

Abstract
Cancer vaccines, which activate the immune system against a target antigen, are attractive for prostate cancer, where multiple upregulated protein targets are identified. However, many clinical trials implementing peptides targeting these proteins have yielded suboptimal results. Using spherical nucleic acids (SNAs), we explore how precise architectural control of vaccine components can activate a robust antigen-specific immune response in comparison to clinical formulations of the same targets. The SNA vaccines incorporate peptides for human prostate-specific membrane antigen (PSMA) or T-cell receptor γ alternate reading frame protein (TARP) into an optimized architecture, resulting in high rates of immune activation and cytolytic ability in humanized mice and human peripheral blood mononuclear cells (hPBMCs). Specifically, administered SNAs elevate the production and secretion of cytokines and increase polyfunctional cytotoxic T cells and effector memory. Importantly, T cells raised from immunized mice potently kill targets, including clinically-relevant cells expressing the whole PSMA protein. Treatment of hPBMCs increases co-stimulatory markers and cytolytically active T cells. This work demonstrates the importance of vaccine structure and its ability to reformulate and elevate clinical targets. Moreover, it encourages the field to reinvestigate ineffective peptide targets and repackage them into optimally structured vaccines to harness antigen potency and enhance clinical outcomes.
Keywords: prostate cancer, vaccine immunotherapy, nanotechnology, spherical nucleic acids (SNAs), humanized mice models
Graphical Abstract
Using modular spherical nucleic acids (SNAs), we show how vaccine architecture dramatically impacts immune responses (i.e., cytolytic T cell priming) when incorporating clinical peptide targets against prostate cancer. By considering structure in vaccine design, we present a way one can reformulate failed clinical targets and enhance their potency.
1. Introduction
Cancer immunotherapy improves survival in patients with metastatic castration-resistant prostate cancer (mCRPC), where chemotherapy, radiotherapy, hormone therapy, or prostatectomy are less effective.[1] Indeed, immunotherapy is particularly promising in prostate cancer (PCa) due to the indolent nature of the disease, the large number of identified targets which have relatively few mutations,[2] and the lack of these targets having a presence in other essential organs, which reduces off-target effects.[3] While immune checkpoint blockade (ICB), consisting of monoclonal antibodies which block immune checkpoints and effectively remove the “brakes” of the immune system, has achieved a level of success in other cancers (i.e., melanoma, lung cancer, renal cell carcinoma),[4,5] clinical trials in advanced PCa have shown less positive response rates.[6–12] Another key immunotherapy implemented for PCa patients, Sipuleucel-T, where a patient’s peripheral blood mononuclear cells (PBMCs) are exposed to the target antigen and granulocyte-macrophage colony-stimulating factor (GM-CSF) and then reinfused back into the patient, is limited in scope and efficacy; it does not lower prostate-specific antigen (PSA) or delay disease progression, and only extends median survival by four months.[13] Immunotherapeutic vaccines that “turn tumors hot” towards target antigens are thus attractive, as PCa vaccines can specifically target well-established upregulated proteins, such as PSA,[14–16] prostate-specific membrane antigen (PSMA),[17–20] prostatic acid phosphatase (PAP),[21–23] or T-cell receptor (TCR) γ alternate reading frame protein (TARP),[24–26] and raise potent immune responses.[27] These proteins have been targets of various clinical trials with suboptimal levels of success.[28–31] Indeed, a major challenge in this field is the delivery of crucial vaccine components in a way that enhances immune responses.[32]
Certain nanostructures are valuable tools for delivering vaccine components and initiating immune responses, as they provide the opportunity to package the immune activator (adjuvant) and target (antigen) with precise structural control over the placement, orientation, and chemical connectivity of each component, resulting in synergistic immune responses. Indeed, such structures advance therapeutic development, as they allow one to both: 1) manipulate and deliver immunologically active components to target sites,[33–40] and 2) program the pharmacokinetics and co-delivery of these compounds.[41–47]
Spherical nucleic acids (SNAs) are nanoscale architectures particularly well suited for immune modulation due to their modularity, tunability, and facile synthesis from chemical building blocks. Indeed, they have been shown to elicit robust immune responses in mice, as well as in an ongoing Phase 1b/2 clinical trial.[44,48–50] SNAs are comprised of highly oriented and densely packed oligodeoxynucleotides (ODNs) radially arranged around a nanoparticle core. SNAs are internalized by cells more rapidly and to greater extents than linear structures, and they do not require toxic ancillary transfection reagents. They have high biocompatibility, are less susceptible to nuclease degradation, and drain to the lymph nodes upon subcutaneous injection.[44,51–55] The SNA’s modularity allows for the generation of libraries of distinct structures with minor nanoscale changes (e.g. the placement of antigen within the SNA), which has enabled the identification of structural features that best stimulate and enhance antitumor immune responses.[44,56] Implementing the findings gained from vaccine structure-function relationships in a new approach termed rational vaccinology allows one to leverage vaccine structure to enhance the potency of the components. Using this approach, vaccine structure has been shown to have a tremendous impact on the resulting efficacy and potency of the induced immune response.[44,48,49,56–58]
The consideration of structure in vaccine design postulates that previously unsuccessful clinical trials may not have failed due to the selected target, but rather because the vaccine did not deliver the components in a structure that elevated their immunogenicity. The ability to enhance vaccine efficacy through structure provides the field with an opportunity to analyze previously unsuccessful clinical candidates and reformulate them as potent vaccines. By enhancing the delivery, signaling kinetics, and intracellular trafficking of vaccine components, rational vaccinology may provide an avenue towards more successful clinical trial outcomes.
In this work, we have employed SNAs in an immunogenically heightened structural arrangement, termed the “hybridized SNA” (Scheme 1), to repurpose previously discovered PCa PSMA and TARP antigens with affinity for human leukocyte antigen serotype A2 (HLA-A2). These antigens have been extensively used in clinical trials with varying success, but previous formulations do not introduce structural control in the vaccine formulation, relying only on the simple mixing of antigen and adjuvant with no control over the placement, orientation, and chemical connectivity of the components. Our work capitalizes on the modularity of the SNA architecture to employ human clinical targets as the antigen source and demonstrate the translatability of rational vaccinology against clinical formulations. Here, we incorporated immunostimulatory unmethylated cytosine-phosphate-guanine (CpG) motif DNA as a toll-like receptor 9 (TLR9) agonist and co-functionalized the SNA with peptide antigen from one of the two aforementioned protein targets. We evaluated the immunostimulatory activity of these structures compared to the clinically-administered “free” peptide and assessed their ability to raise cytolytically active CD8+ T cells in both humanized mouse models and human PBMCs. We demonstrate herein a direct enhancement of numerous antitumor immunological parameters when the epitopes are delivered within this SNA architecture compared to a simple mixture (admix) formulation of free adjuvant and antigen. Specifically, their ability to increase pro-inflammatory cytokine production and secretion, enhance T cell memory formation, and activate human PBMCs such that the raised T cells can kill double the percentage of a clinically-relevant PCa cell line, demonstrates the important role that nanoscale structure can play in impacting future PCa formulations.
Scheme 1.
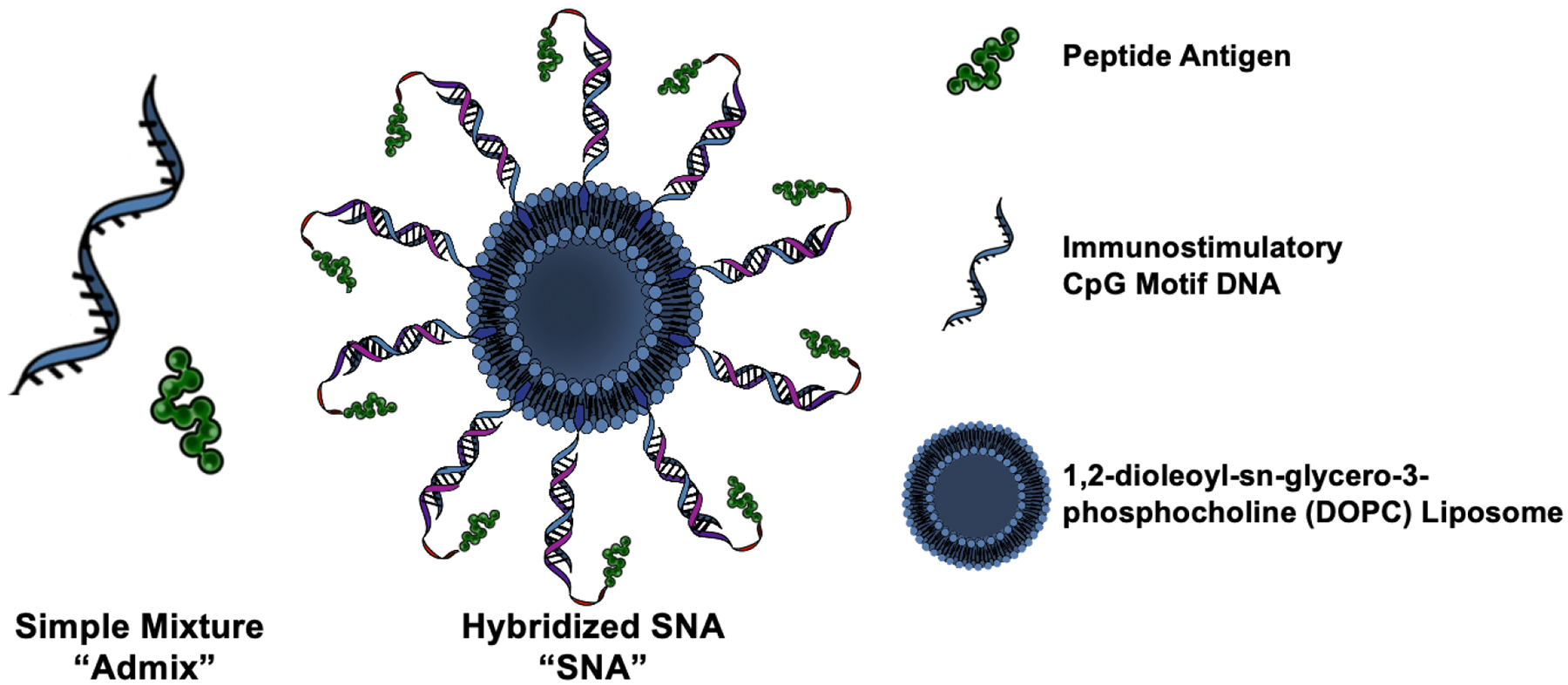
Schematic of the Spherical Nucleic Acid (SNA) hybridized vaccine structure and a simple mixture “admix” formulation.
2. Results and Discussion
2.1. Synthesis of SNAs and Enhancement of Antigen Delivery
PCa vaccines were designed to incorporate human antigens that were previously, or are being, targeted in clinical trials. In an effort to improve antigenicity and subsequently raise immune responses, we explored the role of vaccine structure in the SNA context. Previous work using model or syngeneic antigens has demonstrated that vaccine structure can influence the co-delivery of adjuvant and antigen components, the intracellular trafficking and cross-presentation of antigens, and the kinetics of antigen processing signals.[44,48] However, herein, this work capitalizes on the translational promise of rational vaccinology by utilizing targets that allow for vaccine structure to be benchmarked and validated against clinical formulations. For these studies, we selected an SNA architecture that optimizes the efficacy of the three parameters: co-delivery, trafficking, and kinetics. This “hybridized SNA” (Scheme 1) was synthesized by first conjugating a peptide antigen to a strand complementary to the CpG motif adjuvant DNA shell (“CpG complement”) (Supplementary Table S1). This was done through disulfide bond formation through a cysteine on the peptide’s N-terminus and a 3’-thiol functionalized to the CpG complement using previously published methods.[48] The peptides incorporated in the SNA, PSMA711–719 or TARP29-37-9V (Supplementary Table S2), come from two different PCa upregulated proteins and were selected to illustrate the translatability of this platform across antigen selection. In particular, the TARP29-37-9V mutated epitope-enhanced peptide was selected due to its improved immunogenicity and cytolytic capability.[24] In addition, both peptides have affinity for human HLA-A2*01,[59,60] a common allele present in multiple demographics.[61,62] Incorporating these antigens within the SNA structure offers a direct comparison to clinically formulated simple mixtures of adjuvant and peptide epitope, as they have both been used in clinical trials.[19,20,25,26,63,64] Thus, we have the opportunity to directly assess the benefit obtained through the implementation of SNA vaccine structure, as opposed to the same components in an admix form.
The formation of the two peptide-DNA conjugates was confirmed using electrospray ionization mass spectrometry (ESI-MS, Supplemental Figure S1 and S2). To obtain the duplex, the purified product and a 3’-cholesterol-terminated CpG strand were slow-cooled together. SNAs were synthesized by first preparing ~59 nm 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) liposomes using previously published methods.[44,48] The duplex was added at a 75:1 ratio to DOPC liposomes to form SNAs, equating to a surface density of ~1.1 pmol/cm2 for the duplex containing peptide. Size change measured through dynamic light scattering (DLS) confirmed the SNA product (Supplemental Figure S3), the stability of the structure was quantified in both PBS and serum-containing solution (Supplemental Figure S4), and biocompatibility was verified (Supplemental Figure S5).
We evaluated the ability of the SNA to effectively deliver peptide antigen in vitro and in vivo, as well as the ability of the SNA to protect the antigen from degradation. These parameters are important in understanding the inherent differences between the SNA and previously employed simple mixtures in the presentation of peptide in a vaccine. The SNA increases dendritic cell uptake in vitro after just one hour of incubation (Supplemental Figure S6), and also enhances in vivo accumulation to sites of large immune cell populations (lymph node, spleen). Within splenic DCs, the SNA enhances the number of cells containing antigen and the total amount of antigen in cells (Supplemental Figure S7). Moreover, the SNA is capable of improving peptide stability, as free peptide in the presence of an enzyme is degraded within 5 min, while peptide as part of the SNA remains present for longer during enzymatic incubation (Supplemental Figure S8). These results demonstrate how the SNA vaccine improves peptide antigen delivery.
2.2. Generation of Antigen-specific cytotoxic T lymphocyte responses in humanized mice through SNA Immunization
We evaluated the ability of these SNA vaccines to raise antigen-specific responses in vivo. Since these peptides present on HLA-A2 and not on H2 major histocompatibility complexes (MHCs), we employed humanized immunocompetent AAD transgenic mice.[65] These mice, which express an interspecies hybrid class I MHC gene with alpha-1 and alpha-2 domains of the human HLA-A2.1 gene and murine H-2Dd alpha-3 transmembrane and cytoplasmic domains, are capable of effectively modeling the raised human T cell response to HLA-A2 antigens in an immunocompetent organism. In this case, since the murine immune system is propagating the immune response, we utilized CpG 1826 DNA in the SNA architecture, as this CpG motif is specific for murine TLR9 activation. Using these constructs, mice were vaccinated every other week for three total injections (24 nmol or 30 nmol by DNA and peptide for TARP29-37-9V and PSMA711-719, respectively). On day 35, splenocytes were harvested to determine the ability of these vaccines to raise specific immune responses towards the peptide antigen (Figure 1A). This schedule was utilized based on previous work.[44,48] SNAs containing either the PSMA or TARP epitope elevated the production of the pro-inflammatory cytokine, IFN-γ, and generated a larger percentage of polyfunctional CD8+ T cells, as quantified by the double-positive CD8+ population of IFN-γ and CD107a, a marker of T cell degranulation, following ex vivo stimulation with the peptide (Figure 1B–C). Polyfunctional T cells, which produce more than one cytokine simultaneously, have been considered potent effector T cells against chronic infections and tumors.[66–68] The increase in the proportion of these T cells ranged from 2 to 5-fold. In addition, the SNA increased IFN-γ cytokine secretion, as measured by spot forming cells (SFCs) through enzyme-linked immune absorbent spot (ELISpot), by either ca. 11-fold for PSMA711-719 or 85-fold for TARP29-37-9V compared to the simple mixture (Figure 1D–E). SNAs formulated with either peptide enhanced CD8+ effector memory phenotypes compared to naïve mice, and when formulated with PSMA711-719, CD8+ effector memory (CD62L−CD44+) for the SNA was enhanced compared to the simple mixture (Figure 1F). Moreover, antigen presentation of the PSMA711-719 epitope was enhanced as a result of SNA immunization, as measured by the percentage of antigen-specific T cells capable of differentiating as a direct result of enhanced DC antigen presentation (Figure 1G). SNA immunization enhanced this PSMA antigen-specific CD8+ T cell population >3-fold compared to immunization with the simple mixture. In general, greater CD8+ responses were observed with PSMA711-719, likely a consequence of greater inherent immunogenicity of the peptide.
Figure 1.
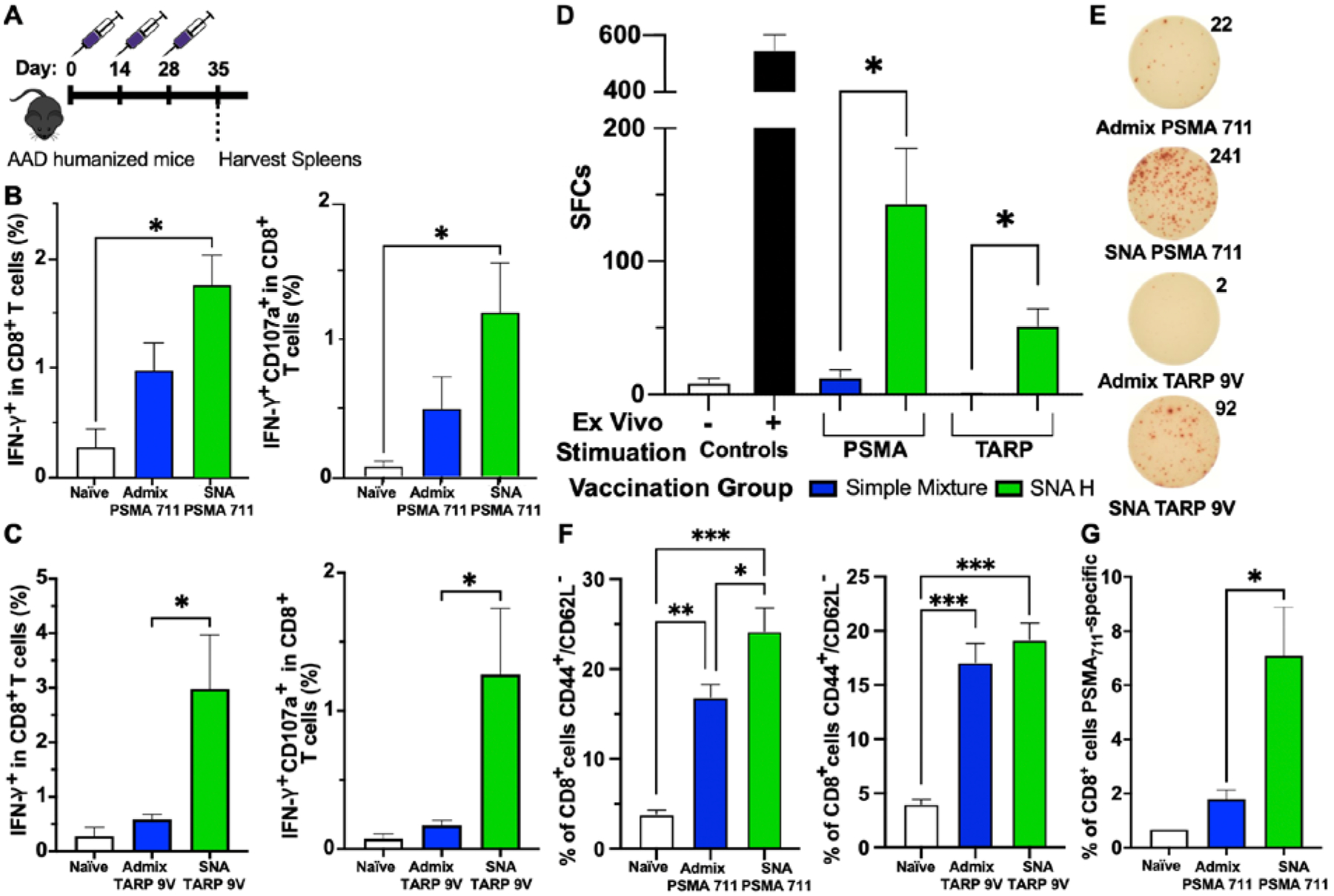
Vaccination of humanized mice with SNAs formulated with PSMA or TARP antigens. A) Injection timeline for immunizations. Male (PSMA) or female (TARP) AAD humanized mice were immunized subcutaneously three times with either a 1:1 simple mixture of peptide (PSMA or TARP) and adjuvant, or SNA. One week after the third injection, splenocytes were harvested and analyzed for antigen-specific responses. B and C) Immunization with SNA containing either antigen increased the percentage of CD8+ T cells that were positive for IFN-γ cytokine production (left) and double positive for IFN-γ and CD107a (degranulation marker, right). D) Secretion of IFN-γ by splenic CD8+ T cells was assessed via ELISpot assay 48 h after restimulation ex vivo with either the PSMA or TARP antigen. E) Representative ELISpot images and counts from each group. F) Effector memory CTL phenotype (CD44+ and CD62L−) was measured within CD8+ T cells. G) Percentage of CD8+ T cells that are PSMA711–719-specific, measured through staining with an antigen-specific pentamer. All data is presented as mean ± SEM with n = 5 – 10 per treatment group. Significance between groups was determined (B, C, F, G) by one-way ANOVA with Tukey’s multiple comparison test and (D) using two-tailed unpaired t tests. *p<0.05; **p<0.01; ***p<0.001.
2.3. Cytolytic Capabilities of CD8+ T cells raised from SNA Immunization
We next assessed if SNA immunization provided enhanced cytolytic capabilities for raised CD8+ T cells towards cells which display the peptide antigen. The capacity of raised T cells to kill antigen-displaying target cells ex vivo at 3 or 4 ratios of effector to target (E/T) was quantified through either the percentage of target cells displaying apoptotic marker Annexin V or the combination apoptotic and necrotic markers Annexin V and 7-AAD after 24 h incubation with isolated splenic CD8+ T cells from vaccinated mice. In peptide-pulsed T2 cells, which can only present exogenous peptides due to TAP deficiency,[69] the differences between SNA and simple mixture raised T cells was most pronounced (Figure 2A–B). SNA formulated with PSMA711-719 exhibited at minimum a 3-fold increase in cytotoxicity (late apoptosis) at all three tested ratios (50:1, 25:1, and 12.5:1) compared to the simple mixture raised T cells (Figure 2A). SNA formulated with TARP29-37-9V significantly increased the killing ability by 43% at the highest ratio tested (100:1) to a final percentage of 23% cells in the T2 target population that expressed both apoptotic and necrotic markers, and by 29% at the lowest ratio tested (12.5:1) to a final percentage of 17% cells in the T2 target population (Figure 2B). This indicates that while the vaccine formulated with TARP29-37-9V is not as potent as the one with the PSMA711-719 peptide in this cytolytic assay, there is still an enhancement through immunization using SNA structure.
Figure 2.
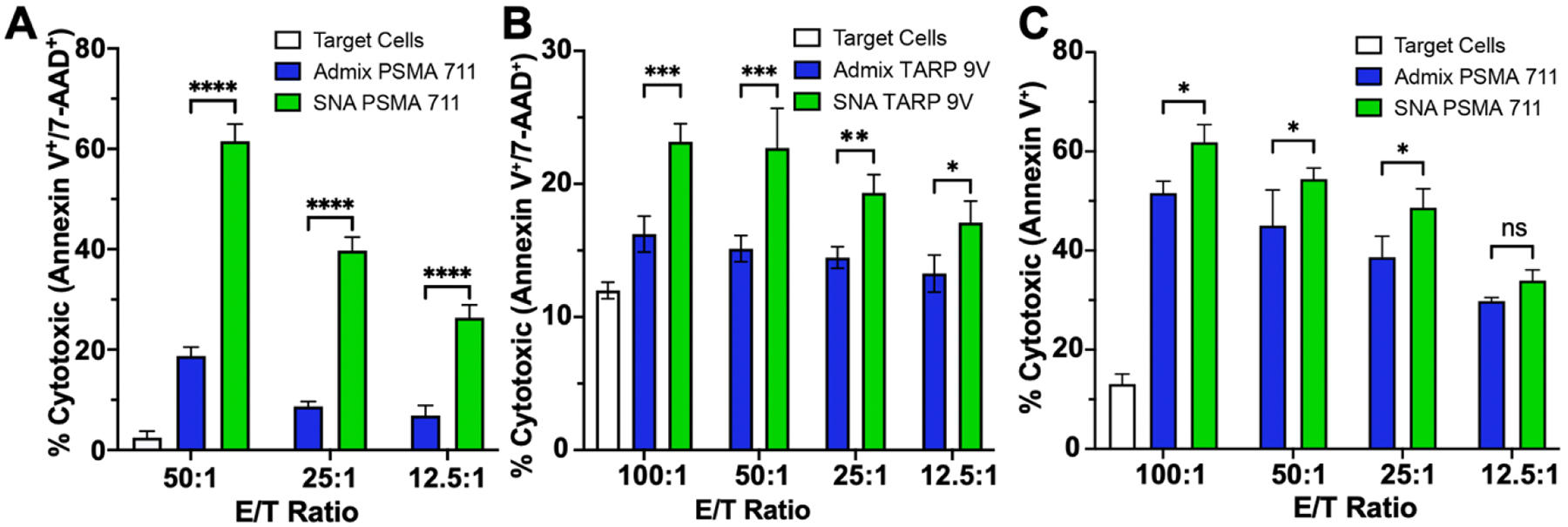
Antigen-specific killing by splenic CD8+ cytotoxic T lymphocytes (CTLs) raised from vaccination against A) PSMA peptide pulsed T2 cells, B) TARP peptide pulsed T2 cells, or C) PC3-PSMA cells at different effector to target (T cell/target cells, E/T) ratios. Apoptosis and/or necrosis quantified via Annexin V and 7-aminoactinomycin D (7-AAD) stain after 24 h co-culture. All data is presented as mean ± SD with n = 3 per treatment group. Significance between groups is shown and was analyzed using two-way ANOVA with Sidak’s multiple comparisons test to analyze differences between treatment groups at each ratio. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; ns=non-significant.
Due to the higher potency of raised T cells from PSMA711-719 immunized mice, we targeted these T cells against a clinically-relevant antigen-presenting cancer cell line, PC3-PSMA (Figure 2C). This human PCa cell line was transfected to express the full PSMA protein,[70] and thus represents a realistic clinical target. Even though humanized mice were only vaccinated with formulations containing one epitope, this epitope’s immunogenicity was significant enough to induce expression of the Annexin V apoptosis marker in ca. 30–60% of the population of PC3-PSMA target cells across all tested ratios. Importantly, CD8+ T cells raised from SNA-immunized mice enhanced the killing ability significantly for the three highest tested ratios compared to the simple mixture formulation. T cells raised from vaccination with TARP29-37-9V were also tested against a clinically relevant cell line natively expressing the TARP protein, MCF-7. Trends were similar to those observed for the PC3-PSMA system but with lower overall killing percentages, which is expected due to the decreased overall targeting ability of the TARP29-37-9V raised T cells to kill pulsed T2 cells expressing a single epitope (Supplementary Figure S9). Overall, these results demonstrate that: 1) antigen immunogenicity can be enhanced by the architecture of a vaccine and 2) optimal vaccine architecture can improve responses compared to those that were previously observed in clinical trials.[20,71] The enhancement of immunogenicity across both peptides indicates the generalizability of the SNA platform and supports the notion that structure can be leveraged to elicit more potent cytolytic T cell immune responses. This ultimately provides the opportunity to question whether previously unsuccessful clinical targets have the incorrect epitope or simply the incorrect vaccine structure, and it motivates the reexamination of these targets in a clinical setting where ideal vaccine structures can be realized.
2.4. SNA Activation in vitro with human peripheral blood mononuclear cells (hPBMCs)
Due to the robust activation of splenocytes and CD8+ T cell responses in humanized mice, we applied and assessed the ability of SNA structures to activate essential human professional HLA-DR+ antigen presenting cells (APCs), such as CD123+ plasmacytoid dendritic cells (pDCs). pDCs have recently been implicated in antitumor immunity[72] due to their role as major producers of predominately type I interferons (IFNs).[73] Translation of findings to human donor cells is essential in assessing the SNA’s ability to robustly activate human pathways, as those observed in humanized mice still rely on CpG 1826 to activate murine TLR9. Validating our in vivo findings with characterization of human activation using CpG 7909 provides a realistic picture of the ability of the SNA to enhance previously targeted clinical peptide formulations for PCa patients. For these analyses, we employed the PSMA SNA formulations as these induced higher levels of T cell killing.
Three different treatment groups were incubated in vitro with human PBMCs: a simple mixture of CpG 7909 and PSMA peptide, the SNA employed herein containing PSMA peptide, and an SNA containing only the TLR9 adjuvant shell as a comparison for the adaptive nature of the response (termed SNATLR9). At two tested concentrations (25 and 100 nM by DNA—and peptide for the simple mixture and SNA groups) and two time points (24 and 48 h), the SNA induced significantly greater expression of co-stimulatory marker CD83 in pDCs as well as secretion of pro-inflammatory cytokines interleukin-1β (IL-1β) and tumor necrosis factor (TNF) (Figure 3A–B, Supplementary Figure S10–12). After 24 h at the low concentration, the simple mixture and SNATLR9 elevated the percentage of pDCs expressing CD83 compared to the untreated control to final values of 15% and 14% of the population, compared to 22% of the pDC population that expressed the CD83 co-stimulatory marker with SNA incubation. The same trend was observed at the higher concentration, where the average percentage of the population expressing CD83 is 19%, 18%, and 26% for the admix, SNATLR9, and SNA-containing PSMA, respectively. After 48 h of incubation with these different formulations, similar trends hold with the SNA elevating the percentage of the population that expresses the CD83 co-stimulatory marker at both tested concentrations (final levels of 32% and 37% at 25 and 100 nM, respectively). Of particular note is that only the SNA significantly increased CD83 expression from 24 to 48 h (Supplementary Figure S13), demonstrating the durability and prolonged effect of the hPBMC response to SNA treatment compared to other formulations.
Figure 3.
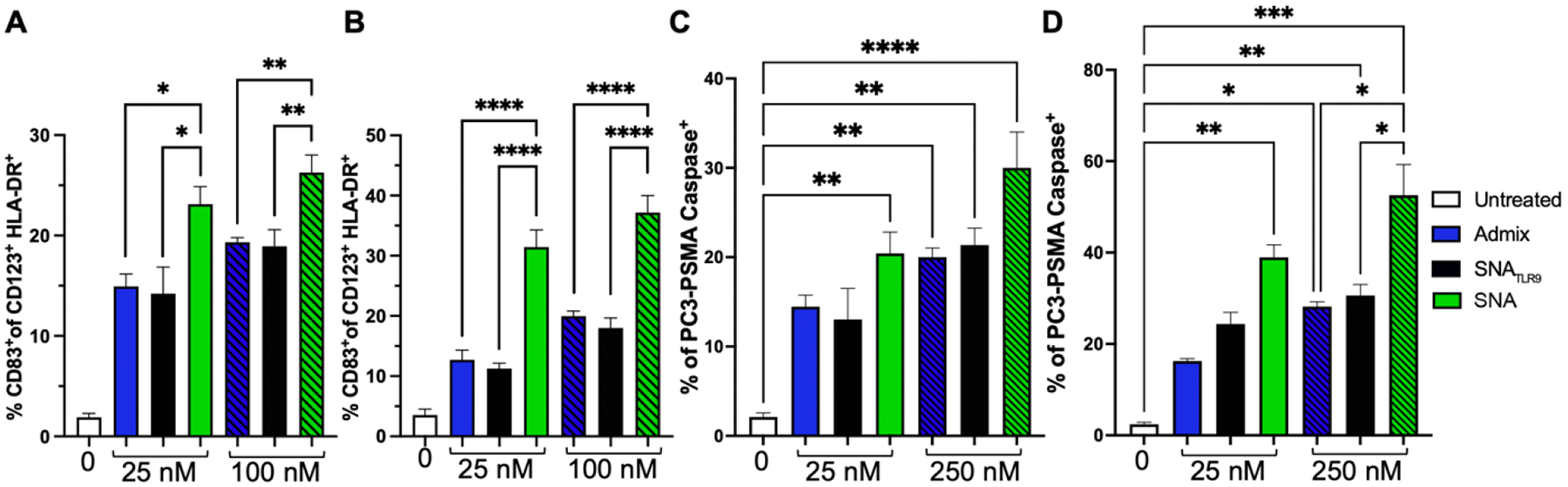
SNAs are capable of activating human PMBCs to raise robust T cell responses. A) Activation of professional APCs (CD123+ pDCs) was quantified through presence of co-stimulatory marker CD83 at 24 h and B) 48 h after incubation with admix, SNATLR9, or SNA targeting PSMA. CD8+ T cells are raised from incubation of hPBMCs with treatment groups for 24 h C) or 48 h D) and are targeted against human PCa cells, PC3-PSMA, for 2 h. The expression of the apoptotic marker, Caspase-3 was measured. The percentage of PCa tumor cells expressing Caspase is shown. All data is presented as mean ± SEM with n = 3 – 4 per treatment group. Significance between groups is shown and was analyzed using one-way ANOVA with Sidak’s multiple comparisons test to analyze differences between treatment groups at each concentration. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
2.5. Antigen-specific cytotoxic T lymphocyte (CTL) responses from human PBMCs
To assess the ability of the different treatments to trigger antigen-specific CTL responses in hPBMCs, we compared the activation of CTLs after treatment with the same three groups as above: a simple mixture, SNA-containing PSMA, and SNATLR9. After 24 or 48 h, T cells were selected from the PBMC population and co-cultured separately for 2 h with PC3-PSMA target cells. Expression of Caspase-3, an enzyme indicative of cellular apoptosis, was measured by flow cytometry to quantify the extent and specificity of T cell-mediated cytotoxicity. After 24 h of culture with the PBMCs at 25 nM, only the SNA formulated with PSMA711-719 significantly increased both the percentage of cells expressing apoptotic protein Caspase-3 (Figure 3C), as well as the median fluorescence intensity (MFI) of Caspase-3 signal (gating strategy and MFI data in Supplementary Figures S14–15). Increasing the treatment incubation concentration with PBMCs to 100 nM did increase the values for both the simple mixture and SNATLR9. However, the apoptotic target cell population was greatest after SNA incubation, where 30% of the population of PC3-PSMA cells were Caspase positive, ca. 10% more than the other two treatments. These trends became more pronounced after 48 h of incubation, with 25 nM of SNA inducing a ca. 2.5-fold increase (from 16% to 39%) compared to admix and a ca. 1.6-fold increase (from 24% to 39%) compared to SNATLR9 in the percentage of the tumor cell population expressing Caspase-3 (Figure 3D). At 100 nM treatment, the SNA-containing PSMA is capable of prompting >50% of Caspase-3 expression amongst the PC3-PSMA tumor cells. This again demonstrates the superior ability of the SNA to successfully initiate ex vivo tumor cell death in human samples and highlights the translational potential of the SNA construct.
3. Conclusion
Considering and employing structure in vaccine design can profoundly impact the resulting efficacy. This is particularly noteworthy when considering the issues in translating promising initial clinical trial results to significant endpoint efficacies, a dilemma for several completed failed PCa clinical trials. This trend in failing to reach endpoint efficacy, likely due to an inability to generate an effective antitumor immune response, suggests that previous clinical formulations may have simply utilized suboptimal overall vaccine structure for delivery rather than incorrect antigenic targets. In this work, we furthered this notion by directly demonstrating that vaccine structure can repurpose clinically-employed antigen targets for two different PCa upregulated proteins. Using humanized mice that recapitulate the human immune system and human donor cells, we achieved heightened immune responses against compositionally equivalent formulations by packaging antigens into a particular SNA vaccine structure.
Taken together, these data demonstrate the power of vaccine design and structure to reformulate clinical targets through specific chemical arrangements that elevate their potency. The insights gained from this work can be applied broadly to include various other clinically identified antigenic PCa epitopes, combinations of those epitopes in structured-informed vaccine design, or antigens from other proteins across cancers. We reveal the direct enhancement of antitumor potency possible when human antigens are delivered with SNA architecture compared to clinically-used free simple mixtures. Thus, this work has important implications on vaccine development and translation, and urges the field to consider vaccine structure in addition to composition when identifying targets, testing them preclinically, and translating them to clinical trials.
4. Experimental Section/Methods
Materials and Animals:
Unless otherwise noted, all reagents were purchased commercially and were used as received. Oligonucleotides were synthesized as described below. Peptides were purchased from Northwestern’s Peptide Synthesis core. Chemicals were purchased from suppliers listed in parentheses. B6.Cg-Immp2lTg(HLA-A/H2-D)2Enge/J (AAD) mice, hemizygous male or female, and C57BL/6 mice, female, 8–12 weeks old, were purchased from Jackson Laboratory and bred in house. Mice were used in accordance with all national and local guidelines and regulations and protocols performed were approved by the institutional animal use committee at Northwestern University (IUCAC). T2 and MCF-7 cells were purchased from ATCC. PC3-PSMA cells were kindly provided by Dr. Timothy Kuzel. Human PBMCs were obtained from Zen-bio (#SER-PBMC-200P-F). Antibodies were purchased from Biolegend or BD and clones are provided in parentheses following the antibody, with the exception of the fixable live/dead which was purchased from Invitrogen (L23105).
Oligonucleotide Synthesis and Purification:
Oligonucleotides were synthesized on an ABI 394 synthesizer using standard phosphoramidite chemistry with phosphate or phosphorothioate backbones, as indicated in the table (Supplementary Table S1). Following synthesis, strands were deprotected using 1:1 solution of 37% Ammonium hydroxide/40% methylamine at 55 °C for 35 min. Strands were then purified using a C18 or C4 (for strands containing cholesterol) column on reverse phase HPLC chromatography and peaks were collected as fractions for subsequent lyophilization. The dimethoxytrityl (DMT) group was removed from the product strands by incubation in 20% aqueous acetic acid at RT for 1 h, followed by 3 washes with ethyl acetate to remove DMT. The final product, in the acetic acid phase, was lyophilized and resuspended in deionized water (diH2O). Oligonucleotide concentration was measured using UV-Vis absorption at 260 nm with an extinction coefficient calculated through the IDT OligoAnalyzer online tool. These coefficients are listed in Supplementary Table S1.
Oligonucleotide-peptide Conjugate Synthesis and Purification:
Thiol-functionalized oligonucleotides in diH2O (3’thiol-CpG complement) were reduced to generate a free thiol for future reactions. Reduction was done using dithiothreitol (DTT, Sigma) dissolved in phosphate buffered saline (PBS) pH 8–8.5 at a final concentration of 100 mM at RT for 40 min. This solution was then washed in a 3 kDa molecular weight cut off (MWCO) spin filter (Amicon) 4x with diH2O. Reduced DNA (<1 mM) was reacted overnight at 50 °C with 0.5 M 2,2’-dithiodipyridine (Aldrithiol-2, Sigma, 143049) in ethanol, in a final solvent ratio of diH2O:ethanol = 90:10. The next day, solutions were centrifuged at 17,000 × g for 2 min to pellet unreacted aldrithiol-2 and the supernatant was transferred to 3 kDa MWCO spin filters for 4 washes with diH2O. After washing, the concentration was measured using UV-Vis absorption at 260 nm using the same extinction coefficient as the oligonucleotide (Supplementary Table S1). Cysteine-containing peptides were reacted in excess with the activated oligonucleotides in a molar ratio ranging from 8:1 to 5:1, in diH2O containing 100 mM tris pH 8. This reaction was run overnight at 50 °C. The solution was then centrifuged at 17,000 × g for 2 min to pellet any crashed out peptide, and the supernatant was transferred to 3 kDa MWCO spin filters for 3 washes with diH2O. The volume was concentrated to <500 μL and the solutions were purified by preparatory scale denaturing (8 M urea) 15% PAGE gels (no more than 0.5 μmol by DNA loaded onto a single gel). Gels were run for 30 min at 175 V, and 3–4 h at 350 V, and subsequently imaged using a UV lamp to cut out desired bands. Cut-out gel bands were crushed and product was collected by 3–4 washes with 1x TBE buffer. Product mass was confirmed by electrospray ionization mass spectrometry (ESI-MS), and concentrations were measured by UV-Vis at 260 nm assuming an extinction coefficient of only the DNA.
SNA Synthesis:
SNAs were synthesized as reported previously.[44,48] Briefly, dried lipid films of 50 mg of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC, Avanti Polar Lipids) were hydrated with 3 mL of PBS, and subjected to 10 freeze-thaws in liquid nitrogen and then sonication. Liposomes were then extruded using sequential high-pressure extrusion using polycarbonate filters with pore sizes of 200, 100, 80, and 50 nm; liposomes were passed through each pore size 3 times. Following extrusion, liposomes were concentrated down to ~1–2 mL using 100 kDa MWCO spin filters. Concentration was determined using a phosphatidylcholine (PC) assay kit (Sigma, MAK049–1KT), assuming a 50 nm liposome contains 18140 lipids per liposome.[52] Size was determined using dynamic light scattering (DLS). Purified oligonucleotide-peptide conjugates were mixed in a 1:1 molar ratio with complementary 3’-cholesterol terminated CpG DNA and centrivapped overnight. The next day, ~20–30 μL of duplex buffer (IDT) was added and the solution was slow cooled to duplex the strands following the program: 70 °C for 10 min, 23 °C for 1.5 h, 4 °C for at least 1 h. Duplex was added to a solution of synthesized liposomes at a ratio of 75 duplexes per liposome, which has been previously established as the maximum loading for 50 nm liposomes.[52] This mixture was incubated at 37 °C overnight, and subsequently stored at 4 °C.
SNA Stability measured through Förster resonance energy transfer (FRET):
SNAs were synthesized as described above, except when preparing FRET liposomes, 1 mol % of the lipid film was comprised of 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl) (Avanti) to fluorescently label the liposome core and allow for FRET between a rhodamine-labeled liposome and Cy5-labeled DNA as the shell (Supplemental Table S1). These SNAs were incubated in either PBS or 10 % normal fetal bovine serum (FBS) in PBS and the fluorescence was monitored over time on a Cytation5 Multi-Mode Reader (FRET exc. λ = 550 nm, em. λ = 660 nm; rhodamine λ = 550 nm, em. λ = 610 nm). FRET % was calculated by dividing the FRET signal by the rhodamine signal and converting this value percentage. After 8 hours, 0.1% SDS was added which destroys liposome structures, to provide the signal at which all dissociation has occurred.
Peptide Degradation Analysis:
Vaccines were assessed for resistance to peptide degradation through Native PAGE gels using fluorescently-labeled peptide. Either free peptide or peptide as part of the SNA (100 pmol) was incubated for different timepoints with 100 μg/mL Proteinase K enzyme (ThermoFisher) in PBS at at 37 °C. At selected timepoints, samples were immediately frozen in liquid nitrogen and stored in a −80 °C freezer until all time points were collected to stop enzyme degradation of the peptide. Time points were compared by 15% Native PAGE gels which were run at 80 V at 4 °C for ~1.5 h. Upon completion, gels were imaged using a ChemiDoc Gel Scanner (BioRad). Densitometry analysis using ImageJ software was conducted on the gels to determine the extent of degradation over time. All timepoints contained at least three replicates.
In Vivo Immunization:
AAD male or female mice, containing the α−1 and α−2 domains of the human HLA-A2.1 gene, and the α−3 transmembrane and cytoplasmic domains of the mouse H-2Dd gene, all under the direction of the human HLA-A2.1 promoter,[65] were subcutaneously immunized fortnightly 3 times with different treatments. Treatments included, for males, simple mixture (admix, 30 nmol peptide (PSMA) and 30 nmol CpG 1826 DNA)), or SNA (30 nmol by PSMA peptide and CpG 1826), and for females, simple mixture (admix, 24 nmol peptide (TARP) and 24 nmol CpG 1826 DNA), or SNA (24 nmol by TARP peptide and CpG 1826). Volume of treatment injected was kept below 200 μL. One week after the final immunization, mice were sacrificed, and spleens were harvested for subsequent immune assessment.
Harvest Procedure:
Removed spleens were temporarily stored in 3–5 mL of RPMI containing 10% heat-inactivated fetal bovine serum (HI-FBS) and 1% Penicillin-Streptomycin (denoted herein as RPMI +/+) until all spleens were collected and brought up from the animal facility. Spleens were passed through a 70 μm cell strainer with a flow of PBS. The cells were then centrifuged at 1200 rpm for 5 min, after which supernatant was removed, and the cells were resuspended in 2 mL ACK lysing buffer (Gibco) for 4 min. To dilute the lysing buffer, PBS was then added to a final volume of 30 mL, and the cells were counted prior to centrifugation to resuspend in RPMI +/+ media at a concentration of 1×108 cells mL−1.
Ifn-γ cytokine Production:
CD8+ T cells were restimulated ex vivo to assess antigen-specific intracellular IFN-γ production. 4 × 106 splenocytes were cultured for 4 h at 37 °C in a 5% CO2 incubator with a 450 μL solution in RPMI+/+ media containing: peptide (10 μg/mL), monensin (2 μM), brefeldin A (5 μg/mL), and CD107a (clone LAMP-1) antibody (0.5 μL). After the 4 h incubation, cells were centrifuged at 1200 rpm for 5 min, aspirated, and washed with 600 μL PBS, prior to 15 min staining with surface antibodies (0.5 μL per sample each of: fixable live/dead-UV, CD8 (clone 53–6.7) -PE, CD4 (clone GK1.5) -APC) at 4 °C. Cells were washed with 600 μL PBS, centrifuged at 1200 rpm for 5 min, aspirated, and resuspended in 100 μL of Cytofix Fixation and Permeabilization solution (BD, 554722) for 20 min at 4 °C. Cells were then washed with 600 μL of Perm/Wash Buffer (BD, 554723), centrifuged at 1200 rpm for 5 min, aspirated, and resuspended in 100 μL of Perm/Wash Buffer containing the intracellular antibody IFN-γ (clone XMG1.2) -PE/Cy7 (0.5 μL per sample). Samples were stored at 4 °C prior to flow cytometry analysis.
T cell Memory Phenotyping:
CD8+ T cells were assessed for effector memory status through staining with antibodies against CD44 and CD62L. Briefly, 3 × 106 splenocytes were washed with 600 μL PBS, and stained for 15 min with surface antibodies (0.5 μL per sample each of: fixable live/dead -UV, CD8 (clone 53–6.7) -APC, CD4 (clone GK1.5) -FITC, CD44 (clone IM7) -BV421, and CD62L (clone MEL14) -PE/Cy7) at 4 °C. Cells were washed with 600 μL PBS, centrifuged at 1200 rpm for 5 min, aspirated, and resuspended in 100 μL of Fixation buffer (Biolegend, 420801). Samples were stored at 4 °C prior to flow cytometry analysis.
ELISpot Assay:
The ELISpot assay was carried out using the commercially available BD Mouse INF-γ ELISPOT Set (#551083) following the instructions from the manufacturer. Briefly, the plate was coated overnight at 4 °C with capture antibody. After this incubation, the plate was washed once with RPMI+/+ media and subsequently blocked for 2 h with 200 μL of RPMI+/+ media. The blocking buffer was washed away and replaced with 2 × 105 splenocytes in 100 μL RPMI+/+. To each well, an additional 100 μL of either antigen, non-specific peptide, media (negative control), or positive control solutions were added (antigen and non-specific peptide were added to a final concentration of 5 μg/mL; positive control = mixture of anti-CD3 and anti-CD28 antibodies at a final concentration of 2 μg/mL each) and left in an incubator at 37 °C in 5% CO2 for 48 h. After this incubation, the plate was washed, and detection antibody, enzyme conjugate, and chromogenic substrate were added according to the manufacturer’s instructions. The dried plate was imaged and analyzed using a CTL Immunospot imager.
Antigen-specific Pentamer Staining:
Splenic CD8+ T cells were assessed for antigen-specificity for the PSMA711 epitope after immunizations and harvesting. 1–2 × 106 splenocytes were washed with 600 μL PBS, and stained following the manufacturer’s instructions (ProImmune) for pentamers. Surface antibodies included fixable live/dead -UV, CD8 (clone 53–6.7) -APC, CD19 (clone 6D5) -FITC, and Pentamer PSMA711–719 - PE. After staining, cells were fixed with 100 μL of Fixation buffer (Biolegend, 420801) and samples were stored at 4 °C prior to flow cytometry analysis.
Ex vivo Antigen-Specific T cell Killing Assay:
CD8+ T cells were magnetically isolated from splenocyte solutions following the protocol provided for the murine CD8a Positive Selection Kit II (StemCell Technologies, #18953). After isolation, cells were resuspended in RPMI+/+ media, and counted. Target cells (T2, PC3-PSMA, or MCF-7) were stained with efluor450 (eBioscience) following their protocol. After staining, T2 cells were pulsed with peptide (either PSMA or TARP) at a concentration of 10 μg/mL in RPMI+/+ media (total volume = 3 mL) for 2 h. Cells were counted after staining (for PC3-PSMA or MCF-7 cells) or pulsing (for T2 cells) and were plated in a 96-well round bottom plate with 5000 cells per well in a volume of 100 μL. Cells were allowed to recover for 30 min in the incubator at 37 °C in 5% CO2 prior to plating of isolated T cells at specified ratios of T cells (effector) to target cells. Cells were co-cultured together for ~20–24 h in the incubator at 37 °C in 5% CO2. The following day, cells were collected into tubes, first by pipetting and for adherent cells, then using 50 μL of trypsin followed by 200 μL of RPMI+/+ media. Samples were washed once with 600 μL of PBS, centrifuged at 1200 rpm for 5 min, aspirated, and resuspended in 100 μL of Annexin V Binding Buffer (Biolegend, 422201) containing 0.5 μL each of 7-AAD (Fisher, 50169259) and Annexin V (Biolegend, 640906). Cells were stained for 15 min at RT prior to flow cytometry analysis.
Cell culture:
All cells were maintained at 37 °C in a 5% CO2 incubator. T2 cells were cultured in complete Iscove’s Modified Dulbecco’s Medium supplemented with 20% HI-FBS and 1% Penicillin-Streptomycin. PC3-PSMA cells were cultured in Kaighn’s Modification of Ham’s F-12 Medium (F-12K) supplemented with 10% HI-FBS and 1% Penicillin-Streptomycin. MCF-7 cells were cultured in Minimum Essential Medium with 10% HI-FBS, 1% Non-Essential Amino Acids (NEAA), and 1% Penicillin-Streptomycin.
In Vivo Biodistribution:
C57BL/6 female mice were subcutaneously immunized with 100 μL volume of Cy5-labeled treatments: simple mixture (admix, 6 nmol peptide (PSMA) and 6 nmol CpG 1826 DNA), or SNA (6 nmol by PSMA peptide and CpG 1826). After 24 h, mice were sacrificed, and lymph nodes and spleens were harvested. Removed organs were assessed for biodistribution through measurements of organ fluorescence using an IVIS Spectrum (PerkinElmer) in vivo imaging system. Resulting images were subsequently analyzed using Living Image software (PerkinElmer). Measurements were acquired from three mice per group (n=3). After imaging, splenocytes were isolated as described in the Harvest Procedure. 30 μL of splenocytes at a concentration of 1×108 cells mL−1 were put into flow tubes and the samples were washed with PBS, centrifuged at 1200 rpm for 5 min, after which supernatant was aspirated and samples were stained at 4 °C for 15 min in 100 μL PBS containing 0.5 μL each of: fixable live/dead -UV and CD11c (clone N418) -BV421. Cells were then washed with 600 μL PBS, centrifuged at 1200 rpm for 5 min, aspirated, and resuspended in 100 μL of fixation buffer (Biolegend, 420801). Samples were stored at 4 °C prior to flow cytometry analysis.
In vitro human PBMC Viability:
Human PBMCs were thawed from storage in liquid nitrogen in a water bath. Cells were mixed and added to 10 mL of RPMI +/+. The solution was centrifuged at 300 × g for 10 min to pellet the cells. The supernatant was aspirated, and the cells were resuspended in 4 mL of media to count. 3 × 105 cells in 100 μL volume were added to flow tubes, and cells were left in a 37 °C in a 5% CO2 incubator to recover while samples were prepared. Samples were diluted in RPMI +/+ media to 2× the concentration of the final desired concentration, and 100 μL of each was added to the respective tube with cells. After 4 h incubation with either admix or SNA, 600 μL of PBS was added to each tube and the tubes were spun at 1200 rpm for 5 min, after which the supernatant was aspirated, and the samples were stained at 4 °C for 15 min in 100 μL PBS containing a fixable live/dead antibody (0.5 μL of: fixable live/dead -UV). Cells were then washed with 600 μL PBS, centrifuged at 1200 rpm for 5 min, aspirated, and resuspended in 100 μL of fixation buffer (Biolegend, 420801). Samples were stored at 4 °C prior to flow cytometry analysis.
In vitro human PBMC Uptake:
Human PBMCs were thawed from storage in liquid nitrogen as described in In vitro human PBMC Viability. 3 × 105 cells in 100 μL volume were added to flow tubes, and cells were left in a 37 °C in a 5% CO2 incubator to recover while samples were prepared. Samples were diluted in RPMI +/+ media to 2× the concentration of the final desired concentration, and 100 μL of each was added to the respective tube with cells. After 1 and 4 h incubation with either admix or SNA, 600 μL of PBS was added to each tube and the tubes were spun at 1200 rpm for 5 min, after which the supernatant was aspirated, and the samples were stained at 4 °C for 15 min in 100 μL PBS containing: 0.5 μL of: fixable live/dead -UV, CD11c (clone B-ly6) -BV421. Cells were then washed with 600 μL PBS, centrifuged at 1200 rpm for 5 min, aspirated, and resuspended in 100 μL of fixation buffer (Biolegend, 420801). Samples were stored at 4 °C prior to flow cytometry analysis.
In vitro human PBMC activation:
Human PBMCs were thawed from storage in liquid nitrogen as described in In vitro human PBMC Viability. 2 × 105 cells in 100 μL volume were added to each well of a 96 well round bottom plate, and cells were left in a 37 °C in a 5% CO2 incubator to recover while samples were prepared. Samples were diluted in RPMI +/+ media to 2× the concentration of the final desired concentration, and 100 μL of each was added to the respective well with cells. After 24 or 48 h incubation with either admix, SNATLR9, or SNA, the plate was centrifuged at 300 × g for 5 min and 180 μL of supernatant was collected in a separate 96 well flat bottom plate for Cytokine Assessment. For Supplementary Figure S12, a treatment group of admix supplemented with 59 nm DOPC liposomes was used at the same concentration of components as the SNA. The cells were washed with PBS and the entire volume was transferred to flow tubes. The tubes were spun at 1200 rpm for 5 min, after which the supernatant was aspirated, and the samples were stained in 100 μL PBS containing surface antibodies (0.5 μL per sample each of: fixable live/dead -UV, CD83 (clone HB15e) -BV421, CD123 (clone 6H6) -APC, HLA-DR (clone L243) -PerCP-Cy5.5) at 4 °C for 15 min. Cells were washed with 600 μL PBS, centrifuged at 1200 rpm for 5 min, aspirated, and resuspended in 100 μL of fixation buffer (Biolegend, 420801). Samples were stored at 4 °C prior to flow cytometry analysis.
Cytokine Assessment from PBMC activation:
Collected supernatant from the In vitro human PBMC activation was used in the purchased Human Inflammatory Cytokine Kit (BD, 551811). The manufacturer’s instructions were followed, and samples were diluted 3× or 8× for supernatant coming from the 25 or 100 nM treatment, respectively. Analysis was run on an A3 Symphony flow cytometer and data were fit to the standard curve following the manufacturer’s protocol.
In vitro human PBMC co-culture and killing of PC3-PSMA:
Human PBMCs were thawed from storage in liquid nitrogen in a water bath. Cells were mixed and added to 10 mL of RPMI +/+. The solution was centrifuged at 300 × g for 10 min to pellet the cells. The supernatant was aspirated, and the cells were resuspended in 4 mL of media to count. 1 × 106 cells in 450 μL volume were added to each well of a 24 well plate, and cells were left in a 37 °C in a 5% CO2 incubator to recover while samples were prepared. Samples were diluted in RPMI +/+ media to 2× the concentration of the final desired concentration, and 450 μL of each was added to the respective well with cells. After 24 or 48 h incubation, cells were transferred to tubes and washed with PBS. Cells were resuspended in 100 μL MojoSort Buffer 1X (BioLegend, 480017), and human CD8+ T cells were magnetically isolated following the protocol provided for the human CD8 Positive Selection Kit II (StemCell Technologies, # 17853). After isolation, cells were resuspended in RPMI +/+ media, and counted. Cell concentration for each was adjusted to 3 × 105 cells/mL and 100 μL was added to wells in a 96 well round bottom plate. Concurrently, PC3-PSMA cells grown in a T75 flask were washed with PBS and trypsinized to remove from the surface, and were collected and resuspended in 4 mL of RPMI +/+ media and counted. Cell concentration was adjusted to 3 × 104 cells/mL and 100 μL was added to the wells with human CD8+ T cells raised from different treatment conditions in a 96 well round bottom plate. Samples were co-cultured for 2 h at 37 °C, after which they were transferred using trypsin to flow tubes and washed with 600 μL PBS. The tubes were spun at 1200 rpm for 5 min, after which the supernatant was aspirated, and the samples were stained in 100 μL PBS containing surface antibodies (0.5 μL per sample each of: CD45 (clone HI30) -BUV661, CD3 (clone SP34-2) -PE-Cy7, at 4 °C for 15 min. Cells were washed with 600 μL PBS, centrifuged at 1200 rpm for 5 min, aspirated, and resuspended in 100 μL of Cytofix Fixation and Permeabilization solution (BD, 554722) for 20 min at 4 °C. Cells were then washed with 600 μL of Perm/Wash Buffer (BD, 554723), centrifuged at 1200 rpm for 5 min, aspirated, and resuspended in 100 μL of Perm/Wash Buffer with an additional 20 μL of Caspase-3 antibody (BD, BDB550914). Samples were left for 30 min at RT, after which they were washed with 600 μL of Perm/Wash Buffer (BD, 554723), centrifuged at 1200 rpm for 5 min, aspirated, and resuspended in 100 μL of Perm/Wash Buffer. Samples were stored at 4 °C prior to flow cytometry analysis.
Statistical Analysis:
All values shown in graphs are mean ± standard deviation (SD) or standard error of the mean (SEM), as described in each figure caption. Statistical Analysis was performed using GraphPad Prism 9 software, and the test and group sample size used for each comparison is described in each figure caption. Comparisons between two groups utilized an unpaired t-test. Comparisons assessing more than two groups used an ANOVA with a posthoc test specified in the figure caption for multiple comparisons analysis between individual groups. No specific pre-processing of data was performed prior to statistical analyses. Significance was defined as p<0.05 (*p<0.05; **p<0.01; ***p<0.001; ****p<0.0001; ns=non-significant).
Supplementary Material
Acknowledgements
Research reported in this publication was supported by the National Cancer Institute of the National Institutes of Health under award U54CA199091. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This project was also supported by the Prostate Cancer Foundation and the Movember Foundation under award 17CHAL08, Convergence Science Medicine Institute seed award, and the Polsky Urologic Cancer Institute of the Robert H. Lurie Comprehensive Cancer Center of Northwestern University at Northwestern Memorial Hospital. M.H.T. acknowledges support from Northwestern University’s Cancer Nanotechnology Training Program Award T32CA186897. J.W.D and S.W. acknowledge support from the Chemistry of Life Processes Predoctoral Training Program at Northwestern University. M.E. was partially supported by the Dr. John N. Nicholson Fellowship. The content is solely the responsibility of the authors and does not necessarily represent the official views of Northwestern University. Peptide Synthesis was performed at the Peptide Synthesis Core Facility of the Simpson Querrey Institute at Northwestern University, with special thanks to Dr. Mark Karver, which has current support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-2025633). This work made use of the IMSERC MS facility at Northwestern University, with special thanks to Mr. Saman Shafaie, which has received support from the Soft and Hybrid Nanotechnology Experimental (SHyNE) Resource (NSF ECCS-2025633), the State of Illinois, and the International Institute for Nanotechnology (IIN). We thank Dr. Timothy Kuzel for the PC3-PSMA cells.
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflict of interest.
Contributor Information
Michelle H. Teplensky, Department of Chemistry and the International Institute for Nanotechnology, Northwestern University, Evanston, IL 60208, United States
Jasper W. Dittmar, Department of Biomedical Engineering, Northwestern University, Evanston, IL 60208, United States
Lei Qin, Department of Medicine, Division of Hematology and Oncology, Northwestern University, Chicago, IL 60611, United States.
Shuya Wang, Interdisciplinary Biological Sciences Program, Northwestern University, Evanston, IL 60208, United States.
Michael Evangelopoulos, Department of Biomedical Engineering, Northwestern University, Evanston, IL 60208, United States.
Bin Zhang, Department of Medicine, Division of Hematology and Oncology, Northwestern University, Chicago, IL 60611, United States.
Chad A. Mirkin, Department of Chemistry and the International Institute for Nanotechnology, Northwestern University, Evanston, IL 60208, United States Department of Biomedical Engineering, Northwestern University, Evanston, IL 60208, United States.
References
- [1].Thakur A, Vaishampayan U, Lum L, Cancers (Basel) 2013, 5, 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Frank S, Nelson P, Vasioukhin V, F1000Research 2018, 7, 1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bilusic M, Heery C, Madan RA, Vaccine 2011, 29, 6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Topalian SL, Drake CG, Pardoll DM, Cancer Cell 2015, 27, 450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Postow MA, Callahan MK, Wolchok JD, J. Clin. Oncol 2015, 33, 1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].de Almeida DVP, Fong L, Rettig MB, Autio KA, Am. Soc. Clin. Oncol. Educ. book. Am. Soc. Clin. Oncol. Annu. Meet 2020, 40, 1. [DOI] [PubMed] [Google Scholar]
- [7].Beer TM, Kwon ED, Drake CG, Fizazi K, Logothetis C, Gravis G, Ganju V, Polikoff J, Saad F, Humanski P, et al. , J. Clin. Oncol 2017, 35, 40. [DOI] [PubMed] [Google Scholar]
- [8].Kwon ED, Drake CG, Scher HI, Fizazi K, Bossi A, van den Eertwegh AJMM, Krainer M, Houede N, Santos R, Mahammedi H, et al. , Lancet Oncol 2014, 15, 700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Slovin SF, Higano CS, Hamid O, Tejwani S, Harzstark A, Alumkal JJ, Scher HI, Chin K, Gagnier P, McHenry MB, et al. , Ann. Oncol 2013, 24, 1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Maeng HM, Berzofsky JA, F1000Research 2019, 8, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Madan RA, Gulley JL, in ASCO-SITC Clin. Immuno-Oncology Symp., 2018. [Google Scholar]
- [12].Bonaventura P, Shekarian T, Alcazer V, Valladeau-Guilemond J, Valsesia-Wittmann S, Amigorena S, Caux C, Depil S, Front. Immunol 2019, 10, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kantoff PW, Higano CS, Shore ND, Berger ER, Small EJ, Penson DF, Redfern CH, Ferrari AC, Dreicer R, Sims RB, et al. , N. Engl. J. Med 2010, 363, 411. [DOI] [PubMed] [Google Scholar]
- [14].Madan RA, Gulley JL, Arlen PM, Expert Rev. Vaccines 2006, 5, 199. [DOI] [PubMed] [Google Scholar]
- [15].Ferraro B, Cisper NJ, Talbott KT, Philipson-Weiner L, Lucke CE, Khan AS, Sardesai NY, Weiner DB, Hum. Vaccin 2011, 7, 120. [DOI] [PubMed] [Google Scholar]
- [16].Clinicaltrials.gov, “PSA Vaccine Therapy in Treating Patients With Advanced Prostate Cancer,” can be found under https://clinicaltrials.gov/ct2/show/NCT00003871, n.d.
- [17].Kübler H, Scheel B, Gnad-Vogt U, Miller K, Schultze-Seemann W, Dorp F, Parmiani G, Hampel C, Wedel S, Trojan L, et al. , J. Immunother. Cancer 2015, 3, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Durso RJ, Andjelic S, Gardner JP, Margitich DJ, Donovan GP, Arrigale RR, Wang X, Maughan MF, Talarico TL, Olmsted RA, et al. , Clin. Cancer Res 2007, 13, 3999. [DOI] [PubMed] [Google Scholar]
- [19].Murphy GP, Tjoa BA, Simmons SJ, Rogers MK, Kenny GM, Jarisch J, Prostate 2000, 43, 59. [DOI] [PubMed] [Google Scholar]
- [20].Murphy G, Tjoa B, Ragde H, Kenny G, Boynton A, Prostate 1996, 29, 371. [DOI] [PubMed] [Google Scholar]
- [21].Kantoff PW, Schuetz TJ, Blumenstein BA, Glode LM, Bilhartz DL, Wyand M, Manson K, Panicali DL, Laus R, Schlom J, et al. , J. Clin. Oncol 2010, 28, 1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kantoff PW, Gulley JL, Pico-Navarro C, J. Clin. Oncol 2017, 35, 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Wargowski E, Johnson LE, Eickhoff JC, Delmastro L, Staab MJ, Liu G, McNeel DG, J. Immunother. Cancer 2018, 6, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Oh SK, Terabe M, Pendleton CD, Bhattacharyya A, Bera TK, Epel M, Reiter Y, Phillips J, Linehan WM, Kasten-Sportes C, et al. , Cancer Res 2004, 64, 2610. [DOI] [PubMed] [Google Scholar]
- [25].ClinicalTrials.gov, “Multi-Epitope TARP Peptide Autologous Dendritic Cell Vaccination in Men With Stage D0 Prostate Cancer,” can be found under https://clinicaltrials.gov/ct2/show/record/NCT02362451, n.d.
- [26].Wood LV, Fojo A, Roberson BD, Hughes MSB, Dahut W, Gulley JL, Madan RA, Arlen PM, Sabatino M, Stroncek DF, et al. , Oncoimmunology 2016, 5, e1197459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Westdorp H, Sköld AE, Snijer BA, Franik S, Mulder SF, Major PP, Foley R, Gerritsen WR, de Vries IJM, Front. Immunol 2014, 5, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Small EJ, in Proc. Genitourin. Cancer Symp., 2009. [Google Scholar]
- [29].Higano CS, in Proc. 2009 Genitourin. Cancer Symp. Am. Soc. Clin. Oncol., 2009. [Google Scholar]
- [30].Gulley JL, Borre M, Vogelzang NJ, Ng S, Agarwal N, Parker CC, Pook DW, Rathenborg P, Flaig TW, Carles J, et al. , J. Clin. Oncol 2019, 37, 1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Comiskey MC, Dallos MC, Drake CG, Curr. Oncol. Rep 2018, 20, 75. [DOI] [PubMed] [Google Scholar]
- [32].Coulie PG, Van den Eynde BJ, van der Bruggen P, Boon T, Nat. Rev. Cancer 2014, 14, 135. [DOI] [PubMed] [Google Scholar]
- [33].Zhang Y, Dosta P, Conde J, Oliva N, Wang M, Artzi N, Adv. Healthc. Mater 2020, 9, 1901101. [DOI] [PubMed] [Google Scholar]
- [34].Wang S, Duan Y, Zhang Q, Komarla A, Gong H, Gao W, Zhang L, Small Struct 2020, 1, 2000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Jiang Y, Krishnan N, Zhou J, Chekuri S, Wei X, Kroll AV, Yu CL, Duan Y, Gao W, Fang RH, et al. , Adv. Mater 2020, 32, 2001808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].He X, Zhou S, Quinn B, Jahagirdar D, Ortega J, Abrams SI, Lovell JF, Small 2021, 17, 2007165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].He X, Zhou S, Huang WC, Seffouh A, Mabrouk MT, Morgan MT, Ortega J, Abrams SI, Lovell JF, ACS Nano 2021, 15, 4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Dosta P, Cryer AM, Prado M, Dion MZ, Ferber S, Kalash S, Artzi N, Adv. NanoBiomed Res 2021, 2100006. [Google Scholar]
- [39].Shah NJ, Najibi AJ, Shih TY, Mao AS, Sharda A, Scadden DT, Mooney DJ, Nat. Biomed. Eng 2020, 4, 40. [DOI] [PubMed] [Google Scholar]
- [40].Wang H, Sobral MC, Zhang DKY, Cartwright AN, Li AW, Dellacherie MO, Tringides CM, Koshy ST, Wucherpfennig KW, Mooney DJ, Nat. Mater 2020, 19, 1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Smith DM, Simon JK, Baker JR, Nat. Rev. Immunol 2013, 13, 592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Goldberg MS, Nat. Rev. Cancer 2019, 19, 587. [DOI] [PubMed] [Google Scholar]
- [43].Irvine DJ, Hanson MC, Rakhra K, Tokatlian T, Chem. Rev 2015, 115, 11109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Wang S, Qin L, Yamankurt G, Skakuj K, Huang Z, Chen P-C, Dominguez D, Lee A, Zhang B, Mirkin CA, Proc. Natl. Acad. Sci 2019, 116, 10473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Mehta NK, Pradhan RV, Soleimany AP, Moynihan KD, Rothschilds AM, Momin N, Rakhra K, Mata-Fink J, Bhatia SN, Wittrup KD, et al. , Nat. Biomed. Eng 2020, 4, 636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Tsai SJ, Amerman A, Jewell CM, Front. Immunol 2021, 11, 3340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Tostanoski LH, Eppler HB, Xia B, Zeng X, Jewell CM, Biomater. Sci 2019, 7, 798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Qin L, Wang S, Dominguez D, Long A, Chen S, Fan J, Ahn J, Skakuj K, Huang Z, Lee A, et al. , Front. Immunol 2020, 11, 1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Callmann CE, Cole LE, Kusmierz CD, Huang Z, Horiuchi D, Mirkin CA, Proc. Natl. Acad. Sci 2020, 117, 17543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Exicure, “Exicure Presents Promising Interim Results from Ongoing Phase 1b/2 Trial of Cavrotolimod at Virtual KOL Event Today,” can be found under https://investors.exicuretx.com/news/news-details/2020/Exicure-Presents-Promising-Interim-Results-from-Ongoing-Phase-1b2-Trial-of-Cavrotolimod-at-Virtual-KOL-Event-Today/default.aspx, 2020.
- [51].Cutler JI, Auyeung E, Mirkin CA, J. Am. Chem. Soc 2012, 134, 1376. [DOI] [PubMed] [Google Scholar]
- [52].Banga RJ, Chernyak N, Narayan SP, Nguyen ST, Mirkin CA, J. Am. Chem. Soc 2014, 136, 9866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Radovic-Moreno AF, Chernyak N, Mader CC, Nallagatla S, Kang RS, Hao L, Walker DA, Halo TL, Merkel TJ, Rische CH, et al. , Proc. Natl. Acad. Sci 2015, 112, 3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Choi CHJ, Hao L, Narayan SP, Auyeung E, Mirkin CA, Proc. Natl. Acad. Sci 2013, 110, 7625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Rosi NL, Giljohann DA, Thaxton CS, Lytton-Jean AKR, Han MS, Mirkin CA, Science 2006, 312, 1027. [DOI] [PubMed] [Google Scholar]
- [56].Yamankurt G, Berns EJ, Xue A, Lee A, Bagheri N, Mrksich M, Mirkin CA, Nat. Biomed. Eng 2019, 3, 318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Skakuj K, Wang S, Qin L, Lee A, Zhang B, Mirkin CA, J. Am. Chem. Soc 2018, 140, 1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Stano A, Scott EA, Dane KY, Swartz MA, Hubbell JA, Biomaterials 2013, 34, 4339. [DOI] [PubMed] [Google Scholar]
- [59].Lodge PA, Jones LA, Bader RA, Murphy GP, Salgaller ML, Cancer Res 2000, 60, 829. [PubMed] [Google Scholar]
- [60].Garetto S, Sizzano F, Brusa D, Tizzani A, Malavasi F, Matera L, Cytotherapy 2009, 11, 1090. [DOI] [PubMed] [Google Scholar]
- [61].Ellis JM, Henson V, Slack R, Ng J, Hartzman RJ, Katovich Hurley C, Hum. Immunol 2000, 61, 334. [DOI] [PubMed] [Google Scholar]
- [62].Chen KY, Liu J, Ren EC, Immunol. Res 2012, 53, 182. [DOI] [PubMed] [Google Scholar]
- [63].Berzofsky JA, Terabe M, Trepel JB, Pastan I, Stroncek DF, Morris JC, Wood LV, Cancer Immunol. Immunother 2018, 67, 1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Berzofsky JA, Wood LV, Terabe M, Expert Rev. Vaccines 2013, 12, 1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Jackson Laboratory, “B6.Cg-Immp2l/J Mouse Strain Datasheet,” can be found under https://www.jax.org/strain/004191, n.d.
- [66].Wimmers F, Aarntzen EHJG, Duiveman-deBoer T, Figdor CG, Jacobs JFM, Tel J, de Vries IJM, Oncoimmunology 2016, 5, e1067745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Spranger S, Koblish HK, Horton B, Scherle PA, Newton R, Gajewski TF, J. Immunother. Cancer 2014, 2, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].De Groot R, Van Loenen MM, Guislain A, Nicolet BP, Freen-Van Heeren JJ, Verhagen OJHM, Van Den Heuvel MM, De Jong J, Burger P, Van Der Schoot CE, et al. , Oncoimmunology 2019, 8, e1648170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Tram C, Hrytsenko O, Stanford M, J. Immunol 2019, 202. [Google Scholar]
- [70].Zhang Q, Helfand BT, Carneiro BA, Qin W, Yang XJ, Lee C, Zhang W, Giles FJ, Cristofanilli M, Kuzel TM, Eur. Urol 2018, 73, 648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Tjoa BA, Simmons SJ, Bowes VA, Ragde H, Rogers M, Elgamal A, Kenny GM, Cobb OE, Ireton RC, Troychak MJ, et al. , Prostate 1998, 36, 39. [DOI] [PubMed] [Google Scholar]
- [72].Poropatich K, Dominguez D, Chan WC, Andrade J, Zha Y, Wray B, Miska J, Qin L, Cole L, Coates S, et al. , J. Clin. Invest 2020, 130, 3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Colonna M, Trinchieri G, Liu Y-J, Nat. Immunol 2004, 5, 1219. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.