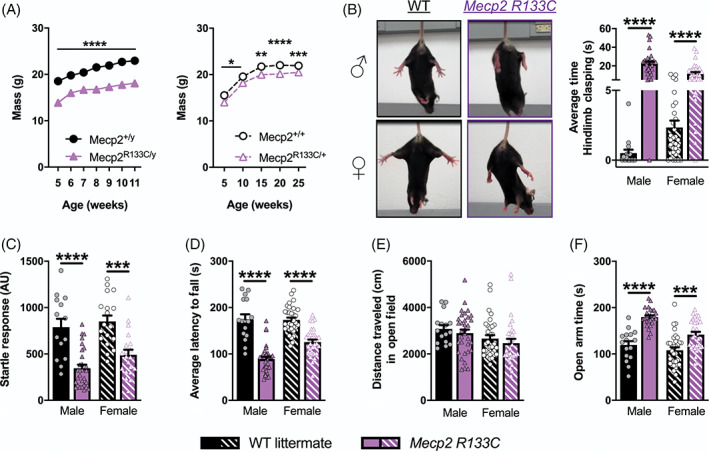
Rett syndrome (RTT)‐like neurological phenotypes are observed in male and female Mecp2 R133C mice. (A) Attenuated weight across all ages (5–11 week‐old males [n = 11–28 per genotype], and 5–25 week‐old females, n = 14–25 per genotype)). (B–F) Mecp2 R133C mice exhibited hindlimb clasping (representative images shown, (B), attenuated acoustic startle response to a 120 dB stimulus (C), decreased latency to fall on an accelerated rotarod (D), normal spontaneous locomotor activity in the open field task (E), and increased time spent in the open arms of an elevated zero maze (F). n = 15–33 per genotype in males, n = 18–32 per genotype in females. Mixed‐effects analysis with Sidak's post‐hoc test, or unpaired t‐test. *within‐genotype comparison. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. WT = filled or patterned black bars or closed or open black circles. Mecp2 R133C = filled or patterned purple bars or closed or open purple triangles. Male = filled, closed. Female = patterned, open
We then conducted a behavioral battery that incorporates evaluation of a series of phenotypes that have been reported to be abnormal in various mouse models of RTT 35 , 36 and that we have used for previous studies. 33 , 37 , 38 Given the prior observation that Mecp2 R133C mice exhibited milder severity compared to Mecp2 null mice, 22 these studies used 6–12 week old male and 20–26 week old female mice. This battery tested for the following behaviors: Acoustic startle response, motor coordination, spontaneous locomotor activity, anxiety, social behavior and social recognition/preference, contextual fear conditioning, and breathing abnormalities. As assessments of sensorimotor gating, we evaluated the acoustic startle response as well as PPI. Both sexes of Mecp2 R133C mice exhibited an attenuated acoustic startle response to a 120 dB stimulus (Figure 1C, t‐test, male: t (41) = 5.287, p < 0.0001, female: t (36) = 4.326, p < 0.001). However, no difference in percentage of PPI was observed in male or female Mecp2 R133C mice compared to WT littermates (Supplementary Figure 2A, ANOVA, male: F (1, 41) = 1.226, p > 0.05, female: F (1, 35) = 1.275, p > 0.05). These data suggest that, while basal startle reactivity is impaired, Mecp2 R133C mice display normal sensorimotor gating.
We next measured performance in an accelerated rotarod to determine motor dysfunction. Both sexes of Mecp2 R133C mice displayed decreased average latency to fall, indicative of attenuated motor coordination (Figure 1D, t‐test, male: t (47) = 8.055, p < 0.0001, female: t (60) = 6.345, p < 0.0001). Given this deficit, we assessed spontaneous locomotor activity in an open field, as gross motor deficits could impact more subtle phenotypic measures of anxiety, cognition, and sociability. We observed no locomotor activity changes in Mecp2 R133C mice compared to WT littermates (Figure 1E, t‐test, male: t (40) = 0.7135, p > 0.05, female: t (60) = 0.8408, p > 0.05), suggesting that the rotarod deficit is specific to motor coordination and not gross motor function. We then utilized an elevated zero maze to evaluate anxiety‐related behavior. Although male Mecp2 R133C/y mice exhibited decreased total distance traveled (Supplementary Figure 2B, t‐test, male: t (35) = 2.434, p < 0.05, female: t (60) = 1.692, p > 0.05), both male and female Mecp2 R133C mice spent more time in the open arms compared to the WT littermates, indicative of decreased anxiety behavior (Figure 1F, t‐test, male: t (35) = 6.785, p < 0.0001, female: t (60) = 3.821, p < 0.001).
To evaluate social behavior as well as learning and memory phenotypes, we utilized a three‐chamber discrimination task. In this assay, both Mecp2 R133C/+ and Mecp2 R133C/y mice were comparable to sex‐matched WT littermates, spending more time with the stranger mouse (“Stranger 1”) over an inanimate object (in this case, an empty cup) (Supplementary Figure 2C, ANOVA, male: F (2, 70) = 257.0, p < 0.0001, female: F (2, 96) = 179.9, p < 0.0001), which suggests normal sociability. However, when presented with a novel mouse (“novel”) in addition to the familiar mouse (“familiar”) from the sociability phase, both sexes of Mecp2 R133C mice failed to demonstrate the same preference for the novel stranger as littermates, spending equal time with both mice (Figure 2A, ANOVA, male: F (2, 68) = 108.8, p < 0.0001 (Mecp2 +/y ), p > 0.05 (Mecp2 R133C/y ), female: F (2, 96) = 135.3, p < 0.05 (Mecp2 +/+ ), p > 0.05 (Mecp2 R133C/+ )). This result is indicative of a deficit in social preference and/or impaired social memory.
FIGURE 2.
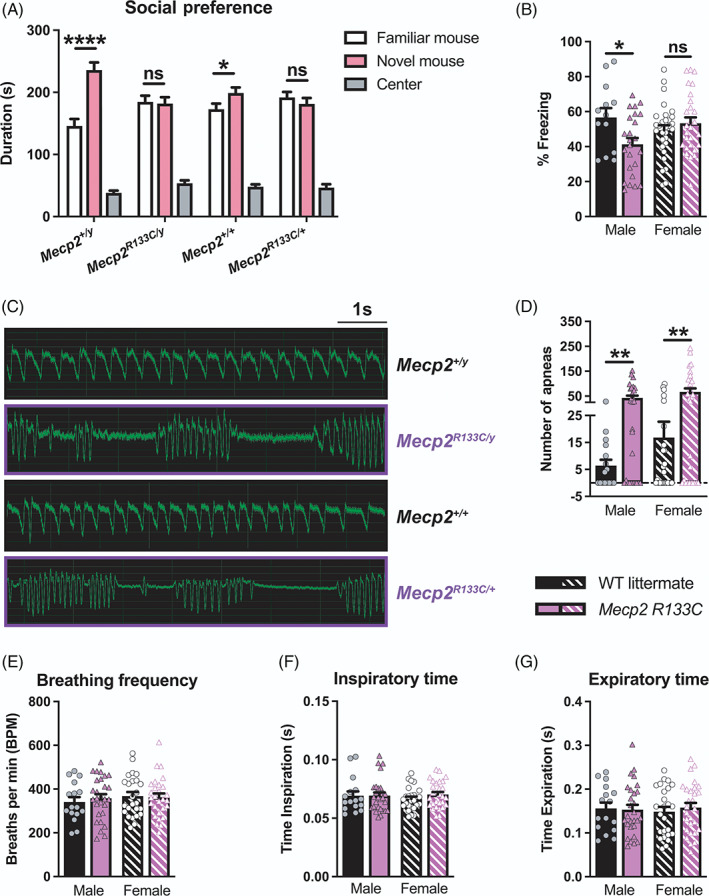
Male and female Mecp2 R133C mice exhibit differential phenotypes in social preference, contextual fear conditioning, and respiratory function. (A) Contrary to wild‐type (WT) littermates that spend more time with novel (pink) than familiar (white) mice in a three‐chamber social preference task, Mecp2 R133C mice did not show preference for the novel mouse. (B) Only male Mecp2 R133C/y displayed decreased percent freezing in contextual fear conditioning 24 h after training. (C,D) Increased apneas were observed in Mecp2 R133C mice, both quantitatively and qualitatively as shown in representative whole‐body plethysmography traces. (E–G) Mecp2 R133C mice exhibited normal respiratory parameters for breathing frequency, inspiratory time and expiratory time. n = 13–29 per genotype in males, n = 21–30 per genotype in females. Two‐way analysis of variance (ANOVA) with t‐test post‐hoc, or unpaired t‐test. *within‐genotype comparison. ns (not significant), *p < 0.05, **p < 0.01, ****p < 0.0001. WT = filled or patterned black bars. Mecp2 R133C = filled or patterned purple bars. Male = filled, closed. Female = patterned, open
We further assessed learning and memory using a contextual fear conditioning test, which measures freezing to a previously aversive stimulus as a proxy of memory. On Day 1, the mice were trained to associate their environment with a mild foot shock. Irrespective of genotype, male and female mice responded to the aversive stimulus (Supplementary Figure 2D, ANOVA, male: F (2, 70) = 34.37, p < 0.0001, female: F (2, 110) = 138.8, p < 0.0001), suggesting normal sensory and short‐term memory acquisition. To evaluate the long‐term memory component, mice were placed back in the same context 24 h after shock administration (training day), and only male Mecp2 R133C/y mice exhibited decreased freezing behavior (Figure 2B, t‐test, male: t (35) = 2.465, p < 0.05, female: t (55) = 0.9306, p > 0.05). In contrast, female Mecp2 R133C/+ mice showed contextual freezing at levels comparable to littermate controls.
Lastly, we investigated the presence of breathing abnormalities, particularly apneas, which are observed in RTT mouse models and patients. Apneas, characterized as “breath‐holding” or hypoventilation, are preceded by periods of hyperventilation 39 and can be quantified using whole‐body plethysmography. We observed that both male and female Mecp2 R133C mice exhibited apneas (Figure 2C–D, t‐test, male: t (42) = 3.067, p < 0.01, female: t (51) = 3.108, p < 0.01) without significant changes in other breathing parameters such as frequency and times of inspiration or expiration (Figure 2E–G, t‐test, all p > 0.05, male: t (42) = 0.5755 [breathing frequency], t (42) = 0.04122 [inspiration time], t (42) = 0.1622 [expiration time], female: t (51) = 0.1999 [breathing frequency], t (51) = 1.253 [inspiration time], t (51) = 0.5968 [expiration time]).
3.2. MeCP2 protein expression in Mecp2 R133C mice with and without MECP2 transgene is brain region‐specific
The milder phenotypes in Mecp2 R133C mice and RTT patients have been linked to some preserved MeCP2 expression and function, as had been previously characterized both in vitro and ex vivo. 22 , 31 , 40 Therefore, we hypothesized that the mutant MeCP2 protein is stably expressed in Mecp2 R133C mice. Using Western blotting, we detected both WT MeCP2 and GFP‐tagged R133C MeCP2 (Supplementary Figure 3A‐B). Interestingly, in male Mecp2 R133C/y mice, the total level of MeCP2 protein was dependent on the brain region, with unchanged expression in the cortex and cerebellum, but decreased in the hippocampus compared to WT littermates (Figure 3A, t‐test, t (8) = 0.4178, p > 0.05 [cortex], t (9) = 2.330, p > 0.05 [cerebellum], t (10) = 2.966, p < 0.05 [hippocampus]). In female Mecp2 R133C/+ mice, we quantified WT and mutant MeCP2, and observed that total MeCP2 expression was similar to WT littermates regardless of brain region (Figure 3B, t‐test, all p > 0.05, t (10) = 1.456 [cortex], t (9) = 0.1474 (cerebellum), t (10) = 1.700 [hippocampus]). Moreover, by comparing the relative expression of these two forms of MeCP2 in female mice, we also observed variability in heterogeneity both between samples and across brain regions, with some mice expressing more of the R133C mutant than WT MeCP2, more of WT MeCP2 than the R133C mutant protein, or equal amounts of both proteins (Supplementary Figure 4).
FIGURE 3.
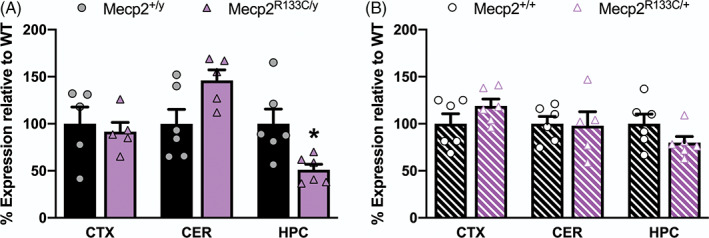
Basal MeCP2 protein expression in Mecp2 R133C mice varies by brain region in Mecp2 R133C/y mice and is unchanged in Mecp2 R133C/+ mice. (A) Total MeCP2 protein is unchanged in the cortex and cerebellum of Mecp2 R133C/y mice but is decreased in the hippocampus. (B) Compared to wild‐type (WT) littermates, Mecp2 R133C/+ mice express normal total MeCP2 levels in the cortex (CTX), cerebellum (CER), and hippocampus (HPC). Total expression is a sum of WT and R133C mutant MeCP2 levels. n = 5–6 per genotype. Unpaired t‐test. *p < 0.05. WT = filled or patterned black bars. Mecp2 R133C = filled or patterned purple bars. Male = filled, closed. Female = patterned, open
We posited that introduction of a WT human MECP2 transgene would further increase protein expression in Mecp2 R133C animals and might elicit unique effects in male versus female mice. To address our hypothesis, we took advantage of a well‐characterized MDS mouse model, MECP2 Tg1/o , 10 which expresses a WT human MECP2 transgene, and have now been bred to the same C57Bl6 congenic background as the R133C mutation. 27 Similar to the approach in Reference 27, we then bred MDS mice with Mecp2 R133C mice to genetically introduce the human MECP2 transgene (Supplementary Figure 5). This breeding strategy led to our experimental mice, male MECP2 Tg1/o ; Mecp2 R133C/y and female MECP2 Tg1/o ; Mecp2 R133C/+ animals, which we compared to the following controls: Mecp2 +/y or Mecp2 +/+ , MECP2 Tg1/o and Mecp2 R133C/y or Mecp2 R133C/+ . Again, we assessed MeCP2 protein expression in the cortex, hippocampus and cerebellum from these mice (representative western blot images in Supplementary Figure 6A‐B). Compared to WT littermates, total MeCP2 protein was increased in MDS mice, MECP2 Tg1/o , regardless of sex and brain region (Figure 4, ANOVA, overall p‐value indicated, male: F (3, 18) = 7.584, p < 0.01 [cortex], F (3, 19) = 7.894, p < 0.01 [cerebellum], F (3, 20) = 3.654, p < 0.05 [hippocampus], female: F (3, 20) = 37.58, p < 0.0001 [cortex], F (3, 19) = 8.113, p < 0.01 [cerebellum], F (3, 20) = 21.24, p < 0.0001 [hippocampus]). A similar observation was seen in the cortex and cerebellum of MECP2 Tg1/o ; Mecp2 R133C/y and MECP2 Tg1/o ; Mecp2 R133C/+ mice, which was not surprising given the stable expression in the Mecp2 R133C mice alone. Interestingly, hippocampal MeCP2 expression in female MECP2 Tg1/o ; Mecp2 R133C/+ mice (Figure 4F) was comparable to that of WT littermates despite Mecp2 R133C/+ mice expressing MeCP2 protein at WT levels. Nonetheless, regardless of brain region, addition of the MECP2 transgene increased WT MeCP2 levels in Mecp2 R133C/+ mice as indicated by a rightward shift in the expression of the WT protein in MECP2 Tg1/o ; Mecp2 R133C/+ mice compared to that in Mecp2 R133C/+ mice (Supplementary Figure 7). Additionally, since MeCP2 protein expression was similar between MECP2 Tg1/o and MECP2 Tg1/o ; Mecp2 R133C/y or MECP2 Tg1/o ; Mecp2 R133C/+ mice in most brain areas, we posited that the introduction of an MECP2 transgene in Mecp2 R133C mice could cause the development of MDS‐like phenotypes.
FIGURE 4.
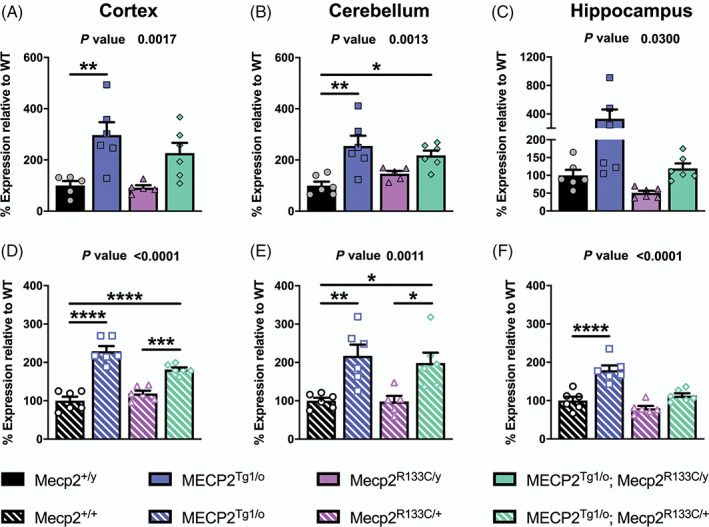
Impact of MECP2 transgene on MeCP2 protein expression in Mecp2 R133C mice. (A–F) Compared to wild‐type (WT) littermates, male and female MECP2 Tg1/o mice demonstrate increased MeCP2 expression in the cortex, cerebellum and hippocampus. Addition of the MECP2 transgene in Mecp2 R133C/y and Mecp2 R133C/+ mice increased total MeCP2 protein in the cortex and cerebellum, but not in the hippocampus. Total expression is a sum of WT and R133C mutant MeCP2 levels. n = 5–6 per genotype. One‐way analysis of variance (ANOVA) with Tukey's post‐hoc test. Overall p‐value indicated at the top of each graph. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. WT = black bars or circles. MECP2 Tg1/o = blue bars or squares. Mecp2 R133C = purple bars or triangles. MECP2 Tg1/o ; Mecp2 R133C = green bars or diamonds. Male = filled or closed. Female = patterned or open
3.3. Phenotypic reversal in male Mecp2R133C /y mice with a WT MECP2 transgene
We first investigated the phenotypic consequences of expressing a WT MECP2 allele in the male global mutant R133C context mice by conducting the aforementioned behavioral battery. We observed reversal of Mecp2 R133C/y ‐associated phenotypes, including the weight deficit and hindlimb clasping phenotype, at all ages (Figure 5A,B, Supplementary Figure 8A, p‐value indicated for MECP2 Tg1/o ; Mecp2 R133C/y vs. Mecp2 R133C/y comparison, [weight] mixed‐effects analysis, F (3, 99) = 53.56, p < 0.0001, [hindlimb clasping] ANOVA, F (3, 95) = 58.71, p < 0.0001 [overall], F (3, 95) = 58.71, p < 0.0001 [at each age range]). Moreover, the addition of WT MECP2 in Mecp2 R133C/y mice reversed acoustic startle response deficiency (Figure 5C, ANOVA, F (3, 89) = 14.05, p < 0.0001 [MECP2 Tg1/o ; Mecp2 R133C/y vs. Mecp2 R133C/y ]), abnormal social preference (Figure 5D, ANOVA, F (2, 160) = 200.9, p < 0.001 [MECP2 Tg1/o ; Mecp2 R133C/y familiar mouse vs. novel mouse]), and increased apneas (Figure 5E, ANOVA, F (3, 93) = 10.56, p < 0.001 [MECP2 Tg1/o ; Mecp2 R133C/y vs. Mecp2 R133C/y ]). The effect on respiratory function was restricted to the apnea phenotype as no changes were observed in breathing frequency as well as inspiratory and expiratory times (Supplementary Figure 8B–D, ANOVA, all p > 0.05, F (3, 93) = 0.8194 [breathing frequency], F (3, 93) = 0.3889 [inspiratory time], F (3, 93) = 0.6070 [expiratory time]).
FIGURE 5.
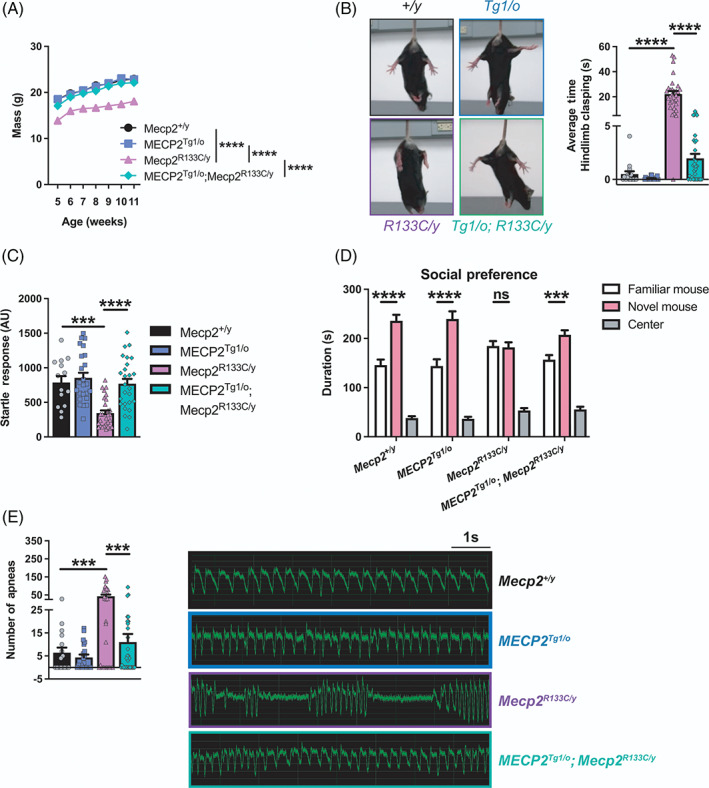
Wild‐type (WT) MECP2 corrects abnormal behaviors observed in Mecp2 R133C/y mice. (A–E) Weight deficit (n = 11–32 per genotype) (A), hindlimb clasping (n = 16–31 per genotype) (B), attenuated acoustic startle response (14–29 per genotype) (C), lack of social preference (n = 15–30 per genotype) (D), and increased apneas (n = 16–32 per genotype) (E) in Mecp2 R133C/y mice were normalized in MECP2 Tg1/o ; Mecp2 R133C/y mice. Male MECP2 Tg1/o mice also exhibited normal behavior in these assays when compared to WT littermates. Representative hindlimb clasping and apnea traces are shown. Mixed‐effects analysis, or one‐ or two‐way analysis of variance (ANOVA) with Tukey's or t‐test post‐hoc. ns (not significant), **p < 0.01, ***p < 0.001, ****p < 0.0001. WT = filled black bars or circles. MECP2 Tg1/o = filled blue bars or squares. Mecp2 R133C/y = filled purple bars or triangles. MECP2 Tg1/o ; Mecp2 R133C/y = filled green bars or diamonds
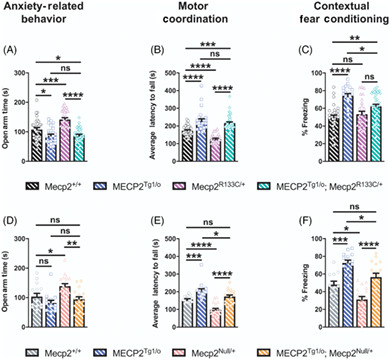
When comparing the phenotype of RTT and MDS mice, several phenotypes consistently present in a bi‐directional manner. These phenotypes include anxiety, motor coordination and associative learning and memory in the form of contextual fear freezing. 10 , 41 , 42 , 43 We observed the bi‐directionality of these phenotypes, in which Mecp2 R133C/y mice exhibited attenuated anxiety, motor coordination and contextual freezing, whereas MECP2 Tg1/o mice displayed contrasting phenotypes of increased anxiety, and abnormally enhanced performance in the accelerated rotarod and contextual fear conditioning tasks (Figure 6A–C, ANOVA, F (3, 83) = 43.22, p < 0.01 [Mecp2 +/y vs. MECP2 Tg1/o ], p < 0.0001 [Mecp2 +/y vs. Mecp2 R133C/y ] [anxiety], F (3, 99) = 49.10, p < 0.0001 [Mecp2 +/y vs. MECP2 Tg1/o and Mecp2 +/y vs. Mecp2 R133C/y ] [motor coordination], F (3, 80) = 13.69, p < 0.05 [Mecp2 +/y vs. MECP2 Tg1/o and Mecp2 +/y vs. Mecp2 R133C/y ] [contextual fear conditioning]). Introduction of an MECP2 transgene in the Mecp2 R133C/y mice reversed these deficits to WT levels, which further established phenotypic reversal in male Mecp2 R133C/y mice (Figure 6A–C, ANOVA, p‐valued indicated for MECP2 Tg1/o ; Mecp2 R133C/y vs. Mecp2 R133C/y comparison, p < 0.0001 [anxiety], p < 0.0001 [motor coordination], p < 0.01 [contextual fear conditioning]).
FIGURE 6.
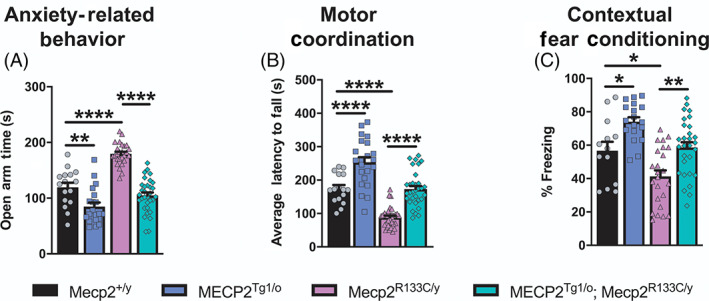
Bi‐directionally affected phenotypes are reversed in Mecp2 R133C/y mice after expression of an MECP2 transgene. (A–C) Opposing phenotypes were observed in MECP2 Tg1/o and Mecp2 R133C/y mice in anxiety‐related behavior (n = 16–32 per genotype) (A), motor coordination (n = 16–33 per genotype) (B), contextual fear learning and memory (n = 13–29 per genotype) (C). Introducing the MECP2 transgene in Mecp2 R133C/y mice normalized these phenotypes to wild‐type (WT) levels. One‐way analysis of variance (ANOVA) with Tukey's post‐hoc test. *p < 0.05, **p < 0.01, ****p < 0.0001. WT = filled black bars or circles. MECP2 Tg1/o = filled blue bars or squares. Mecp2 R133C/y = filled purple bars or triangles. MECP2 Tg1/o ; Mecp2 R133C/y = filled green bars or diamonds
3.4. Phenotype‐specific effect in female Mecp2R133C /+ mice with WT MECP2 transgene
The beneficial effects observed with the introduction of an MECP2 transgene in male Mecp2 R133C/y mice were anticipated, as global MeCP2 disruption would be predicted to result in increased severity, and consequently, a larger window before MDS‐like phenotypes are observed. We next investigated if this reversal would also hold true in female Mecp2 R133C/+ mice as they (1) are mosaic for mutant and WT MeCP2, (2) exhibit milder phenotypes than male mice, and (3) show normal baseline MeCP2 protein levels in the cortex, hippocampus and cerebellum (Figure 3B). Furthermore, the MeCP2 protein expression in MECP2 Tg1/o ; Mecp2 R133C/+ mice was increased and comparable to that in MECP2 Tg1/o mice in most brain regions (Figure 4D–F).
General physical characteristics, specifically weight and hindlimb clasping, were similar in MECP2 Tg1/o ; Mecp2 R133C/+ mice and WT littermates across age (Figure 7A,B, Supplementary Figure 8E, p‐value indicated for MECP2 Tg1/o ; Mecp2 R133C/+ vs. Mecp2 R133C/+ comparison, (weight) mixed‐effects analysis, F (3, 135) = 11.36, p < 0.01 [5, 10, and 25 weeks old], p < 0.0001 [15 and 20 weeks old], [hindlimb clasping] ANOVA, F (3, 125) = 21.23, p < 0.0001 [overall], F (3,125) = 21.54, p < 0.001 [22–25 weeks old], p < 0.0001 [20–23 and 24–28 weeks old]), suggesting that increasing MECP2 dosage corrected weight deficits and the clasping phenotype. This reversal effect with the MECP2 transgene was also observed in the attenuated acoustic startle response and lack of social preference in Mecp2 R133C/+ mice (Figure 7C,D, ANOVA, F (3, 74) = 9.971, p < 0.0001 [MECP2 Tg1/o ; Mecp2 R133C/+ vs. Mecp2 R133C/+ ] [acoustic startle response], F (2, 204) = 239.6, p < 0.01, [MECP2 Tg1/o ; Mecp2 R133C/+ familiar mouse vs. novel mouse] [social preference]). As illustrated in the representative traces and number of apneas, MECP2 Tg1/o ; Mecp2 R133C/+ mice showed comparable apneas to WT littermates, again without changes in other breathing parameters (Figure 7E, Supplementary Figure 8F‐H, ANOVA, p‐value indicated for MECP2 Tg1/o ; Mecp2 R133C/+ vs. Mecp2 R133C/+ comparison, F (3, 107) = 12.67, p < 0.0001 [apnea], F (3, 107) = 1.007, p > 0.05 [breathing frequency], F (3, 107) = 1.353, p > 0.05 [inspiratory time], F (3, 107) = 0.3095, p > 0.05 [expiration time]).
FIGURE 7.
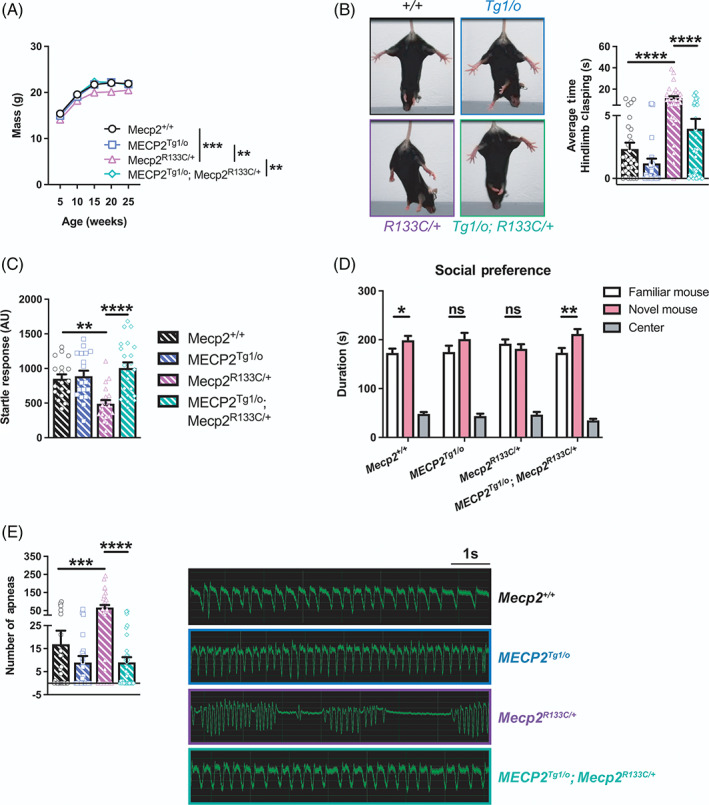
Additional wild‐type (WT) MECP2 normalizes a subset of phenotypes in Mecp2 R133C/+ mice. (A–E) MECP2 Tg1/o ; Mecp2 R133C/+ mice exhibited similar weight (n = 14–26 per genotype) (A), hindlimb clasping behavior (n = 26–40 per genotype) (B), acoustic startle response (n = 18–22 per genotype) (C), social preference (n = 25–31 per genotype) (D), and number of apneas (n = 25–33 per genotype) (E) as WT littermates, illustrating a reversal of abnormal phenotypes observed in Mecp2 R133C/+ mice. Representative hindlimb clasping and apnea traces are shown. Mixed‐effects analysis, or one‐ or two‐way analysis of variance (ANOVA) with Tukey's or t‐test post‐hoc. ns (not significant), *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. WT = patterned black bars or open circles. MECP2 Tg1/o = patterned blue bars or open squares. Mecp2 R133C/+ = patterned purple bars or open triangles. MECP2 Tg1/o ; Mecp2 R133C/+ = patterned green bars or open diamonds
We next evaluated the performance of MECP2 Tg1/o ; Mecp2 R133C/+ mice in the bi‐directionally affected symptom domains of anxiety, motor coordination and associative learning and memory. Similar to their male counterparts, female MECP2 Tg1/o mice displayed increased anxiety, enhanced performance in the accelerated rotarod, and an excessive contextual fear conditioning response (Figure 8A–C, ANOVA, p‐value indicated for Mecp2 +/+ vs. MECP2 Tg1/o comparison, F (3, 123) = 21.46, p < 0.05 [anxiety], F (3, 125) = 38.86, p < 0.0001 [motor coordination], F (3, 111) = 12.63, p < 0.0001 [contextual fear conditioning]). Notably, these phenotypes were also observed in MECP2 Tg1/o ; Mecp2 R133C/+ mice, which although not as robust as MECP2 Tg1/o mice alone, were significantly different from WT littermates (ANOVA, p‐value indicated for Mecp2 +/+ vs. MECP2 Tg1/o ; Mecp2 R133C/+ comparison, F (3, 123) = 21.46, p < 0.05 [anxiety], F (3, 125) = 38.86, p < 0.001 [motor coordination], F (3, 111) = 12.63, p < 0.01 [contextual fear conditioning]). To confirm that these MDS‐like phenotypes were linked to preserved function of the R133C allele, we applied a similar approach in introducing the MECP2 transgene in Mecp2 Null/+ mice. In contrast to the Mecp2 R133C/+ mice, MECP2 Tg1/o ; Mecp2 Null/+ animals were not significantly different from their WT counterparts in all three assays (Figure 8D–F, all p > 0.05 for Mecp2 +/+ vs. MECP2 Tg1/o ; Mecp2 Null/+ comparison, F (3, 60) = 7.374 [anxiety], F (3, 67) = 30.82 [motor coordination], F (3, 58) = 21.01 [contextual fear conditioning]). Aside from the anxiety‐related behavior, wherein MECP2 Tg1/o mice did not spend less time in the open arms, MECP2 Tg1/o ; Mecp2 Null/+ were significantly different from MECP2 Tg1/o mice in motor coordination and contextual freezing (ANOVA, p‐value indicated for MECP2 Tg1/o vs. MECP2 Tg1/o ; Mecp2 R133C/+ comparison, F (3, 60) = 7.374, p > 0.05 [anxiety], F (3, 67) = 30.82, p < 0.05 [motor coordination], F (3, 58) = 21.01, p < 0.05 [contextual fear conditioning]). Altogether, these results suggest that expression of the MECP2 transgene in the clinically relevant Mecp2 R133C/+ genetic background may result in “MDS‐like” phenotypes, and potentially points to a narrower therapeutic window for MeCP2‐targeted therapeutics when used with mild hypomorphic mutations.
FIGURE 8.
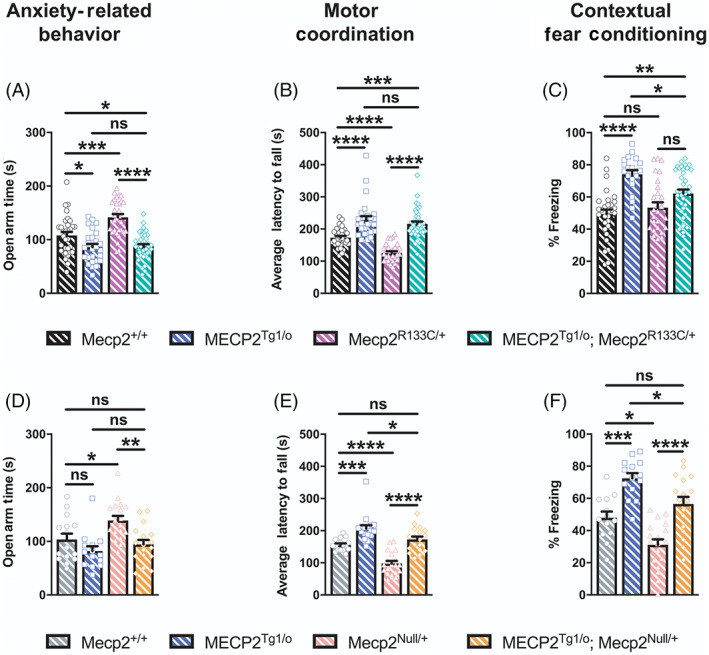
Mecp2 R133C/+ mice with additional wild‐type (WT) MECP2 develop MDS‐like phenotypes in anxiety, motor and cognitive assays. MECP2 Tg1/o and Mecp2 R133C/+ or Mecp2 Null/+ mice exhibited contrasting phenotypes in anxiety‐related behavior (A, D), motor coordination (B, E), and contextual fear learning and memory (D, F). (A–C) MECP2 Tg1/o ; Mecp2 R133C/+ mice phenocopied MECP2 Tg1/o mice and significantly differed from WT littermates, with enhanced anxiety (A), motor coordination (B) and contextual freezing (C). n = 21–40 per genotype. (D–F) In contrast, MECP2 Tg1/o ; Mecp2 Null/+ mice were indistinguishable from WT littermates and/or significantly different from MECP2 Tg1/o . n = 13–19 per genotype. One‐way analysis of variance (ANOVA) with Tukey's post‐hoc test. ns (not significant), *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. WT = patterned black or gray bars or open circles. MECP2 Tg1/o = patterned blue bars or open squares. Mecp2 R133C/+ = patterned purple bars or open triangles. MECP2 Tg1/o ; Mecp2 R133C/+ = patterned green bars or open diamonds. Mecp2 Null/+ = patterned pink bars or open triangles. MECP2 Tg1/o ; Mecp2 Null/+ = patterned orange bars or open diamonds
4. DISCUSSION
In the past decade, gene therapy research for RTT has significantly increased, emerging as a feasible and exciting treatment option. 4 , 5 , 6 , 7 , 8 , 9 Most recently, a report described the efficacy and safety of a self‐complementary AAV9 (scAAV9) encoding for the human MECP2 in male Mecp2 null mice and non‐human primate models. 7 While encouraging, the majority of preclinical studies regarding gene therapy for RTT have not examined safety in the context of mosaic females (with the notable exception of 9 and 44 ). Furthermore, these preclinical studies do not address the impact of hypomorphic MeCP2 mutations on therapeutic index, except for the study by, 23 which illustrated the effects of increasing MeCP2 expression in male mice that were hemizygous for the missense mutation R306C. It is widely accepted that many missense MECP2 mutations are correlative with milder clinical severity as a result of preserved protein function and stability. In this study, we sought to complement existing efforts to generate MeCP2‐based therapeutics by addressing aspects of this diverse clinical landscape. Specifically, we asked whether a 1x increase in MeCP2 dosage from conception would rescue phenotypes in Mecp2 R133C/y and Mecp2 R133C/+ mice without evoking MDS‐like adverse effects.
The R133C mutation is one of most common mutations in patients but it also leads to a mild presentation of the disease, similar to R306C. 19 , 21 , 29 Here, we behaviorally validated the Mecp2 R133C mice as a hypomorphic mutant mouse model of RTT. Both male and female Mecp2 R133C mice exhibited RTT‐like phenotypes, with the exception of a normal contextual fear response in female Mecp2 R133C/+ mice, which differs from what is commonly observed in Mecp2 Null/+ animals. 38 , 42 , 45 , 46 , 47 This further supports the clinical observation that the R133C mutation is not a functional null allele, but rather is hypomorphic and conveys a milder phenotype. Additionally, our data are in agreement with previous reports of Mecp2 R133C and Mecp2 R306C mice, as well as constitutive hypomorphic Mecp2 flox/y mice, which have a 50% reduction in MeCP2 expression. 22 , 23 , 36 , 48 , 49 The lack of abnormal PPI contrasted from previous findings, wherein enhanced PPI was observed in Mecp2 tm1.1Jae heterozygous females, Mecp2 flox/y mice, and glutamatergic‐ or GABAergic‐specific Mecp2 knockout mice. 36 , 48 , 50 , 51 However, compared to these studies, which utilized older mice of different background strains, we evaluated PPI at 6–7 weeks of age for both male and female Mecp2 R133C mice to account for the hearing loss that mice of the C57Bl6 background strain begin to experience at 8 weeks of age. Therefore, it is possible that older Mecp2 R133C mice could exhibit altered PPI.
As has been previously posited, 22 the milder phenotypes seen in the Mecp2 R133C animals are likely attributed to partial functional loss of MeCP2. This hypothesis was supported by our protein expression data demonstrating that expression of the R133C allele was comparable to WT MeCP2 in two brain regions, cortex and cerebellum, in these mice. Interestingly, we observed a decrease in hippocampal MeCP2 protein expression in male Mecp2 R133C/y mice that was not observed in female Mecp2 R133C/+ mice. While mosaicism would be predicted to dilute the mutant allele's aggregate impact on hippocampal MeCP2 expression, this sex‐specific disconnect could explain the associative learning and memory phenotype that was distinct between the male and female Mecp2 R133C animals, where only male mice exhibited a contextual fear conditioning deficit.
To model an optimal 1x rescue of MeCP2 protein, we genetically introduced a human MECP2 transgene into Mecp2 R133C mice. We demonstrated that all of the abnormal phenotypes observed in male Mecp2 R133C/y mice were reversed to that of wild‐type behavior when the MECP2 transgene was expressed, which is highly encouraging. These reversal effects support the conclusion that global MeCP2 mutation or loss broadens the range in which MeCP2 can be increased before MDS‐like phenotypes emerge. Our results are similar to previous studies illustrating that genetically increasing MeCP2 expression in male Mecp2 R306C mice corrected several RTT‐like phenotypes such as deficits in motor function and contextual fear learning and memory. 23 In addition, other MeCP2‐targeting genetic strategies, such as postnatal reactivation of Mecp2, neuronal‐specific germline introduction of Tau‐Mecp2, and viral delivery of human MECP2 in Mecp2 null mice improved phenotypes. 2 , 3 , 4 , 5 , 6 , 7 , 9 , 52 Collectively, these findings support the potential use of MeCP2‐targeted approaches for male RTT patients with the R133C mutation.
To address the impact of a hypomorphic mutation in a mosaic context, which would occur in female patients, we evaluated the consequences of this genetic strategy in female Mecp2 R133C/+ mice. Encouragingly, the majority of the RTT‐like phenotypes in Mecp2 R133C/+ mice were normalized with the additional MECP2 transgene. These findings are in agreement with previous reports of phenotypic reversal with systemic delivery of full‐length MeCP2 in female Mecp2 Null/+ mice, increased MeCP2 expression in Mecp2 T158M mice or germline introduction of human MECP2 in mice harboring the R255X mutation. 9 , 26 , 27 , 44 While increased expression of MECP2 was able to correct a number of deficits in female Mecp2 R133C mice, three phenotypes, anxiety, motor coordination and contextual fear conditioning, were mildly yet significantly “over‐corrected”, and the resulting phenotypes mirrored those of MDS model animals. These data are different from a previous report where genetically increasing MeCP2 protein expression in female Mecp2 T158M or R255X mice reversed motor coordination and contextual freezing to WT behavior levels. 26 , 27 Interestingly, despite lacking an anxiolytic phenotype, the female Mecp2 R255X mice were unaffected with addition of the MECP2 transgene. However, the R255X mutation is different from the R133C mutation in that no protein is produced in Mecp2 R255X mice. 27 Similarly, the T158M mutation decreases protein expression and renders the protein nonfunctional, causing a more severe form of the disorder, similar to Mecp2 null animals. 22 Furthermore, the motor coordination phenotype observed in the Mecp2 R133C/+ mice with the MECP2 transgene was different from a previous study showing that virally‐delivered MeCP2 post‐symptom onset improved attenuated motor behavior in Mecp2 Null/+ mice, 9 as well as a recent report that pre‐symptom onset delivery of viral MeCP2 did not affect motor coordination in Mecp2 Null/+ mice. 44 Nonetheless, these previous studies are all in agreement with our results showing that, in Mecp2 Null/+ mice, addition of the MECP2 transgene reversed attenuated anxiety and performance in motor coordination and contextual freezing tasks to levels seen in WT littermates. Altogether, our data suggest that the development of MDS‐like phenotypes is due to the impact that the R133C MeCP2 mutation has on therapeutic index.
We believe that this study provides important information regarding the feasibility of increasing MeCP2 expression in mosaic female RTT model mice engineered to have a common hypomorphic mutation in MeCP2. Our findings illustrate the promise of MeCP2‐targeted therapies for male and female RTT patients with an R133C MECP2 mutation. However, our data suggest that careful consideration may be required to avoid adverse effects when considering dosage of prospective MeCP2‐based therapeutics in female R133C RTT patients, as preserved function may result in a narrowing of the therapeutic window. One caveat to our studies is that our genetic approach increases MeCP2 levels from conception, which is different from current MeCP2‐targeted treatment strategies that deliver viral MeCP2 after disease onset. It has been hypothesized that post‐mitotic neurons may be more forgiving with regard to the amount of MeCP2 that can be delivered before adverse effects present. It is unknown whether this same caveat can also be applied to efficacy, where post‐mitotic neurons could be less responsive to a 1x increase in MeCP2 protein. Future experiments will be required to determine if either scenario is true, and, if so, what impact this has on therapeutic development. In summary, this study contributes valuable safety data to ongoing efforts to develop MeCP2‐targeted therapeutics that are both safe and effective. Furthermore, these studies highlight the need for similar investigations in other pathological hypomorphic mutations of RTT, and their inclusion in preclinical development efforts.
CONFLICT OF INTEREST
The authors have declared no conflicts of interest exist.
ETHICS STATEMENT
These studies have been approved by Vanderbilt's IACUC committee. Additionally, all authors have approved their authorship and contributions.
AUTHOR CONTRIBUTIONS
Sheryl Anne D. Vermudez, Rocco G. Gogliotti, and Colleen M. Niswender designed the experiments. Sheryl Anne D. Vermudez, Aditi Buch, Bright Arthur, Clarissa Morales, Yuta Moxley, and Hemangi Rajpal performed the behavioral assays and subsequent analyses. Sheryl Anne D. Vermudez conducted the molecular experiments and data analyses. Sheryl Anne D. Vermudez, Rocco G. Gogliotti, and Colleen M. Niswender wrote and edited the manuscript with input from all authors.
Supporting information
Appendix S1: Supplementary Information
ACKNOWLEDGMENTS
We thank Dr. Jeffrey Neul at the Vanderbilt Kennedy Center for providing the MECP2 Tg1/o (C57Bl6 background) mice, Dr. John Allison at the Vanderbilt Mouse Neurobehavioral Core for his assistance in the execution of the behavioral assays, and the Vanderbilt Medical Center Molecular Cell Biology Resource (MCBR) Core for providing the immunoblot imaging system. We also extend our gratitude to Drs. Jeffrey Neul and Lisa Monteggia at Vanderbilt University for providing input throughout the preparation of this manuscript.
Vermudez SAD, Gogliotti RG, Arthur B, et al. Profiling beneficial and potential adverse effects of MeCP2 overexpression in a hypomorphic Rett syndrome mouse model. Genes, Brain and Behavior. 2022;21(1):e12752. 10.1111/gbb.12752
Funding information National Institute of Mental Health, Grant/Award Numbers: F31MH119699, K01MH112983; National Institute of Neurological Disorders and Stroke, Grant/Award Number: R01NS112171; National Institutes of Health, Grant/Award Number: T32 GM007628; Rettsyndrome.org, Grant/Award Number: 3511
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1. Amir RE, van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X‐linked MECP2, encoding methyl‐CpG‐binding protein 2. Nat Genet. 1999;23(2):185‐188. 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 2. Guy J, Gan J, Selfridge J, Cobb S, Bird A. Reversal of neurological defects in a mouse model of Rett syndrome. Science. 2007;315(5815):1143‐1147. 10.1126/science.1138389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Luikenhuis S, Giacometti E, Beard CF, Jaenisch R. Expression of MeCP2 in postmitotic neurons rescues Rett syndrome in mice. Proc Natl Acad Sci U S A. 2004;101(16):6033‐6038. 10.1073/pnas.0401626101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Gadalla KKE, Bailey MES, Spike RC, et al. Improved survival and reduced phenotypic severity following AAV9/MECP2 gene transfer to neonatal and juvenile male Mecp2 knockout mice. Mol Ther. 2013;21(1):18‐30. 10.1038/mt.2012.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sinnett SE, Hector RD, Gadalla KKE, et al. Improved MECP2 gene therapy extends the survival of MeCP2‐null mice without apparent toxicity after intracisternal delivery. Mol Ther Methods Clin Dev. 2017;5:106‐115. 10.1016/j.omtm.2017.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Gadalla KKE, Vudhironarit T, Hector RD, et al. Development of a novel AAV gene therapy cassette with improved safety features and efficacy in a mouse model of Rett syndrome. Mol Ther Methods Clin Dev. 2017;5:180‐190. 10.1016/j.omtm.2017.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Powers S, Miranda C, Dennys‐Rivers C, et al. Rett syndrome gene therapy improves survival and ameliorates behavioral phenotypes in MeCP2 null (S51.002). Neurology. 2019;92(15). [Google Scholar]
- 8. Tillotson R, Selfridge J, Koerner MV, et al. Radically truncated MeCP2 rescues Rett syndrome‐like neurological defects. Nature. 2017;550(7676):398‐401. 10.1038/nature24058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Garg SK, Lioy DT, Cheval H, et al. Systemic delivery of MeCP2 rescues behavioral and cellular deficits in female mouse models of Rett syndrome. J Neurosci. 2013;33(34):13612‐13620. 10.1523/JNEUROSCI.1854-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Collins AL, Levenson JM, Vilaythong AP, et al. Mild overexpression of MeCP2 causes a progressive neurological disorder in mice. Hum Mol Genet. 2004;13(21):2679‐2689. 10.1093/hmg/ddh282. [DOI] [PubMed] [Google Scholar]
- 11. Na ES, Nelson ED, Adachi M, et al. A mouse model for MeCP2 duplication syndrome: MeCP2 overexpression impairs learning and memory and synaptic transmission. J Neurosci. 2012;32(9):3109‐3117. 10.1523/JNEUROSCI.6000-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Augui S, Nora EP, Heard E. Regulation of X‐chromosome inactivation by the X‐inactivation Centre. Nat Rev Genet. 2011;12(6):429‐442. 10.1038/nrg2987. [DOI] [PubMed] [Google Scholar]
- 13. Zhang Q, Zhao Y, Bao X, et al. Familial cases and male cases with MECP2 mutations. Am J Med Genet B Neuropsychiatr Genet. 2017;174(4):451‐457. 10.1002/ajmg.b.32534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Sirianni N, Naidu S, Pereira J, Pillotto RF, Hoffman EP. Rett syndrome: confirmation of X‐linked dominant inheritance, and localization of the gene to Xq28. Am J Hum Genet. 1998;63(5):1552‐1558. 10.1086/302105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Knudsen GPS, Neilson TCS, Pedersen J, et al. Increased skewing of X chromosome inactivation in Rett syndrome patients and their mothers. Eur J Hum Genet. 2006;14(11):1189‐1194. 10.1038/sj.ejhg.5201682. [DOI] [PubMed] [Google Scholar]
- 16. Ribeiro MC, MacDonald JL. Sex differences in Mecp2‐mutant Rett syndrome model mice and the impact of cellular mosaicism in phenotype development. Brain Res. 1729;2020:146644. 10.1016/j.brainres.2019.146644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Young JI, Zoghbi HY. X‐chromosome inactivation patterns are unbalanced and affect the phenotypic outcome in a mouse model of rett syndrome. Am J Hum Genet. 2004;74(3):511‐520. 10.1086/382228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Braunschweig D, Simcox T, Samaco RC, LaSalle JM. X‐chromosome inactivation ratios affect wild‐type MeCP2 expression within mosaic Rett syndrome and Mecp2−/+ mouse brain. Hum Mol Genet. 2004;13(12):1275‐1286. 10.1093/hmg/ddh142. [DOI] [PubMed] [Google Scholar]
- 19. Neul JL, Fang P, Barrish J, et al. Specific mutations in methyl‐CpG‐binding protein 2 confer different severity in Rett syndrome. Neurology. 2008;70(16):1313‐1321. 10.1212/01.wnl.0000291011.54508.aa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Young D, Bebbington A, de Klerk N, Bower C, Nagarajan L, Leonard H. The relationship between MECP2 mutation type and health status and service use trajectories over time in a Rett syndrome population. Res Autism Spectr Disord. 2011;5(1):442‐449. 10.1016/j.rasd.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cuddapah VA, Pillai RB, Shekar KV, et al. Methyl‐CpG‐binding protein 2 (MECP2) mutation type is associated with disease severity in Rett syndrome. J Med Genet. 2014;51(3):152‐158. 10.1136/jmedgenet-2013-102113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Brown K, Selfridge J, Lagger S, et al. The molecular basis of variable phenotypic severity among common missense mutations causing Rett syndrome. Hum Mol Genet. 2016;25(3):558‐570. 10.1093/hmg/ddv496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Heckman LD, Chahrour MH, Zoghbi HY. Rett‐causing mutations reveal two domains critical for MeCP2 function and for toxicity in MECP2 duplication syndrome mice. Elife. 2014;3:e02676. 10.7554/eLife.02676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Merritt JK, Collins BE, Erickson KR, Dong H, Neul JL. Pharmacological readthrough of R294X Mecp2 in a novel mouse model of Rett syndrome. Hum Mol Genet. 2020;29(5):2461‐2470. 10.1093/hmg/ddaa102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Adegbola AA, Gonzales ML, Chess A, LaSalle JM, Cox GF. A novel hypomorphic MECP2 point mutation is associated with a neuropsychiatric phenotype. Hum Genet. 2009;124(6):615‐623. 10.1007/s00439-008-0585-6. [DOI] [PubMed] [Google Scholar]
- 26. Lamonica JM, Kwon DY, Goffin D, et al. Elevating expression of MeCP2 T158M rescues DNA binding and Rett syndrome‐like phenotypes. J Clin Invest. 2017;127(5):1889‐1904. 10.1172/JCI90967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pitcher MR, Herrera JA, Buffington SA, et al. Rett syndrome like phenotypes in the R255X Mecp2 mutant mouse are rescued by MECP2 transgene. Hum Mol Genet. 2015;24(9):2662‐2672. 10.1093/hmg/ddv030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fyfe S, Cream A, de Klerk N, Christodoulou J, Leonard H. InterRett and RettBASE: international Rett syndrome association databases for Rett syndrome. J Child Neurol. 2003;18(10):709‐713. 10.1177/08830738030180100301. [DOI] [PubMed] [Google Scholar]
- 29. Leonard H, Colvin L, Christodoulou J, et al. Patients with the R133C mutation: is their phenotype different from patients with Rett syndrome with other mutations? J Med Genet. 2003;40(5):e52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bebbington A, Anderson A, Ravine D, et al. Investigating genotype‐phenotype relationships in Rett syndrome using an international data set. Neurology. 2008;70(11):868‐875. 10.1212/01.wnl.0000304752.50773.ec. [DOI] [PubMed] [Google Scholar]
- 31. Ghosh RP, Horowitz‐Scherer RA, Nikitina T, Gierasch LM, Woodcock CL. Rett syndrome‐causing mutations in human MeCP2 result in diverse structural changes that impact folding and DNA interactions. J Biol Chem. 2008;283(29):20523‐20534. 10.1074/jbc.M803021200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Foster DJ, Wilson JM, Remke DH, et al. Antipsychotic‐like effects of M4 positive allosteric modulators are mediated by CB2 receptor‐dependent inhibition of dopamine release. Neuron. 2016;91(6):1244‐1252. 10.1016/j.neuron.2016.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fisher NM, Gogliotti RG, Vermudez SAD, Stansley BJ, Conn PJ, Niswender CM. Genetic reduction or negative modulation of mGlu7 does not impact anxiety and fear learning phenotypes in a mouse model of MECP2 duplication syndrome. ACS Chem Nerosci. 2018;9(9):2210‐2217. 10.1021/acschemneuro.7b00414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cheval H, Guy J, Merusi C, De Sousa D, Selfridge J, Bird A. Postnatal inactivation reveals enhanced requirement for MeCP2 at distinct age windows. Hum Mol Genet. 2012;21(17):3806‐3814. 10.1093/hmg/dds208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li W, Pozzo‐Miller L. Beyond widespread Mecp2 deletions to model Rett syndrome: conditional Spatio‐temporal knockout, single‐point mutations and transgenic rescue mice. Autism Open Access. 2012;2012(suppl 1):5. 10.4172/2165-7890.S1-005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chao H‐T, Chen H, Samaco RC, et al. Dysfunction in GABA signalling mediates autism‐like stereotypies and Rett syndrome phenotypes. Nature. 2010;468(7321):263‐269. 10.1038/nature09582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gogliotti RG, Senter RK, Rook JM, et al. mGlu5 positive allosteric modulation normalizes synaptic plasticity defects and motor phenotypes in a mouse model of Rett syndrome. Hum Mol Genet. 2016;25(10):1990‐2004. 10.1093/hmg/ddw074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gogliotti RG, Senter RK, Fisher NM, et al. mGlu7 potentiation rescues cognitive, social, and respiratory phenotypes in a mouse model of Rett syndrome. Sci Transl Med. 2017;9(403):eaai7459. 10.1126/scitranslmed.aai7459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ramirez J‐M, Ward CS, Neul JL. Breathing challenges in Rett syndrome: lessons learned from humans and animal models. Respir Physiol Neurobiol. 2013;189(2):280‐287. 10.1016/j.resp.2013.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kucukkal TG, Alexov E. Structural, dynamical, and energetical consequences of rett syndrome mutation R133C in mecp2. Comput Math Methods Med. 2015;2015:746157. 10.1155/2015/746157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stearns NA, Schaevitz LR, Bowling H, Nag N, Berger UV, Berger‐Sweeney J. Behavioral and anatomical abnormalities in Mecp2 mutant mice: a model for Rett syndrome. Neuroscience. 2007;146(3):907‐921. 10.1016/j.neuroscience.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 42. Samaco RC, McGraw CM, Ward CS, Sun Y, Neul JL, Zoghbi HY. Female Mecp2(+/−) mice display robust behavioral deficits on two different genetic backgrounds providing a framework for pre‐clinical studies. Hum Mol Genet. 2013;22(1):96‐109. 10.1093/hmg/dds406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Santos M, Silva‐Fernandes A, Oliveira P, Sousa N, Maciel P. Evidence for abnormal early development in a mouse model of Rett syndrome. Genes Brain Behav. 2007;6(3):277‐286. 10.1111/j.1601-183X.2006.00258.x. [DOI] [PubMed] [Google Scholar]
- 44. Matagne V, Borloz E, Ehinger Y, Saidi L, Villard L, Roux J‐C. Severe offtarget effects following intravenous delivery of AAV9‐MECP2 in a female mouse model of Rett syndrome. Neurobiol Dis. 2020;105235. 105235. 10.1016/j.nbd.2020.105235. [DOI] [PubMed] [Google Scholar]
- 45. Gogliotti RG, Fisher NM, Stansley BJ, et al. Total RNA sequencing of rett syndrome autopsy samples identifies the M4 muscarinic receptor as a novel therapeutic target. J Pharmacol Exp Ther. 2018;365(2):291‐300. 10.1124/jpet.117.246991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Moretti P, Levenson JM, Battaglia F, et al. Learning and memory and synaptic plasticity are impaired in a mouse model of Rett syndrome. J Neurosci. 2006;26(1):319‐327. 10.1523/JNEUROSCI.2623-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pelka GJ, Watson CM, Radziewic T, et al. Mecp2 deficiency is associated with learning and cognitive deficits and altered gene activity in the hippocampal region of mice. Brain. 2006;129(pt 4):887‐898. 10.1093/brain/awl022. [DOI] [PubMed] [Google Scholar]
- 48. Samaco RC, Fryer JD, Ren J, et al. A partial loss of function allele of methyl‐CpG‐binding protein 2 predicts a human neurodevelopmental syndrome. Hum Mol Genet. 2008;17(12):1718‐1727. 10.1093/hmg/ddn062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kerr B, Alvarez‐Saavedra M, Sáez MA, Saona A, Young JI. Defective body‐weight regulation, motor control and abnormal social interactions in Mecp2 hypomorphic mice. Hum Mol Genet. 2008;17(12):1707‐1717. 10.1093/hmg/ddn061. [DOI] [PubMed] [Google Scholar]
- 50. Kron M, Howell CJ, Adams IT, et al. Brain activity mapping in Mecp2 mutant mice reveals functional deficits in forebrain circuits, including key nodes in the default mode network, that are reversed with ketamine treatment. J Neurosci. 2012;32(40):13860‐13872. 10.1523/JNEUROSCI.2159-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Meng X, Wang W, Lu H, et al. Manipulations of MeCP2 in glutamatergic neurons their contributions to Rett and other neurological disorders. Elife. 2016;5:e14199. 10.7554/eLife.14199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Robinson L, Guy J, McKay L, et al. Morphological and functional reversal of phenotypes in a mouse model of Rett syndrome. Brain. 2012;135(pt 9):2699‐2710. 10.1093/brain/aws096. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary Information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.