

Abstract
The multifunctional glycoprotein fibronectin influences several crucial cellular processes and contributes to multiple pathologies. While a link exists between fibronectin-associated pathologies and the receptor tyrosine kinase EphA2, the mechanism by which EphA2 promotes fibronectin matrix remodeling remains unknown. We previously demonstrated that EphA2 deletion reduces smooth muscle fibronectin deposition and blunts fibronectin deposition in atherosclerosis without influencing fibronectin expression. We now show that EphA2 expression is required for contractility-dependent elongation of tensin- and α5β1 integrin-rich fibrillar adhesions that drive fibronectin fibrillogenesis. Mechanistically, EphA2 localizes to integrin adhesions where focal adhesion kinase mediates ligand-independent Y772 phosphorylation, and mutation of this site significantly blunts fibrillar adhesion length. EphA2 deficiency decreases smooth muscle cell contractility by enhancing p190RhoGAP activation and reducing RhoA activity, whereas stimulating RhoA signaling in EphA2 deficient cells rescues fibrillar adhesion elongation. Together, these data identify EphA2 as a novel regulator of fibrillar adhesion elongation and provide the first data identifying a role for EphA2 signaling in integrin adhesions.
Keywords: EphA2 receptor tyrosine kinase, fibrillar adhesions, fibronectin deposition, focal adhesion kinase, RhoA contractility, p190RhoGAP
Introduction:
Interaction between cells and the extracellular matrix (ECM) represent a critical regulator of cell and tissue homeostasis. Multi-protein complexes, like focal adhesions and fibrillar adhesions, mediate cell adhesion to the matrix, link the extracellular matrix to the cytoskeleton, and facilitate cell-mediated matrix remodeling [1]. Beyond providing a substrate for cell adhesion, the ECM influences numerous cellular functions, including cell survival, proliferation, migration, and differentiation, through signaling proteins located within and in close proximity to the adhesion complex [1]. During tissue remodeling, dynamic matrix deposition changes the local microenvironment to regulate local cell phenotype and enable the remodeling response. Of particular importance, the glycoprotein fibronectin shows enhanced deposition during tissue remodeling and plays a critical role in regulating cell migration and proliferation to facilitate the remodeling response [2]. In addition, fibronectin serves as a scaffold to facilitate the deposition and organization of other matrix proteins [3] and exacerbates fibrosis within the contexts of numerous diseases, like cancer, atherosclerosis, arthritis, and asthma [4–6].
Rather than passive secretion, matrix deposition and assembly requires active cellular processes. During fibronectin fibrillogenesis, fibronectin dimers are first secreted in a globular, soluble conformation, whereupon interactions with cell integrins, primarily α5β1 integrins, mediate their assembly into insoluble fibrils [7]. The integrin complex α5β1 mediates fibronectin matrix assembly by transmitting actomyosin tensile forces to extend the fibronectin conformation and to expose cryptic binding sites [8–12]. The α5β1-mediated stretching of fibronectin requires α5β1 to traffic from the peripheral adhesions into elongated fibrillar adhesion structures rich in α5β1 and the cytoplasmic adaptor protein tensin that links the β1 integrin to the actin cytoskeleton [13, 14]. These fibrillar adhesions align fibronectin into fibrils that serve as a scaffold for cell migration and for deposition of other matrix proteins [15, 16].
Eph receptor tyrosine kinases, the largest mammalian subfamily of receptor tyrosine kinases, act as guidance molecules to regulate development and tissue remodeling by affecting cell adhesion, migration, and proliferation [17–19]. Eph receptors mediate these effects through both ligand-dependent and ligand-independent mechanisms, with ephrinA ligands activating EphA receptors and ephrinB ligands activating EphB receptors. While the EphA2 receptor has garnered interest for its roles in carcinogenesis and proinflammatory diseases [20–22], EphA2 also appears to augment fibrotic diseases, such as atherosclerosis [21], lung injury [23, 24], and myocardial infarction [25]. Although multiple studies identify EphA2 as a regulator of focal adhesion kinase (FAK) signaling [26, 27] and cytoskeletal remodeling through effects on RhoA GTPase signaling [28], the mechanisms by which EphA2 affects fibrosis remain largely unknown. Previous work in our lab found that EphA2 deletion reduces fibronectin levels in experimental models of atherosclerosis and is associated with reduced plaque size and progression [21]. In vitro, depletion of EphA2 in vascular smooth muscle cells (VSMCs) reduces fibronectin deposition, while exogenous expression of EphA2 enhances fibronectin deposition [21, 29]. In VSMCs, EphA2 expression does not regulate fibronectin mRNA transcript or protein levels, nor does it significantly alter the expression or activity of proteases [21]. Given these observations, we hypothesize that EphA2 regulates fibronectin deposition and assembly. Therefore, we sought to determine the mechanisms by which EphA2 mediates fibronectin deposition.
Results:
EphA2 depletion reduces tensin localization and fibrillar adhesion length.
We previously showed that EphA2 knockout mice exhibit reduced plaque fibrosis and EphA2-depleted vascular smooth muscle cells show blunted fibronectin deposition in vitro [21]. Interestingly, EphA2 depletion in human dermal fibroblasts also reduces fibronectin deposition (Supplemental Figure I), suggesting that the EphA2 expression requirement for fibronectin deposition extends beyond a niche function in vascular cells. Fibronectin interacts with cells primarily through RGD-binding integrins (αvβ3, α5β1) [30]. However, EphA2 depletion does not reduce the total expression of αv or β1 integrins (Supplemental Figure IIa/b). While α5 mRNA is enhanced following EphA2 depletion, protein expression remains unchanged (Supplemental Figure IIa/b). The assembly of fibronectin fibrils involves formation of α5β1-rich fibrillar adhesions [7], characterized by long adhesion structures enriched in α5β1 integrin and the cytoskeletal adaptor protein tensin [14, 31]. To assess whether EphA2 affects fibrillar adhesions, we assessed fibrillar adhesion components by Western blotting of isolated integrin adhesion fractions and by immunocytochemistry. This fraction does not contain components from the plasma membrane (TFR1), nucleus (HDAC3), mitochondria (TOMM20), endoplasmic reticulum (Calnexin), or cytoskeleton (β-tubulin), but contains multiple components associated with integrin adhesions, such as various integrins, integrin-linked kinase (ILK), and cytoskeletal adaptor proteins (Supplemental Figure IIIa/b). Following EphA2 knockdown, human VSMCs show significantly reduced tensin and α5 localization to integrin adhesions (Figure 1A–C). Similarly, EphA2 knockdown diminishes tensin staining in fibrillar adhesions (Figure 1D). In contrast, EphA2 overexpression in human VSMCs enhances tensin localization to the integrin adhesion fraction (Supplement IVa/b), suggesting EphA2 is sufficient to promote fibrillar adhesions.
Figure 1: EphA2 depletion reduces tensin localization and fibrillar adhesion length.
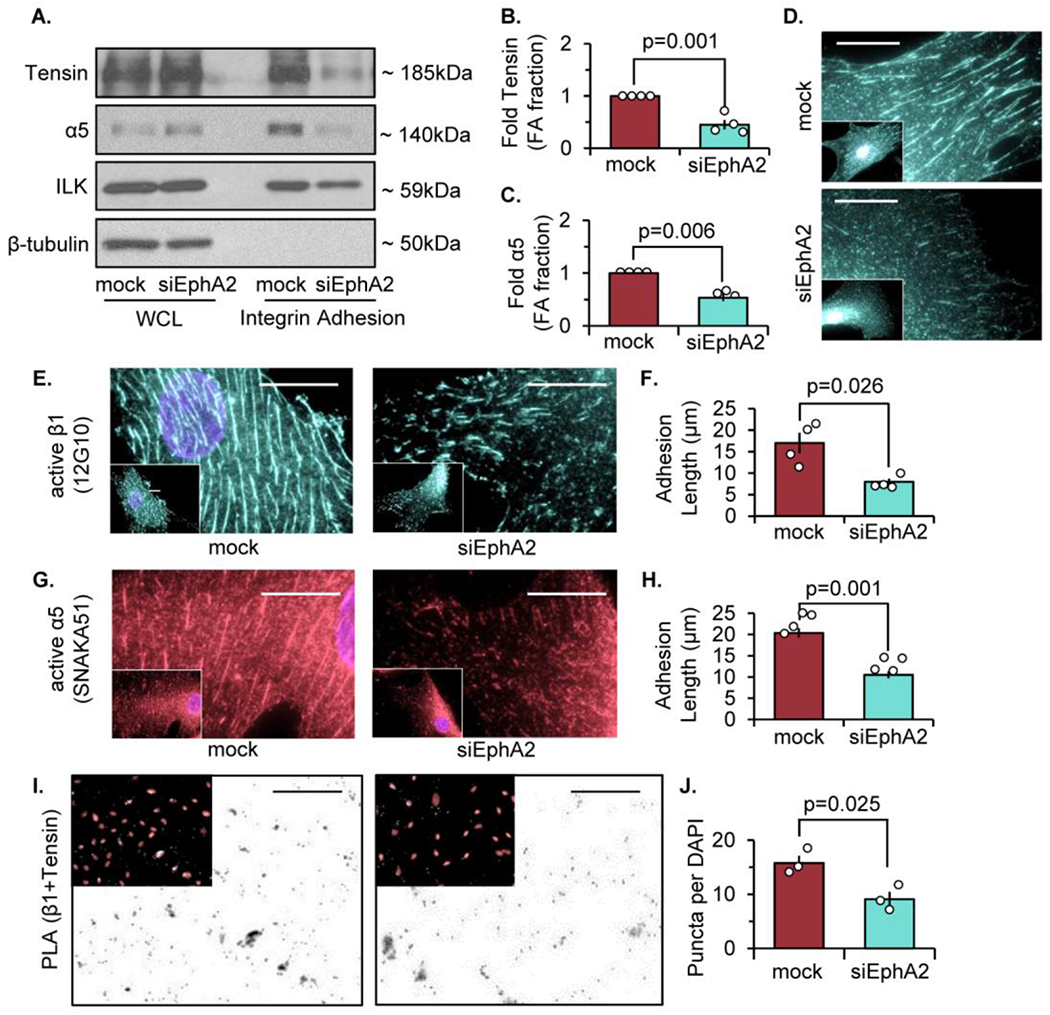
Human vascular smooth muscle cells (VSMCs) were transfected with either mock or siRNA targeted against EphA2 for 24 hours. A-C) Cells were plated onto Matrigel-coated glass slides overnight in 1% serum, and integrin adhesion isolation was performed. Protein expression was measured by immunoblot and normalized to integrin-linked kinase (ILK). β-tubulin from the whole cell lysate (WCL) fraction was shown for integrin adhesion isolation purity. B,C) Tensin and α5 integrin from the integrin adhesion fraction were quantified. D) Cells were stained for tensin. E,F) Cells were stained for active β1 integrin (12G10) and adhesion length was quantified in microns. G,H) Cells were stained for active α5 integrin (SNAKA51) and adhesion length was quantified in microns. (I) Proximity ligation assay (PLA) was performed for β1 and tensin interactions, and counterstained with DAPI (pink). Scale bar = 25μm. J) PLA puncta were quantified per high powered field and normalized to number of DAPI per high powered field. Scale bar = 25um. n=3-4. Data are expressed as mean ±SEM. Statistical comparisons were made using Student’s T-test (B,F,H,J,L). A p-value less than 0.05 is considered significant.
Integrins exist in either an inactive (closed) or active (open) conformation, where active integrins exhibit high affinity binding to the extracellular matrix [32]. We previously demonstrated that fibronectin deposition in endothelial cells requires α5β1 transition to an open, active conformation [33]. However, EphA2 depletion does not affect serum-induced α5β1 integrin activation (Supplemental Figure V). Immunocytochemistry with antibodies targeting ligand-inducible binding sites (LIBS) in α5 integrins (SNAKA51) and β1 integrins (12G10) show that EphA2 knockdown reduces the length of α5β1 adhesions in human VSMCs (Figure 1E–H), consistent with reduced fibrillar adhesion elongation [34, 35]. Furthermore, the association between tensin and β1 integrins, assessed by proximity ligation assay, shows a significant reduction following EphA2 knockdown (Figure 1I–J). However, EphA2 knockdown does not affect the total number of integrin adhesions positive for α5 or β1 (Supplemental Figure VIa/b) and does not affect the number or size of focal adhesions containing the cytoskeletal adaptor proteins paxillin and vinculin (Supplemental Figure VIc–f), suggesting EphA2 most likely affects fibrillar adhesion elongation rather than focal adhesion formation. Taken together, these results demonstrate that EphA2 expression is required for efficient fibrillar adhesion elongation and fibronectin deposition.
Serum treatment induces ligand-independent EphA2 phosphorylation.
While our current data clearly demonstrate a requirement for EphA2 in fibrillar adhesion elongation, the mechanisms responsible for this effect remain unknown. Interaction of EphA2 with its ligand ephrinA1 activates EphA2 kinase activity, inducing phosphorylation at multiple tyrosine residues (e.g. Y588, Y772), yielding binding sites for downstream signaling partners [27, 36–38]. However, EphA2 can also signal in a ligand-independent manner, associated with S897 phosphorylation by Akt and other serine/threonine kinases [39–42]. Since we observe differences in VSMC fibronectin deposition plated overnight in fetal bovine serum, we characterized the EphA2 signaling response to serum treatment. As expected, serum stimulation robustly enhances S897 phosphorylation (Figure 2A/B) but does not affect the ligand-dependent Y588 phosphorylation. In contrast, treatment with ephrinA1 ligand results in enhanced Y588 and Y772 phosphorylation but not S897 phosphorylation (Figure 2C). Unexpectedly, serum also induces EphA2 phosphorylation at the ligand-dependent Y772 site (Figure 2A/B). To assess where EphA2 signaling is occurring in serum-stimulated VSMCs, we performed immunocytochemistry for phospho-EphA2 Y772 and phospho-EphA2 S897. While phospho-EphA2 S897 shows diffuse staining (Figure 2D), phospho-EphA2 Y772 staining co-localizes with integrin adhesions (Figure 2E), as visualized by staining for phospho-FAK Y397. Furthermore, examination of the integrin adhesion protein isolates show EphA2 and phospho-EphA2 Y772 but not phospho-EphA2 S897 (Figure 2F). This data is consistent with a recent proteomic analysis of integrin adhesion complexes showing EphA2 localization to adhesion sites [43].
Figure 2: Serum treatment induces ligand-independent EphA2 phosphorylation.
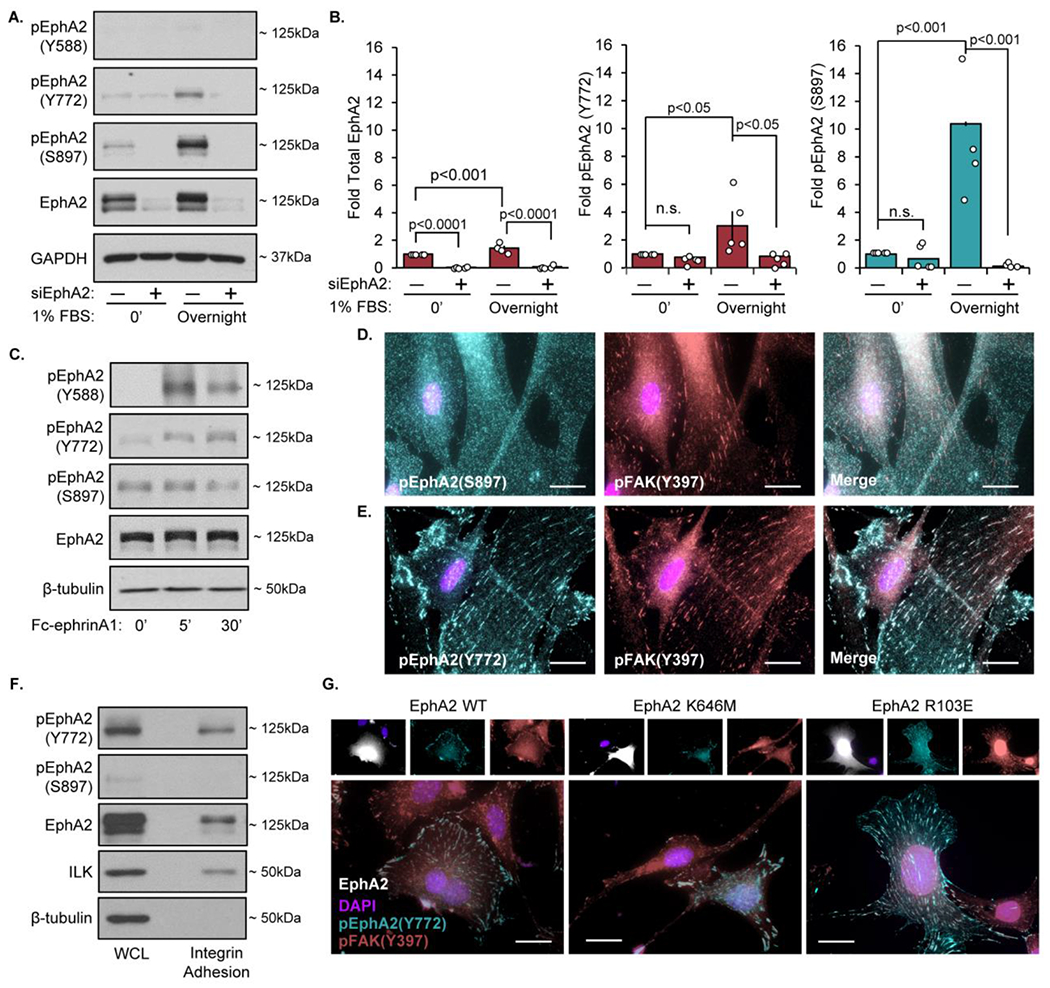
Human VSMCs were transfected with either mock or siRNA targeted against EphA2 for 24 hours. Cells were plated onto Matrigel coated plates overnight in either serum-free or 1% serum. Protein was isolated and analyzed via immunoblotting (A) Representative immunoblots and (B) densitometry analysis of phospho-Y772, phospho-S897, and total EphA2. Total EphA2 was normalized to GAPDH, pEphA2 Y772 and pEphA2 S897 were normalized to total EphA2. Values expressed as fold change from baseline (mock 0’). C) Human VSMCs were serum-starved for four hours, treated with 1μg/mL Fc-EphrinA1 for 5 or 30 minutes, followed by immunoblotting for phosphorylated (Y588, Y772, and S897) and total EphA2. D-E) Human VSMCs were seeded onto Matrigel-coated glass coverslips overnight in 1% serum-containing media and stained for (D) S897-phosphorylated EphA2 (teal), (E) Y772-phosphorylated EphA2 (teal), (D,E) Y397-phosphorylated FAK (red), and DAPI (nuclei). F) Cells were plated onto Matrigel-coated glass slides overnight in 1% serum, followed by integrin adhesion isolation and protein expression was measured via immunoblot. G) EphA2 KO mouse aortic VSMCs cells were transfected with EphA2-WT, K646M, or R103E, plated overnight in 1% serum, and stained for phospho-EphA2 Y772 (teal), Y397-pFAK (pink), EphA2 (white) and DAPI (nuclei). n=4. Data are expressed as mean ±SEM. Statistical comparisons were made using 2-way ANOVA with Bonferroni post-test. A p-value less than 0.05 is considered significant.
While EphA2 Y772 is canonically phosphorylated in response to ephrinA1 ligand (Figure 2C), the lack of Y588 phosphorylation, the more pronounced ligand-induced phosphorylation site, is inconsistent with ligand-dependent EphA2 activation. In addition, serum treatment decreases ephrinA1 expression in VSMCs (Supplemental Figure VIIa). Previous studies suggest EphA2 is capable of signaling by the non-ephrin ligand, progranulin [44] and progranulin stimulates cell migration through alterations in focal adhesion proteins [45]. However, progranulin levels in vascular smooth muscle cells are unaffected by serum stimulation and knockdown of progranulin does not affect EphA2 Y772 phosphorylation (Supplemental Figure VIIb/c). To assess whether Y772 phosphorylation in the integrin adhesions occurs independent of ligand binding, we transfected mouse EphA2 knockout VSMCs with a ligand-binding-deficient EphA2 mutant (R103E), which lacks ephrinA1-induced EphA2 Y772 phosphorylation (Supplemental Figure VIIIa–d) [46]. Consistent with ligand-independent EphA2 Y772 phosphorylation, phospho-EphA2 Y772 co-localizes with integrin adhesions in the ligand-binding-deficient EphA2 R103E mutant, suggesting ligand interaction is not required for localization of EphA2 to the integrin adhesions (Figure 2G). In addition, transfecting mouse EphA2 knockout VSMCs with a kinase-dead EphA2 mutant (K646M) shows similar Y772 phosphorylation in the focal adhesion as observed in the wildtype and ligand-binding-deficient mutants (Figure 2G, Supplemental Figure IXa), suggesting that EphA2 does not mediate its own phosphorylation in this context. Consistent with this, treatment with an ATP-competitive inhibitor of EphA2 kinase activity (ALW-II-41-27) does not block Y772 phosphorylation (Supplemental Figure IXb), suggesting EphA2 may undergo ligand-independent tyrosine transphosphorylation within integrin adhesions.
Re-Expression of EphA2 R103E but not EphA2 Y772F increases fibrillar adhesion elongation in EphA2 knockout cells.
To determine if Y772 phosphorylation is necessary for fibrillar adhesion elongation, EphA2 expression in EphA2 knockout (KO) VSMCs was transiently restored by transfection with either wild-type (WT) EphA2 or a mutated EphA2 in which tyrosine 772 was mutated to phenylalanine (Y772F). While re-expression of both EphA2 constructs was similar both in protein concentration and transfection efficiency, only EphA2-WT was capable of phosphorylation at Y772 (Supplemental Figure Xa/b). Wildtype EphA2 (EphA2-WT; EphA2-positive cells, inset) expression enhances tensin-associated fibrillar adhesion length compared to EphA2 KO VSMCs. However, rescue with EphA2-Y772F does not improve fibrillar adhesion length (Figure 3A/B). Furthermore, rescue with EphA2-Y772F fails to improve fibronectin deposition in EphA2 KO VSMCs, whereas re-expression of wildtype EphA2 enhances fibronectin fibrillogenesis (Figure 3C/D). To verify that ligand-independent EphA2 signaling drives fibrillar adhesion elongation and fibronectin deposition, EphA2 knockout cells were transfected with the ligand binding-deficient EphA2 mutant, EphA2-R103E. Consistent with EphA2-R103E supporting Y772 phosphorylation in focal adhesions (Figure 2G), rescuing EphA2 KO VSMCs with EphA2-R103E is sufficient to restore fibrillar adhesion length and fibronectin deposition similar to wildtype EphA2 (Figure 3E–H). Together these data suggest that ligand-independent phosphorylation of EphA2 at Y772 within focal adhesions promotes fibrillar adhesion elongation and fibronectin deposition.
Figure 3: EphA2-dependent fibrillar adhesion elongation requires Y772 phosphorylation but not EphA2-ligand interactions.
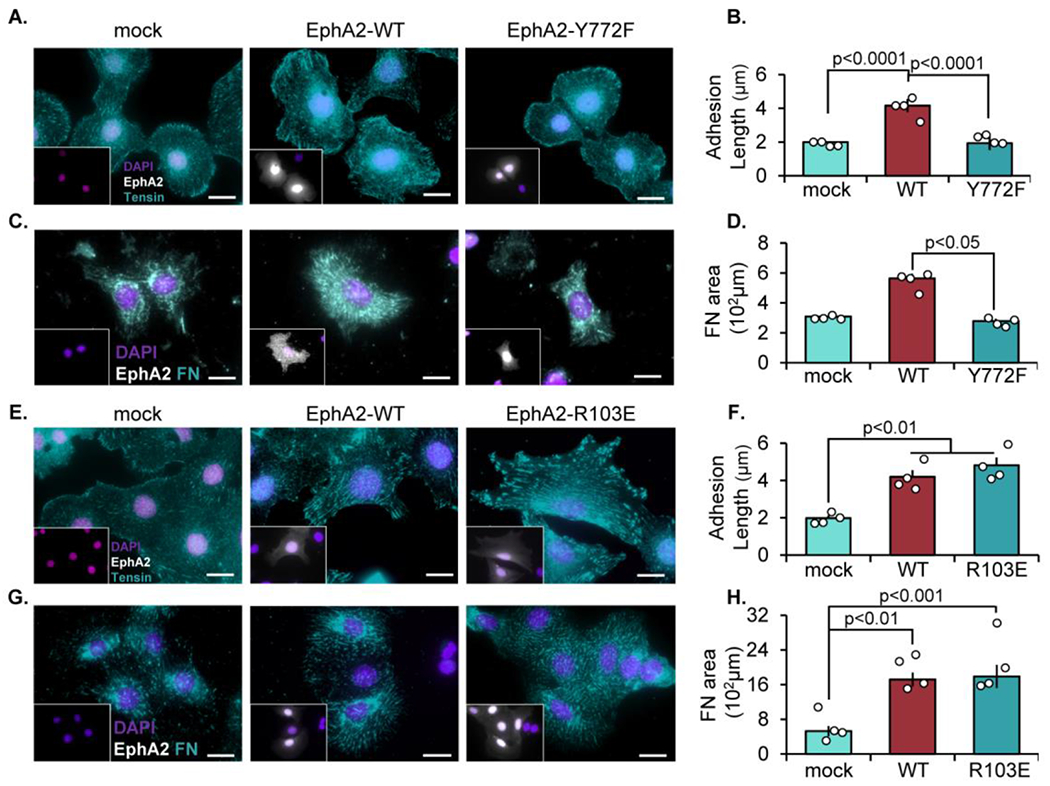
EphA2 KO mouse aortic VSMCs were transfected with either mock, EphA2-WT, (A-D) EphA2-Y772F, or (E-H) EphA2-R103E constructs for 24 hours, then plated onto Matrigel-coated coverslips overnight in 1% serum. Cells were stained for EphA2 (white), and EphA2-positive cells were quantified. A-B,E-F) Cells were stained fortensin (teal) and adhesion length was measured in microns. C-D,G-H) Cells were stained for fibronectin (teal) and quantified as fibronectin-positive area in microns. Scale bar = 25μm. n=4-5. Data are expressed as mean ±SEM. Statistical comparisons were made using One-way ANOVA with Bonferroni post-test. A p-value less than 0.05 is considered significant.
FAK is required for EphA2 phosphorylation within the focal adhesion.
We next sought to assess the signaling mechanisms mediating EphA2 Y772 phosphorylation within focal adhesions. FAK co-localizes with EphA2 in PC-3 cells [27], and ephrinA1 binding to EphA2 can result in either FAK activation or FAK inactivation depending upon the experimental conditions [26, 27]. Since EphA2 appears to be transphosphorylated on Y772 and phospho-EphA2 Y772 co-localizes with active FAK (Figure 2E–G), we sought to determine if FAK regulates EphA2 at Y772. Depletion of FAK with siRNA in human VSMCs significantly reduces Y772 phosphorylation at baseline and in response to serum treatment (Figure 4A/B). Treatment with the ATP-competitive FAK inhibitor PF-573228 similarly blunts both FAK activation and EphA2 Y772 phosphorylation (Figure 4C). Lastly, VSMCs isolated from mice expressing wildtype FAK (FAK WT) or kinase-dead FAK (FAK KD) [47] show significantly reduced Y772 phosphorylation in the absence of FAK kinase activity (Figure 4D). Taken together, these data suggest that FAK activity within integrin adhesions is required for both basal EphA2 Y722 phosphorylation and serum-induced EphA2 Y772 phosphorylation. However, this effect may be indirect through other FAK-associated kinases. Compared with FAK WT VSMCs, FAK KD cells show reduced fibrillar adhesion elongation, as assessed by tensin and α5 localization to the integrin adhesion fraction (Supplemental Figure XIa–c) and by immunocytochemistry for fibrillar adhesion length (Supplemental Figure XId/e). Together these data suggest that FAK activity is required for both focal adhesion EphA2 Y772 phosphorylation and fibrillar adhesion elongation.
Figure 4: FAK is required for EphA2 phosphorylation within the focal adhesion.
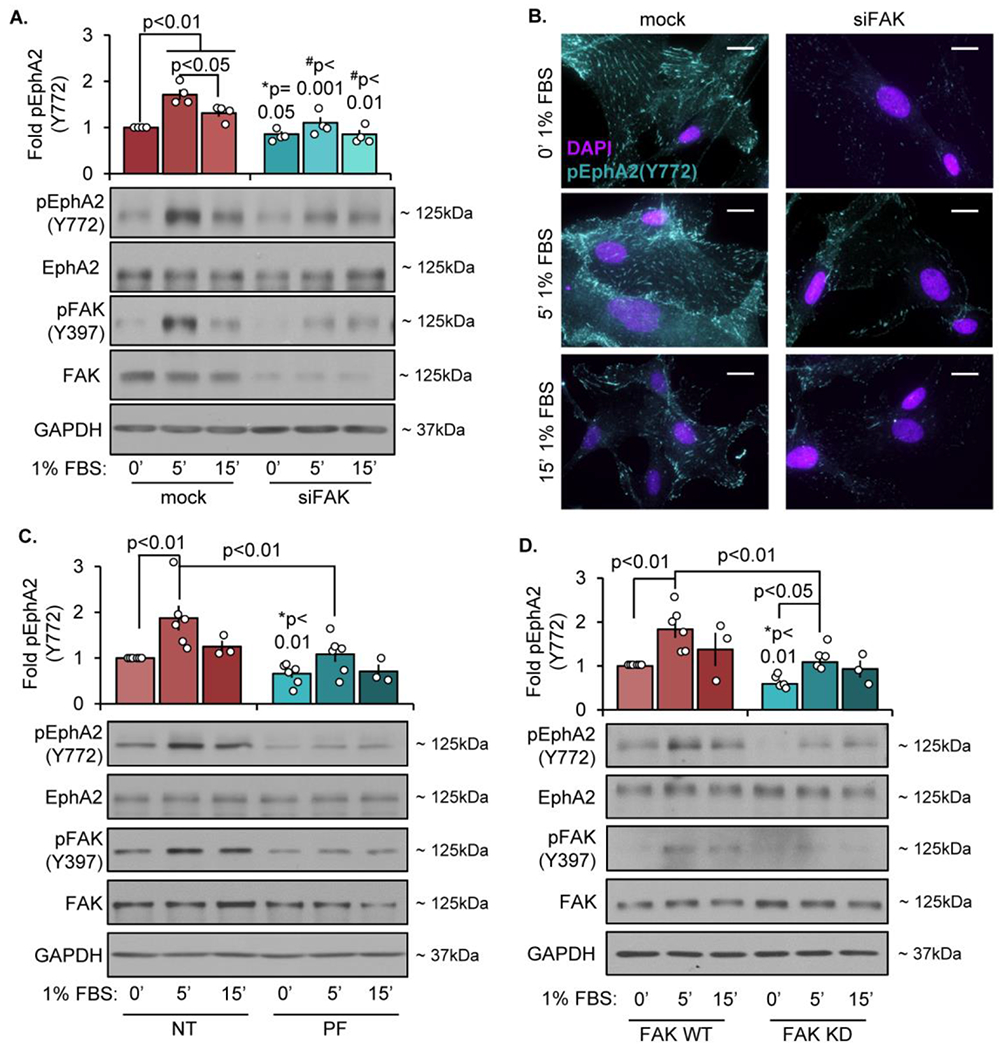
Human VSMCs cells were transfected with either mock or siRNA targeting FAK for 24 hours. Cells were then plated onto Matrigel overnight, serum starved for 4 hours, and treated with 1% serum at the indicated timepoints. A) Phospho-EphA2 was analyzed by immunoblot and normalized to total EphA2 or (B) stained for Y772-phospho EphA2 (teal) and DAPI (nuclei). C) Cells were treated with PF-573228 (PF, 4μM) for 30 minutes, then treated with 1% serum at the indicated timepoints. Phospho-EphA2 was analyzed by immunoblot and normalized to total EphA2. D) FAK WT or FAK KD cells were serum-starved for 4 hours, then treated with 1% serum at the indicated timepoints. Phospho-EphA2 was measured by immunoblot and normalized to total EphA2. Fold changes are relative to mock/NT/WT 0’ timepoint. n=4. Data are expressed as mean ±SEM. Statistical comparisons were made using 2way ANOVA with Bonferroni post-test. #p-values compare between conditions for each treatment (mock vs. siFAK). *p-values compare baseline (0’ 1%FBS) between conditions using Student’s T-test. A p-value less than 0.05 is considered significant.
EphA2 signals through RhoA to promote fibrillar adhesion elongation and fibronectin deposition.
The process of fibronectin fibril assembly is an active cellular process, in which tensin-bound α5β1 integrins traffic centripetally inward towards the center of the cell [48]. This movement requires actomyosin contractility, which facilitates both trafficking of the fibrillar adhesion complexes as well as the mechanical forces necessary for fibronectin unfolding [7]. To measure contractility of human VSMCs, EphA2-expressing and -depleted cells were embedded in a collagen gel generated from rat-tail collagen and permitted to contract the gel overnight. While EphA2-expressing VSMCs contract the gel nearly 63%, EphA2-depleted cells show a reduced ability to contract the gel, with only a 52% reduction (Figure 5A). This effect may be due to reduced VSMC contractility; however, we cannot rule out the possibility that altered proliferation affects cell numbers and thus the ability to contract the collagen gel. Indeed, we and others previously demonstrated the proliferative effects of EphA2 expression [21, 41, 49]. Therefore, we examined phosphorylation and activation of regulatory myosin light chain (MLC)-2, which promotes myosin contractility. Consistent with reduced collagen gel contraction, EphA2 depletion significantly reduces MLC phosphorylation in VSMCs (Figure 5B/C). MLC phosphorylation is regulated by calcium-dependent activation of MLC kinase or calcium-independent activation of the RhoA-Rho kinase (ROCK) pathway, which can both phosphorylate MLC directly and inactivate MLC phosphatase [50]. While EphA2 depletion does not affect serum-induced calcium signaling (Figure 5D–F), EphA2 knockdown blunts serum-induced RhoA activation (Figure 5G). To verify that EphA2 Y772 phosphorylation mediates these effects, MLC phosphorylation was assessed in EphA2 KO cells transfected with EphA2-WT or EphA2-Y772F constructs. While EphA2-WT rescues MLC phosphorylation in EphA2 KO VSMCs, EphA2-Y772F expression fails to enhance MLC phosphorylation (Figure 5H/I). Consistent with EphA2 ligand-independent signaling mediating these effects, MLC phosphorylation is rescued similarly in cells expressing wildtype EphA2 and the ligand binding-deficient EphA2-R103E mutant (Figure 5J/K). Taken together, these data suggest ligand-independent EphA2 Y772 phosphorylation in the focal adhesion regulates RhoA-mediated MLC phosphorylation to influence cell contractility.
Figure 5: EphA2 promotes VSM cell contractility independent of calcium signaling.
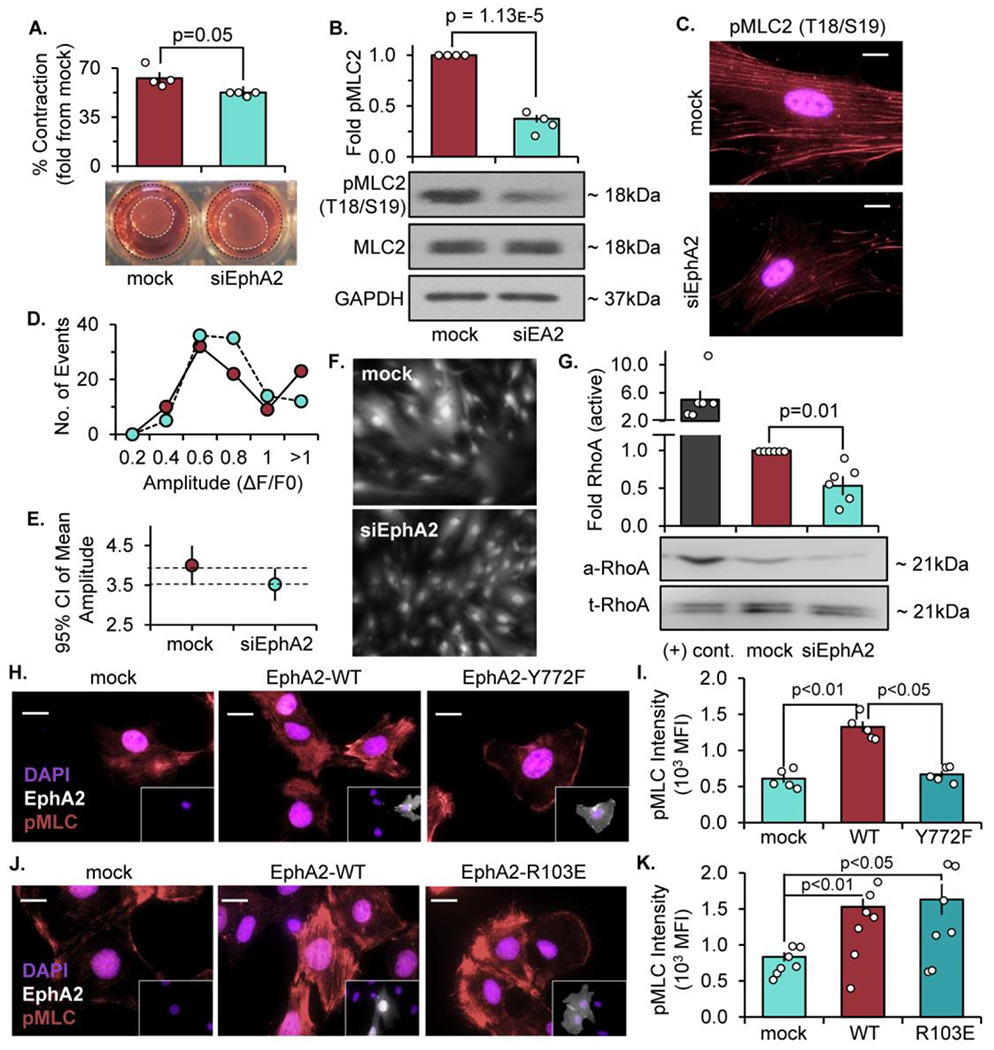
Human VSMCs were transfected with either mock or siRNA targeted against EphA2 for 24 hours. A) Cells were embedded in 0.8% collagen isolated from rat tail and allowed to polymerize at 37°C. The cells and collagen gels were incubated in 1% serum overnight, and collagen gel diameter was measured and expressed as a percent contraction from the diameter of the well. B,C) Cells were plated onto Matrigel overnight in 1% serum and phospho-MLC was measured by B) Western blot and normalized to total MLC or C) staining (pink) with DAPI (nuclei). D-F) Cells were labeled with 1μM Fluo4-AM 45 minutes prior to treatment with 1% serum. Fluo4-AM fluorescence was visualized by microscopy every 15 seconds over a duration of 15 minutes. D) Fluorescence intensity (amplitude, ΔF/F0) per each cell was represented as a histogram. E) Mean amplitude of fluorescence intensity shown as 95% confidence interval, with dashed lines representing overlapping SEM. F) Representative image of Fluo4-AM-labeled cells. G) Cells were plated onto Matrigel overnight in 1% serum and RhoA activity was measured by Rho activation assay. Active RhoA was measured by Western blot and normalized to total RhoA. Non-hydrolyzable GTPγS was utilized as a positive control ((+) cont). N=6 (H-K) EphA2 KO mouse VSMCs transiently expressing EphA2-WT, EphA2-Y772F, and EphA2-R103E were seeded overnight on Matrigel in 1% serum, then (H,J) stained: phospho-MLC (pink), EphA2 (white), and DAPI (nuclei), and (l,K) quantified as mean fluorescence intensity (MFI). Scale bar = 25um. n=4-7. Data are expressed as mean ±SEM. Statistical comparisons were made using Student’s T-test (A,B,G) or Two-way ANOVA with Bonferroni post-test (I,K). A p-value less than 0.05 is considered significant.
Since RhoA activation was blunted by EphA2 depletion (Figure 5G), we next sought to determine if altered RhoA signaling mediates the effect of EphA2 expression on fibrillar adhesion elongation. EphA2 WT and KO mouse VSMCs were transfected with either dominant-negative (N19) or constitutively active (Q63L) RhoA constructs (Supplemental Figure XII) [51]. In EphA2-expressing VSMCs, blocking RhoA signaling by dominant-negative RhoA N19 expression is sufficient to reduce fibrillar adhesion length (Figure 6A/B) and fibronectin deposition (Figure 6C/D), whereas expressing wildtype RhoA has no effect. In contrast, restoring RhoA signaling in EphA2 KO VSMCs by expression of constitutively active RhoA-Q63L rescues fibrillar adhesion length (Figure 6E/F) and fibronectin deposition (Figure 6G/H). Taken together, these data suggest that enhanced RhoA-mediated MLC phosphorylation associated with EphA2 Y772 phosphorylation influences cell contractility, fibrillar adhesion elongation, and fibronectin deposition.
Figure 6: EphA2 signals through RhoA to promote fibrillar adhesion elongation and fibronectin deposition.
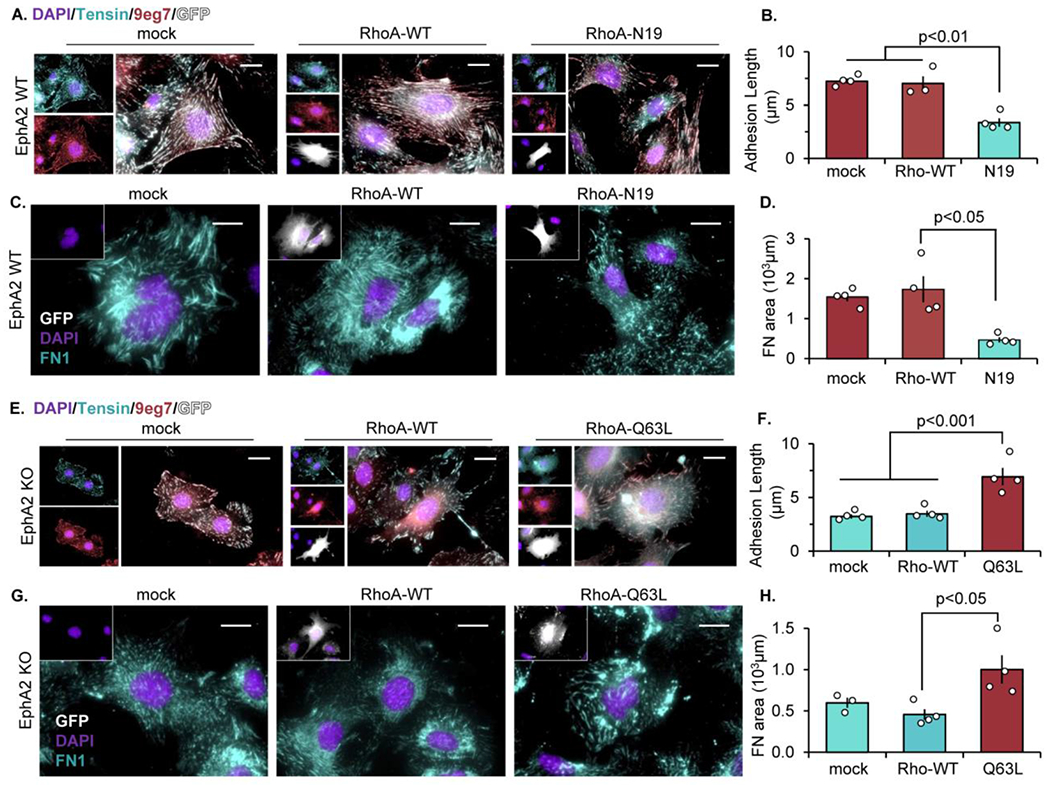
A-D) EphA2 WT mouse VSMCs were transfected with either mock, RhoA-WT, or dominant-negative RhoA (RhoA-N19) for 24 hours. (E-H) EphA2 KO mouse VSMCs were transfected with mock, RhoA-WT, or constitutively active RhoA (RhoA-Q63L) for 24 hours. Cells were plated onto Matrigel overnight post-transfection, and GFP-positive cells (white) were quantified. A/B,E/F) Cells were stained for tensin (teal), active β1 integrin (9eg7, pink), and DAPI (purple) and fibrillar adhesion length was measured in microns. C/D,G/H) Cells were stained for fibronectin (teal) and DAPI (purple) and fibronectin area was measured in microns. Scale bar = 25μm. n=4. Data are expressed as mean ±SEM. Statistical comparisons were made using One-way ANOVA with Bonferroni post-test A p-value less than 0.05 is considered significant.
Deletion of EphA2 inhibits fibronectin deposition through activation of p190 RhoGAP.
In response to ephrinA1, EphA2 signaling can promote RhoA activation by activating multiple RhoGEFs and by inhibiting p190RhoGAP [52–54]. To determine if EphA2 regulates fibrillar adhesion elongation and fibronectin deposition through p190RhoGAP inhibition, we assessed p190RhoGAP phosphorylation on Y1087 and Y1105, associated with p190RhoGAP activation. Activation of p190RhoGAP was enhanced in both EphA2 KO VSMCs (Figure 7A/B) and in VSMCs treated with ALW-II-41-27 (Figure 7C/D). Furthermore, p190RhoGAP depletion in EphA2 KO cells enhances fibrillar adhesion elongation (Figure 7E/F) and fibronectin deposition (Figure 7G). While these data are consistent with focal adhesion EphA2 signaling regulating RhoA, this data does not provide a direct link between EphA2 Y772 phosphorylation and p190RhoGAP inactivation, and low transfection efficiency and the inability of phospho-p190RhoGAP to function in immunocytochemistry limits our ability to strengthen this association.
Figure 7: Deletion of EphA2 enhances p190Rho-GAP phosphorylation to inhibit fibronectin fibrillogenesis.
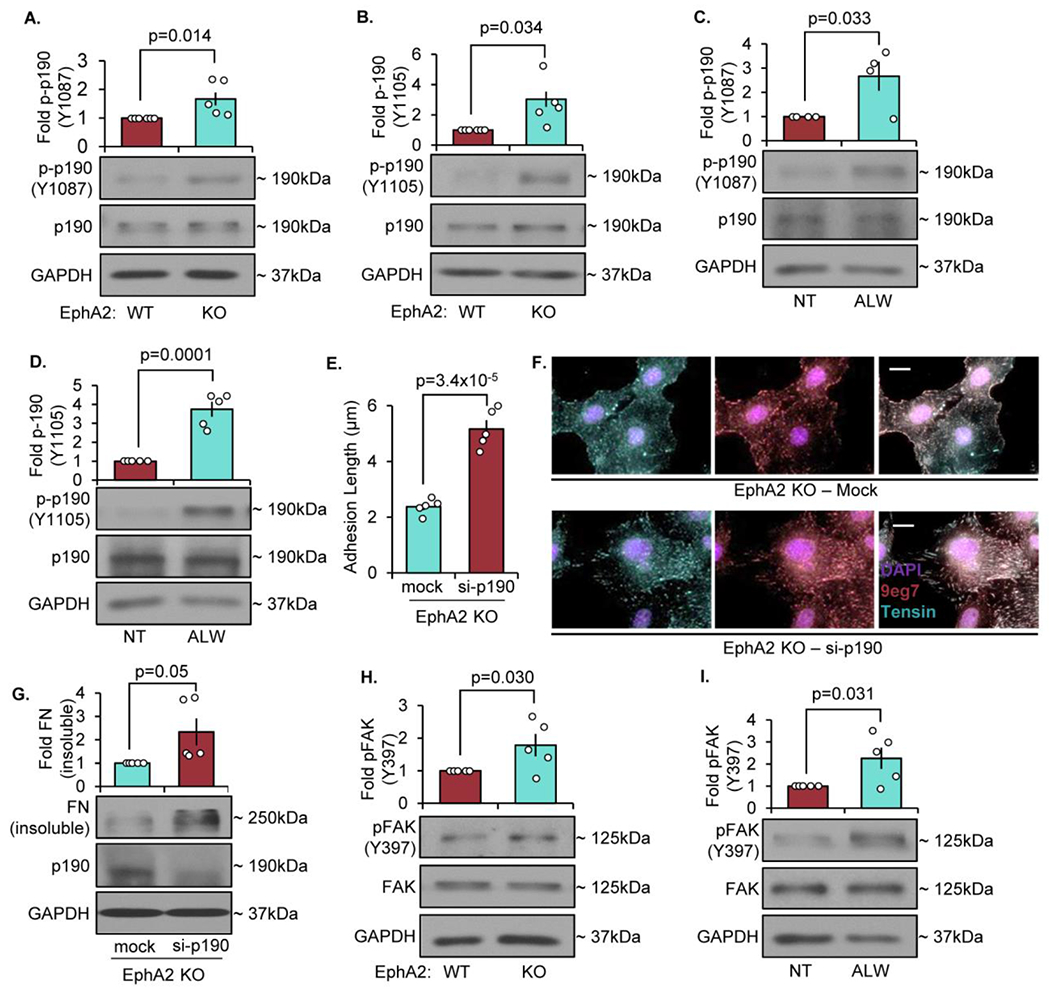
A/B) EphA2 WT or EphA2 KO mouse VSMCs were plated onto Matrigel overnight in 1% serum, and phospho-p190RhoGAP was quantified by Western blot and normalized to total p190RhoGAP. C/D) EphA2 WT mouse VSMCs were plated onto Matrigel overnight in 1% serum, then treated with ALW-II-41-27 (0.5μM) for 30 minutes. Phospho-p190RhoGAP was quantified by Western blot and normalized to total p190RhoGAP. E-G) EphA2 KO mouse VSMCs were transfected with either mock or siRNA against p190Rho-GAP for 24 hours, then plated onto Matrigel overnight. G,H) Cells were stained for tensin (teal), active β1 integrin (9eg7, pink), and DAPI (purple), and fibrillar adhesion length was measured in microns. Scale bar = 25μm. n=4-5. G) Cells were analyzed for fibronectin deposition with deoxycholate extraction. Deoxycholate-insoluble (deposited) fibronectin was normalized to deoxycholate-soluble GAPDH. H/I) EphA2 WT or EphA2 KO mouse VSMCs were plated onto Matrigel overnight in 1% serum, and phospho-FAK Y397 was quantified by Western blot and normalized to total FAK. Data are expressed as mean ±SEM. Statistical comparisons were made using Student’s T-test. A p-value less than 0.05 is considered significant.
In contrast to EphA2 signaling, FAK signaling can activate p190RhoGAP to inactivate RhoA [55, 56], ephrinA1-bound EphA2 can inhibit FAK signaling in other systems [27]. While FAK kinase activity is required for EphA2 Y772 phosphorylation, local EphA2 signaling within the focal adhesion could provide a negative feedback regulation on FAK activation similar to ligand-bound EphA2. Consistent with this reciprocal regulation, FAK phosphorylation is enhanced in EphA2 KO VSMCs (Figure 7H) and in VSMCs treated with the EphA2 kinase inhibitor ALW-II-41-27 (Figure 7I). Therefore, EphA2 signaling may induce p190RhoGAP inactivation directly or indirectly by reducing FAK-mediated p190RhoGAP activation. Together, these data suggest that EphA2 expression reduces FAK activation and p190RhoGAP phosphorylation to enhance actomyosin contractility-driven fibrillar adhesion elongation and fibronectin deposition (Figure 8).
Figure 8: Schematic diagram of proposed signaling pathway:
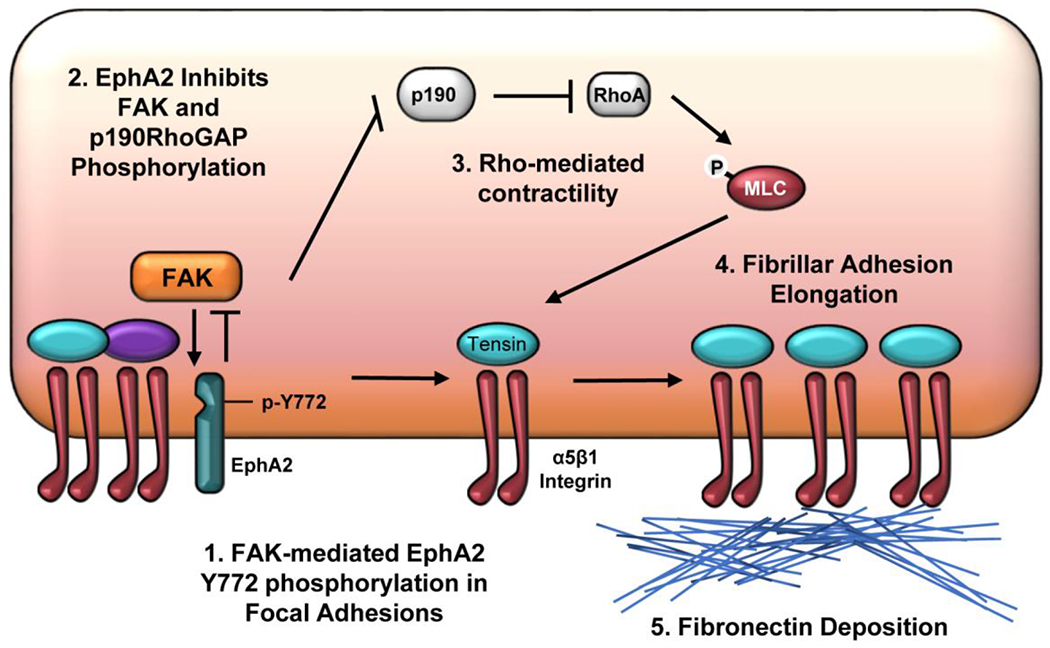
Schematic summary of the proposed mechanism of EphA2-regulated fibronectin deposition. 1) EphA2 within the focal adhesion undergoes ligand-independent phosphorylation on Y772 by focal adhesion kinase (FAK). EphA2 expression limits FAK activity, suggesting a novel reciprocal relationship between FAK and EphA2 signaling in the focal adhesion. 2) Elevated EphA2 signaling and reduced FAK signaling are associated with reduced phosphorylation and activation of p190RhoGAP. 3) Elevated RhoA activity due to inactive p190RhoGAP mediates myosin light chain phosphorylation and cellular contractility. 4) RhoA-driven contractility supports the recruitment of α5β1 integrins into tensin-rich fibrillar adhesions that drive 5) fibronectin deposition.
Discussion:
Previous studies showed forced overexpression of EphA2 enhances fibronectin in cancer cell models [29], and we demonstrated EphA2 depletion reduces fibronectin deposition in vitro and fibroproliferative remodeling in vivo [21]. However, EphA2 expression does not affect matrix gene expression [21], protease expression [21], or the expression or activation of the matrix-binding integrin receptors (Supplemental Figure II), suggesting that EphA2 may regulate the cellular machinery for matrix deposition. In this manuscript, we elucidate a novel reciprocal relationship between FAK and EphA2 signaling within integrin adhesions that promotes fibrillar adhesion elongation and fibronectin deposition. We show that efficient fibrillar adhesion elongation requires ligand-independent EphA2 phosphorylation on Y772 within integrin adhesions, and we demonstrate that FAK inhibition ameliorates EphA2 Y772 phosphorylation (Figures 3/4). RhoA signaling critically regulates fibrillar adhesion elongation [11, 57].
We show that maximal serum-induced RhoA activation requires EphA2 expression, and EphA2 depletion significantly blunts MLC phosphorylation and collagen gel contraction (Figure 5A/B). Consistent with a role for EphA2 Y772 phosphorylation, rescuing EphA2 knockout cells with wildtype EphA2 or ligand binding-deficient EphA2 restores MLC phosphorylation, whereas rescue with EphA2 lacking Y772 phosphorylation (Y772F) does not (Figure 5H/I). Furthermore, inhibiting RhoA blunts fibrillar adhesion elongation and fibronectin deposition in EphA2-expressing cells, whereas activating RhoA restores these processes in EphA2 knockout cells (Figure 6). While FAK mediates EphA2 Y772 phosphorylation in the focal adhesion, EphA2 depletion or inhibition enhances FAK signaling, suggesting EphA2 serves as an endogenous negative feedback pathway for FAK signaling in the focal adhesion. This enhanced FAK activity correlates with increased phosphorylation and activation of the RhoA-inactivating p190RhoGAP, whereas depleting p190RhoGAP promotes fibrillar adhesion elongation and fibronectin deposition in EphA2 KO VSMCs (Figure 7). These results define a novel mechanism by which EphA2 expression in remodeling tissues enhances ECM deposition through regulation of the RhoA signaling that drives fibrillar adhesion elongation and fibronectin deposition (Figure 8).
Interactions between Eph receptors and ephrin ligands classically regulate integrin function and signaling through Rho family GTPases to guide cell motility during tissue patterning and remodeling [19, 58]. Therefore, it is not surprising that multiple studies implicate Eph-ephrin interactions in matrix deposition in a variety of pathological conditions. For example, scleroderma-associated skin fibrosis enhances ephrinB2 expression, and reducing ephrinB2 expression prevents fibroblast matrix deposition in vitro as well as skin fibrosis in scleroderma and cardiac fibrosis in heart failure [59, 60]. The receptors mediating this effect may differ between models, as stellate cell EphB2 critically mediates hepatic fibrosis in cirrhosis models whereas EphA4 mediates fibronectin deposition at somite boundaries [61, 62]. Similarly, EphA2 overexpression drives fibronectin deposition, whereas antibody-mediated EphA2 depletion reduces fibronectin [29]. EphA2 knockout mice show reduced fibrosis associated with atherosclerotic plaques and cardiac myocardial infarction [21, 63], and treatment with all trans-retinoic acid reduces EphA2/ephrinA1 expression associated with diminished fibrosis in bleomycin-induced lung injury [64]. However, these studies generally assume that Eph-ephrin interactions facilitate these processes, and the subsequent downstream signaling mediators have not been assessed. Our data provide the first evidence that ligand-independent EphA2 signaling within the focal adhesions affects fibronectin matrix deposition by controlling tension-driven formation of fibrillar adhesions.
While our data are the first to show an effect for EphA2 signaling within the focal adhesion, other studies show similar EphA2 localization to integrin adhesions [27, 65]. Quantitative proteomics studies of the focal adhesome revealed EphA2 localization to integrin adhesions [43, 66], with spatial proteomics identifying β1 integrins and kindlin2 as proximal interacting proteins [43]. Although EphA2 tyrosine phosphorylation predominantly involves interaction with ephrinA1 and transphosphorylation by the kinase domain [36], focal adhesion EphA2 showed ligand-independent Y772 phosphorylation, which may mediate the basal Y772 phosphorylation observed under unstimulated conditions (Figure 2A–C). Similarly, basal levels of Y772 phosphorylation have been reported in keratinocytes and cancer cell lines [67–69]. Previous studies suggest EphA2 is capable of signaling by the non-ephrin ligand, progranulin [44] and progranulin stimulates cell migration through alterations in focal adhesion proteins [45]. However, progranulin levels in vascular smooth muscle cells are unaffected by serum stimulation and knockdown of progranulin does not affect EphA2 Y772 phosphorylation (Supplemental Figure VII) further suggesting these effects occur independently of ligand binding to EphA2. In addition, the guidance molecules ROBO1 and Slit 2 bind with EphA2; however, whether this interaction enhances Y772 phosphorylation remains unknown [70]. We show that FAK regulates serum-dependent Y772 phosphorylation within focal adhesions (Figure 4); however, whether EphA2 is a direct target of FAK remains unknown. The non-receptor tyrosine kinase Src can mediate growth factor transphosphorylation [71], and phosphoproteomic analysis of Src-expressing cells identified enhanced phosphorylation of both FAK and EphA2 [72]. Additional studies are necessary to determine the potential role of Src in FAK-dependent EphA2 phosphorylation.
Consistent with ligand-independent EphA2 signaling driving fibronectin deposition, a previous report showed that ligand-independent EphA2 S897 phosphorylation on stiff substrates was associated with fibronectin matrix deposition [73]. However, this report did not explore Y772 phosphorylation. While the classic ligand-independent EphA2 S897 phosphorylation site does not show localization to focal adhesions, rescuing EphA2 KO VSMCs with an EphA2 S897A mutant fails to restore fibrillar adhesion elongation and shows a trend toward reduced fibronectin deposition, suggesting that EphA2 signaling outside of focal adhesions also affects these processes (Supplemental Figure XIII). However, we focused our subsequent analysis on the role of EphA2 Y772 signaling due to its predominant localization within integrin adhesion complexes.
While we demonstrate that FAK promotes EphA2 Y772 phosphorylation, multiple lines of evidence suggest EphA2 signaling may also affect local integrin signaling. Local EphA2 activation by ephrinA1 can promote contractility-dependent focal adhesion dynamics through enhancing Src and FAK phosphorylation [26]. While EphA2 activation by ligand binding has been ascribed both positive and negative effects on cell adhesion [27, 74], our results failed to identify a role for EphA2 in regulating integrin expression (Supplemental Figure II) or serum-induced α5β1 integrin activation (Supplemental Figure V). EphA2 could mediate changes in cell adhesion through effects on FAK signaling, although reports describing the effect of ligand-dependent EphA2 signaling on FAK are conflicting. While some studies suggest EphA2 and other Eph receptors’ kinase activity suppress FAK and ILK signaling [27, 75], others demonstrated enhanced FAK phosphorylation following treatment with the EphA2 ligand ephrinA1 [76]. These contradictory findings may reflect differential spatial regulation, since isolating Eph-ephrin interactions into basally-restricted clusters may alter cellular responses [26, 77]. Our results suggest that ligand-independent EphA2 tyrosine phosphorylation in the focal adhesion limits FAK activity (Figure 7H/I), presumably through recruitment of a tyrosine phosphatase or through enhanced integrin clustering due to elevated contractility.
Our data show a clear defect in fibrillar adhesion elongation in cells lacking EphA2 expression correlating with loss of fibronectin fibrillogenesis. Fibrillar adhesion elongation requires contractility-driven translocation of α5β1/tensin complexes out of peripheral integrin adhesions and into long fibrillar adhesions [78]. This localized contractility, regulated by RhoGTPases, unravels globular fibronectin to expose cryptic binding sites on fibronectin to facilitate assembly [10]. Activation of RhoA is required for fibrillar adhesion elongation in a variety of cell models [11, 57, 79]. For example, fibroblasts lacking the RhoA effectors ROCK1/2 are unable to deposit fibronectin despite similar levels of fibronectin section and α5β1 integrin function [11]. Furthermore, von Hippel-Lindau-negative cancer cells show reduced RhoA signaling and defective fibronectin deposition despite normal levels of fibronectin expression [79]. Interestingly, these cells also show elevated expression of p190RhoGAP consistent with a negative role for p190RhoGAP in regulating fibronectin matrix deposition (Figure 7E–G). Peripheral α5β1 integrin signaling may drive this RhoA-dependent fibrillar adhesion elongation, as α5β1 integrins support a high level of RhoA activation following initial cell adhesion [80, 81]. Furthermore, the association between EphA2 and β1 in spatial proteomics analysis may indicate a preferential role for EphA2 in mediating β1-dependent RhoA signaling that drives fibrillar adhesion elongation and fibronectin matrix deposition [43].
Ligand-independent EphA2 signaling can affect RhoA signaling through a variety of mechanisms. Following ephrin binding, Eph receptor tyrosine phosphorylation provides scaffolding sites to recruit SH2 domain-containing RhoGEFs [36]. Ligand-bound EphA receptor recruits the RhoGEF ephexin, which becomes phosphorylated and shows enhanced activity for RhoA [53, 82]. Furthermore, ligand-bound EphA2 activates RhoA through the RhoGEF Vav2 to mediate cell-cell repulsion [54]. The other major EphA receptor in VSMCs, EphA4, interacts with the vascular smooth muscle-specific VsmRhoGEF to regulate Rho-mediated MLC phosphorylation [83]. EphA2 overexpression can also enhance RhoA activity by reducing p190RhoGAP phosphorylation through the recruitment of low molecular weight protein tyrosine phosphatase (LMW-PTP) [28]. Consistent with this model, deletion or inhibition of EphA2 augments p190RhoGAP phosphorylation (Figure 7A–D), whereas p190RhoGAP depletion is sufficient to rescue fibrillar adhesion elongation and fibronectin deposition in EphA2 KO VSMCs (Figure 7E–G). EphA2 may also affect p190RhoGAP through the regulation of FAK signaling. EphA2 expression limits FAK phosphorylation (Figure 7H/I), and FAK can activate p190RhoGAP to decrease RhoA activity during cell spreading and polarized cell migration [55, 56]. Therefore, the increase in p190RhoGAP phosphorylation following EphA2 inhibition could be due to either the enhanced FAK-dependent phosphorylation of p190RhoGAP or the reduced EphA2-mediated p190RhoGAP dephosphorylation (Figure 8).
In conclusion, we identify a novel role for EphA2 signaling within integrin adhesion complexes that limits local FAK activation and p190RhoGAP phosphorylation to enhance RhoA activity, fibrillar adhesion elongation, and fibronectin deposition. These data provide the first insight into the function of EphA2 within integrin adhesions and provide the first mechanistic insight into how EphA2 signaling contributes to tissue fibrosis. In addition, these data suggest that the enhanced EphA2 expression associated with a variety of fibrosis disease models may promote matrix deposition independently of ligand-dependent receptor activation, thereby indicating that therapeutic mechanisms targeting ligand-bound EphA2 or kinase activity may have limited efficacy in comparison to treatments that reduce EphA2 expression or promote EphA2 degradation.
Methods and Materials:
Cell culture:
Human coronary artery or aortic vascular smooth muscle cells (VSMCs) were purchased from Lonza (cats #cc-2583, cc-2571) and Cell Applications (cat #350p-05a) and cultured as described previously [21]. Briefly, cells were maintained in MCDB131 Medium (GenDEPOT #CM034-350) supplemented with 10% fetal bovine serum (FBS) (Genesee #25-514), 10 U/mL penicillin/10 mg/mL streptomycin (Genesee #25-512), GlutaMAX™ (Gibco #35050061) L-Glutamine supplement, 5 ng/mL human recombinant EGF (Peprotech AF-100-15A), 5 ng/mL human recombinant FGF-basic (Peprotech #100-18B), and 5 μg/mL human recombinant insulin (Sigma #91077C). Adult human dermal fibroblasts were purchased from Cascade Biologics, (cat #C0135C0) and maintained in DMEM (Genessee #25-500N) supplemented with 10% FBS (Genesee #25-514), 10U/mL penicillin/10mg/mL streptomycin (Genessee #25-512), and GlutaMAX™ L-Glutamine supplement (Gibco #35050061). Human VSMCs were used between passages 4-6. Cells were plated on either Matrigel (1:50, Corning) or fibronectin (10 ug/mL). The ATP-competitive FAK inhibitor PF-573228 (Tocris #3097) was used at 4 μM compared to DMSO vehicle for 30 minutes prior to serum stimulation. Mouse aortic vascular smooth muscle cells (mouse VSMCs) were cultured in DMEM/F12 (Genesee #25-502) supplemented with 10% FBS and 10 U/mL penicillin/10 mg/mL streptomycin. Cells were plated on either 10 μg/mL human plasma fibronectin or 1:50-diluted Matrigel (Corning #354234) as indicated. For all experiments, cells were plated overnight to establish sufficient adhesion complexes. In addition, cells were maintained in either 1% FBS-supplemented media or serum-starved for 4 hours prior to stimulation with 1% FBS, 1 μg/mL fc-EphrinA1 (R&D Systems #602-A1-200), or 0.5 μM ALW-II-41-27 (a kind gift from Nathanael Grey, Harvard University) as indicated.
EphA2 Wildtype and Knockout Mouse Aortic VSMC Isolation:
Animal protocols were approved by the LSU Health Sciences Center-Shreveport Institutional Animal Care and Use Committee (IACUC). EphA2 wild-type or EphA2 knockout mice were euthanized with isoflurane and pneumothorax and perfused with approximately 10mL warmed HBSS with calcium and magnesium from the apex of the heart through the right atrium. The aorta was cleaned of surrounding fat tissue and excised between the distal aortic arch and the diaphragm. Aortas from three mice per group were digested in HBSS with enzymes (1 mg/mL Collagenase type II (Worthington Biochemical LS004174), 1 mg/mL Soybean trypsin inhibitor (Worthington Biochemical LS003570), 180 μg/mL Elastase (Worthington Biochemical LS002279), and 50 U/mL Penicillin/50 μg/mL Streptomycin for 10 minutes at 37°C. Aortas were dissected longitudinally with scissors and the adventitia was removed. The intimal side was gently wiped with a sterile cotton swab to remove endothelial cells. Medial layers were minced with scissors and further digested in HBSS with enzymes for 1 hour at 37°C. Tissue was triturated through an 18g needle 5 times to create a cell suspension. Cells were pelleted and rinsed twice with 20% FBS-supplemented DMEM/F12 containing 1 U/mL penicillin/1 mg/mL streptomycin. Cells were re-suspended in 20% DMEM/F12 with antibiotics and plated on 0.1% gelatin-coated tissue culture plates. Cells were weaned onto 10% FBS-supplemented media at passage 3. To deplete any remaining endothelial cells, cells were serum-starved for three days. Cells were immortalized using a temperature-sensitive SV40-TAg, which suppresses SV40 expression when cultured at 37°C but not at 33°C.
FAK Wildtype and Kinase-Dead Mouse Aortic VSMC Isolation:
Animal experiments were approved by and performed in accordance with the guidelines of the University of South Alabama Institutional Animal Care and Use Committee. To obtain genetic FAK wild-type (WT) and kinase-dead (KD) VSMCs, FAK flox/flox and FAK WT/KD mice were crossed to produce FAK flox/WT and FAK flox/KD offspring [84]. Mouse aortic SMCs were isolated as previously described with some modifications [85, 86]. Mice were anesthetized with 100 mg/kg ketamine/10m g/kg xylazine via intraperitoneal injection. The aorta was isolated, and the surrounding tissue was excised using a surgical microscope. A 24-gauge syringe was inserted into one end of the aorta, and the lumen was then washed with PBS. The distal end of the aorta was ligated shut, and aorta was filled with 2 mg/mL Collagenase II (Worthington, #LS004174) for 30 minutes, and detached endothelial cells were flushed out three times with PBS. Aortas were dissected into smaller pieces and embedded in Matrigel (Corning, #356230). VSMCs migrated out of the aorta in 20% DMEM. VSMCs were then expanded and maintained in 10% DMEM. Isolated SMCs were checked for expression of SMC markers such as α-smooth muscle actin (α-SMA) by immunostaining. Isolated FAK flox/WT and FAK flox/KD SMCs were treated with Cre adenovirus (Ad-Cre) to generate FAK null/WT and FAK null/KD SMCs. Successful deletion of FAK flox allele was confirmed via PCR and immunoblotting.
Transfections and transductions:
For siRNA treatments, cells were transfected using lipofectamine2000 (Invitrogen #11668019) as per manufacturers’ instructions and 50 nM siRNA targeted against human granulin (Dharmacon SMARTpool #L-009285-00-0005), EphA2 (Dharmacon SMARTpool #L-003116-00-0005), human FAK (Dharmacon SMARTpool #L-003164-00-0005), or mouse p190RhoGAP (Dharmacon SMARTpool #L-042292-01-005). For DNA plasmid treatments, cells were transfected using lipofectamine3000 (Invitrogen #L3000015) as per manufacturer’s instructions and 1 μg/mL DNA plasmid was used for each construct. Cells were used 48 hours post-transfection for all experiments. The human full-length EphA2-WT human plasmid was produced by VectorBuilder and driven by the cytomegalovirus (CMV) promoter with a green fluorescence protein (GFP) reporter gene. The human EphA2-Y772F, S897A, K646M, and R103E mutant plasmids were produced and sequence-validated by the LSUHSC-Shreveport COBRE Molecular Signaling Core utilizing QuickChange II Site-directed mutagenesis kit (Agilent #200523) as per manufacturer’s instructions. The human RhoA-WT (Addgene plasmid # 12965), dominant negative RhoA-TN19 (Addgene plasmid # 12967), and constitutively active RhoA-Q63L (Addgene plasmid # 12968) were a kind gift from Gary Bokoch. The human Tensin-C-mCherry was kind gift from Michael Davidson (Addgene plasmid # 55143). For viral overexpression, human vascular smooth muscle cells were transduced with either GFP control or EphA2 adenoviral constructs (Vectorbuilder) with 6 μg/mL Polybrene (Santa Cruz #sc-134220) for 24 hours (MOI = 3). 24 hours post-transduction, fresh media was exchanged and incubated an additional 24 hours, after which point human VSMCs were serum-starved for 72 hours.
Integrin Activation Assay:
Integrin activation assay was used as previously described [87, 88]. Briefly, a glutathione S-transferase (GST)-tagged fibronectin mimetic of the 9th-11th type III repeat (GST-FNIII9-11) was used to bind active α5β1 integrin. Following serum-starvation for 4 hours, cells were stimulated with serum for 30 minutes. GST-FNIII9-11 (20 μg/mL) in PBS containing 1 mM MgCI2 was added to human VSMCs and incubated for 30 minutes. 0.5 mM MnCI2 was used as a positive control. Unbound GST-FNIII9-11 was rinsed two times with fresh PBS, and cells were lysed in 2x Laemlli buffer. Active α5β1 integrin was measured as the relative of GST detected with Western blot and normalized to GAPDH.
Proximity Ligation Assay:
Human VSMCs were fixed in 3.7% neutral-buffered formaldehyde and permeabilized with 0.5% Triton-X100. The proximity ligation assay (Sigma #DUO92102) was performed per manufacturer’s instructions using mouse anti-β1 integrin (1:200, Santa Cruz #sc-374429) and rabbit anti-tensin (1:500, Sigma #SAB420028). Nuclei were labeled with DAPI (Life Technologies #D3571) and coverslips were mounted onto microscope slides using FluoromountG (SouthernBiotech #0100-01). Cells were quantified for puncta per cell and averaged from at least 5 high-powered field images for each condition.
Western Blotting and Immunoprecipitation:
Western blot was performed as described [21]. Membranes were labeled with rabbit anti-paxillin (1:1000, CST #2542), mouse anti-vinculin (1:1000, Sigma V9131), rabbit anti-TFR1 (1:4000, CST #13113), mouse anti-HDAC3 (1:1000, CST #3949), rabbit anti-TOMM20 (1:4000, Abcam ab186735), mouse anti-calnexin (1:1000, Abcam ab31290), granulin (1:2000, Abcam #ab208777), rabbit anti-tensin (1:1000, Sigma SAB4200283), rabbit anti-μ5 integrin (1:1000, Abcam #ab150361; 1:4000, Santa Cruz Biotechnology sc-10729), rabbit anti-αv integrin (1:1000, Abcam #ab117611; 1:3000, CST #4711), rabbit anti-β1 integrin (1:1000, CST #9699), rabbit anti-β3 integrin (1:1000, CST #4702), rabbit anti-EphA2 (1:1000, CST #6997), rabbit anti-phospho-EphA2 Y588 (1:1000, CST #12677), rabbit anti-phospho-EphA2 Y772 (1:1000, CST #8244), rabbit anti-phospho-EphA2 S897 (1:1000, CST #6347), rabbit anti-ILK1 (1:5000, CST #3856), rabbit anti-β-tubulin (1:5000, CST #2128), rabbit anti-GAPDH (1:5000, CST #2118), rabbit anti-MLC2 (1:1000, CST #3672), rabbit anti-phospho-MLC T18/S19 (1:1000, CST #3674), rabbit anti-RhoA (1:1000, CST #2117), rabbit anti-phospho-FAK Y397 (1:1000, CST #8556), rabbit anti-FAK (1:1000, CST #3285), rabbit anti-phospho-p190RhoGAP Y1087 (1:1000, Thermo #PA540199), rabbit anti-phospho-p190RhoGAP Y1105 (1:1000, Thermo #PA537788), rabbit anti-p190RhoGAP (1:1000, CST #2513), mouse anti-GST (1:1000, Santa Cruz #sc-138), rabbit anti-fibronectin (1:5000, Abcam #ab2413) and rabbit anti-ephrinA1 (1:1000, Abcam #ab133598). Primary antibodies were visualized with horseradish peroxidase AffiniPure secondary antibodies from Jackson ImmunoResearch (goat anti-mouse #115-035-062, goat anti-rabbit #111-035-003). Densitometry was performed using ImageJ Software, and relative protein was normalized to ILK (for integrin adhesion isolation) or GAPDH and beta-tubulin (for all other experiments). For immunoprecipitation, human vascular smooth muscle cells were rinsed with ice-cold PBS and lysed in ice-cold immunoprecipitation lysis buffer containing 20 mM TrisCl, 150 mM NaCI, 1 mM EDTA, 1 mM EGTA, 0.5% Triton, 0.5% NP-40, 1X protease inhibitor cocktail (RPI Corporation #P50900-1.0) and 1X phosphatase inhibitor cocktail (RPI Corporation #P52104-1) pH 7.5. Lysates were immunoprecipitated with mouse anti-β1 integrin (1:50, Santa Cruz #sc-374429) for 4 hours and incubated with GammaBind G Sepharose (GE Healthcare #17-0885-02) for 3 hours. Bound protein was rinsed with immunoprecipitation lysis buffer three times and lysed in 2x Laemmli buffer. Sepharose beads and lysates without antibodies were used as a negative control, with a whole cell lysate used to validate immunoblotting.
Integrin Adhesion Isolation:
Integrin adhesion isolation was performed as described previously [89]. Hydrodynamic force with PBS was applied using a Conair Interplak water pic set at 4. Cells were plated onto Matrigel-coated glass slides at 500,000 cells per slide and incubated in 1%FBS-supplemented media overnight. Matrigel was diluted 1:50, which creates a thin coat rather than a soft gel. Cells were rinsed briefly with PBS and incubated with 2.5 mM triethanolamine pH 7.0 for 3 minutes. Cell bodies were removed with two second passes of hydrodynamic force five times, and integrin-associated proteins remaining on the slide were lysed in 2x Laemlli buffer. Cells from 3 slides were pooled each condition. For whole cell lysate control, cells were plated onto glass slides as described above, and lysed directly in 2x Laemmli buffer, β-tubulin was shown as a negative control.
Collagen Gel Contraction assay:
Rat-tail collagen was isolated, and the collagen gel contraction assay was performed as described previously [90]. Briefly, 200,000 human vascular smooth muscle cells per condition were suspended in 500 μL 0.8% collagen and incubated at 37°C for 1 hour to allow for collagen polymerization. Following polymerization, collagen gels were gently released from the walls of the culture dish with a sterile spatula, and 1%FBS-supplemented media was added overnight to allow for gel contraction. Collagen gel diameter was measured using ImageJ Software. Two gels were measured for each condition, and an average was obtained using ImageJ Software. Data shown as percent contraction from well diameter.
Calcium Labeling Assay:
Human vascular smooth muscle cells were measured for calcium signaling as described previously [91]. Briefly, VSMCs were treated with 1 μM Fluo4-AM (Invitrogen #F14201) in phenol red-free HBSS for 45 minutes at room temperature, then exchanged with fresh HBSS for 15 minutes at 37C. Intracellular calcium was detected following serum stimulation with FITC excitation using a single high-powered field with a Nikon TiE microscope, while cells were maintained at 37°C and 5% CO2 on a stage-mounted environment chamber (Bioscience Tools). Calcium signaling was captured at 15 second intervals for 15 minutes using a Nikon monochrome cooled digital camera (DS-Qi1). Calcium signaling of each cell over time was quantified using the ImageJ SparkMaster Plugin [92] and expressed as intensity (amplitude, ΔF/F0). Cell debris was excluded from the analysis.
Rho Activation Assay:
Cells were transfected as described previously and serum-starved for 4 hours prior to FBS stimulation for 15 minutes. Rho activation was measured using the Active RhoA Detection Kit (Cell Signaling Technologies #8820) and performed per manufacturer’s protocol. Briefly, cells were lysed and incubated with GST-tagged Rhotekin-RBD-conjugated agarose beads, which bind only GTP-bound RhoA [93]. Protein-bound beads were lysed in 2xLaemlli buffer. Samples were analyzed by Western blot, and active RhoA was normalized to total RhoA from whole cell lysates. A positive control was treated with GTPγS (a non-hydrolyzable GTP analogue).
Immunostaining and Microscopy:
Cells were stained for immunofluorescence as described previously [21]. Cells were plated at 40,000 cells/cm2 on glass coverslips coated with Matrigel and incubated overnight in 1% FBS-supplemented media. Cells were fixed in 3.7% neutral buffered formaldehyde, and permeabilized in 0.5% Triton-X100. Coverslips were blocked with 10% horse serum (Life Technologies #16050122) in 1% denatured bovine serum albumin (RPI Corporation #A30075) with 0.5% sodium azide for at least 1 hour prior to incubation overnight with primary antibodies at 4°C. Primary antibodies were used as follows: rabbit anti-Tensin (1:500, Sigma SAB420028), mouse anti-12G10 (1:400, a kind gift from Martin Humphries, University of Manchester), SNAKA51 (1:400, a kind gift from Martin Humphries, University of Manchester), rabbit anti-phospho-MLC T18/S19 (1:400, CST #3674), rat anti-9EG7 (1:400, BD Pharmingen #550531), rabbit anti-phospho-FAK Y397 (1:200, CST #8556), rabbit anti-phospho-EphA2 Y772 (1:200 CST #8244), rabbit anti-EphA2 (1:400, CST #6997), mouse anti-EphA2 (1:400, CST #12927), mouse anti-vinculin (1:400, Sigma #9131), rabbit anti-paxillin (1:100, Abcam ab32115), or Alexa488-conjugated phalloidin (1:200, Invitrogen #12379). Coverslips were incubated for two hours with secondary antibodies at a 1:1000 dilution with 10% horse serum in 1% denatured bovine serum albumin with 0.5% sodium azide. Secondary antibodies were used as follows: Alexa-546 Donkey anti-Rabbit, Alexa-546 Donkey anti-mouse, Alexa-647 Donkey anti-rat, Alexa-647 Donkey anti-mouse (Invitrogen). All coverslips were nuclear counterstained with DAPI (500ng/mL, Life Technologies #D3571). All stains were visualized with a Nikon Eclipse Ti inverted microscope and images were captured using a Photometries CoolSNAP120 ES2 camera and NIS Elements 3.00, SP5 imaging software. At least five randomized high-powered fields were imaged per coverslip for accurate representation of each treatment. Fibrillar adhesion length was measured using NIS Elements BR software.
Quantitative Real-Time PCR:
mRNA isolation, cDNA synthesis, and qRT-PCR were performed from cells as described previously [21]. Briefly, cells were lysed in TRIzol reagent (Life Technologies #15596026) and RNA was isolated with chloroform precipitation. cDNA was synthesized using the iScript cDNA synthesis kit (Biorad #1708891) per manufacturer’s instructions. qRT-PCR was performed using a CFX96-well iCycler (BioRad) and iQSYBR Green Supermix (BioRad #1708880). Primers were designed and validated by PrimerBank [94]. Primer sequences are as follows: ITA5 forward primer: 5’-CAGGGTGGTGCTGTCTACCT-3’, ITA5 reverse primer: 5’-GCTCAGTGGCTCCTTCTCTG-3’, ITAV forward primer: 5’-GCCTGGAACAGCTCTCAAAG-3’, ITAV reverse primer: 5’-AGATTCATCCCGCAGATACG-3’, ITB1 forward primer: 5’-TTCAGTTTGCTGTGTGTTTGC-3’, ITB1 reverse primer: 5’-TTTCTGGACAAGGTGAGCAA-3’, EphA2 forward primer: ‘5-GCACAGGGAAAGGAAGTTGTT-3’, EphA2 reverse primer: 5’-CATGTAGATAGGCATGTCGTCC-3’, Rpl13a forward primer: 5’-GGGCAGGTTCTGGTATTGGAT-3’, Rpl13a reverse primer: 5’-GGCTCGGAAATGGTAGGGG-3’. Results were normalized to the housekeeping gene Rpl13a and expressed as fold change using the 2-[delta][delta]Ct calculation.
Fibronectin Fibrillogenesis:
Fibronectin deposition was analyzed by Western blot as described previously [21]. Briefly, cells were rinsed in ice-cold PBS and lysed in a 2% deoxycholate buffer containing 2 mM TrisCl, 2 mM PMSF, 2 mM EDTA, 2 mM iodoacetic acid, and 2 mM N-ethylmaleimide pH 8.8. DNA was sheared by passing through a 25-gauge needle 5 times, and the deoxycholate-insoluble fraction was isolated by 16,000g centrifugation for 15 minutes. The supernatant was lysed in 6xLaemmli buffer as the deoxycholate soluble fraction, and the pellet was solubilized in a solubilization buffer containing 2% SDS, 25 mM TrisCl, 2 mM PMSF, 2 mM iodoacetic acid, and 2 mM N-ethylmaleimide, and 2 mM EDTA pH 8.0 and lysed in 6x Laemmli buffer.
Statistical Analyses:
All experiments in main figures were performed at least 3 independent times. P-values less than 0.05% were considered significant. All T-tests were two-tailed distribution with two-sampled unequal variance and performed using Microsoft Excel, and GraphPad Prism was used to perform one-way ANOVA and two-way ANOVA. Analyses utilizing one-way ANOVA performed Bonferroni Multiple Comparison test, while analyses utilizing two-way ANOVA performed Bonferroni posttest. For calcium signaling data, amplitude frequency (ΔF/F0) was shown as a frequency distribution and 95% confidence interval of the mean amplitude, where overlapping standard error bars suggest the amplitude means are not significantly different. Outliers with less than 0.05 significance were calculated using GraphPad Outlier Calculator and excluded from data sets. Mean data were represented as bar graphs, with individual data points displayed as white dots overlaid. All data were shown as the mean ± S.E.M.
Supplementary Material
Highlights:
EphA2 expression is required for efficient elongation of tensin- and (α5β1-rich fibrillar adhesions in vascular smooth muscle cells.
EphA2 undergoes ligand-independent Y772 phosphorylation within integrin adhesions through focal adhesion kinase-dependent transphosphorylation.
EphA2 Y772 phosphorylation, but not EphA2 ligand-binding, promotes fibrillar adhesion elongation and fibronectin deposition by enhancing RhoA-mediated contractility.
EphA2 expression limits p190RhoGAP activation to promote contractility-dependent fibrillar adhesion formation and fibronectin deposition.
Funding and Acknowledgements:
The work herein was funded by a Malcolm Feist Predoctoral Fellowship (to ACF), an AHA predoctoral fellowship 17PRE33440111 (to ACF), an AHA Postdoctoral Fellowship 20POST35220022 (to MLS), an AHA Postdoctoral Fellowship #20POST35120288 (to MAF), an AHA Grant-in-Aid 13GRNT17050093 (to AWO), NIH P20 GM121307 and R01 grants HL098435, HL133497, and HL141155 (to AWO), an R01 grant HL136432 (to STL), and an R01 grant HL139755 to (CBP). The authors thank Mahmoud Al Kofai (collagen contraction assay) (LSUHSC-Shreveport) and Nathaniel Gray (ALW-II-41-27) (Harvard University) for providing reagents and expertise.
Abbreviations:
- VSMC
Vascular smooth muscle cell
- FAK
Focal adhesion kinase
- FN
Fibronectin
- WCL
Whole cell lysate
- EA2
EphA2
- MLC
Myosin light chain
- WT
Wild-type
- KO
Knockout
- ILK
Integrin-linked kinase
- Y
tyrosine
- S
serine
- P
phosphorylated
- NT
no treatment
- GEF
guanine nucleotide exchange factor
- GAP
guanine nucleotide activating protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest: None.
References:
- [1].Lock JG, Wehrle-Haller B, Stromblad S, Cell-matrix adhesion complexes: master control machinery of cell migration, Seminars in cancer biology 18(1) (2008) 65–76. [DOI] [PubMed] [Google Scholar]
- [2].Wight TN, Potter-Perigo S, The extracellular matrix: an active or passive player in fibrosis?, Am J Physiol Gastrointest Liver Physiol 301(6) (2011) G950–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Saunders JT, Schwarzbauer JE, Fibronectin matrix as a scaffold for procollagen proteinase binding and collagen processing, Mol Biol Cell 30(17) (2019) 2218–2226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Valiente-Alandi I, Potter SJ, Salvador AM, Schafer AE, Schips T, Carrillo-Salinas F, Gibson AM, Nieman ML, Perkins C, Sargent MA, Huo J, Lorenz JN, DeFalco T, Molkentin JD, Alcaide P, Blaxall BC, Inhibiting Fibronectin Attenuates Fibrosis and Improves Cardiac Function in a Model of Heart Failure, Circulation 138(12) (2018) 1236–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Wang WY, Twu CW, Liu YC, Lin HH, Chen CJ, Lin JC, Fibronectin promotes nasopharyngeal cancer cell motility and proliferation, Biomed Pharmacother 109 (2019) 1772–1784. [DOI] [PubMed] [Google Scholar]
- [6].Stefanelli VL, Choudhury S, Hu P, Liu Y, Schwenzer A, Yeh CR, Chambers DM, Pesson K, Li W, Segura T, Midwood KS, Torres M, Barker TH, Citrullination of fibronectin alters integrin clustering and focal adhesion stability promoting stromal cell invasion, Matrix Biol 82 (2019) 86–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Mao Y, Schwarzbauer JE, Fibronectin fibrillogenesis, a cell-mediated matrix assembly process, Matrix Biol 24(6) (2005) 389–99. [DOI] [PubMed] [Google Scholar]
- [8].Sechler JL, Corbett SA, Schwarzbauer JE, Modulatory roles for integrin activation and the synergy site of fibronectin during matrix assembly, Mol Biol Cell 8(12) (1997) 2563–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Johnson KJ, Sage H, Briscoe G, Erickson HP, The compact conformation of fibronectin is determined by intramolecular ionic interactions, J Biol Chem 274(22) (1999) 15473–9. [DOI] [PubMed] [Google Scholar]
- [10].Zhong C, Chrzanowska-Wodnicka M, Brown J, Shaub A, Belkin AM, Burridge K, Rho-mediated contractility exposes a cryptic site in fibronectin and induces fibronectin matrix assembly, J Cell Biol 141(2) (1998) 539–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yoneda A, Ushakov D, Multhaupt HA, Couchman JR, Fibronectin matrix assembly requires distinct contributions from Rho kinases I and -II, Mol Biol Cell 18(1) (2007) 66–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fogerty FJ, Akiyama SK, Yamada KM, Mosher DF, Inhibition of binding of fibronectin to matrix assembly sites by anti-integrin (alpha 5 beta 1) antibodies, J Cell Biol 111(2) (1990) 699–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Geiger B, Yamada KM, Molecular architecture and function of matrix adhesions, Cold Spring Harb Perspect Biol 3(5) (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Lo SH, Tensin, Int J Biochem Cell Biol 36(1) (2004) 31–4. [DOI] [PubMed] [Google Scholar]
- [15].Singh P, Carraher C, Schwarzbauer JE, Assembly of fibronectin extracellular matrix, Annu Rev Cell Dev Biol 26 (2010) 397–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Georgiadou M, Lilja J, Jacquemet G, Guzman C, Rafaeva M, Alibert C, Yan Y, Sahgal P, Lerche M, Manneville JB, Makela TP, Ivaska J, AMPK negatively regulates tensin-dependent integrin activity, J Cell Biol 216(4) (2017) 1107–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Boyd AW, Lackmann M, Signals from Eph and ephrin proteins: a developmental tool kit, Sci STKE 2001(112) (2001) re20. [DOI] [PubMed] [Google Scholar]
- [18].Funk SD, Orr AW, Ephs and ephrins resurface in inflammation, immunity, and atherosclerosis, Pharmacol Res 67(1) (2013) 42–52. [DOI] [PubMed] [Google Scholar]
- [19].Flanagan JG, Vanderhaeghen P, The ephrins and Eph receptors in neural development, Annu Rev Neurosci 21 (1998) 309–45. [DOI] [PubMed] [Google Scholar]
- [20].Barquilla A, Pasquale EB, Eph receptors and ephrins: therapeutic opportunities, Annu Rev Pharmacol Toxicol 55 (2015) 465–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Finney AC, Funk SD, Green JM, Yurdagul A Jr., Rana MA, Pistorius R, Henry M, Yurochko A, Pattillo CB, Traylor JG, Chen J, Woolard MD, Kevil CG, Orr AW, EphA2 Expression Regulates Inflammation and Fibroproliferative Remodeling in Atherosclerosis, Circulation 136(6) (2017) 566–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Hong HN, Won YJ, Shim JH, Kim HJ, Han SH, Kim BS, Kim HS, Cancer-associated fibroblasts promote gastric tumorigenesis through EphA2 activation in a ligand-independent manner, Journal of cancer research and clinical oncology 144(9) (2018) 1649–1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Carpenter TC, Schroeder W, Stenmark KR, Schmidt EP, Eph-A2 promotes permeability and inflammatory responses to bleomycin-induced lung injury, Am J Respir Cell Mol Biol 46(1) (2012) 40–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Hong JY, Shin MH, Douglas IS, Chung KS, Kim EY, Jung JY, Kang YA, Kim SK, Chang J, Kim YS, Park MS, Inhibition of EphA2/EphrinA1 signal attenuates lipopolysaccharide-induced lung injury, Clin Sci (Lond) 130(21) (2016) 1993–2003. [DOI] [PubMed] [Google Scholar]
- [25].O’Neal WT, Griffin WF, Kent SD, Faiz F, Hodges J, Vuncannon J, Virag JA, Deletion of the EphA2 receptor exacerbates myocardial injury and the progression of ischemic cardiomyopathy, Frontiers in physiology 5 (2014) 132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Chen Z, Oh D, Biswas KH, Yu CH, Zaidel-Bar R, Groves JT, Spatially modulated ephrinA1:EphA2 signaling increases local contractility and global focal adhesion dynamics to promote cell motility, Proc Natl Acad Sci U S A 115(25) (2018) E5696–E5705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Miao H, Burnett E, Kinch M, Simon E, Wang B, Activation of EphA2 kinase suppresses integrin function and causes focal-adhesion-kinase dephosphorylation, Nat Cell Biol 2(2) (2000) 62–9. [DOI] [PubMed] [Google Scholar]
- [28].Fang WB, Ireton RC, Zhuang G, Takahashi T, Reynolds A, Chen J, Overexpression of EPHA2 receptor destabilizes adherens junctions via a RhoA-dependent mechanism, J Cell Sci 121(Pt 3) (2008) 358–68. [DOI] [PubMed] [Google Scholar]
- [29].Hu M, Carles-Kinch KL, Zelinski DP, Kinch MS, EphA2 induction of fibronectin creates a permissive microenvironment for malignant cells, Mol Cancer Res 2(10) (2004) 533–40. [PubMed] [Google Scholar]
- [30].Finney AC, Stokes KY, Pattillo CB, Orr AW, Integrin signaling in atherosclerosis, Cell Mol Life Sci 74(12) (2017) 2263–2282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].McCleverty CJ, Lin DC, Liddington RC, Structure of the PTB domain of tensin1 and a model for its recruitment to fibrillar adhesions, Protein science : a publication of the Protein Society 16(6) (2007) 1223–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Askari JA, Buckley PA, Mould AP, Humphries MJ, Linking integrin conformation to function, J Cell Sci 122(Pt 2) (2009) 165–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Al-Yafeai Z, Pearson BH, Peretik JM, Cockerham ED, Reeves KA, Bhattarai U, Wang D, Petrich BG, Orr AW, Integrin affinity modulation critically regulates atherogenic endothelial activation in vitro and in vivo, Matrix Biol 96 (2021) 87–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Mould AP, Garratt AN, Askari JA, Akiyama SK, Humphries MJ, Identification of a novel anti-integrin monoclonal antibody that recognises a ligand-induced binding site epitope on the beta 1 subunit, FEBS letters 363(1-2) (1995) 118–22. [DOI] [PubMed] [Google Scholar]
- [35].Clark K, Pankov R, Travis MA, Askari JA, Mould AP, Craig SE, Newham P, Yamada KM, Humphries MJ, A specific alpha5beta1-integrin conformation promotes directional integrin translocation and fibronectin matrix formation, J Cell Sci 118(Pt 2) (2005) 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fang WB, Brantley-Sieders DM, Hwang Y, Ham AJ, Chen J, Identification and functional analysis of phosphorylated tyrosine residues within EphA2 receptor tyrosine kinase, J Biol Chem 283(23) (2008) 16017–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hunter SG, Zhuang G, Brantley-Sieders D, Swat W, Cowan CW, Chen J, Essential role of Vav family guanine nucleotide exchange factors in EphA receptor-mediated angiogenesis, Mol Cell Biol 26(13) (2006) 4830–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Murai KK, Pasquale EB, ‘Eph’ective signaling: forward, reverse and crosstalk, J Cell Sci 116(Pt 14) (2003) 2823–32. [DOI] [PubMed] [Google Scholar]
- [39].Miao H, Li DQ, Mukherjee A, Guo H, Petty A, Cutter J, Basilion JP, Sedor J, Wu J, Danielpour D, Sloan AE, Cohen ML, Wang B, EphA2 mediates ligand-dependent inhibition and ligand-independent promotion of cell migration and invasion via a reciprocal regulatory loop with Akt, Cancer Cell 16(1) (2009) 9–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Barquilla A, Lamberto I, Noberini R, Heynen-Genel S, Brill LM, Pasquale EB, Protein kinase A can block EphA2 receptor-mediated cell repulsion by increasing EphA2 S897 phosphorylation, Mol Biol Cell 27(17) (2016) 2757–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Zhou Y, Yamada N, Tanaka T, Hori T, Yokoyama S, Hayakawa Y, Yano S, Fukuoka J, Koizumi K, Saiki I, Sakurai H, Crucial roles of RSK in cell motility by catalysing serine phosphorylation of EphA2, Nat Commun 6 (2015) 7679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kuo MT, Long Y, Tsai WB, Li YY, Chen HHW, Feun LG, Savaraj N, Collaboration Between RSK-EphA2 and Gas6-Axl RTK Signaling in Arginine Starvation Response That Confers Resistance to EGFR Inhibitors, Translational oncology 13(2) (2020) 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Chastney MR, Lawless C, Humphries JD, Warwood S, Jones MC, Knight D, Jorgensen C, Humphries MJ, Topological features of integrin adhesion complexes revealed by multiplexed proximity biotinylation, J Cell Biol 219(8) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Neill T, Buraschi S, Goyal A, Sharpe C, Natkanski E, Schaefer L, Morrione A, Iozzo RV, EphA2 is a functional receptor for the growth factor progranulin, J Cell Biol 215(5) (2016) 687–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Monami G, Gonzalez EM, Hellman M, Gomella LG, Baffa R, Iozzo RV, Morrione A, Proepithelin promotes migration and invasion of 5637 bladder cancer cells through the activation of ERK1/2 and the formation of a paxillin/FAK/ERK complex, Cancer Res 66(14) (2006) 7103–10. [DOI] [PubMed] [Google Scholar]
- [46].Himanen JP, Goldgur Y, Miao H, Myshkin E, Guo H, Buck M, Nguyen M, Rajashankar KR, Wang B, Nikolov DB, Ligand recognition by A-class Eph receptors: crystal structures of the EphA2 ligand-binding domain and the EphA2/ephrin-A1 complex, EMBO reports 10(7) (2009) 722–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Jeong K, Kim JH, Murphy JM, Park H, Kim SJ, Rodriguez YAR, Kong H, Choi C, Guan JL, Taylor JM, Lincoln TM, Gerthoffer WT, Kim JS, Ahn EE, Schlaepfer DD, Lim SS, Nuclear Focal Adhesion Kinase Controls Vascular Smooth Muscle Cell Proliferation and Neointimal Hyperplasia Through GATA4-Mediated Cyclin D1 Transcription, Circ Res 125(2) (2019) 152–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zamir E, Katz M, Posen Y, Erez N, Yamada KM, Katz BZ, Lin S, Lin DC, Bershadsky A, Kam Z, Geiger B, Dynamics and segregation of cell-matrix adhesions in cultured fibroblasts, Nat Cell Biol 2(4) (2000) 191–6. [DOI] [PubMed] [Google Scholar]
- [49].Chen P, Huang Y, Zhang B, Wang Q, Bai P, EphA2 enhances the proliferation and invasion ability of LNCaP prostate cancer cells, Oncol Lett 8(1) (2014) 41–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Kaneko-Kawano T, Takasu F, Naoki H, Sakumura Y, Ishii S, Ueba T, Eiyama A, Okada A, Kawano Y, Suzuki K, Dynamic regulation of myosin light chain phosphorylation by Rho-kinase, PloS one 7(6) (2012) e39269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Subauste MC, Von Herrath M, Benard V, Chamberlain CE, Chuang TH, Chu K, Bokoch GM, Hahn KM, Rho family proteins modulate rapid apoptosis induced by cytotoxic T lymphocytes and Fas, J Biol Chem 275(13) (2000) 9725–33. [DOI] [PubMed] [Google Scholar]
- [52].Alho I, Costa L, Bicho M, Coelho C, Low molecular weight protein tyrosine phosphatase isoforms regulate breast cancer cells migration through a RhoA dependent mechanism, PloS one 8(9) (2013) e76307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Shamah SM, Lin MZ, Goldberg JL, Estrach S, Sahin M, Hu L, Bazalakova M, Neve RL, Corfas G, Debant A, Greenberg ME, EphA receptors regulate growth cone dynamics through the novel guanine nucleotide exchange factor ephexin, Cell 105(2) (2001) 233–44. [DOI] [PubMed] [Google Scholar]
- [54].Batson J, Maccarthy-Morrogh L, Archer A, Tanton H, Nobes CD, EphA receptors regulate prostate cancer cell dissemination through Vav2-RhoA mediated cell-cell repulsion, Biol Open 3(6) (2014) 453–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Sharma SV, Rapid recruitment of p120RasGAP and its associated protein, p190RhoGAP, to the cytoskeleton during integrin mediated cell-substrate interaction, Oncogene 17(3) (1998) 271–81. [DOI] [PubMed] [Google Scholar]
- [56].Tomar A, Lim ST, Lim Y, Schlaepfer DD, A FAK-p120RasGAP-p190RhoGAP complex regulates polarity in migrating cells, J Cell Sci 122(Pt 11) (2009) 1852–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Chen Z, Givens C, Reader JS, Tzima E, Haemodynamics Regulate Fibronectin Assembly via PECAM, Sci Rep 7 (2017) 41223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Holder N, Klein R, Eph receptors and ephrins: effectors of morphogenesis, Development 126(10) (1999) 2033–44. [DOI] [PubMed] [Google Scholar]
- [59].Lagares D, Ghassemi-Kakroodi P, Tremblay C, Santos A, Probst CK, Franklin A, Santos DM, Grasberger P, Ahluwalia N, Montesi SB, Shea BS, Black KE, Knipe R, Blati M, Baron M, Wu B, Fahmi H, Gandhi R, Pardo A, Selman M, Wu J, Pelletier JP, Martel-Pelletier J, Tager AM, Kapoor M, ADAM10-mediated ephrin-B2 shedding promotes myofibroblast activation and organ fibrosis, Nat Med 23(12) (2017) 1405–1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Su SA, Yang D, Wu Y, Xie Y, Zhu W, Cai Z, Shen J, Fu Z, Wang Y, Jia L, Wang Y, Wang JA, Xiang M, EphrinB2 Regulates Cardiac Fibrosis Through Modulating the Interaction of Stat3 and TGF-beta/Smad3 Signaling, Circ Res 121(6) (2017) 617–627. [DOI] [PubMed] [Google Scholar]
- [61].Mimche PN, Lee CM, Mimche SM, Thapa M, Grakoui A, Henkemeyer M, Lamb TJ, EphB2 receptor tyrosine kinase promotes hepatic fibrogenesis in mice via activation of hepatic stellate cells, Sci Rep 8(1) (2018) 2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Julich D, Mould AP, Koper E, Holley SA, Control of extracellular matrix assembly along tissue boundaries via Integrin and Eph/Ephrin signaling, Development 136(17) (2009) 2913–21. [DOI] [PubMed] [Google Scholar]
- [63].DuSablon A, Kent S, Coburn A, Virag J, EphA2-receptor deficiency exacerbates myocardial infarction and reduces survival in hyperglycemic mice, Cardiovasc Diabetol 13 (2014) 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Leem AY, Shin MH, Douglas IS, Song JH, Chung KS, Kim EY, Jung JY, Kang YA, Chang J, Kim YS, Park MS, All-trans retinoic acid attenuates bleomycin-induced pulmonary fibrosis via downregulating EphA2-EphrinA1 signaling, Biochem Biophys Res Commun 491(3) (2017) 721–726. [DOI] [PubMed] [Google Scholar]
- [65].Makarov A, Ylivinkka I, Nyman TA, Hyytiainen M, Keski-Oja J, Ephrin-As, Eph receptors and integrin alpha3 interact and colocalise at membrane protrusions of U251MG glioblastoma cells, Cell Biol Int 37(10) (2013) 1080–8. [DOI] [PubMed] [Google Scholar]
- [66].Schiller HB, Friedel CC, Boulegue C, Fassler R, Quantitative proteomics of the integrin adhesome show a myosin II-dependent recruitment of LIM domain proteins, EMBO reports 12(3) (2011) 259–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Dunne PD, Dasgupta S, Blayney JK, McArt DG, Redmond KL, Weir JA, Bradley CA, Sasazuki T, Shirasawa S, Wang T, Srivastava S, Ong CW, Arthur K, Salto-Tellez M, Wilson RH, Johnston PG, Van Schaeybroeck S, EphA2 Expression Is a Key Driver of Migration and Invasion and a Poor Prognostic Marker in Colorectal Cancer, Clin Cancer Res 22(1) (2016) 230–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Ventrella R, Kaplan N, Hoover P, Perez White BE, Lavker RM, Getsios S, EphA2 Transmembrane Domain Is Uniquely Required for Keratinocyte Migration by Regulating EphrinA1 Levels, J Invest Dermatol (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Xiang YP, Xiao T, Li QG, Lu SS, Zhu W, Liu YY, Qiu JY, Song ZH, Huang W, Yi H, Tang YY, Xiao ZQ, Y772 phosphorylation of EphA2 is responsible for EphA2-dependent NPC nasopharyngeal carcinoma growth by Shp2/Erk-1/2 signaling pathway, Cell death & disease 11(8) (2020) 709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Srivastava S, Pang KM, Iida M, Nelson MS, Liu J, Nam A, Wang J, Mambetsariev I, Pillai R, Mohanty A, McDaniel N, Behal A, Kulkarni P, Wheeler DL, Salgia R, Activation of EPHA2-ROBO1 Heterodimer by SLIT2 Attenuates Non-canonical Signaling and Proliferation in Squamous Cell Carcinomas, iScience 23(11) (2020) 101692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Hansen K, Johnell M, Siegbahn A, Rorsman C, Engstrom U, Wernstedt C, Heldin CH, Ronnstrand L, Mutation of a Src phosphorylation site in the PDGF beta-receptor leads to increased PDGF-stimulated chemotaxis but decreased mitogenesis, EMBO J 15(19) (1996) 5299–313. [PMC free article] [PubMed] [Google Scholar]
- [72].Leroy C, Fialin C, Sirvent A, Simon V, Urbach S, Poncet J, Robert B, Jouin P, Roche S, Quantitative phosphoproteomics reveals a cluster of tyrosine kinases that mediates SRC invasive activity in advanced colon carcinoma cells, Cancer Res 69(6) (2009) 2279–86. [DOI] [PubMed] [Google Scholar]
- [73].Fattet L, Jung HY, Matsumoto MW, Aubol BE, Kumar A, Adams JA, Chen AC, Sah RL, Engler AJ, Pasquale EB, Yang J, Matrix Rigidity Controls Epithelial-Mesenchymal Plasticity and Tumor Metastasis via a Mechanoresponsive EPHA2/LYN Complex, Developmental cell 54(3) (2020) 302–316 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].de Saint-Vis B, Bouchet C, Gautier G, Valladeau J, Caux C, Garrone P, Human dendritic cells express neuronal Eph receptor tyrosine kinases: role of EphA2 in regulating adhesion to fibronectin, Blood 102(13) (2003) 4431–40. [DOI] [PubMed] [Google Scholar]
- [75].Yamazaki T, Masuda J, Omori T, Usui R, Akiyama H, Maru Y, EphA1 interacts with integrin-linked kinase and regulates cell morphology and motility, J Cell Sci 122(Pt 2) (2009) 243–55. [DOI] [PubMed] [Google Scholar]
- [76].Parri M, Buricchi F, Giannoni E, Grimaldi G, Mello T, Raugei G, Ramponi G, Chiarugi P, EphrinA1 activates a Src/focal adhesion kinase-mediated motility response leading to rho-dependent actino/myosin contractility, J Biol Chem 282(27) (2007) 19619–28. [DOI] [PubMed] [Google Scholar]
- [77].Salaita K, Nair PM, Petit RS, Neve RM, Das D, Gray JW, Groves JT, Restriction of receptor movement alters cellular response: physical force sensing by EphA2, Science 327(5971) (2010) 1380–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Pankov R, Cukierman E, Katz BZ, Matsumoto K, Lin DC, Lin S, Hahn C, Yamada KM, Integrin dynamics and matrix assembly: tensin-dependent translocation of alpha(5)beta(1) integrins promotes early fibronectin fibrillogenesis, J Cell Biol 148(5) (2000) 1075–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Feijoo-Cuaresma M, Mendez F, Maqueda A, Esteban MA, Naranjo-Suarez S, Castellanos MC, del Cerro MH, Vazquez SN, Garcia-Pardo A, Landazuri MO, Calzada MJ, Inadequate activation of the GTPase RhoA contributes to the lack of fibronectin matrix assembly in von Hippel-Lindau protein-defective renal cancer cells, J Biol Chem 283(36) (2008) 24982–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Danen EH, Sonneveld P, Brakebusch C, Fassler R, Sonnenberg A, The fibronectin-binding integrins alpha5beta1 and alphavbeta3 differentially modulate RhoA-GTP loading, organization of cell matrix adhesions, and fibronectin fibrillogenesis, J Cell Biol 159(6) (2002) 1071–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].van der Bijl I, Nawaz K, Kazlauskaite U, van Stalborch AM, Tol S, Jimenez Orgaz A, van den Bout I, Reinhard NR, Sonnenberg A, Margadant C, Reciprocal integrin/integrin antagonism through kindlin-2 and Rho GTPases regulates cell cohesion and collective migration, Matrix Biol 93 (2020) 60–78. [DOI] [PubMed] [Google Scholar]
- [82].Sahin M, Greer PL, Lin MZ, Poucher H, Eberhart J, Schmidt S, Wright TM, Shamah SM, O’Connell S, Cowan CW, Hu L, Goldberg JL, Debant A, Corfas G, Krull CE, Greenberg ME, Eph-dependent tyrosine phosphorylation of ephexin1 modulates growth cone collapse, Neuron 46(2) (2005) 191–204. [DOI] [PubMed] [Google Scholar]
- [83].Ogita H, Kunimoto S, Kamioka Y, Sawa H, Masuda M, Mochizuki N, EphA4-mediated Rho activation via Vsm-RhoGEF expressed specifically in vascular smooth muscle cells, Circ Res 93(1) (2003) 23–31. [DOI] [PubMed] [Google Scholar]
- [84].Chen XL, Nam JO, Jean C, Lawson C, Walsh CT, Goka E, Lim ST, Tomar A, Tancioni I, Uryu S, Guan JL, Acevedo LM, Weis SM, Cheresh DA, Schlaepfer DD, VEGF-induced vascular permeability is mediated by FAK, Developmental cell 22(1) (2012) 146–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Kobayashi M, Inoue K, Warabi E, Minami T, Kodama T, A simple method of isolating mouse aortic endothelial cells, J Atheroscler Thromb 12(3) (2005) 138–42. [DOI] [PubMed] [Google Scholar]
- [86].Ray JL, Leach R, Herbert JM, Benson M, Isolation of vascular smooth muscle cells from a single murine aorta, Methods Cell Sci 23(4) (2001) 185–8. [DOI] [PubMed] [Google Scholar]
- [87].Orr AW, Ginsberg MH, Shattil SJ, Deckmyn H, Schwartz MA, Matrix-specific suppression of integrin activation in shear stress signaling, Mol Biol Cell 17(11) (2006) 4686–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Yurdagul A Jr., Green J, Albert P, McInnis MC, Mazar AP, Orr AW, alpha5beta1 integrin signaling mediates oxidized low-density lipoprotein-induced inflammation and early atherosclerosis, Arterioscler Thromb Vasc Biol 34(7) (2014) 1362–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Kuo JC, Han X, Yates JR 3rd, Waterman CM, Isolation of focal adhesion proteins for biochemical and proteomic analysis, Methods Mol Biol 757 (2012) 297–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Al-Kofahi M, Becker F, Gavins FN, Woolard MD, Tsunoda I, Wang Y, Ostanin D, Zawieja DC, Muthuchamy M, von der Weid PY, Alexander JS, IL-1beta reduces tonic contraction of mesenteric lymphatic muscle cells, with the involvement of cycloxygenase-2 and prostaglandin E2, Br J Pharmacol 172(16) (2015) 4038–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Funk SD, Finney AC, Yurdagul A Jr., Pattillo CB, Orr AW, EphA2 stimulates VCAM-1 expression through calcium-dependent NFAT1 activity, Cell Signal 49 (2018) 30–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Picht E, Zima AV, Blatter LA, Bers DM, SparkMaster: automated calcium spark analysis with ImageJ, Am J Physiol Cell Physiol 293(3) (2007) C1073–81. [DOI] [PubMed] [Google Scholar]
- [93].Pellegrin S, Mellor H, Rho GTPase activation assays, Curr Protoc Cell Biol Chapter 14 (2008) Unit 14 8. [DOI] [PubMed] [Google Scholar]
- [94].Wang X, Spandidos A, Wang H, Seed B, PrimerBank: a PCR primer database for quantitative gene expression analysis, 2012 update, Nucleic Acids Res 40(Database issue) (2012) D1144–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.