

Abstract
Ruxolitinib is an FDA-approved orally administered Janus kinase (JAK 1/2) inhibitor that reduces cytokine-induced inflammation. As part of a randomized, Phase 2, open label trial, ruxolitinib (10 mg, bid) was administered to HIV+, virologically suppressed individuals (33 men, 7 women) on antiretroviral therapy (ART), for 5 weeks. Study objectives were to assess safety, tolerability, pharmacokinetics (PK), and modulation of ongoing inflammation that persists even with viral suppression. Herein, we report the population PK subsequently determined from this study. Plasma concentrations of ruxolitinib (294 samples) and antiretroviral agents were measured at week 1 (wk1, N = 39 participants) and week 4 or 5 (wk4/5, N = 37). Ruxolitinib PK was adequately described with a 2-compartment model with first-order absorption and elimination with distribution volumes normalized to mean body weight (91.5 kg) and a separate typical CL for participants administered efavirenz (a known CYP3A4 inducer). Participants administered an ART regimen with efavirenz had an elevated typical CL/F versus the integrase inhibitor regimen (INSTI) group (22.5 versus 12.9 L/hr; N = 14 versus 25). Post hoc predicted CL/F were likewise, more variable and higher (p < 0.0001) in those administered efavirenz. There was ~ 25% variation in ruxolitinib plasma exposures between wk1 and wk4/5. ART plasma concentrations resembled those from PK studies without ruxolitinib. Therefore, INSTI based ART regimens may be preferred over efavirenz based regimens when ruxolitinib is administered to HIV+ individuals.
Keywords: ruxolitinib, population PK, NONMEM analysis, antiretroviral agents, HIV, drug-drug interactions
Introduction:
Ruxolitinib is an orally administered Janus kinase (JAK 1/2) inhibitor FDA-approved for treatment of patients with myelofibrosis, including primary myelofibrosis, post-polycythemia vera-myelofibrosis and post-essential thrombocythemia-myelofibrosis, and is administered orally bid.1 A mechanistic and ex vivo study suggested that ruxolitinib could modulate immunological events associated with HIV persistence and disease progression, at clinically observed plasma concentrations (adjusted for plasma binding).2 In the AIDS Clinical Trials Group (ACTG) A5336 Phase 2 randomized, open label trial, ruxolitinib tablets (JAKAFI™, Incyte/Novartis), were administered at a dose equivalent to 10 mg of ruxolitinib free-base bid for 5 weeks to virologically suppressed HIV+ participants on standard ART regimens.3 Study objectives were to assess safety, tolerability, pharmacokinetics (PK), maintenance of virologic suppression, reduction in persistent inflammation despite virologic suppression, and to monitor drug-drug interactions. In the trial, baseline IL-2 levels were normal. Nevertheless, ruxolitinib treatment significantly decreased other markers of immune activation. A comprehensive report on the primary outcomes, namely safety, immunological and virological aspects of the study, are recently published.3 Ruxolitinib is metabolized via the cytochrome P450 enzyme system (primarily CYP3A4). FDA prescribing information for ruxolitinib tablets recommend dose reduction with concurrent use of strong CYP3A4 inhibitors and close monitoring and possible dose titration is recommended when co-administered with a strong CYP3A4 inducer, based on reported safety and efficacy data.1,4,5 Certain ART regimens include compounds that are substrates and potential modulators of CYP3A4 (e.g. efavirenz (EFV) and rilpivirine (RIL).6–10 Therefore, a population PK model was fitted to describe the variability of ruxolitinib plasma concentrations observed in this study in virally suppressed individuals on standard ART, for comparison with PK studies in individuals with myelofibrosis, and in healthy individuals without HIV.4, 5,11 Also, here we assessed the potential drug-drug interactions between ruxolitinib and co-administered ART regimens.
Methods:
Human subjects protection.
This multi-site ACTG sponsored Phase 2a study was conducted in clinics at Emory University School of Medicine, Atlanta, GA; Johns Hopkins University School of Medicine, Baltimore, MD; National Institutes of Health/National Institute of Allergy and Infectious Disease, Rockville, MD; Harvard University Brigham and Women’s Hospital, Boston, MA; University of California San Francisco (UCSF), San Francisco, CA; and University of Alabama (UAB) School of Medicine, Birmingham, AL. This study was approved by the IRBs of all sites. Enrollment was from May 16, 2016 to January 10, 2018. All participants gave informed consent prior to enrollment.
Study design.
This PK study was nested within a broader study exploring the safety, tolerability, and immunologic effects of 10 mg ruxolitinib tablets administered orally bid, for five weeks in virologically suppressed individuals on existing ART.3 The proportion of men versus women and the ratio of participants on the various ART regimens was not controlled.3 Briefly, HIV+ individuals who were virologically suppressed and receiving ART were recruited and signed informed consents. Enrolled participants were randomly assigned to open-label ruxolitinib (10 mg bid.) or no intervention (controls), while continuing their stable ART regimen. Participants receiving ruxolitinib were followed on-treatment for 5 weeks and then off-treatment for an additional 7 weeks, controls were observed for 12 weeks.3 Further eligibility criteria and study design have been previously reported (ClinicalTrials.gov identifier NCT02475655).
Blood sampling and assays.
Ruxolitinib was administered for more than 4 days prior to PK sampling at the wk1 study visit and then continually twice per day (bid) for 5 weeks. Participants continued to administer their preexisting ART regimens uninterupted throughout the study. Blood samples were collected during planned days of supervised drug administration and blood plasma sampling on wk1 (after at least 4 days of ruxolitinib 10 mg bid) and on week 4 or 5 (wk4/5 of ruxolitinib administration), On the mornings of supervised dosing and PK blood draws, a baseline sample was collected 0.5 hr before the time of supervised drug administration (t = 0 hr). One plasma sample was subsequently collected in each of the following time intervals: 1–1.5 hr; 2.5–4 hr; and 6.0–8 hr, after the supervised administration. The times of drug (ruxolitinib and antiretrovirals) administration and blood draws (hr and minute) were documented. Timings of doses administered one and two days before the wk1 and wk4/5 clinic visits were obtained from participant reported drug diaries and used to model the baseline plasma concentration. Supervised dosing and blood-draw times were obtained from clinical records. The combined data were used for data interpretation and modeling. Blood was collected in heparinized vacutainers and separated into plasma immediately prior to freezing at the study site. PK samples were shipped on dry ice to the Laboratory of Biochemical Pharmacology for drug measurement. Ruxolitinib concentrations in plasma were determined using a liquid chromatography-tandem mass spectrometry (LC-MS/MS) assay, validated according to FDA guidelines.12 All assays used 100 µL of plasma sample and precipitated with 500 µL methanol. Forty µL supernatant was mixed with 60 µL of 0.1% formic acid (containing 1 mg/mL EDTA) for quantification of ruxolitinib. The calibration range for ruxolitinib was from 2 (the lower limit of quantification) to 1,500 nM. The intraday accuracy was between 93.7% and 105.5%, while inter-day accuracy was between 96.0% and 101.7%. Intraday precision was 1.2% to 9.6%, and inter-day precision was 3.1% to 8.7%. Plasma concentrations of ART drugs were assayed simultaneously after drying 200 µL supernatant and being reconstituted in 0.1% formic acid.13 The calibration range for emtricitabine (FTC) was 20 to 20,000 nM, for tenofovir (TFV) was 10 to 10,000 nM, for EFV was 40 to 40,000 nM, for lamivudine (3TC) was 20 to 10,000 nM, for abacavir (ABC) was 10 to 10,000 nM, for dolutegravir (DTG) was 50 to 10,000 nM, for RIL was 2 to 2,000 nM, and for raltegravir (RAL) was 15 to 15,000 nM.
Population PK.
Software.
Modeling and simulation were performed using nonlinear mixed-effects modeling in the software NONMEM 7.4 (ICON Development Solutions, Gaithersburg, MD, USA), run on a PC with a GFORTRAN compiler.14 Post-processing, diagnostic plots and automation were performed using PLT Tools 6.0 (PLT Soft, San Francisco, CA).15 Statistical tests not computed with PLT tools were computed with the R program (R version 3.6.1; R Statistical Foundation, Vienna, Austria http://www.r-project.org/), with additional graphs plotted using the ggplot2 package.16
Model fitting.
Ruxolitinib plasma concentrations and doses were inputted in units of nM, to conform with previous studies, and models were formulated in terms of clearance and volumes.4,11 Plasma concentrations of ruxolitinib and antiretroviral drugs were assumed to have reached steady-state prior to day minus 1 (−1) drug administration, since ruxolitinib has a ~ 3.5 hr t1/2 and participants were stabilized on their ART.1,4,5 Timings of supervised doses and blood draws were taken from clinic records. Dummy doses corresponding to times of administration (according to participant diaries) starting two days before the clinic visit were included in the data modeled, to account for residual concentrations at baseline (before supervised drug administration in the clinic). The time intervals between the reported evening dose were used to compute times intervals longer than 12 hr which were included in the analysis. Concentration < LOQ (4 of 298 observations, from separate individuals) made up less than 2 % of the data and were not modeled. Structural models assumed first-order input/elimination to/from the plasma compartment and 1- (ADVAN 2) or 2-compartments (ADVAN 4) distribution. The models convergence to more than 3 significant digits using the first-order conditional estimation with interaction method and parameter SE were computed using the NONMEM MATRIX = R (Hessian) covariance option.14
Random effects included inter-individual (IIV), inter-occasion variability (IOV) of parameters, and residual errors. Parameters were assumed to be log-normally distributed. The general form of the parameter equation including IIV and IOV was , where Pi,j = estimated parameter for a given individual i at the jth occasion, PTV = the typical (population value) of the parameter, ηi describes the variation of individual i from the population estimate (IIV), and ηi,j represents the variability on occasion j (IOV). In all cases, η was assumed to be normally distributed: ηi~N(0,ωIIV2) and ηi,j~N(0,ωIOV2). An “occasion” was defined as a group of sequential concentrations on wk1 or wk4/5, and variances were considered as sampled from the same distribution. IIV and IOV were converted to %CV using the formula: .
IIV was estimated for oral clearance , V1/F , and V2 , in the appropriate models. The geometric mean oral bioavailability (F) of each participant was fixed = 1 as ruxolitinib is reported to be > 95% orally absorbed.1 Variations in F between wk1 and wk4/5 (IOVF) were estimated. Residual errors (σ2) were modeled with an additive and a proportional component.
Model selection.
One- and two-compartment disposition models with first-order absorption and elimination were compared statistically and graphically for their ability to describe the plasma concentration versus time curves of ruxolitinib. Statistical assessment of the fit of models to the data included comparison of the respective minimum values of the objective function after convergence. (MVOF, equivalent to −2 of the log likelihood function). A decrease in MVOF of 5.99 was considered a significant improvement of the 2-compartment model (p < 0.05, nested models with 2 additional parameters, likelyhood Ratio Test), together with an improvement in dispersion of the data about predictive curves.
After a base model was identified, the effects of subject covariates (measured on the day each participant entered the study) were assessed. These included normalizing the distribution volume of each by mean body weight , followed by normalizing the systemic clearance (CL) by body surface area (BSA), assuming proportionality between BSA and , where BWi and HTi are the weight (mean = 91.5 kg) and height (mean = 174.8 cm), respectively.17 The proportionality scaling factor for BSA was selected as it was reported to have a low root mean square error for BSA estimation (RMSE = 0.0591).17 The effect of modeling a separate CL for individuals on the ART whose post hoc predicted CL appeared to differ most from the others, was also explored, and a 3.84 decrease in the MVOF was considered significant (α = 0.05, one degree of freedom (df)). Likewise, covariates contributing to > 3.84 reduction in MVOF were considered significant. Improvements in model fit was assessed graphically by comparing plots of observed versus population and post hoc predicted plasma concentrations, and conditional weighted residuals versus time and post hoc predicted concentrations.18–20 A visual predictive check analysis was performed to assess the ability of a model similar to a previously published population PK model to simulate the PK variations using > 5,000 simulated individuals on wk1 and wk4/5 (200 for each body weight). Simulated profiles were used to compute 2.5, 5, 25, 50, 75 95, 97.5 percentile curves, which were overlaid with actual plasma concentrations.15,21
Comparison of post hoc predicted CL/F with noncompartment PK studies:
Typical CL/F fitted with the population PK models assume log-normal distributions, while noncompartment PK papers typically report CL/F using means and %CV. Therefore, means and %CV of post hoc (individual) predictions outputted by the model, on wk1 and wk4/5, were computed and compared with means and %CVs of CL/F from noncompartment PK studies.
Assessment of drug-drug interactions.
Distributions of post hoc model predicted CL/F produced by the selected models were compared graphically by week, sex and ART regimen. Statistical comparisons were made of CL/F versus ART regimen using t-tests on log transformed data (2-sided, equal variances) and p < 0.05 was considered significant. Comparisons between individual ART regimens was performed by ANOVA with a Tukey HSD correction for multiple comparisons. Statistics were computed using the R program. Plasma concentrations versus time of the various antiretroviral drugs 3TC, ABC, DTG, EFV, FTC, RIL, and RAL, as well as TFV administered as the alafenamide fumerate (TAF) or the disoproxil fumerate (TDF), were compared visually with published PK studies in HIV-positive or HIV-negative individuals without ruxolitinib.22–31
Results
Demographics.
Pharmacokinetic data were measured in 40 virologically suppressed participants from the ruxolitinib arm of a randomized Phase 2a trial of ruxolitinib in antiretroviral-treated adults with HIV.3 Participants were administered 10 mg ruxolitinib tablets bid for five weeks, together with their ongoing ART; 55% were taking integrase strand transfer inhibitor-based regimens and 45% were taking non-nucleoside reverse transcription containing regimens. Details on the demographics have now been published.3
Coadministered ART regimens.
Plasma concentrations of coadministered ART agents were measured. Fourteen participants with PK measured on wk1 and wk4/5 were administered an ART regimen comprising 1) EFV, with FTC, and TDF, (EFV/FTC/TDF, Atripla™, Gilead Sciences, Inc., San Francisco, CA). Other participants (25 on wk1 and 23 on wk4/5) were alternatively administered integrase strand inhibitor (INSTI) based regimens including: 2) DTG with ABC and 3TC, (DTG/ABC/3TC, Triumeq™, ViiV Healthcare, Inc., Research Triangle Park, NC); 3) DTG (Tivicay™, ViiV Healthcare. Research Triangle Park, NC) with FTC and TAF (FTC/TAF, Descovy™, Gilead Sciences, Inc., San Francisco, CA), (DTG + FTC/TAF); 4) DTG with FTC and TDF (FTC/TDF, Truvada™, Gilead Sciences, Inc., San Francisco, CA). (DTG + FTC/TDF); 5) FTC/TDF and RAL (Isentris™, Merck& Co, Inc., Kenilworth, NJ), (RAL + FTC/TDF); 6) RAL + FTC/TAF, 7) RIL with FTC/TAF (RIL/FTC/TAF, Odefsey™, Gilead Sciences, Inc, San Francisco, CA); and 8) RIL with FTC and TDF (RIL/FTC/TDF, Complera™, Gilead Sciences, Inc., San Francisco, CA).
PK data.
Plasma samples (N = 298) were assayed for ruxolitinib concentrations, of which only four (2 each from wk1 and wk4/5) were below the limit of quantification for the LC-MS assay (LOQ = 2 nM). They constituted less than 2% of the data and were not included in the analysis. Thirty-nine participants had initial samples drawn on wk1 and 37 had follow up samples collected on wk4/5. Each participant had at least one measurable plasma concentration sampled >7 hr (~2 × ruxolitinib t1/2) after a previous dose. Ruxolitinib plasma concentration versus time since the previou dose sampled at wk1 and wk4/5 are shown in (Fig. 1A and Fig. 1B, respectively).
Figure 1.
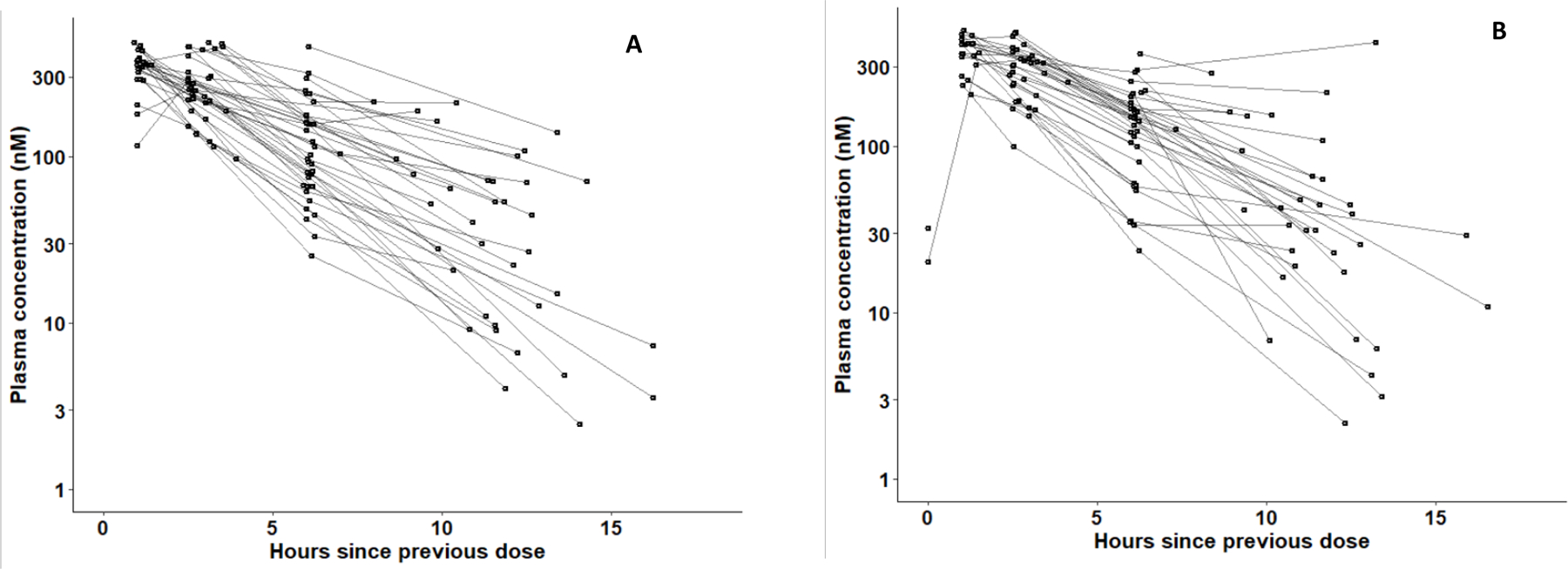
Ruxolitinib (MW = 306.4) plasma concentrations-versus-time since the previous ruxolitinib (10 mg) dose. Plotted are 39 participants (151 observations) on wk1 (A) and 37 participants (143 observations) on wk4/5 (B). Points from the same participant were plotted on the same curve. (To convert nM concentrations to units of ng/ml, multiply by a factor of 0.3064 .
Model development for comparison with previous PK studies.
Ruxolitinib plasma concentrations versus time was modeled using structural 1- and 2-compartment disposition models (Models I and II, respectively, without covariates), assuming first order oral input and elimination from the central (plasma) compartment. Both models converged to more than 3-significant digits and produced SE estimates. However, Model II had less biased goodness-of-fit plots and a significant decrease in the MVOF (ΔMVOF = 11, nested model with 2 extra parameters, p < 0.04, assuming OF is chi-square distributed). Data were insufficient to fit a 3-compartment model. Thus, Model II was selected for further development. Normalizing V1 and V2 by mean body weight (91.5 kg) (Model III) further reduced the MVOF by 10. The PK parameters and variabilities of Model III are summarized in Table 1. Diagnostic plots on wk1 and wk4/5 data demonstrated relatively symmetrical deviations of observed concentrations about the respective lines of identity of population (Fig. 2A and Fig. 2C) and post hoc (individual) predicted concentrations (Fig. 2B and Fig. 2D). Also, the majority of the data and LOESS smoother curves clustered about the line of identity on both linear and logarithmic-concentration scales. Plotting CWRES versus time after the previous dose (Fig. 2E) and population predicted concentrations (Fig. 2F) demonstrated a high degree of symmetry. A visual predictive check analysis demonstrated that Model III was able to simulate the variability in ruxolitinib plasma concentrations versus time observed on wk1 and wk 4/5 of the study (Fig. 3A and Fig. 3B).
TABLE 1:
Ruxolitinib 2-Compartment PK models with V1 and V2 normalized to mean body weights (kg).
| Model III | Model IV (separated CL/F by EFV status) | |||
|---|---|---|---|---|
| Sig. digits | 3.7 | 3.6 | ||
| MVOF | 2,793 | 2,769 | ||
|
| ||||
| Parameter | Typical value | RSE (%) | Typical value | RSE (%) |
| CL/F [L/hr] | 14.7 | 6.8 | 12.9 | 8.3 |
| (with EFV) 22.5 | 8.8 | |||
| V1/F [L] | 59.6 | 7.0 | 47.8 | 18.9 |
| Q/F [L/hr] | 4.94 | 30 | 12.0 | 25.2 |
| V2/F[L] | 7.88 | 49 | 20.7 | 6.0 |
| Ka [1/hr] | 7.05 | 35 | 1.82 | 43.2 |
| F (fixed) | 1 | 1 | ||
|
| ||||
| Error structure | ω2 (CV) |
RSE of ω2
(shrinkage) |
ω2 (CV) |
RSE of ω2
(shrinkage) |
| 0.1182 (35.4%) | 0.032 (8%) | 0.0649 (25.9%) | 0.02439 (9%) | |
| 0.0607 (25.0%) | 0.042 (31%) | NE | ||
| 2.427 (321%) | 2.27 (49%) | 0.957 (127%) | 0.916 (32%) | |
| 0.0567 (24.2%) | 0.018 (24%) | 0.0669 (26.3%) | 0.0344 (18%) | |
| σ2 (proportional) | 0.066 | 0.069 | ||
| σ2 (additive) [nM] | 56.02 | 43.1 | ||
|
| ||||
| Correlations | population | posthoc | population | posthoc |
| r2 (obs vs predicted) | 0.645 | 0.898 | 0.672 | 0.870 |
| med abs pred error | 36% | 13% | 30% | 14% |
CL = apparent clearance from compartment 1 (plasma); Q = apparent clearance between compartments 1 and 2; V1 and V2 = apparent compartment volumes; Ka = first-order absorption rate constant; F= oral bioavailability (fixed = 1) and estimated the variation between wk1 and wk4/5 (IOV). V1 and V2 were normalized to the mean of the participant body weights (91.5 kg). RSE = relative standard error. σ2 are residual errors; MVOF = minimum value of the NONMEM objective function (−2LL). NE = not estimated. %CV of IIV and IOV was calculated , where ω2 is the variance of the log-transformed parameter. r2 = linear regression correlation between predicted and observed concentrations. Med abs pred error = median absolute predicted error.
Figure 2.
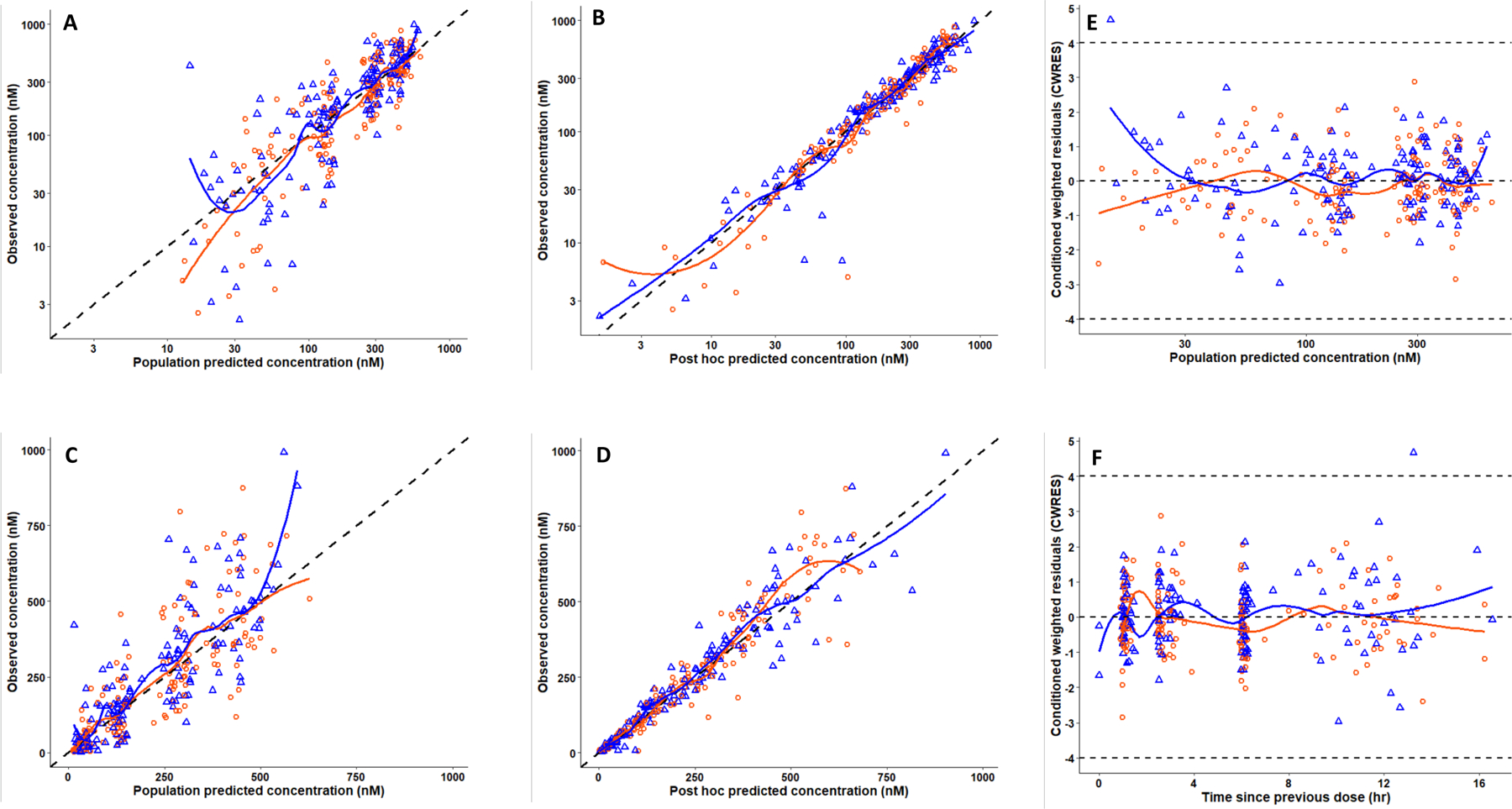
Goodness of fit curves of ruxolitinib population PK Model III. Observed concentrations from wk1 (orange circles) and wk4/5 (blue triangles) versus the population predictions (A) and post hoc (individual) predictions (B) plotted on a log-scale. The same data are depicted on the arithmetic scale (C and D). The black dashed lines represent lines of identities. Plots The right-hand side panels depict conditional weighted residuals (CWRES) versus population predicted concentration (E) and times since the previous dose (F). Orange and blue curves are LOESS regression smoothers. The dashed gray line in panels E and F represents CWRES +4 and −4.
Figure 3.
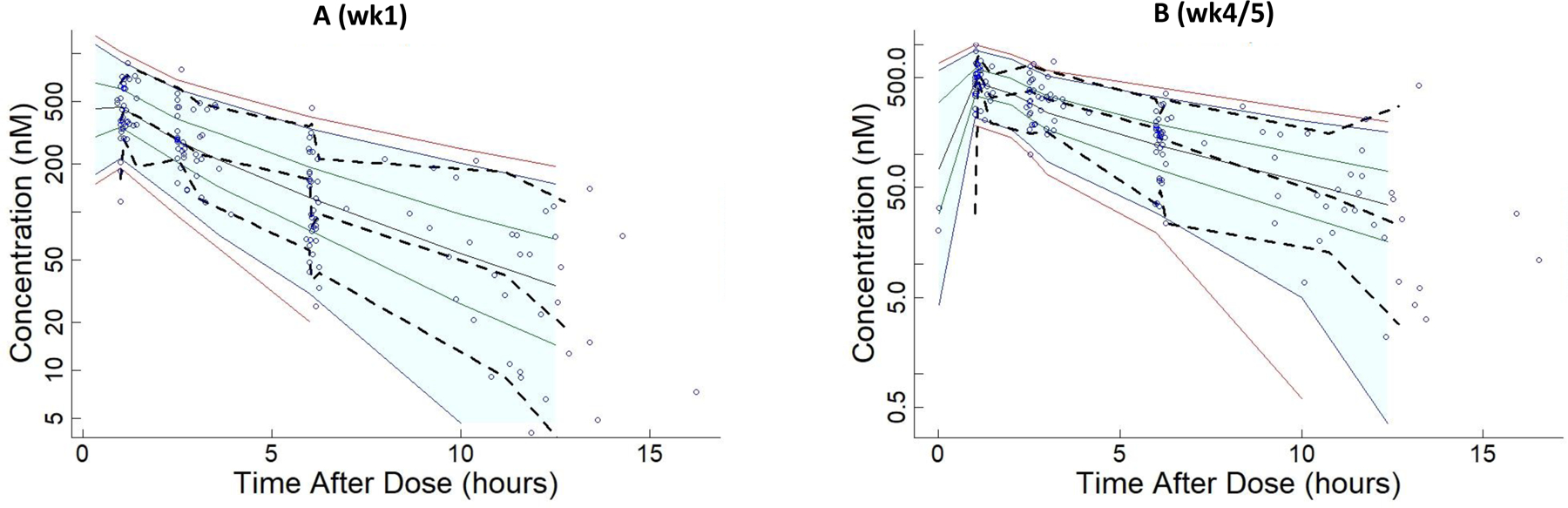
Visual predictive check analysis of ability of Model III to simulate the variations in ruxolitinib plasma concentrations. Solid lines indicate percentiles: 2.5, 97.5 (red); 5, 95 (blue); 25, 75 (green); 50 (black). Dashed lines indicate percentiles 5, 50, and 95 of observations. Predictions are displayed at nominal times; observations are displayed at actual times. 5.3 % of observations were > P95 or < P5 on wk1 (A) and 7.7 % of observations were > P95 or < P5 on wk4/5 (B). Shaded areas indicate 90% prediction intervals and extend to median of input times in the right−most bin. The shaded area is truncated to avoid plotting logs of negative numbers. Any points to right of shaded area are included in the right−most bin.
Boxplots of CL/F for 7 women on wk1 and wk4/5 versus 32 and 30 men on wk1 and wk4/5, suggested similar medians for men and women participants. In addition, participants administered an ART regimen with EFV (EFV/FTC/TDF) generally had higher CL/F (Fig. 4).
Figure 4.
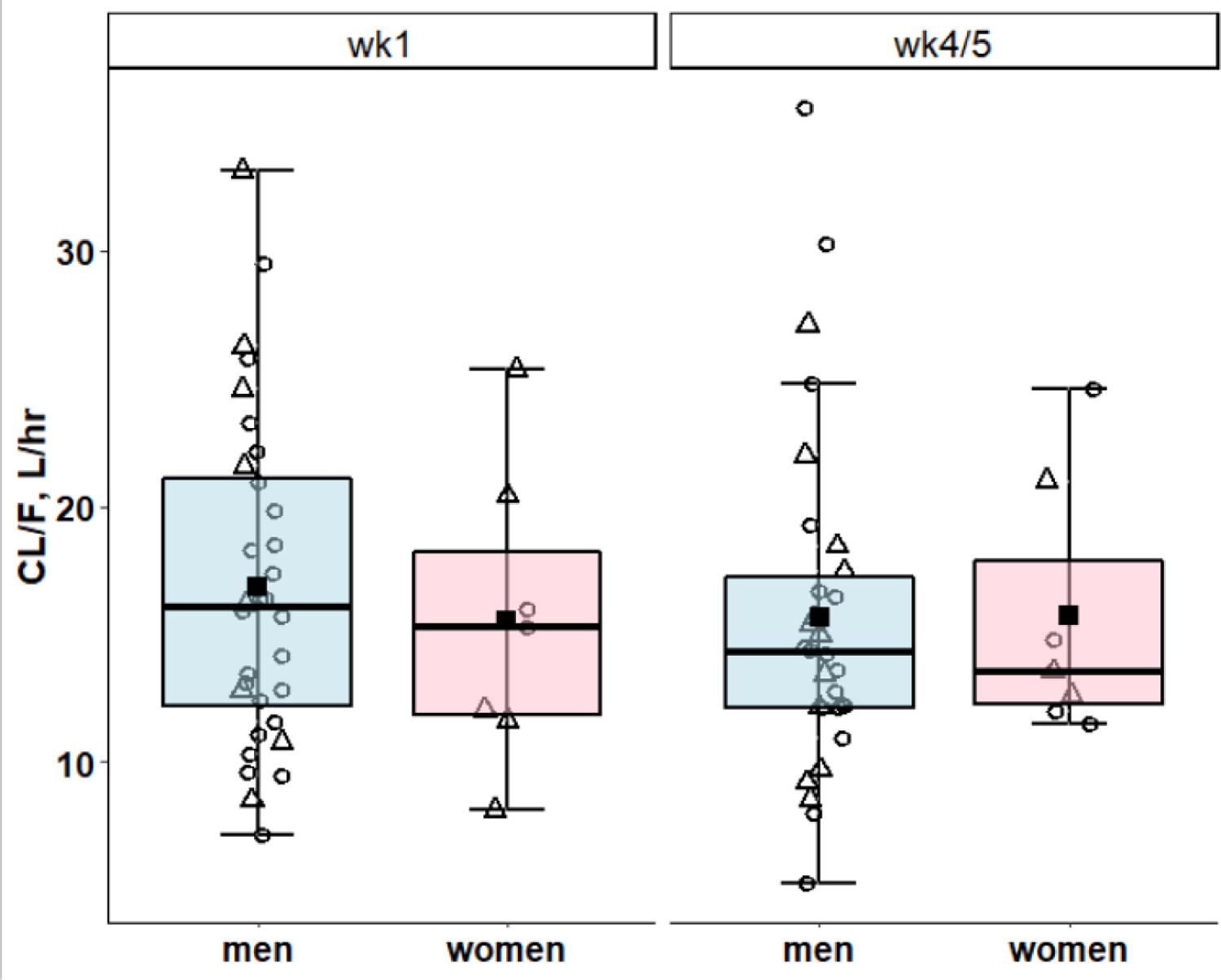
Model IV: Pooled Post hoc predicted CL/F were compared by sex (blue = men, pink = women). Horizontal bars depict medians and quartiles, solid squares are means and open circles and triangles are post hoc predicted ruxolitinib CL/F in the presence of ART with and without EFV, respectively.
The mean and %CV of the post hoc predicted CL/F on wk1 was 16.6 L.hr−1. 37.6% (N = 39) and on wk4/5 was 15.7 L.hr−1, 40.5% (N= 37).
Assessment of drug-drug interactions.
Box plots were used to compare post hoc predicted ruxolitinib CL/F of participants administered individual ART regimens on wk1 and wk4/5, from Model III (Fig. 5A and Fig. 5B). Individuals administered EFV had higher ruxolitinib post hoc predicted CL/F on wk1 and wk4/5 than any of the other ART drugs including RIL (another CYP3A4 substrate). An ANOVA rejected the hypothesis of similar CL/F between regimen (p < 0.03 on wk1 and < 0.01 on wk4/5). However, the number of participants enrolled per INSTI regimen was insufficient to demonstrate statistical significance by ANOVA with a Tukey HSD correction for multiple comparisons.
Figure 5.
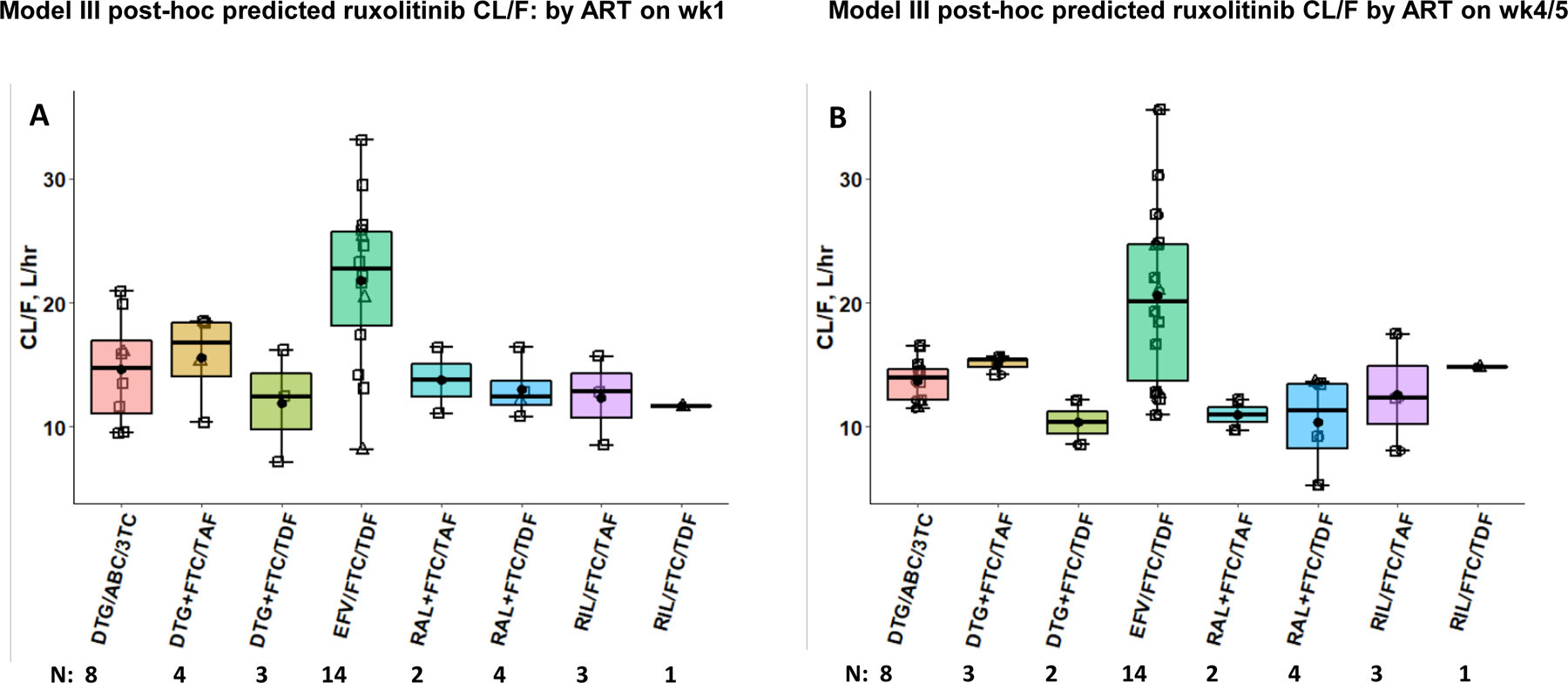
Model III Post-hoc predicted ruxolitinib CL/F: by ART on wk1 (A) and wk4/5 (B). Participants co-administered ruxolitinib and their various ART regimens which included combination of: lamivudine (3TC), emtricitabine (FTC), efavirenz (EFV), tenofovir disoproxil fumarate (TDF), tenofovir alafenamide (TAF), rilpivirine (RIL), abacavir (ABC), dolutegravir (DTG) and raltegravir (RAL). “/” represents ART within the same formulation and “+” ART in co-administered formulations. N = observations/bin. Horizontal bars depict medians and quartiles, closed circles are means, open squares and triangles are post hoc predicted ruxolitinib CL/F of men and women, respectively.
Model III was modified to estimate a separate typical CL parameter for participants administered an ART regimen containing EFV (Model IV). Model IV produced a statistically significant reduction in MVOF (ΔMVOF = 24 one additional parameter, p < 0.00001). However, the IIV of V1 was close to zero (V1 was already normalized to mean body weight), and its exclusion did not alter the MVOF. The typical CL/F was 21.2 L.hr−1 in participants administered EFV (N = 14) and 13.0 in those on an INSTI (N = 25) based ART regimen. The parameters of Model IV are summarized in Table 1. The Ka estimated was smaller than in Model III (1.82 hr−1 versus 7.88 hr−1), but this may have resulted from few data collected during the absorbtion phase. Ruxolitinib plasma concentrations were adequately described by Model IV (Fig. 6A – Fig. 6F).
Figure 6.
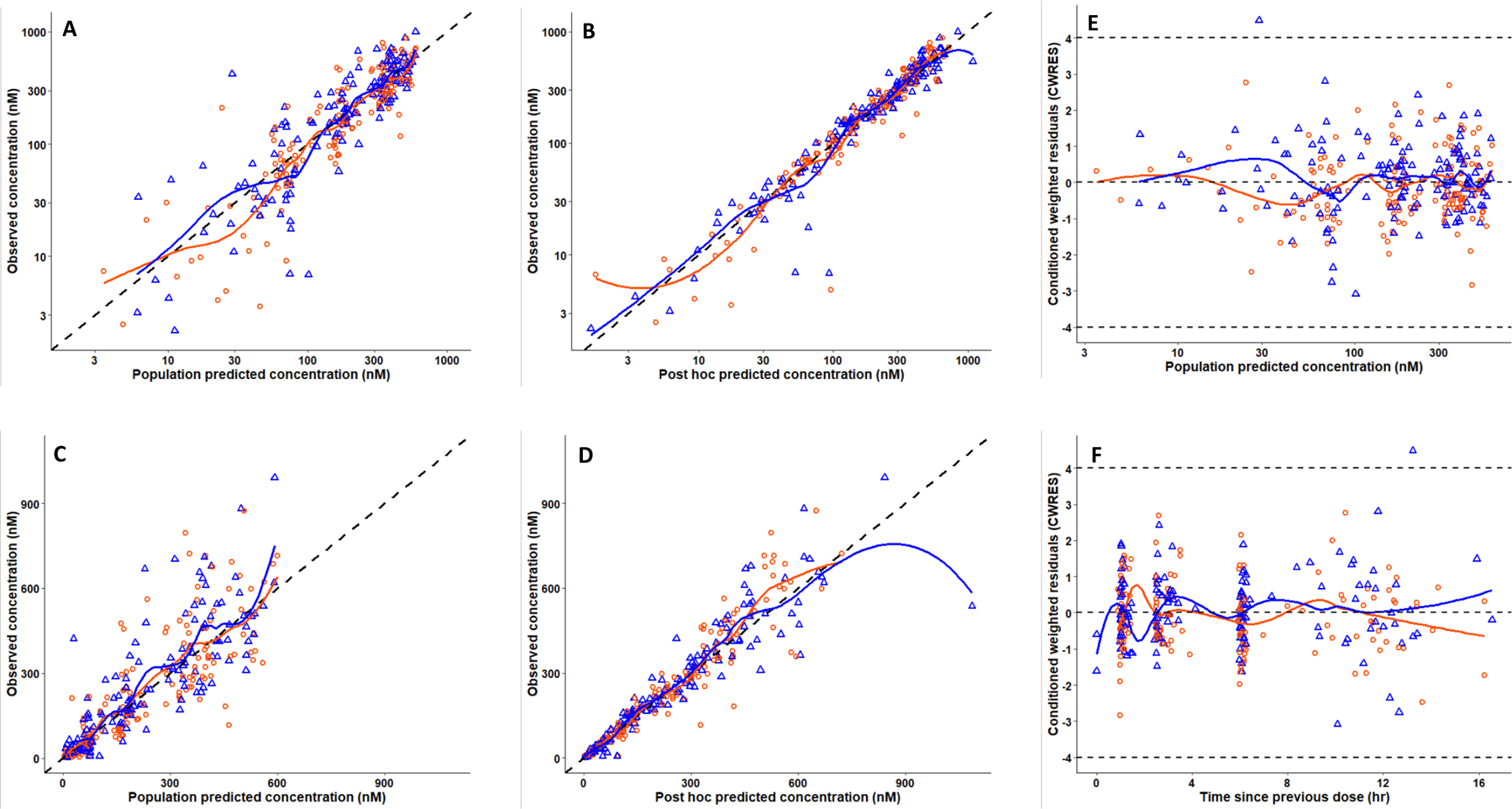
Model IV goodness of fit curves of ruxolitinib population PK (separate CL/F by EFV status). Observed concentrations from wk1 (orange circles) and wk4/5 (blue triangles) versus the population predictions (A) and post hoc (individual) predictions (B) plotted on a log-scale. The same data are depicted on the arithmetic scale (C and D). The black dashed lines represent lines of identities. Plots The right-hand side panels depict conditional weighted residuals (CWRES) versus population predicted concentration (E) and times since the previous dose (F). Orange and blue curves are LOESS regression smoothers. The dashed gray line in panels E and F represents CWRES +4 and −4.
Post-hoc estimates of ruxolitinib CL/F from Model IV, were used in a boxplot analysis of CL/F versus weeks of treatment (wk1 or wk4/5) and co-administered ART regimen (Fig. 7). A pooled analysis of participants administered EFV/FTC/TDF (42% of females and 34% of males on wk1 and wk4/5) versus those administered an INSTI-based regimen demonstrated significantly higher CL/F in the EFV/FTC/TDF treated group. The geometric mean CL/F at wk1 of participants in the INSTI group was 13.8 versus 22.3 L.hr−1 in the EFV/FTC/TDF group (p < 0.0001) and 12.7 versus 20.8 L.hr−1 on wk4/5.
Figure 7.
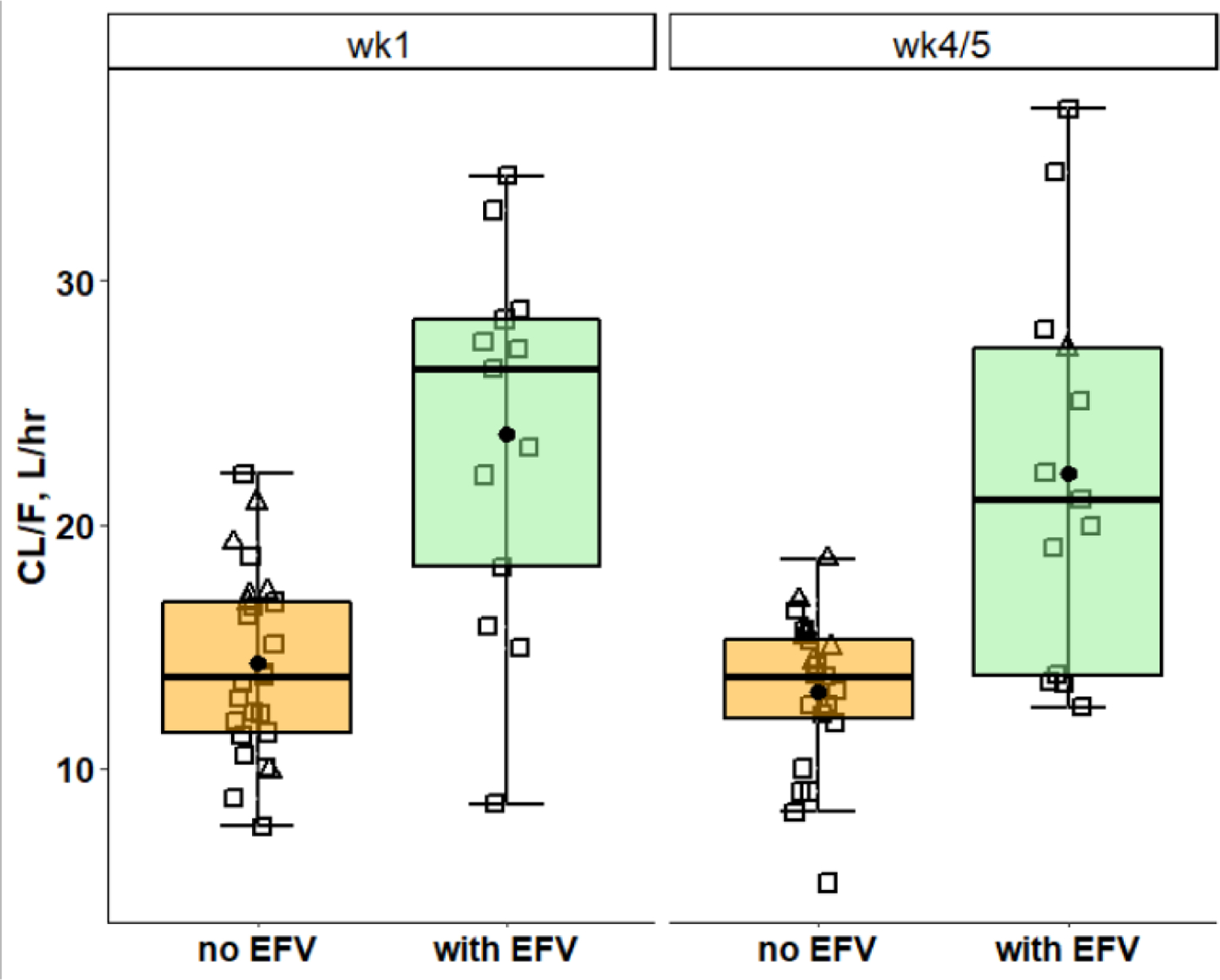
Pooled CL/F of ART without EFV (orange), were compared with those of EFV/FTC/TDF (light green), on wk1 and wk4/5. Horizontal bars depict medians and quartiles, closed circles are means, open squares and triangles are post hoc predicted ruxolitinib CL/F of men and women, respectively.
Plasma concentration of the antiretroviral drugs versus time (since administration) of participants administered EFV, FTC, tenofovir administered as TDF or TAF, DTG, 3TC, ABC, RAL and RIL were similar to PK studies without ruxolitinib (Online supplementary materials).22–31
Discussion.
We report in this paper for the first time the pharmacokinetic profile of Ruxolitinib in HIV infected persons. Ruxolitinib was safely administered orally to 40 ART viral-suppressed participants for five weeks using 10 mg tablets bid. This regimen was selected as it is one of the lower US FDA approved doses for the treatment of myelofibrosis and was deemed safe for this study in HIV+ people with well-controlled viremia.3
Participants received supervised doses of their ART and ruxolitinib on days of PK sampling, and remained in the clinic for less than 8 hr. Samples plotted as times more than 12 hr since the previous dose in Fig. 1A and 1B reflect times between the previous evening dose and the morning “baseline” blood draw which occurred before administering the supervised dose in the clinic on the days of PK sampling. Although these later timings are considered less precise than supervised doses in the clinic, concentrations were, in general, in agreement with expected values on their respective curves. Imprecise timings are expected to have less impact during the elimination phase than during drug absorption and tissue distribution phases.
Comparison of ruxolitinib PK with previous studies.
Ruxolitinib PK was adequately described using a 2-compartment disposition model with first-order absorption and elimination, with body-weight proportional to V1 and V2. (Model III). Chen, et al., fitted a similar model to 272 individuals (56% male and mean body weight of 74.3 kg) with primary myelofibrosis, post-polycythemia vera myelofibrosis, or post-essential thrombocythemia myelofibrosis using data from a Phase 2 and a Phase 3 dose escalating (10 – 200 mg bid.) study and validated using an external dataset of 142 subjects from a separate Phase 3 study.11 The typical value of CL/F estimated in this study in adults living with HIV on ART was 14.7 L.hr−1 , versus 22.1 L.hr−1 (39.1%) in men and 17.7 L.hr−1 in women in the previous study. This study could not discern differences in CL/F between sexes and the medians of post hoc estimated CL/F were only marginally higher in men than women (wk1: 16.0 versus 15.2, wk4/5 14.3 versus 13.6 L.hr−1 (Fig. 4). The typical CL estimated is lower than in the population PK study by Chen, et al., the post hoc predicted CL/F (mean, %CV) on wk1 (16.6 L.hr−1. 37.6%,N = 39) and wk4/5 (15.7 L.hr−1, 40.5%, N = 37) but was similar to CL/F reported in noncompartment PK studies of ruxolitinib tablets (10–200mg) in individuals not on CYP3A4 inducers or inhibitors e.g., 10 mg, (12.6 Lhr−1, 40.6%, N = 16), 25 mg (16.8 L.hr−1, 29.8%, N = 47) or 200 mg (16.9 ± 5.01 L.hr−1, 29.6%, N = 48) ruxolitinib tablets, respectively.4,5 The inability to discern differences in CL/F between sexes in the current study may result from the small number of women in this study (N = 7), of which 42% continued to administer an ART regimen with EFV, a potent inducer of CYP3A4 and the primary CYP enzyme responsible for ruxolitinib metabolism.1,4,5–10 The typical V1 estimate in this study was 59.6 L , (normalized to a mean body weight of 91.5 kg) similar to V1 reported in the study by Chen, et al., (58.6 L, 28%, normalized to a mean body weight of 72.9 kg). The typical V2 estimate was 7.88 L , V2 estimated in the previous study was 11.2 L . The typical estimate of Q/F was 8.2 L.hr−1 versus 2.53 L.hr−1 in the study by Chen, et al. Neither study estimated . Both studies confirmed rapid oral absorption of ruxolitinib, with typical Ka in this study was 7.05 hr−1 compared to 4.12 hr−1 , respectively. There were insufficient data to estimate or a pre-absorption lag time in this study. The limited data in the absorption phase and self-reported dose times > 8 hr, impacted the precision of Ka, Q and V2 estimates. The IOV in oral bioavailability between wk1 and wk4/5 was about 24.8%. A visual predictive check analysis demonstrated that Model III simulated the variability in ruxolitinib plasma concentrations observed in the study (Fig. 3). However, the relatively large shrinkage and SE of the IIV (Table 1) suggest that the IIV estimates warrant confirmation in a larger study.
Assessment of drug-drug interactions.
The large variation in post hoc CL/F in participants administered an ART regimen with EFV (Fig. 5 and Fig. 6) is consistent with a previous study of CYP induction in 77 previously untreated HIV+ individuals following initiation of ART with EFV.8 That study measured EFV PK and changes in β-hydroxycholesterol/cholesterol (4β-OHCC/cholesterol) ratios from baseline for up to 16 weeks following initiation of ART, and the CYP2B6*6 genotypes of the individuals. 4β-OHCC/cholesterol ratios were used as an endogenous biomarker of CYP3A4/5, as 4β-OHCC is a metabolite of cholesterol formed by CYP3A4/5 enzymes.9,10 A wide range of plasma 4β-OHCC/cholesterol ratios was observed, with a median increase in the 4th (257%), 16th (291%) and 48th (165%) week (P < 0.0001). CYP3A4/5 induction was most pronounced in CYP2B6 slow metabolizers.9 Although potentially useful, measurements of 4β-OHCC/cholesterol ratios are not recognized by the FDA as a marker of CYP3A4/5 enzyme activity. Also, CYP genotypes were not measured in the current study.
Although RIL could theoretically induce CYP3A4, ruxolitinib CL/F was not increased with co-administered RIL/FTC/TAF (N = 3) or with RIL/FTC/TDF (N = 1) (Figs. 6A and 6B), consistent with previous study which reported that 25 mg RIL daily did not alter CYP3A-dependent drug metabolism.9 Plasma concentrations of co-administered ARVs resembled concentrations in published studies without ruxolitinib. A study reported by Shi, et al., in healthy participants (N = 12) administered a 30 mg ruxolitinib tablet before and after 10 days of 600 mg rifampin (a strong CYP3A4 inducer) demonstrated a 71% increase in ruxolitinib CL/F.4 Interestingly, the inhibition of interleukin (IL)–6-stimulated STAT3 phosphorylation in whole blood by ruxolitinib decreased by only 10%, possibly due to the accumulation of active metabolites. In contrast, administration of a 10 mg ruxolitinib tablet with 500 mg erythromycin (moderate CYP3A4 inhibitor, N = 15) or 200 mg bid ketoconazole (a strong CYP3A4 inhibitor, N = 16) reduced ruxolitinib CL/F in healthy participants by 21% and 91%, respectively.4 The FDA label of ruxolitinib tablets states that concomitant administration with strong CYP3A4 inducers may decrease ruxolitinib exposure, but recommends no dose adjustment; however, it is advised that patients should be frequently monitored with adjustment of the ruxolitinib dose based on safety and efficacy.1
Conclusions.
The PK parameters and variability of ruxolitinib co-administered orally at 10 mg bid for five weeks in ART-treated adults with HIV were similar to those reported in studies with ruxolitinib alone administered in populations without HIV. However, CL/F of ruxolitinib was significantly higher in individuals administered ART with EFV compared to INSTI based regimens. Meanwhile, ART plasma concentrations were similar to those previously reported in studies without ruxolitinib. Therefore, INSTI based ART regimens may be preferred over efavirenz containing regimens when ruxolitinib is administered to HIV+ individuals.
Supplementary Material
Funding.
This work was supported in part by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number UM1-AI-068634, UM1-AI-068636 and UM1-AI-106701. VCM and RFS received support from the Emory CFAR (P30-AI-050409). RFS provided funding support for the study drug and was also funded in part by R01-MH-116695. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This study was presented in part at the Conference on Retroviruses and Opportunistic infections (CROI) in 2019 in Seattle, WA, USA (Marconi, et al., Abstract #37) and 2020, Boston, MA, USA (Hurwitz, S.J., et al, Abstract # 00452). We are grateful to the patients and clinical staff at the participating sites for their generous contributions to this work and especially the ACTG, SDMC, participating CRSs, and Specialty Laboratories including the Brigham and Women’s Hospital, Harvard Medical School, Case Western Reserve University, and Emory University School of Medicine.
VCM has received investigator-initiated research grants (to the institution) and consultation fees (both unrelated to the current work) from Lilly, Bayer, Gilead Sciences and ViiV. CF received consultation fees unrelated to the current work from Merck, Mylan Pharmaceuticals, and ViiV. MML has consulted for Lilly and he has received competitive grant funding from Gilead (to his institution). JLL has consulted for Gilead and has received grant funding from ViiV (to his institution). All other authors report no potential conflicts. Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Number UM1-AI-068634, UM1-AI-068636 and UM1-AI-106701. RFS is a shareholder of Incyte Corp. VCM and RFS received support from the Emory CFAR (P30-AI-050409). RFS provided funding support for the study drug and was also funded in part by R01-MH-116695.
Footnotes
Data Sharing
Individual participant data and a data dictionary defining each field in the set will be made available to investigators for work that was not initially proposed as part of the parent protocol on a case-by-case basis via request to the AIDS Clinical Trials Group (ACTG) via the link: https://submit.actgnetwork.org/. Completion of an ACTG Data Use Agreement may be required.
References
- 1.JAKAFIi (ruxolitinib) tablets prescribing label - U.S. FDA. https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/202192lbl.pdf Accessed September 29” https://www.accessdata.fda.gov/drugsatfda_docs/label/2011/202192lbl.pdf Accessed September 29, 2020.
- 2.Gavegnano C, Brehm JH, Dupuy FP, et al. Novel mechanisms to inhibit HIV reservoir seeding using Jak inhibitors. PLoS Pathog. 2017;13(12):e1006740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Marconi VC, Moser C, Gavegnano C, et al. Randomized trial of ruxolitinib in antiretroviral-treated adults with HIV. Clin Infect Dis. 10.1093/cid/ciab212/6159796. Published online, March 6, 2021. Accessed 4–14-2021. [DOI] [PMC free article] [PubMed]
- 4.Shi JG, Chen X, Emm T, Scherle PA, et al. The effect of CYP3A4 inhibition or induction on the pharmacokinetics and pharmacodynamics of orally administered ruxolitinib (INCB018424 phosphate) in healthy volunteers. J Clin Pharmacol. 2012; 52(6):809–818. [DOI] [PubMed] [Google Scholar]
- 5.U.S. FDA W, DC. 2011, Clinical pharmacology review of ruxolitinib (JAKAFIi™, Incyte), NDA/BLA Number 202192Orig1s000. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202192Orig1s000ClinPharmR.pdf. Published June, 2011. Accessed December 16, 2020.
- 6.Mouly S, Lown KS, Kornhauser D,et al. Hepatic but not intestinal CYP3A4 displays dose-dependent induction by efavirenz in humans. Clin Pharmacol Ther. 2002;72(1):1–9. [DOI] [PubMed] [Google Scholar]
- 7.Michaud V, Ogburn E, Thong N, et al. Induction of CYP2C19 and CYP3A activity following repeated administration of efavirenz in healthy volunteers. Clin Pharmacol Ther. 2012;91(3):475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Habtewo A, Amogne W, Makonnen E, et al. Pharmacogenetic and pharmacokinetic aspects of CYP3A induction by efavirenz in HIV patients. Pharmacogenomics J. 2013;13(6):484–489. [DOI] [PubMed] [Google Scholar]
- 9.Josephson F, Bertilsson L, Bottiger Y, et al. CYP3A induction and inhibition by different antiretroviral regimens reflected by changes in plasma 4b-Hydroxycholesterol levels. Eur J Clin Pharmacol. 2008;64(8):775–781. [DOI] [PubMed] [Google Scholar]
- 10.Hohmann N, Reinhard R, Schnaidt S, et al. Treatment with rilpivirine does not alter plasma concentrations of the CYP3A substrates tadalafil and midazolam in humans. J Antimicrob Chemother. 2016;71(8):2241–2247. [DOI] [PubMed] [Google Scholar]
- 11.Chen X, Williams WV, Sandor V, Yeleswaram S. Population pharmacokinetic analysis of orally-administered ruxolitinib (INCB018424 Phosphate) in patients with primary myelofibrosis (PMF), post-polycythemia vera myelofibrosis (PPV-MF) or post-essential thrombocythemia myelofibrosis (PET MF). J Clin Pharmacol. 2013;53(7):721–730. [DOI] [PubMed] [Google Scholar]
- 12.U.S. FDA W, DC. 2018. Bioanalytical method validation guidance for industry. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry. Published May, 2018. Accessed September 29, 2020.
- 13.Rimawi BH, Johnson E, Rajakumar A, et al. Pharmacokinetics and placental transfer of elvitegravir, dolutegravir, and other antiretrovirals during pregnancy. Antimicrob Agents Chemother. 2017;61(6):e02213–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Beal SL, Sheiner LB, Boeckmann AJ & Bauer RJ (Eds). NONMEM 7.4 users guides (ICON plc, Gaithersburg, MD, 1989–2018). https://www.iconplc.com/innovation/nonmem/. Accessed February 20, 2020. [Google Scholar]
- 15.Fisher D, Shafer S. 2017. PLT Tools. http://www.pltsoft.com/about_us/index.html. Accessed September 29, 2020.
- 16.Wickham H. ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag; New York. ISBN 978–3-319–24277-4, https://ggplot2.tidyverse.org. Published 2016. Accessed September 10, 2020. [Google Scholar]
- 17.Wang Y, Moss J, Thisted R. Predictors of body surface area. J Clin Anesth. 1992;4(1):4–10. [DOI] [PubMed] [Google Scholar]
- 18.Sheiner LB, Beal SL. Evaluation of methods for estimating population pharmacokinetic parameters. III. Monoexponential model: routine clinical pharmacokinetic data. J Pharmacokinet Biopharm. 1983;11(3):303–319. [DOI] [PubMed] [Google Scholar]
- 19.Karlsson MO, Savic RM. Diagnosing model diagnostics. Clin Pharmacol Ther. 2007;82(1):17–20. [DOI] [PubMed] [Google Scholar]
- 20.Hooker AC, Staatz CE, Karlsson MO. Conditional weighted residuals (CWRES): a model diagnostic for the FOCE method. Pharm Res. 2007;24(12):2187–2197. [DOI] [PubMed] [Google Scholar]
- 21.Post TM, Freijer JI, Ploeger BA, Danhof M. Extensions to the visual predictive check to facilitate model performance evaluation. J Pharmacokinet Pharmacodyn. 2008;35(2):185–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dickinson L, Amin J, Else L, et al. Pharmacokinetic and pharmacodynamic comparison of once-daily efavirenz (400 mg vs. 600 mg) in treatment-naive HIV-infected patients: Results of the ENCORE1 study. Clin Pharmacol Ther. 2015;98(4):406–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valade E, Treluyer JM, Bouazza N, et al. Population pharmacokinetics of emtricitabine in HIV-1-infected adult patients. Antimicrob Agents Chemother. 2014;58(4):2256–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Baheti G, Kiser JJ, Havens PL, Fletcher CV. Plasma and intracellular population pharmacokinetic analysis of tenofovir in HIV-1-infected patients. Antimicrob Agents Chemother. 2011;55(11):5294–5299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Descovy assessment report. EMA/192941/2016 Committee for medicinal products for human use (CHMP). https://www.ema.europa.eu/en/documents/assessment-report/descovy-epar-public-assessment-report_en.pdf. Publised Feb 26, 2016. Accessed September 29, 2020.
- 26.Zhang J, Hayes S, Sadler BM,et al. Population pharmacokinetics of dolutegravir in HIV-infected treatment-naive patients. Br J Clin Pharmacol. 2015;80(3):502–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sabo JP, Lamson MJ. Leitz G, Yong CL, MacGregor TR. Pharmacokinetics of nevirapine and lamivudine in patients with HIV-1 infection. AAPS PharmSci. 2000;2(1):E1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Capparelli EV, Letendre SL, Ellis RJ, Patel P, Holland D, McCutchan JA. Population pharmacokinetics of abacavir in plasma and cerebrospinal fluid. Antimicrob Agents Chemother 2005;49(6):2504–2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Arab-Alameddine M, Fayet-Mello A, Lubomirov R, et al. Population pharmacokinetic analysis and pharmacogenetics of raltegravir in HIV-positive and healthy individuals. Antimicrob Agents Chemother. 2012;56(6):2959–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Aouri M, Barcelo C, Guidi M, et al. Population pharmacokinetics and pharmacogenetics analysis of rilpivirine in HIV-1-infected individuals. Antimicrob Agents Chemother. 2016;61(1):e00899–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang J, Hayes S, Sadler BM,et al. Population pharmacokinetics of dolutegravir in HIV-infected treatment-naive patients. Br J Clin Pharmacol. 2015;80(3):502–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Verstovsek S, Kantarjian H, Mesa RA, et al. Safety and efficacy of INCB018424, a JAK1 and JAK2 inhibitor, in myelofibrosis. N Engl J Med. 2010;363(12):1117–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.