

Abstract
Rearranged during transfection (RET) is involved in the physiological development of some organ systems. Activating RET alterations via either gene fusions or point mutations are potent oncogenic drivers in non-small cell lung cancer, thyroid cancer, and in multiple diverse cancers. RET-altered cancers were initially treated with multikinase inhibitors (MKIs). The efficacy of MKIs was modest at the expense of notable toxicities from their off-target activity. Recently, highly potent and RET-specific inhibitors selpercatinib and pralsetinib were successfully translated to the clinic and FDA approved. We summarize the current state-of-the-art therapeutics with preclinical and clinical insights of these novel RET inhibitors, acquired resistance mechanisms, and future outlooks.
Introduction
RET is the transforming proto-oncogene that encodes a receptor tyrosine kinase and was accidentally discovered in a patient with T cell lymphoma upon transfecting human tumor DNA with rearrangement by Takahashi et al. about three and a half decades ago [1]. While the receptor tyrosine kinase RET is involved in the fetal development of nervous, hematopoietic, gastrointestinal, and genitourinary systems, activating RET aberration is a potent oncogenic driver in many cancer types [2–7]. RET proto-oncogene can be activated aberrantly by two major mechanisms: gene fusion and point mutation. RET gene fusions or rearrangements are the principal aberrations occurring in 1–2% of non-small cell lung cancer (NSCLC) and 10–20% of papillary thyroid carcinoma (PTC). By contrast, RET mutations are the major activating alteration in sporadic medullary thyroid cancer (MTC) and as germline mutation in multiple endocrine neoplasia syndrome (MEN) [8–14]. Advances in next generation sequencing (see Glossary) combined with precision oncology have uncovered RET alterations in several other malignancies, albeit rarely [6,15–20].
Researchers have tried to tackle RET-driven cancers with different approaches and therapeutics. Despite the past decade of effort utilizing various multikinase inhibitors (MKI) with RET inhibitory activity, their efficacy was modest at the expense of noteworthy toxicities and significant impact on health-related quality of life, ultimately leading to their discontinuation due to off-target side effects [21]. Hence, developing RET-specific therapeutics has become paramount. The recent FDA approvals of highly potent and selective RET inhibitors selpercatinib and pralsetinib have stimulated the field of RET-aberrant cancer research. We summarize the utility of precision medicine in addressing RET-driven tumors and depicting the mechanistic insight of the novel selective RET inhibitors, including emerging resistance mechanisms.
RET alterations in cancers
The structure of RET and its fundamentals
The transmembrane receptor tyrosine kinase RET, which is encoded by the proto-oncogene RET located on chromosome 10, plays a role in the early development of kidneys and the enteric nervous system [2–4]. The RET protein comprises an extracellular domain, a transmembrane domain, and an intracellular tyrosine kinase domain [22]. The extracellular domain contains four cadherin-like domains (CLD1–4), a calcium binding site, and a cysteine-rich domain. The intracellular region contains a tyrosine kinase domain and tyrosine phosphorylation sites located next to the C terminal region, where two major isoforms, RET9 and RET51, are positioned due to alternative splicing [23,24]. The latter isoform has stronger tumorigenic activity [14], although both isoforms are coexpressed in many tissues. There are six known RET ligands; artemin, differentiation factor 15 (GDF15), glial cell line-derived neurotrophic factor (GDNF), GDNF receptor α (GFRα)-like protein (GFRAL), neurturin, and persephin, and coreceptors called GDNF family receptor-α (GFRα1–4) [7,25–29]. RET homodimerization, mediated by a ligand-coreceptor complex, leads to the activation of several downstream pathways, including RAS/MAPK, PI3K/AKT, JAK/STAT, PKA, and PKC pathways (Figure 1) [30–34]. Although Y1062 is the important docking site of major pathways, autophosphorylation of certain docking sites specifically gives rise to separate downstream pathways; such as Y1096 leading to RAS/MAPK and PI3K/AKT pathways, Y1015 conferring PLCγ, Y752 and Y928 to JAK/STAT pathway, and Y687 and Y981 binding to Shp2 and Src kinases, respectively [33,35–40].
Figure 1. Rearranged during transfection (RET) pathway and acquired resistance mechanisms (red arrow) to selective RET inhibitors in RET-aberrant cancers.
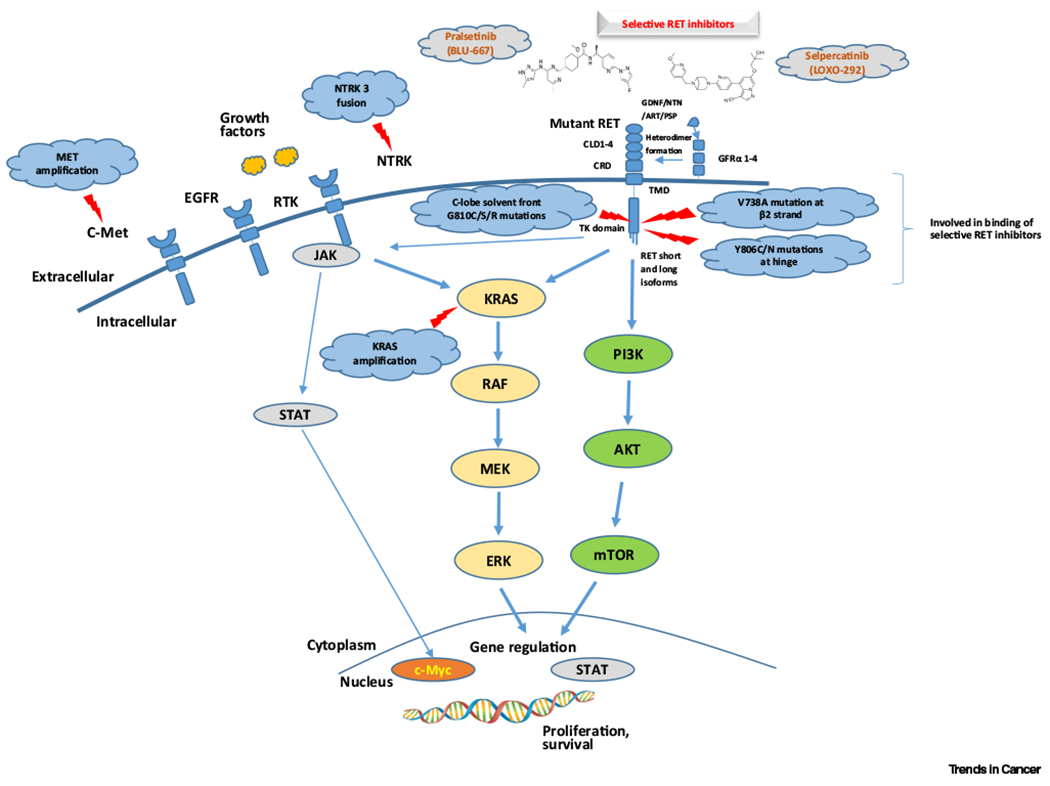
Abbreviations: AKT, protein kinase B; ART, artemin; c-Met, mesenchymal epithelial transition factor; c-Myc, avian myelocytomatosis virus oncogene cellular homolog; CLD 1–4, cadherin-like domain 1–4; CRD, cysteine-rich domain; EGFR, epidermal growth factor receptor; ERK, extracellular signal-regulated kinases; GDNF, glial derived neurotrophic factor family ligands; GFRα 1–4, GDNF family alpha receptors 1–4; JAK, Janus kinase; KRAS, Kirsten rat sarcoma 2 viral oncogene homolog; MAPK pathway (RAF/MEK/ERK), mitogen-activated protein kinase pathway; MEK, MAPK/ERK kinase; mTOR, mammalian target of rapamycin; NTN, neuturin; NTRK, neurotrophic tyrosine receptor kinase; PI3K, phosphatidylinositol 3-kinase; PSP, persephin; RAF, rapidly accelerated fibrosarcoma; RTK, receptor tyrosine kinase; STAT, signal transducer and activator of transcription; TK, tyrosine kinase; TM, transmembrane.
RET fusions
NSCLC and PTC are the most common cancer types harboring RET fusions [10,11,14,41–43]. Adenocarcinomas are the most frequent histology to carry RET rearrangements, followed by adenosquamous, squamous cell, and neuroendocrine cancers. About 2% of patients with NSCLC harbor RET fusions and they tend to occur in relatively younger (<60 years of age), never- or light-smokers, which are similar to those carrying ALK or ROS rearrangements [14,42,44,45]. Meanwhile, 5–10% of PTC carry RET fusions, in which the prevalence is further heightened in patients who had previous radiation exposure [46–54]. Like other common drivers, such as sensitizing EGFR mutations and ALK or ROS1 rearrangements, RET fusions tend to occur in a mutually exclusive fashion, yet they might be acquired in some patients who become resistant to EGFR therapies in EGFR-mutant NSCLC [55,56]. Kinesin family member 5B (KIF5B) from the pericentric inversion of chromosome 10 is the most common somatic rearrangement identified in lung cancer [10,11]. Coiled-coil domain containing 6 (CCDC6)-RET(RET/PTC1) fusion contributes to 10–25% of RET fusions. Other uncommon fusions in lung cancer include NCOA4-RET, TRIM33-RET, ZNF477P-RET, ERCC1-RET, HTR4-RET, CLIP1-RET, FRMD4-RET, and WAC-RET [15,57]. CCDC6-RET and nuclear receptor coactivator 4 (NCOA4)-RET (RET/PTC3) account for the majority (>90%) of rearrangements in PTC [46,58]. More than ten other RET fusions have been reported in PTC, including RET/PTC2, RET/PTC4 through RET/PTC9, ELKS-RET, PCM1-RET, RFP-RET, and HOOK3-RET [57]. RET fusions occur in less than 1% in pancreatic cancer, salivary gland cancer, spitz tumors, colorectal cancer, ovarian cancer, and others [6,16–20]. To date, more than 35 different RET fusion genes have been reported [15]. These fusion partners permit RET kinase expression in cell types in which RET or its coreceptors are not normally expressed and cause dimerization that aberrantly activates the RET kinase [7].
RET point mutations
In contrast to PTC, most MTC carry RET mutations, although a few cases of RET fusions have been reported in MTC [59]. RET point mutations, which occur as germline or somatic mutations, are found in families with multiple endocrine neoplasia type 2 (MEN 2) syndrome. MEN 2 is an inherited cancer syndrome, comprising MTC, pheochromocytoma, and parathyroid hyperplasia and it has three subtypes: MEN 2A, MEN 2B, and familial medullary thyroid cancer (FMTC). Mutations contributing to MEN 2 are clustered in the cysteine-rich domain in exon 10, 11, and the tyrosine kinase domains in exons 13 to 16 of RET [8,12]. RETM918T is the most common and most aggressive mutation out of more than 60 activating RET mutations discovered [15]. RETM918T is located in the kinase domain and has higher ATP-binding affinity and altered RET autophosphorylation trajectory [60].
Treating RET-altered cancers
Response to immunotherapy and chemotherapy
Like patients with NSCLC harboring ALK or ROS1 rearrangements, RET-rearranged lung cancers can respond to pemetrexed-based doublet chemotherapy with an objective response rate (ORR) of 45% and median progression-free survival (PFS) of 19 months [61]. There is emerging data on the response to immunotherapy in RET-altered cancers [62–64]. A study of 74 patients with RET-rearranged lung cancers who were treated with immune checkpoint inhibitors (ICI) either as single- or dual-agent immunotherapy [63] (Table 1) revealed that RET-rearranged lung cancers have low levels of PD-L1 expression and low tumor mutation burden in the majority of patients and poorer responses to immunotherapy. In the MD Anderson Cancer Center cohort, the median time to progression was shorter in patients with RET-aberrant cancers who received ICI compared with non-ICI therapies [62] (Table 1). Retrospective studies including real-world data on ICI in RET-altered cancers are depicted in Table 1. While prospective data on immunotherapy in RET-altered cancers is lacking, the retrospective evidence points to poor response to immunotherapy. RET+ NSCLC seem to be biologically ‘cold’ tumors with low tumor mutational burden (TMB) and low PDL1 expression. RET+ NSCLC usually occurs in patients with a history of no tobacco use or minimal smokers, which could be the link to lower TMB. This lower TMB renders them as ‘cold tumors’ that have an immune-poor microenvironment, rich in regulatory T cells, and poor in activated T cells, responding less to checkpoint inhibitors [65]. However, TMB and tumor microenvironment (TME) are dynamic as T cell-inflamed TME seems to positively correlate with responsiveness to ICIs and adoptive cell therapy. Therefore, future studies should look into opportunities to convert these ‘cold’ oncogene-driven tumors to hot tumors [65].
Table 1.
Immune checkpoint inhibitors (ICI) in RET-altered cancersa
| Study | MSKCC retrospective study [63] | MDACC retrospective study [62] | Flatiron Health-Foundation Medicine NSCLC Clinico-Genomic database (CGD) and Guardant Health database (GHD) [64] |
|---|---|---|---|
| Number of patients | 74 | 70 | 29 in CGD, 40 in GHD |
| Tumor types | RET-rearranged lung cancers | RET-altered solid tumors; NSCLC (n = 29), MTC (n = 32), DTC and other cancers (n = 9) | RET fusion-positive NSCLC |
| Smoking history | 31% tobacco exposure | 29% tobacco exposure | 12/29 tobacco exposure |
| RET fusion partner |
KIF5B (58%) CCDC6 (16%) |
Fusion (49%) - KIF5B (41%) Mutation (51%) - M918T (67%) |
KIF5B (74%) CCDC6 (14%) |
| Immune phenotype findings in available population | PD-L1 expression: zero (58%), 1–49% (23%) Tumor mutational burden (TMB) - RET-rearranged (1.75 Mut/Mb) versus RET wild-type (5.27 Mut/Mb) (P < 0.0001) |
PD-L1 expression: weak (<1%) (56%), 1–49% (22%) Tumor mutational burden (TMB) - All 15 patients have TMB low (≤5 Mut/Mb) |
PD-L1 expression: <1% (7/13), ≥1% (6/13), others missing Tumor mutational burden (TMB) - <6 Mut/Mb (11/14) - ≥6 Mut/Mb (3/14) - Others missing |
| Response in evaluable patients | Response was not observed (n = 13) - SD 3/13 (23%), PD 8/13 (62%), mPFS 3.4 months |
Time to treatment discontinuation (TTD) - Non-ICI group (18 months) versus ICI group (5.2 months) (P = 0.00045) |
Median real-world PFS of 4.2 months and OS of 19.1 months (CGD) |
Abbreviations: DTC, differentiated thyroid cancer; MDACC, MD Anderson Cancer Center; mPFS, median progression-free survival; MSKCC, Memorial Sloan-Kettering Cancer Center; MTC, medullary thyroid cancer; Mut/Mb, mutations per mega base; NA, not available; NSCLC, non-small cell lung cancer; PD, progressive disease; PD-L1, programmed death-ligand 1; SD, stable disease.
Tackling RET proto-oncogene with nonspecific MKIs
The utilization of MKI is one strategy to target RET. Several FDA-approved protein tyrosine kinase inhibitors, including cabozantinib, vandetanib, lenvatinib, sorafenib, and sunitinib, cross-inhibit RET and have been trialed in RET-altered cancers [15,21]. Across different studies, NSCLC patients with RET rearrangements who were treated with cabozantinib, vandetanib, or lenvatinib achieved a modest ORR (16–47%), median PFS (4.9–11.6 months), and median overall survival (OS) (4.9–11.6 months) [66–69] (Table 2). However, ~70% of patients experienced grade 3 and above adverse events (≥G3AEs), leading to dose reduction in the majority of trial participants and discontinuation in some. In contrast to the RET-rearranged NSCLC, some MKIs have been approved in thyroid cancers. Cabozantinib and vandetanib were approved in MTC by the US FDA and the European Medicines Agency (EMA) after the landmark EXAM and ZETA trials showed positive results [70,71] (Table 2). In the EXAM trial, cabozantinib contributed to ORR of 28% and median PFS of 11.2 months compared with ORR/PFS of 0%/4 months in the placebo arm and the activity of cabozantinib was noted regardless of RET mutation status [71,72]. Similar to the study in NSCLC, dose reduction occurred in 79%, while 16% had stopped the trial medication. In the ZETA trial employing vandetanib, the ORR/PFS was 45%/30.5 months in the treatment group versus 13%/19.3 months in the placebo arm [70]. Notably, vandetanib gave rise to greater ORR in M918T-mutant MTC. The pivotal DECISION and SELECT trials led to the approval of sorafenib and lenvatinib in patients with differentiated thyroid cancer (DTC), who were refractory to radioactive iodine [73,74]. Although these MKIs conferred higher ORR and survival benefits in thyroid cancers and modest benefits in NSCLC, they underscored the tolerability issues impacting health-related quality of life from ‘off-target’ toxicities. It is believed that an underlying cause of toxicity is attributed to inhibition of VEGFR1/FLT and VEGFR2/KDR [21].
Table 2.
Safety and efficacy of nonspecific multikinase inhibitors (MKIs) in RET-aberrant malignanciesa
| Cabozantinib | Vandetanib | Lenvatinib | Others | |
|---|---|---|---|---|
| Efficacy and safety in patients with NSCLC | RET + NSCLC (MSKCC study) (n = 26) - ORR = 28%, mPFS = 5.5 months, OS = 9.9 months - ≥G3AEs = 69%, - 73% dose-reduced, - 8% discontinued |
RET + NSCLC (Japan LURET trial) (n = 19) - ORR = 47%, mPFS = 4.7 months, mOS = 11.1 months - 53% dose-reduced, - 21% discontinued RET + NSCLC (Korea trial) (n = 18) - ORR = 18%, - PFS = 4.5 months, - OS = 11.6 months - 27% dose-reduced |
RET + NSCLC (Phase II study) (n = 25) - ORR = 16%, - mPFS = 7.3 months - 64% dose-reduced, 20% discontinued |
GLORY RET registry Cabozantinib in RET + NSCLC (n = 19) - ORR = 37%, - mPFS = 3.6 months, - mOS = 4.9 months Vandetanib in RET + NSCLC (n = 11) - ORR = 18%, - mPFS = 2.9 months, - mOS = 10.2 months Sunitinib in RET + NSCLC (n = 9) - ORR = 22%, - mPFS = 2.2 months - mOS = 6.8 months |
| Efficacy and safety in patients with thyroid cancers | MTC (EXAM trial) - ORR = 32%, - PFS = 14 months, - OS = 44.3 months - ≥G3AEs = 69%, 79% dose-reduced, - 16% discontinued |
MTC (ZETA trial) - ORR = 45%, - PFS 30.5 months, - OS NA - ≥G3AEs = 69%, 35% dose-reduced, - 12% discontinued |
MTC (Phase II study) (n = 59) - ORR = 36%, - mPFS = 9.0 months, - mOS = 16.6 months - 59% dose-reduced, 24% discontinued |
|
| FDA approval timeline | Cabozantinib in metastatic MTC (November 2012) | Vandetanib in metastatic MTC (April 2011) | Lenvatinib in progressive, RAI-refractory DTC (February 2015) | Sorafenib in progressive, RAI-refractory DTC (November 2013) |
Abbreviations: ≥G3AEs, grade 3 and above adverse events; mOS, median overall survival; mPFS, median progression-free survival; MSKCC, Memorial Sloan-Kettering Cancer Center; NA, not available; NSCLC, non-small cell lung cancer; ORR, objective response rate; RAI, radioactive iodine.
RXDX-105 was developed as a VEGFR1/2-sparing dual RET and BRAF MKI [75,76]. In a clinical study, RXDX-105 was efficacious only in non-KIF5B fusion partners [76]. As the clinical virtue has been sparse in patients with NSCLC harboring KIF5B-RET fusion, which remains the most common subtype, more novel approaches and therapeutics are needed. It is unclear why KIF5B-RET fusion-positive NSCLC were nonresponsive to RXDX-105. In a fruit fly model of oncogenic RET activity, KIF5B-RET, but not CCDC6-RET or NCOA4-RET, activated EGFR and FGFR strongly via the kinesin motor domain and RAB GTPase [77]. KIF5B-RET-induced EGFR and FGFR phosphorylation required RET kinase activity. Although KIF5B-RET may have stronger oncogenic activity because of activating multiple signaling hubs, RET kinase activity is a prerequisite; thus, inhibiting RET kinase activity alone is sufficient to silence oncogenic activity. In patient-derived xenograft (PDX) tumor growth experiments, both KIF5B-RET-positive NSCLC PDX and CCDC6-RET-positive PDX responded similarly to RXDX-105 [75]. However, in RET fusion kinase-dependent BaF3 cell models, the RXDX-105 IC50 in BaF3/KIF5B-RET cells was approximately twofold higher than those in BaF3/CCDC6-RET and BaF3/NCOA4-RET cells. The difference in the IC50s could be caused by either different expression levels of the RET fusion kinases or different signaling mechanisms. In the RET fusion-positive NSCLC, the expression level of the RET fusion genes are most likely dictated by the fusion partners located on the 5′ side. Because the mRNA expression data of the RET fusion genes in NSCLC are not available, we examined the mRNA expression levels of KIF5B and the three non-KIF5B fusion partner genes (CCDC6, EML4, PARD3) in lung adenocarcinoma from the TCGA PanCancer Atlas dataset. KIF5B mRNA is expressed at a significantly higher level (P < 0.0001) than the other three genes that are the non-KIF5B RET fusion partners in the clinical study of RXDX-105 (Figure S1 in the supplemental information online) [76], raising the possibility that KIF5B-RET fusion is expressed at a higher level than the other three non-KIF5B-RET fusions reported in the RXDX-105 clinical trial of NSCLC [76]. The higher level of KIF5B-RET would be predicted to generate greater signaling strength and require more drug to inhibit it. This is important because the suboptimal in vivo levels of multikinase RET inhibitors, including RXDX-105 [76], may not be able to adequately suppress the KIF5B-RET kinase activity in NSCLC, resulting in no objective response in KIF5B-RET fusion NSCLC.
While RXDX-105 has potent RET protein kinase inhibitor (TKI) activity and lower VEGFR2/KDR-mediated AEs than other RET MKIs, like cabozantinib and vandetanib [78,79], RXDX-105 was unable to inhibit RETV804M/L gatekeeper mutants [75,80]. Consequently, tumors with RETV804M progressed on RXDX-105 treatment [81]. Structurally, these MKIs bind to the RET kinase domain in a manner that occupies both front and back clefts of the active site by going through the gate that separates these two clefts, which is formed by the gatekeeper V804 and the invariable gatewall K758 residues (Figure 2A) [75,82]. Gatekeeper V804V/M mutants cause steric hindrance that interferes with the binding of these drugs [75,80].
Figure 2. Co-crystal structure of rearranged during transfection (RET)- tyrosine kinase inhibitor (TKI) complex.
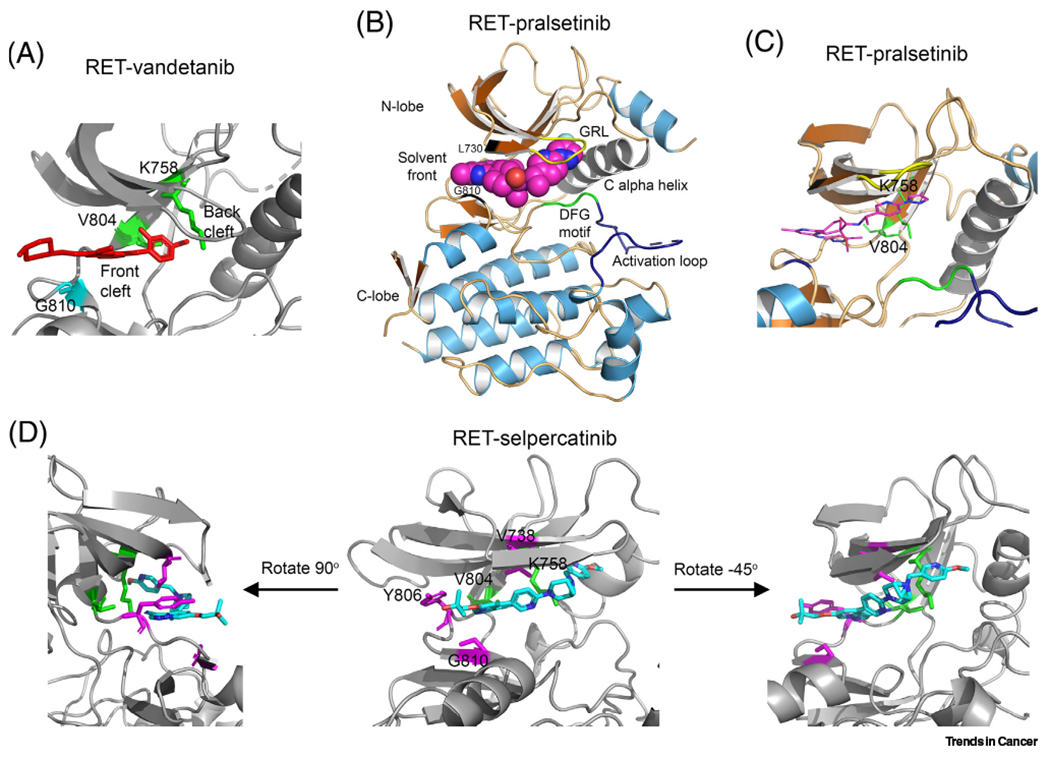
(A) RET-vandetanib complex [Protein Data Bank (PDB): 2IVU] [82]. Red, vandetanib; green, V804 gatekeeper residue and K758 gatewall residue; cyans, G810 solvent front residue. (B,C) Co-crystal structure of RET–pralsetinib complex (PDB: 7JU5) [83]. (D) Co-crystal structure of RET-selpercatinib complex (PDB: 7JU6) [83]. Gatekeeper V804 and gatewall K758 are in green. Magentas denotes residues where selpercatinib-resistant mutations have been identified. Abbreviation: GRL, Gly-rich loop.
RET-specific, gatekeeper mutant-sensitive TKIs
Recently, two RET-specific, gatekeeper mutant-effective TKIs (pralsetinib and selpercatinib) were approved by the US FDA for the treatment of RET-altered NSCLC and thyroid cancers.
Pralsetinib (BLU-667) is an orally bioavailable selective RET inhibitor that is more potent than other RET MKIs. Pralsetinib inhibits the RETV804M/L gatekeeper mutants with a similar potency to RET and RETM918T in an in vitro kinase activity assay [80]. The X-ray crystal structure of the RET-pralsetinib complex reveals that pralsetinib binds to the RET kinase by occupying both the front and back clefts without passing through the gate like vandetanib (Figure 2A). Instead, pralsetinib wraps outside the K758 gatewall residue to access both the front and back clefts (Figure 2B,C) [83]. This binding avoids the interference of gatekeeper mutations while allowing high-affinity binding.
Pralsetinib is also highly selective against VEGFR2 [80], ultimately lessening ‘off-target’ AEs caused by blocking VEGFR2 activity. The findings from the Phase I/II, global, multicenter, registrational ARROW trial (ClinicalTrials.gov Identifier: NCT03037385) (Table 3) revealed that the majority of patients tolerated pralsetinib well, resulting in 4% treatment discontinuation due to treatment-related adverse events (TRAE), and the most common any grade TRAEs were elevated AST (34%), anemia (24%), elevated ALT (23%), and hypertension (22%).
Table 3.
| Selpercatinib (LOXO-292) | Pralsetinib (BLU-667) | |
|---|---|---|
| Clinical trial | LIBRETTO-001 (NCT03157128) | ARROW (NCT03037385) |
| Trial setting | Multicenter, multicohort, Phase I/II | Multicenter, multicohort, Phase I/II |
| RP2D | 160 mg orally twice daily | 400 mg orally once daily |
| Published preliminary efficacy in patients with NSCLC and thyroid cancers |
RET + NSCLC [88] 1. Previously treated (n = 105): ORR 64%, CR 2%, mDOR 17.5 months, mPFS 16.5 months, 1-year PFS 66% 2. Treatment naïve (n = 39): ORR 85%, CR 0%, mDOR NE, mPFS NE, 1-year PFS 75% |
RET + NSCLC [92] 1. Previously treated (n = 87): ORR 61%, CR 6%, mDOR NR, mPFS 17.1 months 2. Treatment naïve (n = 27): ORR 70%, CR 11%, mDOR 9 months, mPFS 9.1 months |
|
RET + thyroid cancers [87] 1. Previously treated RET-mutant MTC (n = 55): ORR 69%, CR 9%, mDOR NE, mPFS NE, 1-year PFS 82% 2. Treatment naïve RET-mutant MTC (n = 88): ORR 73%, CR 11%, mDOR 22 months, mPFS 23.6, 1-year PFS 92% 3. Previously treated RET fusion-positive thyroid cancer (n = 19): ORR 79%, CR 5%, mDOR 18.4, mPFS 20.1, 1-year PFS 64% |
RET + thyroid cancers [93] 1. Previously treated RET-mutant MTC (n = 55): ORR 60%, CR 2%, mDOR NR, mPFS NR 2. Treatment naïve RET-mutant MTC (n = 21): ORR 71%, CR 5%, mDOR NR, mPFS NR 3. Previously treated RET fusion-positive thyroid cancer (n = 9): ORR 89%, CR 0%, mDOR NR, mPFS NR |
|
| Activity in RET + tumors beyond NSCLC and thyroid | AACR 2021 data https://cancerres.aacrjournals.org/content/81/13_Supplement/CT011 |
ASCO 2021 data https://meetinglibrary.asco.org/record/197581/abstract |
| CNS activity | Pre-clinical and clinical data +++/+++ |
Pre-clinical and clinical data +++/++++ |
| FDA approval timeline | May 8, 2020 1. Adult patients with metastatic RET fusion-positive NSCLC; 2. Adult and pediatric patients ≥12 years of age with advanced or metastatic RET- MTC who require systemic therapy; 3. Adult and pediatric patients ≥12 years of age with advanced or metastatic RET fusion-positive thyroid cancer who require systemic therapy and who are RAI-refractory |
September 4, 2020 1. Adult patients with metastatic RET fusion-positive NSCLC December 1, 2020 1. Adult and pediatric patients 12 years of age and older with advanced or metastatic RET-mutant MTC who require systemic therapy or RET fusion-positive thyroid cancer who require systemic therapy and who are RAI-refractory |
Clinical trials with TPX-0046 (NCT04161391), BOS 172738 (NCT03780517), and TAS0953/HM06 (NCT04683250) are ongoing. Early nonclinical studies with BiDAC™ RET inhibitors from C4 therapeutics and KinaRx are ongoing.
Abbreviations: BID, twice daily; G3, grade 3; G4, Grade 4; mDOR, median duration of response; mPFS, median progression-free survival; NCT, National Clinical Trial; NE, could not be evaluated; NR, not reached; RP2D, recommended Phase II dose; TRAE, treatment-related adverse events.
Recently, the FDA approved pralsetinib in patients with RET-altered thyroid cancers [84]. The AcceleRET Lung study of pralsetinib in patients with previously untreated RET fusion-positive NSCLC was recently launched [85]. Concurrently, the preliminary activity of pralsetinib in patients with other RET fusion-positive solid tumors was reported [86]. The cohort included 29 patients: 16 PTC, one undifferentiated thyroid cancer, two cholangiocarcinoma, three pancreatic, three colorectal cancers, and four others. In 11 evaluable patients with thyroid cancer, the ORR was 91%, with all achieving partial response (PR) and the disease control rate (DCR) was 100%. In other RET fusion-positive solid tumors, six out of 12 obtained PR, contributing to an ORR of 50%, whereas the DCR was 92%. Remarkably, all five patients with cholangiocarcinoma and pancreatic cancer (CCDC6-RET fusion, TRIM33-RET and JMJD1C-RET fusions) achieved PR. In a patient with intrahepatic bile duct carcinoma with NCOA4-RET fusion, who progressed after three prior lines of therapy (cisplatin/gemcitabine/nab-paclitaxel, erlotinib/bevacizumab, and osimertinib), showed a deep and durable PR with 64% shrinkage at 20 months of treatment.
Selpercatinib (LOXO-292) is another orally bioavailable, potent RET-specific TKI capable of inhibiting the RETV804M/L gatekeeper mutants [81,83]. Like pralsetinib, selpercatinib docks one end of the ATP-binding pocket without inserting into the gate and wraps outside the K758 gatewall residue to access the back cleft (Figure 2D). In various cell culture models, selpercatinib was >20-fold more potent than cabozantinib, vandetanib, or lenvatinib [78,81,83]. The pivotal multicenter, dose escalation, Phase I/II registrational LIBRETTO-001 trial (ClinicalTrials.gov Identifier: NCT03157128) demonstrated the efficacy of selpercatinib [87,88] (Table 3). The most observed high-grade AEs were hypertension (14–21%), elevated ALT (11–12%), and elevated AST (9–10%). The majority of patients tolerated selpercatinib well, while only 2% had treatment discontinuation due to TRAE. With these promising results, selpercatinib secured FDA approval in patients with RET-altered lung and thyroid cancers in May 2020 [89]. Recently, two randomized Phase III LIBRETTO-431 and LIBRETTO-531 studies in patients with treatment-naïve advanced RET fusion-positive NSCLC and treatment-naïve RET-mutant MTC were launched [90,91].
Both selpercatinib and pralsetinib have been approved for the same indications in RET-aberrant NSCLC and thyroid cancers. Both agents were approved in the treatment-naïve and treatment refractory settings and offer options for patients in the clinic as different drugs have different clinical properties, patient access mechanisms, and coverage. Although mechanism of action and clinical trial results are similar, the trials enrolled different populations with varied entry criteria and eligibility; hence, it may not be appropriate to make cross-trial comparisons. Selpercatinib is dosed orally twice daily, while pralsetinib is given orally once a day. Regarding safety profile, grade 3 or greater QT prolongation and hypersensitivity are safety warnings for selpercatinib, but are not safety warnings for pralsetinib. Pneumonitis, seen in 2% of patients on pralsetinib, is not a safety issue with selpercatinib. Hypertension rates were similar with both drugs [87,88,92,93].
Although RET inhibitors have been approved in RET-altered lung and thyroid cancers, clinical trials for tissue-agnostic and pediatric indication continue and data are emerging. Despite the rare prevalence, the selective RET inhibitors showed robust activity in other RET alteration-positive solid tumors and pediatric populations. The early data from the studies show that RET fusions are oncogenic in multiple tumor types and can be targeted by selective RET inhibition. However, the duration of response and acquired resistance in non-lung and non-thyroid tumors remains unknown. Since RET plays a crucial role in the development of the nervous system and kidney, and in spermatogenesis, the clinical implications of long-term RET inhibition in young children remains to be known. The first experience with selpercatinib in five pediatric patients under institutional review board-approved single patient protocols reported [94] that four out of the five pediatric patients with RET-altered cancers treated with selpercatinib at a starting dose of 90 mg/m2 twice daily (BID) had achieved PR and another patient had stable disease. They included different tumor types, namely PTC (CCDC6-RET), infantile myofibroma/hemangiopericytoma (MYH10-RET), congenital mesoblastic nephroma/infantile fibrosarcoma (SPECC1L-RET), lipofibromatosis (NCOA4-RET), and MTC (RET codon 918 mutation, MEN2B). A Phase I/II LIBRETTO-121 enrolling pediatric patients with RET-altered advanced solid or primary central nervous system (CNS) tumors is currently underway (ClinicalTrials.gov Identifier: NCT03899792).
Preclinical and clinical intracranial response data of selective RET inhibitors
A global RET registry depicted that brain metastases could be found in ~50% of RET-aberrant NSCLC where MKIs conferred suboptimal CNS activity [95]. Hence, it is important that RET-specific inhibitors have CNS coverage. Selpercatinib and pralsetinib have demonstrated intracranial activity in preclinical models. Pralsetinib showed intracranial tumor target engagement in a CCDC6-RET fusion-driven preclinical model, and human DUSP6/SPRY4 transcripts decreased >90% with 10 or 30 mg/kg pralsetinib at 4 hours and indicated full pathway inhibition at doses demonstrating antitumor activity [96]. The negative control, human GSK3B expression was not inhibited with pralsetinib treatment. In the case of selpercatinib, intracranial activity was demonstrated in CCDC6-RET fusion-positive PDX tumor suspensions and treated orally with vehicle, ponatinib (40 mg/kg daily) or selpercatinib (30 mg/kg twice daily) and survival was compared with Kaplan–Meier analysis before and after dose reduction by tenfold on day 52. At proportionately reduced doses, selpercatinib significantly prolonged survival (median not reached) compared with ponatinib (median 18.5 days) [81]. Both selective RET inhibitors cross the blood–brain barrier and demonstrate clinical intracranial responses. In the LIBRETTO-001 trial, intracranial ORR was 82% [95% confidence interval (CI), 60–95], including 23% with complete responses among 22 patients with measurable intracranial efficacy evaluable disease at baseline [97]. In the ARROW trial with pralsetinib, shrinkage of intracranial metastases was seen in all patients with measurable intracranial metastases at baseline and at least one post-baseline intracranial response assessment. Five of nine (56%) patients had an intracranial response, including three with complete response (CR) [92]. Moreover, recent cases have demonstrated the efficacy of RET-specific inhibitor in parenchymal disease and leptomeningeal metastases [98,99]. The case depicted the robust activity of selpercatinib in a patient with RET fusion-positive lung cancer, resulting in a reduction in 65% from baseline at 21 weeks of treatment and subsequent scans showed resolution of leptomeningeal enhancement. A recent anecdotal case report showed subsequent clinical response to selpercatinib in a patient with RET fusion-positive NSCLC, who had a durable extracranial response to pralsetinib, but had leptomeningeal progression [100]. It will be interesting to see if these agents can be used sequentially, especially in these special situations, potentially opening up more options for patients. These RET-specific inhibitors have expanded treatment options where limited options are available and remain an area in dire need of therapeutic innovation. Future studies should explore further clinical CNS activity of both these agents.
TPX-0046, BOS172738, and other RET inhibitors in development
TPX-0046 is a novel orally bioavailable RET/SRC kinase inhibitor and has strong potency on the wild type and RETG810C/S/R solvent-front RET mutants [101]. In addition to RET, TPX-0046 inhibits several other protein tyrosine kinases, such as the SRC family kinases [102] and the clinical trial (ClinicalTrials.gov Identifier: NCT04161391) employing TPX-0046 is underway. BOS172738 (DS-5010) is another novel RET inhibitor with nanomolar potency against RET. BOS172738 showed exquisite potency for the wild type RET, RETV804M/L gatekeeper mutants, and the most common oncogenic RET mutation M918T and had high selectivity against VEGFR2 [103]. The clinical study of BOS172738 is now enrolling patients with RET-altered tumors (ClinicalTrials.gov Identifier: NCT03780517). TAS0953/HM06 is another RET TKI currently in preclinical development. Early nonclinical studies with BiDAC™ (bifunctional degradation activating compounds) RET inhibitor from C4 therapeutics, and second generation selective RET inhibitors from KinaRx and LOXO Oncology (LOX-18228 and LOX-19260) are also ongoing.
Mechanisms of resistance to RET-targeted therapy
De novo resistance and acquired resistance to RET TKIs have been observed in the clinic. The response rates to MKI RET inhibitors such as cabozantinib and vandetanib are low, likely in part to the incomplete suppression of the oncogenic RET kinase. Because off-target AEs, dose-reduction was necessary in 35–79% of MTC patients treated with vandetanib and cabozantinib [21]. Therefore, it is difficult to achieve optimal drug concentrations for RET inhibition with these MKIs [21]. While the more potent pralsetinib and selpercatinib have improved response rates [87,88], over 30% of RET-altered NSCLC and thyroid cancers do not achieve PR to these drugs. Co-occurrence of driver oncogenes, such as RAS and EGFR mutations and MET amplification, are observed in RET-altered cancers [6,104] and as acquired mutations in the laboratory [105]. Conceivably, co-occurrence of other driver oncogenes could by-pass the requirement of the RET oncogene, rendering RET TKIs ineffective. A combination of the MET/ALK/ROS1 TKI crizotinib with selpercatinib in patients who had RET fusion-positive and MET amplification NSCLC resulted in a response of the selpercatinib-resistant tumors to the combinational therapy [106].
Like other protein tyrosine kinase-targeted therapy, acquired secondary RET mutations that cause resistance to multikinase or specific RET TKIs have been identified in preclinical experiments and in patients whose tumors acquire TKI resistance (Figure 1). The gatekeeper RETV804M/L mutants are resistant to vandetanib, cabozantinib, lenvatinib, RXDX-105, and other MKIs used to inhibit RET [21,78,79,107–109]. Other MKI-resistant mutations are located in the hinge Y806, C-lobe solvent front G810, and N-lobe solvent front L730 (also called the roof) sites [78,110,111]. L730V/I roof mutations are resistant to pralsetinib but remain sensitive to selpercatinib in preclinical experiments [110,111], raising the possibility that selpercatinib could be used as a secondary drug when RET roof mutations cause pralsetinib resistance. However, this use has yet to be validated clinically. Mutations located outside the drug binding pockets of the RET kinase domain could cause resistance to RET TKIs [78]. For instance, RETS904F, located in the activating loop of the kinase domain, was found as a secondary mutation in a CCDC6-RET-positive NSCLC patient after the tumors developed vandetanib resistance [112].
Specific RET TKIs, selpercatinib and pralsetinib, circumvent the resistance mechanism of gatekeeper mutations. However, they face new challenges of resistance caused by mutations at non-gatekeeper sites. In a laboratory screening of selpercatinib resistance, G810C/S/R mutations at the C-lobe solvent front, Y806C/N mutations at the hinge, and the V738A mutation at the β2 strand were identified [83]. All five selpercatinib-resistant RET mutants are cross-resistant to pralsetinib. The most substantial resistance is at the C-lobe solvent front G810 site. Co-crystal structures of RET-selpercatinib and RET-pralsetinib show that Y806 and V738 are involved in the binding of these drugs (Figure 2D) and substitution of glycine with an amino acid with a bulky side chain would cause steric clash with these drugs [83].
Substitution of the Gly-810 solvent front residue with Lys, Ser, Cys, or Val was first reported in tissue biopsies and cell-free (cf)DNA from a KIF5B-RET fusion-positive and a CCDC6-RET fusion-positive NSCLC patients, who acquired resistance to selpercatinib after initially responding to the drug [113]. Three of four recurrent tumors in CCDC6-RET PDXs had G810S mutation after treatment with a low dose (3 mg/kg, BID) of selpercatinib.
Tissue and plasma biopsies obtained from patients with RET fusion-positive NSCLC treated with selpercatinib, pralsetinib, or both [111] revealed that some CCDC6-RET fusion-positive NSCLC patients (10%) had acquired G810C/S solvent front mutations during selpercatinib treatment, whereas acquired MET amplification without identifiable RET resistance alterations was observed in three cases (15%). Acquired KRAS amplification was also observed, proposing MET and KRAS amplifications as a more common resistance mechanism in this population. In another report [83], acquired G810C/S and Y806C/N RET mutations were detected in the cfDNA of a patient who had RETM918T+V804M/L MTC, whose liver metastases progressed following initial response to selpercatinib. In addition, acquired G810C mutation was observed in a CCDC6-RETfusion-positive NSCLC, who developed acquired resistance to selpercatinib. Tumor biopsy of another patient with RETM918T-positive metastatic MTC and treated sequentially with vandetanib (2 months), cabozantinib (8 months), selpercatinib (12 months), and pralsetinib (6 weeks) had RETM918T+V804M+G810S mutations at progression on selpercatinib [114]. KHDRBS1-NTRK3 fusion (K8;N14) as an acquired resistance mechanism to selpercatinib in a KIF5B-RET fusion (K15;R12) positive lung cancer and acquired tertiary MET resistance (MET D1228N + LSM8-MET fusion) to selpercatinib and MET inhibitor capmatinib in KIF5B-RET-positive NSCLC with secondary MET amplification as initial resistance to selpercatinib have also been recently reported [115,116]
Concluding remarks
RET-altered cancers occur frequently in MTC and PTC in the forms of oncogenic mutations and gene fusions, respectively. Besides PTC, more than 35 different RET fusion oncogenes have been observed in diverse cancers, which occur most frequently in lung adenocarcinoma. The high response rates of these RET-altered cancers to the highly specific RET TKIs pralsetinib and selpercatinib unequivocally established the oncogenic RET kinase as the therapeutic target in RET-altered cancer. However, several critical issues remain to be addressed, including the incomplete response and relative resistance to selpercatinib and pralsetinib in approximately one-third of RET-altered cancers, and the emergence of acquired resistance to these RET TKIs by secondary on-target mutations and/or acquisition of alternative driver oncogenes. (see Outstanding questions). A combination treatment of amplified MET with crizotinib has illustrated the clinical efficacy of this approach to circumvent co-aberrant oncogenes. The solvent front G810 mutations is emerging as a predominant on-target resistant mechanism to selpercatinib and pralsetinib. A new generation of RET TKI capable of inhibiting G810 solvent front mutants among other on-target resistance mechanisms are needed. Several RET TKIs like TPX-0046, BOS172738, and TAS0953/HM06 have started human clinical studies. Early nonclinical studies with BiDAC™ RET inhibitor from C4 therapeutics and second generation RET inhibitors from KinaRx and LOXO Oncology are in progress.
Outstanding questions.
As patients with NSCLC are at high risk of CNS metastases, can we sustain durable CNS activity from selective RET inhibitors?
Since RET plays a crucial role in the development of the nervous system and kidney, and in spermatogenesis, what would be the clinical implications of long-term RET inhibition in very young children?
What will be the impact of selective RET inhibitors in other RET-altered non-thyroid, non-NSCLC solid tumors and in pediatric population? Although selective RET inhibitors render some promising preliminary activity in other solid tumors, would those RET-specific inhibitors become the next tumor-agnostic indication? The duration of response and acquired resistance in non-lung and non-thyroid tumors is unknown.
In light of new challenge of resistance caused by mutations at non-gatekeeper sites or co-occurrence of other oncogenic drivers, how can we overcome resistance to those newer RET-specific inhibitors? Which combinatorial strategies could be warranted?
Can we achieve more durable and better response with combinatorial approaches utilizing selective RET inhibitors?
The initial results of the global multicenter registrational ARROW and LIBRETTO-001 trials employing the selective RET therapeutics such as LOXO-292 (selpercatinib) and BLU-667 (pralsetinib) are promising. Both agents have received accelerated approval from the FDA. The Phase III randomized trials versus standard of care have been launched.
Postulating and validating the resistance mechanisms is the subject of ongoing studies and second generation RET inhibitors are in clinical development. Furthermore, the intracranial activity of these RET-specific agents has drawn much attention where steering the CNS metastases in the NSCLC remains an arduous therapeutic challenge. Researchers should remain vigilant upon the resurgence of the resistance mechanisms.
Supplementary Material
Highlights.
Rearranged during transfection (RET) is an oncogenic driver activated by either RET fusions or mutations. RET fusions occur predominantly in 2% of lung cancers and 10–20% of thyroid cancers and in low frequency in an increasing number of diverse cancers.
Various nonselective multikinase inhibitors with ancillary RET inhibitory activity have been used in the clinic, despite having modest activity and notable off-target toxicities.
Recently, highly potent and RET-specific inhibitors selpercatinib and pralsetinib have been successfully translated to the clinic and are FDA approved.
Data on acquired resistance to RET-specific inhibitors are rapidly emerging but not fully understood. However recent studies have suggested on-target mutations at non-gatekeeper sites or emergence of off-target alterations such as MET amplification or NTRK fusion as mechanisms of acquired resistance.
Acknowledgments
National Institutes of Health grant R01CA242845 (to B.H.M.M., V.S., and J.W.). V.S. is an Andrew Sabin Family Foundation Fellow at The University of Texas MD Anderson Cancer Center. V.S. acknowledges support of The Jacquelyn A. Brady Fund. MD Anderson Cancer Center Department of Investigational Cancer Therapeutics is supported by the Cancer Prevention and Research Institute of Texas (RP1100584), the Sheikh Khalifa Bin Zayed Al Nahyan Institute for Personalized Cancer Therapy, 1U01 CA180964, NCATS Grant UL1 TR000371 (Center for Clinical and Translational Sciences), and the MD Anderson Cancer Center Support Grant (P30 CA016672).
Declaration of interests
V.S. reports research funding/grant support for clinical trials: Roche/Genentech, Novartis, Bayer, GlaxoSmithKline, Nanocarrier, Vegenics, Celgene, Northwest Biotherapeutics, Berghealth, Incyte, Fujifilm, Pharmamar, D3, Pfizer, Multivir, Amgen, Abbvie, Alfa-sigma, Agensys, Boston Biomedical, Idera Pharma, Inhibrx, Exelixis, Blueprint Medicines, Loxo Oncology, Medimmune, Altum, Dragonfly Therapeutics, Takeda and National Comprehensive Cancer Network, NCI-CTEP and UT MD Anderson Cancer Center, Turning Point Therapeutics, Boston Pharmaceuticals; Travel: Novartis, Pharmamar, ASCO, ESMO, Helsinn, Incyte. Consultancy/advisory board: Helsinn, LOXO Oncology/Eli Lilly, R-Pharma US, INCYTE, QED pharma, Medimmune, Novartis. Other: Medscape. All remaining authors have declared no conflicts of interest.
Glossary
- Acquired resistance:
resistance to therapy that emerges after some length of time after a patient was given a specific therapy. It may include the acquisition of new alterations in the tumor or the expansion of some low level clones that had the plasticity to expand in the face of therapy.
- De novo resistance:
intrinsic (pre-existent) primary resistance where no initial response is observed to a drug in treatment-naïve patients.
- Disease control rate (DCR):
the proportion of patients whose tumors have responded to the therapy and achieved complete response, partial response, and stable disease.
- Driver oncogenes:
genes that promote tumorigenesis when altered by mutations or rearrangements.
- Duration of response (DOR):
the length of time from initial response to subsequent disease progression or relapse or death.
- Fusion genes:
genes that become hybridized and fused from two previously independent genes either via translocation, deletion, or chromosomal inversion.
- Multiple endocrine neoplasia type 2 (MEN 2) syndrome:
an inherited cancer syndrome that comprises medullary thyroid cancer, pheochromocytoma, and parathyroid hyperplasia.
- Next generation sequencing:
a high-throughput parallel deep sequencing method to identify the nucleotide sequence, targeted regions of a genome, or even a whole genome.
- Objective response rate (ORR):
the proportion of patients whose tumors have responded to the therapy either by partial or complete response.
- Progression-free survival (PFS):
the time from randomization of clinical participant to progression of disease or death from any cause.
Footnotes
Supplemental information
Supplemental information associated with this article can be found online https://doi.org/10.1016/j.trecan.2021.07.003.
References
- 1.Takahashi M et al. (1985) Activation of a novel human transforming gene, ret, by DNA rearrangement. Cell 42, 581–588 [DOI] [PubMed] [Google Scholar]
- 2.Chi X et al. (2009) Ret-dependent cell rearrangements in the Wolffian duct epithelium initiate ureteric bud morphogenesis. Dev. Cell 17, 199–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.de Graaff E et al. (2001) Differential activities ofthe RET tyrosine kinase receptor isoforms during mammalian embryogenesis. Genes Dev. 15, 2433–2444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tsuzuki T et al. (1995) Spatial and temporal expression of the ret proto-oncogene product in embryonic, infant and adult rat tissues. Oncogene 10, 191–198 [PubMed] [Google Scholar]
- 5.Kohno T et al. (2020) REToma: a cancer subtype with a shared driver oncogene. Carcinogenesis 41, 123–129 [DOI] [PubMed] [Google Scholar]
- 6.Kato S et al. (2017) RET aberrations in diverse cancers: next-generation sequencing of 4,871 patients. Clin. Cancer Res 23, 1988–1997 [DOI] [PubMed] [Google Scholar]
- 7.Liu X et al. (2020) RET kinase alterations in targeted cancer therapy. Cancer Drug Resist. 3, 472–481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Donis-Keller H et al. (1993) Mutations in the RET proto-oncogene are associated with MEN 2A and FMTC. Hum. Mol. Genet 2, 851–856 [DOI] [PubMed] [Google Scholar]
- 9.Hofstra RM et al. (1994) A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature 367, 375–376 [DOI] [PubMed] [Google Scholar]
- 10.Kohno T et al. (2012) KIF5B-RET lesions in lung adenocarcinoma. Nat. Med 18, 375–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lipson D et al. (2012) Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat. Med 18, 382–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mulligan LM et al. (1993) Germ-line mutations of the RET proto-oncogene in multiple endocrine neoplasia type 2A. Nature 363, 458–460 [DOI] [PubMed] [Google Scholar]
- 13.Romei C et al. (2016) A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma. Nat. Rev. Endocrinol 12, 192–202 [DOI] [PubMed] [Google Scholar]
- 14.Takeuchi K et al. (2012) RET, ROS1 and ALK fusions in lung cancer. Nat. Med 18, 378–381 [DOI] [PubMed] [Google Scholar]
- 15.Subbiah V et al. (2020) State-of-the-art strategies for targeting RET-dependent cancers. J. Clin. Oncol 38, 1209–1221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wiesner T et al. (2014) Kinase fusions are frequent in Spitz tumours and spitzoid melanomas. Nat. Commun 5, 3116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ballerini P et al. (2012) RET fusion genes are associated with chronic myelomonocytic leukemia and enhance monocytic differentiation. Leukemia 26, 2384–2389 [DOI] [PubMed] [Google Scholar]
- 18.Le Rolle AF et al. (2015) Identification and characterization of RET fusions in advanced colorectal cancer. Oncotarget 6, 28929–28937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ogino H et al. (2008) Novel dual targeting strategy with vandetanib induces tumor cell apoptosis and inhibits angiogenesis in malignant pleural mesothelioma cells expressing RET oncogenic rearrangement. Cancer Lett. 265, 55–66 [DOI] [PubMed] [Google Scholar]
- 20.Paratala BS et al. (2018) RET rearrangements are actionable alterations in breast cancer. Nat. Commun 9, 4821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Drilon A et al. (2018) Targeting RET-driven cancers: lessons from evolving preclinical and clinical landscapes. Nat. Rev. Clin. Oncol 15, 151–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishizaka Y et al. (1989) Human ret proto-oncogene mapped to chromosome 10q11.2. Oncogene 4, 1519–1521 [PubMed] [Google Scholar]
- 23.Anders J et al. (2001) Molecular modeling of the extracellular domain of the RET receptor tyrosine kinase reveals multiple cadherin-like domains and a calcium-binding site. J. Biol. Chem 276, 35808–35817 [DOI] [PubMed] [Google Scholar]
- 24.Goodman KM et al. (2014) RET recognition of GDNF-GFRalpha1 ligand by a composite binding site promotes membrane-proximal self-association. Cell Rep. 8, 1894–1904 [DOI] [PubMed] [Google Scholar]
- 25.Amoresano A et al. (2005) Direct interactions among Ret, GDNF and GFRalpha1 molecules reveal new insights into the assembly of a functional three-protein complex. Cell. Signal 17, 717–727 [DOI] [PubMed] [Google Scholar]
- 26.Wang X (2013) Structural studies of GDNF family ligands with their receptors-Insights into ligand recognition and activation of receptor tyrosine kinase RET. Biochim. Biophys. Acta 1834, 2205–2212 [DOI] [PubMed] [Google Scholar]
- 27.Arighi E et al. (2005) RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev. 16, 441–467 [DOI] [PubMed] [Google Scholar]
- 28.Tansey MG et al. (2000) GFRalpha-mediated localization of RET to lipid rafts is required for effective downstream signaling, differentiation, and neuronal survival. Neuron 25, 611–623 [DOI] [PubMed] [Google Scholar]
- 29.Ibanez CF (2013) Structure and physiology of the RET receptor tyrosine kinase. Cold Spring Harb. Perspect. Biol 5, a009134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andreozzi F et al. (2003) Protein kinase C alpha activation by RET: evidence for a negative feedback mechanism controlling RET tyrosine kinase. Oncogene 22, 2942–2949 [DOI] [PubMed] [Google Scholar]
- 31.Fukuda T et al. (2002) Nvel mechanism of regulation of Rac activity and lamellipodia formation by RET tyrosine kinase. J. Biol. Chem 277, 19114–19121 [DOI] [PubMed] [Google Scholar]
- 32.Maeda K et al. (2004) Biochemical and biological responses induced by coupling of Gab1 to phosphatidylinositol 3-kinase in RET-expressing cells. Biochem. Biophys. Res. Commun 323, 345–354 [DOI] [PubMed] [Google Scholar]
- 33.Schuringa JJ et al. (2001) MEN2A-RET-induced cellular transformation by activation of STAT3. Oncogene 20, 5350–5358 [DOI] [PubMed] [Google Scholar]
- 34.Trupp M et al. (1999) Ret-dependent and -independent mechanisms of glial cell line-derived neurotrophic factor signaling in neuronal cells. J. Biol. Chem 274, 20885–20894 [DOI] [PubMed] [Google Scholar]
- 35.Besset V et al. (2000) Signaling complexes and protein-protein interactions involved in the activation of the Ras and phosphatidylinositol 3-kinase pathways by the c-Ret receptor tyrosine kinase. J. Biol. Chem 275, 39159–39166 [DOI] [PubMed] [Google Scholar]
- 36.Hayashi H et al. (2000) Characterization of intracellular signals via tyrosine 1062 in RET activated by glial cell line-derived neurotrophic factor. Oncogene 19, 4469–4475 [DOI] [PubMed] [Google Scholar]
- 37.Liu X et al. (1996) Oncogenic RET receptors display different autophosphorylation sites and substrate binding specificities. J. Biol. Chem 271,5309–5312 [DOI] [PubMed] [Google Scholar]
- 38.Borrello MG et al. (1996) The full oncogenic activity of Ret/ptc2 depends on tyrosine 539, a docking site for phospholipase C gamma. Mol. Cell. Biol 16, 2151–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Perrinjaquet M et al. (2010) Protein-tyrosine phosphatase SHP2 contributes to GDNF neurotrophic activity through direct binding to phospho-Tyr687 in the RET receptor tyrosine kinase. J. Biol. Chem 285, 31867–31875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Encinas M et al. (2004) Tyrosine 981, a novel ret autophosphorylation site, binds c-Src to mediate neuronal survival. J. Biol. Chem 279, 18262–18269 [DOI] [PubMed] [Google Scholar]
- 41.Stransky N et al. (2014) The landscape of kinase fusions in cancer. Nat. Commun 5, 4846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cancer Genome Atlas Research Network (2014) Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yoshihara K et al. (2015) The landscape and therapeutic relevance of cancer-associated transcript fusions. Oncogene 34, 4845–4854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang R et al. (2012) RET fusions define a unique molecular and clinicopathologic subtype of non-small-cell lung cancer. J. Clin. Onco 30, 4352–4359 [DOI] [PubMed] [Google Scholar]
- 45.Tsuta K et al. (2014) RETrearranged non-small-cell lung carcinoma: a clinicopathological and molecular analysis. Br. J. Cancer 110, 1571–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fenton CL et al. (2000) The ret/PTC mutations are common in sporadic papillary thyroid carcinoma of children and young adults. J. Clin. Endocrinol. Metab 85, 1170–1175 [DOI] [PubMed] [Google Scholar]
- 47.Elisei R et al. (2001) RET/PTC rearrangements in thyroid nodules: studies in irradiated and not irradiated, malignant and benign thyroid lesions in children and adults. J. Clin. Endocrinol. Metab 86, 3211–3216 [DOI] [PubMed] [Google Scholar]
- 48.Cheung CC et al. (2001) Analysis of ret/PTC gene rearrangements refines the fine needle aspiration diagnosis of thyroid cancer. J. Clin. Endocrinol. Metab 86, 2187–2190 [DOI] [PubMed] [Google Scholar]
- 49.Nikiforov YE (2002) RET/PTC rearrangement in thyroid tumors. Endocr. Pathol 13, 3–16 [DOI] [PubMed] [Google Scholar]
- 50.Romei C et al. (2012) Modifications in the papillary thyroid cancer gene profile over the last 15 years. J. Clin. Endocrinol. Metab 97, E1758–E1765 [DOI] [PubMed] [Google Scholar]
- 51.Hamatani K et al. (2008) RET/PTC rearrangements preferentially occurred in papillary thyroid cancer among atomic bomb survivors exposed to high radiation dose. Cancer Res. 68, 7176–7182 [DOI] [PubMed] [Google Scholar]
- 52.Hamatani K et al. (2014) Anovel RET rearrangement (ACBD5/RET) by pericentric inversion, inv(10)(p12.1;q11.2), in papillary thyroid cancer from an atomic bomb survivor exposed to high-dose radiation. Oncol. Rep 32, 1809–1814 [DOI] [PubMed] [Google Scholar]
- 53.Leeman-Neill RJ et al. (2013) RET/PTC and PAX8/PPARgamma chromosomal rearrangements in post-Chernobyl thyroid cancer and their association with iodine-131 radiation dose and other characteristics. Cancer 119, 1792–1799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ricarte-Filho JC et al. (2013) Identification of kinase fusion oncogenes in post-Chernobyl radiation-induced thyroid cancers. J. Clin. Invest 123, 4935–4944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Piotrowska Z et al. (2018) Landscape of acquired resistance to osimertinib in EGFR-mutant NSCLC and clinical validation of combined EGFR and RET inhibition with osimertinib and BLU-667 for acquired RET fusion. Cancer Discov. 8, 1529–1539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Offin M et al. (2018) Acquired ALK and RET gene fusions as mechanisms of resistance to osimertinib in EGFR-mutant lung cancers. JCO Precis. Oncol 2, PO.18.00126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Li AY et al. (2019) RET fusions in solid tumors. Cancer Treat. Rev 81, 101911. [DOI] [PubMed] [Google Scholar]
- 58.Cancer Genome Atlas Research Network (2014) Integrated genomic characterization of papillary thyroid carcinoma. Cell 159, 676–690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Grubbs EG et al. (2015) RET fusion as a novel driver of medullary thyroid carcinoma. J. Clin. Endocrinol. Metab 100, 788–793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Plaza-Menacho I et al. (2014) Oncogenic RET kinase domain mutations perturb the autophosphorylation trajectory by enhancing substrate presentation in trans. Mol. Cell 53, 738–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Drilon A et al. (2016) Clinical outcomes with pemetrexed-based systemic therapies in RET-rearranged lung cancers. Ann. Oncol 27, 1286–1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hegde A et al. (2020) Responsiveness to immune checkpoint inhibitors versus other systemic therapies in RET-aberrant malignancies. ESMO Open 5, e000799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Offin M et al. (2019) Immunophenotype and response to immunotherapy of RET-rearranged lung cancers. JCO Precis. Oncol 3, PO.18.00386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bhandari NR et al. (2021) Efficacy of immune checkpoint inhibitor therapy in patients with RET fusion-positive non-small-cell lung cancer. Immunotherapy Published online June 18, 2021. 10.2217/imt-2021-0035 [DOI] [PubMed] [Google Scholar]
- 65.Addeo A et al. (2021) Immunotherapy in non-small cell lung cancer harbouring driver mutations. Cancer Treat. Rev 96, 102179. [DOI] [PubMed] [Google Scholar]
- 66.Drilon A et al. (2016) Cabozantinib in patients with advanced RET-rearranged non-small-cell lung cancer: an open-label, single-centre, phase 2, single-arm trial. Lancet Oncol. 17, 1653–1660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gautschi O et al. (2017) Targeting RET in patients with RET-rearranged lung cancers: results from the global, multicenter RET registry. J. Clin. Oncol 35, 1403–1410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yoh K et al. (2017) Vandetanib in patients with previously treated RET-rearranged advanced non-small-cell lung cancer (LURET): an open-label, multicentre phase 2 trial. Lancet Respir. Med 5, 42–50 [DOI] [PubMed] [Google Scholar]
- 69.Hida T et al. (2019) A phase 2 study of lenvatinib in patients with RET fusion-positive lung adenocarcinoma. Lung Cancer 138, 124–130 [DOI] [PubMed] [Google Scholar]
- 70.ls SA Jr. Wells SA Jr et al. (2012) Vandetanib in patients with locally advanced or metastatic medullary thyroid cancer: a randomized, double-blind phase III trial. J. Clin. Oncol 30, 134–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Elisei R et al. (2013) Cabozantinib in progressive medullary thyroid cancer. J. Clin. Oncol 31,3639–3646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schlumberger M et al. (2017) Overall survival analysis of EXAM, a phase III trial of cabozantinib in patients with radiographically progressive medullary thyroid carcinoma. Ann. Oncol 28, 2813–2819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Brose MS et al. (2014) Sorafenib in radioactive iodine-refractory, locally advanced or metastatic differentiated thyroid cancer: a randomised, double-blind, phase 3 trial. Lancet 384, 319–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schlumberger M et al. (2015) Lenvatinib versus placebo in radioiodine-refractory thyroid cancer. N. Engl. J. Med 372, 621–630 [DOI] [PubMed] [Google Scholar]
- 75.Li GG et al. (2017) Antitumor activity of RXDX-105 in multiple cancer types with RET rearrangements or mutations. Clin. Cancer Res 23, 2981–2990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Drilon A et al. (2019) A phase I/Ib trial of the VEGFR-sparing multikinase RET inhibitor RXDX-105. Cancer Discov. 9, 384–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Das TK and Cagan RL (2017) KIF5B-RET oncoprotein signals through a multi-kinase signaling hub. Cell Rep. 20, 2368–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu X et al. (2018) Drug resistance profiles of mutations in the RET kinase domain. Br. J. Pharmacol 175, 3504–3515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Dagogo-Jack I et al. (2018) Emergence of a RET V804M gatekeeper mutation during treatment with vandetanib in RET-rearranged NSCLC. J. Thorac. Oncol 13, e226–e227 [DOI] [PubMed] [Google Scholar]
- 80.Subbiah V et al. (2018) Precision targeted therapy with BLU-667 for RET-driven cancers. Cancer Discov. 8, 836–849 [DOI] [PubMed] [Google Scholar]
- 81.Subbiah V et al. (2018) Selective RET kinase inhibition for patients with RET-altered cancers. Ann. Oncol 29, 1869–1876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Knowles PP et al. (2006) Structure and chemical inhibition of the RET tyrosine kinase domain. J. Biol. Chem 281, 33577–33587 [DOI] [PubMed] [Google Scholar]
- 83.Subbiah V et al. (2020) Structural basis of acquired resistance to selpercatinib and pralsetinib mediated by non-gatekeeper RET mutations. Ann. Oncol 32, 261–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.US Food and Drug Administration (2020) FDA Approves Pralsetinib for RET-altered Thyroid Cancers, US FDA [Google Scholar]
- 85.Besse B et al. (2020) AcceleRET Lung: a phase III study of first-line pralsetinib in patients (pts) with RET-Usion+advanced/metastatic non-small cell lung cancer (NSCLC). J. Clin. Oncol 38, TPS9633 [Google Scholar]
- 86.Subbiah V et al. (2020) Clinical activity of the RET inhibitor pralsetinib (BLU-667) in patients with RET fusion+ solid tumors. J. Clin. Oncol 38, 109 [Google Scholar]
- 87.Wirth LJ et al. (2020) Efficacy of selpercatinib in RET-altered thyroid cancers. N. Engl. J. Med 383, 825–835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Drilon A et al. (2020) Efficacy of selpercatinib in RET fusion-positive non-small-cell lung cancer. N. Engl. J. Med 383, 813–824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.U.S. Food and Drug Administration (2020) FDA Approves Selpercatinib for Lung and Thyroid Cancers with RET Gene Mutations or Fusions, US FDA [Google Scholar]
- 90.Hernando J et al. (2020) 1927TiP LIBRETTO-531: selpercatinib in patients with treatment (Tx)-naïve RET-mutant medullary thyroid cancer (MTC). Ann. Oncol 31, S1091 [Google Scholar]
- 91.Loong H et al. (2020) 1413TiP LIBRETTO-431: selpercatinib in treatment (Tx)-naïve patients with RET fusion-positive (RET+) non-small cell lung cancer (NSCLC). Ann. Oncol 31, S893 [Google Scholar]
- 92.Gainor JF et al. (2021) Pralsetinib for RET fusion-positive non-small-cell lung cancer (ARROW): a multi-cohort, open-label, phase 1/2 study. Lancet Oncol 22, 959–969 [DOI] [PubMed] [Google Scholar]
- 93.Subbiah V et al. (2021) Pralsetinib for patients with advanced or metastatic RET-altered thyroid cancer (ARROW): a multi-cohort, open-label, registrational, phase 1/2 study. Lancet Diabetes Endocrinol. 9, 491–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ortiz MV et al. (2020) Activity of the highly specific RET inhibitor selpercatinib (LOXO-292) in pediatric patients with tumors harboring RET gene alterations. JCO Precis. Oncol 4, PO.19.00401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Drilon A et al. (2018) Frequency of brain metastases and multikinase inhibitor outcomes in patients with RET-rearranged lung cancers. J. Thorac. Oncol 13, 1595–1601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Evans EK et al. (2019) Pralsetinib (BLU-667) demonstrates robust activity in RET-fusion-driven intracranial tumor models. J. Thorac. Oncol 14, S701 [Google Scholar]
- 97.Subbiah V et al. (2021) Intracranial efficacy of selpercatinib in RET fusion-positive non-small cell lung cancers on the LIBRETTO-001 trial. Clin Cancer Res. 27, 4160–4167 Published online June 4, 2021. 10.1158/1078-0432.CCR-21-0800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Guo R et al. (2019) Response to selective RET inhibition with LOXO-292 in a patient with RET fusion-positive lung cancer with leptomeningeal metastases. JCO Precis. Oncol 3, PO.19.00021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Andreev-Drakhlin A et al. (2020) Systemic and CNS activity of selective RET inhibition with selpercatinib (LOXO-292) in a patient with RET-mutant medullary thyroid cancer with extensive CNS metastases. JCO Precis. Oncol 4, PO.20.00096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tsui DCC et al. (2021) Central nervous system response to selpercartinib in patient with RET-rearranged non-small cell lung cancer after developing leptomeningeal disease on pralsetinib. Clin Lung Cancer Published online June 12, 2021. 10.1016/j.cllc.2021.06.005 [DOI] [PubMed] [Google Scholar]
- 101.Drilon AE et al. (2020) The next-generation RET inhibitor TPX-0046 is active in drug-resistant and naïve RET-driven cancer models. J. Clin. Oncol 38, 3616 [Google Scholar]
- 102.Drilon A et al. (2019) TPX-0046 is novel and potent RET/SRC inhibitor for RET-driven cancers. Ann. Oncol 30, 506P30715156 [Google Scholar]
- 103.Schoffski P et al. (2019) A phase I study of BOS172738 in patients with advanced solid tumors with RET gene alterations including non-small cell lung cancer and medullary thyroid cancer. J. Clin. Oncol 37, TPS3162 [Google Scholar]
- 104.Rich TA et al. (2019) Analysis of cell-free DNA from 32,989 advanced cancers reveals novel co-occurring activating RET alterations and oncogenic signaling pathway aberrations. Clin. Cancer Res 25, 5832–5842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nelson-Taylor SK et al. (2017) Resistance to RET-inhibition in RET-rearranged NSCLC is mediated by reactivation of RAS/MAPK signaling. Mol. Cancer Ther 16, 1623–1633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Rosen EY et al. (2020) Overcoming MET-dependent resistance to selective RET inhibition in patients with RET fusion-positive lung cancer by combining selpercatinib with crizotinib. Clin. Cancer Res 27, 34–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Carlomagno F et al. (2004) Disease associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene 23, 6056–6063 [DOI] [PubMed] [Google Scholar]
- 108.Mologni L et al. (2013) Ponatinib is a potent inhibitor of wild-type and drug-resistant gatekeeper mutant RET kinase. Mol. Cell. Endocrinol 377, 1–6 [DOI] [PubMed] [Google Scholar]
- 109.Huang Q et al. (2016) Preclinical modeling of KIF5B-RET fusion lung adenocarcinoma. Mol. Cancer Ther 15, 2521–2529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Shen T et al. (2021) The L730V/I RET roof mutations display different activities toward pralsetinib and selpercatinib. NPJ Precis. Oncol 5, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lin JJ et al. (2020) Mechanisms of resistance to selective RET tyrosine kinase inhibitors in RET fusion-positive non-small-cell lung cancer. Ann. Oncol 31, 1725–1733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Nakaoku T et al. (2018) A secondary RET mutation in the activation loop conferring resistance to vandetanib. Nat. Commun 9, 625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Solomon BJ et al. (2020) RET solvent front mutations mediate acquired resistance to selective RET inhibition in RET-driven malignancies. J. Thorac. Oncol 15, 541–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Bruce JY et al. (2020) Emergence of resistant clones in medullary thyroid cancer may not be rescued by subsequent salvage highly selective rearranged during transfection-inhibitor therapy. Thyroid 31, 332–333 [DOI] [PubMed] [Google Scholar]
- 115.Subbiah V et al. (2021) Patient-driven discovery and post-clinical validation of NTRK3 fusion as an acquired resistance mechanism to selpercatinib in RET fusion-positive lung cancer. Ann. Oncol 32, 817–819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhu VW et al. (2021) Acquired tertiary MET resistance (MET D1228N and a novel LSM8-MET fusion) to selpercatinib and capmatinib in a patient with KIF5B-RET-positive NSCLC with secondary MET amplification as initial resistance to selpercatinib. J. Thorac. Oncol 16, e51–e54 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.